

Key Points
CXCR4 desensitization dampens B-cell cycle and differentiation after TLR activation.
CXCR4 desensitization limits the strength and length of the extrafollicular immune response.
Abstract
The extrafollicular immune response is essential to generate a rapid but transient wave of protective antibodies during infection. Despite its importance, the molecular mechanisms controlling this first response are poorly understood. Here, we demonstrate that enhanced Cxcr4 signaling caused by defective receptor desensitization leads to exacerbated extrafollicular B-cell response. Using a mouse model bearing a gain-of-function mutation of Cxcr4 described in 2 human hematologic disorders, warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome and Waldenström macroglobulinemia, we demonstrated that mutant B cells exhibited enhanced mechanistic target of rapamycin signaling, cycled more, and differentiated more potently into plasma cells than wild-type B cells after Toll-like receptor (TLR) stimulation. Moreover, Cxcr4 gain of function promoted enhanced homing and persistence of immature plasma cells in the bone marrow, a phenomenon recapitulated in WHIM syndrome patient samples. This translated in increased and more sustained production of antibodies after T-independent immunization in Cxcr4 mutant mice. Thus, our results establish that fine-tuning of Cxcr4 signaling is essential to limit the strength and length of the extrafollicular immune response.
Visual Abstract

Introduction
Following infections, rapid production of neutralizing antibodies (Abs) through the extrafollicular immune response is essential to control infectious agents. Despite the central importance of this pathway in humoral immunity, the molecular mechanisms at play are poorly understood. Early B-cell activation and differentiation initiated through crosslinking of the B-cell receptor (BCR) and Toll-like receptors (TLRs) leads to the production of low-affinity Abs by short-lived extrafollicular plasma cells (PCs), also called plasmablasts (PBs), which provide rapid but transient protection against infection.1 Upon T-cell help, this early response can evolve through the formation of germinal centers (GCs), to the production of high-affinity long-lived PCs that can persist for an extended period of time in the bone marrow (BM) contributing to immunologic memory.2 The chemokine receptor Cxcr4 and its ligand Cxcl12 are important for the T-dependent immune response through regulation of GC B-cell localization and BM homing of PCs.3-8 However, the role of this axis in early extrafollicular B-cell response after antigenic challenge has never been addressed. Following Cxcl12 binding, Cxcr4 signals through the G proteins. Signal duration and strength is tightly regulated by receptor desensitization, which involves phosphorylation of the C-terminal tail of Cxcr4 and the subsequent recruitment of β-arrestins, leading to both uncoupling of the receptor from G proteins and receptor internalization.9 Our group and others have established that this desensitization step is defective in the rare primary immunodeficiency disease warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome (WS),10,11 which is caused by autosomal dominant mutations in the C-tail of CXCR4, resulting in a gain-of-function in response to CXCL12.10,12 Strikingly, similar mutations of CXCR4 have been reported in Waldenström macroglobulinemia, an indolent lymphoplasmacytic B-cell lymphoma characterized by an immunoglobulin M (IgM) spike and BM infiltration of malignant clones with an immunophenotype ranging from activated B cells to PCs.13-15 CXCR4 mutations are observed in ∼30% of Waldenström macroglobulinemia patients in conjunction with a gain of function mutation of the TLR adaptor MYD88 and may be associated with treatment resistance.16,17 Whether and how CXCR4 and MYD88 mutations synergize in Waldenström macroglobulinemia is still unclear; however, their coincidence in this disease prompted our interest in investigating the general importance of Cxcr4 signaling in the context of TLR-induced MyD88 signaling for B cell and PC biology. Here, using a mouse model18 bearing an orthologous Cxcr4 mutation observed both in WS and Waldenström macroglobulinemia,13,14 we demonstrate an unexpected and central role for Cxcr4 desensitization in regulating the early extrafollicular antibody response.
Materials and methods
Mice
Cxcr4+/1013 mice were generated as previously described.18 Blimp1GFP/+ mice were obtained from S. Nutt (The Walter and Eliza Hall Institute of Medical Research, Melbourne, Victoria, Australia)19 and crossed with Cxcr4+/1013 mice. Mice aged 8 to 16 weeks old or 1 year old and cohoused littermates were used in all experiments that were conducted in compliance with the European Union guide for the care and use of laboratory animals, which have been reviewed and approved by an institutional review committee (C2EA-26, Animal Care and Use Committee, Villejuif, France).
Human samples
Cryopreserved BM aspirates from 2 WS patients (1 male, age 47 years; 1 female, age >30 years at sampling, both bearing the CXCR4R334X mutation; collected under National Institutes of Health protocol 09-I-0200) were provided through a Material Transfer Agreement in accordance with National Institutes of Health human subject research policies. BM from hip replacement surgery was obtained from discarded material from 6 patients (4 males and 2 females; 42-58 years old at sampling). All the required ethical and export approvals were obtained from the French Ministries of Health and Agriculture. All individuals provided written informed consent before sampling and data registering. The study was approved by the Comité de Protection des Personnes Ile-de-France VII (protocol 17-030, ID-RCB 2017-A01019-44). Samples were stained with the antibodies indicated in supplemental Table 1 available on the Blood Web site.
Immunization
Mice were immunized intraperitoneally with 25 µg 4-hydroxy-3-nitrophenylacetyl-lipopolysaccharide (NP-LPS) or 25 µg NP-Ficoll (Biosearch Technologies). Blood, spleen, and BM were harvested and analyzed 3, 6, 12, and 45 days after challenge with NP-LPS and 6 days after NP-Ficoll immunization.
Single cell suspension and flow cytometry
Isolation of organs was performed, and single cell suspensions were stained as previously described,7 using the Abs described in supplemental Table 1. Analyses were carried out on an LSRII Fortessa flow cytometer (BD Biosciences), and data were analyzed with the Flowjo software (TreeStar, Ashland, OR).
In vitro functional assays
For functional assays, total splenocytes or B cells isolated using a naïve mouse B-cell (CD43−) isolation kit (Miltenyi Biotec) were cultured using complete medium (RPMI with 10% fetal calf serum, 1% penicillin/streptomycin, 50 µM β-mercaptoethanol, 1 mM sodium pyruvate; Gibco) with 1 U/mL interleukin 4 (IL-4; Miltenyi) and 5 ng/mL IL-5 (Miltenyi) supplemented with 1 µg/mL LPS (Invivogen) or 10 µg/mL CpG oligodeoxynucleotides (Invivogen) and/or 50 nM Cxcl12 (R&D), and/or 10 µM AMD3100 (Sigma Aldrich) for 5 minutes and up to 5 days at 37°C. MK-2206 (Ambeed) and rapamycin (Life Technologies) were used at 1 µM and 50 nM, respectively. Phosphoflow was performed using the PerFix EXPOSE kit (Beckman Coulter). PB differentiation and apoptosis were assessed by flow cytometry (supplemental Table 1). For proliferation, splenocytes were loaded with cell trace violet (CTV) (Thermofisher) according to the manufacturer’s instructions and then stimulated. For cell cycle analysis, cells were permeabilized and fixed with the Foxp3/transcription factor staining buffer set (eBioscience) and labeled with an anti–Ki-67 antibody, and 4′,6-diamidino-2-phenylindole (DAPI) was added just before analysis.
Adoptive transfer experiments
Splenocytes from wild-type (WT) × Blimp1GFP/+ and Cxcr4+/1013 × Blimp1GFP/+ mice were stimulated in vitro with LPS for 4 days. Cells from each genotype were loaded with either CTV or CT yellow (CTY; Thermofisher). A 50:50 mix of cells was made and checked by flow cytometry before injection. A total of 106 cells were transferred intravenously into C57Bl/6J CD45.1 WT recipients. Blood, BM, and spleens were harvested and analyzed at 24 and 48 hours after transfer.
Transwell migration assay
Chemotaxis assays were performed using a Transwell assay as previously described.10 Splenocytes were incubated for 45 minutes at 37°C before migration for 3 hours through a 5-µm pore Transwell insert (Corning) with medium only or with Cxcl12 (R&D) in the lower chamber. Input cells that migrated to the lower chamber were collected, stained, and enumerated by flow cytometry. The fraction of cells migrating across the membrane was calculated as follows: ([number of cells migrating to the lower chamber in response to chemokine or medium]/[number of cells added to the upper chamber at the start of the assay]) × 100.
Immunofluorescence
Spleens were fixed for 4 hours in phosphate-buffered saline (PBS)-paraformaldehyde 4% and incubated overnight in PBS-sucrose 20% before being embedded in optimum cutting temperature compound (TissueTek). Cryosections were permeabilized with PBS Triton X-100 0.5% for 15 minutes, washed twice with PBS, and blocked with PBS-bovine serum albumin (BSA) 5%. Sections were stained with primary Abs and secondary Ab (supplemental Table 1) and Hoechst 33342. Slides were scanned using a NanoZoomer Digital Pathology system using ×40 objective lenses with numerical aperture of 0.75 (Hamamatsu Photonic). BM embedding, sectioning, and staining were performed as described.20 Sections (30 µm) were permeabilized, saturated, and stained as for the spleen. Images were acquired using a TCS SP8 confocal microscope and processed and quantified with Fiji and Imaris.
ELISA and ELISpot
Anti-NP and anti-IgM ELISA were performed as previously described.7 Enumeration of total IgM and NP-specific IgM Ab-secreting cells (ASCs) were performed by incubating spleen or BM cells overnight at 37°C on plates precoated with NP15-BSA or goat anti-mouse IgM at 5 µg/mL and saturated with culture medium containing 10% fetal calf serum. Plates were then washed and incubated with peroxidase-conjugated goat anti-mouse IgM (Southern Biotech). Spots were revealed with 3-amino-9-ethylcarbazole (Sigma-Aldrich) and analyzed using an ispot Reader (Autoimmun Diagnostika GMBH).
Time lapse imaging
B cells were isolated and stimulated with LPS. Cells were either imaged using a Biostation (NIKON) in a 37°C, 5% CO2 supplemented chamber at a concentration of 104 cells/mL or using the Incucyte S3 at a concentration of 104 cells per well. Cells were imaged for phase-contrast and green fluorescent protein (GFP) fluorescence every 15 or 30 minutes, respectively. Images and movies were analyzed with the biostation IM software, the Incucyte S3 software, and Fiji.
Multiplex quantitative polymerase chain reaction
Multiplex quantitative polymerase chain reaction (qPCR) analysis was performed using the Biomark system (Fluidigm). Seventy-five cells per well were sorted directly into reverse transcription/preamplification mix, as previously described21 (supplemental Table 2). Targeted cDNA preamplification was performed before processing with the dynamic array protocol according to the manufacturer’s instructions (Fluidigm). Mean expression of Actb and Gapdh was used for normalization.
Statistical analysis
Data are expressed as means ± standard error of the mean (SEM). The statistical significance between groups was evaluated using the 2-tailed unpaired Mann-Whitney nonparametric test (Prism software, GraphPad) unless indicated otherwise in figure legends. Heatmaps were presented using the Z score, and clustering analyses were performed using the Pearson rank correlation test (http://www.heatmapper.ca). Principal component analysis (PCA) was performed using R ggplot packages.
Results
Cxcr4 desensitization limits plasma cell generation
To better understand the pathophysiology of WS, we previously generated a knock-in mouse model bearing a heterozygous nonsense point mutation common to a subset of patients (mutation C1013G in the open reading frame).18 These mice, referred to as Cxcr4+/1013, phenocopy the pan-leukopenia, a major hematologic feature of the disease.18 Moreover, using this model, we recently showed that the gain of function of Cxcr4 affects in a B-cell intrinsic manner the T-dependent humoral immune response recapitulating vaccination failure reported in some WS patients.7,12,22,23 We took advantage of this model to assess whether Cxcr4 signaling may play a role in the early extrafollicular response and in particular after stimulation with MyD88-dependent TLR ligands. First, we stimulated in vitro splenic B cells from WT and Cxcr4+/1013 mice with the TLR4 and TLR9 ligands, LPS and CpG, respectively, in the presence or absence of Cxcl12. Under these conditions, PB differentiation, defined as formation of B220lowCD138+ cells, was increased for B cells from Cxcr4+/1013 mice (Figure 1A-B; supplemental Figure 1A-B) and was associated with a twofold increase in IgM concentration measured in the culture supernatant (Figure 1B). The addition of Cxcl12 to the TLR agonists in the stimulation medium led to a small increase of PB in both WT and Cxcr4+/1013 B-cell cultures (Figure 1B; supplemental Figure 1A-B). Interestingly, stimulation with Cxcl12 alone promoted some PB differentiation of Cxcr4+/1013 B cells, a phenomenon significantly less marked in WT cultures (Figure 1B; supplemental Figure 1B). This Cxcl12-mediated PC differentiation was abolished by addition of the Cxcr4 antagonist AMD3100 confirming that Cxcr4 signaling promoted it (supplemental Figure 1B). These observations thus hinted toward a role for Cxcr4 signaling in early B-cell activation and differentiation after stimulation with TLR ligands.
Figure 1.
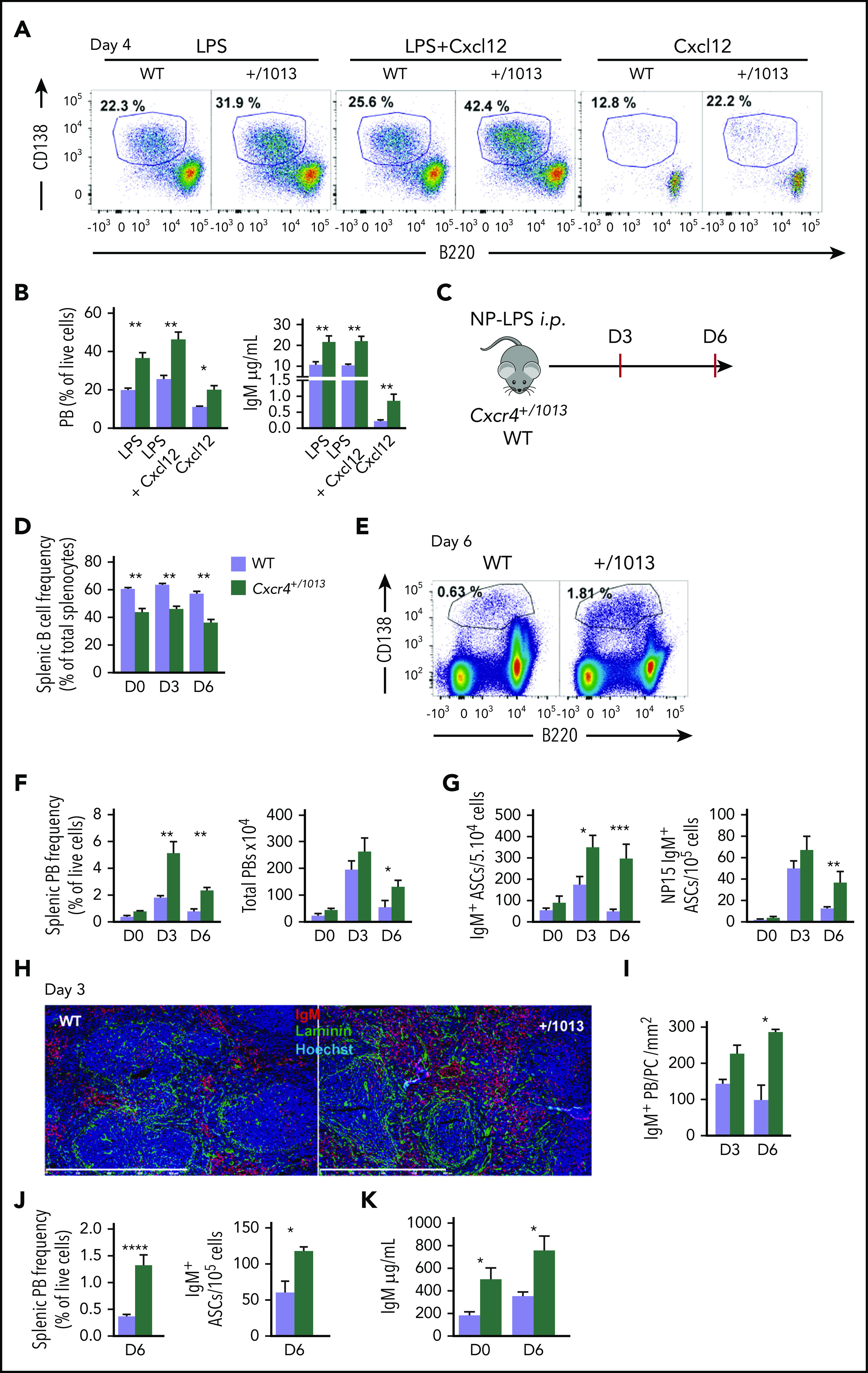
Cxcr4 desensitization limits plasma cell differentiation in vitro and in vivo. (A-B) Splenic B cells were cultured in presence of LPS, LPS+Cxcl12, or Cxcl12 alone for 4 days. (A) Representative dot plots for the gating of PBs (B220loCD138+) generated in vitro after 4 days of culture. (B) The proportion of PBs was determined by FACS and IgM concentrations in the supernatants were determined by ELISA. (C) Schematic diagram for the NP-LPS immunization. (D) Frequency of splenic B cells (CD19+ B220+) during NP-LPS immunization. (E) Representative dot plots for PB gating in the spleen of both WT and Cxcr4+/1013 mice 6 days after NP-LPS immunization. (F) Percentage and total number of PBs in the spleen at days 0, 3 and 6 after immunization. (G) Quantification of total IgM+ and NP15-specific IgM+ ASCs in the spleen by ELISpot at days 0, 3, and 6 after immunization. (H) Representative staining of spleen sections from WT and Cxcr4+/1013 mice at day 3 after immunization. PBs are stained with an anti-IgM Ab (red), basal membrane is stained with an anti-laminin Ab (green), and nuclei are stained with Hoechst 33342 (Blue), Scale bar: 800 µm. (I) Quantification of IgM+ PB accumulation within the spleen at days 3 and 6. (J) Frequency of total PBs and of IgM+ ASCs determined by flow cytometry and ELISpot, respectively, in the spleen 6 days after NP-Ficoll immunization. (K) Serum titers of total IgM from both genotypes were measured by ELISA at days 0 and 6 after NP-Ficoll immunization. Results are from 3 independent experiments (A-G, J-K) or 1 representative experiment of 2 (H-I) (mean ± SEM, n = 2-7). Mann-Whitney U test was used to assess statistical significance except for panel I, where the Student t test was used (*P < .05, **P < .01, ***P < .001).
We next determined whether the gain of function of Cxcr4 could affect T-independent B-cell immune responses in vivo. Immunization with the T-independent type 1 antigen NP-LPS did not correct the severe B-cell lymphopenia observed in the spleens of Cxcr4+/1013 mice18 (Figure 1C-D). However, compared with WT mice, we observed a significant increase in the frequency of splenic PBs in Cxcr4+/1013 mice from days 3 through 6 after immunization (Figure 1E-F). Consistent with this, we detected an increased frequency of IgM+ ASCs by ELISpot in the spleens of Cxcr4+/1013 mice compared with WT mice (Figure 1G), and an increased titer of total IgM in the serum of mice bearing the Cxcr4 gain-of-function mutation (supplemental Figure 1C). Moreover, the frequencies of NP-specific IgM+ ASCs in the spleen were increased at day 6 in Cxcr4+/1013 mice compared with WT mice (Figure 1G). Importantly, the T-independent B cell response to NP-LPS immunization was also more sustained in Cxcr4+/1013 mice compared with WT mice as demonstrated by the still elevated frequency and number of PBs observed at day 6 in the mutant mice (Figure 1F-G). Analysis of the splenic PB population by imaging revealed an increased detection of IgM+ PBs in sections of Cxcr4+/1013 spleens compared with WT tissues (Figure 1H-I). Similarly, immunization with the T-independent type 2 NP-Ficoll led to increased splenic PBs and increased total IgM+ serum titers (Figure 1J-K) in the mutant mice compared with controls. Together, these results show that Cxcr4 desensitization is an important negative regulator of extrafollicular PB generation in the spleen.
Cxcr4 desensitization limits the entry of B cells into cycle
We next addressed the cellular mechanism underlying enhanced PB generation in Cxcr4+/1013 mice. We cultured WT × Blimp1-GFP and Cxcr4+/1013 × Blimp1-GFP splenic B cells with LPS and assessed the kinetics of appearance of GFP as a readout for PB differentiation by video microscopy. As shown in Figure 2A, some GFP+ cells were detectable in Cxcr4+/1013 cultures as early as 46 hours after stimulation, whereas they started appearing in WT cultures 20 hours later (Figure 2A-B). As B-cell and PB survival seemed unaffected (supplemental Figure 2A-B), this accelerated appearance and accumulation of PBs in culture could be caused by an enhanced cycling and/or proliferation of B cells and PBs. B-cell proliferation, measured by CTV dye dilution in vitro after LPS stimulation, was indeed altered in the mutant condition at day 2 both with and without exogenous Cxcl12 addition (Figure 2C-D; supplemental Figure 2C). However, from day 3, B-cell proliferation was equivalent between WT and mutant B cells (Figure 2D), suggesting this was only a transient phenotype. Likewise, proliferation in newly generated PBs was not different between WT and mutant conditions at any of the time points analyzed (Figure 2E-F; supplemental Figure 2D). Previous reports have suggested that B-cell differentiation into PB necessitates at least 3 rounds of cell division.24 Together with our results, this brought us to consider whether it was not the proliferation of B cells per se that was affected, but rather their entry into the cell cycle. Indeed, we observed a significant increase in the percentage of B cells in cycle, especially in the G1 phase, in Cxcr4+/1013 cultures compared with WT ones (Figure 2G-H). Addition of Cxcl12 to LPS in the culture medium led to a similar result, and treatment with AMD3100 had no effect on the exacerbated entry into cell cycle observed in mutant B cells (supplemental Figure 2E-F). As expected, most PBs were cycling,25 and no difference was observed between our 2 experimental groups for this cell type (Figure 2I). This demonstrates that Cxcr4 desensitization is an essential negative regulator of B cell entry into cycle and that deregulation of this process contributes to exacerbated PB differentiation.
Figure 2.
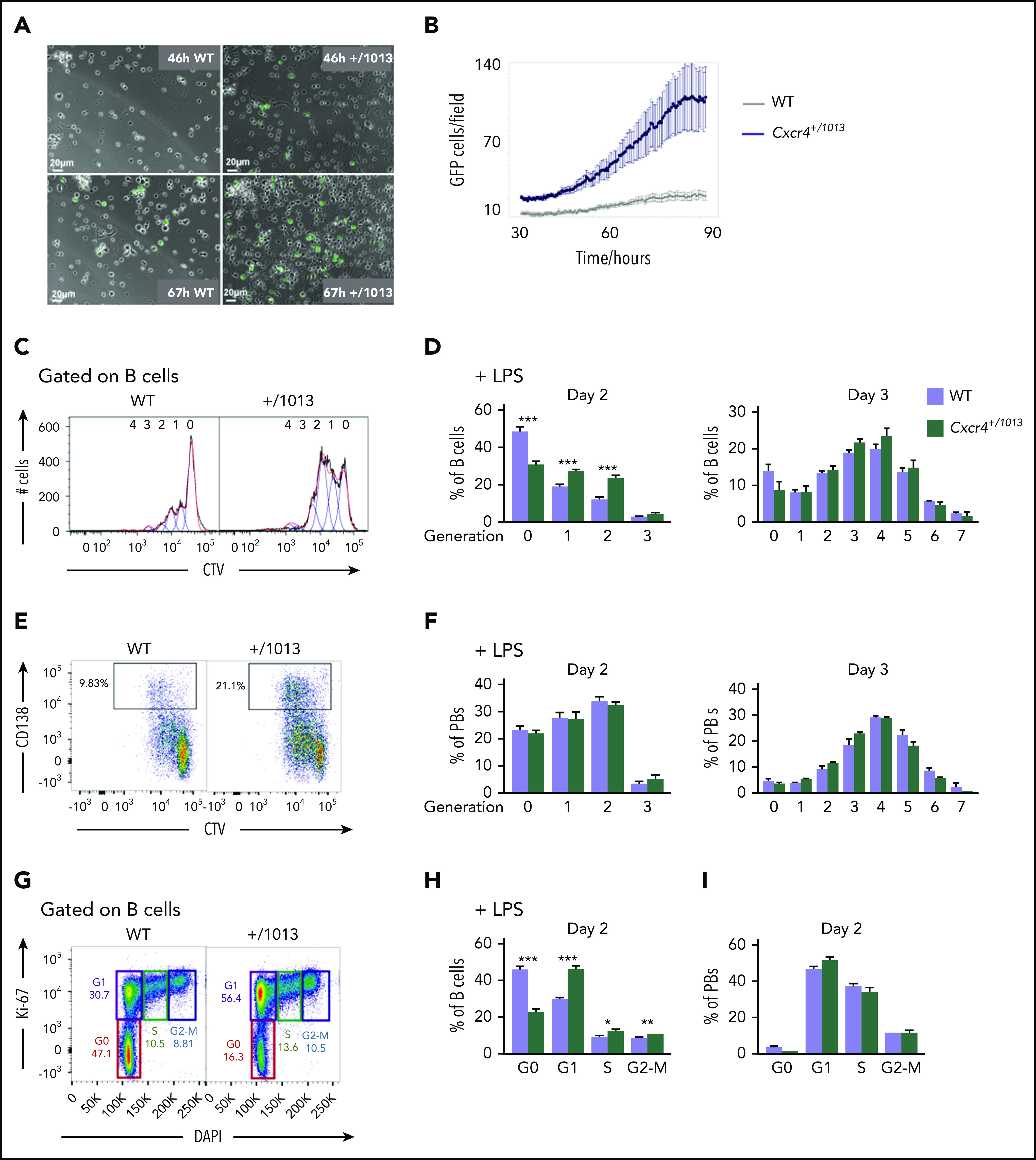
Cxcr4 desensitization limits the entry of B cells into cycle. (A) Representative fields from time-lapse imaging of splenic B cells from WT X Blimp1-GFP and Cxcr4+/1013 X Blimp1-GFP mice cultured in the presence of LPS. Images were taken using the Biostation at 46 and 67 hours after stimulation. Scale bar: 20 µm. (B) Quantification of GFP+ cells/ imaged fields during LPS stimulation of splenic B cells. GFP fluorescence detection was measured up to 90 hours after stimulation using the Incucyte technology. (C-F) Splenocytes from WT and Cxcr4+/1013 mice were loaded with CTV and cultured in presence of LPS for 3 days. (C) Representative histograms for CTV dilution in LPS stimulated B cells at day 2. (D) Frequency of B cells present in each CTV dilution generation at days 2 and 3. (E) Representative dot plots for CTV dilution during PB (CD138+) differentiation at day 2 after LPS stimulation. (F) Frequency of PBs present in each CTV dilution generation at days 2 and 3. (G) Representative plots showing the different phases of the cell cycle (G0, G1, S, G2-M) for splenic B cells at day 2 after LPS stimulation stained with DAPI and for Ki-67. (H-I) Frequency of splenic B cells and PBs in each cell cycle phase at day 2 after LPS stimulation. Results are from 2 independent experiments (C-I) or 1 representative experiment of 2 (A-B) (mean ± SEM, n = 2-7 for C-H; n = 2 for A-B). Mann-Whitney U test was used to assess statistical significance (*P < .05, **P < .01, ***P < .001).
Exacerbated mTORC1 signaling in Cxcr4+/1013 mutant B cells
A wealth of literature reports that, on TLR crosslinking, B-cell activation and proliferation are dependent on phosphatidylinositol 3-kinase activation and downstream Akt and mechanistic target of rapamycin (mTOR) signaling.26-29 Moreover, several works including ours showed that crosslinking of mutant Cxcr4 on B cells promoted aberrant activation of the Akt and mTORC1 pathways.7,30,31 We confirmed ex vivo that the gain of function of Cxcr4 led to an exacerbated Akt phosphorylation on Cxcl12 stimulation in the presence or absence of TLR crosslinking. However, inhibition of Akt did not impact B cell cycle entry in this system (Figure 2H; supplemental Figure 3C). In parallel, we observed that Cxcl12 stimulation led to a transient increase of the mTORC1 target pS6 in B cells, whereas LPS stimulation promoted strong and sustained S6 activation. Importantly, pS6 signaling was significantly increased in mutant B cells compared with WT controls in all conditions and that was even more marked when both Cxcl12 and LPS were used conjointly (Figure 3A-B). Moreover, AMD3100 preincubation was reducing the frequency of pS6+ mutant B cells after Cxcl12 stimulation, hence supporting the role of Cxcr4 gain of function in this process (Figure 3B). These results suggest that TLR4 and the mutant Cxcr4 synergize to promote exacerbated and sustained mTORC1 signaling in Cxcr4+/1013 B cells. Furthermore, treatment with the mTORC1 inhibitor rapamycin significantly reduced LPS-induced cell cycle entry in WT and Cxcr4+/1013 B cells (Figure 3C). In line with this, when rapamycin was added, mutant B-cell differentiation was down to the level observed for untreated WT B cells (Figure 3D). Hence, our data suggest that mutant Cxcr4 and TLR4 signaling synergize to promote stronger mTORC1 signaling, leading to increased B cell entry into cycle and subsequently enhanced PB differentiation.
Figure 3.
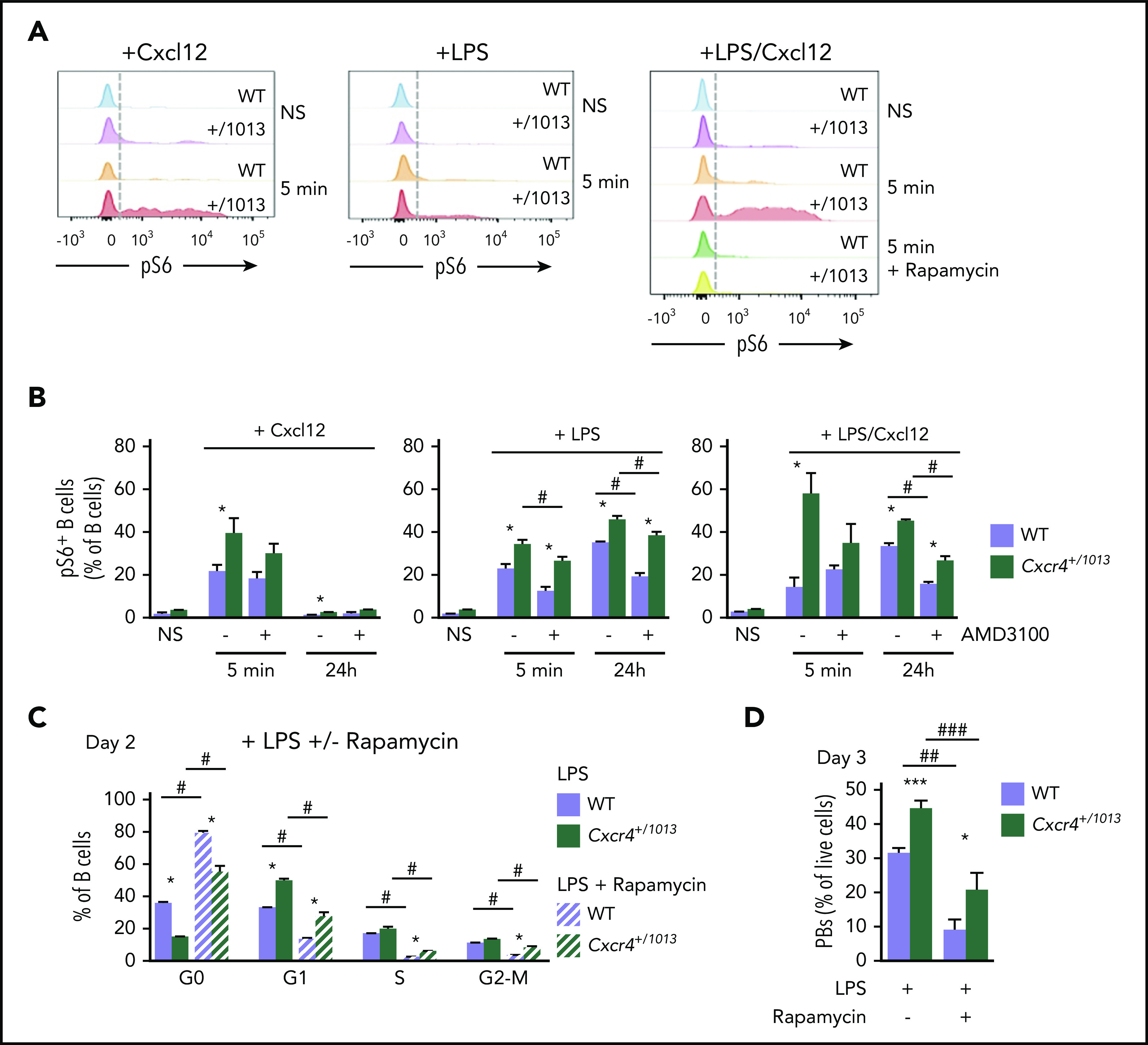
Exacerbated mTORC1 signaling in Cxcr4+/1013mutant B cells. (A) Representative histograms for pS6 in splenic B cells nonstimulated (NS) or stimulated for 5 minutes with Cxcl12, LPS, or a combination of both. The impact of rapamycin on LPS+Cxcl12 stimulated cells is also shown. (B) Quantification of the frequency of pS6+ B cells among splenocytes nonstimulated or stimulated for 5 minutes or 24 hours with Cxcl12, LPS, or a combination of both in the presence or absence of AMD3100. (C) Frequency of splenic B cells in each cell cycle phase at day 2 after LPS stimulation in the presence or absence of rapamycin. (D) Frequency of PBs determined by FACS 3 days after LPS stimulation in presence or in absence of rapamycin. Results are from 1 representative experiment of 2 to 3 (A-C) or 2 pooled experiments (D) (mean ± SEM, n = 4 for A-C; n = 6-18 for D). Mann-Whitney U test was used to assess statistical significance (WT vs Cxcr4+/1013: *P < .05, **P < .01; untreated vs AMD3100: #P < .05 [3B]; LPS alone vs LPS + rapamycin: #P < .05, ##P < .01 [3C-D]).
Fine-tuning of Cxcr4 signaling regulates plasmablast settling within the BM
The T-independent extrafollicular B-cell response mainly leads to the generation of short-lived PBs, although it has been reported that some may be able to home to and persist in the BM.32-34 We therefore investigated whether the enhanced extrafollicular response observed in the spleen in Cxcr4+/1013 mice was mirrored by changes in BM homing. After NP-LPS immunization, we observed an increased detection of total IgM+ ASCs from day 3 and of NP-specific IgM+ ASCs from day 6 in the BM of Cxcr4+/1013 X Blimp1-GFP mice compared with WT mice (Figure 4A-D). This was associated with a fivefold increase in PBs (Blimp1-GFPhi CD138+) in the BM of mutant mice compared with WT mice from day 3 after immunization (Figure 4E). Moreover, fully differentiated PCs (Blimp1-GFPlow CD138+) were significantly increased from day 6 in the BM of the Cxcr4+/1013 mice compared with their WT littermates (Figure 4E). Consistently, we enumerated in situ significantly more IgM+ PB/PCs per mm2 at day 6 after NP-LPS immunization in the BM sections of Cxcr4+/1013 mice compared with WT mice (Figure 4F-G). Thus, Cxcr4 desensitization is essential to limit BM accumulation of PBs generated through the extrafollicular response.
Figure 4.
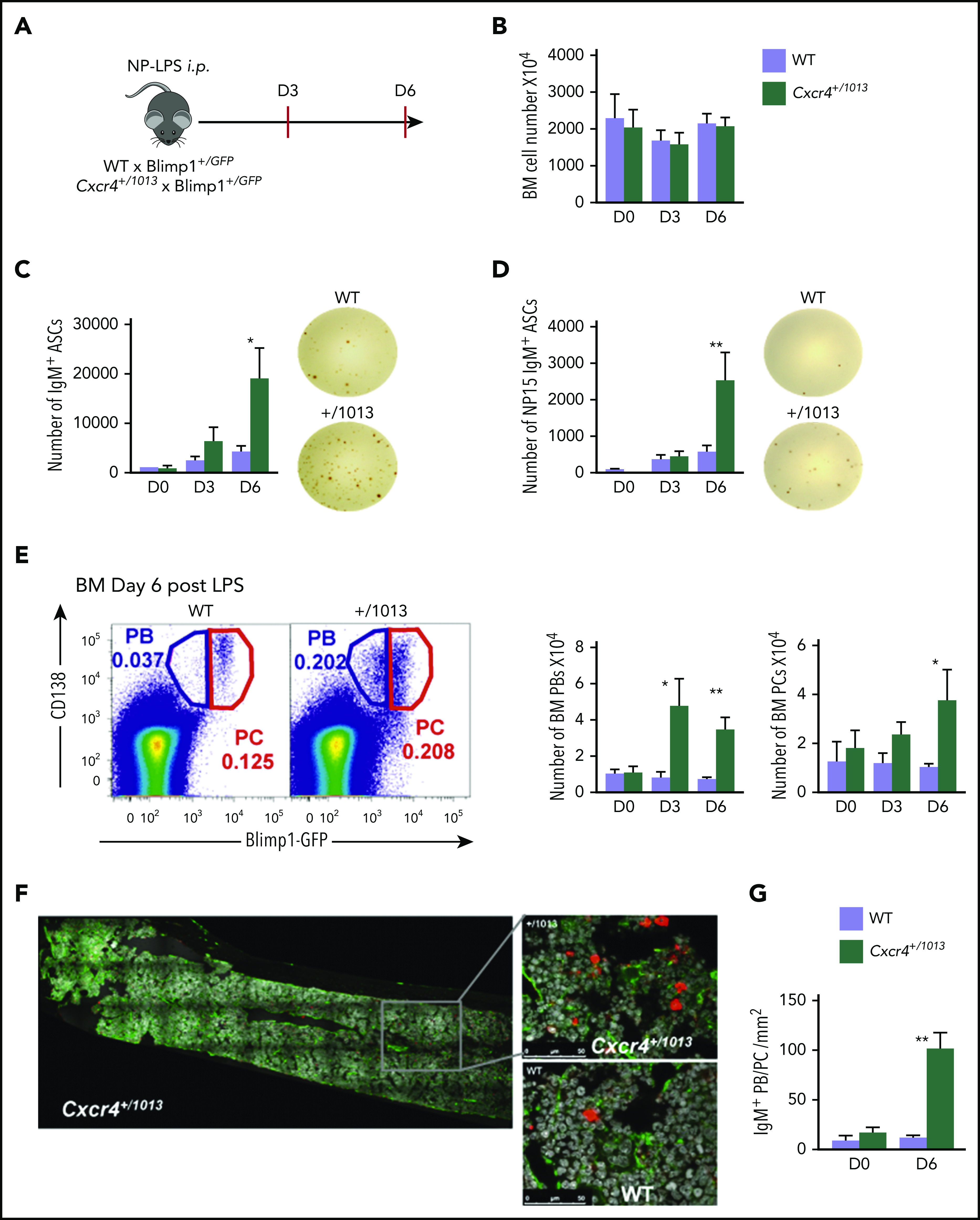
Fine-tuning of Cxcr4 signaling is required to limit plasmablast accumulation within the BM. (A) Schematic diagram describing the immunization protocol. Blimp1-GFP expression was used to distinguish PBs (Blimp1-GFPlow CD138+) from PCs (Blimp1-GFPhi CD138+). (B) Total BM cellularity of both WT and Cxcr4+/1013 mice during NP-LPS immunization. (C-D) Quantification and representative spots for total IgM+ ASCs (C) and NP-specific IgM+ ASCs (D) in the BM determined by ELISpot at days 0, 3, and 6 after immunization. (E) Representative dot plots and total cell numbers for PCs and PBs in the BM of both WT and Cxcr4+/1013 mice at days 0, 3, and 6 after immunization. (F) Representative images of IgM+ PB/PC staining in BM sections (femur) from WT and Cxcr4+/1013 mice at day 6 after immunization. Whole BM imaging is shown only for Cxcr4+/1013 mice. Sections were stained with Ab against laminin (green) and IgM (red), and nuclei were counterstained with Hoechst 33342 (gray). Scale bars: 50 µm. (G) Quantification of the number of IgM+ PB/PC per mm2 fields at days 0 and 6 after immunization. Results are from 2 to 4 independent experiments (mean ± SEM, n = 5-9 [A-E] or n = 3 [G]). Mann-Whitney U test was used to assess statistical significance (*P < .05, **P < .01).
Plasmablasts spontaneously accumulate in the BM in the absence of Cxcr4 desensitization in mice and humans
Previous reports have suggested that PB/PCs increase with age in mice.35 Accordingly, 1-year-old middle-aged Cxcr4+/1013 mice display a very significant increase in the frequency and number of PBs compared with WT mice at steady state that was particularly marked for BM PBs (Figure 5A-D). Thus, the spontaneous and ongoing immune response observed in mice as they age appears exacerbated by the Cxcr4 gain-of-function mutation, and this translates into an abberant accumulation of PBs within the BM.
Figure 5.
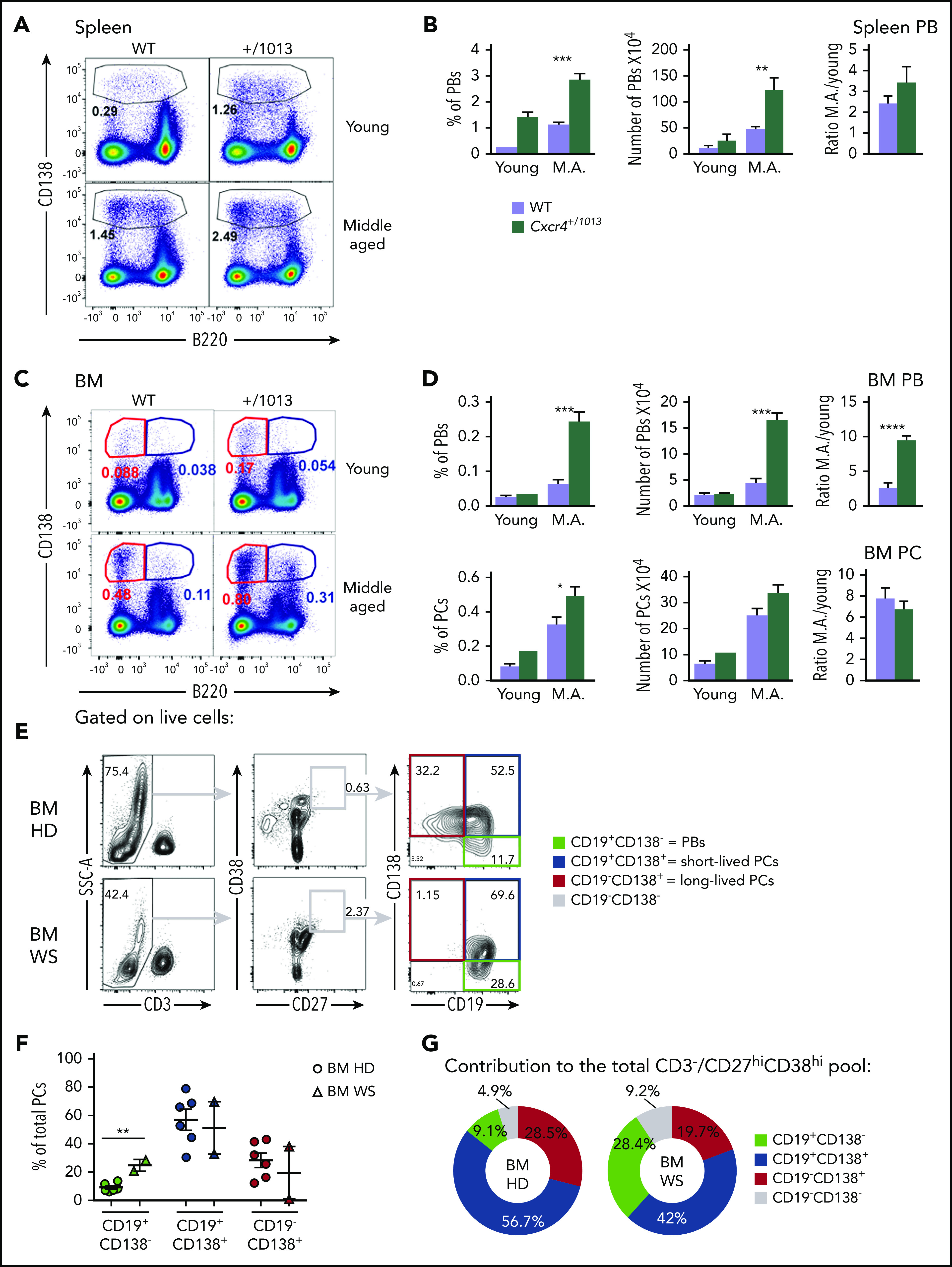
Plasmablasts spontaneously accumulate in the BM in the absence of Cxcr4 desensitization in mice and humans. (A) Representative dot plots showing the gating strategy for PBs in the spleen of young and middle-aged (M.A) mice of both genotypes. (B) Frequency and total number of PBs in the spleen of young and M.A. mice. The ratio M.A./young is also represented. (C) Representative dot plots for PBs (B220lo CD138+) and PCs (B220- CD138+) in the BM of young and M.A. mice from both genotypes. (D) Frequency and total number of PBs and PCs in the BM of young and M.A. mice from both genotypes. The ratio M.A./young is also represented. Results are from 2 independent experiments (mean ± SEM, n = 3-12). Mann-Whitney U test was used to assess statistical significance (*P < .05, **P < .01, ***P < .001, ****P < .0001). (E) Representative dot plots for the gating strategy of the different PB/PC subsets in the BM of control healthy donor (HD) and WS patients. Mononuclear cells were gated on FSC-A/SSC-A, doublets, and dead cells were excluded. On the live cells, T cells were excluded, and total PB/PC were gated as CD27hiCD38hi. The 3 PB/PC subsets were gated based on the expression of CD19 and CD138 from the less to the more mature: CD19+CD138− (green box), CD19+CD138+ (blue box), and CD19−CD138+ (red box). (F) Flow cytometry–based quantification of the frequency of the 3 subsets among total PCs for control and WS BM. (G) Pie chart representation of the contribution of each PB/PC subset to the total BM pool for the control and WS samples. The average contribution of the 3 subsets to the total PC pool for control and WS BM is shown. n = 6 for the control BM and n = 2 for the WS BM. Unpaired t test was used to assess statistical significance (**P < .01).
We next investigated whether CXCR4 gain-of-function mutations might also distort the basal distribution of PCs in BM from WS patients. To this end, we compared BM samples from 2 WS patients carrying the heterozygous CXCR4R334X mutation with aged-matched control BMs. When we compared WS and control BMs, we observed that the frequencies of PBs (CD19+CD138−) were significantly increased in patients compared with control BM (Figure 5E-F), with the 2 less mature fractions representing most of the BM PB/PCs in the patient samples (Figure 5G). Altogether, these results suggest that gain-of-function mutations of CXCR4 can be associated with spontaneous aberrant accumulation of PBs within the BM in middle-aged mice and humans.
Cxcr4 desensitization intrinsically regulates splenic plasmablast homing properties
We then explored whether the exacerbated PB accumulation within the BM observed in absence of Cxcr4 desensitization could be accounted for by a cell intrinsic advantage in terms of BM homing. Mutant splenic PBs express slightly but significantly more surface Cxcr4 than their WT counterpart (Figure 6A-B). Moreover, in a transwell-based migration assay, mutant splenic PBs migrated more toward Cxcl12 in vitro than their WT counterparts, and this was abrogated by the addition of AMD3100 (Figure 6C). Moreover, by cotransfer experiments of in vitro–generated PBs loaded with either CTV or CTY and mixed at a 1:1 ratio (Figure 6D-E), we showed that Cxcr4+/1013 mutant PBs had an intrinsic advantage for homing to the BM compared with WT PBs at all time points considered, whereas the reverse was true for the spleen (Figure 6F). This experiment demonstrates that, on top of their enhanced generation in secondary lymphoid organs, mutant PBs also have an intrinsic advantage in terms of BM homing.
Figure 6.
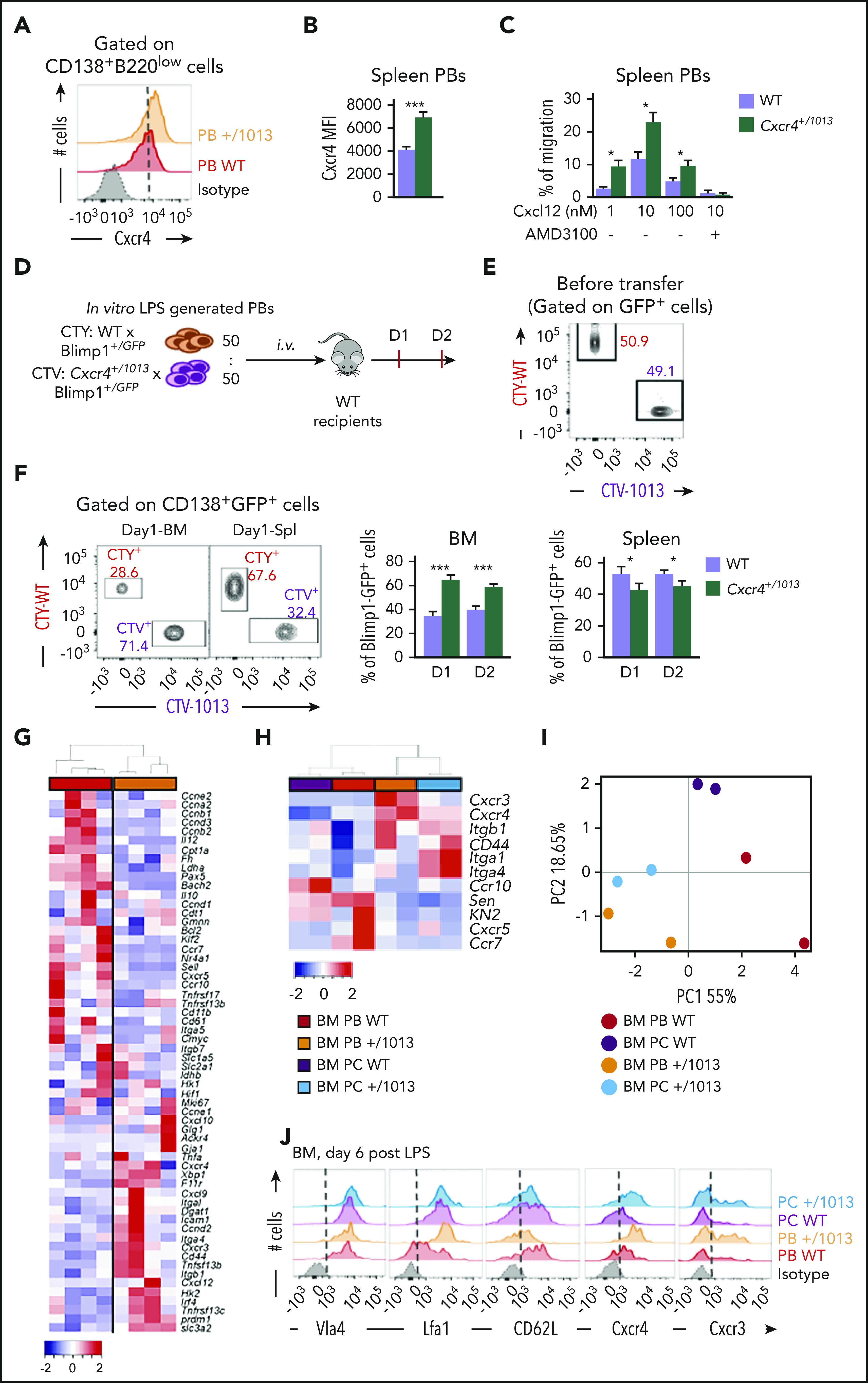
Cxcr4 desensitization intrinsically regulates splenic plasmablast homing properties. (A) Representative FACS plot for Cxcr4 surface expression on splenic PBs. The dashed vertical line is placed at the peak of the Cxcr4 staining on WT PBs. (B) Quantification of the geometrical mean of Cxcr4 at the surface of splenic PBs (MFI: geometrical mean fluorescence intensity). (C) Migration index of splenic PBs to Cxcl12 determined by transwell-based assay. Cxcl12 was added to the lower well at the indicated concentrations with or without AMD3100. (D) Schematic diagram describing the transfer experiment protocol. Blimp1GFP/+ PBs were generated in vitro from splenic B cells and labeled with CTV for Cxcr4+/1013 cells and with CTY for WT cells. Labeled WT and mutant cells were mixed at a 1:1 ratio and transferred into CD45.1 WT recipients. (E) Representative dot plot of the cell mix (1:1) prior transfer. (F) Representative dot plots and frequency of CTV+ (Cxcr4+/1013 × Blimp1GFP/+) and CTY+ (WT × Blimp1GFP/+) cells in the BM and the spleen of recipient mice at days 1 and 2 after transfer. (G) Heatmap showing the relative expression of 60 transcripts from sorted BM PBs from both WT and Cxcr4+/1013 mice determined by Biomark multiplex qPCRs at day 6 after immunization with NP-LPS. Data are presented by applying a column Z score based on (2−ΔΔCt). (H) Heatmap showing the relative expression of 11 selected transcripts from BM PBs and PCs from both WT and Cxcr4+/1013 mice determined by Biomark multiplex qPCRs at day 6 after immunization. Data are presented by applying a column Z score based on (2−ΔΔCt). (I) PCA was performed on BM-PBs and BM-PCs from both groups using a correlation test. (J) Representative histograms for the indicated proteins from BM PBs and PCs from both WT and Cxcr4+/1013 mice are shown. Results are from 2 to 3 independent experiments (mean ± SEM, n = 4-10). Mann-Whitney U test was used to assess statistical significance (*P ≤ .05, **P < .01, ***P < .001).
Transcriptional profiling of WT and Cxcr4+/1013 BM PBs revealed significant differences with a notable alteration in the expression of several genes involved in cell adhesion and migration (Figure 6G). We thus further assessed the impact of Cxcr4 signaling on the migratory and adhesion properties of BM PBs and PCs. We showed that mutant PBs and PCs had a unique transcriptional profile characterized by increased expression of Cxcr4, Cxcr3, Itgal, Cd44, and Itgb1 and decreased expression of Ccr7, Cxcr5, Sell, and Klf2 compared with WT PBs and PCs (Figure 6H). These differences were sufficient to differentially cluster WT and Cxcr4+/1013 BM PB/PCs in a PCA (Figure 6I). Moreover, these altered expressions were confirmed by classical reverse transcriptase-qPCR (supplemental Figure 4A-C) and flow cytometry (Figure 6J; supplemental Figure 4D-E). In summary, our data support that Cxcr4 desensitization is a key control mechanism that intrinsically limits the homing of PBs to the BM, and that once there, this gain of function may confer them with specific localization and/or cell-cell interaction potential.
Cxcr4 desensitization is essential to limit plasmablast maturation and persistence within the BM
We last questioned the fate of these aberrant PBs within the BM. The number of PBs in the BM of Cxcr4+/1013 mice peaked at day 6 and then rapidly declined and was back to the baseline WT level 12 days after NP-LPS immunization (Figure 7A). In parallel, the number of more mature B220− PCs progressively and markedly increased in the BM of Cxcr4+/1013 mice, reaching a peak between 8 and 12 days after immunization, and then slowly declined (Figure 7A). The distinct kinetics for PBs and PCs suggest that PBs reaching the BM of Cxcr4+/1013 mice may be able to differentiate into PCs. We exploited our transfer experiments (Figure 6D) and observed that at day 1 in the BM, most of the transferred cells were found in the PB gate (CD138hiB220+), and very few were detected within the PC gate (CD138hiB220−; Figure 7B). At day 2, however, most of the transferred cells were detected among the CD138hiB220− PCs (Figure 7B), and approximately 80% of them came from the Cxcr4+/1013 cells, whereas the reverse was true for PBs (Figure 7C). In addition, we analyzed the expression of key developmental factors of PCs and observed that Cxcr4+/1013 BM PBs had a transcriptional profile corresponding to more mature cells than their WT counterparts, albeit less mature than fully differentiated PCs, and characterized by decreased expression of the B cell transcription factors Pax5 and Bach2 and enhanced expression of the PC transcription factors Prdm1, Xbp1, and Irf4 (Figure 7D). Finally, we observed that total and NP-specific IgM+ ASCs were increased in the BM of Cxcr4+/1013 mice compared with the WT ones, and this increase was still observed 45 days after immunization (Figure 7E). Thus, Cxcr4+/1013 PBs may differentiate into PCs and persist for more than 1 month in the BM. Altogether, our results demonstrate that Cxcr4 desensitization limits both PB maturation into PCs and their persistence within the BM.
Figure 7.
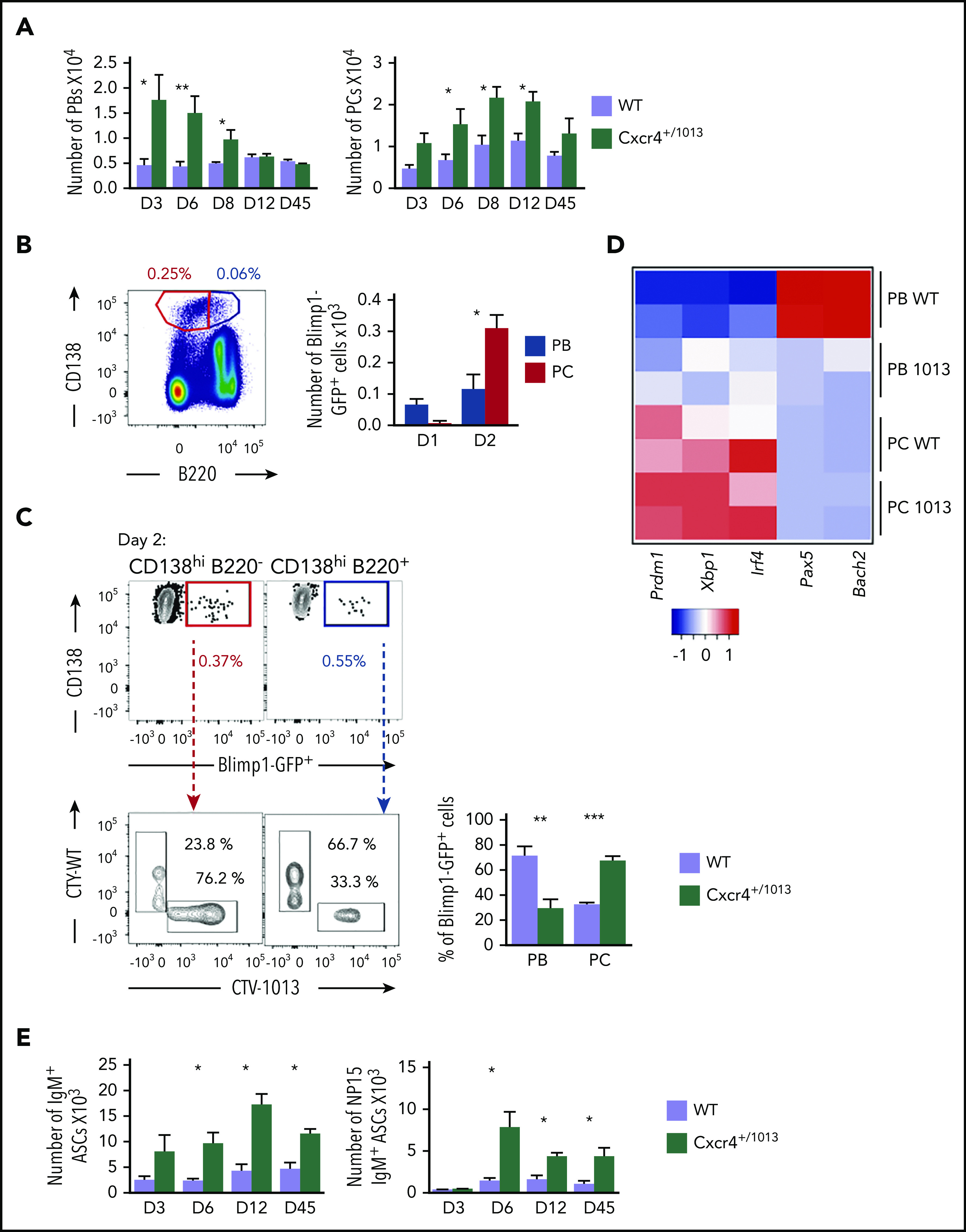
Cxcr4 desensitization is essential to limit plasmablast maturation and persistence within the BM. (A) PB (B220loCD138+) and PC (B220−CD138+) numbers in the BM of both WT or Cxcr4+/1013 mice after NP-LPS immunization at the indicated time points. (B) Blimp1GFP/+ PBs were generated in vitro from splenic B cells and labeled with CTV for Cxcr4+/1013 cells and with CTY for WT cells as previously described in Figure 6D and transferred into WT recipient mice. Representative dot plots of PC (red) and PB (blue) gating and total number of transferred Blimp1GFP/+ PBs and PCs in the BM of recipient mice at days 1 and 2 after transfer. (C) Representative dot plots and quantification of the frequency of CTV+ (Cxcr4+/1013 × Blimp1-GFP) and CTY+ (WT × Blimp1-GFP) cells in both the PC (CD138+B220−) and PB (CD138+B220low) compartments in recipient mice at day 2 after transfer. (D) Heatmap showing the relative expression of B and PC master regulator genes presented as (2−ΔΔCt) in BM PCs and PBs from both genotypes at day 6 after NP-LPS immunization. Data are presented by applying a column Z score. (E) Number of total ASCs IgM+ and NP15-IgM+ in the BM assessed by ELIspot at the indicated time points after NP-LPS immunization. Results are from at least 2 representative experiments (A-D,E) or from 2 independent experiments (C) (mean ± SEM, n = 2-9). Mann-Whitney U test was used to assess statistical significance (*P < .05, **P < .01, ***P < .001).
Discussion
Despite its essential role in host protection, how the extrafollicular immune response is regulated remains mysterious. Here we demonstrate that Cxcr4 desensitization is an essential regulatory mechanism controlling the duration and the strength of Ab production after induction of a T-independent immune response by finely modulating the pool of extrafollicular-derived PCs.
Our data support a critical role for Cxcr4 desensitization during this process at multiple checkpoints of B-cell activation and PC differentiation. First, we observed that impaired Cxcr4 desensitization led to increased TLR-driven B-cell differentiation into PB with, as consequences, more PBs generated and increased IgM production in the course of an extrafollicular response. This is associated with enhanced entry into cycle and is consistent with previous studies that have linked B-cell division to their capacity to differentiate into PCs.25,36,37 In addition, we showed that Cxcr4 gain of function following Cxcl12 stimulation and in synergy with TLR signaling promoted induction of the mTORC1 pathway that drives cell cycle entry and PB differentiation.
Second, PBs and PCs bearing the Cxcr4 gain-of-function mutation display an enhanced BM tropism that may not only be caused by the gain of function of Cxcr4 per se as the expression of a whole range of migration/adhesion factors are modified in PBs in Cxcr4+/1013 mice, including integrins that can be activated by Cxcr4 signaling.38,39
Third, differentiation of PBs into PCs and their persistence are enhanced in the BM of Cxcr4+/1013 mice compared with WT mice. This could be through direct effects of the enhanced Cxcr4 signaling on the PC maturation transcriptional program. Alternatively or conjointly, different environmental cues specific to mutant PB/PCs and linked with their altered adhesion/migration potential might contribute to this phenomenon. Importantly, some insights gained from the study of a Cxcr4 gain-of-function mouse model were validated in the BM of WS patients in whom we observed an over-representation of PBs.
Conceptually our results suggest an important role for Cxcr4 signaling in limiting both the number of PBs generated and the lapse of time during which Abs generated in the course of a TLR-dependent extrafollicular response are produced. Indeed, Cxcr4 desensitization not only regulates the early generation of PBs in secondary lymphoid organs after immunization but also their homing to and maturation within the BM, hence ensuring that only a few PCs generated during this response persist in the BM. As such, Cxcr4 desensitization seems to act as a gatekeeper, ensuring that the Abs produced by PBs during the extrafollicular response are restricted to a transient wave of protection. Limiting PB homing to the BM in this way may also advert the filling of BM niches normally dedicated to other cell types, including GC-derived high affinity PCs that start arising around day 9 after immunization.2 Thus, our results, taken together with our previously published data,7 suggest that defective long-term vaccine protection, previously reported in WS patients,12,22,23 may be the consequence of defective PC homeostasis with an over-representation of low-affinity extrafollicular PBs and reduced contribution to the humoral response of GC-derived high affinity PCs.
Somatic CXCR4 mutations are observed in ∼30% of Waldenström macroglobulinemia patients, most of the time in conjunction with a gain-of-function mutation of the TLR adaptor MYD88,16,17,40,41 and the presence of the 2 mutations is associated with shorter time to treatment and earlier progression with ibrutinib.13,15,31,42 Our data thus support that CXCR4 nonsense mutations leading to premature stop codon may promote BM homing of malignant cells and niche-mediated therapy resistance previously reported in this disease.43,44 Moreover, they suggest the paradigm-shifting hypothesis of a potential CXCR4-dependent extrafollicular origin for Waldenström macroglobulinemia malignant clones. Whether CXCR4 frameshift mutations, also observed in some Waldenström macroglobulinemia patients, may have similar impact remains to be evaluated.45,46
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank M.-L. Aknin, H. Gary, F. Gaudin, V. Nicolas, B. Lecomte (Ingénierie et Plateformes au Service de l’Innovation Thérapeutique, technical facilities Plateforme de Cytométrie en Flux [PLAIMMO], Plateforme d’Histologie Expérimentale [PHIC], Plateforme d'Imagerie Cellulaire [MIPSIT] and AnimEx, Clamart), J. Megret, C. Cordier (Flow Cytometry Core Facility, Structure Fédérative de Recherche Necker, Paris, France), P. Chappert, A. Vandenberghe (Institut Necker-Enfants Malades, Paris, France), C. Doliger, and S. Duchez (Flow Cytometry Core Facility, Institut de Recherche Saint Louis, Paris, France) for technical assistance and M.A. Linterman, S. Luther, and S. Peebles for comments on our manuscript.
The study was supported by the Laboratory of Excellence in Research on Medication and Innovative Therapeutics (LabEx LERMIT) (M.E. and K.B.); an Agence Nationale de la Recherche (ANR) Jeunes Chercheurs/Jeunes Chercheuses grant (ANR-19-CE15-0019-01), an ANR @RAction grant (ANR-14-ACHN-0008), a Fondation Arthritis grant, a grant from IdEx Université de Paris (ANR-18-IDEX-0001), and a “Fondation ARC pour la recherche sur le cancer” grant (P JA20181208173) (M.E.); and an ANR Projets de Recherche Collaborative grant (ANR-17-CE14-0019), an Institut National du Cancer grant (PRT-K 2017), and the Association Saint Louis pour la Recherche sur les Leucémies (K.B.). D.H.M and P.M.M. are supported by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases, National Institutes of Health. N.A. and M.K. were supported by a fellowship from the French Ministry for education and by a fourth-year fellowship from the “Fondation ARC pour la recherche sur le cancer” (N.A.). V.R. was supported by a fellowship from the Fondation pour la Recherche Médicale and a fourth-year fellowship from the Ligue Nationale Contre le Cancer. R.H.-A. was supported by a master fellowship from the “Fondation ARC pour la recherche sur le cancer.” The EMiLy U1160 INSERM unit is a member of the OPALE Carnot Institute, The Organization for Partnerships in Leukemia.
Footnotes
All data are available from the corresponding author upon reasonable request.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: N.A. designed and performed experiments, analyzed data, and wrote the manuscript; A.B. performed experiments, analyzed data, and wrote the manuscript; V.R. and R.H.-A. performed experiments and analyzed data; J.N., V.B., M.K., E.C., N.S., N.D., and M.M. helped with experiments; P.M.M. and D.H.M. provided essential human reagents and reviewed the manuscript; K.B. contributed to the project design and to the manuscript redaction; and M.E. designed the project, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Marion Espéli, Institut de Recherche Saint Louis, INSERM U1160, 1 Av Claude Vellefaux, 75010 Paris, France; e-mail: marion.espeli@inserm.fr.
REFERENCES
- 1.MacLennan ICM, Toellner K-MM, Cunningham AF, et al. Extrafollicular antibody responses. Immunol Rev. 2003;194(1):8-18. [DOI] [PubMed] [Google Scholar]
- 2.Weisel FJ, Zuccarino-Catania GV, Chikina M, Shlomchik MJ. A temporal switch in the germinal center determines differential output of memory B and plasma cells. Immunity. 2016;44(1):116-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bannard O, Horton RM, Allen CDC, An J, Nagasawa T, Cyster JG. Germinal center centroblasts transition to a centrocyte phenotype according to a timed program and depend on the dark zone for effective selection [published correction appears in Immunity. 2013;39(6):P1182]. Immunity. 2013;39(5):912-924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allen CDC, Ansel KM, Low C, et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol. 2004;5(9):943-952. [DOI] [PubMed] [Google Scholar]
- 5.Hargreaves DC, Hyman PL, Lu TT, et al. A coordinated change in chemokine responsiveness guides plasma cell movements. J Exp Med. 2001;194(1):45-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nie Y, Waite J, Brewer F, Sunshine M-J, Littman DR, Zou Y-R. The role of CXCR4 in maintaining peripheral B cell compartments and humoral immunity. J Exp Med. 2004;200(9):1145-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biajoux V, Natt J, Freitas C, et al. Efficient plasma cell differentiation and trafficking require Cxcr4 desensitization. Cell Rep. 2016;17(1):193-205. [DOI] [PubMed] [Google Scholar]
- 8.Victora GD, Schwickert TA, Fooksman DR, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell. 2010;143(4):592-605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore CAC, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. 2007;69(1):451-482. [DOI] [PubMed] [Google Scholar]
- 10.Balabanian K, Lagane B, Pablos JL, et al. WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood. 2005;105(6):2449-2457. [DOI] [PubMed] [Google Scholar]
- 11.Kawai T, Choi U, Whiting-Theobald NL, et al. Enhanced function with decreased internalization of carboxy-terminus truncated CXCR4 responsible for WHIM syndrome. Exp Hematol. 2005;33(4):460-468. [DOI] [PubMed] [Google Scholar]
- 12.Gulino AV, Moratto D, Sozzani S, et al. Altered leukocyte response to CXCL12 in patients with warts hypogammaglobulinemia, infections, myelokathexis (WHIM) syndrome. Blood. 2004;104(2):444-452. [DOI] [PubMed] [Google Scholar]
- 13.Treon SP, Cao Y, Xu L, Yang G, Liu X, Hunter ZR. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood. 2014;123(18):2791-2796. [DOI] [PubMed] [Google Scholar]
- 14.Poulain S, Roumier C, Venet-Caillault A, et al. Genomic landscape of CXCR4 mutations in Waldenström macroglobulinemia. Clin Cancer Res. 2016;22(6):1480-1488. [DOI] [PubMed] [Google Scholar]
- 15.Gustine JN, Xu L, Tsakmaklis N, et al. CXCR4 S338X clonality is an important determinant of ibrutinib outcomes in patients with WaldenströmWaldenström macroglobulinemia. Blood Adv. 2019;3(19):2800-2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Treon SP, Xu L, Hunter Z. MYD88 mutations and response to ibrutinib in Waldenström’s macroglobulinemia. N Engl J Med. 2015;373(6):584-586. [DOI] [PubMed] [Google Scholar]
- 17.Leblond V, Kastritis E, Advani R, et al. Treatment recommendations from the Eighth International Workshop on Waldenström’s Macroglobulinemia. Blood. 2016;128(10):1321-1328. [DOI] [PubMed] [Google Scholar]
- 18.Balabanian K, Brotin E, Biajoux V, et al. Proper desensitization of CXCR4 is required for lymphocyte development and peripheral compartmentalization in mice. Blood. 2012;119(24):5722-5730. [DOI] [PubMed] [Google Scholar]
- 19.Kallies A, Hasbold J, Tarlinton DM, et al. Plasma cell ontogeny defined by quantitative changes in blimp-1 expression. J Exp Med. 2004;200(8):967-977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kusumbe AP, Ramasamy SK, Starsichova A, Adams RH. Sample preparation for high-resolution 3D confocal imaging of mouse skeletal tissue. Nat Protoc. 2015;10(12):1904-1914. [DOI] [PubMed] [Google Scholar]
- 21.Thai LH, Le Gallou S, Robbins A, et al. BAFF and CD4+ T cells are major survival factors for long-lived splenic plasma cells in a B-cell-depletion context. Blood. 2018;131(14):1545-1555. [DOI] [PubMed] [Google Scholar]
- 22.Handisurya A, Schellenbacher C, Reininger B, et al. A quadrivalent HPV vaccine induces humoral and cellular immune responses in WHIM immunodeficiency syndrome. Vaccine. 2010;28(30):4837-4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mc Guire PJ, Cunningham-Rundles C, Ochs H, Diaz GA. Oligoclonality, impaired class switch and B-cell memory responses in WHIM syndrome. Clin Immunol. 2010;135(3):412-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hasbold J, Corcoran LM, Tarlinton DM, Tangye SG, Hodgkin PD. Evidence from the generation of immunoglobulin G-secreting cells that stochastic mechanisms regulate lymphocyte differentiation. Nat Immunol. 2004;5(1):55-63. [DOI] [PubMed] [Google Scholar]
- 25.Fairfax KA, Kallies A, Nutt SL, Tarlinton DM. Plasma cell development: from B-cell subsets to long-term survival niches. Semin Immunol. 2008;20(1):49-58. [DOI] [PubMed] [Google Scholar]
- 26.Wicker LS, Boltz RC Jr., Matt V, Nichols EA, Peterson LB, Sigal NH. Suppression of B cell activation by cyclosporin A, FK506 and rapamycin. Eur J Immunol. 1990;20(10):2277-2283. [DOI] [PubMed] [Google Scholar]
- 27.Donahue AC, Fruman DA. Proliferation and survival of activated B cells requires sustained antigen receptor engagement and phosphoinositide 3-kinase activation. J Immunol. 2003;170(12):5851-5860. [DOI] [PubMed] [Google Scholar]
- 28.Yusuf I, Zhu X, Kharas MG, Chen J, Fruman DA. Optimal B-cell proliferation requires phosphoinositide 3-kinase-dependent inactivation of FOXO transcription factors. Blood. 2004;104(3):784-787. [DOI] [PubMed] [Google Scholar]
- 29.Kay JE, Kromwel L, Doe SE, Denyer M. Inhibition of T and B lymphocyte proliferation by rapamycin. Immunology. 1991;72(4):544-549. [PMC free article] [PubMed] [Google Scholar]
- 30.Roselli G, Martini E, Lougaris V, Badolato R, Viola A, Kallikourdis M. CXCL12 mediates aberrant costimulation of B lymphocytes in warts, hypogammaglobulinemia, infections, myelokathexis immunodeficiency. Front Immunol. 2017;8(SEP):1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roccaro AM, Sacco A, Jimenez C, et al. C1013G/CXCR4 acts as a driver mutation of tumor progression and modulator of drug resistance in lymphoplasmacytic lymphoma. Blood. 2014;123(26):4120-4131. [DOI] [PubMed] [Google Scholar]
- 32.Bortnick A, Chernova I, Quinn WJ III, Mugnier M, Cancro MP, Allman D. Long-lived bone marrow plasma cells are induced early in response to T cell-independent or T cell-dependent antigens. J Immunol. 2012;188(11):5389-5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taillardet M, Haffar G, Mondière P, et al. The thymus-independent immunity conferred by a pneumococcal polysaccharide is mediated by long-lived plasma cells. Blood. 2009;114(20):4432-4440. [DOI] [PubMed] [Google Scholar]
- 34.Foote JB, Mahmoud TI, Vale AM, Kearney JF. Long-term maintenance of polysaccharide-specific antibodies by IgM-secreting cells. J Immunol. 2012;188(1):57-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han S, Yang K, Ozen Z, et al. Enhanced differentiation of splenic plasma cells but diminished long-lived high-affinity bone marrow plasma cells in aged mice. J Immunol. 2003;170(3):1267-1273. [DOI] [PubMed] [Google Scholar]
- 36.Tangye SG, Hodgkin PD. Divide and conquer: the importance of cell division in regulating B-cell responses. Immunology. 2004;112(4):509-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barwick BG, Scharer CD, Bally APR, Boss JM. Plasma cell differentiation is coupled to division-dependent DNA hypomethylation and gene regulation. Nat Immunol. 2016;17(10):1216-1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glodek AM, Honczarenko M, Le Y, Campbell JJ, Silberstein LE. Sustained activation of cell adhesion is a differentially regulated process in B lymphopoiesis. J Exp Med. 2003;197(4):461-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petty JM, Lenox CC, Weiss DJ, Poynter ME, Suratt BT. Crosstalk between CXCR4/stromal derived factor-1 and VLA-4/VCAM-1 pathways regulates neutrophil retention in the bone marrow. J Immunol. 2009;182(1):604-612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hunter ZR, Xu L, Tsakmaklis N, et al. Insights into the genomic landscape of MYD88 wild-type Waldenström macroglobulinemia. Blood Adv. 2018;2(21):2937-2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cao X, Meng Q, Cai H, et al. Detection of MYD88 L265P and WHIM-like CXCR4 mutation in patients with IgM monoclonal gammopathy related disease. Ann Hematol. 2017;96(6):971-976. [DOI] [PubMed] [Google Scholar]
- 42.Cao Y, Hunter ZR, Liu X, et al. The WHIM-like CXCR4(S338X) somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom’s macroglobulinemia. Leukemia. 2015;29(1):169-176. [DOI] [PubMed] [Google Scholar]
- 43.Jalali S, Ansell SM. Bone marrow microenvironment in Waldenstrom’s Macroglobulinemia. Best Pract Res Clin Haematol. 2016;29(2):148-155. [DOI] [PubMed] [Google Scholar]
- 44.Ghobrial IM, Maiso P, Azab A, et al. The bone marrow microenvironment in waldenstrom macroglobulinemia. Ther Adv Hematol. 2011;2(4):267-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Castillo JJ, Xu L, Gustine JN, et al. CXCR4 mutation subtypes impact response and survival outcomes in patients with Waldenström macroglobulinaemia treated with ibrutinib. Br J Haematol. 2019;187(3):356-363. [DOI] [PubMed] [Google Scholar]
- 46.Hunter ZR, Xu L, Yang G, et al. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014;123(11):1637-1646. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.