

Abstract
Monoclonal antibodies (mAb) have had a transformative impact on treating cancers and immune-disorders. However, their use is limited by high development time and monetary cost, manufacturing complexities, suboptimal pharmacokinetics, and availability of disease-specific targets. To address some of these challenges, we developed an entirely synthetic, multivalent, Janus nanotherapeutic platform, called Synthetic Nanoparticle-Antibodies (SNAbs). SNAbs, with phage-display identified cell-targeting ligands on one “face”, and Fc-mimicking ligands on the opposite “face”, were synthesized using a custom, multi-step, solid-phase chemistry method. SNAbs efficiently targeted and depleted myeloid-derived immune-suppressor cells (MDSCs) from mouse-tumor and rat-trauma models, ex-vivo. Systemic injection of MDSC-targeting SNAbs efficiently depleted circulating MDSCs in a mouse triple-negative breast cancer model, enabling enhanced T cell and Natural Killer cell infiltration into tumors. Our results demonstrate that SNAbs are a versatile and effective functional alternative to mAbs, with advantages of a plug-and-play, cell-free manufacturing process, and high-throughput screening (HTS)-enabled library of potential targeting ligands.
Keywords: monoclonal antibody, Janus nanoparticle, myeloid-derived suppressor cell, antibody-dependent cellular responses, immunotherapy
Graphical Abstract
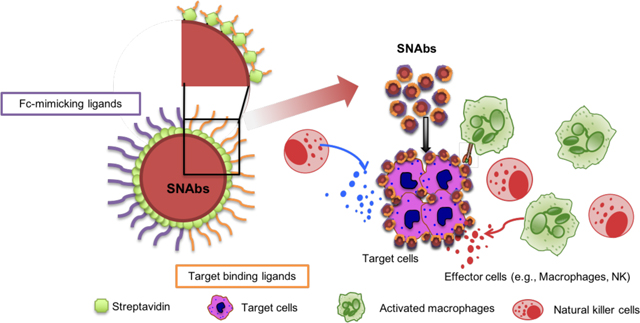
Immunotherapies based on monoclonal antibodies (mAb) are effective in improving the survival rate of patients with hematological malignancies and immune-disorders.1,2 Despite such success, there are significant challenges that hinder the development and broader use of mAbs. These include a complex and expensive development and manufacturing process, limited solid tissue (e.g., tumor) penetration and retention, and a narrow range of available targets. Current development methods for producing mAbs either select high-affinity candidates from a repertoire of clones in transgenic animals,3 or employ human antibody (scFv) phage display with antigens or cells.4 Following selection, their production requires sophisticated eukaryotic machinery in cells. This process is time-consuming, labor-intensive, and expensive.3 Moreover, the efficacy of mAbs can be hampered by their limited penetration and retention in solid tissues, such as tumors.3,5 Existing alternatives to mAbs, including diabodies, minibodies and peptibodies, have more homogenous intratumoral distribution but suffer from faster clearance from the body.6 In addition, for cancer and immune disorders, the lack of unique surface antigens on important target cell types, such as myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs), impedes the development of specific mAbs.7,8
To address some of these shortcomings, we developed a fully synthetic, multivalent nanoparticle-antibody (SNAbs) technology with advantages of a cell-free, animal-free, purely chemical synthesis method; tailorable properties (e.g., size, shape, valency, and surface chemistry) that can be potentially modulated to enhance tissue penetration and retention; and a flexible, plug-and-play platform amenable to targeting a wide variety of cell types. SNAbs consist of multivalent, bifunctional, Janus gold (Au) nanoparticles modified with cell-targeting ligands on one “face” and antibody-Fc-mimicking ligands on the other “face” (Fig. 1a). Janus particles are particles that have anisotropic surface chemistry and have previously been reported for ex vivo activation of T-cell receptors9,10, drug delivery11,12, cell tracking13 and bio-imaging.14–18 Leveraging the bifunctionality of the Janus structure, SNAbs have the capability, like mAbs, to pair specific target cells with effector cells (e.g., macrophages or Natural Killer (NK) cells), thereby triggering target cell killing (Fig. 1b).
Figure 1.

Schematic illustration of SNAbs and their hypothetical mechanism of action. (a) SNAbs are Janus nanoparticles bearing two distinct chemically modified faces. One of the two faces presents targeting ligands to perform the function of Fab domains in mAbs, and the other displays Fc-mimicking ligands to crosslink Fc receptors on the effector cells as Fc fragments in mAbs. (b) Once administered into patients or animals with diseases, the SNAbs circulate and recognize target cells in blood or organs of interest by binding onto their surface proteins and engaging with effector cells (e.g. macrophages, NK cells) to induce antibody-like cellular cytotoxicity or phagocytosis.
To generate SNAbs, first, Janus Au nanoparticles were synthesized using solid-phase chemistry (Fig. 2a).19 Briefly, streptavidin (SA)-coated Au nanoparticles (SA-AuNP-SAs, 30 nm spheres, with an average hydrodynamic diameter of 70 nm) were bound onto aminomethyl resin (200–300 μm) via a reducible crosslinker, sulfo-NHS-S-S-Biotin (Supplementary Fig. 1a). After washing off the unbound SA-AuNP-SAs, the crosslinkers’ disulfide bonds were cleaved with tris(2-carboxyethyl)phosphine (TCEP), resulting in the release of Janus nanoparticles (SA-AuNP-SH) with asymmetric surface chemistry (Supplementary Fig. 1b, c). One face of the Janus nanoparticles contained free-streptavidin (SA) for binding to biotin, and the other face provided biotin-NH-(CH2)2-SH, for reaction with maleimide group, which allowed spatially separated conjugation and presentation of two different ligands. Scanning transmission electron microscopy (STEM) bight-field (BF) imaging, high-angle annular dark-field (HAADF) imaging, and X-ray Energy Dispersive Spectroscopy (EDS) mapping of the SA-AuNP-SHs and SA-AuNP-SAs with biotinylated quantum dots (QDs) (Fig. 2b, Supplementary Fig. 2a,b), or maleimide-Au nanoprobes (Supplementary Fig. 2c) confirmed the asymmetric distribution of available biotin binding sites or free SH groups on the Janus nanoparticles, respectively. Upon incubation with Alexa-Fluor C2-maleimide and subsequent washing, the SA-AuNP-SH particles showed increased fluorescence compared to nonJanus SA-AuNP-SA (Fig. 2c), which further validated the successful thiol-substitution of biotin-binding sites and accessibility of free thiol groups for maleimide-terminated ligands reaction. Fluorescence quantification of sulfo-maleimide Cy5- or biotin Cy5-labeled SA-AuNP-SHs confirmed the presence of 13 active biotin-binding sites for biotinylated ligand conjugation and 16 active SH groups for maleimide-SH reaction on each Janus nanoparticles (Supplementary Table 2).
Figure 2.

Fabrication and characterization of the Janus Au nanoparticles and molecular dynamic modeling of the interaction between G3/G3* and S100A8/A9. (a) Aminomethyl resins were functionalized with sulfo-NHS-S-S-biotin crosslinker (step 1) and then bound with streptavidin-coated, 30nm Au nanoparticles (step 2). The cleavage of the disulfide bonds in the crosslinker by TCEP releases the Janus Au nanoparticles (step 3), which has a streptavidin face with open biotin-binding pockets and a thiol face with available free thiols. (b) STEM-BF, STEM-HAADF, and EDS mapping images of 10–12 nm biotinylated quantum dot (QD)-conjugated nonJanus (SA-AuNP-SA, left column) and Janus (SA-AuNP-SH, right column) gold nanoparticles demonstrates the asymmetric distribution of open biotin-binding sites on the Janus nanoparticles fabricated using the method described in a. Colors: red-gold (nanoparticles), blue-oxygen (streptavidin), cyan blue- Cd (biotinylated QDs). (c) Available thiol groups on either Janus nanoparticles or unmodified nanoparticles (to compare here, labeled as SA-AuNP-SA in the graph, named as AuNP in other parts of the paper) shown by conjugation of Alexa Fluor 647-maleimide dye, where signal is normalized to the concentration of nanoparticles. Data are presented as mean ± s.d. N=2 independent samples with n=3 technical replicates. (d-g), Molecular dynamics simulation of G3 or G3* binding onto human S100A8/A9 heterotetramer. Ten short (100 ns each) molecular dynamics simulations with human S100A8/A9 heterotetramer demonstrate the capability of both G3-biotin and G3*-biotin to interact with the S100A8/A9 proteins. (d-e), The superposition of the final frames of the five 100-ns G3 simulations (d) and G3* simulations (e). f-g, The contact probability (defined as coming within 3.5 Å) determined from all five 100 ns G3 simulations (f) and G3* simulations (g). The sequence along the x axis is that of each peptide.
To generate SNAbs and show mAb-like functionality, we selected MDSCs as target cells. MDSCs are heterogeneous, immunosuppressive cells of myeloid origin that are upregulated in cancer, chronic inflammation, tuberculosis and immune-dysregulation arising from trauma, burn, or sepsis.20–26 MDSCs decrease proliferation and survival of effector T cells, recruit Tregs, and suppress cytotoxic T cell (CTL) functions.27,28 Depleting MDSCs could restore immune-homeostasis and provide a potential immunotherapeutic strategy for cancers and immune-disorders.28,29 MDSCs have been treated using small molecule chemotherapies (e.g., doxorubicin30, 5-fluorouracil31) and inhibitors (e.g., cyclooxygenase-2 inhibitor,32 Silibinin33), and broadly-targeted mAbs (e.g., anti-mouse Gr-1), all of which either have high systemic toxicity and undesired off-target response, or only work in mouse models.29,34–37 Therefore, we decided to develop MDSC-targeting SNAbs as a potential therapeutic-platform to deplete MDSCs.
To target MDSCs, we first selected the G3 peptide (WGWSLSHGYQVK), a 12-mer peptide previously identified through phage-display against murine MDSCs with specific binding affinity to S100A8/A9 proteins.38 During our studies, we also discovered a related sequence G3* (KSLWVQWSGGHY), with similar binding capacity to MDSCs. S100A8/A9 is pro-inflammatory proteins that participate in local intracellular communications and regulates MDSC recruitment in the tumors.39 MDS
Cs have high levels of surface-receptors that bind soluble S100A8/A9 proteins, and also express 10-fold higher cell-surface S100A8/A9 proteins compared to other cell types in tumor and inflammation.22,40–42
We found that free G3 peptides bind strongly to both polymorphonuclear (PMN) and monocytic MDSCs (M-MDSCs) isolated from tumor-bearing mice compared to an irrelevant control peptide, while the free G3* bound more strongly to PMN-MDSCs than M-MDSCs (Supplementary Fig. 3). Free G3 and G3* also showed some binding to mouse macrophages (Supplementary Fig. 3), dendritic cells (DCs), and NK cells, but not to B cells and T cells (Supplementary Fig. 4). To verify the binding of peptides to their protein targets in silico, we performed molecular dynamic (MD) simulations to study the binding of G3 and G3* to human S100A8/A9 heterotetramers (Fig. 2d–g). G3 peptides interacted with two predominant areas on human S100A8/A9 tetramers through hydrophobic interactions and hydrogen bonds, while G3* peptides associated with many different regions of S100A8/A9 in multiple conformations through hydrophobic interactions, hydrogen bonds and electrostatic interactions (Supplementary Fig. 5–6). These results indicate that both G3 and G3* can be used to target human S100A8/A9 over-expressing cells, such as MDSCs.
To make MDSC-targeting SNAbs, we proceeded to functionalize the streptavidin “face” of the Janus SA-AuNP-SH with C-terminal-biotinylated G3 or G3* peptides (Fig. 3a). On the opposite thiol (SH) “face”, we conjugated the cp33 peptide, a human IgG1 Fc-mimicking ligand for binding to Fc gamma receptors (FcγRs) on immune effector cells, through the succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC) group on the C-terminal of cp33 (Fig. 3a).43,44 We confirmed that the Fc-mimicking ligand, cp33, could bind specifically and more strongly to the FcγR-expressing effector cells, i.e.,CD11b+F4/80+ macrophages, than control peptides in a mixture of mouse splenocytes (Supplementary Fig. 3c).
Figure 3.

Ligand modification on Janus Au nanoparticles and evaluation of the binding of SNAbs onto mouse MDSCs and macrophages by photoacoustic (PA) imaging. (a) Following fabrication of Janus nanoparticles, the Fc-mimicking ligands, cp33, was conjugated onto the thiol face of the Janus nanoparticles via thiol-maleimide reaction, and the targeting ligands, G3, was modified onto the streptavidin face via biotin-streptavidin interaction. (b,c) Alexa Fluor 680-NHS-estertagged MDSC-targeting peptide (G3) and Alexa Fluor 680-NHS-ester-tagged Fc-mimicking peptide (cp33) were reacted with SA-AuNP-SH or SA-AuNP-SA. The conjugation level of each peptide was assessed by measuring fluorescence of the samples and normalizing against nanoparticle concentrations. Background fluorescence of the untagged-peptide-modified nanoparticles was subtracted from the measured values of the samples. Data are presented as mean ± s.d. N=2 (left) or 3 (right) independent samples with n=3 or n=6 technical replicates. (d) Hydrodynamic sizes of nonJanus SA-AuNP-SA, Janus nanoparticles SA-AuNP-SH and SNAbs after modification with ligands were measured by dynamic light scattering on a Malvern Zetasizer. (e) The photoacoustic (PA) and ultrasound (US) images of nanoparticle-treated samples of mouse MDSCs. Top: PA images of the cell inclusions of SNAb, NonJanus AuNP-cp33, NonJanus AuNP-cp33 and AuNP at a wavelength of 532 nm. Bottom: US images of the cell inclusions of SNAb and AuNP. (f,g) The relative amount of nanoparticles bound to mouse MDSCs (f) or RAW 264.7 macrophages (g) based on the average PA signals of each cell inclusion (0.5 million cells/40𝜇L). PA signals shown in the graphs were normalized against the laser energy and backscattered ultrasound signals. Data are presented as mean ± s.d. of at least six cross-section images of two or more technical replicates of corresponding independent samples.
Using fluorophore-labeled G3 and Cp33 peptides, we confirmed the successful surface functionalization of ligands on Janus AuNPs. As expected, the fluorescence intensity following G3-conjugation to nonJanus SA-AuNP-SA was almost twice of that of G3-conjugated SA-AuNP-SH (Fig. 3b). Also, fluorescent cp33-SMCC peptides were effectively conjugated to SA-AuNP-SH or G3-AuNP-SH, but not to SA-AuNP-SA (Fig. 3c). The hydrodynamic size of the nanoparticles increased from ~70nm to ~100nm after modification (Fig. 3d, Supplementary Table 3). The fully functionalized Janus G3-AuNPcp-33 and G3*-AuNP-cp33 are termed G3-SNAbs and G3*-SNAbs, respectively, throughout our discussion, and they are collectively called MDSC-SNAbs. In contrast, non-Janus streptavidin-coated gold nanoparticles are named with the peptide ligands on them from hereon (Table 1). For example, AuNP-cp33 is non-Janus streptavidin-coated gold nanoparticles conjugated with cp33 peptides uniformly on the nanoparticles
Table 1.
Summary of the nanoparticles used in this paper.
| Nanoparticle Type | Janus (J) or NonJanus (N) | Ligand 1 | Ligand 2 | Other names |
|---|---|---|---|---|
| SA-AuNP-SH | J | N/A | N/A | Janus nanoparticles |
| G3-SNAb | J | G3-biotin | Cp33-SMCC | MDSC-SNAb, Janus G3-AuNP-cp33 |
| G3*-SNAb | J | G3*-biotin | Cp33-SMCC | MDSC-SNAb, Janus G3*-AuNP-cp33 |
| IrrelPep-SNAb | J | scAHNP-biotin | Cp33-SMCC | scAHNP-AuNP-cp33 |
| G3-AuNP-Fc | J | Fc-biotin | G3-SMCC | N/A |
| Biotin-AuNP-NEM | J | Biotin | NEM | Janus control NP |
| SA-AuNP-SA | N | N/A | N/A | nonJanus nanoparticles, AuNP |
| AuNP-cp33 | N | Cp33-biotin | N/A | N/A |
| AuNP-G3/cp33 | N | G3-biotin | Cp33-biotin | N/A |
Photoacustic (PA) imaging was used to quantify Au nanoparticle binding to cells. More traditional methods, like FACS, could not detect the AuNPs due to small particle size and lack of fluorescence without particle modification. However, gold nanoparticles are a well-established contrast agent in PA imaging due to strong optical absorption and surface plasmon resonance. Because PA signal is directly proportional to nanoparticle concentration and absorption coefficient, gold nanoparticles have been used for PA cell tracking applications.45–47 For these reasons, PA imaging is a valid approach for in vitro assessment of nanoparticle-cell binding. We used PA signal to evaluate the binding of G3-SNAbs to target cells. The PA images of cell samples treated with G3-SNAbs or control SA-AuNP-SA (for short, AuNP) showed specific binding of the G3-SNAbs to mouse tumor-associated MDSCs and to RAW 264.7 macrophages (Fig. 3e–g), indicating that G3-SNAbs can recognize both target cells (MDSCs) and effector cells (macrophages). NonJanus AuNP-cp33 also bound to both cell types because of their expression of FcγRs.
Ex-vivo experiments were performed to test the mAb-like cell killing capability of the MDSC-SNAbs. Incubation of MDSCs, isolated from the spleens of 4T1 tumor-bearing mice, with G3-SNAbs or control nanoparticles in the absence of effector cells did not trigger apoptosis or necrosis of the cells (Supplementary Fig. 7). To test SNAb-mediated MDSC-specific killing, we conducted splenocyte suspension assays (Supplementary Fig. 8a), in which a mixture of splenocytes (containing MDSCs, macrophages, DCs, NK, T and B cells, etc.) isolated from the spleens of tumor-bearing mice were treated with G3-SNAbs or G3*-SNAbs, and several controls, including nonJanus AuNP-G3/cp33 (both peptides biotinylated and randomly conjugated to AuNP), nonJanus AuNP-cp33 (cp33-modified AuNP), AuNP, and PBS. The percentage and viability of the major cell populations in the culture were measured using flow cytometry after 24 hours. This assay reflects the in vivo microenvironment with a mixture of various types of immune cells and varied effector cell-to-target cell ratios dictated by disease progression. SNAbs significantly reduced total MDSCs in the whole splenocyte populations by 10.2% in the G3-SNAb group and by 16.2% in the G3*-SNAb group (Fig. 4a) compared to the PBS-treated group. This SNAb-specific reduction is remarkable, considering that the macrophage to MDSC ratio (1:4.5) in the splenocyte assay was very low. The viability of total MDSCs, PMN-MDSCs and M-MDSCs were also drastically reduced by SNAb treatment (Fig. 4b–d). The same effect was achieved by Janus G3-AuNP-Fc nanoparticles (replacing cp33 with Fc fragments of human IgG1 antibody on the Janus AuNPs) (Supplementary Fig. 8c–d). Interestingly, despite binding to both target and effector cells (Fig. 3e–g), the nonJanus particles (including nonJanus AuNP-G3/cp33 and AuNP-cp33) did not reduce MDSC percentage or viability (Supplementary Fig. 8c–d), suggesting that the polarized presentation of both G3/G3* and cp33 peptides in a Janus structure is essential to induce efficient killing of MDSCs. The orientation and localization of the targeting and Fc-mimicking ligands on the Janus nanoparticles may bind to and activate receptors more effectively on effector cells.18,48–50
Figure 4.

MDSC-SNAbs induce antibody-like killing of mouse and rat MDSCs in the presence of effector cells, such as macrophages. (a-h) In a mouse splenocyte-suspension assay, the single cell suspension from the spleens of 4T1-tumor-bearing mice were treated with equal concentration of various nanoparticle formulations for 24 hours. The cells were then stained with fluorescent antibodies and fixable viability dye and analyzed by flow cytometry for the total percentage (a) of live and dead MDSC (PMN-MDSC and monocytic MDSCs), the percentage of dead MDSCs (b), dead PMN-MDSCs (CD11b+Ly6G+Ly6Clow) (c) or dead M-MDSCs (CD11b+Ly6G-Ly6Chigh) (d) out of corresponding cell populations, and the percentages of CD11b+F4/80+macrophages (e), CD3-B220+B cells (f), CD11b+CD11c+DCs (g), and CD3+T cells (h) out of total cells. (i) Rat MDSC and CD11b+ monocytes sorted from peripheral blood mononuclear cell (PBMC) of rats with femoral segmental defects were cultured ex vivo at 1:10 ratio and treated with MDSC-SNAbs or control treatments for 24 hours. The percentages of MDSCs (CD11b+His48+) in the co-culture after treatment were measured by flow cytometry. Data are presented in box plots (n=6) or presented by individual values with mean and standard deviation (n=6). Significance was determined using one-way ANOVA with Tukey post-hoc test (**** p<0.0001, *** p<0.0002, ** p<0.0021, * p<0.0332). AuNPs are nonJanus streptavidin coated Au nanoparticles. NonJanus AuNP-G3/cp33 nanoparticles are AuNPs coated with randomly distributed G3 and cp33. G3-SNAbs and G3*-SNAbs are Janus G3-AuNP-cp33 or G3*-AuNP-cp33 nanoparticles targeting MDSCs.
Apart from MDSCs, treatment with G3- and G3*-SNAbs resulted in higher percentages of F4/80+ cells, CD11c+ DC cells, CD3+ T cells (CD3+CD4+, CD3+CD8+), and B220+ B cells (Fig. 4e–h), likely due to the reduction of MDSCs. The only exception was CD49b+ NK cells whose percentage decreased slightly following SNAb treatment (Supplementary Fig. 8b). Narumi et al. showed that S100A8/A9 could induce NK cell expansion and activation (e.g., IFN-γ production) by binding to receptors on NK cells. Therefore, the reduction of S100A8/A9-expressing MDSCs may result in decreased stimulatory signaling to NK cells and thus a decrease in the NK cell percentage.51 Altogether, we found that G3 and G3* SNAbs were efficient and fairly-specific for MDSCs when evaluated in antibody-like depletion assays.
Besides cancer, the prevalence of MDSCs has also been reported in trauma and has been associated with poor healing outcomes, sepsis, and increased risk of multi-organ failure due to altered immune homeostasis and increased susceptibility to bacterial and viral infections.52,53,54 We used PA imaging to show that the peptide-modified nanoparticles (AuNP-G3, AuNP-G3*, and AuNP-cp33) could also bind to circulating MDSCs and macrophages isolated from a rat infection-trauma model (Supplementary Fig. 9). In an effector-target co-culture assay (trauma-associated MDSCs:CD11b+ effector cells = 1:10 ratio), 24 hours of G3-SNAb treatment significantly reduced the percentage of MDSCs compared to control treatments, showing the cross-reactivity of the G3-SNAbs in rat (Fig. 4i). These data suggest that MDSC-SNAbs can work in multiple species and different disease scenarios.
Next, to study the biodistribution of SNAbs, we selected a mouse model of 4T1 breast cancer. This model recapitulates some properties of human metastatic triple-negative breast cancer, an aggressive cancer that lacks effective treatment and in which MDSCs accumulate in blood, lymphoid organs, and tumors, and promote metastasis.40–42 Six hours after systemic administration of G3-SNAbs via tail vein injection, a high concentration was detected in the blood, which then decreased over time with a half-life of 26 hours (Fig. 5a). Over 30% of the SNAbs were accumulated in the liver after 6 hours (Fig. 5b). At all time-points (i.e., 6, 24, 48 hrs), the spleen had the highest SNAb concentration among all organ types tested (Fig. 5a, Supplementary Fig. 10). SNAbs did not accumulate in the lung or kidney, but ~3.4% of the injected SNAbs accumulated in the tumor after 48 hours (Fig. 5b, Supplementary Fig. 10f). This is in line with prior literature that nanoparticles of 30–100 nm in size preferentially penetrate and retain in the liver, the spleen, and vascularized tumors and their draining lymph nodes over 48 hours in mice.55,56
Figure 5.

Biodistribution and therapeutic effects of MDSC-SNAbs in a 4T1 mouse breast cancer model. (a,b) Biodistribution of SNAbs after intravenous injection in tumor-bearing mice. G3-SNAbs were administered through tail vein injection on day 9 post tumor inoculation. Lung, liver, spleen, kidney, tumor and blood were harvested after 6, 24, or 48 hours for ICP-MS analysis. Changes in the biodistribution over time, presented as concentration of Au per ug of tissue (a) and calculated percentage of SNAbs in each organ out of total injected amount of nanoparticles (b), were plotted with mean ± s.d. of 3 biological samples (n = 3). (c-g) Therapeutic effects of MDSCs in the 4T1 murine model. (c) The schedule of tumor inoculation and systemic injection (i.v. via the tail vein) of G3-SNAbs (Janus G3-AuNP-cp33), IrrelPep-SNAb (Janus scAHNP-AuNP-cp33), Janus control NP (Janus biotin-AuNP-NEM), AuNP and PBS (n=7, 7.5×1010/20𝜇L per injection). The percentage of total MDSCs (PMN-MDSCs and M-MDSCs) out of total cells in the blood (d) and spleens (e), the percentage of CD3+CD8+ cytotoxic T cells (f) and CD3-CD49b+NK cells (g) infiltrated in the tumors after treatment on day 11 are presented in box plots with box showing median, 25 and 75 percentile and whiskers showing min and max. After removing outliers, n=6 for untreated groups, n=6 in (d) and n=7 in (e) and (f-g) for AuNP group, N=7 for Janus control NP, n=4 in (e) and (g), n=5 in (d) and n=6 in (f) for IrrelPep-SNAb group, and n=7 for G3-SNAb single injection group, n=4 in (d),(e),(g) and n=5 in (f) and for G3-SNAb 3-injection groups. Significance was determined using one-way ANOVA with Tukey post-hoc test (**** p<0.0001, *** p<0.0002, ** p<0.0021, * p<0.0332).
Finally, to test the therapeutic effects of SNAbs in pre-established tumor-bearing mice in vivo, we administered G3-SNAbs, along with control nanoparticles (irrelevant peptide-modified SNAbs (IrrelPep-SNAbs), biotin-and-N-ethylmaleimide-modified Janus nanoparticles (Janus control NPs with biotin on one face and a malemide methyl group on the other side – biotin-AuNP-NEM), and streptavidin-coated SA-AuNP-SA (referred to as AuNPs), at 0.75×1010 nanoparticles per mouse, systemically, via tail vein injection, 10 days after orthotropic inoculation of 4T1 tumor cells (Fig. 5c). We also tested if three injections of G3-SNAbs (day 6, 8, and 10) could enhance the anti-tumor effect over a single injection (day 10). The rationale behind systemic MDSC depletion in metastatic aggressive cancers is to ensure that the MDSC-mediated immunosuppressive “brake” on endogenous anti-tumor immunity is released, at least partially, and more effector cells, e.g. T cells infiltrate into tumors to attack the malignant cells.
In these in vivo experiments, we observed a small but statistically significant reductions in the percentages of total MDSCs with G3-SNAb injection, in both single- and three-injection groups. In the blood, the average decrease of circulating MDSCs was by 7.0% and 6.7%, and in the spleen by 54.6% and 66.1% respectively, compared to the untreated (tumor-bearing, but no treatment) group (Fig. 5d–e). Control AuNPs (SA-AuNP-SA) and IrrelPep-SNAbs did not have any effect on MDSCs in the blood, while the Janus control NPs (biotin-AuNP-NEM) did bring down the MDSC percentage by 4.3% on average compared to the untreated group. In the spleen, AuNPs and Janus control NPs did not have any significant effect on MDSCs, while IrrelPep-SNAb showed non-specific reduction of MDSCs.
Specifically, among the MDSC subgroups, the PMN-MDSC percentages decreased in blood with G3-SNAb treatment, while the M-MDSC percentage increased in the G3-SNAb single injection group, but remained the same in the three-injection group (Supplementary Fig. 11a–d and Fig. 12, 15). In the spleen, the G3-SNAb-treated groups showed reduced PMN-MDSCs while M-MDSCs remained the same, compared to the untreated group (Supplementary Fig. 11c–d and Fig. 12, 15). These results suggest that PMN-MDSCs are significantly more depleted by SNAb treatment in both blood and spleen compared to M-MDSCs, which results in the M-MDSC percentages remaining the same or slightly increased in the SNAb-treated animals.
In the tumor, reduction of the total MDSCs and PMN-MDSCs by G3-SNAbs were not statistically significantly from the controls, but the percentages of M-MDSCs had much less variance among mice in the G3-SNAb single-injection and three-injection groups and were comparably lower than those of the control groups (untreated, AuNPs, Janus control NP and IrrelPep-SNAb groups) (Supplementary Fig. 11e–g).
Most importantly, the reduction in MDSCs after G3-SNAb single- and multi-injection treatments resulted in a strong increase in CD3+CD8+ T cell infiltration into the primary tumor by 89.6% and 107.2% respectively, compared to the untreated group. In contrast, the control particle treatments (IrrelPep-SNAbs, Janus control NPs, and AuNPs) did not elicit significant T cell infiltration (Fig. 5f, Supplementary Fig. 13 and 16), even though some of these groups appear to show non-specific effect on MDSCs, as discussed above. This is a critical result, since effector T cell infiltration is the targeted therapeutic effect for MDSC depletion.
These observations demonstrated the potential of G3-SNAbs to restore immune surveillance in solid tumors. Single treatment of G3-SNAb also remarkably increased the percentage of NK cells in tumors compared to the untreated group (Fig. 5g, Supplementary Fig. 14 and 16), which potentiates stronger innate immune attack against the tumor. Additionally, although not statistically significant, a trend of fewer immunosuppressive Tregs were observed in the G3-SNAb single-injection group compared to the untreated group (Supplementary Fig. 11h and 13). Interestingly, G3-SNAb three-injection treatment didn’t increase the NK cell percentage nor reduce Tregs in the tumors as much as the single-injection treatment of G3-SNAb.
In summary, we have designed, fabricated, and characterized fully-synthetic Janus nanoparticle antibodies (SNAbs) that can target and kill specific cells, like mAbs. The G3 and cp33 ligands were identified through peptide-phage display techniques, one of the many high-throughput molecular evolution tools. The targeting and activation ligands of SNAbs can also be identified and developed through other high-throughput screening tools, like aptamer screening. These techniques can identify ligands with comparable affinity as conventional mAbs to relevant antigens through iterative screening of a library of peptide (or aptamer.etc) sequences against a selected protein, cell, or tissue of interest without the need to know cell-specific surface markers.57 The identified peptides (or aptamer.etc) have the advantages of small physical size, flexible structure, and low immunogenicity compared to mAbs and can be easily chemically synthesized at lower cost. They can be readily used to functionalize onto the surface of Janus nanoparticles to generate a multitude of SNAbs targeting a wide range of cell types.57,58 Unlike mAbs or peptibodies, SNAbs use nanoparticles as scaffolds which have a high surface-area-to-volume ratio for the presentation of a high density of ligands, leading to high binding efficiency and increased targeting specificity.59,60 As shown by the PA images and ex vivo killing experiments, MDSC-SNAbs possess strong binding capability on both target cells (MDSCs) and effector cells.
Besides targeting capability, we also showed that MDSC-SNAbs were able to induce specific killing of MDSCs ex vivo in two disease-relevant animal models, i.e., the murine 4T1 breast cancer model and the rat infected-trauma model. These results demonstrated that the synthetic Janus nanoparticles modified with both targeting and Fc-mimicking ligands can trigger effective antibody-like innate immune responses in the presence of effector cells. The Fc-mimicking ligand, cp33, could be replaced by Fc fragments to achieve a similar effect, showing the flexibility in ligand types for SNAbs. NonJanus AuNP-cp33 nanoparticles also bound as well as SNAbs to both target cells and effector cells, but failed to induce specific killing of MDSCs, suggesting that the mechanism of action of SNAbs is more than simply pairing target and effector cells by binding. Taking the in silico simulations results into account, MDSC-SNAbs may have wide applicability in multiple species, including human, and in various diseases, such as cancers and immune disorders.
The dose of SNAbs used in our in vivo studies was ~104 times lower than that of anti-Gr-1 mAb doses reported in literature.61,62 This is a remarkable dose-sparing response, which could be attributed to a combination of biodistribution (accumulate in the spleen, the location of MDSC expansion after tumor onset28,63), tissue retention, the Janus nature of the SNAb, and the multivalency of SNAb. IgG-coated immune complexes must cluster multiple Fc receptors on the same effector cell to induce antibody-dependent immune responses.64 Studies have also shown that increase in IgG-Fc valency augments Fc receptor signaling, such as calcium fluxes and Syk phosphorylation,65 and the IgG density on beads positively correlates with the early signaling in phagocytosis.66 The multivalent presentation of the Fc-mimicking peptide cp33 may enhance the therapeutic response of SNAbs in vivo compared to mAbs, which will be investigated in future studies.
In addition to the direct depletion of MDSCs, MDSC-SNAbs were the only groups that elicited significantly enhanced cytotoxic T cell and NK cell infiltration in tumors, which is a promising and essential sign of a successful tumor immunotherapy because such infiltrations could promote a tumor immune microenvironment that potentiates stronger anti-tumor immunity. As 4T1 breast cancer model is an aggressive model featuring fast progression to death, other tumor models, such as lymphoma models, should be considered when evaluating the long-term survival benefit offered by MDSC depletion using SNAbs. Future investigations should also study the anti-tumor effects of SNAb-based depletion of MDSCs, especially as an adjuvant therapy to checkpoint inhibitors, CAR-T cells, chemo/radiotherapies, and tumor vaccines. In addition to tumors, MDSCs are also a potential target in many other diseases, such as trauma, sepsis, and tuberculosis, since the increase of MDSCs suppresses adaptive immune responses and leads to poor outcomes.23,24,26,67,68 Therefore, MDSC-SNAbs could be a promising immunotherapy in multiple other diseases.
As a synthetic, functional alternative to mAbs, the potential of SNAbs is not limited to MDSC depletion. As a platform nanotechnology, its flexibility lies in the tailorability of physical and chemical properties, e.g., particle material, size, and shape; ligand valency; ligand types (aptamer, peptides, protein domain, complementary RNA or DNA sequence). Such design flexibility could allow for improved biodistribution and targeting efficiency, and the plug-and-play facile conjugation of ligands would allow rapid development of a wide variety of specific cell-depleting nanotherapeutics.58 In conclusion, our results demonstrated the synthesis and characterization of a novel class of nanotherapeutics, SNAbs, that functions as multivalent mAbs, and offer a promising platform tool for treating malignancies, infectious diseases, and other immune disorders.
Supplementary Material
Acknowledgements:
The authors acknowledge financial support from the Georgia Tech Foundation through the Robert A. Milton Chaired Professorship to Krishnendu.Roy., start-up funds from the Georgia Institute of Technology, funds from the Children and Athletes Regenerative Medicine Foundation (12456J4), and Giglio Breast Cancer Innovation Fund (1255d06). For TEM imaging, the authors acknowledge the Institute for Electronics and Nanotechnology at the Georgia Tech, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (ECCS-2025462) and the Center for Nanoscale Materials, an Office of Science user facility supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. For molecular dynamic simulations, the authors acknowledge support from the National Institutes of Health (R01-GM123169). Computational resources were provided through the Extreme Science and Engineering Discovery Environment (XSEDE; allocation TG-MCB130173), which is supported by the National Science Foundation.
Footnotes
Supplementary Information Available:
Material and methods; Additional data on the validation of the Janus nanoparticle fabrication method; Additional STEM-EDS images of Janus and nonJanus nanoparticles with biotinylated QDs and 1.8 nm maleimide-Au nanoprobes; Additional data on free peptide binding on MDSCs, macrophages, DCs, T and B cells in a single-cell suspension of splenocytes; Data on Molecular dynamics simulation of G3 and G3* binding on human S100A8/A9 heterotetramer; Extended data of mouse splenocyte suspension assay of SNAbs; Data on the toxicity of SNAbs on MDSCs isolated from 4T1-tumor-bearing mice; MDSC isolation from blood in rat infection trauma model; Photoacoustic imaging data on the binding of peptide-modified nanoparticles on rat MDSCs and macrophages; Additional data on biodistribution of SNAbs in vivo; Additional data of in vivo MDSC depletion by SNAbs in 4T1 murine tumor model; MDSC, T and NK cell gating strategy from 4T1 murine tumor model; Representative flow plots of in vivo animal experiments; Supplementary tables, including summary of peptides, and antibody types used in the study, modification level quantification of Janus nanoparticles, and typical hydrodynamic size and zeta potential of the nanoparticles.
Ethics declarations:
Competing interests:
The authors declare no competing interests.
Data availability:
The data that support the plots within this paper and other findings of this study are available from the corresponding author upon reasonable request.
References:
- (1).Hwu W; Ph D; Topalian SL; Hwu P; Chen S; Ph D; Salay TM; Alaparthy S; Ph D; Grosso JF; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366 (26), 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Scott AM; Wolchok JD; Old LJ Antibody Therapy of Cancer. Nat. Rev 2012, 12 (4), 278–287. 10.1038/nrc3236. [DOI] [PubMed] [Google Scholar]
- (3).Chames P; Van Regenmortel M; Weiss E; Baty D. Therapeutic Antibodies: Successes, Limitations and Hopes for the Future. Br. J. Pharmacol. 2009, 157 (2), 220–233. 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hammers CM; Stanley JR Antibody Phage Display: Technique and Applications. J. Invest. Dermatol. 2014, 134 (2), e17. 10.1038/jid.2013.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Xiang D; Zheng C; Zhou SF; Qiao S; Tran PHL; Pu C; Li Y; Kong L; Kouzani AZ; Lin J; et al. Superior Performance of Aptamer in Tumor Penetration over Antibody: Implication of Aptamer-Based Theranostics in Solid Tumors. Theranostics 2015, 5 (10), 1083–1097. 10.7150/thno.11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Xenaki Katerina T., O. S. and van B. en H PMP. Antibody or Antibody Fragments: Implications for Molecular Imaging and Targeted Therapy of Solid Tumors. Front. Immunol. 2017, 8. 10.3389/fimmu.2017.01287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kumar V; Patel S; Tcyganov E; Gabrilovich DI The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends in Immunology. 2016. 10.1016/j.it.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Quail DF; Joyce JA Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19 (11), 1423–1437. 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Chen B; Jia Y; Gao Y; Sanchez L; Anthony SM; Yu Y. Janus Particles as Artificial Antigen-Presenting Cells for T Cell Activation. ACS Appl. Mater. Interfaces 2014, 6 (21), 18435–18439. 10.1021/am505510m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Yi Y; Sanchez L; Gao Y; Lee K; Yu Y. Interrogating Cellular Functions with Designer Janus Particles. Chem. Mater. 2017, 29 (4), 1448–1460. 10.1021/acs.chemmater.6b05322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Fu J; An D; Song Y; Wang C; Qiu M; Zhang H. Janus Nanoparticles for Cellular Delivery Chemotherapy: Recent Advances and Challenges. Coord. Chem. Rev. 2020, 422, 213467. 10.1016/j.ccr.2020.213467. [DOI] [Google Scholar]
- (12).Shaghaghi Behrad, Khoee Sepideh, B. S. Preparation of Multifunctional Janus Nanoparticles on the Basis of SPIONs as Targeted Drug Delivery System. Int. J. Pharm. 2019, 559 (25), 1–12. 10.1016/j.ijpharm.2019.01.020. [DOI] [PubMed] [Google Scholar]
- (13).Song Guosheng, Chen Min, Zhang Yanrong, Cui Liyang, Qu Haibo, Zheng Xianchuang, Wintermark Max, Liu Zhuang, and R. J. Janus Iron Oxides @ Semiconducting Polymer Nanoparticle Tracer for Cell Tracking by Magnetic Particle Imaging. Nano Lett 2018, 18 (1), 182–189. 10.1021/acs.nanolett.7b03829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Espinosa A; Reguera J; Curcio A; Muñoz-Noval Á; Kuttner C; Van de Walle A; Liz-Marzán LM; Wilhelm C. Janus Magnetic-Plasmonic Nanoparticles for Magnetically Guided and Thermally Activated Cancer Therapy. Small 2020, 16 (11), 1–14. 10.1002/smll.201904960. [DOI] [PubMed] [Google Scholar]
- (15).Yi Y; Sanchez L; Gao Y; Yu Y. Janus Particles for Biological Imaging and Sensing. Analyst 2016, 141 (12), 3526–3539. 10.1039/c6an00325g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hu J; Zhou S; Sun Y; Fang X; Wu L. Fabrication, Properties and Applications of Janus Particles. Chem. Soc. Rev. 2012, 41 (11), 4356–4378. 10.1039/c2cs35032g. [DOI] [PubMed] [Google Scholar]
- (17).Kaewsaneha C; Tangboriboonrat P; Polpanich D; Eissa M; Elaissari A. Janus Colloidal Particles : Preparation, Properties, and Biomedical Applications. 2013. [DOI] [PubMed] [Google Scholar]
- (18).Tang JL; Schoenwald K; Potter D; White D; Sulchek T. Bifunctional Janus Microparticles with Spatially Segregated Proteins. Langmuir 2012, 28 (26), 10033–10039. 10.1021/la3010079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Peiris PM; Schmidt E; Calabrese M; Karathanasis E. Assembly of Linear Nano-Chains from Iron Oxide Nanospheres with Asymmetric Surface Chemistry. PLoS One 2011, 6 (1). 10.1371/journal.pone.0015927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Tsiganov Evgeny N. ; Verbina EM; Radaeva TV; Sosunov VV; George; Kosmiadi A; Nikitina IY; Lyadova, and V. I. Gr-1dimCD11b+ Immature Myeloid-Derived Suppressor Cells but Not Neutrophils Are Markers of Lethal Tuberculosis Infection in Mice. J Immunol 2014, 192 (10), 4718–4727. 10.4049/jimmunol.1301365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Daker S. El; Sacchi A; Tempestilli M; Carducci C; Goletti D; Vanini V; Colizzi V; Lauria FN; Martini F; Martino A. Granulocytic Myeloid Derived Suppressor Cells Expansion during Active Pulmonary Tuberculosis Is Associated with High Nitric Oxide Plasma Level. PLoS One 2015, 10 (4), 1–10. 10.1371/journal.pone.0123772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Suzanne Ostrand-Rosenberg and Pratima Sinha. Myeloid-Derived Suppressor Cells: Linking Inflammation and Cancer. J Immunol 2009, 182 (8), 4499–4506. 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mathias B, Delmas AL, Ozrazgat-Baslanti T, Vanzant EL, Szpila BE, Mohr AM, Moore FA, Brakenridge SC, Brumback BA, Moldawer LL, E. P. Human Myeloid-Derived Suppressor Cells Are Associated with Chronic Immune Suppression after Severe Sepsis/Septic Shock. Ann Surg. 2017, 265 (4), 827–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Schrijver IT; Théroude C; Roger T. Myeloid Derived Suppressor Cells in Sepsis. Front. Immunol 2019, 10 (FEB), 1–10. 10.3389/fimmu.2019.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Cuenca AG; Delano MJ; Kelly-Scumpia KM; Moreno C; Scumpia PO; LaFace DM; Heyworth PG; Efron PA; Moldawer LL A Paradoxical Role for Myeloid-Derived Suppressor Cells in Sepsis and Trauma. Mol. Med. 2011, 17 (3–4), 281–292. 10.2119/molmed.2010.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Magcwebeba T; Dorhoi A; Du Plessis N. The Emerging Role of Myeloid-Derived Suppressor Cells in Tuberculosis. Front. Immunol. 2019, 10 (APR). 10.3389/fimmu.2019.00917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Nagaraj Srinivas, Schrum Adam G., Cho Hyun-Il, Celis Esteban, and D. I.; Gabrilovich. Mechanism of T-Cell Tolerance Induced by Myeloid-Derived Suppressor Cells. J Immunol. 2010 2011, 184 (6), 3106–3116. 10.4049/jimmunol.0902661.Mechanism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wesolowski R; Markowitz J; Carson WE Myeloid Derived Suppressor Cells - a New Therapeutic Target in the Treatment of Cancer. J. Immunother. cancer 2013, 1 (1), 10. 10.1186/2051-1426-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Brudecki Laura, Ferguson Donald A., McCall Charles E., G ME. Myeloid-Derived Suppressor Cells Evolve during Sepsis and Can Enhance or Attenuate the Systemic Inflammatory Response. Infect. Immun. 2012, 80 (6), 2026–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Alizadeh D; Trad M; Hanke NT; Larmonier CB; Janikashvili N; Bonnotte B; Katsanis E; Larmonier N. Doxorubicin Eliminates Myeloid-Derived Suppressor Cells and Enhances the Efficacy of Adoptive T-Cell Transfer in Breast Cancer. Cancer Res. 2014, 74 (1), 104–118. 10.1158/0008-5472.CAN-13-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Vincent J; Mignot G; Chalmin F; Ladoire S; Bruchard M; Chevriaux A; Martin F; Apetoh L; Rebe C; Ghiringhelli F. 5-Fluorouracil Selectively Kills Tumor-Associated Myeloid-Derived Suppressor Cells Resulting in Enhanced T Cell-Dependent Antitumor Immunity. Cancer Res. 2010, 70 (8), 3052–3061. 10.1158/0008-5472.CAN-09-3690. [DOI] [PubMed] [Google Scholar]
- (32).Obermajer N; Muthuswamy R; Lesnock J; Edwards RP; Kalinski P. Positive Feedback between PGE2 and COX2 Redirects the Differentiation of Human Dendritic Cells toward Stable Myeloid-Derived Suppressor Cells. Blood 2011, 118 (20), 5498–5506. 10.1182/blood-2011-07-365825.The. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Forghani P; Khorramizadeh MR; Waller EK Silibinin Inhibits Accumulation of Myeloid-Derived Suppressor Cells and Tumor Growth of Murine Breast Cancer. Cancer Med. 2014, 1–10. 10.1002/cam4.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ma C; Kapanadze T; Gamrekelashvili J; Manns MP; Korangy F; Greten TF Anti-Gr-1 Antibody Depletion Fails to Eliminate Hepatic Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. J. Leukoc. Biol. 2012, 92 (6), 1199–1206. 10.1189/jlb.0212059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Xing Y-F; Zhou Y-Q; Ma G-W; Feng D-Y; Cai X-R; Li X. Issues with Anti-Gr1 Antibody-Mediated Myeloid-Derived Suppressor Cell Depletion. Ann. Rheum. Dis. 2016, 75 (8), e49–e49. 10.1136/annrheumdis-2016-209786. [DOI] [PubMed] [Google Scholar]
- (36).Sukhatme, Vikas P; Husain Z. Cancer Therapy Targeting Tetraspanin 33 (Tspan33) IN Myeloid Derived Suppressor Cells. (43) International Publication Date WO 2016/210241 Al, 2016. [Google Scholar]
- (37).Luu VP; Hevezi P; Vences-Catalan F; Maravillas-Montero JL; White CA; Casali P; Llorente L; Jakez-Ocampo J; Lima G; Vilches-Cisneros N; et al. TSPAN33 Is a Novel Marker of Activated and Malignant B Cells. Clin. Immunol. 2013, 149 (3 PB), 388–399. 10.1016/j.clim.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Qin H; Lerman B; Sakamaki I; Wei G; Cha SC; Rao SS; Qian J; Hailemichael Y; Nurieva R; Dwyer KC; et al. Generation of a New Therapeutic Peptide That Depletes Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. Nat. Med. 2014, 20 (6), 676–681. 10.1038/nm.3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bresnick Anne R., Weber David J., and Z DB. S100 Proteins in Cancer. Nat. Rev. Cancer 2015, 15 (2), 96–109. 10.1038/nrc3893.S100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Bronte V; Brandau S; Chen SH; Colombo MP; Frey AB; Greten TF; Mandruzzato S; Murray PJ; Ochoa A; Ostrand-Rosenberg S; et al. Recommendations for Myeloid-Derived Suppressor Cell Nomenclature and Characterization Standards. Nature Communications. 2016. 10.1038/ncomms12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Sinha P; Okoro C; Foell D; Freeze HH; Ostrand-Rosenberg S; Srikrishna G. Proinflammatory S100 Proteins Regulate the Accumulation of Myeloid-Derived Suppressor Cells. J Immunol. 2008, 181 (7), 4666–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zhao F; Hoechst B; Duffy A; Gamrekelashvili J; Fioravanti S; Manns MP; Greten TF; Korangy F. S100A9 a New Marker for Monocytic Human Myeloid-Derived Suppressor Cells. Immunology 2012, 136 (2), 176–183. 10.1111/j.1365-2567.2012.03566.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Bonetto Stephane, Spadola Loredana, Buchanan Andrew G., Jermutus Lutz, and J. L. Identification of Cyclic Peptides Able to Mimic the Functional Epitope of IgG1-Fc for Human FcγRI. FASEB J. 2009, 23 (2), 575–585. 10.1096/fj.08-117069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Mcenaney PJ; Fitzgerald KJ; Zhang AX; Douglass EF; Shan W; Balog A; Kolesnikova MD; Spiegel DA Chemically Synthesized Molecules with the Targeting and Effector Functions of Antibodies. J. Am. Chem. Soc. 2014, 136, 18034–18043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Kubelick KP; Snider EJ; Ross Ethier C; Emelianov S. Development of a Stem Cell Tracking Platform for Ophthalmic Applications Using Ultrasound and Photoacoustic Imaging. Theranostics 2019, 9 (13), 3812–3824. 10.7150/thno.32546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Donnelly EM; Kubelick KP; Dumani DS; Emelianov SY Photoacoustic Image-Guided Delivery of Plasmonic-Nanoparticle-Labeled Mesenchymal Stem Cells to the Spinal Cord. Nano Lett. 2018, 18 (10), 6625–6632. 10.1021/acs.nanolett.8b03305. [DOI] [PubMed] [Google Scholar]
- (47).Luke GP, Yeager D, E. S. Biomedical Applications of Photoacoustic Imaging with Exogenous Contrast Agents. Annu. Biomed. Eng. 2012, 40 (2), 422–437. 10.1007/s10439-011-0449-4. [DOI] [PubMed] [Google Scholar]
- (48).Lee K; Yu Y. Janus Nanoparticles for T Cell Activation: Clustering Ligands to Enhance Stimulation. J. Mater. Chem. B 2017, 5 (23), 4410–4415. 10.1039/c7tb00150a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Pacheco PM; Le B; White D; Sulcheck T. Tunable Complement Activation By Particles With Variable Size and Fc Density. Nano Life 2013, 03 (02), 1341001. 10.1142/s1793984413410018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Pacheco P; White D; Sulchek T. Effects of Microparticle Size and Fc Density on Macrophage Phagocytosis. PLoS One 2013, 8 (4), 1–9. 10.1371/journal.pone.0060989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Narumi K; Miyakawa R; Ueda R; Hashimoto H; Yamamoto Y; Yoshida T; Aoki K. Proinflammatory Proteins S100A8/S100A9 Activate NK Cells via Interaction with RAGE. J. Immunol. 2015, 194 (11), 5539–5548. 10.4049/jimmunol.1402301. [DOI] [PubMed] [Google Scholar]
- (52).Vantucci Casey E., Ahn Hyunhee, M LS; Pradhan P; , Wood Levi B., Guldberg RE; Roy K; Willett NJ Development of Systemic Immune Dysregulation in a Rat Trauma Model with Biomaterial-Associated Infection. bioRxiv 2020, 1–36. 10.1101/2020.01.10.901769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Tschoeke SK & Ertel W. Immunoparalysis after Multiple Trauma. Injury 2007, 38, 1346–1357. [DOI] [PubMed] [Google Scholar]
- (54).Cheng L; Xu J; Chai Y; Wang C; Han P. Dynamic Changes in Trauma-Induced Myeloid-Derived Suppressor Cells after Polytrauma Are Associated with an Increased Susceptibility to Infection. Int. J. Clin. Exp. Pathol. 2017, 10 (11), 11063–11068. [PMC free article] [PubMed] [Google Scholar]
- (55).Matsumoto Y; Nichols JW; Toh K; Nomoto T; Cabral H; Miura Y; Christie RJ; Yamada N; Ogura T; Kano MR; et al. Vascular Bursts Enhance Permeability of Tumour Blood Vessels and Improve Nanoparticle Delivery. Nat. Nanotechnol. 2016, 11 (6), 533–538. 10.1038/nnano.2015.342. [DOI] [PubMed] [Google Scholar]
- (56).Cabral H; Makino J; Matsumoto Y; Mi P; Wu H; Nomoto T; Toh K. Systemic Targeting of Lymph Node Metastasis through the Blood Vascular System by Using Size-Controlled. 2015, No. 5, 4957–4967. [DOI] [PubMed] [Google Scholar]
- (57).Molek P; Strukelj B; Bratkovic T. Peptide Phage Display as a Tool for Drug Discovery: Targeting Membrane Receptors. Molecules 2011, 16 (1), 857–887. 10.3390/molecules16010857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Zhou J; Rossi J. Aptamers as Targeted Therapeutics: Current Potential and Challenges. Nat. Rev. Drug Discov. 2017, 16 (3), 181–202. 10.1038/nrd.2016.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Nixon AE; Sexton DJ; Ladner RC Drugs Derived from Phage Display From Candidate Identification to Clinical Practice Drugs Derived from Phage Display. MAbs 2014, 6 (1), 73–85. 10.4161/mabs.27240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Martinez-Veracoechea FJ; Frenkel D. Designing Super Selectivity in Multivalent Nano-Particle Binding. Proc. Natl. Acad. Sci. 2011, 108 (27), 10963–10968. 10.1073/pnas.1105351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Srivastava MK; Zhu L; Harris-White M; Kar U; Huang M; Johnson MF; Lee JM; Elashoff D; Strieter R; Dubinett S; et al. Myeloid Suppressor Cell Depletion Augments Antitumor Activity in Lung Cancer. PLoS One 2012, 7 (7). 10.1371/journal.pone.0040677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Thaci B, Ahmed AU, Ulasov IV, Wainwright DA, Nigam P, Auffinger B, Tobias AL, Han Y, Zhang L, Moon K-S, and L M. Depletion of Myeloid-Derived Suppressor Cells during Interleukin-12 Immunogene Therapy Does Not Confer a Survival Advantage in Experimental Malignant Glioma. Cancer Gene Ther. 2014, 21 (1), 38–44. 10.1038/cgt.2013.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Tang L; Yang X; Yin Q; Cai K; Wang H; Chaudhury I; Yao C; Zhou Q; Kwon M; Hartman J. a.; et al. Investigating the Optimal Size of Anticancer Nanomedicine. Proc. Natl. Acad. Sci. 2014, 111 (43), 15344–15349. 10.1073/pnas.1411499111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Goodridge HS; Underhill DM; Touret N. Mechanisms of Fc Receptor and Dectin-1 Activation for Phagocytosis. Traffic 2012, 13, 1062–1071. 10.1111/j.1600-0854.2012.01382.x. [DOI] [PubMed] [Google Scholar]
- (65).Ortiz DF; Lansing JC; Rutitzky L; Kurtagic E; Prod T; Choudhury A; Washburn N; Bhatnagar N; Beneduce C; Holte K; et al. Elucidating the Interplay between IgG-Fc Valency and Fc g R Activation for the Design of Immune Complex Inhibitors. Sci. Transl. Med. 2016, 8, 13. [DOI] [PubMed] [Google Scholar]
- (66).Zhang Y; Hoppe AD; Swanson J. a. Coordination of Fc Receptor Signaling Regulates Cellular Commitment to Phagocytosis. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (45), 19332–19337. 10.1073/pnas.1008248107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Lai D; Qin C; Shu Q. Myeloid-Derived Suppressor Cells in Sepsis. Biomed Res. Int 2014, 2014, 598654. 10.1155/2014/598654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Salminen A; Kaarniranta K; Kauppinen A. Immunosenescence: The Potential Role of Myeloid-Derived Suppressor Cells (MDSC) in Age-Related Immune Deficiency. Cell. Mol. Life Sci. 2019, 76 (10), 1901–1918. 10.1007/s00018-019-03048-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.