

Abstract
We report the development of in vivo subcellular high-resolution mass spectrometry (HRMS) for proteo-metabolomic molecular systems biology in complex tissues. With light microscopy, we identified the left-dorsal and left-ventral animal cells in cleavage-stage non-sentient Xenopus laevis embryos. Using precision-translated fabricated microcapillaries, the subcellular content of each cell was double-probed, each time swiftly (<5 s/event) aspirating <5% of cell volume (~10 nL). The proteins and metabolites were analyzed by home-built ultrasensitive capillary electrophoresis electrospray ionization employing Orbitrap or time-of-flight HRMS. Label-free detection of ~150 metabolites (57 identified) and 738 proteins found proteo-metabolomic networks with differential quantitative activities between the cell types. With spatially and temporally scalable sampling, the technology preserved the integrity of the analyzed cells, the neighboring cells, and the embryo. 95% of the analyzed embryos developed into sentient tadpoles that were indistinguishable from their wild-type siblings based on anatomy and visual function in a background color preference assay.
Keywords: single cell, mass spectrometry, protein, metabolite, embryo
Entry for the Table of Contents
Single-cell mass spectrometry was advanced to enable dual characterization of proteins and metabolites in identified cells in Xenopus laevis embryos. The analyzed embryos developed to behaving tadpoles with indistinguishable anatomy and visual function from their control siblings in a back-ground color preference assay. In vivo single-cell mass spectrometry expands the analytical toolbox of molecular systems biology.
Introduction
Unbiased measurement of transcripts, proteins, and metabolites in a live cell promises to revolutionize investigations in molecular systems cell biology. Even today, after the invention of single-cell transcriptomics[1], there exists no single technology capable of the unbiased characterization of both proteins and metabolites in the same single cell in vivo to enable studies at the level of the functioning organisms. While tools of molecular biology and high/super-resolution optical microscopy empowered systems biology for live organisms, they only work for a limited number of gene products at a time that also have to be known ahead of time, thus limiting research scope to targeted studies, typically building on prior knowledge.
Single-cell high-resolution mass spectrometry (HRMS) emerged as a powerful alternative for targeted and untargeted (discovery) studies (reviewed in refs. [1a, 1b, 1d, 2]). Specialized technologies in cell handling, microscale sample processing[3], high-efficiency chemical separations[4], and ion generation extended time-of-flight and orbitrap HRMS instruments to sufficient sensitivity and throughput for single-cell analyses. For example, using nanoHPLC, NanoPOTS enabled the detection of 650+ proteins in single HeLa cells,[3f] which was recently increased to 850+ proteins on a new-generation mass spectrometer.[4c] ScoPE-MS also used nanoHPLC to quantify ~750 proteins from a mammalian cell.[3e] The technology was recently advanced to the quantification of ~1,000 proteins/cell.[3e, 5] MALDI-TOF followed by nanoLC-HRMS enabled the characterization of lipids, peptides, and proteins in large numbers of single cells dissociated from a rat root ganglion.[6] The data emerging from these and other single-cell studies can be used to derive new hypotheses for biological investigations to establish the functional implications of the observed molecular cell heterogeneity.
We and others custom-built ultrasensitive capillary electrophoresis (CE) electrospray ionization (ESI) platforms to enable single-cell analyses. These instruments were used to detect transcripts, ~3,000 proteins, or ~100 metabolites in single cells/neurons dissected from Aplysia californica[4a, 7], Xenopus laevis embryos[3a, 8], or the rat[4a]. Using capillary microsampling, we extended these measurements to the direct analysis of metabolites[9] or proteins[3d] in single cells in early developing X. laevis and zebrafish embryos. Although single-cell HRMS technologies usher an emerging era of multi-omic single-cell ‘omics,[10] these tools so far required isolation or sorting of the cells, preventing studies in vivo. We here report the first example of in vivo single-cell HRMS that enables dual proteo-metabolomics of spatiotemporally identified single cells in a live embryo, which freely develops to a normally behaving tadpole post analysis.
Results and Discussion
We developed the bioanalytical technology and demonstrated its use in molecular systems cell biology with compatibility for cell-, neurodevelopmental, and behavioral biology. Figure 1 presents our analytical and biological tracks, essentially connecting the physical and life sciences. To enable molecular network analyses in live embryos, proteins and metabolites were measured by balancing sensitivity and developmental viability. In the context of neurodevelopment, the analysis is considered in vivo if the cell is able to heal its membrane, divide, and form progenies for tissue specification and development to give way to a normally behaving organism, viz. a tadpole. The biological trace in the study determined this biological phenotype on the basis of morphological, survival, and behavioral assays. By design, the integration of these tracks generates multi-dimensional metadata to open a window into molecular systems biology and help develop new knowledge and support hypothesis-driven studies.
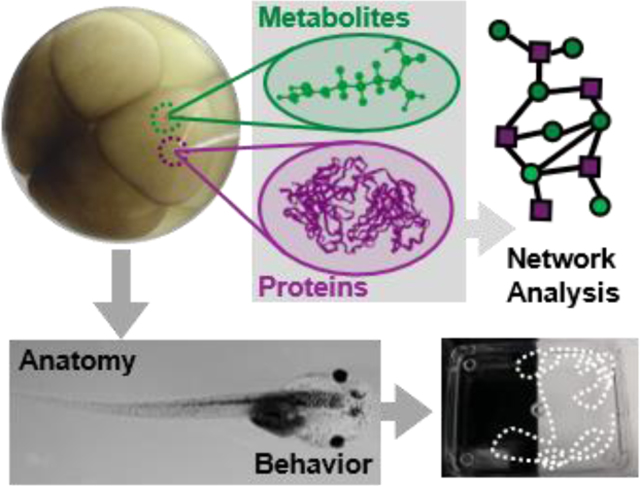
Figure 1.

Interdisciplinary strategy enabling in vivo subcellular proteo-metabolomic systems biology with demonstrated compatibility for cell-, neurodevelopmental, and behavioral biology using Xenopus laevis as the biological model. As an example, the left dorsal-animal (L-D1) and left ventral-animal (L-V1) cells were identified and the content of each cell rapidly microsampled twice using clean capillaries each time. The collected protein and metabolite samples were analyzed using custom-built ultrasensitive capillary electrophoresis (CE) nano/micro-flow electrospray ionization (ESI) high-resolution orbitrap and time-of-flight (TOF) mass spectrometry (HRMS). The tadpoles developing from the embryos were characterized for survival, anatomy, and behavior. Scale bars = 250 μm (black), 1 mm (white)
The vertebrate South African clawed frog (Xenopus laevis) was an ideal biological model for technology development, validation, and application in this study. Each cell in the cleavage-stage frog embryo is identifiable based on pigmentation and location and has reproducible tissue fates.[11] Figure 1 exemplifies the identification of the left dorsal-animal (L-D1) and left ventral-animal (L-V1) cells in a live 8-cell embryo, which respectively form the neural and epidermal tissues. X. laevis embryos develop externally to the mother and contain considerably large cells (180 nL volume/cell in the 8-cell embryo), permitting direct inspection and access to the cells. However, the cells populate the embryo in a complex three-dimensional morphology (spherical arrangement) and divide rapidly, in ~15–25 min per cell cycle between the 2- and 32-cell stages.[12] These physical and characteristics combined with the transient nature of an “embryonic body” fundamentally challenge existing single-cell HRMS technologies in terms of scalability for spatial, temporal, and in vivo operation.
We recognized spatiotemporally scalable sampling indispendisible for study (Fig. 1). While dissociation[13] and manual dissection[3a, 8a, 14] permit cell isolation from the embryo, the former loses spatial information on cell heterogeneity, and the latter requires substantial dexterity and lacks scalability; these features both have important implications in cell and developmental biology. With analyses occurring ex vivo, the current tools exclude the possibility of biological studies over critical developmental stages, ranging from gastrulation and neurulation to organogenesis and metamorphosis. Likewise, these strategies are incompatible with assessing function and behavior at an organismal level, such as the sentient tadpole. Currently, there exists no single-cell HRMS technology capable of determining the proteo-metabolome of single cells embedded in complex tissues or organisms in vivo with scalability.
We proposed double capillary microsampling as a potential solution for analyzing the cells in vivo. To serve as a micropipette for sample collection, a borosilicate capillary (0.75/1 mm inner/outer diameter) was flame-pulled, then cleaved to a 20-μm tip diameter (see SI Methods), and connected to a microinjector delivering negative pressure pulses. The tip of the capillary was inserted into the cell of interest under translation by a precision micromanipulator and guidance by real-time stereomicroscopy. In each of N = 20 stereotypical 8-cell embryos (experimental group), the L-D1 and L-V1 cells were each microsampled twice in a random order with both cells from the same embryo and one sampling for each ‘omic analysis. By performing the single-cell analyses exclusively on one side of the embryo (the left, experimental), tissues and organs arising from the nonsampled cells on the other side (right D1, R-D1 and right V1, R-V1) served as the internal control in each embryo to facilitate analysis of tadpole anatomy (see later). For the control group (Ctrl.), N = 20 stereotypical 8-cell embryos were cultured under identical conditions, without microaspiration. This approach was inspired by in situ microprobe sampling that we recently developed for cells in zebrafish and X. laevis embryos, albeit functioning only ex vivo and exclusively for metabolomics[3c, 9] or proteomics[3d], but not both ‘omes at the same time or on the same cell. The technical details are discussed in the SI document. We experimentally tested that double microsampling using larger capillaries (e.g., ~80 μm tips) and/or longer aspiration times (scalability) allowed us to aspirate ~100 nL, viz. 50% of the cell volume. These amounts were sufficient for single-cell HRMS; however, the embryos failed to survive the analysis due to substantial damage by the large capillary (see Fig. S1).
To enable cell sampling in vivo in the live embryo, we scaled the approach with assistance from survival assays (Fig. 2). Figure S1 presents significant membrane damage on the microsampled cells in a 16-cell embryo before refinement of this approach. In a series of experiments (data not shown), the tip size of the microprobe, the volume collected, and the speed of microsampling were tailored to the cells while monitoring the success of cell divisions after sampling under the stereomicroscope. Each sampling employed a clean and unused capillary to aid cell and embryonic survival by avoiding accidental contamination. Figure 2A tracks the percentage of embryos surviving after ~10 nL, or ~5% of the cell volume, was microaspirated twice from the L-D1 and L-V1 cells (N = 20 embryos). Double microsampling took less than 30 s per cell. By using 20-μm-tip capillaries and conducting two independent sampling events in each cell instead of a single one, we were able to promote cell viability and microhandling of viscous cell contents for downstream sample processing. Embryonic survival was quantified over 6 key stages of vertebrate development: cleavage (Nieuwkoop-Faber, NF 6), gastrula (NF 10.5, Fig. 2B), neurula (NF 13, Fig. 2B), early tailbud (NF 22), late tailbud (NF 36 and 41), and tadpole (NF 45, Fig. 2B). Compared to 100% of tadpoles successfully developing in the control group (unperturbed wild-type, WT) at all these stages of development, 100% of the embryos developed to the neurula stage and 95% survived to tadpoles in the experimental group. Therefore, capillary microsampling was successfully scaled to preserve viability.
Figure 2.

Developmental and behavioral impact of the technology. (A) Survival analysis (Surv.) at key developmental stages (NF, Nieuwkoop-Faber) for N = 20 embryos in the control (Ctrl.) group and N = 20 embryos in the experimental. (B) Representative (top panel) Ctr. gastrulae and Ctrl. neurula and (bottom) Ctr. and experimental (Exp.) tadpoles. Shown: midline separating left (L)-right (R) axis, body length, brain area, and distance between eyes and olfactory (Olf.) organs. (C) Comparison of tadpole anatomy. (Top) Close-up image of a tadpole showing the experimental (left) and control (right) side. Right tissues are labeled. (Middle) Analysis of organ size on the left (experimental side) between N = 5 Ctrl. (C) and N = 6 Exp. (E) surviving tadpoles randomly selected. Key: n.s., non-significant, *p < 0.05, Mann-Whitney test. (Bottom) Analysis of tissue areas for total brain (Ctr. vs. Exp. group), the eyes and, the olfactory organs in the 6 Exp. tadpoles. Key: n.s., non-significant, Wilcoxon signed rank, paired). (D) Comparison of visual function in a background-color preference assay for N = 16 Ctrl. vs. N = 15 Exp. tadpoles. Assay validation via double axotomy of the optic nerves in N = 4 positive Ctrl. tadpoles (+Ctrl.). Tadpole movies in Videos S1 (Ctrl.) and S2 (+Ctrl.). Key: n.s., non-significant; *p < 0.05, **p < 0.005 (Mann-Whitney). Scale bars, 250 μm (white) and 1 mm (black). Box-whisker plots: Box, 1×standard error of the mean (SEM), whiskers: 1.5×SEM.
Anatomy was also analyzed for the surviving tadpoles. Figure 2B presents typical examples for tadpoles from the control and experimental groups. Their morphologies were characterized in terms of whole-body length as well as the sizes of the eyes, olfactory organs (Olf.), and total brain, which partially derive from the L-D1 and L-V1 cells (Fig. 2B). Body symmetry was also analyzed for the eyes and olfactory tissues along the midline separating the left (experimental) side and the right (control) side (see close-up in Fig. 2C). As shown in Figure 2C, the body length (p = 0.315, Mann-Whitney) as well as the center-to-center distances between the eyes (p = 0.411, Mann-Whitney) and the olfactory tissues (p = 0.523, Mann-Whitney) were indistinguishable, as was the total area of the brain (p = 0.121, Mann-Whitney). The size (area) of the eyes (p = 0.142, Wilcoxon signed rank, paired) and olfactory organs (p = 0.142, Wilcoxon signed rank, paired) were also indistinguishable between the experimental and control sides in the tadpoles. Therefore, double subcellular capillary microsampling of the L-D1 and L-V1 in the cleavage-stage embryo led to no detectable impairment on tadpole development, morphologically.
Physical appearance, however, does not equate to performance; therefore, we further evaluated the animals based on behavior. The visual (also including motor, sensory, and neural processing) function was compared at stages 45–50, when tadpoles display a robust preference for lighter background in a color preference assay[15] (see setup in Fig. 1). The technical details are available in the SI document. Indeed, as shown in Figure 2D, when presented with a dark vs. light background in a tank, the control (WT) tadpoles resided ~91% of the time over the light background (N = 16). These tadpoles recapitulated this robust behavioral phenotype at this stage,[15] thus confirming the robustness of the assay in our hands. We further validated the sensitivity of the assay to detect visual impairment using a positive control (+Ctrl). Tadpoles that underwent double axotomy of their optic nerves (N = 4) explored the light background only at ~56% of the time, in a significant difference from the control group (p = 1.64×10–5). Indeed, without vision, tadpoles are anticipated to make random explorations, spending ~50% on either side of the tank. These results also agreed with results from an independent investigation[15], thus confirming that the assay was sufficiently sensitive to detect visual function in our experiments. Swimming ~89% time over the light area, the experimental group (N = 15) presented indistinguishable visual behavior from the control tadpoles (p = 0.41, Mann Whitney). Representative behaviors can be viewed for a Ctrl. and +Ctrl. tadpole in Videos S1 and S2, respectively. Double microsampling of each of the 2 cells in the same embryo preserved embryonic viability and organismal behavior. The approach opened, for the first time in single-cell HRMS, the possibility of dual ‘omics of single cells in a live organism, which also undergoes dynamic spatial and temporal reorganization to form a behaving animal, as demonstrated here for the X. laevis embryo and its tadpole in vivo in this work.
Detection of the limited amounts of proteins and metabolites from the cells necessitated ultrasensitive analysis. From each cell type in the same embryo (N = 4), the double aspirates were independently processed via a proteomic and metabolomic workflow (recall Fig. 1). To eliminate potential systematic biases during sample collection, we randomized the location of subcellular sampling and the order of aspirating the protein and metabolite samples. Each embryo, cell, and processed protein and metabolite sample was assigned a unique identifier in our study, although this information was purposefully not used during data analysis to eliminate potential (conscious or subconscious) biases during sample preparation, processing, or instrumental measurement. This information was only revealed after data collection to aid results interpretation. As detailed in the SI document, each subcellular sample was processed to yield metabolic and proteomic microextracts. For bottom-up proteomics, potential protein losses on vial and pipette tip surfaces were minimized by eliminating the traditional steps of desalting, alkylation, and reduction. The final subcellular samples contained the tryptic digest in 2 μL for bottom-up proteomics and 4 μL for metabolomics. To avoid systematic bias during instrumental measurement, the subcellular ‘omes were blinded for cell type and analyzed in a randomized order.
With ~100–10,000-times smaller amounts of material collected from the cells in this study than those typically processed/measured by standard HPLC-HRMS, we turned to our ultrasensitive CE-ESI-HRMS instruments for assistance. These platforms provided ~700 zmol sensitivity for peptides on a tribrid quadrupole-orbitrap-ion trap[3a, 3d] and ~60 amol sensitivity for metabolites on a quadrupole time-of-flight high-resolution[8a, 9a] mass spectrometer. The technical details are in the SI document. To facilitate the instrumental measurements, the metabolomic analytical track was limited to cationic electrophoresis in this study. Dual cationic-anionic metabolomics is also possible on these instruments to deepen the detectable portion of the single-cell metabolome.[16] Measurement of ~0.5% (v/v) of the protein extract, thus ~0.03% of the cell’s volume, identified 738 proteins (false discovery rate < 1%), listed in Table S1A. Similarly, analysis of ~0.25% (v/v) of the metabolite sample (volume), viz. ~0.01% of the cell’s volume, produced ~150 nonredundant metabolic molecular features, 57 of which were identified with a high Level-1[17] confidence, listed in Table S1B. Notably, detection of these ~800 proteins and metabolites was achieved in an untargeted (discovery) manner, requiring no prior knowledge of cellular composition or the use of functional probes; antibodies or antisense oligos were unnecessary for our technology.
These multi-omics data also opened a window into (sub)cellular biochemistry, directly in the live embryo. Statistical enrichment analysis using the Kyoto Encyclopedia of Genes and Genomes[18] revealed significant coverage of several known pathways (Fig. 3A). For example, the arginine-proline pathway, TCA cycle, glycolysis, and pyruvate metabolism were represented with statistical significance, whereas several other amino acid pathways had high pathway impact. Notably, these proteins and metabolites assessed cellular biochemistry in complementary performance. For example, the biosynthesis of aminoacyl-tRNA and valine-leucine-isoleucine was only represented by metabolites, whereas glycolysis/gluconeogenesis was solely enriched for by proteins (see statistics and fold changes in Table S2). Further, other pathways, such as arginine biosynthesis and arginine-proline metabolism, were represented by both ‘omes (Table S2). Figure 3B annotates arginine-proline metabolism based on the complementary detection of proteins and metabolites in the cells. The observed coverage of the network raises a potential for studies targeting particular ‘omic processes. By also including anionic metabolites during CE-ESI-HRMS in future experiments, we anticipate many other cellular pathways to be detectable by this approach, such as the TCA cycle and energy production as well as drug metabolism. These results illustrate the benefit of measuring more than one ‘ome in single cells toward a holistic understanding of cellular systems biology at the molecular level.
Figure 3.

Qualitative and quantitative proteo-metabolomic systems biology on the L-D1 and L-V1 cell in the live embryo. (A) KEGG pathway analysis of the identified proteins (Table S1) and metabolites (Table S2). Pathway data are in Table S3. (B) Example showing proteins and metabolites with complementary enrichment in the arginine-proline pathway. (C) Unsupervised (blinded) hierarchical cluster–heat map analysis of the detected protein and metabolite quantities revealing reproducible, systematic, and significant differences between the cell types. Top 40 most significantly dysregulated molecules shown (p < 0.05, see quantitative statistics in Table S3).
Finally, we applied the technology to ask whether the pluripotent cells harbored detectable differences in proteo-metabolomic activity at such an early stage of development. Although embryonic cells have reproducible tissue fates in the cleavage-stage embryo of Xenopus, unbiased and parallel characterization of their proteome and metabolome has not been possible due to a lack of sufficiently sensitive and specific molecular tools. We relatively quantified ~450 proteins and ~80 metabolites between the L-D1 and L-V1 cells in each embryo (N = 4) based on the subcellular HRMS data obtained in this study. Quantification was based on signal abundances in the HRMS datasets (see Methods, SI), which offer an effective and robust proxy for concentration in CE-ESI-HRMS.[3a, 4a] Quantified protein and metabolite signals are presented in Table S3. As highlighted earlier, the phenotypes of the cells or the identifier of each embryo were hidden to ensure bias-free data analysis. Figure 3C presents the unsupervised hierarchical cluster analysis (HCA) and intensity heat map of the data based on the top 40 statistically most significantly dysregulated molecules, including 13 metabolites and 27 proteins.
The pathway data are listed in Table S2. The top dendrogram clusters the samples into two groups in the HCA plot. Upon revealing the identity of the samples, we learned that the groups corresponded to the D1 and V1 cell types. Further, the heatmap revealed notable quantitative molecular differences between the cell types. The left dendrogram organized the proteins and metabolites into two groups in the HCA plot. The HCA heatmap readily captures reproducible abundance differences between the cell types. Specifically, in this map, 20 molecules, containing 10 metabolites and 10 proteins, were more abundant in the L-D1, whereas 20 molecules, encompassing 3 metabolites and 17 proteins were enriched in the L-V1. Table S3 tabulates the proteins and metabolites with statistically (p ≤ 0.05) and biologically significant dysregulation (|fold change| ≥ 1.5). These intriguing molecular differences would have been lost due to averaging across a large number of cells pooled for classical HRMS approaches.
Conclusion
We enabled in vivo single-cell proteomics and metabolomics in the same single cell in live chordate embryos using X. laevis as the biological model. The technology revealed quantitative proteo-metabolomic differences between cells at the cleavage stage. These results afford previously unavailable insights into the establishment of cell heterogeneity during embryogenesis; they also challenge our current understanding of the underlying molecular processes. Differential production of these molecules reveals that asymmetry along the dorsal-ventral, one of the three primary axes of the vertebrate body plan, is set up rather early during embryonic development, when transcriptional heterogeneity is not detectable along this axis based on sequencing of the respective single-cell transcriptomes[19] in Xenopus. These findings support our earlier discovery of molecules, such as metabolites[8a], affecting tissue fates via noncanonical mechanisms of action. Although determining the exact biological significance of these findings goes beyond the scope of this study, the data generated in this work may be used to develop hypotheses for experiments targeting specific proteins and metabolites and their functions.
Supporting future investigations, our technology is scalable in space and time to other types of cells and different biological models. Subcellular HRMS is compatible with complex tissues and live development, as demonstrated for the live embryo here. It does not escape our attention that our technology can be used to perform multi-’omics on subcellular organelles. Further, the approach is also adaptable to classical and modern tools of biology and health studies, such as optical microscopy and behavioral assays (as demonstrated in this study) to characterize phenotypes as well as established or contemporary tools of molecular biology, including expression/translation-blocking morpholinos or gene editing by CRISPR-Cas9, to probe function. We anticipate in vivo proteo-metabolomic subcellular CE-ESI-HRMS to expand the contemporary toolbox of cell and developmental biology, neuroscience, and health research. This technology realizes new strengths in the molecular study of the building block of life, the cell, and processes governing the formation of tissues, organs, and entire organisms with complex behavior.
Supplementary Material
Acknowledgements
This research was partially supported by an Arnold and Mabel Beckman Foundation Beckman Young Investigator award (to P.N.), the National Science Foundation under awards DBI-1826932 (to P.N.) and IOS-1832968 CAREER (to P.N.), and the National Institutes of Health under award 1R35GM124755 (to P.N.). The content presented in this work and the conclusions drawn are solely the responsibility of the authors and do not necessarily represent the official views of the sponsors. We are grateful to Dr. Sally Moody (The George Washington University, Washington, DC) for supplying X. laevis embryos for a pilot experiment.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Dr. Camille Lombard-Banek, Department of Chemistry & Biochemistry, University of Maryland, 0107 Chemistry Building, 8051 Regents Drive, College Park, MD 20742 (USA).
Jie Li, Department of Chemistry & Biochemistry, University of Maryland, 0107 Chemistry Building, 8051 Regents Drive, College Park, MD 20742 (USA).
Dr. Erika P. Portero, Department of Chemistry & Biochemistry, University of Maryland, 0107 Chemistry Building, 8051 Regents Drive, College Park, MD 20742 (USA)
Dr. Rosemary M. Onjiko, Department of Chemistry, The George Washington University, 800 22nd St NW, Washington, DC 20052 (USA)
Chase D. Singer, Department of Chemistry & Biochemistry, University of Maryland, 0107 Chemistry Building, 8051 Regents Drive, College Park, MD 20742 (USA)
David O. Plotnick, Department of Chemistry, The George Washington University, 800 22nd St NW, Washington, DC 20052 (USA)
Reem Q. Al Shabeeb, Department of Chemistry, The George Washington University, 800 22nd St NW, Washington, DC 20052 (USA)
Prof. Dr. Peter Nemes, Department of Chemistry & Biochemistry, University of Maryland, 0107 Chemistry Building, 8051 Regents Drive, College Park, MD 20742 (USA); Department of Chemistry, The George Washington University, 800 22nd St NW, Washington, DC 20052 (USA); Department of Anatomy & Cell Biology, School of Medicine and Health Sciences, The George Washington University, 2300 I Street NW, Washington, DC 20037 (USA).
References
- [1](a).Comi TJ, Do TD, Rubakhin SS, Sweedler JV, J. Am. Chem. Soc 2017, 139, 3920–3929; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang L, Vertes A, Angew. Chem. Int. Edit 2018, 57, 4466–4477; [DOI] [PubMed] [Google Scholar]; (c) Couvillion SP, Zhu Y, Nagy G, Adkins JN, Ansong C, Renslow RS, Piehowski PD, Ibrahim YM, Kelly RT, Metz TO, Analyst 2019, 144, 794–807; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kelly RT, Mol. Cell. Proteomics 2020, 19, 1739–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2](a).Zenobi R, Science 2013, 342, 1243259; [DOI] [PubMed] [Google Scholar]; (b) Levy E, Slavov N, in Systems Biology, Vol. 62 (Eds.: Kolch W, Fey D, Ryan CJ), Portland Press Ltd, London, 2018, pp. 595–605; [Google Scholar]; (c) Specht H, Slavov N, J. Proteome Res 2018, 17, 2565–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3](a).Lombard-Banek C, Moody SA, Nemes P, Angew. Chem. Int. Edit 2016, 128, 2500–2504; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Virant-Klun I, Leicht S, Hughes C, Krijgsveld J, Mol. Cell. Proteomics 2016, 15, 2616–2627; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Onjiko RM, Portero EP, Moody SA, Nemes P, J Vis Exp 2017, e56956; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lombard-Banek C, Moody SA, Manzin MC, Nemes P, Anal. Chem 2019, 91, 4797–4805; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Budnik B, Levy E, Harmange G, Slavov N, Genome Biol. 2018, 19, 12; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zhu Y, Clair G, Chrisler WB, Shen Y, Zhao R, Shukla AK, Moore RJ, Misra RS, Pryhuber GS, Smith RD, Ansong C, Kelly RT, Angew. Chem. Int. Edit 2018, 57, 12370–12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4](a).Nemes P, Rubakhin SS, Aerts JT, Sweedler JV, Nat. Protoc 2013, 8, 783–799; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li S, Plouffe BD, Belov AM, Ray S, Wang X, Murthy SK, Karger BL, Ivanov AR, Mol. Cell. Proteomics 2015, 14, 1672–1683; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cong YZ, Liang YR, Motamedchaboki K, Huguet R, Truong T, Zhao R, Shen YF, Lopez-Ferrer D, Zhu Y, Kelly RT, Anal. Chem 2020, 92, 2665–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Specht H, Emmott E, Petelski AA, Huffman RG, Perlman DH, Serra M, Kharchenko P, Koller A, Slavov N, bioRxiv 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Do TD, Ellis JF, Neumann EK, Comi TJ, Tillmaand EG, Lenhart AE, Rubakhin SS, Sweedler JV, ChemPhysChem 2018, 19, 1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7](a).Lapainis T, Rubakhin SS, Sweedler JV, Anal. Chem 2009, 81, 5858–5864; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nemes P, Knolhoff AM, Rubakhin SS, Sweedler JV, Anal. Chem 2011, 83, 6810–6817; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu JX, Aerts JT, Rubakhin SS, Zhang XX, Sweedler JV, Analyst 2014, 139, 5835–5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8](a).Onjiko RM, Moody SA, Nemes P, Proc. Natl. Acad. Sci. U. S. A 2015, 112, 6545–6550; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lombard-Banek C, Reddy S, Moody SA, Nemes P, Mol. Cell. Proteomics 2016, 15, 2756–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9](a).Onjiko RM, Portero EP, Moody SA, Nemes P, Anal. Chem 2017, 89, 7069–7076; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Onjiko RM, Plotnick DO, Moody SA, Nemes P, Anal. Methods 2017, 9, 4964–4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10](a).Macaulay IC, Ponting CP, Voet T, Trends Genet. 2017, 33, 155–168; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ma AJ, McDermaid A, Xu J, Chang YZ, Ma Q, Trends Biotechnol. 2020, 38, 1007–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hirose G, Jacobson M, Dev. Biol 1979, 71, 191–202. [DOI] [PubMed] [Google Scholar]
- [12].Karimi K, Fortriede JD, Lotay VS, Burns KA, Wang DZ, Fisher ME, Pells TJ, James-Zorn C, Wang Y, Ponferrada VG, Chu S, Chaturvedi P, Zorn AM, Vize PD, Nucleic Acids Res. 2018, 46, D861–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Briggs JA, Weinreb C, Wagner DE, Megason S, Peshkin L, Kirschner MW, Klein AM, Science 2018, 360, 980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sun L, Dubiak KM, Peuchen EH, Zhang Z, Zhu G, Huber PW, Dovichi NJ, Anal. Chem 2016, 88, 6653–6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Viczian AS, Zuber ME, J Vis Exp 2014, e51726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Portero EP, Nemes P, Analyst 2019, 144, 892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schrimpe-Rutledge AC, Codreanu SG, Sherrod SD, McLean JA, J. Am. Soc. Mass Spectrom 2016, 27, 1897–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K, Nucleic Acids Res. 2017, 45, D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19](a).Flachsova M, Sindelka R, Kubista M, Sci Rep 2013, 3, 2278; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) De Domenico E, Owens NDL, Grant IM, Gomes-Faria R, Gilchrist MJ, Dev. Biol 2015, 408, 252–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.