

Abstract
Introduction
Antibodies are an essential research tool for labeling surface proteins but can potentially influence the behavior of proteins and cells to which they bind. Because of this, researchers and clinicians are interested in the persistence of these antibodies, particularly for live-cell applications. We developed an easily adoptable method for researchers to characterize antibody removal timelines for any cell–antibody combination, with the benefit of studying broad, hypothesized mechanisms of antibody removal.
Methods
We developed a method using four experimental conditions to elucidate the contributions of possible factors influencing antibody removal: cell proliferation, internalization, permanent dissociation, and environmental perturbation. This method was tested on adipose-derived stem cells and a human lung fibroblast cell line with anti-CD44, CD90, and CD105 antibodies. The persistence of the primary antibody was probed using a fluorescent secondary antibody daily over 10 days. Relative contributions by the antibody removal mechanisms were quantified based on differences between the four culture conditions.
Results
Greater than 90% of each antibody tested was no longer present on the surface of the two cell types after 5 days, with removal observed in as little as 1 day post-labeling. Anti-CD90 antibody was primarily removed by environmental perturbation, anti-CD105 antibody by internalization, and anti-CD44 antibody by a combination of all four factors.
Conclusions
Antibody removal mechanism depended on the specific antibody tested, while removal timelines for the same antibody depended more on cell type. This method should be broadly relevant to researchers interested in quantifying an initial timeframe for uninhibited use of antibody-labeled cells.
Supplementary Information
The online version contains supplementary material available at 10.1007/s12195-021-00670-3.
Keywords: Immunolabeling, Internalization, Degradation, Adipose-derived stem cells, CD44, CD90, CD105, Surface markers
Introduction
Antibodies are widely used to label specific proteins of interest on the surface of cells. Applications of antibody binding to live cells include immunofluorescence imaging for visualization and protein behavior studies, cell sorting based on protein expression, and targeted drug delivery.38 Often, antibody-labeled cells are desirable for downstream applications such as enrichment and cellular expansion, high cell yield assays, or implantation in vivo. However, the presence of antibodies on live cells can have myriad effects including protein internalization for recycling or degradation, upregulation of other proteins in the cell, blocking of functional surface receptor protein binding sites, and interference with expansion and differentiation of stem cells.6, 8, 12, 33, 35, 41
The specific effects of antibody presence are unpredictable and only able to be elucidated experimentally. These properties are often exploited by researchers in the selection of antibodies for various applications including tumor targeting, drug internalization by macrophages, upregulation of genes, induction of tau uptake in neuronal cells, internalization of viral surface proteins and insulin receptors, and activation or apoptosis of T cells.1, 5, 7, 20, 25, 29, 32, 40, 43, 44 However, these effects could be detrimental if antibodies are only intended for cell type identification. Furthermore, if antibody-labeled cells are destined for implantation in vivo, the presence of large quantities of mouse or other foreign antibodies on the surface can cause negative immune responses in humans.15,36 It is therefore critical to determine the timeframe of influence for specific antibodies used to label surface proteins before any in vitro or in vivo downstream applications, a step commonly overlooked by researchers. Stem cell researchers in particular may benefit from such information as stem cells are commonly identified and sorted via surface marker profiles prior to further expansion, in vitro differentiation, or injection for in vivo stem cell therapy.26,33 Making this process as fast and efficient as possible is a central goal for stem cell therapies, which is why knowing the exact timeframe for antibody removal after labeling would benefit this work.10, 11, 46 Methods have been developed to remove antibodies from the cell surface more quickly, including low pH buffers, heat, and cleavable conjugates, like the avidin–biotin system.2, 9, 14, 25 However, these approaches have been shown to be problematic due to their damaging effects on the cell, incomplete antibody removal, or conjugate molecules remaining on the cell surface, thereby proving inadequate and compromising the studies in which they were employed.28,42 Therefore, in many cases a reliance on native antibody removal from live cells could be the most effective option for ensuring downstream use of the cells is unaffected by any antibody-induced behavioral changes, immune responses, or other ill effects of forced removal.
While there has been significant effort focused on the fate and internalization timeframes of antibodies used for therapeutic treatments, very little has been explored in this area involving antibodies used strictly for labeling purposes.21 In the early 1990s, a pair of studies investigated antibody internalization and degradation timeframes by tumor cells using radiolabeling in live cells.18,37 However, due to the experimental design, they could only hypothesize the mechanisms at play rather than determine them conclusively. Another study used a lysosomal inhibitor to probe the trafficking pathways for internalization of radiolabeled antibodies on neuroblastoma cells, with similar limitation to having a timeframe but no contribution of mechanisms besides internalization via several pathways.29 Since these earlier works, fluorophores have grown to replace radioisotopes, and researchers have developed molecular tools to aid in the study of antibody internalization, like fluorophore-quencher conjugates and nanocarrier liposomes containing fluorophores and metal chelators.21,28 To date, however, these tools have primarily been used for antibody screening for rapid internalization, not for the study of surface marker immunolabeling.
In this manuscript, we present a method for elucidating the duration and mechanism of antibody removal from a cell surface. We consider four primary factors that play a role in this process. First is the effect of proliferation of live cells, which causes the antibody concentration on their surface to be diluted with each cell division. Next is internalization by the cell via recycling or degradation pathways, which has previously been shown to be antibody-dependent with some antibodies following typical endosome recycling protein pathways, others triggering a different lysosomal degradation, and some failing to trigger any internalization at all.5,28,39 Next, permanent dissociation of the whole primary antibody from the protein target can occur, which differs from the thermally-driven binding and unbinding events that occur every fraction of a second between antibody and antigen that are used to determine the dissociation constant of an antibody.17,23 The final factor is environmental conditions, which we can hypothesize based on supporting literature include factors like mechanical perturbation, temperature, pH, and glucose exposure that may cause partial degradation or removal of the bound antibodies.9,30
The goal of this study was to develop a method to quantify the timeline and ascribe the hypothesized mechanisms of antibody removal summarized above and in Table 1. As seen in the third column, the means of primary antibody loss are successive, with each treatment eliminating another possible mechanism that could influence antibody loss. This allows for quantification of the contribution of each mechanism based on the difference in fluorescence intensity relative to the subsequent experimental condition. We chose to test this method on stem cells as they are commonly identified and sorted based on antibody labeling of a defined, surface marker expression profile before further experiments or in vivo implantation. Therefore, we used adipose-derived stem cells (ASCs) labeled with antibodies for three characteristic stem cell markers: CD44, CD90, and CD105.26 To compare the removal properties of these three antibodies when bound to a different cell type, we chose WI-38 human lung fibroblasts, which exhibit similar antigen profiles to mesenchymal stem cells, specifically including our three representative proteins.3
Table 1.
Summary of factors that contribute to primary antibody removal in each of the experimental conditions. Moving from live cells to fixed cells, each culture condition eliminates a potential mechanism of removal, which allows for approximate quantification of the overall contribution of each mechanism to antibody removal based on the difference between subsequent conditions
| Experimental condition | Description | Means of primary antibody loss |
|---|---|---|
| Live proliferative | Cells left untreated |
1. Environmental removal 2. Permanent dissociation 3. Internalization 4. Dilution by proliferation |
| Live non-proliferative | Cells treated with mitomycin C to arrest proliferation |
1. Environmental removal 2. Permanent dissociation 3. Internalization |
| Fixed cell | Cells fixed before primary antibody addition, allowing natural antibody detachment |
1. Environmental removal 2. Permanent dissociation |
| Fixed cell/antibody | Cells fixed after primary antibody addition, inhibiting natural antibody detachment | 1. Environmental removal |
Materials and Methods
Cells, Culture Reagents, and Antibodies
Primary human adipose-derived stem cells pooled from the subcutaneous fat tissue of five healthy, female donors 29–57 years of age (ASCs, passage 5, superlot #36, Zen-Bio Inc.) and WI-38 human lung fibroblast cell line (ATCC, #CCL-75.1) were used to demonstrate this antibody removal characterization method. Primary antibodies included anti-CD44 (Thermo Fisher Scientific, MA5-13890, 1:800 for ASCs and WI-38 cells), anti-CD90 (Thermo Fisher Scientific, MA5-16671, 1:2000 for ASCs, 1:400 for WI-38 cells), and anti-CD105 (Thermo Fisher Scientific, MA5-11854, 1:400 for ASCs and 1:100 for WI-38 cells). Secondary antibodies (anti-mouse IgG, labeled with AlexaFluor 488, Thermo Fisher Scientific, A-21121 at 1:1000 and A-11001 at 1:2000 dilution) were used to probe for the presence of the primary antibodies. All antibody concentrations were optimized via serial dilution and fluorescence imaging prior to experimental use.
Cells were grown to confluence in T-75 flasks prior to the start of experiments. ASCs were expanded and maintained in medium containing DMEM/F12 (HyClone, SH30023.01), 1X penicillin/streptomycin/Amphotericin B (HyClone, Cytiva, SV30079.01), 10% fetal bovine serum (FBS) (Zen-Bio Inc., SER-500), 1 ng/mL fibroblast growth factor (R&D Systems, 233-FB), 5 ng/mL epidermal growth factor (R&D Systems, 236-EG), and 0.25 ng/mL transforming growth factor-β1 (R&D Systems, 240-B). WI-38 cells were expanded and maintained in medium containing MEM (Hyclone, Cytiva, SH30265.01), 1X penicillin/streptomycin (Hyclone, Cytiva, SV30010), and 10% FBS (GenClone, Genessee Scientific, 25-550).
Setup of Experimental Conditions
Two days prior to primary antibody treatment, ASCs and WI-38 cells were seeded into eight wells per time point in separate 96-well plates for the “live non-proliferative” and “fixed cell” conditions (Fig. 1, top two rows). This seeding procedure was repeated at one day prior to primary antibody treatment for the “live proliferative” and “fixed cell/antibody” conditions (Fig. 1, bottom two rows). All wells were seeded at a density of 6000–7000 cells/well. For a 10-day time course, this resulted in plates containing 11 rows with 8 wells/condition (n = 5 wells for evaluation and n = 3 wells for matched secondary antibody-only controls). Prior to the start of experiments, cells were incubated overnight at 37 °C, 5% CO2 in culture medium to allow for adhesion and re-equilibration. It is important to note that the two fixed cell conditions were kept in separate plates from the live cell conditions to avoid inadvertent cell death from evaporating fixative in adjacent wells.
Figure 1.

Schematic of experimental setup, seeding, and treatment of cells in each set of wells for the four conditions. This two-day seeding process prior to antibody addition was designed for ease of execution, though it could be condensed into one day if necessary while still allowing 24 h for cell attachment.
To prepare the experimental conditions, subsets of cells were either treated with chemical fixatives, antimitotic, or left as is. The live, non-proliferative condition included cells that were treated for 150 min with mitomycin C (Sigma, M4287), a chemotherapeutic agent that arrests cell division, in culture medium at concentrations of 10 μg/mL for ASCs or 1.25 μg/mL for WI-38 cells based on optimization experiments (Supp. Fig. 1). The fixed cell condition included cells that were fixed for 10 min in 10% buffered formalin phosphate. The fixed cell/antibody condition followed the same protocol, only this was done after primary antibody treatment was completed. The live proliferative condition included cells that were kept in culture medium only. On the designated day, all samples were blocked for 1 h at 37 °C with 3% bovine serum albumin (BSA) in Hanks' Balanced Salt Solution (HBSS) to decrease non-specific binding. The primary antibody to be studied was diluted in 1% BSA/HBSS and then added to the first 5 wells of each condition and timepoints. The remaining 3 wells were kept in 1% BSA/HBSS to serve as secondary-only controls. After 40 min at 37 °C, primary antibody was removed from the samples and washed three times with HBSS. Most protocols suggest a 30–60 min incubation for immunofluorescence labeling of cell monolayers as opposed to thicker tissues. A 40-min timeframe at 37 °C, which accelerates molecular interactions, should be more than sufficient to saturate surface antigen binding to our thin cell monolayer even with static incubation. Due to the high binding affinity of antibodies, wash steps are performed primarily to remove unbound antibody to reduce background fluorescence. An optimal wash protocol of three, 3-min wash steps has been previously reported, with more extensive wash times up to 1 h having no significant reduction of background fluorescence.27 Though we acknowledge the possibility for limited additional antibody removal with more washes, the goal of the study was to mimic a typical protocol used for antibody staining which commonly includes three washes. Following primary antibody addition and washing, culture medium was added to all wells, and plates were maintained in a 37 °C, 5% CO2 incubator for the full time-course experiment.
Monitoring Primary Antibody Persistence
Binding of secondary antibody to the cell surface was used as a positive indicator of primary antibody presence. For each daily time point, the fluorescence intensity of bound secondary antibody was assessed via optical imaging (Supp. Fig. 2). Cells were blocked with 3% BSA/HBSS for 1 h. Then, fluorescent secondary antibody was added in 1% BSA/HBSS (8 wells per condition: 5 experimental wells treated with primary antibody previously and 3 secondary-only control wells to assess non-specific binding). Secondary antibody was incubated on the samples at 37 °C for 40 mins protected from light. After washing with HBSS, a 1:500 dilution of Hoechst dye (Invitrogen, H3570) in HBSS was added for 15 mins for nuclear staining. Following several washes in HBSS, sample wells were imaged using a Biotek Cytation™ 3 Cell Imaging Multi-Mode Reader (Biotek, Agilent) with 12 images obtained per well. This procedure of blocking, secondary antibody labeling, Hoechst staining, and imaging was repeated daily for the entire time course.
Image Analysis
Antibody fluorescence within each captured field-of-view was quantified as a ratio of the foreground to the background (FBR) using a MATLAB script with manual identification and selection of a representative background region. For each image analyzed, the mean pixel value of a cell-free region of interest (ROI), or image background, was manually identified. The mean intensity of the ROI was subtracted from the entire image, and negative values were set to zero. Otsu’s method was then used to calculate the global image threshold for generating the foreground and background binary masks from the ROI-subtracted image.31 These masks were used to then identify raw foreground and background pixel values of the original image. The pixel intensity averages of the isolated foreground and background regions were divided to produce the FBR for each image. FBR values were used to represent the abundance of primary antibody on the sample, with high FBR values indicating more antibody. In the time course graphs, fluorescence is presented as the average FBR of five antibody-treated wells for each timepoint with the average FBR of three secondary-only control wells subtracted, and then all timepoints were normalized to the time 0 value for their series.
Statistics and Calculations
FBRs for each captured field-of-view were averaged across the 12 images taken per well with images containing fewer than two cells or auto-fluorescent debris removed from the analysis. Statistical significance between experimental (n = 5) and secondary-only control (n = 3) wells was determined using a t-test at p < 0.05 in single iterations of each experiment. When this comparison was no longer statistically significant, we considered the primary antibody added at Time 0 to be fully removed from the cell surface. Of note, the fluorescence intensity at Time 0 does impact calculations of how rapidly the live cell conditions reach what is considered full removal due to imaging limits of detection, though the overall trends remained the same. Time course experiments for anti-CD90 and anti-CD105 antibodies on ASCs were repeated in two separate iterations by two researchers to evaluate the reproducibility of the general methodology (Supp. Fig. 3).
Results
The presence of anti-CD44, anti-CD90, and anti-CD105 antibodies on labeled ASCs and WI-38 cells was interrogated by the extent of secondary antibody binding. This was quantified by measuring fluorescence intensity over 10 days for the four cellular conditions devised in this method (Fig. 2). The observed persistence trends were found to be more closely associated within antibody type than cell type (Fig. 3). The anti-CD44 and anti-CD105 antibodies remained present on both fixed cell types for the 10-day experiment while anti-CD90 antibody only persisted on the fixed antibody condition. The non-proliferative condition exhibited a delayed fluorescence decrease in all experiments, ranging from two to six days behind the live proliferative condition, which was expected due to the lack of antibody dilution via cell division. The live proliferative condition, which is the most useful for extrapolating information for research applications, showed full antibody removal ranging from 1 to 8 days following initial labeling as determined by a lack of statistical difference between control and experimental groups (Fig. 3). Persistence times for partial removal percentages are noted in Table 2, since not all downstream applications may require full removal of bound antibodies. For instance, many in vitro applications may be more tolerant to the presence of foreign antibodies compared to in vivo applications in which the innate immune system plays a role. While the persistence trends aligned with the antibody used, the removal timepoints did show cell type dependence, with ASCs exhibiting the same or longer persistence than WI-38 cells labeled with the same antibody (Table 2). WI-38 cells had lower initial fluorescence intensity for anti-CD90 and CD105 antibodies, indicating lower levels of those surface proteins and therefore a faster drop below the fluorescent limit of detection. Two iterations of this method with anti-CD90 and anti-CD105 antibodies on ASCs were performed by two different researchers to ensure reproducibility. The time course traces for all four conditions followed similar trajectories between the two iterations (Supp. Fig. 3). The removal timepoints were identical for anti-CD105 antibody and only different by one day for anti-CD90 antibody.
Figure 2.

Representative images of each condition for anti-CD105 antibody on ASCs over 5 days. Similar datasets were collected for each antibody-cell type pairing for semi-quantitative analysis. Brightness and contrast have been uniformly enhanced here for visual clarity and publication purposes only.
Figure 3.

Normalized antibody fluorescence over 10 days for ASCs and WI-38 cells labeled with anti-CD44, anti-CD90, and anti-CD105 antibodies applied at Time 0 for all four conditions. Colored bars above each graph indicate antibody persistence, which ends when there is no statistically significant difference between experimental and secondary-only control groups within each condition for all subsequent timepoints (p > 0.05).
Table 2.
Days post-antibody labeling for each antibody and cell type combination when the primary antibody is diminished to 90, 80, and 70% of the starting fluorescence levels in the live proliferative condition.
| Days post-labeling | CD44 ASCs | CD44 WI-38 s | CD90 ASCs | CD90 WI-38 s | CD105 ASCs | CD105 WI-38 s |
|---|---|---|---|---|---|---|
| Live Cell Ab > 90% Gone | 4 | 5 | 4 | 1 | 2 | 3 |
| Live Cell Ab > 80% Gone | 3 | 4 | 2 | 1 | 2 | 1 |
| Live Cell Ab > 70% Gone | 2 | 3 | 1 | 1 | 2 | 1 |
Since the factors contributing to antibody removal considered in this study are additive among the four experimental conditions, the percentage contribution of each factor can be calculated based on the difference between the relative antibody persistence values, or the normalized FBRs, at each timepoint (Fig. 4, Supplementary Calculations). The resulting removal factors were calculated up to the timepoint of cellular confluence before which all potential mechanisms could still have an effect. ASCs reached confluence in the live proliferative condition on day 5 and WI-38 cells on day 7 (difference due to relative size of the cell types).
Figure 4.
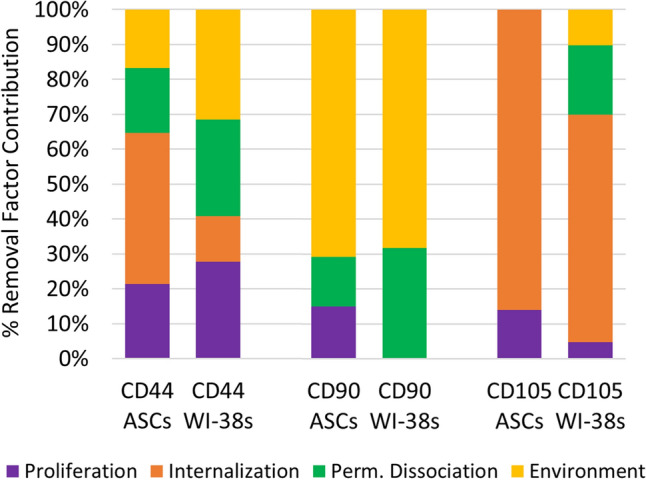
Percent contribution due to the considered removal mechanisms for antibody and cell type combinations. Removal factor profiles were more similar for like antibodies than like cell types, indicating that for this set of comparisons antibody characteristics played a larger role in determining their removal than cell type behavior.
The percentage contribution of each factor in antibody removal aligns predominantly with antibody type rather than cell type. All four factors played a role in the removal of anti-CD44 antibody, in similar percentages (20–40%). Anti-CD90 antibody was predominantly affected by environmental factors over the first few days in culture (~ 75%), contributing to its sharp decline after initial labeling. Anti-CD105 antibody was primarily affected by internalization, with the majority of its removal (65–85%) attributed to this factor.
Discussion
In this study, we developed a simple protocol to investigate timelines and contributions of the primary mechanisms of surface antibody removal post-labeling of cells. Results demonstrated that this technique could identify differences in antibody persistence across a small, representative selection of three antibodies and two cell types. The antibodies tested were > 90% removed by Day 5 in culture, with this removal occurring over a range of 1–5 days post-labeling, congruent with other studies.18,25 We observed that removal timelines for antibodies were determined primarily by the antibody-cell pairing, with the mechanisms of removal being more closely tied to the specific antibody alone. Anti-CD44 antibody was eliminated most slowly with contributions from all four removal mechanisms. Anti-CD90 antibody was removed most rapidly due to initial environmental impacts, presumably via washing off of weakly bound antibodies and partial antibody degradation. Removal of anti-CD105 antibody occurred quickly in the live cell conditions in the first few days in culture but remained mostly bound in both fixed conditions, indicating that internalization was the major mechanism removing it from the cell surface. Though these findings are antibody-specific, they demonstrate the wide range of results observable with our method.
Though our methodology was designed for any researcher to study the labeling persistence properties of their antibody of interest, the findings from our specific experiments are particularly relevant for stem cell applications due to the use of these antibodies to detect characteristic stem cell surface markers. Results indicated that a week in culture after labeling would be more than sufficient for antibody removal without need for additional measures. Antibody labeling can affect expression behavior of the labeled surface protein, other affiliated proteins, and potentially cause an immune response if antibody-bound cells are used in vivo.15,36,39 We developed this assessment protocol in large part because it is quicker and more efficient to study antibody removal as opposed to any collateral effects on gene and protein expression in cells or each antibody’s immune response in various animal species. There are no methods for removing antibodies from cells that are without risks to cell viability. Additionally, negative selection protocols, where the cell type of interest remains unlabeled while other cells are removed via a targeting antibody, are impractical for many applications.28,42 This is true in cases where the cells need to be sorted based on a gradient of protein levels, not just a binary signal, which is what is done in negative selection protocols.34 Negative cells in a sample also rarely express a common protein that can be targeted for omission from the purified population. It is more common to use multiple negative markers in these cases, but this is only feasible if positive markers have been identified for all “negative” cells, which is often impractical for highly heterogeneous or undefined cell populations, subpopulations, or phenotypes (e.g., primary cell isolates).
The four experimental conditions for this method were designed to elucidate the removal mechanisms contributing to loss of antibody from the cell surface, which include proliferative dilution of antibody, internalization, permanent dissociation, and environmental removal. In comparing the two fixed conditions, fixing cells before antibody addition allows for permanent dissociation to occur while fixing after antibody addition crosslinks the antibody to the surface, and thus it cannot undergo natural dissociation from the antigen. The fixed cells with the freely bound antibody can experience environmental removal and permanent dissociation while the antibody fixed to the cells can only be removed by environmental factors. The difference in removal time and amount of lost fluorescence between the two conditions would therefore be just the contribution of permanent dissociation of the antibody from the antigen. These differences between conditions are propagated through all four experiments, as one potential mechanism of removal is added for each condition. While the mechanisms cannot be considered completely independent (e.g., proliferating cells may also have increased surface protein internalization), we have assumed each was the primary factor for differences observed between successive experimental conditions. For the antibodies/cells studied, results indicated that environmental factors and protein/antibody internalization were the most prevalent removal contributors (Fig. 4). Dilution from cell division only occurs while the cells are proliferating, with this contributor acting exponentially over a short time before the cells reach confluence. Internalization is the most widely studied antibody elimination mechanism and often depends on native protein turnover rates, although in some cases antibody binding triggers protein degradation or recycling.4,6,18,20,39,40 For example, it has been shown that the binding of antibodies to insulin receptors, which are proteins with an already fast innate turnover rate, initiate endocytosis of the antibody-protein complex in less than 15 min.40 Other studies more closely match the antibody internalization times we observed of 70–76 h.18,25 Though antibody binding can drive faster protein turnover, the innate protein turnover rates likely play a role in antibody internalization as well. A study of thousands of proteins in five primary, non-dividing cell types showed a range of 4 to > 100,000 h half-lives.24 In this study, CD105 exhibited a 17 h half-life on monocytes, CD90 had a 35 h half-life on neurons, and CD44 had 31 h, 52 h, 58 h, and 71 h half-lives across four cell types. This demonstrates the variability in turnover of each protein as well as the same protein across substantially different cell types. Comparing our results for anti-CD105 antibody, which exhibited the greatest removal from internalization, a half-life of 17 h on monocytes (~ 56 h for 90% turnover) is comparable to the 2- or 3-day 90% removal timeframe we observed on ASCs and WI-38s (Table 2). Our internalization results align more closely within each antibody/antigen pair across the two cell types, with large variability in percentage contribution to antibody elimination overall. Additionally, Kyriakos and colleagues concluded that internalization was the predominant mechanism for antibody removal via normal, non-clathrin-dependent endocytosis of surface proteins compared to whole antibody dissociation.18 This corroborates our finding that half of our experiments exhibited internalization as the predominant removal mechanism (Fig. 4).
Permanent dissociation and environmental factors are likely interrelated for a given antibody and were observed to generally influence removal shortly after antibody addition. Researchers have shown that permanent dissociation of whole, intact antibodies bound to surface proteins on tumor cells could be observed after only 4 h post-labeling out of the total 76 h necessary for full antibody removal.18 That initial 4 h time span accounted for all of the dissociated antibodies released during the experiment but only 20% of the total antibody elimination. The average contribution of permanent dissociation across all six of our experiments was 19%, aligning well with these previous results. This could be coincidental, as we would not expect the three antibodies we tested to have the same binding affinity and dissociation kinetics as the tumor-targeting antibodies previously referred to. Lastly, environmental factors played a role predominantly in the first 24 h of our experiments, the most drastic removal being 50–70% of anti-CD90 antibody during that first day. We hypothesize that this mechanism encompasses degradation from the physical culture environment as well as mechanical agitation of the antibody during wash steps. In terms of environmental effects on degradation, longitudinal antibody stability is typically determined by manufacturers only for potential storage conditions, including 4 °C (recommended) and room temperature (20–25 °C), which is considered an accelerated stability study condition.30 The antibodies in the current study would therefore be considered to be in a high stress environment at 37 °C and thus degrade more quickly than the 3–12 months noted by the vendor for storage at 4 °C. Antibodies are typically stored in phosphate buffered saline (PBS) with antimicrobial sodium azide and often bovine serum albumin (BSA) for protein stabilization, which would be lacking once introduced into the cell culture medium used in our experiments.13,19 Antibodies in our method would also be incidentally removed or prematurely degraded by mechanical agitation from pipetting, photo degradation when exposed to ambient light or during imaging of adjacent wells, and exposure to high levels of glucose in culture medium which may trigger advanced degradation due to excessive antibody glycation.16,30,45 In the current work, these aggregated effects appeared to be antibody-dependent as all three antibodies studied were subjected to the same conditions, but only the anti-CD90 antibody was drastically affected. The anti-CD90 experiments did not undergo any differential treatment due to researcher performance or culture conditions that would have caused these differences, leading us to believe that variability in antibody stability is the most plausible explanation.
Proper implementation of the described protocol requires an understanding of several key characteristics for any given antibody and cell type. First, cellular proliferation rates need to be managed appropriately. In the current study, overgrowth of cells over time in the live proliferative condition complicated image analysis due to limited background visibility for normalization. Furthermore, ASCs in regions that reached confluence tended to condense and detach at later time points (e.g., Day 7). For the presented data, we observed no significant correlation between cell number and normalized fluorescence intensity of the images nor the raw background fluorescence, and image analysis methods were designed to be independent of cell number except for the exclusion of images containing less than two cells. Another characteristic to consider is the strength of cell adhesion to the plate surface over time for both live and fixed cells. Ideally, only the live proliferative condition should exhibit changing cell numbers; however, we observed a decrease in cell number for fixed conditions, particularly noticeable for ASCs. Upon further exploration, we determined that storage of fixed cells at 37 °C, as opposed to a standard 4 °C, can cause cells to uplift over time (Supp. Fig. 4). We chose to maintain all samples at the same temperature over the course of the experiment to avoid adding a confounding variable that affects antibody persistence. Lastly, the magnitude of fluorescence signal at Time 0, which varies based on the antibody used, can influence the absolute persistence times measured using our method. An antibody with a high starting signal will take longer to deteriorate to the level of noise than one with low starting signal, especially if the signal strength is indicative of more extensive antibody binding across the cell surface. Our two fixed conditions started with lower fluorescence intensities than the live conditions, an observation commonly reported that is due to the crosslinking of surface proteins that end up blocking potential binding sites on the antigens and, in this case, on the primary antibodies when already bound to the cells in the fixed cell/antibody condition.22 This uncontrolled variable may have contributed to the fluctuations in fluorescence signal of those conditions in different sets of wells over time. Relatedly, we determined that labeling with higher primary antibody concentration caused slightly increased initial fluorescence intensity but did not influence removal timelines (Supp. Fig. 5). These observations increased confidence in our model methodology since the number of antibodies on the cells did not affect their persistence.
Overall, we observed that the properties of antibody removal were consistent for each antibody on two cell types, with mechanisms being driven by antibody-antigen pairings and timelines varying across all combinations studied. The demonstrated methodology is suitable for studying any antibody-antigen pair on a given cell type, providing useful data on the persistence of the antibody over time. This will allow researchers to use marker-sorted, or otherwise antibody-labeled, cells with more confidence in downstream studies or clinical applications sensitive to the presence of foreign antibodies. In conclusion, this method provides a simple way to address the gap in research protocols surrounding a lack of investigation into antibody persistence on cell surfaces prior to experimental use.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgments
The authors would like to acknowledge Ryan Dubay for creating the MATLAB image analysis script used in the study. Funding support was provided by NIH/NIAMS (R01 AR063642, EMD) and Brown University’s Undergraduate Teaching and Research Award (OWB).
Author Contributions
MED, OWB, and EMD designed all experiments. OWB conducted preliminary optimization experiments, initial antibody removal iterations, and antibody dilution/concentration experiments. MED carried out all remaining experiments and iterations as well as conducted final analysis and interpretation of data. MED and EMD wrote the manuscript with figure contributions from OWB.
Conflict of interest
Megan E. Dempsey, Olivia Woodford-Berry, and Eric M. Darling declare that they have no conflicts of interest.
Ethical Standards
No human or animal studies were carried out by the authors for this article.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ackerman ME, Pawlowski D, Wittrup KD. Effect of antigen turnover rate and expression level on antibody penetration into tumor spheroids. Mol. Cancer Ther. 2008;7:2233–2240. doi: 10.1158/1535-7163.MCT-08-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alon R, Bayer EA, Wilchek M. Affinity cleavage of cell surface antibodies using the avidin-biotin system. J. Immunol. Methods. 1993;165:127–134. doi: 10.1016/0022-1759(93)90114-m. [DOI] [PubMed] [Google Scholar]
- 3.Alt E, Yan Y, Gehmert S, Song YH, Altman A, Gehmert S, Vykoukal D, Bai X. Fibroblasts share mesenchymal phenotypes with stem cells, but lack their differentiation and colony-forming potential. Biol. Cell. 2011;103:197–208. doi: 10.1042/BC20100117. [DOI] [PubMed] [Google Scholar]
- 4.Audran R, Drenou B, Wittke F, Gaudin A, Lesimple T, Toujas L. Internalization of human macrophage surface antigens induced by monoclonal antibodies. J. Immunol. Methods. 1995;188:147–154. doi: 10.1016/0022-1759(95)00213-8. [DOI] [PubMed] [Google Scholar]
- 5.Belleudi F, Marra E, Mazzetta F, Fattore L, Giovagnoli MR, Mancini R, Aurisicchio L, Torrisi MR, Ciliberto G. Monoclonal antibody-induced ErbB3 receptor internalization and degradation inhibits growth and migration of human melanoma cells. Cell Cycle. 2012;11:1455–1467. doi: 10.4161/cc.19861. [DOI] [PubMed] [Google Scholar]
- 6.Bergtold A, Desai DD, Gavhane A, Clynes R. Cell surface recycling of internalized antigen permits dendritic cell priming of B cells. Immunity. 2005;23:503–514. doi: 10.1016/j.immuni.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 7.Congdon EE, Gu J, Sait HB, Sigurdsson EM. Antibody uptake into neurons occurs primarily via clathrin-dependent Fcgamma receptor endocytosis and is a prerequisite for acute tau protein clearance. J. Biol. Chem. 2013;288:35452–35465. doi: 10.1074/jbc.M113.491001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davies OG, Cooper PR, Shelton RM, Smith AJ, Scheven BA. Isolation of adipose and bone marrow mesenchymal stem cells using CD29 and CD90 modifies their capacity for osteogenic and adipogenic differentiation. J. Tissue. Eng. 2015;6:1–10. doi: 10.1177/2041731415592356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.des Roziers NB, Squalli S. Removing IgG antibodies from intact red cells: comparison of acid and EDTA, heat, and chloroquine elution methods. Transfusion. 1997;37:497–501. doi: 10.1046/j.1537-2995.1997.37597293880.x. [DOI] [PubMed] [Google Scholar]
- 10.Dominici MLBK, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini FC, Krause DS, Deans RJ, Keating A, Prockop DJ, Horwitz EM. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 11.Francis SL, Duchi S, Onofrillo C, Di Bella C, Choong PFM. Adipose-derived mesenchymal stem cells in the use of cartilage tissue engineering: the need for a rapid isolation procedure. Stem Cells Int. 2018;2018:8947548. doi: 10.1155/2018/8947548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gossett DR, Weaver WM, Mach AJ, Hur SC, Tse HTK, Lee W, Amini H, Di Carlo D. Label-free cell separation and sorting in microfluidic systems. Anal. Bioanal. Chem. 2010;397:3249–3267. doi: 10.1007/s00216-010-3721-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiskoot W, Beuvery EC, de Koning AA, Herron JN, Crommelin DJ. Analytical approaches to the study of monoclonal antibody stability. Pharm. Res. 1990;7:1234–1241. doi: 10.1023/a:1015925519154. [DOI] [PubMed] [Google Scholar]
- 14.Jones AR, Stutz CC, Zhou Y, Marks JD, Shusta EV. Identifying blood–brain-barrier selective single-chain antibody fragments. Biotechnol. J. 2014;9:664–674. doi: 10.1002/biot.201300550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khazaeli MB, Conry RM, LoBuglio AF. Human immune response to monoclonal antibodies. J. Immunother. 1994;15:42–52. doi: 10.1097/00002371-199401000-00006. [DOI] [PubMed] [Google Scholar]
- 16.Kiese S, Papppenberger A, Friess W, Mahler HC. Shaken, not stirred: mechanical stress testing of an IgG1 antibody. J. Pharm. Sci. 2008;97:4347–4366. doi: 10.1002/jps.21328. [DOI] [PubMed] [Google Scholar]
- 17.Kulin S, Kishore R, Hubbard JB, Helmerson K. Real-time measurement of spontaneous antigen-antibody dissociation. Biophys. J . 2002;83:1965–1973. doi: 10.1016/S0006-3495(02)73958-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kyriakos RJ, Shih LB, Ong GL, Patel K, Goldenberg DM, Mattes MJ. The fate of antibodies bound to the surface of tumor cells in vitro. Cancer Res. 1992;52:835–842. [PubMed] [Google Scholar]
- 19.Le Basle Y, Chennell P, Tokhadze N, Astier A, Sautou V. Physicochemical stability of monoclonal antibodies: a review. J. Pharm. Sci. 2020;109:169–190. doi: 10.1016/j.xphs.2019.08.009. [DOI] [PubMed] [Google Scholar]
- 20.Leemans A, De Schryver M, Van der Gucht W, Heykers A, Pintelon I, Hotard AL, Moore ML, Melero JA, McLellan JS, Graham BS, Broadbent L, Power UF, Caljon G, Cos P, Maes L, Delputte P. Antibody-induced internalization of the human respiratory syncytial virus fusion protein. J. Virol. 2017;91:e00184. doi: 10.1128/JVI.00184-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Liu PC, Shen Y, Snavely MD, Hiraga K. A cell-based internalization and degradation assay with an activatable fluorescence-quencher probe as a tool for functional antibody screening. J. Biomol. Screen. 2015;20:869–875. doi: 10.1177/1087057115588511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mason JT, O’leary TJ. Effects of formaldehyde fixation on protein secondary structure: a calorimetric and infrared spectroscopic investigation. J. Histochem. Cytochem. 1991;39:225–229. doi: 10.1177/39.2.1987266. [DOI] [PubMed] [Google Scholar]
- 23.Mason DW, Williams AF. The kinetics of antibody binding to membrane antigens in solution and at the cell surface. Biochem. J. 1980;187:1–20. doi: 10.1042/bj1870001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mathieson T, Franken H, Kosinski J, Kurzawa N, Zinn N, Sweetman G, Poeckel D, Ratnu VS, Schramm M, Becher I, Steidel M. Systematic analysis of protein turnover in primary cells. Nat. Commun. 2018;9:1–10. doi: 10.1038/s41467-018-03106-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matzku S, Bröcker EB, Brüggen J, Dippold WG, Tilgen W. Modes of binding and internalization of monoclonal antibodies to human melanoma cell lines. Cancer Res. 1986;46:3848–3854. [PubMed] [Google Scholar]
- 26.Mildmay-White A, Khan W. Cell surface markers on adipose-derived stem cells: a systematic review. Curr. Stem Cell Res. Ther. 2017;12:484–492. doi: 10.2174/1574888X11666160429122133. [DOI] [PubMed] [Google Scholar]
- 37.Mosedale DE, Metcalfe JC, Grainger DJ. Optimization of immunofluorescence methods by quantitative image analysis. J. Histochem. Cytochem. 1996;44:1043–1050. doi: 10.1177/44.9.8773570. [DOI] [PubMed] [Google Scholar]
- 28.Nielsen UB, Kirpotin DB, Pickering EM, Drummond DC, Marks JD. A novel assay for monitoring internalization of nanocarrier coupled antibodies. BMC Immunol. 2006;7:24. doi: 10.1186/1471-2172-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Novak-Hofer I, Amstutz HP, Morgenthaler JJ, Schubiger PA. Internalization and degradation of monoclonal antibody chCE7 by human neuroblastoma cells. Int. J. Cancer. 1994;57:427–432. doi: 10.1002/ijc.2910570322. [DOI] [PubMed] [Google Scholar]
- 30.Nowak C, Cheung JK, Dellatore SM, Katiyar A, Bhat R, Sun J, Ponniah G, Neill A, Mason B, Beck A, Liu H. Forced degradation of recombinant monoclonal antibodies: a practical guide. MAbs. 2017;9:1217–1230. doi: 10.1080/19420862.2017.1368602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Otsu N. A threshold selection method from gray-level histograms. IEEE Trans. Syst. Man Cybernet. B. 1979;9:62–66. [Google Scholar]
- 32.Qing X, Pitashny M, Thomas DB, Barrat FJ, Hogarth MP, Putterman C. Pathogenic anti-DNA antibodies modulate gene expression in mesangial cells: involvement of HMGB1 in anti-DNA antibody-induced renal injury. Immunol. Lett. 2008;121:61–73. doi: 10.1016/j.imlet.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roda B, Reschiglian P, Zattoni A, Alviano F, Lanzoni G, Costa R, Di Carlo A, Marchionni C, Franchina M, Bonsi L, Bagnara GP. A tag-less method of sorting stem cells from clinical specimens and separating mesenchymal from epithelial progenitor cells. Cytom Part B Clin Cy. 2009;76:285–290. doi: 10.1002/cyto.b.20472. [DOI] [PubMed] [Google Scholar]
- 34.Sarnik SA, Sutermaster BA, Darling EM. Mass-added density modulation for sorting cells based on differential surface protein levels. Cytom. A. 2020 doi: 10.1002/cyto.a.24192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schaffar L, Dallanegra A, Breittmayer JP, Carrel S, Fehlmann M. Monoclonal antibody internalization and degradation during modulation of the CD3/T-cell receptor complex. Cell. Immunol. 1988;116:52–59. doi: 10.1016/0008-8749(88)90209-2. [DOI] [PubMed] [Google Scholar]
- 36.Sears HF, Bagli DJ, Herlyn D, DeFreitas E, Suzuki H, Steele G, Koprowski H. Human immune response to monoclonal antibody administration is dose-dependent. Arch. Surg. 1987;122:1384–1388. doi: 10.1001/archsurg.1987.01400240030004. [DOI] [PubMed] [Google Scholar]
- 37.Shih LB, Thorpe SR, Griffiths GL, Diril H, Ong GL, Hansen HJ, Goldenberg DM, Mattes MJ. The processing and fate of antibodies and their radiolabels bound to the surface of tumor cells in vitro: a comparison of nine radiolabels. J. Nucl. Med. 1994;35:899–908. [PubMed] [Google Scholar]
- 38.Specht EA, Braselmann E, Palmer AE. A critical and comparative review of fluorescent tools for live-cell imaging. Annu. Rev. Physiol. 2017;79:93–117. doi: 10.1146/annurev-physiol-022516-034055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.St-Pierre CA, Leonard D, Corvera S, Kurt-Jones EA, Finberg RW. Antibodies to cell surface proteins redirect intracellular trafficking pathways. Exp. Mol. Pathol. 2011;91:723–732. doi: 10.1016/j.yexmp.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trischitta V, Wong KY, Brunetti A, Scalisi R, Vigneri R, Goldfine ID. Endocytosis, recycling, and degradation of the insulin receptor. Studies with monoclonal antireceptor antibodies that do not activate receptor kinase. J. Biol. Chem. 1989;264:5041–5046. [PubMed] [Google Scholar]
- 41.Trowbridge IS, Lopez F. Monoclonal antibody to transferrin receptor blocks transferrin binding and inhibits human tumor cell growth in vitro. PNAS. 1982;79:1175–1179. doi: 10.1073/pnas.79.4.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsaltas G, Ford CH. Cell membrane antigen-antibody complex dissociation by the widely used glycine-HCL method: an unreliable procedure for studying antibody internalization. Immunol. Invest. 1993;22:1–12. doi: 10.3109/08820139309066189. [DOI] [PubMed] [Google Scholar]
- 43.Tsoukas CD, Landgraf B, Bentin J, Valentine M, Lotz M, Vaughan JH, Carson DA. Activation of resting T lymphocytes by anti-CD3 (T3) antibodies in the absence of monocytes. J. Immunol. 1985;135:1719–1723. [PubMed] [Google Scholar]
- 44.Wallberg M, Recino A, Phillips J, Howie D, Vienne M, Paluch C, Azuma M, Wong FS, Waldmann H, Cooke A. Anti-CD 3 treatment up-regulates programmed cell death protein-1 expression on activated effector T cells and severely impairs their inflammatory capacity. Immunology. 2017;151:248–260. doi: 10.1111/imm.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wei B, Berning K, Quan C, Zhang YT. Glycation of antibodies: modification, methods and potential effects on biological functions. MAbs. 2017;9:586–594. doi: 10.1080/19420862.2017.1300214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yokota N, Hattori M, Ohtsuru T, Otsuji M, Lyman S, Shimomura K, Nakamura N. Comparative clinical outcomes after intra-articular injection with adipose-derived cultured stem cells or noncultured stromal vascular fraction for the treatment of knee osteoarthritis. Am. J. Sports Med. 2019;47:2577–2583. doi: 10.1177/0363546519864359. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.