

Abstract
Objectives:
Congenital diarrheal disorders is a group of inherited enteropathies presenting in early life and requiring parenteral nutrition. In most cases, genetics may be the key for precise diagnosis. We present an infant girl with chronic congenital diarrhea that resolved after introduction of fructose-based formula but had no identified mutation in the SLC5A1 gene. Using whole exome sequencing (WES) we identified other mutations that better dictated dietary adjustments.
Methods:
WES of the patient and her parents was performed. The analysis focused on recessive model including compound heterozygous mutations. Sanger sequencing was used to validate identified mutations and to screen the patient’s newborn sister and grandparents. Expression and localization analysis were performed in the patient’s duodenal biopsies using immunohistochemistry.
Results:
Using WES we identified a new compound heterozygote mutation in sucrase-isomaltase (SI) gene; a maternal inherited known V577G mutation, and a novel paternal inherited C1531W mutation. Importantly, the newborn offspring carried similar compound heterozygous mutations. Computational predictions suggest that both mutations highly destabilize the protein. SI expression and localization studies determined that the mutated SI protein was not expressed on the brush border membrane in the patient’s duodenal biopsies, verifying the diagnosis of congenital sucrase-isomaltase deficiency (CSID).
Conclusions:
The novel compound heterozygote V577G/C1531W SI mutations lead to lack of SI expression in the duodenal brush border, confirming the diagnosis of CSID. These cases of CSID extend the molecular spectrum of this condition, further directing a more adequate dietary intervention for the patient and newborn sibling.
Keywords: compound heterozygous, congenital sucrase-isomaltase deficiency, sucrase-isomaltase
Congenital diarrheal disorders (CDD) is a rare enteropathy usually presenting in the first weeks of life with failure to gain weight, severe dehydration, and electrolytes disorder, requiring parenteral fluids and nutrition. There are many etiologies for congenital diarrhea; most are related to specific genetic defects inherited as autosomal recessive traits leading to enzymatic deficiencies, carbohydrates, lipid or protein transport disorders, enterocyte polarization disorders, and hormonal, immunological, or metabolic disorders (1). When congenital diarrhea is suspected, careful history, physical examination, routine blood tests and stool culture, and endoscopic procedures are required. If the diagnosis is still not clear, a genetic evaluation with whole exome sequencing may, however, lead to the correct diagnosis.
Within the scope of CDD, congenital sucrase-isomaltase deficiency (CSID) is characterized by the reduction or absence of the disaccharidases sucrase and isomaltase activity that results in malabsorption of carbohydrates and osmotic diarrhea. CSID has variable clinical presentations. It can present in infants and toddlers as severe diarrhea and failure to thrive (FTT), along with irritability, abdominal distention, diaper rash, and vomiting. The clinical investigation reveals steatorrhea due to partial villus atrophy, transient hypoglycemia, acidosis and dehydration, and clinical diagnosis can rely on responses to elimination diets. CSID can present as chronic diarrhea with normal growth in children and as chronic nonspecific diarrhea, or irritable bowel syndrome in adolescents and adults. In older children and adults, the investigation can include sucrose breath hydrogen test (2). Endoscopy in all cases reveals normal or nonspecific macroscopic and histologic findings and disaccharide enzymatic activity, if available, is impaired.
Sucrase-isomaltase (SI) is an enzymatic complex, composed of 2 highly similar subunits, originating from a single polypeptide precursor. SI is essential for the digestion of dietary carbohydrates including starch, sucrose, and isomaltose. SI is a type II transmembrane glycoprotein expressed in the intestinal brush border membrane. There are 5 phenotypes of SI deficiency that lead to abnormal intracellular transport and/or function and are associated with specific SI gene mutations; in phenotypes I and II SI accumulates in the endoplasmic reticulum and the Golgi, respectively (3298A → C p.Q1098P (3) and 1021 T → C p.L340P (4)), in Phenotype III SI is enzymatically inactive, in Phenotype IV SI is mis-sorted to the basal rather than apical membrane (Q117R mutation (5)), and in Phenotype Vonly the isomaltase subunit is correctly targeted to the brush border membrane (3,6,7). Several SI compound heterozygous mutations were previously documented including V577G and G1073D and C1229Y and F1745C (8,9). The c.273_274delAG mutation is a novel frame shift mutation, which is responsible for the high prevalence of CSID among people of Inuit descent (10). Other SI mutations S594P, T694P, and R1367G were reported in patients with clinical symptoms of CSID, but their molecular phenotype is unknown (5,8,11).
Glucose-galactose malabsorption (GGM) is another autosomal recessive disorder caused by a defect in glucose and galactose transport (SLC5A1 gene) across the intestinal brush border. GGM causes a severe form of neonatal watery diarrhea and life-threatening dehydration (12). Although there are several major differences between CSID and GGM, patients of both disorders can tolerate fructose. In the present study, whole exome sequencing (WES) identified novel compound heterozygote mutations in SI gene supporting a diagnosis of CSID rather the GGM as was initially suspected in an infant girl with severe CDD and clinical tolerance of fructose-based formula. CSID diagnosis was further validated by an abnormal protein expression on the brush border in the patient’s duodenal biopsies. The diagnosis enabled less restricted nutritional recommendation for the patient and her newborn sibling carrying similar compound heterozygote mutation.
METHODS
Patients
The patient came for second opinion and subsequent follow-up at the gastroenterology unit in the Edmond and Lily Safra Children’s Hospital, Sheba Medical Center, after a long hospitalization at another hospital. Written informed consent was obtained, and the institutional review board approved the genetic studies.
Exome Sequencing
WES was performed as previously described (13). One microgram of dsDNA was sheared by sonication (Covaris M220 instrument) to an average size of 200 bp. Library construction was performed on a Wafergen Apollo324 that size-selects fragments by double-solid phase reversible immobilization binding with different concentrations of PEG for a high cut and a low cut. After 9 cycles of polymerase chain reaction amplification using the Clonetech Advantage II kit, 350 ng of genomic library was recovered. Three libraries with different barcodes were pooled before exome enrichment (3-plex) using the NimbleGen EZ Exome V2 kit. Library pools were enriched according to the manufacturer’s recommendations and sequenced on an Illumina HiSeq2500, generating around 30 million paired end reads per samples 125 bases long each equivalent to 7.5 Gb of usable high-quality sequence per sample. We used the BWA mem algorithm (version 0.7.12) (14), for alignment of the sequence reads to the human reference genome (hg19). The HaplotypeCaller algorithm of GATK version 3.4 was applied for variant calling, as recommended in the best practice pipeline (15). KGG-seq v.08 (16) was used for annotation of detected variants and in-house scripts were applied for filtering based on family pedigree, local dataset of variants detected in previous sequencing projects, and for detection of suspected compound heterozygous variants appropriately inherited from both parents.
ImmunohistochemistryFormalin-fixed tissues were dehydrated, embedded in paraffin, and sectioned at 4 μm. Slides were warmed up to 60°C for 1 hour, dewaxed in xylene, and rehydrated. Antigen retrieval was performed by heating up to 110°C for 7 minutes and for another 2 minutes at that temperature in a 10 mmol/L citrate buffer pH 6 using a pressure cooker in a microwave (Milestone, Microwave Laboratory Systems, Shelton, CT). After 10 minutes of cooling, the slides were rinsed in trisbuffered saline (TBS) buffer and exposed to 10 minutes endogenous peroxidase block in 3% H2O2/phosphate-buffered saline. After rinses in TBS, sections were incubated overnight at 4°C with primary antibody anti-SI (HPA011897, Sigma-Aldrich, St. Louis, MO, 1:1000). Detection was performed with anti-rabbit Envision+ System-HRP labeled polymer (Dako, K4001). Sections were incubated for 60 minutes at room temperature with the Envision + System-HRP labeled polymer. After TBS rinses, antibody binding was visualized with the substrate-chromogen AEC. Sections were counterstained with hematoxylin and coverslipped with an aqueous mounting fluid (Glycergel Mounting Medium, Dako, Agilent Technologies, Glostrup, Denmark). Images were captured using a light microscope and digitally recorded at ×200 and ×400 magnification.
In Silico Mutagenesis Modeling
We used the FoldX algorithm to assess ΔΔG between wild type and mutations. FoldX is based on empirical energy terms that were compared and correlated with experimental data of a large number of mutations in monomeric proteins and protein-protein complexes (17,18). FoldX (version 3.0b4) was applied to study residues V577 and C1531 in the following manner: the 3D structures that contain the mutated locations (V577 and C1531) were extracted from the Protein Data Bank (pdb id 3lpo for V577 and 3top for C1531) and optimized using the FoldX repairPDB function; structures corresponding to single-point mutants (V577G and C1531W) were generated using the BuildModel protein mutagenesis function; the stability of the wild-type and the mutated structures was calculated using the stability function; and ΔΔG values (ΔGmutant − ΔGwild-type) were obtained. Mutations having ΔΔG < −1 kcal/mol are considered destabilizing and those with ΔΔG ≥3 kcal/mol are considered highly destabilizing.
Solvent Accessibility and Contact Analysis Model
The solvent accessibility and contacts of wild type V577 and C1531 and the mutated residues G577 and W1531 were calculated based on the structure of the wild-type and mutant proteins, using analytic Voronoi tessellation (19,20). The procedure allocates contact surfaces between neighboring atoms. The solvent accessible surface is the remaining surface of each atom that was not assigned to contact other atoms.
RESULTS
Case Presentation
The proband (patient III, 7; Fig. 1A), a 6-week-old girl, born to nonconsanguineous parents of mixed Jewish descent (Afghanistan, India, Iraq, Morocco, and Ashkenaz, Fig. 1), presented with chronic watery diarrhea (up to 13 times a day) dehydration, and FTT since the age of 3 weeks, while fed with a cow’s milk– based formula. Changing to hydrolyzed and amino acid–based formulas showed no improvement and even worsening of symptoms. Her history revealed no signs of infection, and there was no family history of immunodeficiency, or other cases of diarrhea. The father experienced symptoms of lactose intolerance, the mother had no GI symptoms. Physical examination showed no structural abnormalities. Laboratory tests including complete blood count, blood gasses, serum electrolytes, albumin, pancreatic enzymes, thyroid function tests, lipid profile, and vitamin levels were normal. Blood, urine, and stool cultures were negative. Urine vanillylmandelic acid and homovanillic acid, sweat test, fecal elastase, and α1 antitrypsin were normal. T-cell repertoire and immunoglobulins levels were also normal. HIV serology was negative. Abdominal x-ray and ultrasonography showed slight, nonspecific thickening of the intestinal wall. Gastric and intestinal biopsies showed no evidence of microvillus inclusion disease or tufting enteropathy, when examined under light and electron microscopy and staining for neuroendocrine cells was normal. Disaccharide enzymatic activity of duodenal biopsies was unavailable. Diarrhea resolved with fasting and supplementation of total parenteral nutrition, and upon initiation of enteral fructose–based formula (Galactomin 19, Nutricia) the stools remained normal. Based on the onset and severity of symptoms and her tolerance to fructose-based formula, the patient was suspected to have GGM. Sequencing of SLC5A1 coding regions, however, revealed no mutations. The girl was discharged at the age of 5 months, with exclusively enteral fructose-based formula, free of diarrhea, with normal weight gain (5.97 kg, 10th percentile), and development.
FIGURE 1.
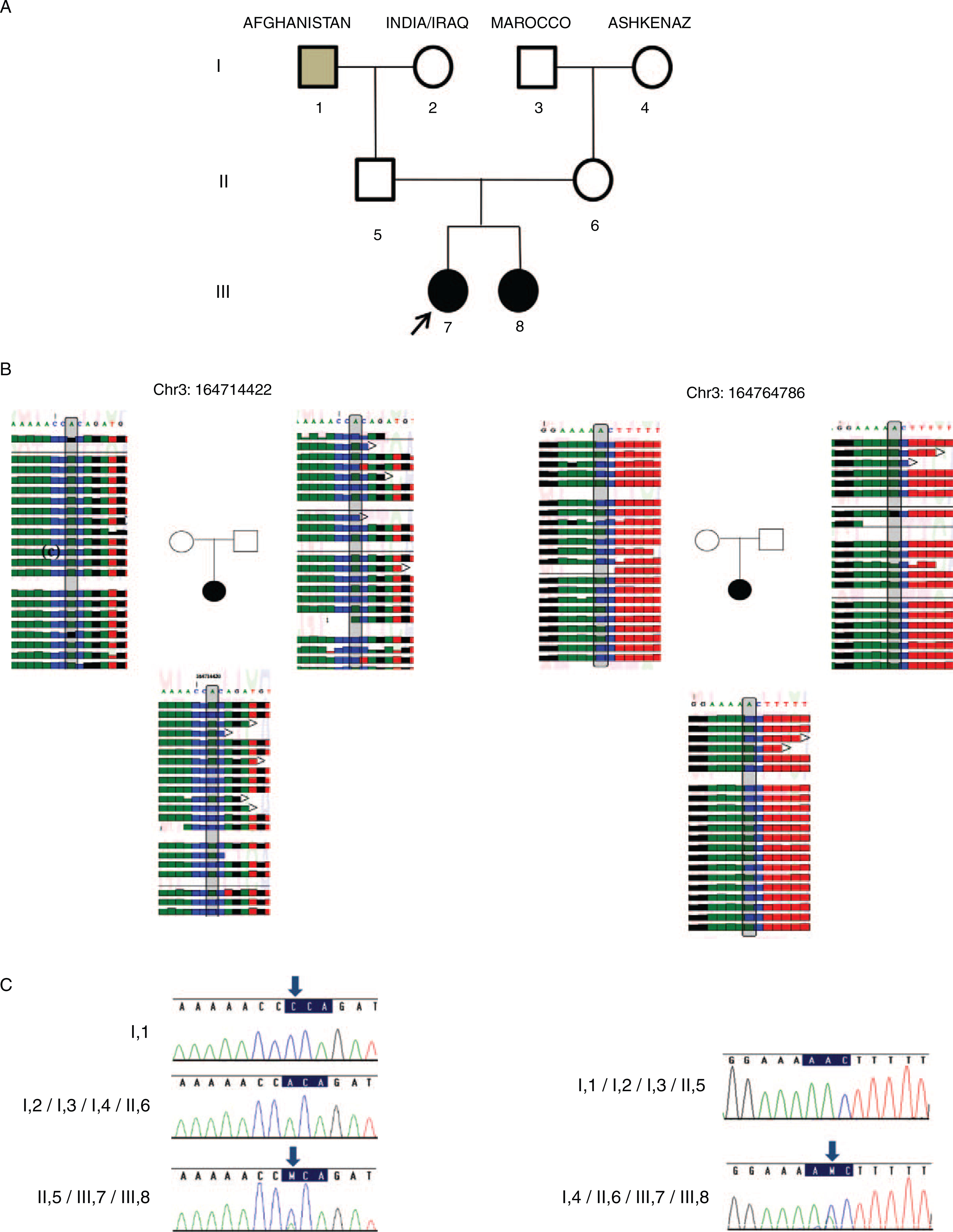
Sucrase-isomaltase (SI) mutations in our family. A, Pedigree of the family. Affected patients are depicted in solid shape. Arrow points to the proband. Grey shape indicates SI homozygous state and black shapes indicate SI compound heterozygous state. B, Exome sequencing of the proband (III,7) and her parents II,5 & II,6. The proband (III,7) is a compound heterozygote for both the mutations; 422A → C p.164714422 and 486A → C p.164764786, father (II,5) is heterozygote for 422A → C p.164714422 mutation and mother (II,6) is heterozygote for 486A → C p.164764786 mutation. C, Chromatograms represent Sanger sequencing of the mutations for each family member. I,1 is homozygote and II,5 heterozygotes for 422A → C mutation. I,4 and II,6 are heterozygotes for 486A → C mutation. Patients III,7 and her sibling (III, 8) are compound heterozygous for 422A → C p.164714422 and 486A → C p.164764786.
Genetic Evaluation
WES was performed for patient III,7 and her parents. WES yielded 15,386 variants, which affect protein sequence. This list was subsequently reduced to 838 rare variants, by filtering out variants with minor allele frequency ≥0.01, those present in our in-house exomes (n = 361), in the 1000 Genomes Project (1KG; http://browser.1000genomes.org/index.html), in dbSNP 135 database, or in the NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/). After filtering, no potential homozygous variants were detected, but using in-house scripts to extract compound heterozygote inherited variants, we found 15 variants (Supplemental Digital Content 1, Table 1, http://links.lww.com/MPG/A810) in 7 genes that did not include SLC5A1. The SI gene was pursued as mutations in this gene were reported to cause CSID (8), and may potentially explain the chronic diarrhea that resolved with fructose-based formula. The SI gene encodes 2 enzymatic subunits that form the SI complex, where amino acids 110–1007 encode the isomaltase and amino acids 1008–1827 encode the sucrose subunits. The maternal SI missense mutation (NM_001041:C.1730T>G:P.V577G, Table 1) is present in both ClinVar and HGMD databases (CM060473) (8), whereas the paternal mutation (NM_001041:C.4593T>G:P.C1531W, Table 1) has not been described before, reveling a novel compound heterozygous mutation (Fig. 1B). Sanger sequencing showed that paternal grandfather (I1) is homozygote for the newly identified C1531W mutation (Fig. 1C). Moreover, Sanger sequencing revealed that the patient’s newborn sibling (III,8) is also compound heterozygote of the same C1531W and the V577G mutations (III,8). The sibling is free of symptoms on an infant formula that is free of the carbohydrates sucrose and isomaltose.
TABLE 1.
Population frequencies of the V577G and C1531 mutations and their damaging effect predictions
| Position | Inheritance | Variant | Prediction | MAF | ClinVar | HGMD |
|---|---|---|---|---|---|---|
| chr3: 164764786 | Mother | Missense (A>C) V577G | Damaging (7/7 programs) | IHD: 2/361 EA: 0.0014 C (12): A (8588) AA: 0.0002 C (1): A (4405) ExAC: 226/114952 (1 homozygoes) 1000 genomes: 0 |
Y | CM060473;PHEN = “SUCRASE ISOMALTASE DEFICIENCY” |
| chr3: 164714422 | Father | Missense (A>C) C1531W | Damaging (7/7 programs) | IHD: 0 EA: 0 AA: 0 ExAC: 1/119970 1000 genomes: 0 |
N | N |
Damaging effect of the mutation was assessed by SIFT, 2 × Polyphen2, LRT, MutationTaster, MutationAssessor, and FATHMM.
AA = African Americans; EA = European Americans; ExAC = exome aggregation consortium; HGMD = human gene mutation database; IHD = in-house database (n = 361); MAF = minor allele frequency.
The 2 mutations identified in patient III,7 are located in different SI protein domains (Fig. 2A); V577G is located within the isomaltase domain and C1531W is located within the sucrase domain. Moreover, both mutations (V577G and C1531W) segregate with the disease and are highly conserved in vertebrate species (Fig. 2B). Seven commonly used prediction programs (SIFT, 2 × Polyphen2, LRT, MutationTaster, MutationAssessor, and FATHMM) unanimously predict that this amino acid substitution has a damaging effect on the protein function (21) (Table 1).
FIGURE 2.
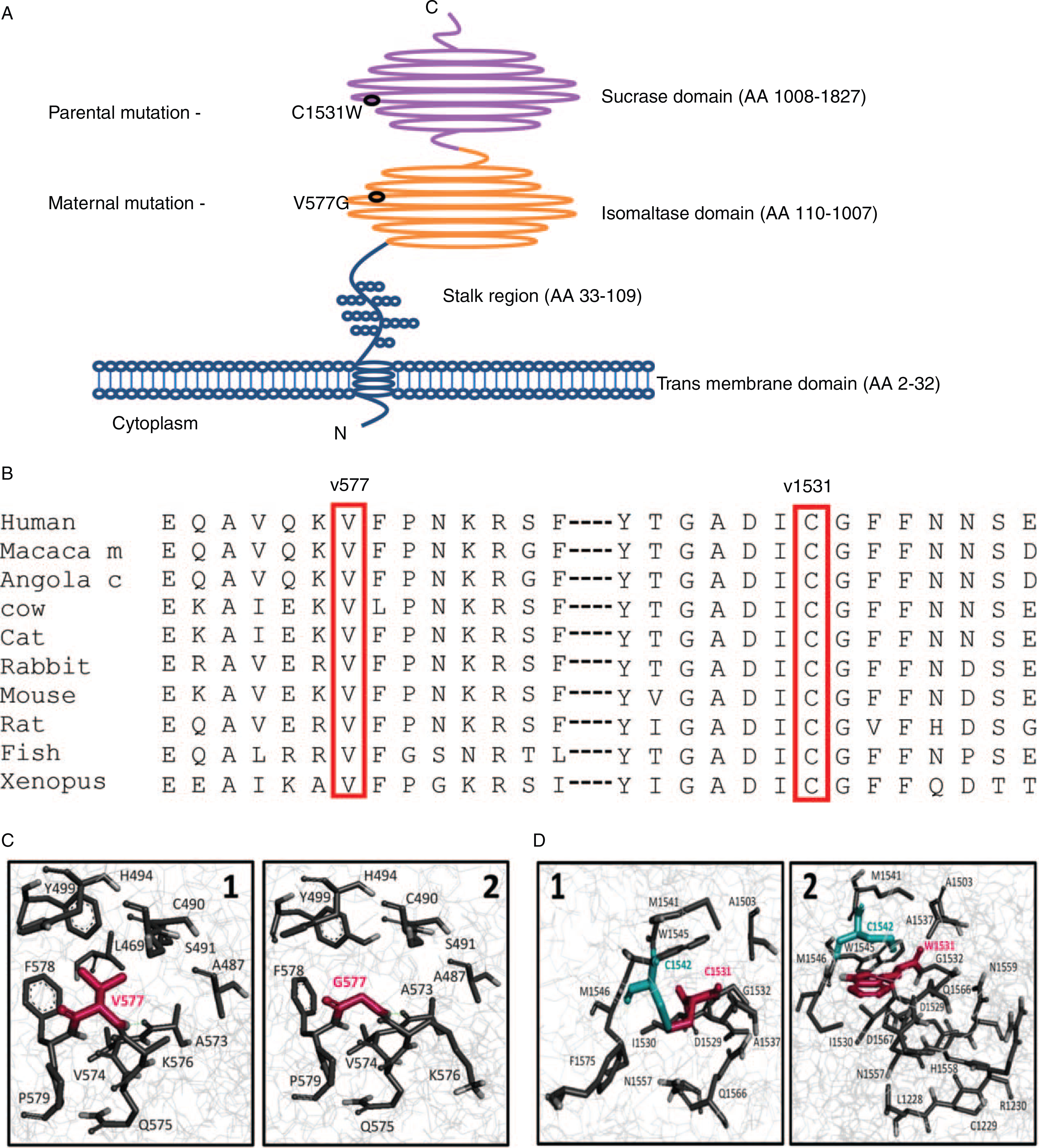
Conservation and modeling of sucrase-isomaltase (SI) wild type and mutated protein. A, Schematic illustration of the SI protein domains with the mutations positions (V577G within the isomaltase domain and C1531W within the sucrase domain). B, Conservation of valine at position 577 and cysteine at position 1531, among different species across evolution. C, SI 3D protein model of wild type (WT) (1) and V577G mutation (2). V577G mutation (pdb id: 3lpo). Position 577 in the context of the SI structure (wireframe representation). Residues predicted to be in contact with the residue in position 577 are presented as sticks. The residue in position 577 is depicted in magenta. Upon mutation to Gly, position 577 no longer interacts with L502. D, SI 3D protein model of WT (1) and C1531W mutation (2). C1531W mutation (pdb id: 3top). Position 1531 in the context of the SI structure (wireframe representation). Residues that are predicted to be in contact with the residue in position 1531 are presented as sticks. The residue in position 1531 is depicted in magenta. C1542, which forms a disulfide bond with C1531, is depicted in cyan. Trp is unable to form the disulfide bond formed between C1531 and C1542. Furthermore, W1531 contacts seven residues that C1531 does not interact with (ie, L1228, C1229, R1230, H1558, N1559, D1567, or F1568).
To assess the contribution of V577 and C1531 to the structural stability of SI, we performed a computational in silico mutagenesis analyses in which we separately mutated V577 to glycin (G) and C1531 to tryptophan (W) and calculated the change in protein stability. Furthermore, we analyzed the solvent accessibility and contacts of V577, C1531 and the mutated residues G577 and W1531. As previously described the V557G variant gives rise to a misfolded protein and intracellular arrest (8,22) (Fig. 2C). The results of our in silico mutagenesis analysis also indicate that the V557G variant is a highly destabilizing mutation (ΔΔGV577g = 4.08 kcal/mol). Gly is much smaller relative to Val and therefore while V577 is essentially buried in the WT structure, G577 is partially exposed to the solvent in the mutant protein. For most of the residues with which residue 577 interacts, the contact area between the mutant (G577) decreases relative to the contact area in the WT structure (V577). Supplemental Digital Content 2, Table 2, http://links.lww.com/MPG/A811 depicts the residues with which position 577 forms contacts and its distance from each contacting residue. Residue C1531 is located within a loop region of sucrase. By virtue of its ability to form inter- and intrachain disulfide bonds with other cysteine residues, cysteine plays an important role in protein structure. This sulfur-containing residue is replaced in the mutant by the larger aromatic and more hydrophobic Trp residue. The thiol group of C1531 is indeed involved in a disulfide bond, which is abolished in the C1542W mutant. Hence, the mutant protein lacks the covalent bond formed between C1531 and C1542 (Fig. 2C). Our in silico mutagenesis analysis indicated a highly destabilizing effect upon mutation of C1531 to Trp (ΔΔG C1531W = 21.2 kcal/mol), which can be partially accounted by the loss of the disulfide bond. Supplemental Digital Content 2, Table 3, http://links.lww.com/MPG/A811 lists the residues with which position 1531 is in contact with and its distance from each contacting residue. It can be seen that because C1531 is fully buried and W1531 is slightly exposed to the solvent, there are some differences regarding their ability to contact adjacent residues. Some of these contacts are highly unfavorable as they represent severe steric clashing. Under these conditions, it can well be that the protein actually cannot fold to the near native state, and folds (if at all) to a significantly different nonfunctional fold. Thus, the C1531W point mutation induces a highly unstable conformation with steric clashes due to its bulky aromatic rings and the absence of the disulfide bond, or alternatively folds to a nonfunctional significantly altered structure.
Sucrase-isomaltase Expression and Localization
To verify that the compound heterozygote mutations effect SI expression and localization, we used anti-SI antibodies and duodenal biopsies taken from patient III,7 and control (Fig. 3). Immunohistochemistry of patient’s and control duodenal biopsies with anti-SI antibody showed the expected expression and localization of SI protein on the brush border membrane of the small intestine in the control duodenum biopsy, whereas no SI can be detected in the brush border membrane of patient III,7 biopsies (Fig. 3).
FIGURE 3.
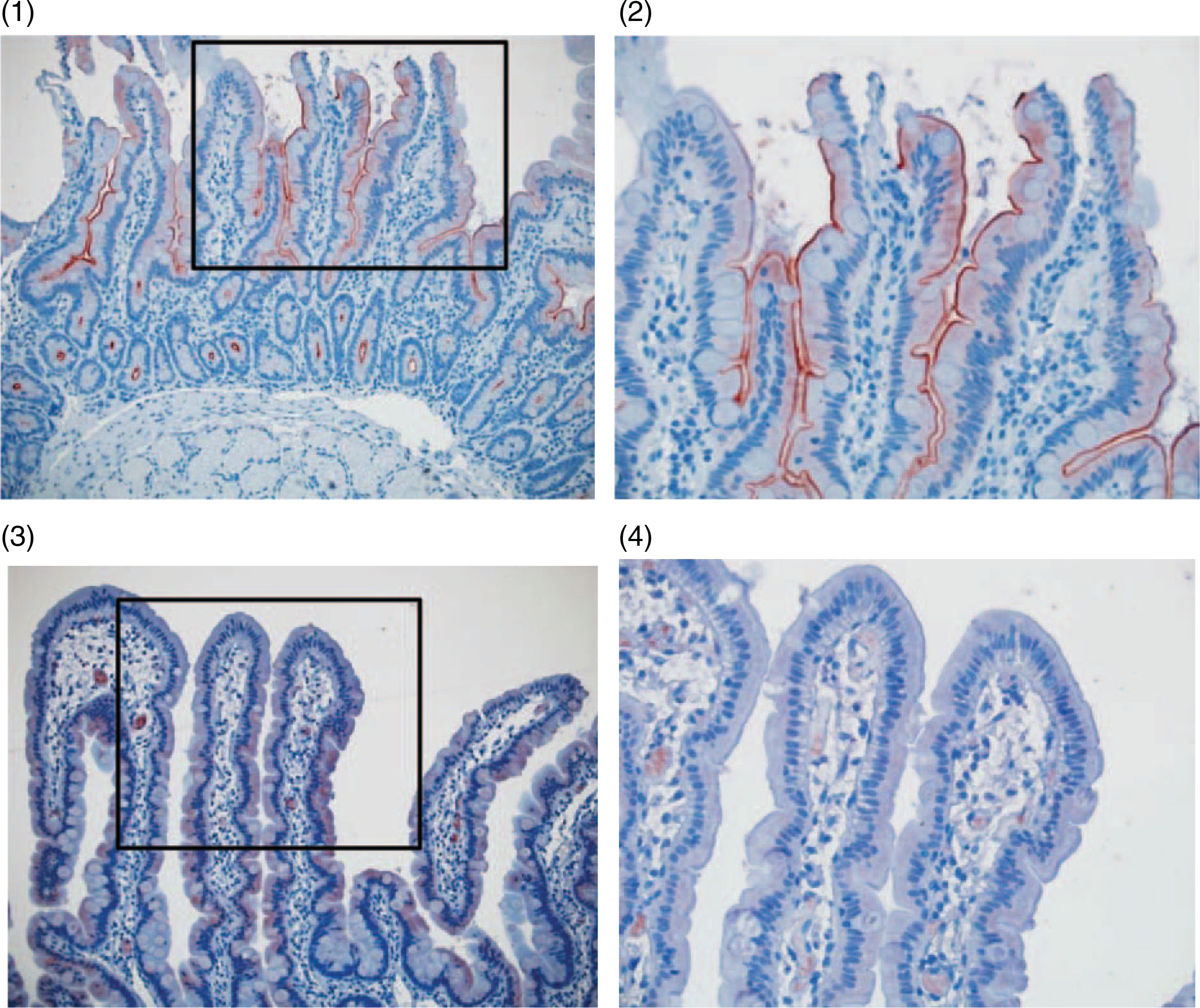
Abnormal sucrase-isomaltase (SI) protein expression and intracellular localization in duodenal biopsies of patient III-7. Immunohistochemistry staining for SI in the control intestine biopsy X200 (1) and X400 (2) and in the patient III,7 intestine biopsy X200 (3) and X400 (4).
DISCUSSION
Several SI complex mutations were previously reported in inherited enteropathies. The clinical presentation of SI deficiency in infancy is severe, with diarrhea and FTT, but later in life, in children and adults, chronic nonspecific diarrhea or irritable bowel syndrome, without growth failure, are the predominant symptoms (2). Clinical phenotypes were previously associated with several homozygous mutations and compound heterozygosity states (V577G and G1073D, C1229Y and F1745C) (9). We identified a new compound heterozygous state, composed of the known V577G mutation and a novel C1531W mutation, which has not been previously described (Table 1) in association with CSID. We further show that both the known V577G and the novel C1531W mutations are highly conserved across vertebrate species and that both mutations are predicated to be deleterious in all databases tested including SIFT, Polyphen2, LRT, MutationTaster, MutationAssessor, and FATHMM. Importantly, the in silico mutagenesis modeling suggested that these mutations likely destabilize the protein native confirmation and likely leading to impaired trafficking of SI and/or nonfunctional SI structure. Finally, we verify a lack of SI protein expression on the brush border membrane in the patient’s duodenal biopsies.
Patient III,7 showed severe and less typical early presentation of CSID. Such severe and early onset presentation of CSID was probably due to rapid recurrent changes of the infant’s formula to formulas, which contain higher amounts of maltose and/or sucrose (ie, Similac Comfort, Abbott & Neocate, Nutricia). Unfortunately, patient III,7 developed restrictive eating pattern and food aversion, possibly due to her protracted clinical course and food restriction before the diagnosis. She is currently treated by the feeding team to expand her intake. Interestingly, we identified the paternal grandfather, originally from Afghanistan and a son of consanguineous parents, as being homozygous for the C1531W mutation. Further investigation revealed that he also voluntary restricted his diet and eliminated different types of sweets since childhood. We also identified the patient’s newborn sibling as having similar compound heterozygous mutations. Unlike the older sibling, she developed mild vomiting and diarrhea at the age of 4 weeks while fed a milk protein-based formula. She is maintained on a formula containing minimal amounts of sucrose and maltose (Enfamil AR, Mead Johnson Nutrition) and her symptoms completely resolved.
CONCLUSIONS
Our findings expand the molecular spectrum of CSID and emphasize the need for early diagnosis and treatment of children with CDD. Fructose -free formulas can be used as a “therapeutic trial” in infants with an unclear etiology of CDD, and if resolution of symptoms occurs, further investigation of the disaccharidase deficiency can be conducted.
Supplementary Material
What Is Known
Diagnosis of chronic congenital diarrhea can be challenging. Genetics may be the key for precise diagnosis.
Congenital sucrase-isomaltase deficiency can present as severe diarrhea and failure to thrive in infancy and as chronic nonspecific diarrhea, or irritable bowel syndrome in children and adults.
What Is New
Identification of new compound heterozygote mutations in sucrase-isomaltase gene; a known V577G mutation and a novel C1531W mutation, in an infant that presented with severe diarrhea and failure to thrive.
Verification that the compound heterozygote mutations cause congenital sucrase-isomaltase deficiency by showing lack of brush border sucrase-isomaltase protein expression in the patient’s duodenal biopsies.
ACKNOWLEDGMENTS
The authors wish to thank the patients and their family for their kind assistance. This work was supported in part by the I-CORE program (grants no. 41/11), the Gene and Protein Expression core of the NIH-supported Cincinnati Children’s Hospital Research Foundation Digestive Health Center (1P30DK078392–01), and the Israel Science Foundation (grant no 908/15).
This work was supported in part by the I-CORE program (grants no. 41/11), the Gene and Protein Expression core of the NIH-supported Cincinnati Children’s Hospital Research Foundation Digestive Health Center (1P30DK078392–01), and the Israel Science Foundation (grant no. 908/15).
Footnotes
The authors report no conflicts of interest.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text, and links to the digital files are provided in the HTML text of this article on the journal’s Web site (www.jpgn.org).
REFERENCES
- 1.Canani RB, Terrin G. Recent progress in congenital diarrheal disorders. Curr Gastroenterol Rep 2011;13:257–64. [DOI] [PubMed] [Google Scholar]
- 2.Treem WR. Clinical heterogeneity in congenital sucrase-isomaltase deficiency. J Pediatr 1996;128:727–9. [DOI] [PubMed] [Google Scholar]
- 3.Ouwendijk J, Moolenaar CEC, Peters WJ, et al. Congenital sucrase-isomaltase deficiency: identification of a glutamine to proline substitution that leads to a transport block of sucrase-isomaltase in a pre-Golgi compartment. J Clin Invest 1996;97:633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacob R, Zimmer KP, Schmitz J, et al. Congenital sucrase-isomaltase deficiency arising from cleavage and secretion of a mutant form of the enzyme. J Clin Invest 2000;106:281–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spodsberg N, Jacob R, Alfalah M, et al. Molecular basis of aberrant apical protein transport in an intestinal enzyme disorder. J Biol Chem 2001;276:23506–10. [DOI] [PubMed] [Google Scholar]
- 6.Hauri HP, Roth J, Sterchi EE, et al. Transport to cell surface of intestinal sucrase-isomaltase is blocked in the Golgi apparatus in a patient with congenital sucrase-isomaltase deficiency. Proc Natl Acad Sci 1985;82:4423–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fransen JA, Hauri HP, Ginsel LA, et al. Naturally occurring mutations in intestinal sucrase-isomaltase provide evidence for the existence of an intracellular sorting signal in the isomaltase subunit. J Cell Biol 1991;115:45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sander P, Alfalah M, Keiser M, et al. Novel mutations in the human sucrase-isomaltase gene (SI) that cause congenital carbohydrate malabsorption. Hum Mutat 2006;27:119. [DOI] [PubMed] [Google Scholar]
- 9.Alfalah M, Keiser M, Leeb T, et al. Compound heterozygous mutations affect protein folding and function in patients with congenital sucrase-isomaltase deficiency. Gastroenterology 2009;136:883–92. [DOI] [PubMed] [Google Scholar]
- 10.Marcadier JL, Boland M, Scott R, et al. Congenital sucrase-isomaltase deficiency: identification of a common Inuit founder mutation. CMAJ 2015;187:102–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ritz V, Alfalah M, Zimmer KP, et al. Congenital sucrase-isomaltase deficiency because of an accumulation of the mutant enzyme in the endoplasmic reticulum. Gastroenterology 2003;125:1678–85. [DOI] [PubMed] [Google Scholar]
- 12.Abdullah AMA, El-Mouzan MI, El-Shiekh OK, et al. Congenital glucose-galactose malabsorption in Arab children. J Pediatr Gastroenterol Nutr 1996;23:561–4. [DOI] [PubMed] [Google Scholar]
- 13.Stephen J, Viboux T, Haberman Y, et al. Congenital protein losing enteropathy: an inborn error of lipid metabolism due to DGAT1 mutations. Eur J Hum Genet 2016;24:1268–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li MX, Gui HS, Kwan JS, et al. A comprehensive framework for prioritizing variants in exome sequencing studies of Mendelian diseases. Nucleic Acids Res 2012;40:e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol 2002;320:369–87. [DOI] [PubMed] [Google Scholar]
- 18.Schymkowitz J, Borg J, Stricher F, et al. The FoldX web server: an online force field. Nucleic Acids Res 2005;33:W382–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McConkey BJ, Sobolev V, Edelman M. Quantification of protein surfaces, volumes and atom-atom contacts using a constrained Voronoi procedure. Bioinformatics 2002;18:1365–73. [DOI] [PubMed] [Google Scholar]
- 20.McConkey BJ, Sobolev V, Edelman M. Discrimination of native protein structures using atom-atom contact scoring. Proc Natl Acad Sci U S A 2003;100:3215–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X, Wu C, Li C, et al. dbNSFP v3.0:a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat 2016;37:235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Discovery Studio Modeling Environment. Release 3.5. San Diego: Accelrys Software Inc; 2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.