

Key Points
NFAT5 amplifies TLR-induced antipathogen transcription responses.
NFAT5 induces chromatin accessibility and H3K27me3 demethylation.
NFAT5 facilitates recruitment of p65/NF-κB and c-Fos to promoters.
Visual Abstract
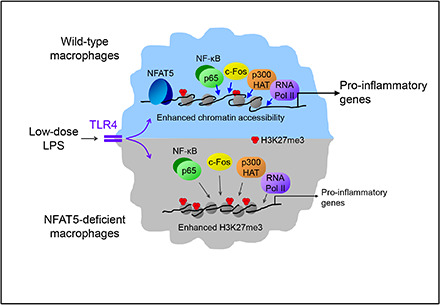
Abstract
The ability of innate immune cells to respond to pathogen-associated molecular patterns across a wide range of intensities is fundamental to limit the spreading of infections. Studies on transcription responses to pathogen-activated TLRs have often used relatively high TLR ligand concentrations, and less is known about their regulation under mild stimulatory conditions. We had shown that the transcription factor NFAT5 facilitates expression of antipathogen genes under TLR stimulation conditions corresponding to low pathogen loads. In this study, we analyze how NFAT5 optimizes TLR-activated responses in mouse macrophages. We show that NFAT5 was required for effective recruitment of central effectors p65/NF-κB and c-Fos to specific proinflammatory target genes, such as Nos2, Il6, and Tnf in primary macrophages responding to low doses of the TLR4 ligand LPS. By contrast, NFAT5 was not required for p65/NF-κB recruitment in response to high LPS doses. Using the transposase-accessible chromatin with high-throughput sequencing assay, we show that NFAT5 facilitated chromatin accessibility mainly at promoter regions of multiple TLR4-responsive genes. Analysis of various histone marks that regulate gene expression in response to pathogens identified H3K27me3 demethylation as an early NFAT5-dependent mechanism that facilitates p65 recruitment to promoters of various TLR4-induced genes. Altogether, these results advance our understanding about specific mechanisms that optimize antipathogen responses to limit infections.
Introduction
The ability of sentinel cells such as macrophages to detect reduced loads of microbes allows immune defenses to limit the pathogen burden and curtail infections. This capacity depends on specific sensors such as TLRs, which recognize a variety of pathogen-associated molecular patterns in a wide range of concentrations and activate proinflammatory and antipathogen responses that are for the most part regulated by gene transcription (1–4). Although different transcription factors participate in the response to TLR and considerable progress has been made in identifying their respective target genes, less is known about mechanisms involved in optimizing transcription responses to reduced loads of pathogens, a capacity that is fundamental in the defense against infections.
Early work identified C/EBPδ as a transcription factor particularly relevant in the response to sustained high pathogen loads (5) and described that NF-κB–induced C/EBPδ expression amplifies the inflammatory response in genes such as Il6. More recently, we found that NFAT5, which is upregulated by NF-κB, optimizes gene expression in primary macrophages particularly in response to mild TLR stimulation conditions that correspond to a reduced pathogen burden (6). NFAT5 shows structural and functional similarities with other members of the family of Rel homology domain-containing factors, which comprises the NF-κB and calcineurin-dependent NFATc proteins (7, 8). NFAT5 regulates diverse proinflammatory and antimicrobial macrophage functions, supporting the classical polarization of macrophages and their ability to shape inflammatory T helper responses (9), modulating specific gene expression patterns downstream different TLRs (6, 10), and sustaining macrophage MHC class II expression (11). As result of these, NFAT5 enhances the direct antibacterial activity of macrophages (9) and their ability to control Leishmania infection in vivo (6), attenuates antiviral responses by macrophages and dendritic cells (10), and also strengthens macrophage antitumor capacity (9) and ability to promote allograft rejection (11). NFAT5 is not only activated by pathogen-derived products, but also by sodium hypertonicity (12), and the combination of both stimuli enhances NFAT5-dependent antimicrobial macrophage responses in skin and kidney (13, 14).
In response to TLR, NFAT5 promotes the induction of multiple genes, including those encoding for the inducible NO synthase (iNOS) and cytokines and chemokines such as IL-6, TNF-α, and CCL2, which overall confer macrophages inflammatory and antimicrobial capacity (6, 9). As described above, this role is more relevant under conditions of low-intensity TLR stimulation, as NFAT5-deficient macrophages exhibit a marked impairment in the expression of iNOS, IL-6, and TNF-α upon stimulation with low doses of the TLR4 agonist LPS, but less pronounced defects with a stronger stimulation (6). This allows NFAT5 to optimize the expression of genes that have different transcription requirements, as it is readily recruited in a TLR-dependent manner to the promoters of secondary response genes such as Nos2, Il6, and Ptgs2 but constitutively bound to promoters of primary response target genes such as Tnf, Il1a, Ccl2, and Traf1 already in steady-state macrophages (6, 15). Constitutive association with some inflammatory genes is a characteristic of NFAT5 that differs from other TLR-responsive transcription regulators, overall suggesting that NFAT5 is poised for the rapid induction of antipathogen responses.
Our previous work led us to propose that elucidating how NFAT5 amplifies expression of proinflammatory genes could allow us to better understand how macrophages can optimize transcription responses to reduced pathogen burdens. In this study, we show that under mild LPS stimulation, NFAT5 facilitates the recruitment of transcription regulators of antipathogen responses such as p65/NF-κB and c-Fos to target genes and enhances chromatin accessibility around the promoter region of numerous TLR-responsive genes. Analysis of different histone modifications in NFAT5-deficient macrophages uncovered a specific defect in LPS-induced H3K27me3 demethylation in NFAT5-target genes. Our results suggest that this mechanism is relevant for p65/NF-κB recruitment to target gene promoters because inhibition of H3K27me3 demethylases impaired p65/NF-κB recruitment in wild-type macrophages down to the reduced level of NFAT5-deficient cells. Altogether, our findings show how NFAT5 endows macrophages with an amplification mechanism that optimizes their responsiveness to pathogens.
Materials and Methods
Mice
NFAT5-deficient mice have a deletion in the Nfat5 gene in the region encoding the first exon of its DNA-binding domain (16). Deletion of this genomic region leads to an essentially complete loss of NFAT5 protein in multiple tissues, including leukocytes (6, 12, 16). This mouse model was generated by targeting Nfat5 in embryonic stem cells of the 129/sv background, which were then used to produce blastocysts that were implanted in C57BL/6 mice. After confirming germline transmission, mixed-background 129/sv/C57BL/6 mice were backcrossed with 129/sv mice for more than 10 generations to obtain the Nfat5 mutant allele in the 129/sv background (16). Nfat5+/– (heterozygous) mice were maintained in a pure 129/sv background and intercrossed to obtain Nfat5–/– mice and control Nfat5+/+ littermates, which were analyzed between 6 to 10 wk of age (6). Tissue-specific conditional NFAT5-knockout mice were obtained by crossing Nfat5-floxed (Nfat5fl/fl) mice with appropriate Cre driver mice. Nfat5fl/fl mice were generated in a pure C57BL/6 background by targeting the Nfat5 gene in C57BL/6 embryonic stem cells using a vector different from that used above, but that also targeted the region encoding the first exon of NFAT5 DNA-binding domain by inserting flanking LoxP sites (17). C57BL/6 Nfat5fl/fl mice were crossed with LysM-Cre or Csf1r-Cre mice (all C57BL/6) to obtain mice with NFAT5-deficient myeloid cells. LysM-Cre mice were purchased from The Jackson Laboratory (catalog no. 004781) (11), and Csf1r-Cre mice (18) were kindly provided by Toby Lawrence (King’s College London, London, U.K.). All experiments were done using independent pairs of NFAT5-deficient mice and control wild-type littermates of both sexes. Mice were bred and maintained in specific pathogen–free conditions, and animal handling was performed according to institutional guidelines that comply with the Association for Assessment and Accreditation of Laboratory Animal Care and the European Communities Council Directive (86/609/EEC) and were approved by the institutional ethical committee (Barcelona Biomedical Research Park and Pompeu Fabra University Animal Care and Use Committee).
Isolation and culture of macrophages
To obtain bone marrow–derived macrophages (BMDM) (6), mice were sacrificed and the femoral and tibial marrow was flushed from the bones with DMEM supplemented with 2 mM glutamine, 50 µM β-mercaptoethanol, and 1 mM sodium pyruvate plus penicillin/streptomycin (all from Life Technologies) (incomplete medium). Cells were then resuspended in complete DMEM media (incomplete supplemented with 10% heat-inactivated FBS) with 25% (vol/vol) L929-conditioned medium (as the source of macrophage colony–stimulating factor), and incubated for 7 d in polystyrene Petri dishes. Differentiated macrophages were directly used for chromatin immunoprecipitation (ChIP) or were harvested with ice-cold PBS plus 1 mM EDTA by gentle pipetting, washed with PBS, and seeded in tissue culture–treated plates for Western blot and mRNA analysis (0.8 × 106 cells per 3 ml per well). Peritoneal macrophages were isolated 3 h after i.p. injection of LPS (0.05 µg/kg mouse) using a rat anti-Mouse CD11b mAb (M1/70.15 hybridoma) and Dynabeads Sheep anti-rat IgG magnetic beads (11035; Invitrogen).
Peritoneal macrophages were obtained from myeloid cell–specific NFAT5-deficient mice (LysM-Cre Nfat5fl/fl in C57BL/6 background) (10). Peritoneal macrophages used for mRNA analysis were harvested by two consecutive lavages with 5 ml of ice-cold PBS. For obtaining thioglycollate-elicited peritoneal macrophages, mice were injected i.p. with 1 ml of 3% thioglycollate medium (B2551; Sigma-Aldrich), and 5 d later, mice were sacrificed to obtain peritoneal cells. Suspensions of naive or thioglycollate-elicited peritoneal cells were subjected to immunomagnetic depletion to eliminate B cells, eosinophils, and neutrophils. For this, each peritoneum was adjusted to 1 ml in complete DMEM, incubated at 4°C under rotation for 30 min, and then 10 μl of Dynabeads Sheep anti-Mouse IgG (11031; Invitrogen) were added to deplete B cells. The suspension was incubated at 4°C under continuous rotation for another 30 min. Beads and attached B cells were then removed with a magnet. Eosinophils and neutrophils were then depleted by incubation (4°C, 30 min) with 5 μl of rat anti-mouse SiglecF (552125; BD Pharmingen) and 5 μl of rat anti-mouse Ly6G (127602; BioLegend), both previously coated to Dynabeads Sheep anti-rat IgG (11035; Invitrogen). After immunomagnetic depletion, the remaining cell suspension was plated in 12-well (thioglycollate-elicited macrophages) or 24-well (naive peritoneal macrophages) plates for 2 h at 37°C in a 5% CO2 atmosphere to let macrophages adhere. Nonadherent cells were removed by replacing the media and macrophages were left at 37°C in a 5% CO2 atmosphere overnight before stimulation.
Reagents
LPS was from Escherichia coli strain 055:B5 (LPS, L2880; Sigma-Aldrich) or from E. coli serotype EH100 (ultrapure LPS, TLR grade, ALX-581-010-L001; Enzo Life Sciences). Formaldehyde, sodium chloride, Trizma base, glycine, EDTA, iodoacetamide, sodium pyrophosphate (NaPPi), sodium orthovanadate, β-glycerophosphate, PMSF, leupeptin, pepstatin A, aprotinin, NaDodSO4 (SDS), Tween-20, glycine, glycerol, methanol, Triton X-100 (TX-100), Nonidet P-40, sodium deoxycholate, potassium chloride, zinc chloride, and trichostatin A (TSA) were all from Sigma-Aldrich. Sodium fluoride (NaF) was from Merck, magnesium chloride was from Calbiochem, and HEPES was from Lonza. KRN2 was from Sigma-Aldrich (SML2464) or MedChemExpress (HY-112125/HY-112125A), and cells were pretreated for 1 h. Both chloride and bromide salt KRN2 formulations were used with the same results. GSK-J4 (hydrochloride) was from Tocris Bioscience (4594) or from Cayman Chemical (12073). GSK-126 (15415) and UNC1999 (14621) were from Cayman Chemical. These inhibitors were used at 10 µM. Pretreatment with GSK-J4 was for 1 h, and pretreatment with GSK-126 and UNC1999 was for 16 h before stimulating the macrophages. This extended pretreatment was done to inhibit histone methyltransferases overnight prior to stimulating the cells, and thus facilitate the activity of histone demethylases upon LPS stimulation.
ChIP
ChIP was performed as previously described (6). BMDMs were stimulated with LPS as indicated in the figure legends, and fixed with 0.75% formaldehyde for 10 min at room temperature. Formaldehyde was then quenched with glycine (final concentration, 0.326 M) for 5 min. After washing the plates twice with ice-cold PBS, cells were collected with cell scrapers, lysed in 0.5 ml of lysis buffer (50 mM Tris-HCl [pH 8], 10 mM EDTA, 1% SDS, 1 mM PMSF, 5 µg/ml leupeptin/aprotinin, 1 µg/ml pepstatin A, 10 mM NaF, 10 mM sodium orthovanadate, and 20 mM β-glycerophosphate) for 30 min at room temperature. Lysates were split in 2 × 1.5-ml tubes containing 250 μl of lysate and sonicated (Diagenode Bioruptor UCD-200TM-EX), for six to nine cycles (30s on, 30s off) at the high-power setting to obtain DNA fragments between 500–1000 bp. After sonication, samples were centrifuged to remove insoluble debris, supernatants were collected, and 5% of each sample was separated to use as chromatin input for normalization. The rest of the sample was diluted 10 times in dilution buffer (1% TX-100, 20 mM Tris-HCl [pH 8], 2 mM EDTA, 150 mM NaCl, 1 mM PMSF, 5 µg/ml leupeptin, 5 µg/ml aprotinin, 1 µg/ml pepstatin A, 10 mM NaF, 10 mM sodium orthovanadate, and 10 mM β-glycerophosphate) for immunoprecipitation. Samples were precleared with protein A–Sepharose beads (GE Healthcare) that were previously preadsorbed with sheared salmon sperm DNA (Invitrogen) and BSA (New England BioLabs) 4 h to overnight at 4°C. After removing the preclearing beads, specific Abs (detailed below) were added to the lysates and incubated overnight at 4°C with rotation. Preadsorbed protein A–Sepharose beads were then added and incubated for 1–3 h at 4°C, and then washed three times with washing buffer (0.1% SDS, 1% TX-100, 20 mM Tris-HCl [pH 8], 2 mM EDTA, and 150 mM NaCl) and once with final washing buffer (0.1% SDS, 1% TX-100, 20 mM Tris-HCl [pH 8], 2 mM EDTA, and 500 mM NaCl). To elute the DNA, beads were incubated with elution buffer (1% SDS and 100 mM NaHCO3, 200 µl per sample) for 15 min at room temperature in a shaker. To reverse the cross-linking, samples were incubated overnight at 65°C. Samples were then incubated with 3 µg of RNase A (Thermo Fisher Scientific) at 37°C for 30 min, after which 70 µg of proteinase K (Roche Diagnostics) were added and incubated at 50°C for 1 h. DNA was purified by ethanol precipitation. For real-time quantitative PCR (RT-qPCR), LightCycler 480 SYBR Green I Master (Roche Diagnostics), LightCycler 480 Multiwell Plate (Roche Diagnostics), and the LightCycler 480 Real-Time PCR System (Roche Diagnostics) were used following the instructions provided by the manufacturers. Primers used are listed in Supplemental Table I. Immunoprecipitated DNA from each sample was normalized to its respective chromatin input. Abs to p65/RelA (sc-372), c-Fos (sc-7202), p300 (sc-585), and normal rabbit IgG (sc-2027) were from Santa Cruz Biotechnology. In Supplemental Fig. 3B, the Ab to p65/RelA used was from Diagenode (NFKBp65 C15310256), and its control preimmune serum was from Bethyl Laboratories. The control rabbit IgG used in Supplemental Fig. 3C was from Diagenode (C15410206). Abs to RNA polymerase II (RNA pol II) (ab817), total histone H3 (ab1791), total histone H4 (ab7311), H3K4me3 (ab8580), and H4K20me3 (ab9053) were from Abcam; Abs to acetylated histone H3 (lysines 9 and 14, 06-599), acetylated histone H4 (lysines 5, 8, 12, and 16, 06-866), and H3K27me3 (07-449) were from MilliporeSigma; the Ab to EZH2 (pAb-039-050) was from Diagenode; the rabbit polyclonal Ab to JMJD3 was kindly provided by Dr. Gioacchino Natoli (European Institute of Oncology and Humanitas University, Milan, Italy) (19); and the Ab to ubiquitously transcribed tetratricopeptide repeat X chromosome (UTX) was from Bethyl Laboratories (A302-374A).
Transposase-accessible chromatin with high-throughput sequencing
BMDMs were left untreated or stimulated for 2 h with 0.3 ng/ml LPS and then harvested with PBS plus 1 mM EDTA by gentle pipetting, and 50,000 cell pellets were obtained. Nuclei preparation and transposase reactions were done as described in (20) with minor modifications. Briefly, nuclei were obtained with the CelLytic NuCLEAR Extraction system (Sigma-Aldrich), and resuspended in 50 μl 1× TD buffer containing 2.5 μl of transposase (Nextera, Illumina). The transposase reaction was conducted for 30 min at 37°C. Library amplification and barcoding were performed with NEBNext Q5 Hot Start HiFi PCR Master Mix (New England BioLabs) using index primers, designed according to (20) at a final concentration of 1.25 µM. PCR was conducted for 12–13 cycles. Library purification was performed with AgenCourt AMPure XP beads (Beckman Coulter) and library size distribution was assessed using the Bioanalyzer High Sensitivity DNA Kit (Agilent). Assays for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) libraries were quantified before pooling and sequencing using the Real-Time Library Quantification Kit (KAPA Biosystems). Paired-end sequencing was performed on an HiSeq 2500 (Illumina) with 50 cycles for each read.
For ATAC-seq analysis, data were collected using 2 × 50 bp reads from an Illumina HiSeq2500 machine. The quality of sequenced reads was assessed using the FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Raw reads were mapped to the mouse genome (Mus musculus GRCm38 ENSEMBL release 87) (ftp://ftp.ensembl.org/pub/release-87/fasta/mus_musculus) using Bowtie2 version 2.3.0, using options –very-sensitive, –X2000 (fragments up to 2 kb were able to be aligned), and -m1 (only uniquely aligned reads were collected) (21). For all data files, PCR duplicates were removed using Picard (MarkDuplicates method) version 2.10.5 (http://broadinstitute.github.io/picard). Read start sites were adjusted to represent the center of the transposon binding events as follows: all reads aligning to the positive strand were offset by +4 bp, and all reads aligning to the negative strand were offset by –5 bp. Enriched and depleted regions were called using MACS2 version 2.1.1.20160309 (callpeak method using options –f BAMPE, –q 0.05, –nomodel, –extsize 150) (22). Differential binding events were assessed with DiffBind version 2.6.4 (in Bioconductor v3.6 and R v3.4.1 environment) (23), taking into account the biological duplicates; in this study, binding events were considered at false discovery rate ≤ 0.1. The annotation of the differential binding events was done using the HOMER annotatePeaks method. Additional annotation was added with biomaRt (via biomaRt R package) (24, 25). Count per million were calculated using the EdgeR package (26). Mapped reads were visualized as a track hub in the University of California, Santa Cruz genome browser (27). The accession number for the ATAC-seq dataset is GSE111476.
For searching potential NFAT5 binding sites in the differentially accessible regions of the ATAC-seq analysis, we used the consensus NFAT5-binding sequence ([A/C/T][A/G]TGGAAA[C/A/T]A) previously identified by affinity selection of a double-strand oligonucleotide library with the recombinant dimeric DNA-binding domain of NFAT5 (7). We used the findMotif motif search tool (with options fasta, -mknown, -nomotif, -len 5,12 -nogo) from the HOMER suite (http://homer.ucsd.edu/homer/) (24) to look for NFAT5-binding motif enrichment in promoter regions (–1500 bp to +500 bp from the transcription start site [TSS], and –5000 bp to +500 bp from the TSS) whose accessibility was increased or reduced by NFAT5 in LPS-stimulated macrophages. In the –1500 to +500 regions search, NFAT5 motifs were found in 947 of 2482 automatically generated regions (38.15%; p = 1 × 10–51; Benjamini q = 0.0000) showing NFAT5-enhanced accessibility and in 89 of 163 regions (54.6%; p = 1 × 10–3; Benjamini q = 0.0004) with NFAT5-repressed accessibility. For the search in regions between –5000 bp to +500 bp, NFAT5 motifs were found in 2120 of 2680 regions (79.1%; p = 1 × 10–49; Benjamini q = 0.0000) with NFAT5-enhanced accessibility, and in 266 of 303 regions (87.8%; p = 1 × 10–5; Benjamini q = 0.0000) with NFAT5-repressed accessibility.
Identification of intergenic regions with enhancer features that were differentially accessible between wild-type and NFAT5-deficient cells was done based on their content in acetylated K27 in histone H3 (H3K27Ac). Published data (28) were extracted from the Gene Expression Omnibus database (29) (Gene Expression Omnibus accession number GSE119691) in browser extensible data format. We considered unstimulated and 1-h and 2-h LPS-stimulated samples. The mm9 coordinates from the published browser extensible data files were converted to mm10 coordinates with the liftOver online tool from the University of California, Santa Cruz genome browser (27). BEDTools merge (30) was used to retrieve common peaks between replicates. These sets of common peaks between replicates were intersected with those of our ATAC-seq annotated in intergenic regions using BEDTools. Resulting intersections were annotated to the closest TSS using ENSEMBL release 87 annotation.
DNA affinity precipitation assay (DAPA)
Cells were stimulated as indicated and nuclear protein extracts were obtained with CelLytic NuCLEAR Extraction system (Sigma-Aldrich) following the manufacturer’s instructions. A total of 70–100 μg of nuclear protein were precleared with streptavidin-agarose beads (Genscript) in binding buffer (20 mM HEPES [pH 7.9], 10% glycerol, 50 mM KCl, 0.2 mM EDTA, 1.5 mM MgCl2, 10 μM ZnCl2, 1 mM DTT, 0.25% Triton X-100) for 1 h at 4°C. Lysates were incubated with 1 μg of poly(dI)/poly(dC) and 1 μg of poly(dG)/poly(dC) (GE Healthcare) for 10 min at 4°C. A biotinylated probe corresponding to the upstream NF-κB site in Nos2 promoter (–996, see sequence in Supplemental Table I), was added to the lysates and incubated for 50 min at 4°C in rotation. Streptavidin-agarose beads were then added, incubated for 2 h at 4°C, and then washed four times with ice-cold binding buffer and once with ice-cold PBS. DNA-bound protein was eluted by adding reducing 2× Laemmli buffer and heating at 100°C for 10 min. To detect the fraction of p65 bound to the biotinylated probe, Western blot analysis was performed as described below.
Restriction enzyme accessibility assay
Assays were done following the methodology described in (31). BMDMs (5 × 106 to 6 × 106 cells) were stimulated as indicated and lysed in 10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 0.5% NP-40, 0.15 mM spermine (Sigma-Aldrich), 0.5 mM spermidine (Sigma-Aldrich). Nuclei were pelleted by centrifugation at 850 × g for 5 min and washed in digestion buffer (NEBuffer 2; New England BioLabs), then resuspended and divided in two different conditions: a control digestion with 15 U of BglII endonuclease (New England BioLabs) alone, and a digestion with 100 U of HinfI endonuclease (New England BioLabs), which has recognition sites in the proximal promoters of Nos2, Il6, and Tnf, plus 15 U of BglII endonuclease. Digestion with BglII was used to obtain a starting pool of DNA fragments of comparable length between all samples. Digestions were incubated for 1 h at 37°C, and then samples were treated with proteinase K (Roche Diagnostics) at 56°C overnight. DNA was purified with phenol/chloroform (Sigma-Aldrich) and subjected to RT-qPCR using LightCycler 480 SYBR Green Master (Roche Diagnostics) and primers that amplified regions containing HinfI restriction sites in the proximal promoters of Nos2, Tnf, and Il6 (Supplemental Table I). To normalize the amplified DNA across samples, RT-qPCR quantification of the Gapdh gene amplified with primers in a region with no HinfI restriction sites was used as reference.
Quantification of mRNA levels
Total RNA from BMDM was isolated using the High Pure RNA Isolation Kit (Roche Diagnostics), quantified in a NanoDrop (ND-1000) spectrophotometer, and 200 ng to 2 μg of total RNA was retrotranscribed to cDNA using Transcription First Strand cDNA Synthesis Kit (Roche Diagnostics). cDNA was then subjected to RT-qPCR using LightCycler 480 SYBR Green Master (Roche Diagnostics), and the primers listed in Supplemental Table I. Samples were normalized to L32 (L32 ribosomal protein gene) mRNA levels using the LightCycler Software, version 1.5. For peritoneal macrophages, total RNA was isolated using the RNeasy Mini Kit (QIAGEN), quantified in a NanoDrop spectrophotometer (ND-1000), and 100 ng of RNA were retrotranscribed to cDNA using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems).
Immunoblot assays
For protein detection by Western blotting, BMDMs were lysed in Triton X-100 lysis buffer (0.5 × 106 to 1 × 106 cells in 100–200 µl) (1% TX-100, 40 mM HEPES [pH 7.4], 120 mM NaCl, 1 mM EDTA, 10 mM NaPPi, 10 mM β-glycerophosphate, 1 mM PMSF, 5 µg/ml leupeptin, 5 µg/ml aprotinin, 1 µg/ml pepstatin A, 1 mM NaF, and 1 mM sodium orthovanadate). Protein concentration in the lysates was quantified using the BCA assay (Pierce; Thermo Fisher Scientific) to ensure loading the same amount of protein per sample in the gels. For the fractionation of nuclear and cytoplasmic extracts, lysates were prepared with CelLytic NuCLEAR Extraction Kit (Sigma-Aldrich). In all cases, lysates were boiled in reducing 1× Laemmli buffer, and 10–50 µg of total protein were subjected to SDS-PAGE and transferred to PROTRAN membranes (Schleicher & Schuell) in 25 mM Tris, (pH 8.4), 192 mM glycine, and 20% methanol. After blocking the membranes with 5% dry milk in TBS, the following primary Abs were used: rabbit polyclonal NFAT5-specific Ab was from Affinity Bioreagents (PA1-023); mouse mAb to β-tubulin (sc-32293) and rabbit polyclonal Abs to p65/RelA and to IκBα were from Santa Cruz Biotechnology (sc-372 and sc-371); to detect PARP-1, a mix of equal volumes of three independent mouse anti-human PARP-1 monoclonal Abs, kindly provided by Dr. José Yélamos (Hospital del Mar Medical Research Institute, Barcelona, Spain), was used (32). Secondary Abs to mouse IgG (NA931V) and to rabbit IgG (NA934V) coupled to HRP were from Amersham. Protein bands were visualized by ECL, using Supersignal West Pico Chemiluminescent Substrate (Pierce; Thermo Fisher Scientific).
Immunocytochemistry
BMDMs were cultured for 24 h on sterile glass coverslips coated with 0.01% w/v poly-l-lysine (Sigma-Aldrich). Cells were stimulated with 1 ng/ml LPS for the indicated times and then fixed with 3% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4), washed and permeabilized with wash buffer (0.5% NP-40 in PBS), and blocked with 10% FCS in wash buffer (33). Cells were then incubated with an Ab to p65/RelA (sc-372; Santa Cruz Biotechnology) in blocking solution, then with goat anti-rabbit IgG-FITC (F0382; Sigma-Aldrich). Preparations were mounted on slides with the antifading agent Slowfade (Invitrogen Molecular Probes), and cells were visualized by confocal laser microscopy (Leica TCS SP2). Images were acquired using the Leica Confocal Software v.2.6.1 Build 1537.
Coimmunoprecipitation
BMDMs were stimulated for the indicated time points and lysed in lysis buffer (2 mM EDTA, 2 mM EGTA, 30 mM NaF, 30 mM Na pyrophosphate, 20 mM Tris-HCl [pH 7.4], 2 mM sodium orthovanadate, 0.5% saponin, and the protease inhibitors 1 mM PMSF, 5 µg/ml leupeptin, 5 µg/ml aprotinin, and 1 µg/ml pepstatin A). Lysates (0.5–1 mg of protein) were precleared with protein A–Sepharose beads that were previously preadsorbed with fish sperm DNA and BSA for 1 h at 4°C. After removing the preclearing beads, Abs (rabbit polyclonal anti-p65 or normal rabbit IgG control) were added to the lysates and incubated overnight at 4°C under rotation. Preadsorbed protein A–Sepharose beads were then added, incubated for 90 min at 4°C, and then washed four times in lysis buffer. Beads were finally boiled in reducing 1× Laemmli buffer and the resulting supernatants were analyzed by Western blot as described above.
Statistical analysis
Statistical analysis was done with the GraphPad Prism 6 software. When Gaussian distribution and normality could be determined, samples were analyzed with parametric (Gaussian distribution) or nonparametric (not Gaussian distribution) tests. In most figures, results comprise three to seven independent experiments, which does not allow to determine normality, and in this case, Gaussian distribution was assumed and parametric t tests were used. Data in (Fig. 1A were from 12–13 samples, which allowed us to determine normality. In this case, some sample sets (wild-type Il6 and Tnf and Nfat5–/– Nos2) were found to follow a abnormal distribution, and the data were analyzed with a Mann–Whitney (nonparametric) test. Significance was determined using a paired t test when analyzing successive independent experiments in each of which macrophages from one wild-type and a littermate NFAT5-deficient mouse were compared, or when comparing different treatments in the same experiment and type of macrophage (Figs. 1B, 1D, 2C, 3A, 5, 6; Supplemental Fig. 1C, 1D), with an unpaired t test (Fig. 2A, 2B; Supplemental Fig. 1A, 1C, 1D) or with a one-sample t test when comparing groups of experiments in which the respective controls were assigned a reference value of 1-fold or 100% (Figs. 1E, 2A, 2B; Supplemental Fig. 3).
FIGURE 1.

Recruitment of p65/RelA to the promoter of LPS-responsive genes in NFAT5-deficient macrophages. (A) In vivo induction of iNOS, IL-6, and TNF-α by LPS in wild-type (Nfat5+/+) and NFAT5-deficient (Nfat5–/–) mice. RT-qPCR measurements of iNOS, IL-6, and TNF-α mRNAs in purified peritoneal macrophages obtained from LPS-treated mice. Nfat5+/+ (n = 12) and Nfat5–/– (n = 13) mice were injected i.p. with a low dose of LPS (0.05 µg/kg of body weight) diluted in PBS, and CD11b-positive cells were collected and lysed 3 h after injection. mRNA values were normalized to L32 mRNA. (B) Induction of LPS-responsive and NFAT5-dependent genes in wild-type and NFAT5-deficient BMDMs. mRNA for the indicated genes was measured by RT-qPCR in samples from Nfat5+/+ or Nfat5–/– BMDMs either left untreated (Unst) or stimulated during 90 min with 0.3 or 100 ng/ml LPS. mRNA values are shown normalized to L32 RNA, and represent four independent experiments, each comparing one pair of Nfat5+/+ and Nfat5–/– BMDM from littermate mice. (C) Analysis of the NFAT5-dependent recruitment of p65 to various target gene promoters in BMDM treated with different concentrations of LPS (0.1–100 ng/ml) for 2 h. Immunoprecipitated chromatin in each sample was normalized to its respective chromatin input. Rabbit IgG was included as a negative control. Values shown are from three to four independently performed experiments, each comparing one pair of NFAT5-deficient and control littermate mice. (D) ChIP of p65/RelA in various promoters in Nfat5+/+ or Nfat5–/– BMDM left untreated (0) or stimulated with LPS (1 ng/ml) for 1 to 8 h. Values shown are the mean ± SEM from three (Trem1, Serpinb2, Upp1), four (Nos2, Il6, Tnf, Ccl2, Ticam1), and six (Nfkbia) independently performed experiments, each comparing one culture of Nfat5+/+ BMDM with Nfat5–/– BMDM from a littermate mouse. (E) Recruitment of p65 to Edn1 and Lcn2 gene promoters in Nfat5+/+ or Nfat5–/– BMDMs left untreated (t = 0) or stimulated with LPS (1 ng/ml) as indicated. Values shown are the mean ± SEM from three independently performed experiments each comparing one pair of NFAT5-deficient and control littermate mice. Immunoprecipitated chromatin in each sample was normalized to its respective chromatin input and then values were normalized to the wild-type 2 h sample (which was given an arbitrary value of 1). (F) ChIP of p65 and a negative control IgG in the promoters of two genes (Actb and Cd3e) that are not induced by NF-κB in LPS-stimulated macrophages. Statistical significance was determined with a Mann–Whitney U test (A), a paired t test (B–D), and a one-sample t test (*p < 0.05) (E). *p < 0.05, **p < 0.01. The p values between 0.05 and 0.1 are indicated.
FIGURE 2.

Recruitment of c-Fos, p300, and RNA pol II to target genes in response to LPS in NFAT5-deficient macrophages. ChIP of c-Fos (A), p300 (B), and RNA pol II (Pol II) (C) in promoter regions of different genes in Nfat5+/+ or Nfat5–/– BMDM left untreated or stimulated with 1 ng/ml of LPS for the indicated times. Rabbit IgG was included as a negative control. Analysis of RNA Pol II in Tnf in (C) included the promoter (pTnf) and coding region (exon 4). Values shown are the mean ± SEM from four independently performed experiments (except the 4-h time point with two experiments, and Actb and Cd3e with three experiments). Each experiment included the comparison of one culture of Nfat5+/+ BMDM with Nfat5–/– BMDM from a littermate mouse. In (A) and (B), immunoprecipitated chromatin in each sample was normalized to its respective chromatin input and then values were normalized to the wild-type 1-h sample (A) and 2-h sample (B), which were given an arbitrary value of 1. Statistical significance in (A) and (B) was assessed with a one-sample t test for comparison with the reference 1-h (A) and 2-h (B) samples, and comparisons between the rest of the samples were done with an unpaired t test (*p < 0.05). Statistical significance in (C) was assessed with a paired t test (*p < 0.05). The p values between 0.05 and 0.1 are also indicated.
FIGURE 3.

Genome-wide defects in chromatin accessibility in NFAT5-deficient macrophages. (A) Restriction enzyme accessibility (REA) analysis to NFAT5 target genes in LPS-stimulated NFAT5-deficient macrophages. Nuclei from wild-type (WT) (Nfat5+/+) and NFAT5-deficient (Nfat5–/–) BMDM untreated (–) or stimulated with 0.3 ng/ml of LPS for 0.5 or 2 h were treated with HinfI restriction enzyme, and the isolated chromatin was analyzed by RT-qPCR using primers flanking the HinfI restriction site located before the TSS of each gene. Values are represented as percentage of uncut DNA after normalization with respect to the Gapdh promoter in the same samples. The reference sample for 100% uncut DNA corresponds to nuclei of Nfat5+/+ macrophages not treated with HinfI. Values shown are the mean ± SEM of three independent experiments (*p < 0.05, paired t test). (B) Expression of NFAT5, iNOS, and TNF-α mRNA in the two biological replicates of WT and NFAT5-deficient BMDM used for ATAC-seq analysis. (C) Amplification by qPCR of Nos2, Il6, and Tnf promoter regions in the ATAC-seq genomic libraries prepared from WT and NFAT5-deficient BMDM. Greater qPCR amplification of specific regions in the libraries indicates that these regions accumulated more transposon insertions and thus were more accessible. (D) Correlation of the ATAC-seq signal from individual replicates in 2 pairs of WT and NFAT5-deficient (KO) BMDM in unstimulated and LPS stimulation conditions. (E) ATAC-seq signal (averaged from two independent biological replicates) in WT and NFAT5-deficient macrophages (KO) left unstimulated (Unst) or stimulated with LPS (0.3 ng/ml, 2 h). Each dot represents an ATAC-seq peak identified in the bioinformatics analysis. The number of regions more accessible in WT than in KO cells (blue), and more accessible in KO than WT cells (red) are indicated. (F) Bar graphs and pie charts show the annotation of genomic regions with ATAC-seq peaks from (E) compared with the mouse genome (mm10). (G) Increased accessibility in putative intergenic enhancers in LPS-stimulated WT macrophages compared with NFAT5-deficient cells. qPCR, quantitative PCR.
FIGURE 5.

Analysis of histone modifications in NFAT5 target genes in macrophages. (A and B) ChIP analysis of acetylated histone H3 (lysines 9 and 14, AcH3) (A) and acetylated histone H4 (lysines 5, 8, 12, and 16, AcH4) (B) in the promoters of Nos2, Il6, and Tnf in Nfat5+/+ (wild-type) and Nfat5–/– (NFAT5-deficient) BMDM untreated (t = 0) or stimulated with LPS [0.3 ng/ml in (A) and 1 ng/ml in (B)] from 30 min to 2 h. Results are shown as the ratio of acetylated histone to its respective total histone after normalizing immunoprecipitated chromatin in each sample to its respective total chromatin input. Values shown are the mean ± SEM from four independent experiments, each comparing one culture of Nfat5+/+ BMDM with Nfat5–/– BMDM from a littermate mouse. (C) Recruitment of p65 to the promoters of Nos2, Il6, and Tnf in Nfat5+/+ or Nfat5–/– BMDM pretreated (5 h) or not with the HDAC inhibitor TSA (500 nM) and then stimulated with 0.3 ng/ml LPS for 1 h. Values show the mean ± SEM from four to five independent experiments, each comparing one or two pairs of Nfat5+/+ and Nfat5–/– BMDM from littermate mice (*p < 0.05). (D) Abundance of histone H3, H3K4me3, H3K27me3, and H4K20me3 in the promoters of Nos2, Il6, and Tnf in wild-type BMDM in basal unstimulated conditions. Immunoprecipitated chromatin in each sample is shown relative to its respective total chromatin input. Values shown are the mean ± SEM from three independently performed experiments (*p < 0.05). (E) ChIP analysis of H3, H3K4me3, and H3K27me3 in the promoter and enhancer of Nos2 and the promoters of Il6 and Tnf in wild-type (Nfat5+/+) and NFAT5-deficient (Nfat5–/–) BMDM untreated (t = 0) or stimulated with LPS (0.3 ng/ml, 0.5–2 h). Immunoprecipitated chromatin in each sample was normalized to its respective total chromatin input. Values shown are the mean ± SEM from four independent experiments, each comparing one culture of Nfat5+/+ BMDM with Nfat5–/– BMDM from a littermate mouse (#p < 0.05 or §p < 0.05 between unstimulated and LPS-treated Nfat5+/+ (#) or Nfat5–/– (§) BMDM. *p < 0.05, **p < 0.01 between Nfat5+/+ and Nfat5–/– BMDM). (F) ChIP analysis of H4K20me3 in the promoters of Nos2, Il6, or Tnf in wild-type (Nfat5+/+) and NFAT5-deficient (Nfat5–/–) BMDM untreated (t = 0) or stimulated with LPS (0.3 ng/ml, 1–2 h). Immunoprecipitated chromatin in each sample is shown relative to its respective total chromatin input. Values shown are the mean ± SEM from three independent experiments, each comparing one culture of Nfat5+/+ BMDM with Nfat5–/– BMDM from a littermate mouse (#p < 0.05 between unstimulated and LPS-treated Nfat5+/+ BMDM). Statistical significance in (C)–(F) was assessed with a paired t test. The p values between 0.05 and 0.1 are indicated.
FIGURE 6.

Effect of H3K27 trimethylation on the expression of NFAT5 target genes and the recruitment of p65/RelA to their promoters. (A) Binding of the H3K27 methyltransferase PRC2 component EZH2 to two regions of Nos2 was assessed by ChIP in wild-type (Nfat5+/+) and NFAT5-deficient (Nfat5–/–) BMDM untreated (t = 0) or stimulated with LPS (0.3 ng/ml, 0.5–2 h). Immunoprecipitated chromatin in each sample is shown relative to its respective total chromatin input. Background levels of chromatin immunoprecipitated with a control IgG are shown. Values are the mean ± SEM from three independently performed experiments, each comparing one culture of Nfat5+/+ BMDM with Nfat5–/– BMDM from a littermate mouse. (B) and (C) Binding of JMJD3 (B) and UTX (C) to the proximal promoter of Nos2 assessed by ChIP in Nfat5+/+ or Nfat5–/– BMDM untreated (t = 0) or stimulated with 1 ng/ml LPS (0.5–2 h). Values are the mean ± SEM from three independently performed experiments. (D) ChIP of H3K27me3 in the Nos2, Il6, or Tnf promoters in wild-type BMDMs pretreated or not with the JMJD3 and UTX inhibitor GSK-J4 (J4, 10 μM, 1 h) or with the EZH2 inhibitor UNC1999 (U, 10 μM, 16 h), and then stimulated with 0.3 ng/ml of LPS for 1 h. Immunoprecipitated chromatin in each sample was normalized to its respective chromatin input and then values were normalized to the unstimulated control, which was given an arbitrary value of 1. Values show the mean ± SEM from four BMDM cultures that included all conditions and nine additional experiments testing the effect of GSK-J4. (E) Induction of iNOS, IL-6, and TNF-α mRNA in Nfat5+/+ or Nfat5–/– BMDM pretreated or not with the EZH2 inhibitors GSK-126 (126) and UNC-1999 (UNC) or with the JMJD3 and UTX inhibitor GSK-J4 (J4) and then stimulated with 0.3 ng/ml LPS for 90 min. All inhibitors were used at 10 μM. Values show the mean ± SEM from three (GSK-126 and UNC-1999) or four (GSK-J4) independent experiments, each comparing one or two pairs of Nfat5+/+ and Nfat5–/– BMDM from littermate mice. (F) Binding of p65 to the Nos2, Il6, and Tnf promoters in Nfat5+/+ or Nfat5–/– BMDM pretreated or not with the JMJD3 and UTX inhibitor GSK-J4 (J4, 10 μM) and then stimulated with 0.3 ng/ml LPS for 1 h. Background level of chromatin immunoprecipitated with a control rabbit IgG is shown. Values show the mean ± SEM from four to eight independent experiments, each comparing one or two pairs of Nfat5+/+ and Nfat5–/– BMDM from littermate mice. Statistical significance in (C)–(F) was assessed with a paired t test for comparisons between untreated and inhibitor-treated cells or between Nfat5+/+ and Nfat5–/– cells (*p < 0.05, ** p < 0.01). The p values between 0.05 and 0.1 are indicated.
Results
NFAT5 optimizes p65/RelA recruitment to a subset of TLR-responsive genes
Consistent with our previous work (6), different concentrations of the TLR4 agonist LPS elicited a significant induction of the mRNA of main NFAT5 target genes (Nos2, Il6, and Tnf) in vivo and in vitro, but their dependence on NFAT5 was greater under milder stimulation (Fig. 1A, 1B and Supplemental Fig. 1A). This was observed in BMDMs (Fig. 1B) as well as in peritoneal macrophages from two different NFAT5-deficient mouse models (Fig. 1A, and Supplemental Fig. 1A), in all of which lack of NFAT5 reduced the induction of iNOS and TNF-α by LPS. We also observed that the dependence of some genes on NFAT5 appeared to vary under different stimulatory conditions, and for instance IL-6 was NFAT5-independent in peritoneal macrophages stimulated ex vivo with LPS, and IL-6, TNF-α, and iNOS were induced normally in thioglycollate-elicited NFAT5-deficient macrophages (Supplemental Fig. 1A, 1B). Besides testing NFAT5-deficient BMDM from two different mouse models, we also considered using a recently described NFAT5 inhibitor, the KRN2 compound. KRN2 inhibits the LPS-induced accumulation of NFAT5 by preventing its NF-κB–dependent transcription (34). We found that KRN2 inhibited IL-6 and iNOS induction by LPS and that it did so similarly in wild-type and NFAT5-deficient macrophages (Supplemental Fig. 1C, 1D), indicating that KRN2 could inhibit some LPS response genes independently of NFAT5. This observation raises some caution about interpreting results with KRN2 at short stimulation times because preexisting NFAT5 will not be affected by it (34), and at the same time, this compound can have NFAT5-independent effects. Because many of the NFAT5-regulated genes identified in our previous study are known to be direct targets of p65/RelA (NF-κB) (35, 36), a master transcription factor in antipathogen responses, we explored whether NFAT5 could facilitate recruitment of p65 to these genes. For this, we did ChIP assays with an anti-p65 Ab in a set of promoters that we had previously shown to bind NFAT5 (6), and compared wild-type and NFAT5-deficient macrophages stimulated with different concentrations of LPS. Recruitment of p65 to the promoters of the NFAT5-regulated genes Nos2, Tnf, and Il6 was reduced in NFAT5-deficient macrophages in a range of LPS doses (0.3–10 ng/ml) but not when stimulated with a higher dose (100 ng/ml) (Fig. 1C). Time-course experiments with an extended panel of NFAT5-dependent (Nos2, Il6, Tnf, Ccl2, Trem1, Serpinb2, Edn1, Lcn2, Upp1), and independent (Nfkbia and Ticam1) genes (6) revealed that lack of NFAT5 impaired the recruitment of p65 to the promoters of Nos2, Il6, Tnf, Ccl2, Serpinb2, and Trem1 in macrophages stimulated with 1 ng/ml LPS and had a milder effect for Edn1 and Lcn2 (Fig. 1D, 1E). This defect occurred since early time points of stimulation (1 h) and was more noticeable during the time window of maximal p65 recruitment to each gene (Fig. 1D). By contrast, lack of NFAT5 did not affect p65 recruitment to the promoters of the NFAT5-independent Nfkbia and Ticam1 and the NFAT5-dependent Upp1 (6) (Fig. 1D). In these experiments, we included two genes, Actb and Cd3e, as controls that were not induced by LPS nor are NF-κB dependent in macrophages, and found no significant binding of p65 to their promoters (Fig. 1F). These results show that NFAT5 lowered the stimulus threshold needed to recruit p65/RelA to different target genes, thus enhancing p65 responsiveness to low-intensity TLR stimulation.
Our results also suggested that lack of NFAT5 did not cause a general impairment of p65 recruitment to genes, as illustrated by its normal LPS-induced binding to promoters of Upp1, Nfkbia, and Ticam1 in NFAT5-deficient cells. Consistent with this, NFAT5 did not influence p65 nuclear translocation or its intrinsic DNA-binding capacity in LPS-stimulated macrophages (Supplemental Fig. 2A, 2B). Previous work had reported that NFAT5 could interact with p65 in nonmacrophage cell lines in basal conditions and in response to hyperosmotic stress (37, 38). We tested this interaction in coimmunoprecipitation assays in primary macrophages, but did not detect an association between endogenous NFAT5 and p65 in basal conditions or after LPS stimulation (Supplemental Fig. 2C). As a positive control, p65 bound its repressor IκBα in unstimulated macrophages, and this association was dynamically regulated as IκBα was degraded and then reinduced upon LPS stimulation. Altogether, these findings indicate that although NFAT5 cooperated with p65/RelA in the induction of a set of TLR-responsive genes, it was not an obligate cofactor for its activation in macrophages.
NFAT5 facilitates the inducible recruitment of c-Fos, p300, and RNA pol II to a set of TLR-responsive genes
We next asked whether NFAT5 could also influence recruitment of other transcription regulators to TLR-responsive genes. For this, we analyzed the AP-1 family member c-Fos, as it is recruited to LPS-inducible genes in a stimulus-dependent manner but activated by signaling mechanisms different from those controlling p65 (39). We found that NFAT5-deficient macrophages exhibited defective recruitment of c-Fos to Nos2, Il6, Tnf, and Ccl2 promoters in response to LPS, but not to the NFAT5-independent gene Ticam1 (Fig. 2A). As controls, we did not observe c-Fos recruitment to the promoters of Actb or Cd3e. These findings and our previous work (6) indicate that NFAT5 optimizes TLR-induced gene expression by enhancing access of other transcription factors to common target genes.
Impaired recruitment of p65/RelA and c-Fos to the promoters of several TLR-responsive genes in NFAT5-deficient cells suggested the possibility that other transcription regulators might also be affected. We therefore analyzed p300 and RNA pol II, which are core transcriptional regulators downstream multiple signaling pathways including TLRs (19, 40). LPS-induced recruitment of p300 to Nos2 and Tnf was attenuated in NFAT5-deficient macrophages (Fig. 2B). This was already noticeable at the peak of the NFAT5-dependent recruitment of p65 and c-Fos and also at later time points. Because p300 is recruited to target genes through its binding to NF-κB and AP-1 (41–43), our results suggest that impaired p300 accumulation in promoters of NFAT5-deficient macrophages could be associated with defective recruitment of p65/RelA and c-Fos. Regarding RNA pol II, its constitutive association with the Tnf promoter (44) and LPS-induced recruitment to transcribed regions of the Tnf gene (shown in this study for exon 4) were not affected, but its LPS-induced binding to the promoter of Nos2 was reduced in NFAT5-deficient macrophages (Fig. 2C).
NFAT5 influences chromatin accessibility in macrophages
The finding that NFAT5 optimizes the recruitment of different transcription regulators to various target gene promoters raised the question of whether it might influence chromatin configuration. We performed chromatin accessibility assays in isolated nuclei (31) and analyzed the influence of NFAT5 in the sensitivity of native chromatin to the restriction enzyme HinfI. These assays revealed that the proximal promoters of Nos2, Tnf, and Il6 presented lower LPS-induced accessibility in NFAT5-deficient macrophages, detected by their increased proportion of uncut DNA (Fig. 3A). Although differences were more clearly observed for the three promoters after 2 h of stimulation, reduced accessibility in Nos2 and Tnf promoters was already detected after 30 min. In view of this result, we analyzed whether NFAT5 could facilitate chromatin decondensation in a larger scale using the assay for ATAC-seq. We used two independent biological replicates of untreated and LPS-stimulated (0.3 ng/ml, 2 h) wild-type and NFAT5-deficient BMDMs, the latter showing reduced expression of NFAT5-target genes Nos2 and Tnf (Fig. 3B). In line with our results with the restriction enzyme accessibility assays, we observed that Nos2, Il6, and Tnf promoters were less represented in the ATAC libraries from two independent cultures of LPS-stimulated NFAT5-deficient macrophages (Fig. 3C), confirming that these regions had lower accessibility in NFAT5-deficient macrophages. Pairwise comparisons of the ultrasequencing data of the ATAC libraries showed a high correlation between each pair of biological replicates (Fig. 3D). LPS stimulation induced substantial changes in chromatin accessibility in both macrophage genotypes in multiple regions (Fig. 3E, 3F). We identified more than 7,000 genomic regions showing different accessibility (false discovery rate value of 0.1) between wild-type and NFAT5-deficient macrophages under LPS stimulation, and only ∼800 in unstimulated conditions (Fig. 3E). Whereas NFAT5 facilitated chromatin accessibility in multiple genomic regions of basal and LPS-treated macrophages, its influence was particularly noticeable upon LPS stimulation in regions enriched in gene promoters, between –5 kb to +500 bp around the TSS, with 2,705 promoters displaying reduced accessibility in NFAT5-deficient macrophages and only 308 being more accessible (Fig. 3F). We also noticed that a high percentage (63.7%) of intergenic regions more accessible in LPS-stimulated wild-type cells coincided with previously identified enhancer regions, enriched in H3K27Ac (28). By contrast, the proportion of H3K27Ac-enriched intergenic regions with increased accessibility in NFAT5-deficient macrophages was much lower, 18.4% (Fig. 3G). These results indicated that NFAT5 facilitated chromatin accessibility in numerous promoters and enhancers in LPS-stimulated macrophages. Among the promoters with NFAT5-dependent accessibility identified in the ATAC-seq analysis, we again found Nos2, Il6, and Tnf, which had been our diagnostic NFAT5-regulated genes (Fig. 4A, see blue boxes). The ATAC-seq analysis revealed more extensive accessibility defects for Tnf and other NFAT5-dependent genes such as Serpinb2 and Trem1 (6) than for Nos2 and Il6, which showed a more modest and localized defect (Fig. 4A). We next used the consensus NFAT5-binding sequence obtained by affinity selection of an oligonucleotide library with the recombinant DNA-binding domain of NFAT5 (7) to infer potential binding sites within regions displaying NFAT5-modulated accessibility. We detected consensus NFAT5 binding sites in 79.1% of the regions between –5 kb to +500 bp around the TSS that showed NFAT5-enhanced accessibility and in 87.8% of those repressed by NFAT5. Shortening the search in the TSS upstream region from –5 kb to –1.5 kb still identified the NFAT5 site in 38.15% of regions that displayed NFAT5-dependent accessibility and in 54.6% of NFAT5-repressed ones. This observation suggested that NFAT5-regulated changes in accessibility might involve its ability to influence proximal and more distal gene regulatory elements, as we have shown for its regulation of a distal Ciita enhancer in macrophages (11).
FIGURE 4.

Reduced chromatin accessibility in gene promoters of NFAT5-deficient macrophages. (A) Genome browser snapshots showing the ATAC-seq signals in promoter regions of NFAT5-induced genes (Nos2, Il6, Tnf, Trem1, Serpinb2). Regions differentially accessible between LPS-treated wild-type and NFAT5-deficient macrophages are highlighted by blue boxes. False discovery rate (FDR), fold change, and statistical significance of differential accessibility between LPS-stimulated wild-type and NFAT5-deficient macrophages for the respective regions are indicated. (B) Comparison of chromatin regions differentially accessible (ATAC-seq) with transcriptomic analysis of gene expression between LPS-stimulated wild-type and NFAT5-deficient macrophages (GSE26343). The upper panel shows all genes identified in the microarray analysis for which differentially accessible regions were identified in the ATAC-seq. The bottom panel shows the subset of genes with >2-fold expression difference between wild-type and NFAT5-deficient macrophages. Genes showing a direct correlation between expression and chromatin accessibility are marked in red in the bottom panel (see also Supplemental Table I for the respective lists of genes: ATAC versus RNA top DE genes and ATAC versus RNA all DE genes).
Comparison of the genes whose accessibility was modulated by NFAT5 with a set of LPS-responsive NFAT5-target genes (6) (dataset accession number GSE26343) showed that 32% of the genes (261 out of 807) regulated by LPS in an NFAT5-dependent manner also showed NFAT5-dependent changes in accessibility in one or more regions (Supplemental Table I). We observed a direct correlation between NFAT5-dependent accessibility and expression in many genes, but also found genes with NFAT5-enhanced accessibility whose expression was repressed by it (Fig. 4B upper panel, Supplemental Table I), suggesting that NFAT5-induced chromatin accessibility could associate with gene expression or repression. However, when we focused on genes whose expression was strongly influenced by NFAT5 [either induced or repressed more than 2-fold in wild-type versus NFAT5-deficient cells (6)] we found a stronger positive correlation between NFAT5-dependent accessibility in promoters and/or transcribed regions and NFAT5-induced gene expression (Fig. 4B bottom panel, Supplemental Table I). Altogether, different assays showed that NFAT5 facilitates chromatin accessibility, being particularly relevant in the promoters of numerous TLR-responsive genes.
Lack of NFAT5 impairs H3K27me3 demethylation in the promoters of target genes
In view of the effect of NFAT5 in macrophage chromatin accessibility and the observation that this was more noticeable at promoter regions (Fig. 3F), we analyzed the NFAT5-regulated Nos2, Il6, and Tnf promoters for their distribution of histone marks that are modulated in response to pathogens. As we had observed defective LPS-induced recruitment of the histone acetyl transferase p300 to these promoters in NFAT5-deficient macrophages, we first analyzed their levels of acetylated histone H3 (lysines 9 and 14, AcH3) and acetylated histone H4 (lysines 5, 8, 12, and 16, AcH4), but we did not detect a consistent pattern of histone acetylation changes that distinguished NFAT5-deficient and control macrophages (Fig. 5A, 5B). Nonetheless, as histone acetyl transferases can target histones in residues other than those tested in this study [reviewed in (45)], we explored whether boosting histone acetylation by inhibiting histone deacetylases (HDAC) could rescue the binding of p65 to Nos2, Il6, and Tnf promoters in NFAT5-deficient macrophages. We found that the HDAC inhibitor TSA did not enhance the recruitment of p65 to Nos2, Il6, and Tnf promoters in NFAT5-deficient macrophages (Fig. 5C). We also noticed that TSA even impaired the recruitment of p65 to the Tnf promoter in both wild-type and NFAT5-deficient cells (Fig. 5C). In summary, our results indicate that histone acetylation may not be a main determinant in explaining the reduced recruitment of transcription regulators as p65 to Nos2, Il6, and Tnf in NFAT5-deficient macrophages.
We next explored other histone marks known to influence chromatin accessibility. We got interested in works showing that chromatin compaction can cause transcriptional silencing involving polycomb-group proteins, which have long been known to induce heterochromatin-like structures that hinder the accessibility of transcription regulators (46). In turn, dense chromatin regions activate the polycomb-repressive complex 2 (PRC2) to inhibit genes by catalyzing trimethylation of lysine 27 of histone H3 (H3K27me3) (47, 48) and recruiting the H3K4me3 demethylase Rbp2 (Jarid1a) (49). These two histone modifications and H4K20me3, another repressive mark that induces chromatin compaction (50), have been reported to modulate TLR-induced gene expression in macrophages (19, 51, 52). Initial assessment of wild-type macrophages in unstimulated conditions showed that Nos2, Il6, and Tnf promoters had comparable H3 density and H4K20me3 levels, but varied in H3K27me3- and H3K4me3-relative abundance, with Tnf showing the highest level of H3K4me3 and Nos2 the highest abundance of H3K27me3 (Fig. 5D). Time-course experiments with 0.3 ng/ml LPS showed significant NFAT5-dependent H3K27me3 demethylation in Nos2 enhancer and promoter and Il6 promoter, which was sustained from 30 min to 2 h after stimulation (Fig. 5E). For Tnf, as seen for this early response gene (51), the predominant effect of LPS was a rapid and pronounced eviction of histones paralleled by a decrease in H3K27me3 (Fig. 5E), with NFAT5-deficient macrophages showing normal H3 eviction but attenuated and more transient H3K27me3 demethylation (Fig. 5E). Regarding H3K4me3, its abundance in Nos2 promoter and enhancer was reduced in NFAT5-deficient cells already in basal conditions, and although it increased at 2 h of stimulation, it still remained below the level of wild-type macrophages (Fig. 5E). In the Il6 and Tnf promoters, LPS did not induce marked changes in the relative abundance of H3K4me3 between NFAT5-deficient and wild-type cells (Fig. 5E). Finally, we did not observe H4K20me3 differences in any of these genes between LPS-treated wild-type and NFAT5-deficient macrophages (Fig. 5F). The result that p65 recruitment and H3K27me3 demethylation were partially impaired in LPS-activated NFAT5-deficient macrophages was also observed in BMDMs of an independent conditional NFAT5-knockout model, Csf1r-Cre Nfat5fl/fl mice (Supplemental Fig. 3). Altogether, this set of results analyzing five types of histone modifications in the regulatory regions of Nos2, Il6, and Tnf genes identify impaired LPS-induced demethylation of H3K27me3 as a defect common to these genes in NFAT5-deficient macrophages.
Regulation of p65/RelA recruitment to promoters by local NFAT5-dependent H3K27me3 demethylation
Defective stimulation-dependent clearance of H3K27me3 could reflect an imbalance between eviction of PRC2 complexes, which perform this repressive trimethylation (53), and recruitment of H3K27me3 demethylases Jumonji domain-containing 3 (JMJD3) and UTX, which induce proinflammatory genes in macrophages (19, 51, 54). We focused the analysis of these enzymes in the Nos2 gene, as it exhibited a marked NFAT5-dependent H3K27me3 demethylation in LPS-stimulated macrophages and also recruits JMJD3 in a stimulus-dependent manner (19). NFAT5-deficient macrophages did not show increased binding of the PRC2 catalytic subunit EZH2 to the Nos2 gene promoter nor to its CpG-rich intronic region, which displays LPS-sensitive H3K27me3 marks (19) (Fig. 6A). NFAT5-deficient cells also showed normal stimulus-dependent recruitment of JMJD3 to the Nos2 promoter (Fig. 6B). Regarding UTX, its LPS-induced recruitment to Nos2 was mildly impaired in NFAT5-deficient macrophages after 2 h of stimulation, but not at 1 h, when differences in H3K27me3 between wild-type and NFAT5-deficient cells were already evident in this gene (Figs. 5E, 6C). These results indicate that defective H3K27me3 demethylation of target genes in NFAT5-deficient macrophages would not be explained by altered recruitment of EZH2, JMJD3, or UTX and suggested that these cells might have an imbalance in the activity of the enzymes that control H3K37me3 levels.
We next inhibited the activity of EZH2 with the compound UNC1999 (55), and JMJD3 and UTX demethylases with GSK-J4. GSK-J4 has been used for interrogating the contribution of these demethylases to TNF-α induction in LPS-activated human primary macrophages (51), as well as their role in stem cells and tumors (56–59). Although basal H3K27me3 levels in Nos2, Il6, and Tnf promoters were not affected by these inhibitors in unstimulated macrophages, they were in LPS-treated cells (Fig. 6D). GSK-J4 reduced H3K27me3 demethylation of these promoters whereas UNC1999 enhanced it, confirming that Nos2, Il6, and Tnf promoters were simultaneously controlled by the opposed activities of H3K27me3 demethylases and methyltransferases in LPS-stimulated macrophages. We therefore analyzed the effect of inhibiting H3K27 methyltransferases and demethylases on the expression of Nos2, Il6, and Tnf upon mild LPS stimulation. Neutralization of the H3K27 methyltransferase EZH2 with two independent inhibitors GSK-126 (60) and UNC1999 (55) greatly enhanced iNOS and IL6 mRNA expression in wild-type macrophages (Fig. 6E). These inhibitors also enhanced induction of both genes in NFAT5-deficient cells, although without rescuing them to the levels of wild-type macrophages. TNF-α mRNA was not further induced by EZH2 inhibitors in either cell type (Fig. 6E). Conversely, inhibition of JMJD3 and UTX demethylases with GSK-J4 essentially cancelled LPS-induced expression of iNOS, IL-6, and TNF-α mRNA in NFAT5-deficient and control macrophages (Fig. 6E). Altogether, these findings suggest that expression of iNOS and IL-6 was controlled by a balance between simultaneously active H3K27me3 demethylases and methyltransferases; whereas induction of TNF-α was mainly controlled by H3K27me3 demethylation.
We then asked whether H3K27me3 demethylases could facilitate the binding of p65/RelA and tested the effect of inhibiting their activity on its recruitment to the promoter regions of Nos2, Il6, and Tnf. GSK-J4 attenuated the LPS-induced recruitment of p65 to Nos2, Il6, and Tnf promoters in wild-type macrophages (Fig. 6F). By contrast, the reduced recruitment of p65 to these promoters in NFAT5-deficient cells was not further repressed by GSK-J4, which suggested that the ability of NFAT5 to enhance p65 recruitment to the Nos2, Il6, and Tnf promoters required active H3K27me3 demethylases. These results above, together with the H3K27me3 demethylation analysis in (Fig. 5E, suggest that impaired H3K27me3 demethylation of Nos2, Il6, and Tnf promoters in NFAT5-deficient cells led to reduced recruitment of p65 and subsequently to attenuated gene expression. Altogether, our combined analyses suggest that the mechanism by which NFAT5 amplifies gene expression in TLR-stimulated macrophages involves its facilitating chromatin accessibility and H3K27 demethylase-dependent recruitment of other transcription regulators.
Discussion
In this study, we show that NFAT5 optimizes the recruitment of transcription regulators to specific genes, acting as an amplifier of low-input signals from pathogen receptors. NFAT5 enhanced chromatin accessibility and H3K27me3 demethylation in target genes and facilitated the recruitment of transcription regulators as NF-κB to their promoters. An earlier work had found that sustained NF-κB responses during persistent TLR signaling were modulated by the balance between C/EBPδ, which acted as an NF-κB amplifier, and ATF3, which opposed it after prolonged TLR stimulation (5). These previous findings and our current results on the regulation of NF-κB and c-Fos by NFAT5 illustrate how the coordination of a limited number of transcription factors may allow macrophages to use diverse mechanisms to optimize gene expression under different intensities of pathogen burden.
Our analysis of different histone marks known to regulate chromatin compactness and gene expression in macrophages revealed that NFAT5 facilitated H3K27me3 demethylation in the regulatory regions of Nos2, Il6, and Tnf. Although we could also detect discrete defects in H3K4me3 methylation in regulatory regions of Nos2 in NFAT5-deficient cells, only H3K27me3 demethylation was consistently affected in the three promoters analyzed. We also found that NFAT5 facilitated H3K27me3 demethylation in different gene promoters without affecting histone eviction. Because previous work showed that combined LPS and IFNγ stimulation of macrophages caused both H3K27 demethylation and H3 eviction in different promoters (19), our current data expand these observations and suggest that macrophages can use diverse chromatin regulatory mechanisms to respond to different combinations of stimuli.
Our results suggest that H3K27me3 levels in the regulatory regions of Nos2, Il6, and Tnf reflected a balance between simultaneously active methyltransferases and demethylases. Intriguingly, our analysis of the NFAT5 target gene Nos2 in NFAT5-deficient macrophages did not reveal apparent defects in the early recruitment of demethylases JMJD3 and UTX or the PRC2 methyltransferase catalytic subunit EZH2. This could suggest that defective H3K27me3 demethylation at specific promoters in NFAT5-deficient cells might involve an imbalance in the local activity of demethylases and methyltransferases rather than a defect in their recruitment. This possibility would be consistent with earlier works showing that condensed chromatin can enhance the H3K27 methyltransferase activity of PRC2 (47, 48), and it can be speculated that the greater chromatin condensation we found in promoters of NFAT5-deficient macrophages could favor the activity of H3K27 methyltransferases over demethylases. Differences between wild-type and NFAT5-deficient macrophages in both chromatin accessibility (restriction enzyme accessibility assay) and H3K27me3 were detectable early after LPS stimulation (30 min), pointing that they occur in a close time window. It remains to be determined whether both mechanisms are causally linked, and how NFAT5 ultimately regulates them. Nonetheless, our results suggest that H3K27me3 demethylation is a necessary event by which NFAT5 enhances p65 recruitment to the promoters of Nos2, Il6, and Tnf, as it was impaired by inhibition of H3K27me3 demethylases in wild-type cells but not in NFAT5-deficient macrophages. In the latter, the already reduced recruitment of p65 to these genes was not further downregulated upon inhibiting H3K27me3 demethylases. Altogether, our overall results are consistent with the interpretation that NFAT5-facilitated chromatin accessibility and removal of repressive H3K27me3 marks at specific regions are key mechanisms underlying its ability to enhance the recruitment of p65 and other transcription regulators.
Previous work in nonmacrophage cell lines had reported that NFAT5 could interact with p65/Rel A in response to hypertonic stress (38), and another study with embryonic fibroblasts showed that NFAT5 enhanced LPS-induced NF-κB activity by facilitating its interaction with the histone acetyltransferase p300 (37). Our results in primary macrophages indicate that attenuated recruitment of p65 to target genes in NFAT5-deficient BMDM is independent of histone acetyltransferase function, and suggests that NFAT5 can modulate NF-κB function through mechanisms different from those described earlier. Our current results are in line with findings by Kruidenier et al. on the regulation of the TNFA gene by H3K27me3 demethylases in human macrophages (51). These authors showed that LPS-induced recruitment of RNA pol II to the TNFA TSS and TNF-α expression were controlled by JMJD3/UTX-mediated local demethylation of the TSS region (51). Similar to our findings with the Tnf promoter in mouse macrophages, Kruidenier et al. showed that, although the TSS of human TNFA has low basal H3K27me3 levels that were further reduced by JMJD3/UTX upon LPS stimulation, H3K27me3 density in a 1.5 kb upstream region was much less affected by LPS and JMJD3/UTX. In another context, the group of Steven Kunkel showed that inhibition of JMJD3 in IL-4–stimulated macrophages reduced H3K27me3 demethylation by 25–60% in the promoters of Chi3l3, Retnla, and Arg1 and caused a proportional decrease in their expression (61). Our results and those by others (51, 61) support the notion that localized modulation of H3K27me3 levels in discrete genomic regions can control recruitment of transcription activators and gene expression in macrophages responding to different stimuli.
NFAT5 influenced chromatin accessibility in multiple genomic regions, with its greatest impact in promoters. We found a positive correlation between gene sets showing NFAT5-regulated chromatin accessibility in promoters or transcribed regions and those whose expression was enhanced by this factor in a previous transcriptome study (6). Our ATAC-seq assay also uncovered NFAT5-regulated accessibility sites in genes whose expression has not been found to be NFAT5-dependent and also in intergenic regions. Although the potential impact of these changes is yet to be investigated, several interpretations can be discussed. Apart from the possibility that not all chromatin accessibility changes may necessarily be reflected at the gene expression level, our earlier transcriptome study (6) used a single time point of 6 h, which could have missed potential differences between wild-type and NFAT5-deficient macrophages at earlier or later stimulation times. Also, some chromatin regions showing accessibility changes might not influence immediate nearby genes but act as distal regulatory elements, as we showed recently for an NFAT5-regulated remote enhancer of the MHC class II transactivator Ciita embedded in the Tvp23a gene (11). The number of regions showing NFAT5-regulated chromatin accessibility was much higher in LPS-activated macrophages, but we also identified NFAT5-regulated regions in unstimulated cells, a result consistent with our previous finding that NFAT5 can be bound to chromatin and regulate gene expression in unstimulated macrophages (6, 11). These results suggest the possibility that the landscape of genes in which chromatin accessibility could be facilitated by NFAT5 might vary depending on microenvironment signals sensed by the macrophage. Effective defense against infections requires macrophages to be able to respond to wide gradients of pathogen-derived stimuli. It can be anticipated that genes whose induction is facilitated by NFAT5 would mediate diverse functions in pathogen-activated macrophages. These might include among others, proinflammatory and migratory activity involving iNOS, IL-6, CCL2, and CCL5 (RANTES), control of extracellular matrix degradation by SerpinB2 (PAI-2), or the modulation of the intensity of innate immune responses through receptors such as TREM-1. Thus, by enhancing the responsiveness of central transcriptional mechanisms to low concentrations of pathogen products NFAT5 can help maintaining the robustness of macrophage antipathogen function.
Supplementary Material
Acknowledgments
We thank Andrea Cerutti, Irene Puga, Lluís Espinosa, Angel Corbí, Antonio Castrillo and members of our laboratory for comments on the manuscript and helpful discussions. We thank Gioacchino Natoli (European Institute of Oncology, Milan, Italy) for discussion and helpful suggestions to the manuscript, and together with Elena Prosperini (also European Institute of Oncology, Milan, Italy) for providing the polyclonal Ab to JMJD3, Toby Lawrence for the Csf1r-Cre–transgenic mice, F. Jeffrey Dilworth for advice on Abs to UTX, and José Yélamos (Hospital del Mar Medical Research Institute, Barcelona, Spain) for providing the monoclonal anti-PARP1 Ab. Alba Deyà is acknowledged for excellent technical assistance with mouse genotyping. We thank Jochen Hecht and Núria Andreu of the Centre for Genomic Regulation Genomics unit for the ATAC-seq.
This work was supported by the Spanish Ministry of Science and Innovation and European Fund for Regional Development (RTI2018-095902-B-I00), Fundació la Marató TV3 (201619-30), and the Marie Curie International Reintegration Programme of the European Union (MCIRG516308 to C.L.-R.). Research in the laboratory of C.L.-R. and J.A. was also supported by the Generalitat de Catalunya (2017SGR888) and the Spanish Ministry of Science Innovation and Universities through the “Unidad de Excelencia María de Maeztu” program funded by the Agencia Estatal de Investigación-AEI (CEX2018-000792-M). The Centre for Genomic Regulation acknowledges support of the Spanish Ministry of Economy and Competitiveness, “Centro de Excelencia Severo Ochoa,” and the Centres de Recerca de Catalunya Programme of Generalitat de Catalunya. Additional funding to C.L.-R. was from the ICREA Acadèmia Award of Institució Catalana de Recerca i Estudis Avançats (Generalitat de Catalunya).
G.L. performed and analyzed most of the experimental work and drafted the figures. M.B. performed the in vivo peritoneal macrophage experiments, various chromatin immunoprecipitation (ChIP) assays, and experiments with H3K27me3 inhibitors, and set up the protocols to prepare macrophage nuclei for tagmentation. M.R.-B., L.H., and D.R.-G. did additional ChIP and gene expression experiments during the revision of the manuscript. S.B., L.C., and C.C. performed the bioinformatics analysis of the assay for transposase-accessible chromatin with high-throughput sequencing experiment and the NFAT5 motif search analysis. H.H.E. set up the ChIP assays with anti-UTX Abs. S.G. contributed and analyzed the histone acetylation experiments. J.P. supervised the bioinformatics analysis, provided feedback on data analysis interpretation, and reviewed the manuscript. J.A. provided critical advice, interpreted the data, prepared the figures, and edited the manuscript. C.L.-R. designed and supervised the work, interpreted the data, prepared the figures, and wrote the manuscript.
The sequences presented in this article have been submitted to the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE111476.
The online version of this article contains supplemental material.
- ATAC-seq
- assay for transposase-accessible chromatin with high-throughput sequencing
- BMDM
- bone marrow–derived macrophage
- ChIP
- chromatin immunoprecipitation
- HDAC
- histone deacetylase
- iNOS
- inducible NO synthase
- PRC2
- polycomb-repressive complex 2
- RNA pol II
- RNA polymerase II
- RT-qPCR
- real-time quantitative PCR
- TSA
- trichostatin A
- TSS
- transcription start site
- UTX
- ubiquitously transcribed tetratricopeptide repeat X chromosome
Disclosures
The authors have no financial conflicts of interest.
References
- 1. Morris M. C., Gilliam E. A., Button J., Li L.. 2014. Dynamic modulation of innate immune response by varying dosages of lipopolysaccharide (LPS) in human monocytic cells. J. Biol. Chem. 289: 21584–21590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morris M. C., Gilliam E. A., Li L.. 2015. Innate immune programing by endotoxin and its pathological consequences. Front. Immunol. 5: 680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smale S. T., Natoli G.. 2014. Transcriptional control of inflammatory responses. Cold Spring Harb. Perspect. Biol. 6: a016261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zak D. E., Tam V. C., Aderem A.. 2014. Systems-level analysis of innate immunity. Annu. Rev. Immunol. 32: 547–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Litvak V., Ramsey S. A., Rust A. G., Zak D. E., Kennedy K. A., Lampano A. E., Nykter M., Shmulevich I., Aderem A.. 2009. Function of C/EBPdelta in a regulatory circuit that discriminates between transient and persistent TLR4-induced signals. Nat. Immunol. 10: 437–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buxadé M., Lunazzi G., Minguillón J., Iborra S., Berga-Bolaños R., Del Val M., Aramburu J., López-Rodríguez C.. 2012. Gene expression induced by Toll-like receptors in macrophages requires the transcription factor NFAT5. J. Exp. Med. 209: 379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lopez-Rodríguez C., Aramburu J., Rakeman A. S., Rao A.. 1999. NFAT5, a constitutively nuclear NFAT protein that does not cooperate with Fos and Jun. Proc. Natl. Acad. Sci. USA 96: 7214–7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. López-Rodríguez C., Aramburu J., Jin L., Rakeman A. S., Michino M., Rao A.. 2001. Bridging the NFAT and NF-kappaB families: NFAT5 dimerization regulates cytokine gene transcription in response to osmotic stress. Immunity 15: 47–58. [DOI] [PubMed] [Google Scholar]
- 9. Tellechea M., Buxadé M., Tejedor S., Aramburu J., López-Rodríguez C.. 2017. NFAT5-regulated macrophage polarization supports the proinflammatory function of macrophages and T lymphocytes. J. Immunol. 200: 305–315. [DOI] [PubMed] [Google Scholar]
- 10. Huerga Encabo H., Traveset L., Argilaguet J., Angulo A., Nistal-Villán E., Jaiswal R., Escalante C. R., Gekas C., Meyerhans A., Aramburu J., López-Rodríguez C.. 2020. The transcription factor NFAT5 limits infection-induced type I interferon responses. J. Exp. Med. 217: e20190449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Buxadé M., Huerga Encabo H., Riera-Borrull M., Quintana-Gallardo L., López-Cotarelo P., Tellechea M., Martínez-Martínez S., Redondo J. M., Martín-Caballero J., Flores J. M., et al. 2018. Macrophage-specific MHCII expression is regulated by a remote Ciita enhancer controlled by NFAT5. J. Exp. Med. 215: 2901–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aramburu J., López-Rodríguez C.. 2019. Regulation of inflammatory functions of macrophages and T lymphocytes by NFAT5. Front. Immunol. 10: 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jantsch J., Schatz V., Friedrich D., Schröder A., Kopp C., Siegert I., Maronna A., Wendelborn D., Linz P., Binger K. J. J., et al. 2015. Cutaneous Na+ storage strengthens the antimicrobial barrier function of the skin and boosts macrophage-driven host defense. Cell Metab. 21: 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Berry M. R., Mathews R. J., Ferdinand J. R., Jing C., Loudon K. W., Wlodek E., Dennison T. W., Kuper C., Neuhofer W., Clatworthy M. R.. 2017. Renal sodium gradient orchestrates a dynamic antibacterial defense zone. Cell 170: 860–874.e19. [DOI] [PubMed] [Google Scholar]
- 15. Ramirez-Carrozzi V. R., Nazarian A. A., Li C. C., Gore S. L., Sridharan R., Imbalzano A. N., Smale S. T.. 2006. Selective and antagonistic functions of SWI/SNF and Mi-2beta nucleosome remodeling complexes during an inflammatory response. Genes Dev. 20: 282–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. López-Rodríguez C., Antos C. L., Shelton J. M., Richardson J. A., Lin F., Novobrantseva T. I., Bronson R. T., Igarashi P., Rao A., Olson E. N.. 2004. Loss of NFAT5 results in renal atrophy and lack of tonicity-responsive gene expression. Proc. Natl. Acad. Sci. USA 101: 2392–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Drews-Elger K., Ortells M. C., Rao A., López-Rodriguez C., Aramburu J.. 2009. The transcription factor NFAT5 is required for cyclin expression and cell cycle progression in cells exposed to hypertonic stress. PLoS One 4: e5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Deng L., Zhou J. F., Sellers R. S., Li J. F., Nguyen A. V., Wang Y., Orlofsky A., Liu Q., Hume D. A., Pollard J. W., et al. 2010. A novel mouse model of inflammatory bowel disease links mammalian target of rapamycin-dependent hyperproliferation of colonic epithelium to inflammation-associated tumorigenesis. Am. J. Pathol. 176: 952–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. De Santa F., Narang V., Yap Z. H., Tusi B. K., Burgold T., Austenaa L., Bucci G., Caganova M., Notarbartolo S., Casola S., et al. 2009. Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. EMBO J. 28: 3341–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Buenrostro J. D., Wu B., Chang H. Y., Greenleaf W. J.. 2015. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 109. 21.29.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Langmead B., Salzberg S. L.. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., Liu X. S.. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ross-Innes C. S., Stark R., Teschendorff A. E., Holmes K. A., Ali H. R., Dunning M. J., Brown G. D., Gojis O., Ellis I. O., Green A. R., et al. 2012. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481: 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., Glass C. K.. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38: 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Durinck S., Spellman P. T., Birney E., Huber W.. 2009. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 4: 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Robinson M. D., McCarthy D. J., Smyth G. K.. 2009. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kent W. J., Sugnet C. W., Furey T. S., Roskin K. M., Pringle T. H., Zahler A. M., Haussler D.. 2002. The human genome browser at UCSC. Genome Res. 12: 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Comoglio F., Simonatto M., Polletti S., Liu X., Smale S. T., Barozzi I., Natoli G.. 2019. Dissection of acute stimulus-inducible nucleosome remodeling in mammalian cells. Genes Dev. 33: 1159–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Edgar R., Domrachev M., Lash A. E.. 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30: 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Quinlan A. R., Hall I. M.. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wei J., Guo H., Gao C., Kuo P. C.. 2004. Peroxide-mediated chromatin remodelling of a nuclear factor kappa B site in the mouse inducible nitric oxide synthase promoter. Biochem. J. 377: 809–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aoufouchi S., Yélamos J., Milstein C.. 1999. Inhibition of apoptosis of a PARP(-)/(-)cell line transfected with PARP DNA-binding domain mutants. J. Mol. Biol. 290: 943–949. [DOI] [PubMed] [Google Scholar]
- 33. Estrada-Gelonch A., Aramburu J., López-Rodríguez C.. 2009. Exclusion of NFAT5 from mitotic chromatin resets its nucleo-cytoplasmic distribution in interphase. PLoS One 4: e7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Han E.-J., Kim H. Y., Lee N., Kim N.-H., Yoo S.-A., Kwon H. M., Jue D.-M., Park Y.-J., Cho C.-S., De T. Q., et al. 2017. Suppression of NFAT5-mediated inflammation and chronic arthritis by novel κB-binding inhibitors. EBioMedicine 18: 261–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barish G. D., Yu R. T., Karunasiri M., Ocampo C. B., Dixon J., Benner C., Dent A. L., Tangirala R. K., Evans R. M.. 2010. Bcl-6 and NF-kappaB cistromes mediate opposing regulation of the innate immune response. Genes Dev. 24: 2760–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schreiber J., Jenner R. G., Murray H. L., Gerber G. K., Gifford D. K., Young R. A.. 2006. Coordinated binding of NF-kappaB family members in the response of human cells to lipopolysaccharide. Proc. Natl. Acad. Sci. USA 103: 5899–5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee H. H., Sanada S., An S. M., Ye B. J., Lee J. H., Seo Y.-K., Lee C., Lee-Kwon W., Küper C., Neuhofer W., et al. 2016. LPS-induced NFκB enhanceosome requires TonEBP/NFAT5 without DNA binding. Sci. Rep. 6: 24921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roth I., Leroy V., Kwon H. M., Martin P.-Y. Y., Féraille E., Hasler U.. 2010. Osmoprotective transcription factor NFAT5/TonEBP modulates nuclear factor-kappaB activity. Mol. Biol. Cell 21: 3459–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ogawa S., Lozach J., Jepsen K., Sawka-Verhelle D., Perissi V., Sasik R., Rose D. W., Johnson R. S., Rosenfeld M. G., Glass C. K.. 2004. A nuclear receptor corepressor transcriptional checkpoint controlling activator protein 1-dependent gene networks required for macrophage activation. Proc. Natl. Acad. Sci. USA 101: 14461–14466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ghisletti S., Barozzi I., Mietton F., Polletti S., De Santa F., Venturini E., Gregory L., Lonie L., Chew A., Wei C.-L. L., et al. 2010. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity 32: 317–328. [DOI] [PubMed] [Google Scholar]
- 41. Zhong H., Voll R. E., Ghosh S.. 1998. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell 1: 661–671. [DOI] [PubMed] [Google Scholar]
- 42. Bannister A. J., Kouzarides T.. 1995. CBP-induced stimulation of c-Fos activity is abrogated by E1A. EMBO J. 14: 4758–4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee J. S., See R. H., Deng T., Shi Y.. 1996. Adenovirus E1A downregulates cJun- and JunB-mediated transcription by targeting their coactivator p300. Mol. Cell. Biol. 16: 4312–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hargreaves D. C., Horng T., Medzhitov R.. 2009. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 138: 129–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Selvi B. R., Mohankrishna D. V., Ostwal Y. B., Kundu T. K.. 2010. Small molecule modulators of histone acetylation and methylation: a disease perspective. Biochim. Biophys. Acta 1799: 810–828. [DOI] [PubMed] [Google Scholar]
- 46. Zink D., Paro R.. 1995. Drosophila Polycomb-group regulated chromatin inhibits the accessibility of a trans-activator to its target DNA. EMBO J. 14: 5660–5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yuan W., Wu T., Fu H., Dai C., Wu H., Liu N., Li X., Xu M., Zhang Z., Niu T., et al. 2012. Dense chromatin activates polycomb repressive complex 2 to regulate H3 lysine 27 methylation. Science 337: 971–975. [DOI] [PubMed] [Google Scholar]
- 48. Beisel C., Paro R.. 2011. Silencing chromatin: comparing modes and mechanisms. Nat. Rev. Genet. 12: 123–135. [DOI] [PubMed] [Google Scholar]
- 49. Pasini D., Hansen K. H., Christensen J., Agger K., Cloos P. A. C., Helin K.. 2008. Coordinated regulation of transcriptional repression by the RBP2 H3K4 demethylase and polycomb-repressive complex 2. Genes Dev. 22: 1345–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Evertts A. G., Manning A. L., Wang X., Dyson N. J., Garcia B. A., Coller H. A.. 2013. H4K20 methylation regulates quiescence and chromatin compaction. Mol. Biol. Cell 24: 3025–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kruidenier L., Chung C. W., Cheng Z., Liddle J., Che K., Joberty G., Bantscheff M., Bountra C., Bridges A., Diallo H., et al. 2012. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 488: 404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stender J. D., Pascual G., Liu W., Kaikkonen M. U., Do K., Spann N. J., Boutros M., Perrimon N., Rosenfeld M. G., Glass C. K.. 2012. Control of proinflammatory gene programs by regulated trimethylation and demethylation of histone H4K20. Mol. Cell 48: 28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Agger K., Christensen J., Cloos P. A., Helin K.. 2008. The emerging functions of histone demethylases. Curr. Opin. Genet. Dev. 18: 159–168. [DOI] [PubMed] [Google Scholar]
- 54. De Santa F., Totaro M. G., Prosperini E., Notarbartolo S., Testa G., Natoli G.. 2007. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 130: 1083–1094. [DOI] [PubMed] [Google Scholar]
- 55. Konze K. D., Ma A., Li F., Barsyte-Lovejoy D., Parton T., Macnevin C. J., Liu F., Gao C., Huang X.-P., Kuznetsova E., et al. 2013. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem. Biol. 8: 1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Carey B. W., Finley L. W. S., Cross J. R., Allis C. D., Thompson C. B.. 2014. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518: 413–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hashizume R., Andor N., Ihara Y., Lerner R., Gan H., Chen X., Fang D., Huang X., Tom M. W., Ngo V., et al. 2014. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med. 20: 1394–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lane A. A., Chapuy B., Lin C. Y., Tivey T., Li H., Townsend E. C., van Bodegom D., Day T. A., Wu S.-C., Liu H., et al. 2014. Triplication of a 21q22 region contributes to B cell transformation through HMGN1 overexpression and loss of histone H3 Lys27 trimethylation. Nat. Genet. 46: 618–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ntziachristos P., Tsirigos A., Welstead G. G., Trimarchi T., Bakogianni S., Xu L., Loizou E., Holmfeldt L., Strikoudis A., King B., et al. 2014. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature 514: 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. McCabe M. T., Ott H. M., Ganji G., Korenchuk S., Thompson C., Van Aller G. S., Liu Y., Graves A. P., Della Pietra A. III, Diaz E., et al. 2012. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492: 108–112. [DOI] [PubMed] [Google Scholar]
- 61. Ishii M., Wen H., Corsa C. A. S., Liu T., Coelho A. L., Allen R. M., Carson W. F. IV, Cavassani K. A., Li X., Lukacs N. W., et al. 2009. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 114: 3244–3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.