

Abstract
Glypican-1 and its heparan sulfate (HS) chains play important roles in modulating many biological processes including growth factor signaling. Glypican-1 is bound to a membrane surface via a glycosylphosphatidylinositol (GPI)-anchor. In this study, we used all-atom molecular modeling and simulation to explore the structure, dynamics, and interactions of GPI-anchored glypican-1, three HS chains, membranes, and ions. The folded glypican-1 core structure is stable, but has substantial degrees of freedom in terms of movement and orientation with respect to the membrane due to the long unstructured C-terminal region linking the core to the GPI-anchor. With unique structural features depending on the extent of sulfation, high flexibility of HS chains can promote multi-site interactions with surrounding molecules near and above the membrane. This study is a first step toward all-atom molecular modeling and simulation of the glycocalyx, as well as its modulation of interactions between growth factors and their receptors.
Keywords: heparan sulfates, glycosaminoglycan, glycocalyx, molecular dynamics simulation, glycosylphosphatidylinositol
INTRODUCTION
Heparan sulfate proteoglycans (HSPGs) are proteins with heparin-like glycosaminoglycan (GAG) chains that can be found on cell surfaces and in extracellular matrices. Evidence supporting their roles in modulating growth factor signaling has accumulated for more than 30 years. Specific interactions between HS chain and growth factors have been meticulously studied (Li et al. 2016, Schlessinger et al. 2000). Membrane-associated HSPGs are either transmembrane proteins (syndecans) or glycosylphosphatidylinositol (GPI)-linked proteins (glypicans) (Kramer and Yost 2003). The extended HS chains have been proposed to serve as co-receptors that bind both growth factor receptors and their ligands, a hypothesis further
supported by studies with free heparin or HS chains (Bernfield et al. 1999). Glypicans and syndecans are found across a wide range of organisms. In drosophila, zebrafish, and xenopus, missing these proteins results in specific phenotypes that indicate their significant roles in growth and development. For example, HSPG facilitates growth factor receptor binding and internalization, which modulate the signal transduction (Li et al. 2016, Schlessinger et al. 2000). Recent studies have identified changes in specific glypican expression levels in many different cancer types, indicating their key roles in the growth factor interactions and in many types of cancer (Wang et al. 2019).
In addition to modulating growth factor signaling, specific HSPGs appear to have additional roles. For example, syndecan-4 interacts with the cytoskeleton for cytoplasmic signaling and is critical for cell migration during development of multiple tissues (Morgan et al. 2013). Syndecan-1 and glypican-1 appear to be important in endothelial responses to fluid shear stress (Zeng et al. 2018) that control nitric oxide production (Ebong et al. 2014, Svensson et al. 2012, Tarbell et al. 2014). Syndecans are required for the 24-hr realignment of endothelial cells in response to flow (Ebong et al. 2014). In addition, HSPGs appear to be important in development of synaptic specificity (Condomitti and de Wit 2018).
HS chains are composed of repeating units of disaccharides that vary in length between tissues (Shi and Zaia 2009) and are linked to the proteins with a short linkage region. The sulfate groups are attached in the Golgi by specific enzymes, resulting in HS chains with regions of varying sulfation (Carlsson et al. 2008). Despite similarly modified chains, differences in HS chain length may exist between different proteins in a single cell type (Tumova et al. 2000). Studies of free HS chains indicate some flexibility in the longer chains (Khan et al. 2013). HSPGs on the surfaces of most cell types contribute 50 to 90% of GAGs in the glycocalyx that surrounds single cells in culture (Tarbell et al. 2014), and similar glycocalyx complexes have been studied on the apical surfaces of epithelial and endothelial cells. Besides the HSPGs, the glycocalyx contains chondroitin sulfate chains, hyaluronic acid, and glycoproteins. These components are tissue specific, and the glycocalyx may range between 500 nm and tens of micrometers (Ebong et al. 2011), extending the reach of cells away from the plasma membrane. Intriguingly, many viruses associate with HS chains or other glycocalyx components during the process of initial viral attachment to the cells. Herpes simplex virus I even uses HSPGs to gain internalization (Cagno et al. 2019).
In humans, there are six glypicans divided into two subgroups and four syndecans (type I membrane proteins that connect to the cytoplasm). The best studied glypican is glypican-1, and its N-terminal well-folded core domain structure has been determined to be a cylinder-like shape (Svensson et al. 2012). A disordered C-terminal domain contains the attachment sites of three closely spaced HS (close to the folded core) and the GPI-anchor (at the C-terminal end). GPI-linked proteins have more freedom than transmembrane proteins to move in the plane of the membrane due to the lack of cytoskeletal links (Svensson et al. 2012). However, the HS chains of both HSPG types have been determined to interact with growth factor receptors and other proteins, which may limit their mobility.
Despite some structural information for HS chains and HSPG proteins, our current understanding of their function is largely based on physiological outcomes. More specific structural and dynamic information about HSPG proteins and the HS chains is required to facilitate a better understanding of their functions. This knowledge can be obtained from all-atom simulations of the protein, associated HS chains, and the surrounding membrane. The utility of this approach is demonstrated by a recent investigation showing that syndecan-4, the HSPG required for elongation and alignment of endothelial cells with flow, undergoes conformational changes and mediates the transmission of shear forces to the cell surface (Jiang et al. 2017).
In this work, we have performed all-atom molecular modeling and simulation of GPI-anchored glypican-1 with three HS chains in a lipid bilayer in order to explore the landscape of their possible structure, dynamics, and interactions. We observe high stability of the folded glypican-1 core, and find that the flexible unstructured C-terminal domain linking the core to the membrane anchor gives the core a large amount of freedom to tilt and rotate relative to the membrane. We also find that the HS chains are highly dynamic with local flexibility related to the degree of sulfation of the chain. The flexibility of the GAGs allows for the possibility of multiple interaction sites; during the simulation, they make contact with the neighboring glypican-1 protein core, other HS chains on the same glypican, HS chains on neighboring glypicans, ions in solution, especially the sulfated residues, and surrounding lipid head groups. This GAG flexibility makes it possible to imagine the interactions of these chains with the growth factor and its receptor complexes, as well as cell adhesion structures, viruses, and other players. The simulated system also provides opportunities to begin exploring some of these interactions at the molecular level.
RESULTS AND DISCUSSION
Orientations of the glypican-1 core domain and the C-terminal domain are highly dynamic
To gain insights into the structure, dynamics, and interactions of GPI-anchored glypican-1 carrying three HS chains in a membrane bilayer, we performed 1-µs all-atom molecular dynamics (MD) simulations with three independent replicas. The model structures of the membrane along with N-glycans, HS chains, and GPI-anchor are presented in Figure 1A and 1B. The N-glycan sequences at Asn79 and Asn116 (Figure 1C) were chosen according to the previous experimental work (Awad et al. 2015). A single glypican-1 protein was modeled with the missing unstructured C-terminal domain (Asn474-Ser530) and three HS chains of the same structure (Figure 1D) located at Ser486, Ser488, and Ser490 (Carlsson et al. 2008). The HS sulfation patterns are complicated and cell/tissue specific rather than being specific for the protein cores. Therefore, in our work, we simply followed the HS sequence in reference (Carlsson et al. 2008). The GPI anchor sequence is shown in Figure 1E. A detailed description of the model structures and procedures of system preparation are presented in the Methods section.
Fig. 1.

(A) Snapshot of the glypican-1 system: glypican-1 protein in green, N-glycans in purple sticks, heparan sulfates in sticks with HS486 in red, HS488 in blue, and HS490 in yellow, GPI-anchors at both leaflets in magenta sticks, and membrane bilayers colored as van der Waals spheres with POPC in dark gray, PSM in gray, and Chol in brown. (B) Top view of the glypican-1 system with the primary simulation system in a gray box and surrounding image systems in the X-/Y- directions. (C) N-glycan sequences used in this study: galactose (Gal), N-acetylglucosamine (GlcNAc), mannose (Man), l-fucose (Fuc), and N-acetylneuraminic acid (Neu5Ac). (D) Heparan sulfate sequence in this study: xylose (Xyl), glucuronic acid (GlcA), glucosamine (GlcN), and iduronic acid (IdoA). Sulfation of sugar residues is denoted by the letter ‘S’. (E) GPI-anchor sequence: phosphoethanolamine (EtNP) and myo-inositol 1-phosphate (Ins). Linkages are given by numbers and anomeric configurations by letters, e.g., an α-(1 → 4)-linkage is denoted by ‘14A’ and a β-(1 → 4)-linkage is denoted by ‘14B’.
To investigate the orientation of glypican-1 in a bilayer, tilt (α) and rotation (β) angles of the glypican-1 core domain, as well as tilt angle (ω) of the unstructured C-terminal domain, are characterized (Figure 2A). Part of helices (Glu75-Arg90) and two residues (Ala83 and Pro244) located on one side of the protein were selected to describe α and β. A principal vector (⇀p1) of Glu75-Arg90 was used to define α as the angle between ⇀p1 and ⇀z (unit vector in the Z direction). A vector (⇀p2) from Ala83 to Pro244 was used to define β as the angle between the normal vector (⇀n2) formed by ⇀p1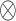 ⇀p2 and ⇀z. The angle ω and the end-to-end distance (DEE) of the C-terminus domain were defined using Gly473 and Ser530.
⇀p2 and ⇀z. The angle ω and the end-to-end distance (DEE) of the C-terminus domain were defined using Gly473 and Ser530.
Fig. 2.

(A) Schematic representation of glypican-1 core tilt (α), rotation (β), and C-terminal tilt (ω) angles with respect to the Z axis, i.e., the membrane normal; see the main text for their definition. The selected residues used to define α and β are shown as van der Waals spheres with pink (Glu75-Arg90), green (Ala83), blue (Pro244), red (Gly473), and orange (Ser530). (B–C) α and β angle distributions. (D) Distributions of root mean square deviation (RMSD) of glypican-1 core helical domain. (E–F) End to end distance (between Gly473 and Ser530) and ω angle distributions. Note that each color in (B–F) represents each replica among three replicas: red for replica-1, green for replica-2, and blue for replica-3. (G–I) Snapshots at 300 ns, 100 ns, and 100 ns for (G) replica-1, (H) replica-2, and (I) replica-3.
Figures 2B and 2C show α and β angle distributions. The distributions of α are ranging from 0° to 120° for replica-2 and from 30° to 120° for replica-1 and 3. For β, the angle distributions are ranging from 0° to 180° for all replicas, indicating that the protein structure undergoes large orientational changes with respect to the membrane. To investigate the structure change of the glypican-1 helical core domain, the root-mean-square deviation (RMSD) is employed (Figure 2D). The RMSD values only fluctuate around 1–3 Å for all replicas, indicating that the glypican-1 core domain undergoes large fluctuations in terms of its orientation relative to the membrane while the structure itself remains stable during the simulation time. This is further confirmed by the secondary structure analysis of glypican-1 for all replicas (Figure S1). No significant changes in the secondary structures were observed during the entire simulation time.
It is noteworthy that the core domain rotates to almost parallel to the membrane normal with the helical vector ⇀p1 pointing upward for replica-1, making α small (14°). On the other hand, for replica-2 and replica-3 with ⇀p1 pointing downward, the α values become very large (~150° for replica-2 and ~ 163° for replica-3). Glypican-1 has a long unstructured C-terminal domain (i.e., Asn474-Ser530), such that the core domain can be located quite far away from the GPI-anchored point, especially when the C-terminal region is extended. Figures 2E and 2F show the dynamics of the C-terminal domain, represented by DEE and ω. To illustrate the magnitude of the fluctuations, snapshots were taken from each replica at 300 ns (Figure 2G) and 100 ns (Figure 2H-I). DEE fluctuates in a range of 20–70 Å in replica-1 (red in Figure 2E and a snapshot in Figure 2G). In Figure 2G, it is evident that the entire core protein with the C-terminal domain extended (DEE ~ 70 Å) moves to one side of the membrane while the GPI anchor stays near the center. On the other hand, by the end of 100 ns of replica-3 (blue in Figure 2E and a snapshot in Figure 2I), DEE is relatively short and close to the initial value. Accordingly, in Figure 2I, the glypican-1 core domain does not move far away from the GPI-anchored point, compared to the starting position (Figure 1A). In Figure 2F, ω shows a large fluctuation between 0° to 90°. Replica-1 in Figure 2G has a ω value of around 70° whereas ω of replica-3 in Figure 2I is only around 10° (see also Supplemental Movie S1-S3 for snapshots of each replica). Svensson et al. (Svensson et al. 2012) predicted that this long C-terminal domain might give the core protein a large freedom in terms of orientation relative to the membrane. Since both the core domain and the HS chains play important roles for brain function, such a large orientational freedom of glypican-1 (core domain as well as HS chains) may enhance the process of binding, activating, or transporting growth factors and enzymes (Jen et al. 2009).
Heparan sulfates show high flexibility
Figure 3 represents the center of mass of the Z coordinates (ZCOM) of each residue in HS chains and the distance (DO-S) between each HS residue and the Ser residue bearing its HS chain. The shaded area colors show different HS domains: a non-sulfated NA domain, a highly-sulfated NS domain, and a less-sulfated transition NS/NA domain. Every residue shows a large range of ZCOM (20–220 Å), especially the ones in the NS and NS/NA domains. Note that rotation of glypican-1 core domain and the extension of the C-terminal domain also influence the ZCOM range. As the glypican-1 core rotates, as shown in Figure 2G, one of the HS chains shows a higher ZCOM than the other two. However, by considering the averaged behaviors of all replicas over the entire simulation time (blue points), there is no significant difference among HS chains. An averaged plateau or slight decrease in ZCOM with a larger fluctuation range toward the terminal HS residues (especially HS486) is due to the fold back of HS chains. Interactions between these terminal HS residules of each chain and the glypican-1 core protein is elaborated below.
Fig. 3.

(A) Center of mass of the Z coordinate (ZCOM) of each residue in heparan sulfate chains HS486, HS488, and HS490. Z = 0 is set to the bilayer center. (B) Distance of each residue in each heparan sulfate chain to its Ser residue. In (A) and (B), the background is shaded to show linker (cyan), non-sulfated NA domain (brown), highly sulfated NS domain (blue), and less-sulfated transition NS/NA domain (pink). Note that in each figure, the averaged values are represented as blue points, the first 25%–75% ascending values in shaded boxes, and remaining values as black lines.
As for DO-S, a much smaller variation is observed from the non-sulfated residues (linker and NA domains). As shown in Figure 3B, these linker and NA domains exhibit quite linear correlations while the remaining parts of the HS chains do not always show this pattern. The last residue on each chain shows the largest DO-S fluctuation varying from 10–250 Å. For example, referring to Figure 2I, the last 20 residues of HS490 fold back to and interact with the glypican-1 core domain, resulting in relatively small DO-S. This particular conformation persists in this chain, showing various modes of interactions between glypican-1 and HS chains.
In order to gain a better understanding of the orientation of each HS chain, we characterize the tilt angle (θ) of each chain as well as the internal angle (γ) between each pair of the chains (Figure 4). The linker and NA domains of each chain roughly preserve a linearly extended polysaccharide conformation (Figure 3B) and thus were used to define θ and γ. Figure 4B shows that each HS chain exhibits a large range of θ fluctuations. HS486 (θ1) shows the largest range of 170° (10–180°), while HS490 (θ3) shows the smallest range of 115° (30–145°). The same trend is observed in ZCOM (Figure 3A), where ZCOM fluctuations in non-sulfated domains of HS486 are the largest. When considering the 50% data around the mean value (light blue boxes in Figure 4B), the fluctuation of each chain is much smaller: 55° (50–105°) for θ1, 45° (70–115°) for θ2, 30° (65–95°) for θ3. Initially, each chain was placed horizontally (90°). Averaged over the entire simulation time, all chains still stay around 90°. HS488, on the other hand, shows slightly larger θ. This is mainly due to multi-site interactions with the glypican-1 core protein in the neighboring system (blue in Figure 2I and see below).
Fig. 4.

(A, C) Schematic representations of tilt angles (θ) of each heparan sulfate chain with respect to the Z axis and internal angles (γ) between each pair of heparan sulfate chains. For the representation of each chain direction, a vector was defined from the Ser residue bearing each heparan sulfate to the center of mass of the linker-NA domains. (B, D) Box plots of angle distributions defined in (A) and (C) with the mean values in blue lines, the first 25%–75% ascending values in light blue boxes and remaining values in black lines. Note  for HS486,
for HS486, 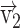 for HS488, and
for HS488, and  for HS490.
for HS490.
Angle γ was employed to represent the angle between the chains (Figure 4C and D). The fluctuations among three γ angles show similar range around 60° (0–120° for γ12, 30–135° for γ23, and 15–120° for γ31). Each HS chain was built based on the sequence in the Figure 1D and equilibrated before the production simulations. Influenced by the initial locations of Ser residues, γ12 is smaller than the other two on average.
Heparan sulfates show diverse structural heterogeneity
Figure 5 represents the glycosidic torsion angle distributions of the disaccharide units in HS486 and the results are similar for HS488 and HS490 (Figure S2 and S3). The central region of the ϕ/ψ scatter plots is highly populated for all glycosidic linkages of the polysaccharides, which corresponds to a syn-conformation of disaccharide elements in a HS chain. This is the main conformational state observed for d-GlcA-(β1 → 3)-d-Gal, which is present in the linker region, although a second state, referred to as an anti-ϕ conformation (Landersjö et al. 1997), is also present to some extent. For d-GlcA-(β1 → 4)-d-GlcNAc and d-GlcA-(β1 → 4)-d-GlcN, some excursions occur to anti-ϕ or anti-ψ conformations (Höög et al. 2001) where ϕ ≈ 180° or ψ ≈ 180°, respectively. In comparison to these GlcA-containing disaccharides, similar torsion angle distributions are observed also for l-IdoA-(α1 → 4)-d-GlcNAc and l-IdoA-(α1 → 4)-d-GlcN, a result which to a first approximation is anticipated since β-d-hexoses and α-l-hexoses show a similar conformational space at their respective glycosidic linkages (Widmalm 2013). In contrast, d-GlcNAc-(α1 → 4)-d-GlcA and d-GlcN-(α1 → 4)-d-GlcA display a band-like region that is populated along the ψ torsion angle. For d-GlcN-(α1 → 4)-l-IdoA, the conformational region corresponds to a syn-conformation. Thus, the HS chains exhibit flexibility at ψ torsion angles, to some degree for the β-linked residues and to a larger extent for the α-linked residues.
Fig. 5.

Distributions of the glycosidic dihedral angles of the disaccharide units in HS486. Data points were colored based on the domain: GlcA (linker) in cyan, GlcA (NA) in light blue, GlcA (NS/NA) in blue, GlcA (NS) in dark blue; IdoA (NS/NA) in orange, IdoA (NS) in brown; GlcNAc (NA) in light purple, GlcNAc (NS/NA) in pink; GlcN (NS/NA) in lime, and GlcN (NS) in dark green. The glycosidic dihedral angles ϕ and ψ are defined by H1'-C1'-O(n)-C(n) and C1'-O(n)-C(n)-H(n), where n is the linkage position, and a prime represents an atom from a sugar (n + 1) in the polysaccharide.
Though there is only a small difference for the glycosidic dihedral angles between α-linked l-IdoA and β-linked d-GlcA residues, their ring conformation shows significant differences (Figure S4 and S5). The most abundant ring conformation of d-GlcA is 4C1 and that of l-IdoA is 1C4 (see ring conformations in Figure S4). Since the 1C4 chair conformation is a low energy state for GlcA, it does not show any notable conformational change during the simulation period. In contrast, the 2So skew conformation can be observed for IdoA for a significant time period (as high as 14% of the entire simulation time in one case) (Hsieh et al. 2016). By clustering IdoA ring conformations by neighboring residue types, one can represent the fraction of different ring conformation occurrence (Figure S5). Specifically, IdoA22 and IdoA26 (red) are followed by sulfated sugar residues; IdoA28, IdoA30, and IdoA32 themselves (blue) are sulfated; IdoA36 and IdoA46 (green) have both neighboring residues that are sulfated; and IdoA50 (cyan) follows a sulfated sugar residue. Note that the numbering of HS chain residues starts from the reducing end (i.e., Xyl as residue number 1). Clearly, IdoA36 and IdoA46 (green) have the highest probability of having 2So conformations and IdoA22 and IdoA26 (red) have some. Transitions from 1C4 to 5S1 or 2So and between 5S1 and 2So took place during the simulations (Figure S4). Furthermore, IdoA residues having 2So conformations also show a similar fraction of time in 5S1 conformations (Oborský et al. 2013), which in these simulations are populated to a larger extent than the more commonly observed 2So conformation.
Heparan sulfates show multi-site interactions with surrounding molecules
As shown in Figure 3 and 4, each HS chain shows high dynamic fluctuations in ZCOM and the internal angle θ/γ. Thus, the polysaccharides are very likely to interact with various components in the system. Figure 6 shows the averaged interaction patterns of each residue in the HS chains over the entire simulation time. Clearly, sugar residues near glypican-1 protein (i.e., the linker and most part of NA domains) interact with glypican-1 (cyan color bars) for a significant portion of the simulation time. Furthermore, some linker residues also show interactions with other HS chains due to proximities between Ser486, Ser488, and Ser490. Specifically, there is a notable interaction of HS490 with the glypican-1 core protein (cyan color bars). This represents the fold-back behavior of the chain.
Fig. 6.

Averaged interaction patterns of each HS residue with its environment over the entire simulation time of three replicas. The interaction pattern graph shows the frequency of occurrence within 4 Å from each of N-glycan 116 (pink), N-glycan 79 (brown), glypican-1 in the primary system (cyan), glypican-1 in the periodic image system (red), heparan sulfates in the primary system (yellow), heparan sulfates in the image system (gray), lipids (orange), Cl− (green), K+ (purple), and water molecules (blue). Bottom panel shows the schematic of the linker, NA, NS/NA, and NS domains.
Clearly, the terminal residues of each chain are able to interact with other HS chains from the neighboring periodic image system. As shown in Figure 1B, terminal residues of HS488 can interact with the linker domain of HS488 from the periodic image system with a more or less stretch conformation. Interactions of HS chains with each other are highly stochastic due to their high flexibility and the relatively large system size. It is important to note that, due to the selected membrane bilayer size in this work, HS chain interactions with neighboring glypican-1 protein has been observed in all chains, with the probability of no more than 10% of the time on average. Interacting with the neighboring protein mostly happened near the ending residue on each chain, while HS488 also show some central residues (NS domains) contributing to this interaction pattern. As also shown in Figure 4B, this central NS domain-neighboring protein interaction leads to a relatively higher internal angle of chain HS488 (blue in Figure 2I). As indicated by other groups (Clausen et al. 2020, Jiang et al. 2020), these interactions are predominantly electrostatic by nature. Therefore, if glypican-1 density were higher than the current system or HS chains were longer than the current one, more interactions of HS with neighboring HS and glypican-1 (or core protein) would be expected, perhaps forming dense HS chain networks.
Besides some interactions with glypican-1, other HS chains, and water molecules, each charged sugar residue is mostly interacting with K+ ions. The more sulfated sites each residue has, the higher the interaction possibility is with K+ ions. For example, the NS domains that contain most charged sugar residues show up to 50% probability of interacting with K+, and NA domains that have the least charged sugar residues have only around 10% probability of interacting with K+. This trend is the same for different HS chains. In addition to this, some residues also interact with lipid head groups (Figure 2H and Figure 6 of HS488). Sugar residues in NS and NS/NA domains show a small probability (less than 2%) of interacting with lipid acyl chains. In addition, as seen in Figure 3A, these residues infrequently fall into the interaction range of lipid head groups (< 30 Å).
CONCLUSIONS
In this study, we describe the structure and dynamics of glypican-1 and its three HS chains and their interactions with the surrounding environment. The glypican-1 core domain structure remains stable during the simulation period (RMSD ~ 2 Å), but exhibits large variations in terms of its distance and orientation with respect to the membrane. The unstructured C-terminal domain connecting the core domain to the GPI anchor is relatively long and contains more than 50 residues (Asn474-Ser530) including the three HS sites (Ser486, Ser488, and Ser490). It is the conformal flexibility of the unstructured C-terminal domain that gives the glypican-1 core domain a significant freedom for its movement and orientation. The observed extension of the unstructured domain also offers accessibility of enzymes to add and modify HS chains. We speculate that such conformational plasticity in the unstructured domain may enhance membrane association by decoupling the small GPI anchor from large movements of the larger core domain. The effectiveness of this mechanism could be explored in future experiments by measuring membrane dissociation rates.
Another possible function of the unstructured domain may be to modify the mechanical response of the protein to shear stress. One important result from this simulation is the value of the average distance between the core domain and the lipid membrane. This quantity is of interest to experimentalists and theorists investigating how shear flow may affect glycocalyx molecules. Calculations and experiments show that the size and shape of the aqueous part of lipid-anchored proteins determine how much force shear flow can apply to the lipid anchor (Jönsson et al. 2011, Jönsson and Jönsson 2015). Hu et al. compared the drift velocity of streptavidin bound to biotinylated lipids with or without a 4-carbon acyl chain link between the lipid head group and the biotin, and found that the presence of the link allowed the complex to move twice as fast. The link increased the height of the avidin molecule above the membrane, and also reduced the friction between the avidin and surrounding lipids (Hu et al. 2016). Further experiments and simulations are required to explore this possibility.
It has been shown that HSPG serves as a receptor or attachment factor for a number of viruses, such as herpes simplex virus, Dengue virus, and human papillomavirus (see Cagno et al. (Cagno et al. 2019) for a complete list). Typically, viruses interact with the negative charges of HSPG through the basic residues of viral surface proteins. Such interactions increase the concentration of viruses on the surface of host cells. The viruses then interact with more specific entry receptors to achieve cell entry. Therefore, HSPG has been proposed as a broad-spectrum antiviral target. Several HSPG-mimicking compounds are currently under clinical trials to assess their antiviral efficacy. Notably, recent studies suggest that severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus causing the coronavirus disease 2019 (COVID-19) pandemic, can interact with HSPG through its surface spike protein (Clausen et al. 2020, Hao et al. 2020, Liu et al. 2020). SARS-CoV-2 spike protein was shown to bind heparin with an affinity (Kd) of 55 nM and that the interaction can be inhibited by a HS oligosaccharide (Liu et al. 2020). Another study showed that the spike protein binds HS in a sulfation-dependent manner, and it does not bind sialic acid residues. Moreover, HS binds the polybasic residues near the S1/S2 junctional region of SARS-CoV-2 spike. This interaction can stabilize the S protein trimer in the open conformation to facilitate the binding between the receptor binding domain of spike and angiotensin-converting enzyme 2, the major entry receptor of SARS-CoV-2 (Hao et al. 2020). Therefore, the computational model presented in the current study can be potentially used to elucidate the dynamic interactions between viruses (including SARS-CoV-2) and HSPG in a more physiological setting, as well as to aid in the development of broad spectrum antivirals to block virus entry.
Furthermore, observed variations in the position, end-to-end distance, and orientation of each HS chain reflect its flexibility and high dynamic nature. Residues on each HS chain can interact with multiple chemical moieties in the environment, including ions, lipid head groups, and the glypican-1 core protein and HS chains in the primary system, as well as neighboring periodic image systems. We find that more sulfated residues show a higher probability to interact with K+ ions and lipid groups.
Such variability and mobility of HS chain orientations along with the range of interactions indicate that the glypican HS chains can clearly interact with growth factors, their receptors, adhesion molecules, and more at the cell surface. Furthermore, it is clear that even a single glypican HSPG can bridge such molecules over fairly significant distances due to the variable extensions and orientations of the HS chains and the unstructured C-terminal domain. This study provides an important step to begin analysis of the specific interactions that are involved in HSPG linking of growth factors with their receptors as well as other HSPG-facilitated interactions on the surface. Observed differences in K+ interactions make it clear that interactions with HS binding sites in other proteins, e.g. growth factors, could be examined in future studies. The data here also support that virus particles interacting with multiple HS chains from different HSPGs could facilitate clustering and thereby induce internalization. It will be interesting to see how the high dynamic fluctuation and interaction pattern change with a HS chain sequence with different sulfation patterns and lengths.
METHODS
All simulations were performed with GROMACS (Abraham et al. 2015). The CHARMM36(m) force field was employed for lipids (Klauda et al. 2012, Klauda et al. 2010), protein (Huang et al. 2017), and carbohydrates (Guvench et al. 2008, Guvench et al. 2009, Hatcher et al. 2009). A crystal structure of glypican-1 core domain (PDB ID: 4YWT, Asp24-Gly473) was taken from the Protein Data Bank (Awad et al. 2015). In this work, TIP3P was used as a water model (Jorgensen et al. 1983) with 0.15 M KCl aqueous solution.
We chose to use a lipid composition of POPC:PSM:Chol = 1:1:1 for all systems to represent a neuronal plasma membrane using 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), cholesterol (Chol), and palmitoyl-sphingomyelin (PSM). Based on the sequence from UniProt (Consortium 2019), the missing C-terminal domain of glypican-1 protein was initially built as a linear chain using the internal coordinate information in the CHARMM force field and CHARMM (Brooks et al. 2009). Through a series of simulations, the C-terminal domain became collapsed with Asn474 at Z = 77 Å (to the core domain) and Ser530 at Z = 35 Å (to a GPI linker), where the bilayer is centered at Z = 0 and its normal is along the Z axis. Then, glypican-1 C-terminal residue Ser530 was linked to a GPI (Ferguson et al. 2009) anchored in the membrane. The model structure of the membrane, GPI-anchor, N-glycans, and HS chains were generated through CHARMM-GUI Glycan Reader & Modeler (Jo et al. 2011, Park et al. 2017, Park et al. 2019), Membrane Builder (Jo et al. 2009, Wu et al. 2014), and Glycolipid Modeler (Jo et al. 2011, Lee et al. 2018). The initial system size was 240 Å × 240 Å × 240 Å to have enough space to include all the initial components, yielding a total of 1,080,442 atoms for each replica. All simulation systems and simulation inputs were generated through CHARMM-GUI (Jo et al. 2008, Lee et al. 2016) and CHARMM (Brooks et al. 2009). Visualization was done via VMD and PyMOL (DeLano 2002, Humphrey et al. 1996), and all analyses were carried out by CHARMM (Brooks et al. 2009).
The van der Waals interactions were smoothly switched off over 10–12 Å by a force-based switching function (Dion et al. 2004) and the long-range electrostatic interactions were calculated by the particle-mesh Ewald method (York et al. 1993) with a mesh size of ~ 1 Å. The time-step was set to 3 fs using the hydrogen mass repartitioning technique (Hopkins et al. 2015) and bonds containing hydrogen atoms were constrained by the LINCS algorithm (Hess et al. 1997). Temperature was held at 310.15 K; the constant temperature was controlled by Nosé-Hoover temperature coupling method (Hoover 1985, Nosé 1984). The system in a canonical ensemble (NVT, constant particle number, volume, and temperature) was first relaxed with all the solute atoms subjected to harmonic restraints. An isothermal–isobaric ensemble (NPT, constant particle number, pressure, and temperature) was then applied to adjust the solvent density. The Parrinello-Rahman pressure coupling method was used to control the constant pressure (Nosé and Klein 1983, Parrinello and Rahman 1981). A dihedral restraint force constant was set to 1 kcal/(mol·rad2) to maintain the carbohydrate chair conformation only during the equilibration steps. For the production runs, each replica was simulated for 1 μs where trajectories were saved every 75 ps. The results described herein are based on the analysis of production simulations.
FOUNDING SOURCES
This work was supported in part by grants from NSF DBI-1707207, XSEDE MCB070009 (to WI), Swedish Research Council 2017–03703 (to GW), NIH HL152348 (to XFZ). The authors thank Hongjing Ma for her initial work on this project.
Conflict of interest statement
None declared.
Supplementary Material
Contributor Information
Chuqiao Dong, Department of Mechanical Engineering and Mechanicss, Lehigh University, Bethlehem, PA, 18015, United States.
Yeol Kyo Choi, Department of Biological Sciences, Lehigh University, Bethlehem, PA, 18015, United States.
Jumin Lee, Department of Biological Sciences, Lehigh University, Bethlehem, PA, 18015, United States.
X Frank Zhang, Department of Mechanical Engineering and Mechanicss, Lehigh University, Bethlehem, PA, 18015, United States; Department of Bioengineering, Lehigh University, Bethlehem, PA, 18015, United States.
Aurelia Honerkamp-Smith, Department of Physics, Lehigh University, Bethlehem, PA, 18015, United States.
Göran Widmalm, Department of Organic Chemistry, Stockholm University, S-106 91 Stockholm, Sweden.
Linda J Lowe-Krentz, Department of Biological Sciences, Lehigh University, Bethlehem, PA, 18015, United States.
Wonpil Im, Department of Biological Sciences, Lehigh University, Bethlehem, PA, 18015, United States; Department of Bioengineering, Lehigh University, Bethlehem, PA, 18015, United States; Department of Chemistry, Lehigh University, Bethlehem, PA, 18015, United States.
References
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. 2015. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 1:19–25. [Google Scholar]
- Awad W, Adamczyk B, Örnros J, Karlsson NG, Mani K, Logan DT. 2015. Structural aspects of N-glycosylations and the C-terminal region in human glypican-1. J Biol Chem. 290:22991–23008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernfield M, Götte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. 1999. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 68:729–777. [DOI] [PubMed] [Google Scholar]
- Brooks BR, Brooks CL III, Mackerell AD Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. 2009. CHARMM: The biomolecular simulation program. J Comput Chem. 30:1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagno V, Tseligka ED, Jones ST, Tapparel C. 2019. Heparan sulfate proteoglycans and viral attachment: True receptors or adaptation bias? Viruses. 11:596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson P, Presto J, Spillmann D, Lindahl U, Kjellén L. 2008. Heparin/heparan sulfate biosynthesis Processive formation of N-sulfated domains. J Biol Chem. 283:20008–20014. [DOI] [PubMed] [Google Scholar]
- Clausen TM, Sandoval DR, Spliid CB, Pihl J, Perrett HR, Painter CD, Narayanan A, Majowicz SA, Kwong EM, McVicar RN, et al. 2020. SARS-CoV-2 infection depends on cellular Heparan Sulfate and ACE2. Cell. 183:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condomitti G, de Wit J. 2018. Heparan sulfate proteoglycans as emerging players in synaptic specificity. Front Mol Neurosci. 11:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium U. 2019. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. 2002. Pymol: An open-source molecular graphics tool. CCP4 Newsletter on protein crystallography. 40:82–92. [Google Scholar]
- Dion M, Rydberg H, Schröder E, Langreth DC, Lundqvist BI. 2004. Van der Waals density functional for general geometries. Phys Rev Lett. 92:246401. [DOI] [PubMed] [Google Scholar]
- Ebong EE, Lopez-Quintero SV, Rizzo V, Spray DC, Tarbell JM. 2014. Shear-induced endothelial NOS activation and remodeling via heparan sulfate, glypican-1, and syndecan-1. Integr Biol. 6:338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebong EE, Macaluso FP, Spray DC, Tarbell JM. 2011. Imaging the endothelial glycocalyx in vitro by rapid freezing/freeze substitution transmission electron microscopy. Atertio Thromb Vasc Biol. 31:1908–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson MA, Kinoshita T, Hart GW. 2009. Glycosylphosphatidylinositol anchors. In: Essentials of Glycobiology. 2nd ed: New York, USA: Cold Spring Harbor Laboratory Press. [PubMed] [Google Scholar]
- Guvench O, Greene SN, Kamath G, Brady JW, Venable RM, Pastor RW, Mackerell AD Jr. 2008. Additive empirical force field for hexopyranose monosaccharides. J Comput Chem. 29:2543–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guvench O, Hatcher E, Venable RM, Pastor RW, MacKerell AD Jr. 2009. CHARMM additive all-atom force field for glycosidic linkages between hexopyranoses. J Chem Theory Comput. 5:2353–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W, Ma B, Li Z, Wang X, Gao X, Li Y, Qin B, Shang S, Cui S, Tan Z. 2020. Binding of the SARS-CoV-2 spike protein to Glycans. bioRxiv. 10.1101/2020.05.17.100537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatcher E, Guvench O, MacKerell AD Jr. 2009. CHARMM additive all-atom force field for aldopentofuranoses, methyl-aldopentofuranosides, and fructofuranose. J Phys Chem B. 113:12466–12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B, Bekker H, Berendsen HJ, Fraaije JG. 1997. LINCS: A linear constraint solver for molecular simulations. J Comput Chem. 18:1463–1472. [Google Scholar]
- Höög C, Landersjö C, Widmalm G. 2001. Oligosaccharides display both rigidity and high flexibility in water as determined by 13C NMR relaxation and 1H, 1H NOE spectroscopy: Evidence of anti-ϕ and anti-ψ torsions in the same Glycosidic linkage. Chem A Eur J. 7:3069–3077. [DOI] [PubMed] [Google Scholar]
- Hoover WG. 1985. Canonical dynamics: Equilibrium phase-space distributions. Phys Rev A. 31:1695. [DOI] [PubMed] [Google Scholar]
- Hopkins CW, Le Grand S, Walker RC, Roitberg AE. 2015. Long-time-step molecular dynamics through hydrogen mass repartitioning. J Chem Theory Comput. 11:1864–1874. [DOI] [PubMed] [Google Scholar]
- Hsieh P-H, Thieker DF, Guerrini M, Woods RJ, Liu J. 2016. Uncovering the relationship between sulphation patterns and conformation of iduronic acid in heparan sulphate. Sci Rep. 6:29602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S-K, Huang L-T, Chao L. 2016. Membrane species mobility under in-lipid-membrane forced convection. Soft Matter. 12:6954–6963. [DOI] [PubMed] [Google Scholar]
- Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmüller H, MacKerell AD. 2017. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat Methods. 14:71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. 1996. VMD: Visual molecular dynamics. J Mol Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- Jen Y-HL, Musacchio M, Lander AD. 2009. Glypican-1 controls brain size through regulation of fibroblast growth factor signaling in early neurogenesis. Neural Dev. 4:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang XZ, Gong H, Luo KH, Ventikos Y. 2017. Large-scale molecular dynamics simulation of coupled dynamics of flow and glycocalyx: Towards understanding atomic events on an endothelial cell surface. J R Soc Interface. 14:20170780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang XZ, Guo L, Luo KH, Ventikos Y. 2020. Membrane deformation of endothelial surface layer interspersed with Syndecan-4: A molecular dynamics study. Ann Biomed Eng. 48:357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Iyer VG, Im W. 2008. CHARMM-GUI: A web-based graphical user interface for CHARMM. J Comput Chem. 29:1859–1865. [DOI] [PubMed] [Google Scholar]
- Jo S, Lim JB, Klauda JB, Im W. 2009. CHARMM-GUI membrane builder for mixed bilayers and its application to yeast membranes. Biophys J. 97:50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Song KC, Desaire H, MacKerell AD Jr, Im W. 2011. Glycan reader: Automated sugar identification and simulation preparation for carbohydrates and glycoproteins. J Comput Chem. 32:3135–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jönsson P, Gunnarsson A, Höök F. 2011. Accumulation and separation of membrane-bound proteins using hydrodynamic forces. Anal Chem. 83:604–611. [DOI] [PubMed] [Google Scholar]
- Jönsson P, Jönsson B. 2015. Hydrodynamic forces on macromolecules protruding from lipid bilayers due to external liquid flows. Langmuir. 31:12708–12718. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. 1983. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 79:926–935. [Google Scholar]
- Khan S, Fung KW, Rodriguez E, Patel R, Gor J, Mulloy B, Perkins SJ. 2013. The solution structure of heparan sulfate differs from that of heparin implications for function. J Biol Chem. 288:27737–27751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauda JB, Monje V, Kim T, Im W. 2012. Improving the CHARMM force field for polyunsaturated fatty acid chains. J Phys Chem B. 116:9424–9431. [DOI] [PubMed] [Google Scholar]
- Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD Jr, Pastor RW. 2010. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J Phys Chem B. 114:7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer KL, Yost HJ. 2003. Heparan sulfate core proteins in cell-cell signaling. Annu Rev Genet. 37:461–484. [DOI] [PubMed] [Google Scholar]
- Landersjö C, Stenutz R, Widmalm G. 1997. Conformational flexibility of carbohydrates: A folded conformer at the ϕ dihedral angle of a Glycosidic linkage. J Am Chem Soc. 119:8695–8698. [Google Scholar]
- Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y, et al. 2016. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J Chem Theory Comput. 12:405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Patel DS, Ståhle J, Park S-J, Kern NR, Kim S, Lee J, Cheng X, Valvano MA, Holst O, et al. 2019. CHARMM-GUI membrane builder for complex biological membrane simulations with glycolipids and lipoglycans. J Chem Theory Comput. 15:775–786. [DOI] [PubMed] [Google Scholar]
- Li Y, Sun C, Yates EA, Jiang C, Wilkinson MC, Fernig DG. 2016. Heparin binding preference and structures in the fibroblast growth factor family parallel their evolutionary diversification. Open Biol. 6:150275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Chopra P, Li X, Wolfert MA, Tompkins SM, Boons G-J. 2020. SARS-CoV-2 spike protein binds heparan sulfate in a length-and sequence-dependent manner. bioRxiv. 10.1101/2020.05.10.087288. [DOI] [Google Scholar]
- Morgan MR, Hamidi H, Bass MD, Warwood S, Ballestrem C, Humphries MJ. 2013. Syndecan-4 phosphorylation is a control point for integrin recycling. Dev Cell. 24:472–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosé S. 1984. A molecular dynamics method for simulations in the canonical ensemble. Mol Phys. 52:255–268. [Google Scholar]
- Nosé S, Klein M. 1983. Constant pressure molecular dynamics for molecular systems. Mol Phys. 50:1055–1076. [Google Scholar]
- Oborský P, Tvaroška I, Králová B, Spiwok VC. 2013. Toward an accurate conformational modeling of iduronic acid. J Phys Chem B. 117:1003–1009. [DOI] [PubMed] [Google Scholar]
- Park S-J, Lee J, Patel DS, Ma H, Lee HS, Jo S, Im W. 2017. Glycan reader is improved to recognize most sugar types and chemical modifications in the protein data Bank. Bioinformatics. 33:3051–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S-J, Lee J, Qi Y, Kern NR, Lee HS, Jo S, Joung I, Joo K, Lee J, Im W. 2019. CHARMM-GUI glycan Modeler for modeling and simulation of carbohydrates and glycoconjugates. Glycobiology. 29:320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello M, Rahman A. 1981. Polymorphic transitions in single crystals: A new molecular dynamics method. J Appl Phys. 52:7182–7190. [Google Scholar]
- Schlessinger J, Plotnikov AN, Ibrahimi OA, Eliseenkova AV, Yeh BK, Yayon A, Linhardt RJ, Mohammadi M. 2000. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol Cell. 6:743–750. [DOI] [PubMed] [Google Scholar]
- Shi X, Zaia J. 2009. Organ-specific heparan sulfate structural phenotypes. J Biol Chem. 284:11806–11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson G, Awad W, Håkansson M, Mani K, Logan DT. 2012. Crystal structure of N-glycosylated human Glypican-1 Core protein structure of two loops evolutionarily conserved in vertebrate Glypican-1. J Biol Chem. 287:14040–14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarbell JM, Simon SI, Curry F-RE. 2014. Mechanosensing at the vascular interface. Annu Rev Biomed Eng. 16:505–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumova S, Woods A, Couchman JR. 2000. Heparan sulfate chains from glypican and syndecans bind the hep II domain of fibronectin similarly despite minor structural differences. J Biol Chem. 275:9410–9417. [DOI] [PubMed] [Google Scholar]
- Wang S, Qiu Y, Bai B. 2019. The expression, regulation, and biomarker potential of glypican-1 in cancer. Front Oncol. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmalm G. 2013. A perspective on the primary and three-dimensional structures of carbohydrates. Carbohydr Res. 378:123–132. [DOI] [PubMed] [Google Scholar]
- Wu EL, Cheng X, Jo S, Rui H, Song KC, Dávila-Contreras EM, Qi Y, Lee J, Monje-Galvan V, Venable RM, et al. 2014. CHARMM-GUI membrane builder toward realistic biological membrane simulations. J Comput Chem. 35:1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York DM, Darden TA, Pedersen LG. 1993. The effect of long-range electrostatic interactions in simulations of macromolecular crystals: A comparison of the Ewald and truncated list methods. J Chem Phys. 99:8345–8348. [Google Scholar]
- Zeng Y, Zhang XF, Fu BM, Tarbell JM. 2018. The role of endothelial surface glycocalyx in mechanosensing and transduction. In: Molecular, Cellular, and Tissue Engineering of the Vascular System. New York, USA: Springer. p. 1–27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.