

Abstract
The diagnosis of chromosomal mosaicism in the preimplantation and prenatal stage is fraught with uncertainty and multiple factors need to be considered in order to gauge the likely impact. The clinical effects of chromosomal mosaicism are directly linked to the type of the imbalance (size, gene content and copy number), the timing of the initial event leading to mosaicism during embryogenesis/fetal development, the distribution of the abnormal cells throughout the various tissues within the body as well as the ratio of normal/abnormal cells within each of those tissues. Additional factors such as assay noise and culture artifacts also have an impact on the significance and management of mosaic cases. Genetic counseling is an important part of educating patients about the likelihood of having a liveborn with a chromosome abnormality and these risks differ according to the time of ascertainment and the tissue where the mosaic cells were initially discovered. Each situation needs to be assessed on a case-by-case basis and counseled accordingly. This review will discuss the clinical impact of finding mosaicism through: embryo biopsy, chorionic villus sampling (CVS), amniocentesis, and non-invasive prenatal testing (NIPT) using cell free DNA (cfDNA).
INTRODUCTION
The development of an embryo into a fully formed, healthy baby is a highly complex and regulated process that requires precise temporal transcription and translation of our genetic code. Chromosomal aneuploidy brings with it major changes in gene dosage which typically has a catastrophic effect on this fine-tuned process. Indeed, the primary effect of aneuploidy is embryonic and/or fetal lethality.1–8 The greater the number of protein coding genes involved, the higher the chance the pregnancy will miscarry.1
With few exceptions, every somatic cell in the human body should have the same chromosome number and structure. This concept is based on the supposition that mitotic division errors are rare. In reality, early cell divisions are error-prone and chromosomal mosaicism is the consequence. The clinical effects of chromosomal mosaicism are directly linked to the size of the gene imbalance, the timing of the initial event, and, most likely, the distribution of the abnormal cells in tissues. A chromosome segregation error that creates a normal cell line in an embryo carrying a meiotic error can be viewed as a mechanism that can attenuate the lethality of a chromosomal aneuploidy through a shift of the gene dosage back towards normal. Conversely, the earlier an abnormal cell line arises in a chromosomally normal conception, the greater the proportion of the abnormal cells. An error occurring within the first few cell divisions is likely to be expressed as abnormal cells in extra-embryonic tissues as well as the inner cell mass.
Genetic counseling is strongly recommended for patients who find themselves dealing with a preimplantation/prenatal diagnosis involving chromosomal mosaicism. Each situation needs to be assessed on a case-by-case basis and counseled accordingly. Patients should understand the risk for a liveborn with a mosaic chromosome abnormality.
This review will briefly present the mechanism by which mosaicism arises. We will then discuss the incidence and significance of finding mosaicism through: [1] embryo biopsy, [2] chorionic villus sampling (CVS), [3] amniocentesis and [4] non-invasive prenatal testing (NIPT) using cell free DNA (cfDNA). Clinical impact will mainly focus on reproductive and prenatal diagnostic issues; the role of post-zygotic chromosome segregation in cancer and other post-natal diseases will not be discussed in detail.
MECHANISMS LEADING TO CHROMOSOMAL MOSAICISM
Mosaic aneuploidies are generated by two principal mechanisms: post-zygotic mitotic chromosome segregation errors (Figures 1b–d) and post-zygotic mitotic trisomy/monosomy rescue of a pre-existing aneuploidy of meiotic origin (Figure 2).
Figure 1.
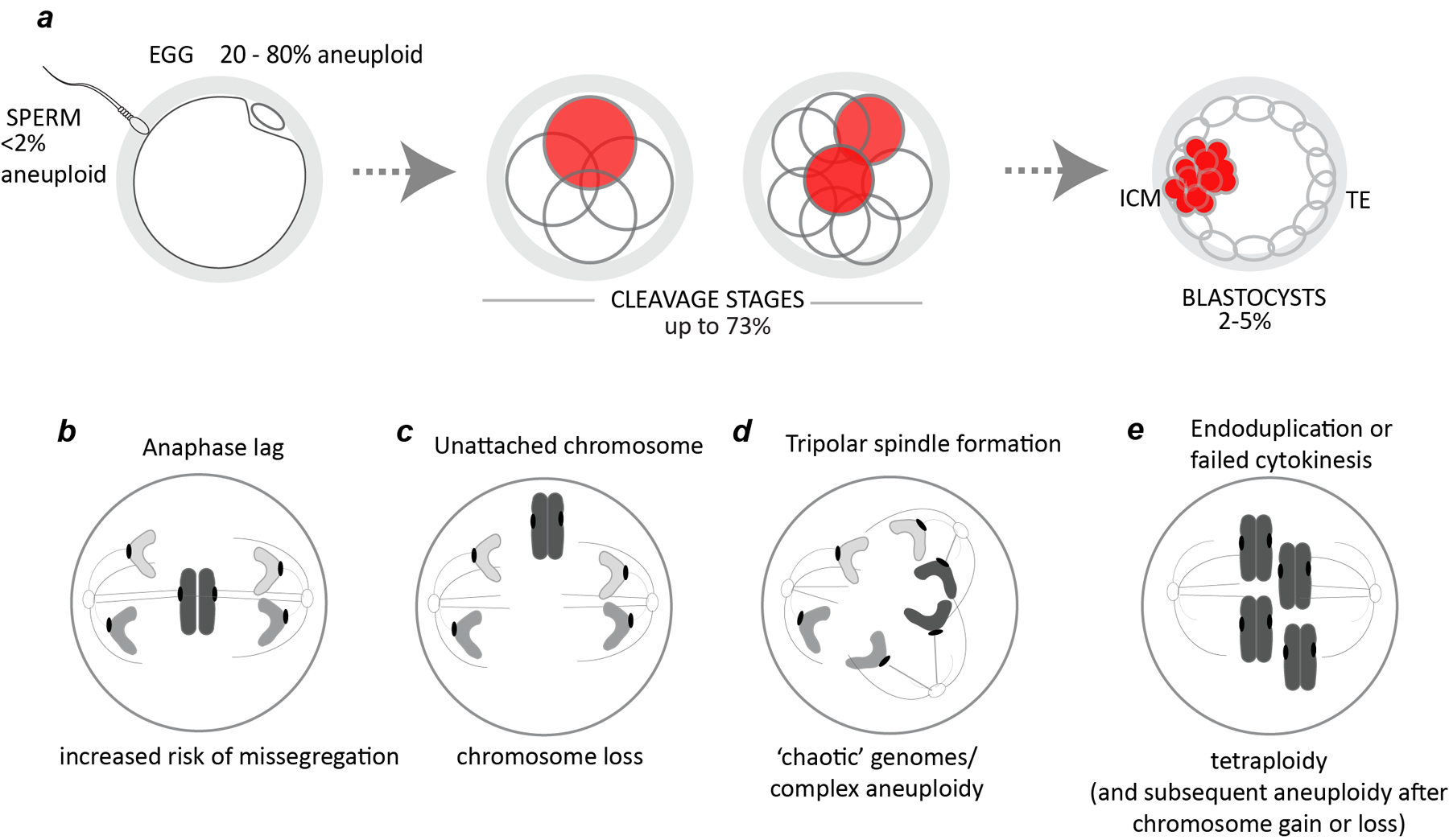
Origins of mosaic preimplantation embryos in human. a, Incidence of aneuploid sperm and eggs (single cells) and mosaic embryos at the cleavage and blastocyst stages (% of cases).14–24 b-e, examples of mechanisms resulting in chromosomally mosaic embryos. b, anaphase lag. c, failed chromosome capture. d, tripolar spindle formation. e, endoduplication or failed cytokinesis result in tetraploid cells.
Figure 2.
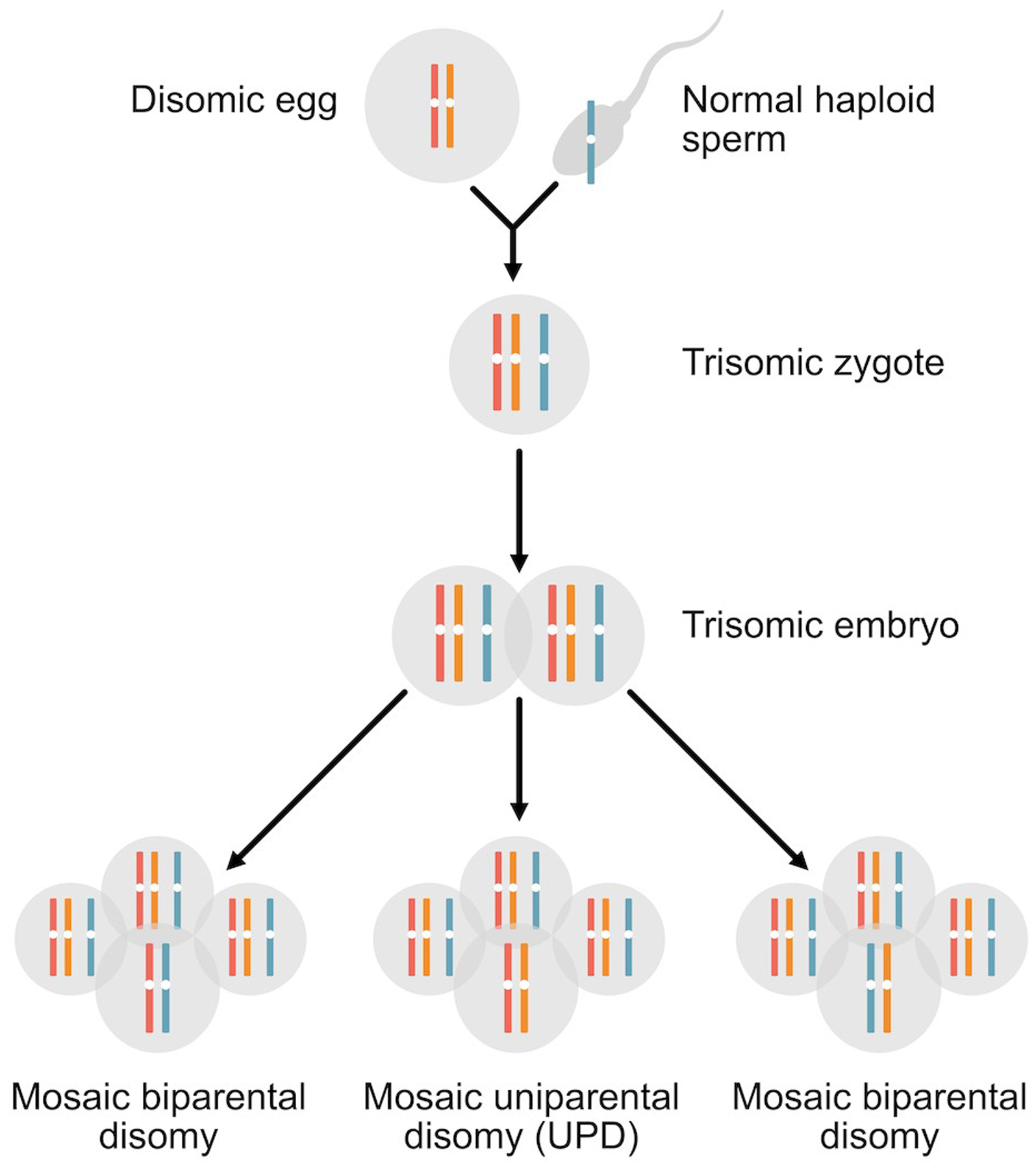
Trisomy rescue. This example illustrates a maternal meiosis I segregation error that has produced a disomic egg. Fertilization by the normal haploid sperm leads to a trisomic embryo. Depending on which chromosome is lost during trisomic rescue, there are three possible outcomes for the rescued disomic homologues. When either of the maternal chromosomes is lost during trisomic rescue, the resulting disomic cell line will contain both a maternal and paternal homologue (biparental inheritance). When the paternal chromosome is lost during the rescue event, the resulting disomic homologue will only derive from one parent, the mother (uniparental disomy, UPD). In all cases, mosaicism for a trisomic and disomic cell line will be present.
Several mechanisms have been proposed to cause errors in chromosome segregation in the embryonic divisions. Anaphase lag refers to chromosomes that remain at the mid-pole after the vast majority of sister chromatids for the other chromosomes have segregated (Fig 1b). This phenomenon generally affects one or only a few chromosomes and can arise due to sister chromatids being under-replicated, entangled or attached to microtubules emanating from both spindle poles. Anaphase lag is associated with elevated segregation error and non-reciprocal forms of aneuploidy (i.e., chromosome loss without corresponding chromosome gain). In some cases, chromosomes may also fail to be captured by the spindle, a phenomenon demonstrated by live cell imaging of human zygotes (Fig 1c).4
During the zygotic division the two parental genomes are captured on a single spindle, a process that is highly error-prone. Abnormal tripolar spindles also occur in preimplantation embryos resulting in massive chromosome loss (‘chaotic’ mosaicism; Fig. 1d).2 Their formation may in part be influenced by a common genetic variant in or near the centrosomal regulator PLK4 that, when maternally transmitted, may predispose to tripolar spindle formation.6 The maternal transmission is important because mRNAs and proteins from the egg drive the initial embryonic divisions until embryonic genome activation. Endoduplication, where chromosomes are re-replicated without an intervening cellular division, failed cytokinesis, and overduplication of centrosomes have been linked to the formation of tetraploid cells (Fig 1e).8 In some cases after failed cytokinesis, this may give rise to binucleate cells.3 Tetraploidy is observed in human conceptuses, however it also induces chromosomal instability, aneuploidy, and tumorogenesis in mouse embryos.9,10 Other mechanisms including those associated with embryonic arrest have been reviewed by Daughtry and colleagues.11
In some cases, a combination of two chromosome segregation errors leads to two copies of a chromosome being inherited from a single parent, a phenomenon called uniparental disomy (UPD). A review of mechanisms for UPD can be found elsewhere in this Special Issue.12 Similar to consanguinity, uniparental isodisomy (two identical [i.e., two homozygous chromosomes as depicted in Figure 2) leads to an increased risk for autosomal recessive diseases in genes in the homozygous region. UPD can also cause aberrant expression levels of genes that are imprinted (only expressed from one parent’s chromosome and silenced on the other). Certain chromosomes contain imprinted regions (chromosomes 6, 7, 11, 14, 15, 20) and UPD for these chromosomes can result in clinical abnormalities.13
MOSAICISM IN HUMAN PREIMPLANTATION EMBRYOS
Incidence
Mosaicism is estimated to affect up to 70% of human preimplantation embryos.14 By the blastocyst stage, an estimated 2% to 50% of embryo biopsies are reported to be mosaic,15–23 with the wide range reflecting a combination of biological mechanisms (true mosaicism)24 together with technological artifacts that stem from differences in assessing mosaicism from a single trophectoderm biopsy of 3–7 cells.25,26 Dissociation of blastocyst embryos show concordance of inner cell mass and trophectoderm biopsies in 95–98% of embryos,17,27–30 suggesting that by the blastocyst stage, high-level mosaicism confined to one of these two respective lineages is relatively rare (2–5%; Figure 1a).
Clinical Significance
The report in 2015 by Greco and colleagues that transfer of some mosaic aneuploid embryos can lead to the birth of apparently healthy babies, has sparked an ongoing debate regarding the appropriate management and clinical significance of mosaicism identified in human preimplantation embryos.16 The exact factors that govern which mosaic embryos will lead to normal outcomes are yet to be determined. It is clear that transfer of a mosaic embryo significantly reduces the clinical pregnancy rate as well as the ongoing/live birth rate while considerably increasing the chance of miscarriage compared to the transfer of apparently euploid embryos only.15,18,19,21–23,31–33 The most recent prospective study by Zhang and coworkers showed a live birth rate of 27.1% in the mosaic embryo group compared to 47.0% in the euploid group (p < 0.001).33
The level of mosaicism identified in trophectoderm biopsies has been a key focus with some studies showing better clinical outcomes in embryos transferred with mosaic aneuploidy proportions <50% compared to those with a higher proportion of abnormal cells (>50%).19 However, this finding is not uniformly supported by all studies. Kushnir et al. found no significant differences in ongoing pregnancy or miscarriage rates among mosaic embryo transfers at any threshold of aneuploidy.34 While Zhang and colleagues did not specifically look at outcome difference in mosaic embryos above and below 50%, they did find that pregnancy outcomes were globally decreased in the <50% abnormal cell embryo group compared with euploid embryos.33 In addition, they found no significant difference in outcome when comparing embryos with <40% aneuploidy to embryos with <50% aneuploidy, further supporting the notion that that the degree of trophectoderm mosaicism is a poor predictor of ongoing pregnancy and miscarriage.33 One major concern with mosaic embryo transfer studies is that the group receiving the putative mosaic embryos is enriched for patients where previous transfer with euploid embryos failed to implant. The first prospective non-selection RCT has now reported that there are no differences in pregnancy and live birth outcomes from transfer of putative mosaic embryos up to 50% compared to euploid embryos.35
There are multiple factors that can affect the detection of mosaicism in preimplantation embryos. A prime consideration is the extent to which a random sampling of five to ten cells accurately reflects the genomic picture in the remainder of the embryo. Random sampling may indicate the level of mosaicism within a specific biopsy but mathematical models indicate that the level will not necessarily be an accurate representation of the entire trophectoderm and/or embryo.36 It is also important to understand that when using preimplantation genetic testing for aneuploidy (PGT-A) methods such as next generation sequencing (NGS), a mosaic diagnosis is based on detection of an intermediate copy number, i.e. between 1 and 2 (mosaic monosomy) or between 2 and 3 (mosaic trisomy), with 2 representing the normal situation. While intermediate copy number can indeed be caused by mosaicism, other factors such as assay noise/artifact, amplification bias, contamination, mitotic state, variation in embryo biopsy technique, embryology laboratory conditions, and laboratory thresholds set for calling mosaicism can all impact the assessed copy number.25,26,37 Marin et al performed a meta-analysis of blastocyst reanalysis and found that the concordance with the original mosaic result was only 42.6%.26 It therefore seems clear that a simple assessment of the mosaic level in a trophectoderm biopsy does not provide a reliable predictor of the reproductive potential of a mosaic embryo.
The introduction of NGS technologies into PGT-A has led to a greater number of partial chromosomal aneuploidies (segmental aneuploidies (SA)) being reported. The significance of finding a SA is challenging even when encountered in routine prenatal and postnatal specimens. Mosaic SAs in preimplantation embryos brings further complexity in assessing clinical significance. Conflicting outcomes have been reported in patients who elected to transfer mosaic SAs embryos with reduced implantation rates being observed in some studies15 while other studies indicated no measurable difference.23 Embryos with mosaic SA’s appear to have lower miscarriage rates compared to mosaic embryos with whole chromosome aneuploidies.23,33 In some instances, embryos diagnosed with mosaic SAs displayed whole chromosome aneuploidy in other sections of the embryo.38 A mosaic SA can also be attributed to assay noise or a temporal artifact caused by asynchrony of DNA replication.39 With data on the clinical predictive value of mosaic SAs severely lacking, the debate on whether to report them and how to manage them if reported will continue.
For patients with no euploid embryos, the transfer of mosaic embryos may be the only possible pathway to achieving a pregnancy. Choosing this pathway is reasonable only when patients are appropriately informed of the potential outcomes. For mosaic embryos, the likelihood of viability depends on the specific chromosome/s involved and the proportion and distribution of the normal and abnormal cells. Based on these principles, various prioritization models have been proposed to guide patients and clinicians dealing with mosaic embryos diagnosed by PGT-A.40,41 Grati et al., based their prioritization model on the incidence of mosaic aneuploidies in CVS samples, the follow-up results of amniocentesis (including UPD), the frequency of mosaic aneuploidies in products of conception, and the likelihood that a given aneuploidy would be observed in a liveborn.41 The higher the score for a given chromosome abnormality, the lower the priority for transfer. The efficacy of prioritization tools remains to be determined. There will be variability in outcomes associated with the transfer of mosaic embryos and therefore careful genetic counseling is needed for patients in this situation. Prenatal diagnosis laboratories must be informed of any prior mosaic PGT-A results so that additional karyotyping and/or FISH studies can be performed, as appropriate.
MOSAICISM IDENTIFIED THROUGH CHORIONIC VILLUS SAMPLES
Chorionic villi (CV) are comprised of two placental layers; the external cytotrophoblast and the internal mesenchymal core. The mesenchyme is considered to be developmentally more closely related to the fetus.42 Cytogenetic analysis of CV can be achieved either by direct preparation (DP) which reflects the rapidly dividing cytotrophoblasts and/or long-term culture (LTC) of the mesenchyme. Most laboratories in the USA analyze only LTC while European laboratories often analyze both DP and LTC. For cases with evidence for mosaicism, recommended practice is to perform amniocentesis and the detection of the abnormality in amniotic fluid cells is considered to be an indication of true fetal mosaicism (TFM) and those not confirmed are considered to be confined placental mosaicism (CPM). Cytogenetic analysis of both layers has provided insights into the incidence and distribution of chromosomal mosaicism in the placenta and fetus.43
Incidence
The presence of chromosomal mosaicism is observed in approximately 1–2% of CV samples in the general prenatal population and usually involves a disomy-trisomy mosaicism due to both meiotic or mitotic errors.43–48 Approximately 87% of the abnormal cell lines in CVS are confined to the placenta with the remaining 13% determined to be true fetal mosaicism. Figures 3 and 4 summarize observed rates for the different types of mosaicism in CVS based on 57,539 CVS analyzed by the TOMA laboratory.49
Figure 3:
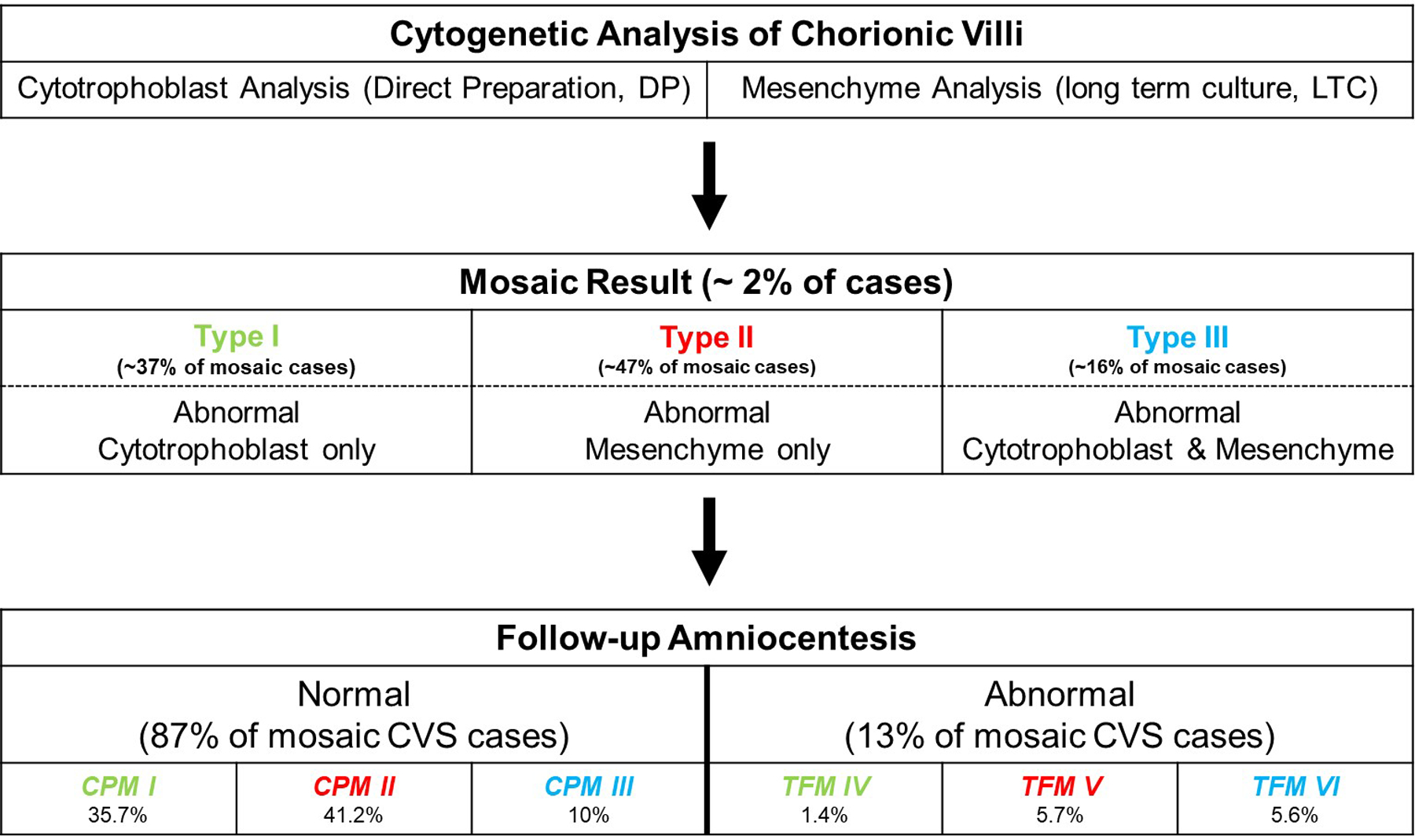
Schematic representation showing the final classification of mosaicism detected at the time of chorionic villus sampling after follow-up confirmatory amniocentesis. CPM, Confined Placental Mosaicism; TFM, True Fetal Mosaicism. If the chromosome abnormality is only detected in the trophoblast and is not confirmed at the time of amniocentesis, it is classified as CPM I. If it is confirmed, it is referred to as TFM IV. Similarly, if the abnormality is only detected in the mesenchyme and is not confirmed at the time of amniocentesis, it is classified as CPM II. If it is confirmed, it is referred to as TFM V. If the abnormality is detected in both layers and not confirmed at the time of amniocentesis, it is classified as CPM III. If confirmed, it is referred to as TFM VI. Data derived from Malvestiti et al.43 and Benn et al.49
Figure 4:
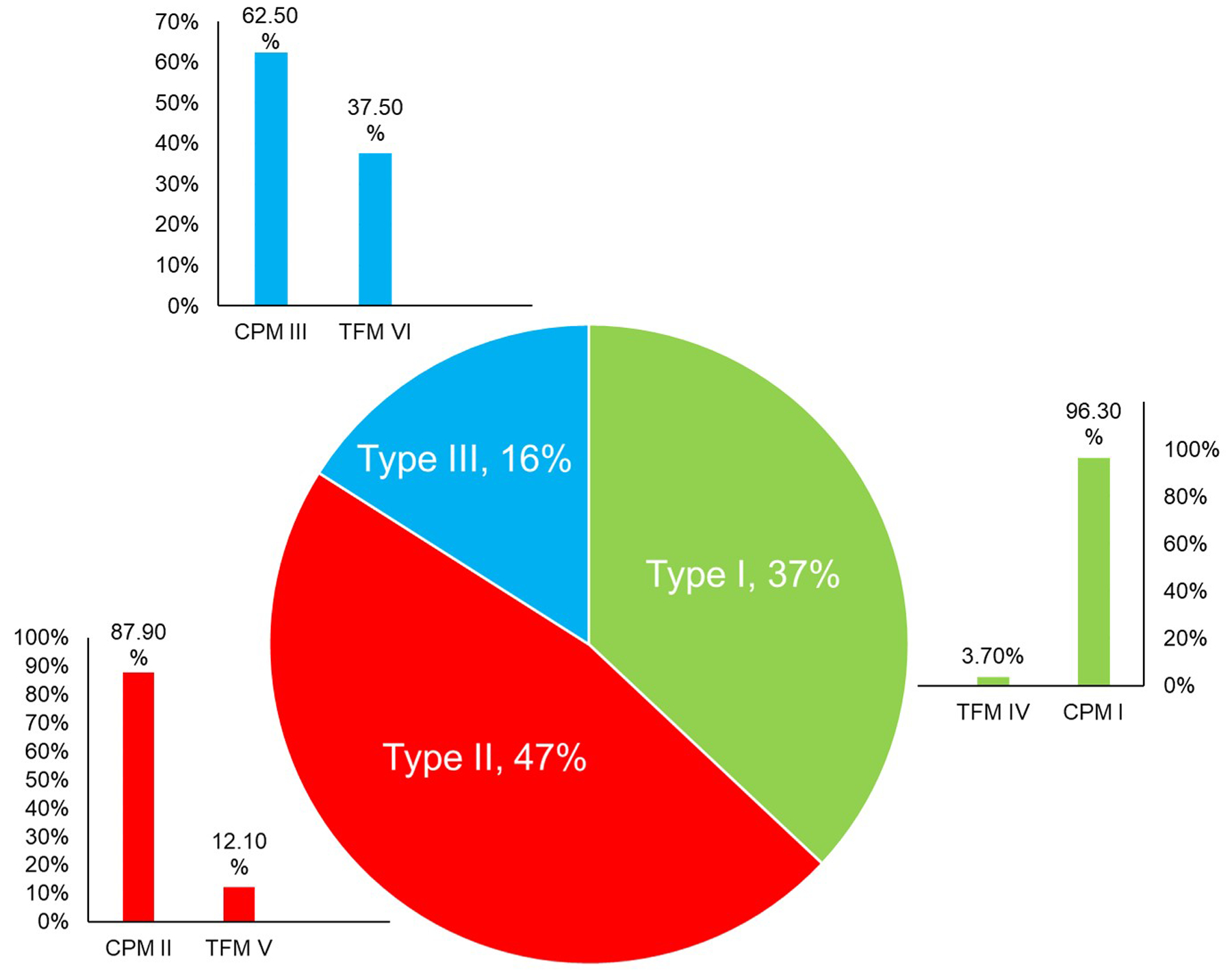
Frequency of the type of mosaicism observed at the time of CVS (Pie Chart) and likelihood of each type being confirmed at the time of amniocentesis (Bar Charts). CPM: Confined Placental Mosaicism. TFM: True Fetal Mosaicism. Data derived from Malvestiti et al.43 and Benn et al.49
Clinical significance
Figures 3 and 4 demonstrate that the lineage(s) in which an abnormality is detected is an important consideration when assessing the likelihood that the abnormality will be confirmed through amniocentesis. It is emphasized that most mosaicisms detected through CVS are not confirmed at amniocentesis.
In addition to the type of mosaicism observed at CVS, other factors influence the likelihood of confirming TFM on amniocytes. The mechanism of formation of the mosaic aneuploidy (mitotic or meiotic) is important. Meiotic abnormalities are maternal age dependent and at the highest risk for fetal abnormalities and UPD. Mosaicism present in both the cytotrophoblast and mesenchyme are mainly meiotic in origin and the absence of any normal cells in a layer also increases the chance that the abnormality is meiotic in origin.50,51 In addition to the common trisomies (13, 18, 21), mosaic trisomies with an increased rate of meiotic origin include those involving chromosomes 9, 15, 16 and 22.50 Conversely, mosaicism involving trisomies 2 and 7 are frequently detected in CVS but almost never confirmed as TFM and are rarely associated with UPD implying a mostly mitotic origin.43,52–55 Mosaic trisomies of chromosomes 4, 8, 9, 12, 14, 15, 16 and 20 show a variable likelihood of being TFM. All other mosaic aneuploidies have a very low chance of being TFM.43,53,56
Figure 5 shows the overall likelihood of confirming TFM for a select group of chromosomes. It should be noted that even with a normal result with high amniocyte cell counts, hidden mosaicism cannot be excluded. The presence of ultrasound anomalies is often an indicator of TFM. However, the absence of fetal abnormalities, while somewhat reassuring, does not completely eliminate the chance of TFM or later clinical consequences. There is never any possibility of absolute assurance that a mosaicism detected through CVS will be inconsequential.
Figure 5:
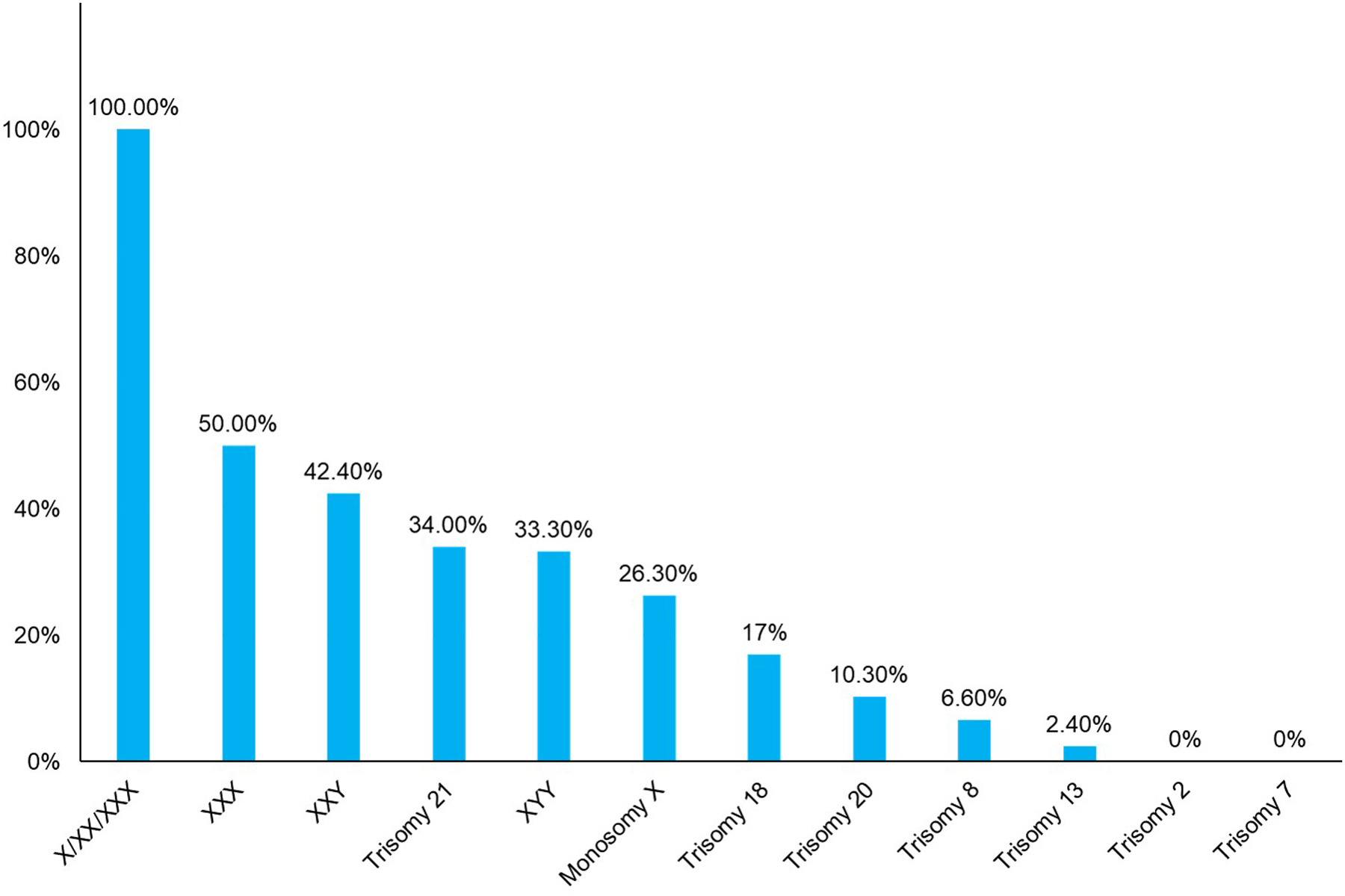
Overall likelihood of confirming true fetal mosaicism at amniocentesis for select chromosomes. Data derived from Malvestiti et al.43 and Benn et al.49
Chromosomes known to be viable in the trisomic state (e.g., trisomy 21) are more worrisome when detected as mosaic at the time of CVS. If the trisomy detected at CVS involves a chromosome that is usually considered to be incompatible with life, such as trisomy 10, the abnormality is more likely to represent confined placental mosaicism. Recommended follow-up studies include amniocentesis as well as high resolution ultrasound to assess the fetal morphology. When amniocentesis is done as a follow-up to a mosaic CVS, the laboratory should be made aware of the results of the CVS as increased cell counts and appropriate FISH studies could be performed to rule out mosaicism in the amniocentesis sample with a higher degree of confidence compared with routine cytogenetic studies. FISH studies using probes specific for the chromosome of interest offer the benefit of directly and rapidly assessing a larger number of uncultured cells, concomitantly eliminating artifacts caused by cell culturing. Chromosomal microarray analysis (CMA) may also be useful for detecting mosaicism in uncultured cells but may fail to detect mosaicism less than 20%.57 A particularly challenging aspect of counseling patients is conveying the message that TFM can never be ruled out with 100% certainly as cryptic mosaicism can still exist in other tissues even if apparently absent in amniocytes.
The possibility that mosaicism that is confined to the placenta might be associated with pregnancy complications has been a long-standing controversy. The most recent data indicates that, except for trisomy 16, for all other CPMs involving a rare autosomal trisomy (RAT, defined as an autosomal trisomy not involving chromosome 13, 18, or 21), birthweight, NICU admission rates, hypertensive disorder frequencies, and spontaneous pre-term birth rates, and apgar scores, are not significantly different from control pregnancies without CPM.58 Notable exception are the small proportion of cases where the abnormality is present in both the cytrophoblast and mesenchymal layers. These cases are associated with low birthweight.59,60 CPM for trisomy 16 prompts special clinical management. In addition to fetal malformations and intrauterine demise, CPM for trisomy 16 carries a risk for fetal intrauterine growth restriction (IUGR), preeclampsia, and preterm birth.61–63 A diagnosis of trisomy 16 mosaicism therefore requires follow-up monitoring for hypertensive disorders and fetal growth restriction. Placental dysfunction and insufficiency are thought to be key factors contributing to both maternal preeclampsia and IUGR.64–66 A 2018 Danish cohort study indicated a higher risk of adverse pregnancy outcome in trisomy 16 CPM cases referred initially for CVS because of notably low PAPP-A levels compared to patients referred initially for CVS for advanced maternal age.62 This dataset highlights the potential differences in outcome according to indication and may explain some of the variance reported in the literature. It is important to note that favorable pregnancy outcomes are observed in a significant proportion of patients. In the Danish study, 32% of trisomy 16 CPM cases included children born at term with normal birthweight and no malformations.62 Sparks and colleagues reported on a similar sized cohort that included both trisomy 16 CPM cases as well as trisomy 16 TFM cases.63 While there was a significantly higher incidence of congenital anomalies, particularly musculoskeletal, in trisomy 16 TFM patients versus those with CPM, the majority of children (TFM + CPM) demonstrated normal neurodevelopmental outcomes and high health-related quality of life scores.63 True fetal mosaic trisomy 16 is associated with a highly variable phenotype with the full spectrum of anomalies including: IUGR, asymmetric craniofacial and body findings, hearing loss, hypospadias, scoliosis, abnormal pigmentation of skin, pre-term delivery, maternal hypertension, two vessel cord, clinodactyly, pulmonary hypoplasia, and congenital heart defect.67,68 The degree of severity is likely related to the ratio of trisomic to disomic cells in the various organ systems.
MOSAICISM IDENTIFIED THROUGH AMNIOTIC FLUID SAMPLES
Amniotic fluid cells derive from various fetal anatomical organs such as urogenital tract, respiratory apparatus, and epithelial system.69–71 Samples can also contain maternal tissue fragments, maternal blood and placental or membrane cells. Laboratory methods include LTC for karyotyping and either DC or LTC for fluorescence in situ hybridization (FISH), chromosome microarray and other cytogenomic technologies. For routine karyotyping, an abnormality can sometimes be confined to a single cell or culture and these are generally dismissed as cell culture or chromosome preparation artifacts or otherwise insignificant (“pseudomaicism”). Specific protocols exist for the work-up of cases to help distinguish these from true mosaicism.72 The diagnosis of true mosaicism is based on the detection of the same abnormality in two or more independent cell cultures.
Incidence
Based on amniocenteses performed mostly because of advanced maternal age, approximately 0.2% of cases show true mosaicism, 0.76% show a multiple cell pseudomosaicism and 3.73% show a single cell pseudomosaicism.72 True mosaicism generally involves a sex chromosome abnormality or either trisomy 21, 18 or 13.
Clinical significance
Although the results from cytogenetic analysis of amniotic fluid cells is considered to be fully diagnostic for the fetal karyotype, it is important to recognize that mosaic findings are often not confirmed in follow-up studies on the baby or abortus.72 This may reflect tissue-specific differences in cell distributions or limitations in the testing. In addition, the likelihood of confirmation is also influenced by which chromosome is involved. Certain chromosomes are more likely to be confirmed (Sex chromosomes and trisomy 21) while others (chromosomes 2 and 7) are almost never seen on follow-up amniocentesis (Figure 5).43,52–55 For cases where mosaicism is diagnosed in amniotic fluid cells, the relative proportion of abnormal cells in amniotic fluid is poorly correlated with that in fetal tissues. Moreover, a low level of mosaicism may not express itself as an obvious abnormality in the baby or fetus. One of the most clear-cut examples of this occurs with loss of a sex chromosome where only a low proportion of mosaic cases show an abnormal phenotype. For RATs, much of the available data on outcomes and confirmation are based on case report compilations that are subject to publication biases.
TFM diagnosed by amniocentesis does not necessarily guarantee a poor outcome as the resulting phenotype would depend on the level of abnormal cells and its distribution in critical tissues such as brain, heart, etc. In a 2018 study of 17 consecutive prenatal trisomy 8 mosaicism cases, an overall positive prognosis was observed for TFM cases with low-level mosaicism in amniotic fluid when no congenital anomalies were apparent by ultrasound inspection.73 In reality, unless ultrasound anomalies are already evident in the fetus, precise predictions about the prognosis in individual cases are not possible. Genetic counseling by appropriately trained geneticists or healthcare providers for patients diagnosed with CPM/TFM at the time of amniocentesis it is crucial to help them understand the complexities of the various possible outcomes and steer them to support group resources such as UNIQUE (www.rarechromo.org).
RELEVANCE OF MOSAICISM TO NON-INVASIVE PRENATAL TESTING USING CELL-FREE DNA
Cell-free DNA (cfDNA) testing is based on quantification of the cell-free DNA fragments circulating in the maternal plasma which derive from both the mother and the apoptotic trophoblasts.74–76 Therefore, results from non-invasive prenatal testing (NIPT) using cfDNA essentially reflect the genetic constitution of the cytotrophoblast and not the fetus itself. NIPT tests are not designed to detect mosaicism although it is sometimes apparent when the laboratory assessment of presence or absence of abnormality appears to be intermediate and incompatible with the level of fetal DNA in the sample.77 Mosaicism can also be strongly suspected based on NIPT results that would be incompatible with viability, for example, for an autosomal monosomy or a RAT. Mosaicism cannot be expected to be detected when there is only a small, or non-existent, change in the overall proportion of fetal DNA fragments (for example, 45,X/47,XXX).
Incidence
There are currently no estimates for the expected proportion of mosaic cases that might be detectable by NIPT. However, there is information on cases where a RAT is detected by genome-wide NIPT in a viable pregnancy and the most likely explanation is mosaicism. The reported incidence of RATs varies considerably from as low as 1:83578 to as high as approximately 1:91.79 Apart from study design differences, the variance appears related to the reason for testing which reflects either a high or low a priori risk.79 The recent TRIDENT-2 NIPT study from the Netherlands showed a RAT incidence of 0.18% (~ 1:556) in a “general risk” population. This contrasts the 1.11% (~1:91) reported in their TRIDENT-1 study which assessed a “high risk” population.79 The latest pooled analysis by Benn and colleagues of 10 genome-wide NIPT studies calculated the average RAT incidence, irrespective of indication, to be approximately 1:310 (0.32%).49 This rate includes RATs attributable to an unrecognized demised twin80 or a maternal malignancy.81,82 The most common RATs detected by NIPT were trisomies 16, 22 and 15 with a frequency of >9% for each.49 The incidence of RATs appears to be statistically higher in CVS trophoblasts (0.41%) compared to NIPT-based studies (0.32%)49.
Clinical significance
There are three primary outcomes following a prediction of a RAT by a NIPT whole genome screen. As with CVS, CPM is the most likely explanation (90–94%%).49,79,83,84 If the RAT is present in a very high proportion of cells, miscarriage is likely.5,85,86 If the pregnancy is viable and the RAT is confirmed by amniocentesis, there may be a variable phenotype ranging from normal to severely affected.43 The challenge comes with the inability to accurately predict which outcome will be present in each specific case. Comprehensive ultrasound is indicated for the detection of anatomic anomalies and growth restriction.58,78,87–92
Benn et al. compiled outcome information from six studies that included 151 cases with RATs identified by whole-genome NIPT. A normal live birth was reported in 41.1%; fetal loss in 27.2%; fetal anomalies in 7.3%, clinically significant UPD in 2.0%; and fetal growth restriction or low birth weight (determined by variable cutoffs used in different studies) in 14.6%.49 Cytogenetic confirmation studies were not performed for every case. The value of reporting RATs from a NIPT whole genome screen remains highly controversial and much work is needed to assess clinical utility of detecting RATs through NIPT.
POSSIBLE SIGNIFICANCE OF UNDETECTED MOSAICISM
From the foregoing discussion it should be clear that progression from embryo to birth is associated with a massive reduction in the proportion of cases with true mosaic chromosome abnormalities. Some of this will be associated with early embryo lethality, failure to implant, spontaneous fetal death, and correction of the aneuploid line. Some may also be associated with a selective disadvantage for the cells with chromosome imbalance. Even though an abnormal cell line may diminish to the point that it is at a low level, the developmental consequences of its presence early in pregnancy may still be apparent. Examples of this could be growth asymmetry and pigmental abnormalities, or other conditions caused by hypoplasia.
What is the consequence of having a low-level residual abnormal cell population that may be substantially confined to specific tissues and undetected in those tissues routinely analyzed when a chromosome abnormality is suspected? Possibilities include recurrence for common autosomal trisomies,93 recurrent pregnancy loss with identical chromosome imbalances, premature ovarian failure,94 and cancer.95 The term “occult mosaicism” has been proposed to describe the situation where mosaicism is hidden but suspected on the basis of clinical findings.96 When ultrasound anomalies are present, there is a greater index of suspicion for occult mosaicism in other tissues when only normal cells are observed in amniocytes. The application of contemporary molecular cytogenomic methods provides opportunities to search for low-level mosaicism in tissues previously not amenable to conventional cytogenetic analysis and to resolve some of these relevant questions. For example, trisomy 8 detected in CVS is rarely identified by karyotype in subsequent amniocyte cultures, likely due to selective growth disadvantage.97,98 However, direct analysis of uncultured amniocytes by fluorescence in situ hybridization (FISH) or chromosomal microarray analysis (CMA) has proven efficacious in identifying trisomy 8 mosaicism in follow-up amniocentesis studies.99,100
CONCLUSION
In the microarray and NGS era, clinicians and patients often remark on the challenges and difficulties of encountering variants of uncertain significance (VUS). In reality, uncertain significance is not a new concept in clinical genetics as we have been dealing with uncertainty in outcome since the dawn of prenatal cytogenetic diagnosis, primarily in the form of marker chromosomes, mosaicism and de novo rearrangements. The significance of mosaicism in a prenatal diagnosis setting is well recognized and rather than refer to it as a FUS (finding of uncertain significance), we should approach mosaicism as a “Finding of Uncertain Outcome” (FUO). Conversely, the clinical utility of actively investigating and reporting VUS/FUO in a screening framework, such as that of NIPT/cfDNA testing, is highly controversial. Our ability to predict the clinical impact is guided by the time of ascertainment and then by the results of follow-up procedures like ultrasound and amniocentesis. Ultimately, unless we biopsy every major fetal tissue (brain, heart, kidney etc…), and, in the absence of apparent fetal anomalies, the prenatal finding of mosaicism will remain an FUO.
What’s already known about this topic?:
Clinical outcome for cases diagnosed with chromosomal mosaicism is fraught with uncertainty.
Patient management requires a comprehensive knowledge of the potential outcomes for each specific mosaic abnormality
What does this study add?:
A review of current knowledge of the origins and clinical impact of chromosomal mosaicism
We discuss how the frequency, spectrum, and clinical signifance of mosaic chromosome abnormalities differ according to how they were ascertained
Footnotes
COI statement: F.R.G., is full-time employee of TomaLab without ownership shares; she is an expert panel member for Roche and consultant for Menarini Silicon. B.L. is a consultant for Igenomix and Natera.
REFERENCES
- 1.Benn P, Grati FR. Aneuploidy in first trimester chorionic villi and spontaneous abortions: Windows into the origin and fate of aneuploidy through embryonic and fetal development. Prenat Diagn. 2020. [DOI] [PubMed] [Google Scholar]
- 2.Chatzimeletiou K, Morrison EE, Prapas N, Prapas Y, Handyside AH. Spindle abnormalities in normally developing and arrested human preimplantation embryos in vitro identified by confocal laser scanning microscopy. Human reproduction. 2005;20(3):672–682. [DOI] [PubMed] [Google Scholar]
- 3.Hardy K, Winston RM, Handyside AH. Binucleate blastomeres in preimplantation human embryos in vitro: failure of cytokinesis during early cleavage. J Reprod Fertil. 1993;98(2):549–558. [DOI] [PubMed] [Google Scholar]
- 4.Holubcova Z, Blayney M, Elder K, Schuh M. Human oocytes. Error-prone chromosome-mediated spindle assembly favors chromosome segregation defects in human oocytes. Science. 2015;348(6239):1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levy B, Sigurjonsson S, Pettersen B, et al. Genomic imbalance in products of conception: single-nucleotide polymorphism chromosomal microarray analysis. Obstet Gynecol. 2014;124(2 Pt 1):202–209. [DOI] [PubMed] [Google Scholar]
- 6.McCoy RC, Newnham LJ, Ottolini CS, et al. Tripolar chromosome segregation drives the association between maternal genotype at variants spanning PLK4 and aneuploidy in human preimplantation embryos. Hum Mol Genet. 2018;27(14):2573–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menasha J, Levy B, Hirschhorn K, Kardon NB. Incidence and spectrum of chromosome abnormalities in spontaneous abortions: New insights from a 12-year study. Genet Med. 2005;7(4):251–263. [DOI] [PubMed] [Google Scholar]
- 8.Yaguchi K, Yamamoto T, Matsui R, et al. Tetraploidy-associated centrosome overduplication in mouse early embryos. Commun Integr Biol. 2018;11(4):e1526605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev. 2007;17(2):157–162. [DOI] [PubMed] [Google Scholar]
- 10.Paim LMG, FitzHarris G. Tetraploidy causes chromosomal instability in acentriolar mouse embryos. Nat Commun. 2019;10(1):4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daughtry BL, Chavez SL. Chromosomal instability in mammalian pre-implantation embryos: potential causes, detection methods, and clinical consequences. Cell Tissue Res. 2016;363(1):201–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benn P. Uniparental disomy: Origin, frequency, and clinical significance. Prenat Diagn. 2020. [DOI] [PubMed] [Google Scholar]
- 13.Grati FR. Chromosomal Mosaicism in Human Feto-Placental Development: Implications for Prenatal Diagnosis. J Clin Med. 2014;3(3):809–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Echten-Arends J, Mastenbroek S, Sikkema-Raddatz B, et al. Chromosomal mosaicism in human preimplantation embryos: a systematic review. Hum Reprod Update. 2011;17(5):620–627. [DOI] [PubMed] [Google Scholar]
- 15.Fragouli E, Alfarawati S, Spath K, et al. Analysis of implantation and ongoing pregnancy rates following the transfer of mosaic diploid-aneuploid blastocysts. Hum Genet. 2017;136(7):805–819. [DOI] [PubMed] [Google Scholar]
- 16.Greco E, Minasi MG, Fiorentino F. Healthy Babies after Intrauterine Transfer of Mosaic Aneuploid Blastocysts. N Engl J Med. 2015;373(21):2089–2090. [DOI] [PubMed] [Google Scholar]
- 17.Huang J, Yan L, Lu S, Zhao N, Qiao J. Re-analysis of aneuploidy blastocysts with an inner cell mass and different regional trophectoderm cells. J Assist Reprod Genet. 2017;34(4):487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Munne S, Blazek J, Large M, et al. Detailed investigation into the cytogenetic constitution and pregnancy outcome of replacing mosaic blastocysts detected with the use of high-resolution next-generation sequencing. Fertil Steril. 2017;108(1):62–71 e68. [DOI] [PubMed] [Google Scholar]
- 19.Spinella F, Fiorentino F, Biricik A, et al. Extent of chromosomal mosaicism influences the clinical outcome of in vitro fertilization treatments. Fertil Steril. 2018;109(1):77–83. [DOI] [PubMed] [Google Scholar]
- 20.Victor AR, Griffin DK, Brake AJ, et al. Assessment of aneuploidy concordance between clinical trophectoderm biopsy and blastocyst. Human reproduction. 2019;34(1):181–192. [DOI] [PubMed] [Google Scholar]
- 21.Victor AR, Tyndall JC, Brake AJ, et al. One hundred mosaic embryos transferred prospectively in a single clinic: exploring when and why they result in healthy pregnancies. Fertil Steril. 2019;111(2):280–293. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L, Wei D, Zhu Y, Gao Y, Yan J, Chen ZJ. Rates of live birth after mosaic embryo transfer compared with euploid embryo transfer. J Assist Reprod Genet. 2019;36(1):165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zore T, Kroener LL, Wang C, et al. Transfer of embryos with segmental mosaicism is associated with a significant reduction in live-birth rate. Fertil Steril. 2019;111(1):69–76. [DOI] [PubMed] [Google Scholar]
- 24.Gruhn JR, Zielinska AP, Shukla V, et al. Chromosome errors in human eggs shape natural fertility over reproductive life span. Science. 2019;365(6460):1466–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Capalbo A, Ubaldi FM, Rienzi L, Scott R, Treff N. Detecting mosaicism in trophectoderm biopsies: current challenges and future possibilities. Human reproduction. 2017;32(3):492–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marin D, Xu J, Treff NR. Preimplantation genetic testing for aneuploidy: A review of published blastocyst reanalysis concordance data. Prenat Diagn. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Girardi L, Serdarogullari M, Patassini C, et al. Incidence, Origin, and Predictive Model for the Detection and Clinical Management of Segmental Aneuploidies in Human Embryos. Am J Hum Genet. 2020;106(4):525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lawrenz B, El Khatib I, Linan A, et al. The clinicians dilemma with mosaicism-an insight from inner cell mass biopsies. Human reproduction. 2019;34(6):998–1010. [DOI] [PubMed] [Google Scholar]
- 29.Navratil R, Horak J, Hornak M, et al. Concordance of various chromosomal errors among different parts of the embryo and the value of re-biopsy in embryos with segmental aneuploidies. Mol Hum Reprod. 2020;26(4):269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sachdev NM, McCulloh DH, Kramer Y, Keefe D, Grifo JA. The reproducibility of trophectoderm biopsies in euploid, aneuploid, and mosaic embryos using independently verified next-generation sequencing (NGS): a pilot study. J Assist Reprod Genet. 2020;37(3):559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lledo B, Morales R, Ortiz JA, et al. Implantation potential of mosaic embryos. Syst Biol Reprod Med. 2017;63(3):206–208. [DOI] [PubMed] [Google Scholar]
- 32.Munne S, Spinella F, Grifo J, et al. Clinical outcomes after the transfer of blastocysts characterized as mosaic by high resolution Next Generation Sequencing- further insights. Eur J Med Genet. 2020;63(2):103741. [DOI] [PubMed] [Google Scholar]
- 33.Zhang YX, Chen JJ, Nabu S, et al. The Pregnancy Outcome of Mosaic Embryo Transfer: A Prospective Multicenter Study and Meta-Analysis. Genes (Basel). 2020;11(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kushnir VA, Darmon SK, Barad DH, Gleicher N. Degree of mosaicism in trophectoderm does not predict pregnancy potential: a corrected analysis of pregnancy outcomes following transfer of mosaic embryos. Reproductive biology and endocrinology : RB&E. 2018;16(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Capalbo A, Poli M, Rienzi L, et al. A prospective double-blinded non-selection trial of reproductive outcomes and chromosomal normalcy of newborns derived from putative low/moderate-degree mosaic IVF embryos. MedRxiv. 2021( 10.1101/2021.02.07.21251201). [DOI] [Google Scholar]
- 36.Gleicher N, Metzger J, Croft G, Kushnir VA, Albertini DF, Barad DH. A single trophectoderm biopsy at blastocyst stage is mathematically unable to determine embryo ploidy accurately enough for clinical use. Reproductive biology and endocrinology : RB&E. 2017;15(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Practice C, Genetic Counseling Professional Group of the American Society for Reproductive Medicine. Electronic address aao. Clinical management of mosaic results from preimplantation genetic testing for aneuploidy (PGT-A) of blastocysts: a committee opinion. Fertil Steril. 2020;114(2):246–254. [DOI] [PubMed] [Google Scholar]
- 38.Magli MC, Albanese C, Crippa A, et al. Permanence of de novo segmental aneuploidy in sequential embryo biopsies. Human reproduction. 2020;35(4):759–769. [DOI] [PubMed] [Google Scholar]
- 39.Van der Aa N, Cheng J, Mateiu L, et al. Genome-wide copy number profiling of single cells in S-phase reveals DNA-replication domains. Nucleic Acids Res. 2013;41(6):e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cram DS, Leigh D, Handyside A, et al. PGDIS Position Statement on the Transfer of Mosaic Embryos 2019. Reprod Biomed Online. 2019;39 Suppl 1:e1–e4. [DOI] [PubMed] [Google Scholar]
- 41.Grati FR, Gallazzi G, Branca L, Maggi F, Simoni G, Yaron Y. An evidence-based scoring system for prioritizing mosaic aneuploid embryos following preimplantation genetic screening. Reprod Biomed Online. 2018;36(4):442–449. [DOI] [PubMed] [Google Scholar]
- 42.Bianchi DW, Wilkins-Haug LE, Enders AC, Hay ED. Origin of extraembryonic mesoderm in experimental animals: relevance to chorionic mosaicism in humans. Am J Med Genet. 1993;46(5):542–550. [DOI] [PubMed] [Google Scholar]
- 43.Malvestiti F, Agrati C, Grimi B, et al. Interpreting mosaicism in chorionic villi: results of a monocentric series of 1001 mosaics in chorionic villi with follow-up amniocentesis. Prenat Diagn. 2015;35(11):1117–1127. [DOI] [PubMed] [Google Scholar]
- 44.Medical Research Council European trial of chorion villus sampling. MRC working party on the evaluation pf chorion villus sampling. Lancet. 1991;337(8756):1491–1499. [PubMed] [Google Scholar]
- 45.Cytogenetic analysis of chorionic villi for prenatal diagnosis: an ACC collaborative study of U.K. data. Association of Clinical Cytogeneticists Working Party on Chorionic Villi in Prenatal Diagnosis. Prenat Diagn. 1994;14(5):363–379. [DOI] [PubMed] [Google Scholar]
- 46.Ledbetter DH, Zachary JM, Simpson JL, et al. Cytogenetic results from the U.S. Collaborative Study on CVS. Prenat Diagn. 1992;12(5):317–345. [DOI] [PubMed] [Google Scholar]
- 47.Vejerslev LO, Mikkelsen M. The European collaborative study on mosaicism in chorionic villus sampling: data from 1986 to 1987. Prenat Diagn. 1989;9(8):575–588. [DOI] [PubMed] [Google Scholar]
- 48.Wolstenholme J, Rooney DE, Davison EV. Confined placental mosaicism, IUGR, and adverse pregnancy outcome: a controlled retrospective U.K. collaborative survey. Prenat Diagn. 1994;14(5):345–361. [DOI] [PubMed] [Google Scholar]
- 49.Benn P, Malvestiti F, Grimi B, Maggi F, Simoni G, Grati FR. Rare autosomal trisomies: comparison of detection through cell-free DNA analysis and direct chromosome preparation of chorionic villus samples. Ultrasound Obstet Gynecol. 2019;54(4):458–467. [DOI] [PubMed] [Google Scholar]
- 50.Robinson WP, Barrett IJ, Bernard L, et al. Meiotic origin of trisomy in confined placental mosaicism is correlated with presence of fetal uniparental disomy, high levels of trisomy in trophoblast, and increased risk of fetal intrauterine growth restriction. Am J Hum Genet. 1997;60(4):917–927. [PMC free article] [PubMed] [Google Scholar]
- 51.Wolstenholme J. Confined placental mosaicism for trisomies 2, 3, 7, 8, 9, 16, and 22: their incidence, likely origins, and mechanisms for cell lineage compartmentalization. Prenat Diagn. 1996;16(6):511–524. [DOI] [PubMed] [Google Scholar]
- 52.Battaglia P, Baroncini A, Mattarozzi A, et al. Cytogenetic follow-up of chromosomal mosaicism detected in first-trimester prenatal diagnosis. Prenat Diagn. 2014;34(8):739–747. [DOI] [PubMed] [Google Scholar]
- 53.Hahnemann JM, Vejerslev LO. European collaborative research on mosaicism in CVS (EUCROMIC)--fetal and extrafetal cell lineages in 192 gestations with CVS mosaicism involving single autosomal trisomy. Am J Med Genet. 1997;70(2):179–187. [DOI] [PubMed] [Google Scholar]
- 54.Sachs ES, Jahoda MG, Los FJ, Pijpers L, Reuss A, Wladimiroff JW. Interpretation of chromosome mosaicism and discrepancies in chorionic villi studies. Am J Med Genet. 1990;37(2):268–271. [DOI] [PubMed] [Google Scholar]
- 55.Sifakis S, Staboulidou I, Maiz N, Velissariou V, Nicolaides KH. Outcome of pregnancies with trisomy 2 cells in chorionic villi. Prenat Diagn. 2010;30(4):329–332. [DOI] [PubMed] [Google Scholar]
- 56.Grati FR, Bajaj K, Zanatta V, et al. Implications of fetoplacental mosaicism on cell-free DNA testing for sex chromosome aneuploidies. Prenat Diagn. 2017;37(10):1017–1027. [DOI] [PubMed] [Google Scholar]
- 57.Levy B, Wapner R. Prenatal diagnosis by chromosomal microarray analysis. Fertil Steril. 2018;109(2):201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grati FR, Ferreira J, Benn P, et al. Outcomes in pregnancies with a confined placental mosaicism and implications for prenatal screening using cell-free DNA. Genet Med. 2020;22(2):309–316. [DOI] [PubMed] [Google Scholar]
- 59.Toutain J, Horovitz J, Saura R. Type 3 confined placental mosaicisms excluding trisomies 16 are also associated with adverse pregnancy outcomes. Genet Med. 2020;22(2):446–447. [DOI] [PubMed] [Google Scholar]
- 60.Benn P, Ferreira J, Grati FR. Response to Toutain et al. Genet Med. 2020;22(2):444–445. [DOI] [PubMed] [Google Scholar]
- 61.Brandenburg H, Los FJ, In’t Veld P. Clinical significance of placenta-confined nonmosaic trisomy 16. Am J Obstet Gynecol. 1996;174(5):1663–1664. [DOI] [PubMed] [Google Scholar]
- 62.Grau Madsen S, Uldbjerg N, Sunde L, Becher N, Danish Fetal Medicine Study G, Danish Clinical Genetics Study G. Prognosis for pregnancies with trisomy 16 confined to the placenta: A Danish cohort study. Prenat Diagn. 2018;38(13):1103–1110. [DOI] [PubMed] [Google Scholar]
- 63.Sparks TN, Thao K, Norton ME. Mosaic trisomy 16: what are the obstetric and long-term childhood outcomes? Genet Med. 2017;19(10):1164–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blair JD, Langlois S, McFadden DE, Robinson WP. Overlapping DNA methylation profile between placentas with trisomy 16 and early-onset preeclampsia. Placenta. 2014;35(3):216–222. [DOI] [PubMed] [Google Scholar]
- 65.Langlois S, Yong PJ, Yong SL, et al. Postnatal follow-up of prenatally diagnosed trisomy 16 mosaicism. Prenat Diagn. 2006;26(6):548–558. [DOI] [PubMed] [Google Scholar]
- 66.Yong PJ, Langlois S, von Dadelszen P, Robinson W. The association between preeclampsia and placental trisomy 16 mosaicism. Prenat Diagn. 2006;26(10):956–961. [DOI] [PubMed] [Google Scholar]
- 67.Rieubland C, Francis D, Houben L, Corrie S, Bankier A, White SM. Two cases of trisomy 16 mosaicism ascertained postnatally. Am J Med Genet A. 2009;149A(7):1523–1528. [DOI] [PubMed] [Google Scholar]
- 68.Yong PJ, Barrett IJ, Kalousek DK, Robinson WP. Clinical aspects, prenatal diagnosis, and pathogenesis of trisomy 16 mosaicism. J Med Genet. 2003;40(3):175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cremer M, Treiss I, Cremer T, Hager D, Franke WW. Characterization of cells of amniotic fluids by immunological identification of intermediate-sized filaments: presence of cells of different tissue origin. Hum Genet. 1981;59(4):373–379. [DOI] [PubMed] [Google Scholar]
- 70.Morris HH, Bennett MJ. The classification and origin of amniotic fluid cells. Acta Cytol. 1974;18(2):149–154. [PubMed] [Google Scholar]
- 71.Tyden O, Bergstrom S, Nilsson BA. Origin of amniotic fluid cells in mid-trimester pregnancies. Br J Obstet Gynaecol. 1981;88(3):278–286. [DOI] [PubMed] [Google Scholar]
- 72.Benn P. Prenatal diagnosis of chromosomal abnormalities through chorionic villus and amniocentesis. In: MAaM JM, ed. Genetic Disorders of the Fetus. 7th ed. Hoboken, NJ: Wiley; 2016. [Google Scholar]
- 73.Cassina M, Calo A, Salviati L, Alghisi A, Montaldi A, Clementi M. Prenatal detection of trisomy 8 mosaicism: Pregnancy outcome and follow up of a series of 17 consecutive cases. Eur J Obstet Gynecol Reprod Biol. 2018;221:23–27. [DOI] [PubMed] [Google Scholar]
- 74.Faas BH, de Ligt J, Janssen I, et al. Non-invasive prenatal diagnosis of fetal aneuploidies using massively parallel sequencing-by-ligation and evidence that cell-free fetal DNA in the maternal plasma originates from cytotrophoblastic cells. Expert Opin Biol Ther. 2012;12 Suppl 1:S19–26. [DOI] [PubMed] [Google Scholar]
- 75.Flori E, Doray B, Gautier E, et al. Circulating cell-free fetal DNA in maternal serum appears to originate from cyto- and syncytio-trophoblastic cells. Case report. Human reproduction. 2004;19(3):723–724. [DOI] [PubMed] [Google Scholar]
- 76.Tjoa ML, Cindrova-Davies T, Spasic-Boskovic O, Bianchi DW, Burton GJ. Trophoblastic oxidative stress and the release of cell-free feto-placental DNA. Am J Pathol. 2006;169(2):400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rafalko JM, Caldwell S, Tynan J, Almasri E, Weinblatt V, McCullough R. Impact of mosaicism ratio on positive predictive value of cfDNA screening. Prenat Diagn. 2021;41(1):28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Scott F, Bonifacio M, Sandow R, Ellis K, Smet ME, McLennan A. Rare autosomal trisomies: Important and not so rare. Prenat Diagn. 2018;38(10):765–771. [DOI] [PubMed] [Google Scholar]
- 79.van der Meij KRM, Sistermans EA, Macville MVE, et al. TRIDENT-2: National Implementation of Genome-wide Non-invasive Prenatal Testing as a First-Tier Screening Test in the Netherlands. Am J Hum Genet. 2019;105(6):1091–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Curnow KJ, Wilkins-Haug L, Ryan A, et al. Detection of triploid, molar, and vanishing twin pregnancies by a single-nucleotide polymorphism-based noninvasive prenatal test. Am J Obstet Gynecol. 2015;212(1):79 e71–79. [DOI] [PubMed] [Google Scholar]
- 81.Benn P, Plon SE, Bianchi DW. Current Controversies in Prenatal Diagnosis 2: NIPT results suggesting maternal cancer should always be disclosed. Prenat Diagn. 2019;39(5):339–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bianchi DW, Chudova D, Sehnert AJ, et al. Noninvasive Prenatal Testing and Incidental Detection of Occult Maternal Malignancies. JAMA. 2015;314(2):162–169. [DOI] [PubMed] [Google Scholar]
- 83.Van Opstal D, Eggenhuizen GM, Joosten M, et al. Noninvasive prenatal testing as compared to chorionic villus sampling is more sensitive for the detection of confined placental mosaicism involving the cytotrophoblast. Prenat Diagn. 2020;40(10):1338–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Van Opstal D, van Maarle MC, Lichtenbelt K, et al. Origin and clinical relevance of chromosomal aberrations other than the common trisomies detected by genome-wide NIPS: results of the TRIDENT study. Genet Med. 2018;20(5):480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Johnson A, Wapner RJ, Davis GH, Jackson LG. Mosaicism in chorionic villus sampling: an association with poor perinatal outcome. Obstet Gynecol. 1990;75(4):573–577. [PubMed] [Google Scholar]
- 86.Wapner RJ, Simpson JL, Golbus MS, et al. Chorionic mosaicism: association with fetal loss but not with adverse perinatal outcome. Prenat Diagn. 1992;12(5):347–355. [DOI] [PubMed] [Google Scholar]
- 87.Baena N, De Vigan C, Cariati E, et al. Prenatal detection of rare chromosomal autosomal abnormalities in Europe. Am J Med Genet A. 2003;118A(4):319–327. [DOI] [PubMed] [Google Scholar]
- 88.Brady PD, Delle Chiaie B, Christenhusz G, et al. A prospective study of the clinical utility of prenatal chromosomal microarray analysis in fetuses with ultrasound abnormalities and an exploration of a framework for reporting unclassified variants and risk factors. Genet Med. 2014;16(6):469–476. [DOI] [PubMed] [Google Scholar]
- 89.Hume RF Jr., Kilmer-Ernst P, Wolfe HM, et al. Prenatal cytogenetic abnormalities: correlations of structural rearrangements and ultrasonographically detected fetal anomalies. Am J Obstet Gynecol. 1995;173(4):1334–1336. [DOI] [PubMed] [Google Scholar]
- 90.Nicolaides KH, Snijders RJ, Gosden CM, Berry C, Campbell S. Ultrasonographically detectable markers of fetal chromosomal abnormalities. Lancet. 1992;340(8821):704–707. [DOI] [PubMed] [Google Scholar]
- 91.Fiorentino F, Bono S, Pizzuti F, et al. The clinical utility of genome-wide non invasive prenatal screening. Prenat Diagn. 2017;37(6):593–601. [DOI] [PubMed] [Google Scholar]
- 92.Pertile MD, Halks-Miller M, Flowers N, et al. Rare autosomal trisomies, revealed by maternal plasma DNA sequencing, suggest increased risk of feto-placental disease. Sci Transl Med. 2017;9(405). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cuckle H, Benn P. Review of epidemiological factors (other than maternal age) that determine the prevalence of common autosomal trisomies. Prenat Diagn. 2020. [DOI] [PubMed] [Google Scholar]
- 94.Lakhal B, Braham R, Berguigua R, et al. Cytogenetic analyses of premature ovarian failure using karyotyping and interphase fluorescence in situ hybridization (FISH) in a group of 1000 patients. Clin Genet. 2010;78(2):181–185. [DOI] [PubMed] [Google Scholar]
- 95.Mark HF. Constitutional trisomy 8 mosaicism and cancer. Cancer Genet Cytogenet. 1996;86(1):87–88. [DOI] [PubMed] [Google Scholar]
- 96.Trisomy Benn P. 16 and trisomy 16 Mosaicism: a review. Am J Med Genet. 1998;79(2):121–133. [PubMed] [Google Scholar]
- 97.Southgate WM, Wagner CL, Shields SM, Cantu ES, Pai GS. Mosaic trisomy 8: a cautionary note regarding missed antenatal diagnosis. J Perinatol. 1998;18(1):78–80. [PubMed] [Google Scholar]
- 98.van Haelst MM, Van Opstal D, Lindhout D, Los FJ. Management of prenatally detected trisomy 8 mosaicism. Prenat Diagn. 2001;21(12):1075–1078. [DOI] [PubMed] [Google Scholar]
- 99.Chen CP, Hsu CY, Chern SR, Wu PS, Chen SW, Wang W. Prenatal diagnosis of mosaic trisomy 8 by amniocentesis in a fetus with ventriculomegaly and dysgenesis of the corpus callosum. Taiwan J Obstet Gynecol. 2020;59(1):127–129. [DOI] [PubMed] [Google Scholar]
- 100.Chen CP, Su YN, Chern SR, et al. Prenatal diagnosis of trisomy 8 mosaicism. Taiwan J Obstet Gynecol. 2012;51(4):666–668. [DOI] [PubMed] [Google Scholar]