

Abstract
Determining chromatin-associated protein localization across the genome has provided insight into the functions of DNA-binding proteins and their connections to disease. However, established protocols requiring large quantities of cell or tissue samples currently limit applications for clinical and biomedical research in this field. Furthermore, most technologies have been optimized to assess abundant histone protein localization, prohibiting the investigation of nonhistone protein localization in low cell numbers. We recently described a protocol to profile chromatin-associated protein localization in as low as one cell: ultra-low-input cleavage under targets and release using nuclease (uliCUT&RUN). Optimized from chromatin immunocleavage and CUT&RUN, uliCUT&RUN is a tethered enzyme-based protocol that utilizes a combination of recombinant protein, antibody recognition and stringent purification to selectively target proteins of interest and isolate the associated DNA. Performed in native conditions, uliCUT&RUN profiles protein localization to chromatin with low input and high precision. Compared with other profiling technologies, uliCUT&RUN can determine nonhistone protein chromatin occupancies in low cell numbers, permitting the investigation into the molecular functions of a range of DNA-binding proteins within rare samples. From sample preparation to sequencing library submission, the uliCUT&RUN protocol takes <2 d to perform, with the accompanying data analysis timeline dependent on experience level.
Introduction
The precise localization of regulatory proteins to chromatin is essential to understanding all DNA-templated processes, such as transcription and DNA replication, and is therefore fundamental to understanding many biological phenomena1–3. One important class of DNA binding proteins, termed transcription factors (TFs), bind in a sequence-dependent manner to a range of chromatin locations, but are enriched at gene regulatory features such as enhancers and promoters1,4–6. Appropriate binding of these proteins is required for many biological processes. For example, in development, lineage-specific TFs modulate cell state and lineage commitment1–3. Within the developing embryo, small populations of fated stem cells give rise to precursor cell types under the direction of the TFs they express2,5,7,8. Recent advances in low-cell-input transcriptomics have identified specific expression patterns of TFs in the early embryo9–12. However, mapping TF occupancies at these critical time points has remained near impossible with described chromatin profiling approaches because of the large number of cells or embryos required.
Here we detail ultra-low-input cleavage under targets and release using nuclease (uliCUT&RUN), the first protein profiling technique adapted to mapping the localization of TFs to chromatin within single embryos at early developmental time points (such as blastocysts) and in single cells. We recently used uliCUT&RUN to determine that the genome-wide occupancy of the TF NANOG is altered in single blastocysts depleted of the ATPase of the nucleosome remodeling complex esBAF13. In addition, we demonstrated that uliCUT&RUN successfully maps occupancy of TFs in single cells and confirmed a central assumption of multicell mapping that higher signals are indicative of increased factor occupancy13. Here, we detail the application of uliCUT&RUN to individual blastocysts and note specific alterations for application to single cells.
Overview of approach
uliCUT&RUN is a variant of CUT&RUN, with key modifications to reduce background signal, increase output and decrease the amount of starting material required to generate high-quality protein occupancy profiles from mammalian cells or embryos13–15. More recently, we have found that uliCUT&RUN is amenable to input material from a range of mammalian cell and tissue types, making it a highly versatile technique to map protein occupancy in many different contexts16.
The protocol begins with nucleus extraction from a single blastocyst, a small population of cells or a single cell (Fig. 1). Lightly permeabilized nuclei are then bound to lectin-coated Concanavalin A magnetic beads (Fig. 2). Next, nuclei are incubated with an antibody directed against a chromatin-associated protein of interest. Many ChIP-grade antibodies are amenable to use with uliCUT&RUN, making the mapping of additional DNA-associated proteins such as nucleosome remodelers, cofactors or modified histones with low-input material possible. In addition, we have found some antibodies that are not deemed ChIP grade by companies amenable to CUT&RUN, perhaps because of the lack of crosslinking in CUT&RUN. Nuclei are incubated with the recombinant fusion construct protein A-micrococcal nuclease (pA-MNase). Importantly, this recombinant protein was described by the Laemmeli group upon their development of chromatin immunocleavage (ChIC) technology, from which CUT&RUN was derived for genome-wide application17. Protein A, a bacterial protein isolated from Staphylococcus aureus, binds the IgG backbone of some antibodies (including rabbit IgG and mouse IgG2a) with high affinity while leaving MNase suspended in the nucleoplasm connected by a polypeptide tether. The addition of divalent calcium activates the endo-exonucleolytic activity of MNase, which then cleaves double-stranded DNA (or single-stranded DNA) directly adjacent to the antibody-bound protein of interest. The relaxed specificity MNase has for double- and single-stranded DNA permits applicability to proteins associating with single-stranded DNA (such as within the transcription bubble). Endonuclease digestion permits solubilization of the protein of interest and the DNA fragments with which the protein is associated. This digestion is followed by centrifugation, which separates these specific protein–DNA complexes from the uncleaved chromatin fraction, and phenol–chloroform isolation followed by alcohol–salt precipitation yields purified, uliCUT&RUN-enriched DNA.
Fig. 1 |. Overview of sample preparation for uliCUT&RUN. uliCUT&RUN is amenable to many sources of low-input material.
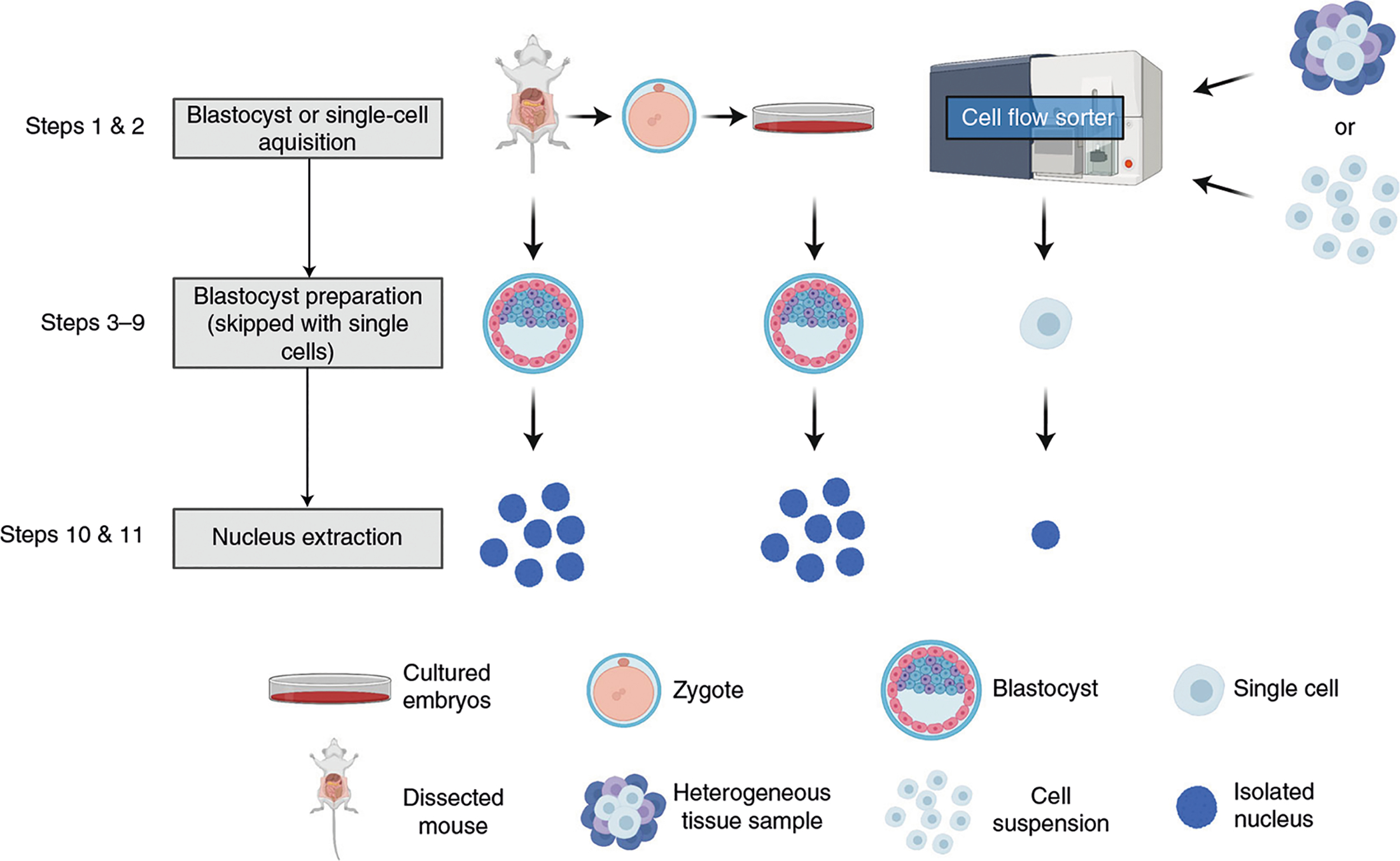
For embryological studies, freshly harvested blastocysts can be assayed directly, or harvested zygotes can be experimentally manipulated and cultured to the blastocyst stage prior to use in a uliCUT&RUN experiment. For single-cell applications, individual cells can be grown in culture or isolated from clinical or animal model samples and processed via cell sorting before experimentation. After individual embryos or cells have been isolated, nuclei are extracted. Figure created with BioRender.com.
Fig. 2 |. Overview of the uliCUT&RUN approach.
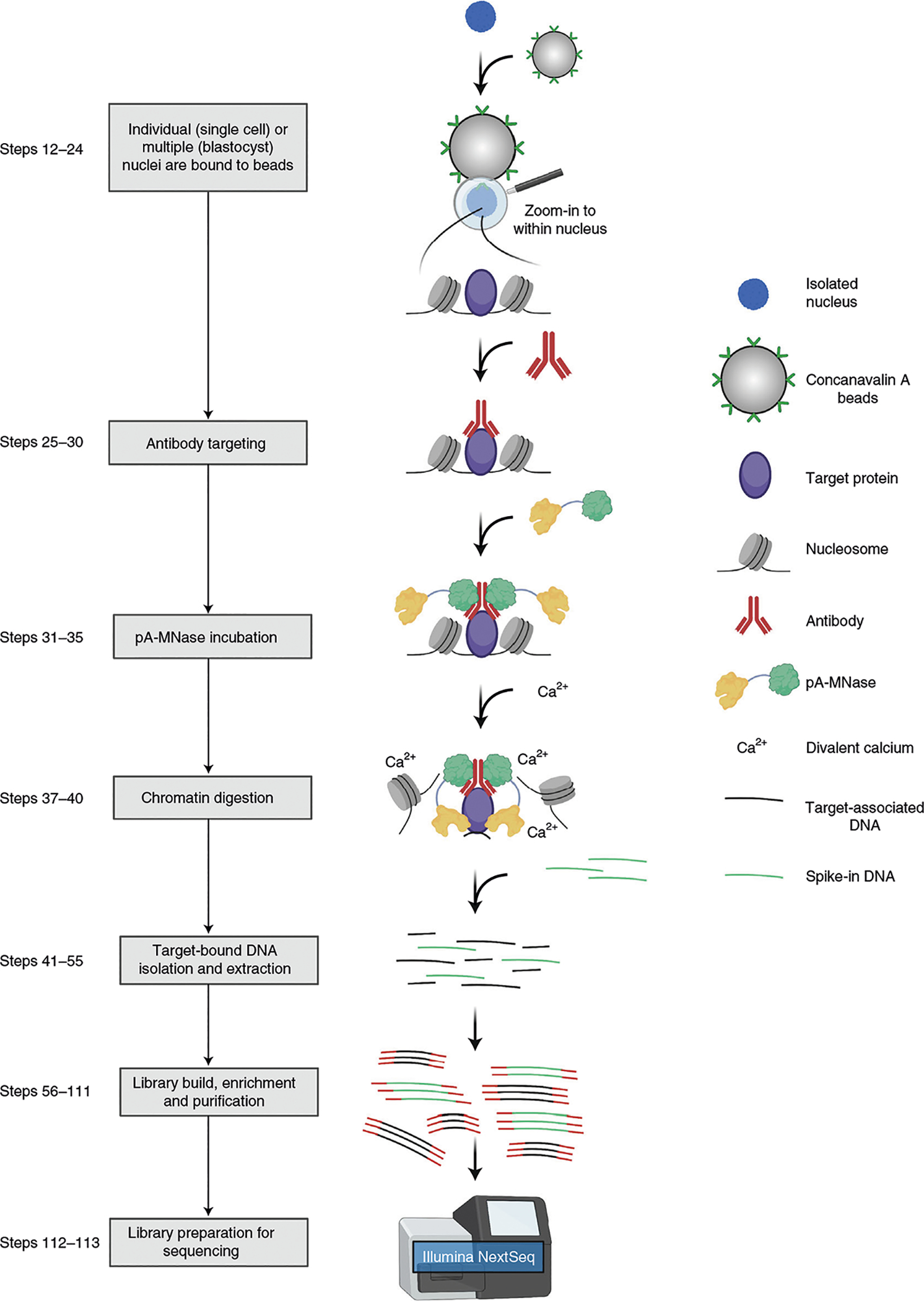
In this technique, isolated cell nuclei are permeabilized in hypotonic buffer and bound to lectin-coated magnetic beads. Next, an antibody directed against the target protein of interest is added, followed by incubation with pA-MNase, a recombinant fusion protein. Protein A (pA) tightly associates with the IgG backbone of the target-anchored antibody, while the addition of divalent calcium induces MNase digestion of DNA adjacent to the target protein. MNase digestion followed by endonuclease cleavage permits release of the target protein–DNA complex into the soluble fraction. Purification steps allow isolation of the target-bound DNA, which is then used to generate a high-quality sequencing library. Figure created with BioRender.com.
A custom, user-sourced library preparation protocol with NEBNext stem-loop adapters and indices provides a cost-effective method to build multiplexed libraries for Illumina sequencing, although kits with all reagents included, such as NEBNext Ultra DNA Library Prep Kit for Illumina, are also suitable. We have found that sequencing uliCUT&RUN libraries at a depth of 100,000 (uniquely) mapped (deduplicated) reads from single cells, and 1,000,000 (uniquely) mapped reads for individual blastocysts yields high-quality sequencing data. This depth requires ~15,000,000 raw reads per sample, and thus, several samples can be multiplexed for a single sequencing run on an Illumina NextSeq (or other compatible sequencing instrument; Table 2). Finally, we detail an easy-to-use yet effective bioinformatic pipeline from open-source programs that can be conducted by any user with basic Unix experience and access to the proper computational resources to process next-generation sequencing data into high-quality protein localization profiles (Fig. 3).
Table 2 |.
uliCUT&RUN sample sequencing statistics
| Sample name | Target | Total reads | mm10 mapped reads | % Reads mm10 mapped | sacCer3 mapped reads | % Reads SacCer3 mapped |
|---|---|---|---|---|---|---|
| NoAb_500,000cells | No-antibody control | 12,519,390 | 7,724,115 | 61.69721528 | 4,386 | 0.035033656 |
| NoAb_50,000cells | No-antibody control | 21,124,738 | 13,619,070 | 64.46976999 | 18,632 | 0.08819991 |
| NoAb_5,000cells | No-antibody control | 22,840,561 | 13,639,479 | 59.716042 | 111,161 | 0.486682442 |
| NoAb_500cells | No-antibody control | 6,721,902 | 2,738,302 | 40.73701164 | 88,437 | 1.315654409 |
| NoAb_50cells | No-antibody control | 31,441,140 | 9,944,171 | 31.62789581 | 348,488 | 1.108382202 |
| NoAb_10cells | No-antibody control | 22,386,707 | 1,528,715 | 6.828672926 | 109,941 | 0.491099473 |
| CTCF_500,000cells | CTCF | 24,622,812 | 16,910,022 | 68.67624218 | 3,001 | 0.012187885 |
| CTCF_50,000cells | CTCF | 28,244,629 | 19,181,790 | 67.91305349 | 9,666 | 0.034222436 |
| CTCF_5,000cells | CTCF | 86,425,016 | 55,116,921 | 63.77426763 | 158,771 | 0.183709541 |
| CTCF_500cells | CTCF | 16,011,197 | 7,590,239 | 47.40581857 | 51,441 | 0.321281413 |
| CTCF_50cells | CTCF | 33,064,993 | 18,592,267 | 56.22945996 | 153,859 | 0.465322947 |
| CTCF_10cells | CTCF | 52,838,179 | 12,155,437 | 23.00502635 | 219,423 | 0.415273585 |
| NoAb_50cells | No-antibody control | 26,177,953 | 3,759,352 | 14.36075616 | 186,217 | 0.711350501 |
| Sox2_50cells | Sox2 | 37,306,970 | 3,788,443 | 10.15478608 | 140,045 | 0.375385618 |
| Nanog_50cells | Nanog | 11,878,529 | 4,291,975 | 36.13220963 | 58,996 | 0.496660823 |
| Oct4_50cells | Oct4 | 54,221,820 | 5,211,784 | 9.611968023 | 169,106 | 0.311878133 |
| NoAb_1cell_rep1 | No-antibody control | 23,265,110 | 74,478 | 0.320127435 | 51,691 | 0.222182487 |
| NoAb_1cell_rep2 | No-antibody control | 27,236,861 | 97,674 | 0.358609606 | 67,396 | 0.24744408 |
| NoAb_1cell_rep3 | No-antibody control | 21,219,173 | 50,829 | 0.239542795 | 43,526 | 0.205125808 |
| NoAb_1cell_rep4 | No-antibody control | 21,366,345 | 226,075 | 1.058089252 | 54,499 | 0.255069363 |
| NoAb_1cell_rep5 | No-antibody control | 64,767,963 | 196,799 | 0.303852385 | 134,532 | 0.207713804 |
| CTCF_1cell_rep1 | CTCF | 8,058,422 | 33,946 | 0.421248726 | 3,158 | 0.039188814 |
| CTCF_1cell_rep2 | CTCF | 29,785,051 | 117,613 | 0.394872582 | 71,249 | 0.239210603 |
| CTCF_1cell_rep3 | CTCF | 39,235,948 | 157,304 | 0.400918056 | 94,497 | 0.240842913 |
| CTCF_1cell_rep4 | CTCF | 25,623,725 | 167,324 | 0.653004198 | 63,394 | 0.247403529 |
| CTCF_1cell_rep5 | CTCF | 27,905,711 | 97,412 | 0.349075499 | 53,672 | 0.19233339 |
| blast_NoAb_rep1 | No-antibody control | 48,402,426 | 134,232 | 0.277324942 | 391,250 | 0.808327252 |
| blast_NoAb_rep2 | No-antibody control | 10,924,378 | 261,010 | 2.38924358 | 130,484 | 1.194429559 |
| blast_CTCF_rep1 | CTCF | 74,269,402 | 556,739 | 0.749620954 | 770,472 | 1.037401648 |
| blast_CTCF_rep2 | CTCF | 81,540,028 | 468,019 | 0.573974539 | 111,8201 | 1.371352239 |
Sample name: the name of the sample that describes the target, the number of cells (1–500,000), or if a blastocyst (blast) was assayed, and the replicate number, if applicable. Target: identity of the targeted chromatin-associated protein Total reads: the total raw sequencing reads for that sample mm10 mapped reads: the number of reads mapped to mm10 reference genome % Reads mm10 mapped: the percentage of total raw reads mapped to the mm10 reference genome sacCer3 mapped reads: the number of reads mapped to sacCer3 reference genome % sacCer3 mapped: the percentage of total raw reads mapped to sacCer3 reference genome
Fig. 3 |. Overview of the uliCUT&RUN bioinformatic analysis pipeline.
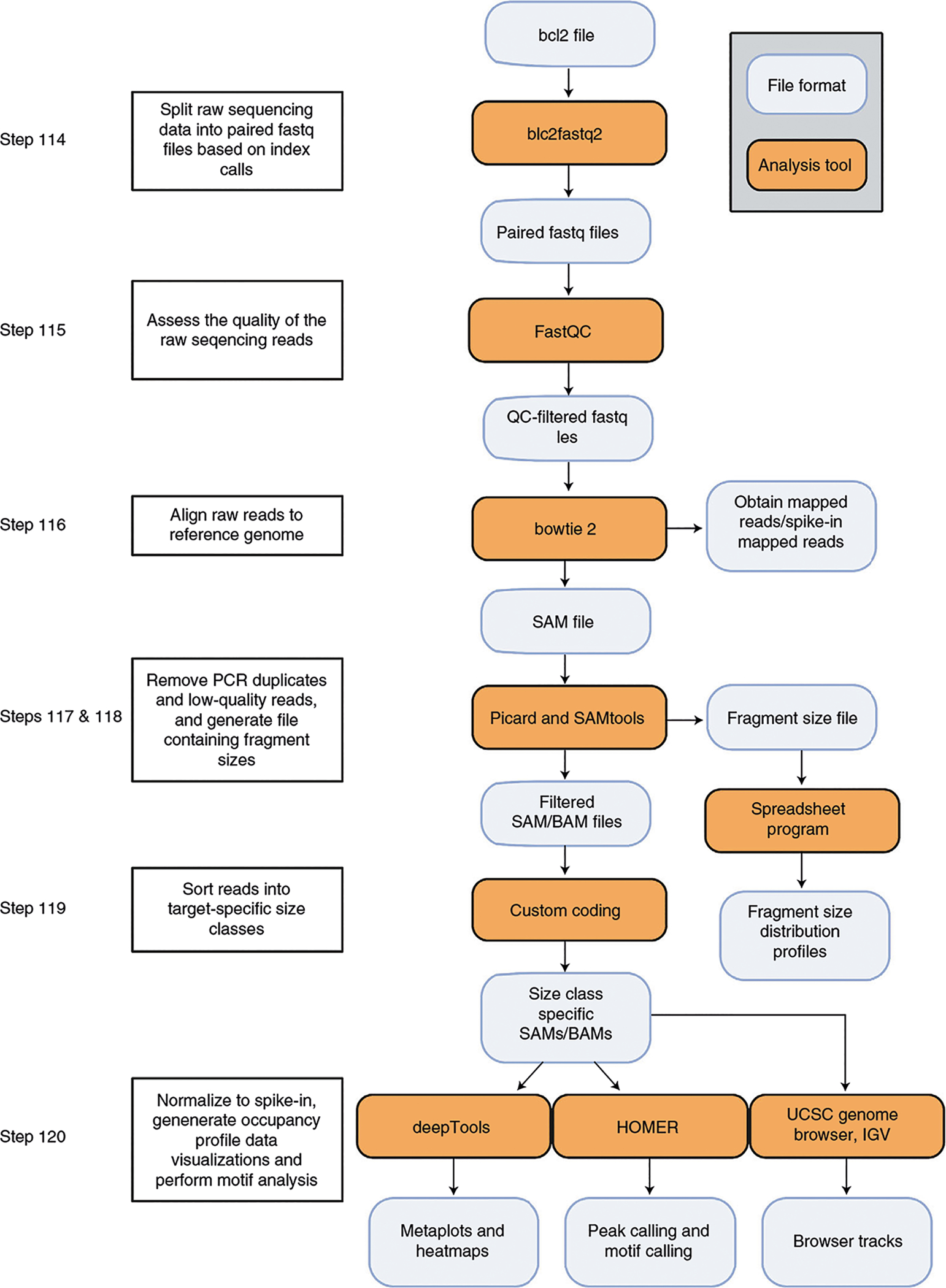
We provide a pipeline that instructs the user to begin with raw sequencing reads and output informative analyses and data visualizations with quality control metrics at various steps. This pipeline can serve as the basis for a more advanced, user-designed uliCUT&RUN analysis. Figure created with BioRender.com.
Development of the technique
uliCUT&RUN follows the core aspects of traditional CUT&RUN but has undergone optimizations specific for low input. For general low-input applications, we have modified the wash steps, the amount of added spike-in and the library amplification, and have integrated the use of NEBNext stem-loop adapters and indices into the library build. We reduced the number of wash steps relative to traditional CUT&RUN to reduce DNA loss and to bolster library yield. In a similar manner, a reduction in the amount of added spike-in yields sufficient reads for normalization without sacrificing a large portion of overall uniquely mapped reads to the spike-in using ultra-low-input samples. Next, we optimized the number of PCR cycles for library amplification and include steps to further optimize the PCR cycle number on a per-sample basis to minimize library overamplification. Additionally, we integrated the use of NEBNext adapters into the library build protocol. We have found that stem-loop adapters dramatically reduced the amount of adapter dimer present in each sample relative to inline Y-shaped adapters (compare Fig. 4a with Supplementary Fig. 1a). This permits a time-efficient method of library purification with AMPure XP beads, increasing the throughput ability of the approach. For single-cell application, we optimized the duration of pA-MNase digestion and further adjusted the amount of added spike-in DNA. These modifications allow this protocol to yield robust, Illumina-compatible sequencing libraries for protein localization mapping in ultra-low-input materials.
Fig. 4 |. Example quality controls for uliCUT&RUN libraries.
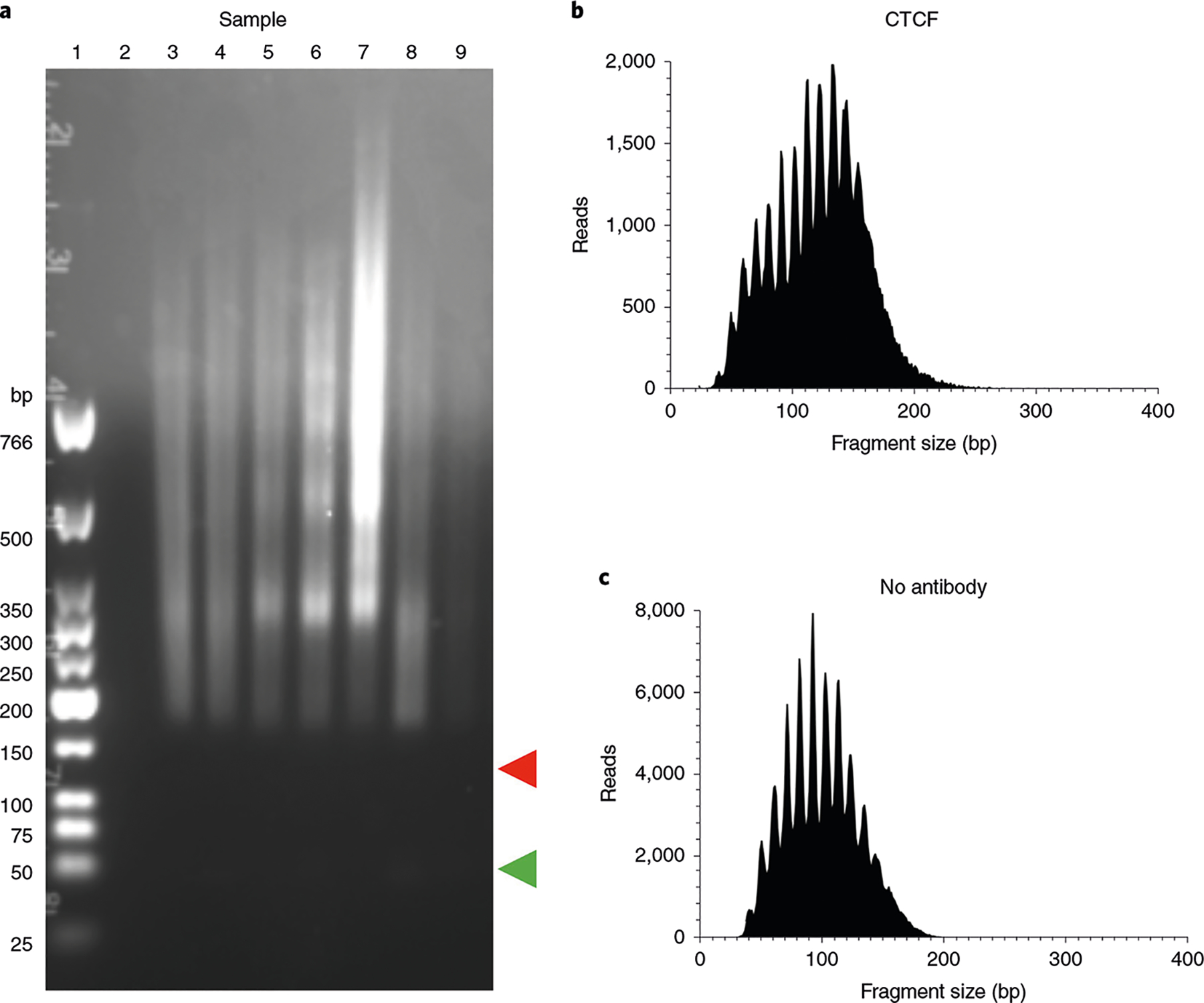
a, Ethidium bromide-stained agarose gel of seven blastocyst uliCUT&RUN samples after library preparation with NEBNext stem-loop adapters. Lane 1 contains the NEB low molecular weight marker, and lanes 3–9 show individual libraries. Lanes 3–8 contain CTCF-enriched libraries, while lane 9 contains the no-antibody control. Lane 2 was intentionally left empty. Lane 3 is an example of a successful library prep: there is a bright smear of the library (175–766+ bp), while the adapter dimer band (red arrow, 120 bp) and primer dimer band (green arrow, 25 bp) are either missing or barely visible. b,c, Postsequencing (adapters trimmed) size distribution plots for uliCUT&RUN on individual blastocysts for CTCF (anti-CTCF Millipore, cat. no. 07–729) (b) and no antibody (c); n = 1 each.
Comparison with other methods
ChIP-based approaches
Several approaches have been developed to map the localization of chromatin-associated proteins, beginning with the original ChIP assay. Developed and refined in the 1980s, ChIP was used to map the occupancy of RNA polymerase II in bacterial and eukaryotic cells18–20. In ChIP, target protein-bound DNA fragments are enriched from fragmented chromatin under native or cross-linked conditions through immunoprecipitation of the DNA–protein complex. The purified DNA can be used in single-locus experiments (such as in ChIP-qPCR21), or DNA libraries can be constructed and submitted for deep sequencing (ChIP-seq22). The use of native or crosslinking conditions for any chromatin profiling assay is dependent upon the target protein, as both carry inherent benefits and drawbacks. For ChIP, target profiling requires chromatin to be fragmented, most often through sonication (when crosslinked) or with MNase digestion (when native). Tightly associated targets such as nucleosomes and histone modifications can be assayed under native conditions, with chromatin fragmentation through MNase digestion. Less stable DNA interactors, such as TFs, often require crosslinking conditions (formaldehyde, UV radiation, etc.) to preserve their transient interactions, and allow for fragmentation by sonication. Importantly, sonication avoids the AT-rich sequence biases associated with MNase-based fragmentation23,24. MNase digestion can achieve higher resolution (~150 bp) than the range of fragmentation obtained when chromatin is sonicated (~150–400 bp), as MNase will digest DNA not shielded by protein interaction. Resolution becomes important in scenarios where distinguishing the precise location of the target protein tells important information about the local chromatin environment. This might include understanding how nucleosome positioning or TF occupancy at specific binding sites around gene regulatory locations influences the underlying transcription. Yet crosslinking might better capture indirect associations, such as the target protein interacting with DNA through an associated protein. Originally, ChIP-seq required ~107 cells as input material for target detection and could map occupancy with a resolution of 150–400 bp25,26. Over the past decade, a range of optimizations for ChIP-seq have permitted for low input (summarized in Table 1).
Table 1 |.
Low-input genome-wide chromatin protein profiling methods
| Technique | Lowest cell number used to profile histones | Histone protein/modification profiled | Lowest cell number to profile nonhistone protein | Nonhistone protein profiled | Applied to embryos? | Reference |
|---|---|---|---|---|---|---|
| Nano-ChIP-seq | 10,000 | H3K4me3 | N/A | N/A | No | Adli et al.68 |
| LinDA ChIP-seq | 10,000 | H3K4me3 | 5,000 | ERα | No | Shankaranarayanan et al.32 |
| Carrier-assisted ChIP-seq | N/A | N/A | 10,000 | ERα | No | Zwart et al.69 |
| ChIPmentation | 10,000 | H3K4me3, H3K27me3 | 100,000 | GATA4, CTCF | No | Schmidl et al.30 |
| cChIP-seq | 10,000 | H3K4me3, H3K4me1, H3K27me3 | N/A | N/A | No | Valensisi et al.70 |
| Small-scale ChIP-seq | 1,000 | H3K27me3 | N/A | N/A | No | Ng et al.71 |
| ULI-N-ChIP-seq | 500 | H3K27me3 H3K4me1 |
N/A | N/A | Yes | Liu et al.72 |
| iChIP-seq | 500 | H3K4me3, H3K4me1 | 10,000 | PU.1 | No | Lara-Astiaso et al.73 |
| Mint-ChIP-seq | 500 | H3, H3K4me3, H3K27me3, H3K27ac | N/A | N/A | No | van Galen et al.74 |
| FARP-ChIP-seq and RP-ChIP-seq | 500 | H3K4me3, H3K27me3 | N/A | N/A | No | Zheng et al.75 |
| μChIP-seq | 500 | H3K4me3, H3K27ac | N/A | N/A | Yes | Dahl et al.76 |
| STAR-ChIP-seq | 200 | H3K4me3 | N/A | N/A | Yes | Zhang et al.77 |
| TCL-ChIP-seq | 200 | H3K4me3, H3K36me3, H3K27me3, H3K27ac | N/A | N/A | No | Zarnegar et al.78 |
| Stacc-seq | 200 | H3K4me3, H3K27me3 | 200 | RNAPII | Yes | Lui et al.43 |
| MOWChIP-seq | 100 | H3K4me3, H3K27ac | N/A | N/A | No | Cao et al.31 |
| ChIL-seq | 100 | H3K4me3, H3K27me3, H3K27ac | 1,000 | CTCF, MyoD | No | Harada et al.79 |
| CUT&RUN | 100 | H3K27me3 | 1,000 | CTCF | No | Skene et al.45,46 |
| Drop-ChIP-seq | 1–200 | H3K4me3, H3K4me2 | N/A | N/A | No | Rotem et al.33 |
| ACT-seq and iACT-seq | 1 | H3K4me3 | 1,000 | Brd4 | No | Carter et al.42 |
| scChIP-seq | 1 | H3K4me3, H3K27me3 | N/A | N/A | No | Grosselin et al.34 |
| scChIC-seq | 1 | H3K4me3, H3K27me3 | N/A | N/A | No | Ku et al.44 |
| CUT&Tag | 1 | H3K27me3 | 60–500,000 | RNAPII, CTCF, Sox2, NPAT | No | Kaya-Okur et al.41 |
| uliCUT&RUN | 10 | H3K4me3 | 1 | CTCF, Sox2, Nanog | Yes | Hainer et al.13 |
Many notable improvements that reduce the required input material for ChIP-seq, related to basic protocol optimizations, sample processing and library construction, have influenced both crosslinking and native ChIP-seq sensitivity for histone modifications and DNA-binding proteins (Table 1). These innovations, and many others26, have opened native and crosslinking ChIP-seq application to ultra-low cell numbers, rare biological specimens and embryological materials. It should be stressed that several ChIP-seq techniques, both native and crosslinking, have been demonstrated to profile histone modifications at or below 1,000 cells (or the chromatin aliquot equivalent), and nonhistone proteins in 5,000 cells or more (Table 1). The integration of transposase technology, pioneered with the hyperactive transposase Tn5 and Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq)27–29, with ChIP-seq resulted in development of ChIPmentation. ChIPmentation involves Tn5 loading sequencing adapters onto crosslinked, fragmented chromatin bound to beads coated with target-specific antibodies30. Similar to other low-input ChIP-based approaches, ChIPmentation excels at histone modification profiling with ~10,000 cells at a resolution of 100–200 bp30. In their current form, low-input ChIP-seq approaches involving crosslinking appear more effective than their native counterparts for histone and nonhistone protein profiling. Crosslinking approaches can profile abundant histone modifications in 100 cells31, and DNA-binding proteins including TFs can be assessed in ~5,000 cell equivalents32. Native ChIP-seq approaches can currently profile histone modifications in single-cell equivalents and embryological material equivalent to 200 cells33,34.
The resolution of ChIP-seq has evolved as well, with two specialized techniques, ChIP-exo35 and ChIP-Nexus36, providing nucleotide resolution, through λ-exonuclease digestion. However, these techniques are not currently suited for low-input material. The base pair resolution of low-input cross-linking and native ChIP-seq approaches has likely improved, however. Fragmentation through sonication can be optimized to 100–800 bp fragments, and to address biases in shearing from heterochromatic or euchromatic regions26,37. MNase digestion could conceivably be calibrated to digest only DNA unprotected from the target protein, suggesting that even higher resolution is possible with low-input native ChIP-seq approaches. The major problems remaining for low-input ChIP-seq are the requirement for microfluidic platforms for single-cell profiling26 and profiling nonhistone proteins below 5,000 cells, which has not been demonstrated32. Furthermore, the available low-input ChIP-seq approaches with single-cell profiling capabilities require larger amounts of starting material than single cells as input to the experimental setup (such as with micro-fluidics)26,33,34. The current low-input ChIP-seq approaches have not been compared systematically side-by-side, and so it is hard to say what approaches can best address these issues. Still, ChIP-seq protocols show potential for further optimization in ultra-low-input application, and it will be interesting to follow these technologies in the near future.
Enzyme/antibody-tethered assays
The generation of recombinant fusion proteins and antibody-tethering approaches led to a family of assays orthogonal to ChIP-seq. In general terms, these approaches use antibody- or target-protein-guided DNA modification activity to determine target protein localization along chromatin. These assays, like ChIP-seq, can be performed on lightly crosslinked cells, native cells or isolated cell nuclei and carry their own inherent benefits and biases. Examples of the earliest techniques in this family of assays include DNA adenine methyltransferase identification (DamID)38,39, chromatin endogenous cleavage (ChEC) and ChIC17,40. DamID fuses a DNA adenine methyltransferase to the target DNA-binding protein. DamID delivers targeted adenine methylation at a GATC consensus sequence in the vicinity of the DNA-binding partner, which can be detected at specific loci with PCR38. ChEC and ChIC implement the fusion of MNase to the protein of interest or to protein A, respectively, with single-locus resolution similar to ChIP-qPCR17. Like ChIP-seq, the integration of next-generation sequencing adapts these techniques for genome-wide application. Though young relative to the over four-decade tenure of ChIP, enzyme/antibody-tethered approaches have expanded in diversity in recent years (Table 1). The most recent additions to this family have utilized transposase-fusion proteins (pA-Tn5) to profile histone modifications and RNA polymerase II in low cell numbers41,42 and mouse embryological material43. Excluding our approach, in their current form, enzyme/antibody-tethered approaches can profile histone modifications in single cells in native44 and crosslinking conditions41,42,44 and nonhistone proteins in as low as 60 cells41.
Enzyme/antibody-tethered techniques are efficient in their recovery of DNA fragments, as they solubilize only the target protein–DNA complex instead of solubilizing chromatin25,26. In addition, enzymatic DNA modification activity (methylation, digestion, tagmentation, etc.) is largely limited to DNA adjacent to the target protein; therefore, a smaller fraction of the nontarget regions of the genome will be represented in the background signal, which reduces potential background signal25,26,41. Because of efficient recovery and low background signal, enzyme-tethered techniques often require lower amounts of starting material and fewer total sequencing reads to map chromatin features in comparison with similar low-input ChIP-seq approaches45–47. Still, background signal does persist in these approaches and needs to be accounted for with the proper controls. Similar to MNase digestion in native ChIP-seq, DNA bound by the target protein is protected from enzymatic DNA modification activity41,45. However, for enzyme-tethered assays, DNA modification activity is largely limited to DNA directly adjacent to the target protein14,41,48. As DNA occupancy signatures are target dependent (<120 bp for nonhistone DNA-binding proteins, ~150 bp for mononucleosomes), the resolution of these approaches is also target dependent (with the exception of DamID, where methylation occurs over relatively large distances regardless of the protein targeted). These fragment sizes are then enriched for in downstream bioinformatic analyses, which enhances the potential resolution of these approaches14,47,49. Like ChIP-seq, enzyme-tethering assays have parameters to be optimized before application and unique limitations. Nonetheless, they represent a group of fundamentally different but complementary approaches to ChIP-seq recently added to the chromatin profiling toolkit.
uliCUT&RUN is a native approach unique in the capacity to profile nonhistone protein localization with low-input mammalian cells, specifically in single cells and blastocysts. Here, we focus on the critical factors and limitations when adapting uliCUT&RUN to new mammalian systems, and how we have found best to address these (see ‘Limitations of approach’). Our protocol is designed to work with native, freshly collected specimen at the bench, with minimal specialized equipment. Here, we detail a typical experiment involving single embryos or individual cells with the information necessary to help new users establish uliCUT&RUN in their laboratory.
Applications of uliCUT&RUN
We optimized protein localization mapping with uliCUT&RUN while envisioning implementation in three settings: experimentally manipulated and/or transgenic embryological work, single-cell studies and limited clinical specimens from patient samples or animal models. For embryological work, uliCUT&RUN would be advantageous when characterizing the occupancies of DNA-binding proteins under various experimental settings or to map changes in the stage-specific TF occupancies over the course of development. Defining the differences in localization of essential TFs during multiple points in development, such as between early- and late-stage embryos, might help to better understand how they drive development. Similarly, uliCUT&RUN could be applied to epigenomic localization studies with embryological material from extremely limited sources. uliCUT&RUN represents an important addition to the ever-growing genomic tool kit for single-cell studies. Used alongside other recently developed single-cell technologies, such as single-cell RNA sequencing (scRNA-seq), single-cell ATAC-seq (scATAC-seq29) and single cell Hi-C50, uliCUT&RUN could be applied to assess the relationship between individual cell TF profiles and cellular heterogeneity within complex metazoan tissues and developing embryos pertaining to transcriptomics and chromatin organization. In these contexts, uliCUT&RUN could reveal novel insights into chromatin-binding protein localization at the single-cell level that larger-scale, cell-averaging approaches can miss. Finally, application of uliCUT&RUN to clinical specimens permits previously unavailable avenues for research, primarily due to the large amount of material required with described methods and limited numbers of cells present in clinical specimens. Detection of differential TF occupancies between experimentally manipulated and control early blastocysts (~30–50 cells) demonstrates that uliCUT&RUN has the capacity to facilitate studies examining the differences between healthy and diseased patient tissues and animal models with limited material input13.
Expertise needed to implement uliCUT&RUN
The protocol outlined below consists of three aspects: blastocyst/single-cell harvest and preparation (Fig. 1), uliCUT&RUN (with the associated deep sequencing library preparation, Fig. 2) and data analysis (Fig. 3). We outline a general overview for how to harvest blastocysts from dissected uteri, or how to harvest zygotes for experimental manipulation and culture to blastocyst stage prior to use (Fig. 1). In practice, this portion of the protocol requires an advanced level of expertise in anatomy, dissection and embryology. In addition, we suggest options for obtaining single cells. However, working closely with the appropriate core facilities is recommended if flow sorting expertise or equipment is not available or established within your laboratory. Furthermore, appropriate single-cell sorting (such as sorting based on a specific marker or by size) must be optimized for cell or tissue type. uliCUT&RUN and the associated library build require standard molecular biology techniques, and therefore can be completed with basic molecular biology experience and careful attention (Fig. 2). The resulting Illumina libraries can be processed by any standard sequencing facility, institutional or commercial, if in-house facilities are not available. The data analysis described below requires a basic understanding of Unix commands, but consists of open-source programs that can be operated on a standard computing cluster or with a personal computer containing appropriate space and processing power. This data analysis pipeline can serve as a starting point for experienced users to develop custom analyses to suit their needs (Fig. 3).
Limitations of uliCUT&RUN
uliCUT&RUN is a powerful technique to study genomic localization of chromatin-bound proteins in various mammalian systems. However, there are limitations to the application of this technique that should be accounted for in experimental design. First, as with any antibody-based technique, uliCUT&RUN application is dependent upon the availability of high-quality, target-specific antibodies. As not all antibodies are equivalent, care must be taken to ensure a high-quality antibody is used51, with possible optimizations required for specific antibodies. While ‘ChIP-grade’ antibodies are available, these antibodies are tested under crosslinking conditions, and so it is not straightforward to expect them to work well in native conditions. The Henikoff group has recommended that antibodies validated as high quality in immunofluorescence should work well47. Second, a downside to conducting uliCUT&RUN under native conditions is that the detection capabilities are dependent upon strong affinity interactions between the target protein and DNA. Transient interactions might not be captured, and so information on the nature of any potential target’s interaction with DNA should be considered. Notably, we and others have considered that crosslinking uliCUT&RUN can be performed, with a crosslinking CUT&RUN approach described previously52. Light-crosslinking CUT&RUN (for example, 0.1% (wt/vol) formaldehyde for 5 min) could stabilize transient protein–DNA interactions that might be difficult to detect in native conditions. Third, untargeted pA-MNase background digestion can occur. Free pA-MNase can digest open regions of chromatin, and preferentially digests AT-rich, nucleosome-free regions of DNA53. However, the use of a no primary antibody (where a primary antibody targeting the protein of interest is not added but pA-MNase is added; referred to as no antibody or No Ab) or an IgG control accounts for background digestion. Fourth, uliCUT&RUN as described below cannot be applied to cells lacking intact nuclei, such as mitotic cells. As CUT&RUN has been adapted previously for application to mitotic cells by replacing use of lectin-coated beads with centrifugation steps54, it is likely that uliCUT&RUN could be modified for mitotic cell application in a similar manner. Finally, the current form of uliCUT&RUN is a low-throughput technique requiring fresh biological samples and thus is limited by access to facilities that supply specimens, the amount of material that can be harvested freshly and/or by how many samples the user can handle. Using the protocol below, we have found that up to 48 samples can be processed in parallel with no observable decrease in data quality. However, as the analogous technique cleavage under target and tagmentation (CUT&Tag) and other forms of CUT&RUN (such as CUT&RUN. Auto) have been optimized for high-throughput studies, we speculate that adaptation of uliCUT&RUN for high-throughput application is possible41,55.
Because of the nature of this protocol, quality-control stopping points are limited. Therefore, the troubleshooting guide included below is designed to help the user identify at which steps in the protocol issues might arise, resulting in either low DNA recovery following DNA isolation, a low-quality library following library recovery or issues arising over the course of data analysis. We have experienced each of the issues mentioned within this table and therefore include an explanation on how we resolved the respective issues. Importantly, there are other possible issues that we have not experienced and therefore may not be detailed.
Experimental design
Input material selection
The uliCUT&RUN protocol outlined below is optimized for input materials ranging from single feeder-free murine embryonic stem cells (E14 mESCs) to early-stage mouse embryos (blastocysts). This protocol has been optimized for application to mammalian cells, and therefore should be generally applicable to various cell types. Included are suggested options for obtaining blastocysts, but later-stage embryos can also be used. In addition, we indicate points in the protocol where experimental manipulation of embryos (drug exposure or microfluidic injection) can be integrated into the protocol as desired. While various methods have been described for obtaining single cells, we have found that flow sorting by cell size or FACS represents a high-throughput option with high specificity and, most critically, maintains cell viability prior to use. Importantly, the exact growth conditions, harvest and FACS process are cell type specific. Below we have detailed a general approach for obtaining single cells from culture, but we recommend consulting with your FACS personnel or core facilities to determine optimal input conditions and parameters for your cell type. It is advised to run a pilot experiment (using higher cell numbers, if possible, and a well-established antibody such as CTCF) first to confirm the methodology can be applied to your system. When possible, we advise to optimize the protocol for application to a new biological material or for a new user with nonprecious biological materials of a similar nature. This would be especially important for working with materials from rare clinical specimens or limited cell populations.
Controls
uliCUT&RUN has a series of integrated control metrics at the experimental and bioinformatic analysis levels that allow the user to assess the quality of the experiment. First, we use heterologous DNA spike-in controls to detect technical variation introduced during the protocol. We routinely use Saccharomyces cerevisiae genomic DNA that has been crosslinked, MNase-digested, agarose gel purified, and diluted to a concentration of 10 ng/μL (Supplementary Methods). DNA from other sources can be used, but it is essential that it is not the same as or closely related to the experimental system and comes from an organism with a well-annotated reference genome. Alternatively, the Henikoff group has described the use of carryover contaminating DNA from the purification of pA-MNase out of E. coli as a valid spike-in ref.49. Second, samples are always performed alongside a no primary antibody (no antibody) or IgG control sample, which provides a metric of comparison for background pA-MNase digestion. This control sample is performed in parallel with every assay conducted, with the same amount of input material from the same biological source as experimental samples. It should be noted that this parallel control sample does not control for sample-to-sample variation, and so care must be taken to ensure differences in sample handling are kept to a minimum. Integral components of the library build protocol are stringent purification steps and the agarose gel check step. The agarose gel step has the inherent benefit of observing whether the library was constructed, amplified and of the appropriate size distribution (see Fig. 4a for an example of well-constructed libraries.) As an additional quality control step, we recommend analyzing each library with a Fragment Analyzer (or Bioanalyzer/TapeStation) to confirm the proper distribution of fragments. Finally, after sequencing and alignment to the proper reference genome, the described bioinformatic pipeline instructs the user to produce a fragment size distribution profile. A fragment size distribution profile shows if the experiment yielded an enrichment of the proper fragment sizes above background. For experiments profiling TFs, an enrichment of fragments <120 bp in size should be expected (Fig. 4b) above the no-antibody control (Fig. 4c). Importantly, as previously shown, CUT&RUN for CTCF can map both CTCF binding sites and CTCF-flanking nucleosomes13,15. Therefore, CTCF profiles show both TF and nucleosome-size enrichment.
Sequencing depth and multiplexing
We have previously found that a sequencing depth of 100,000 uniquely mapped reads is sufficient to map occupancy of highly expressed TFs in single mESCs, and that 1,000,000 uniquely mapped reads are sufficient for murine blastocysts13 (Table 2). The percentage of uniquely mapping reads for single cells is ~0.75% and for blastocysts is ~7%, and so we recommend at least 15,000,000 raw reads per single cell and blastocyst. However, the sequencing depth might need to be optimized for your protein of interest and in your system. Factors to consider when evaluating a potential target for uliCUT&RUN are the class of DNA binding protein (TF, histone modification, etc.), protein abundance in the nucleus, and the anticipated number of genomic locations to which the target can localize. Sequencing depth might need to be adjusted to acquire sufficient coverage for some target proteins. We integrated the use of NEBNext stem-loop adapters into the library preparation protocol, which allows for the multiplexing of sequencing libraries for a single sequencing run with little adapter dimer. Importantly, indices across all four NEBNext stem-loop adapter kits (each containing 12 barcoded primers) can be combined for multiplexing as necessary. In addition, uliCUT&RUN libraries can be added to Illumina sequencing runs containing other compatible libraries (e.g., RNA-seq or ChIP-seq libraries with compatible sequencing barcodes). To this end, the user should determine how many individual libraries need to be sequenced and at what depth before deciding on which Illumina platform to sequence.
Analysis of uliCUT&RUN data
The bioinformatic analysis that we detail shows the user how to visualize the localization of their protein of interest at annotated genomic locations for blastocysts and single cells in the form of heatmaps, metaplots and browser tracks (Fig. 3; also see ‘Code availability’). With minimal modification, this analysis can be applied to a wide variety of database-acquired or user-assembled genomic locations in their experimental system, including subsets of genes, gene regulatory sites, intergenic loci or additional genomic locations (example resulting data shown in Figs. 5 and 6). Once binding locations for the protein of interest have been defined, the provided analysis also instructs how to perform DNA-binding protein motif analysis for datasets from populations of cells, such as blastocysts (Fig. 6c). Analyses of this nature are helpful in identifying de novo motifs for the target protein or in identifying additional DNA-binding proteins with which the target protein interacts.
Fig. 5 |. Expected results for 50 ES cell uliCUT&RUN for TFs.
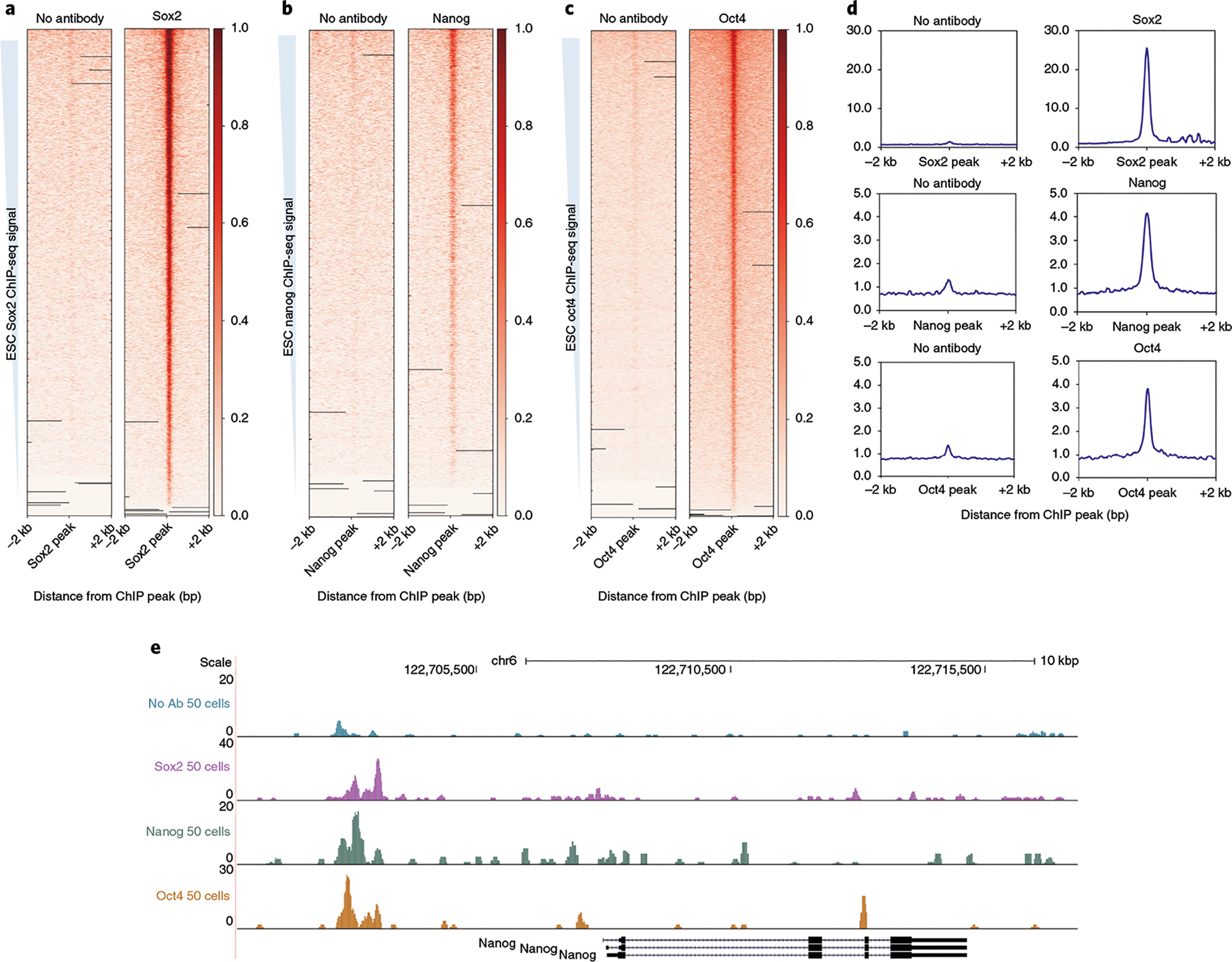
a–c, Heatmaps generated with deepTools for uliCUT&RUN assay of 50 mouse ES cells profiled for the occupancy of the specified TFs over peaks called from bulk ChIP-seq datasets (GSE11724 for Sox2, Nanog and Oct4 datasets), sorted from strongest to weakest peak within the ChIP-seq data. d, Metaplots generated over peaks called from published ChIP-seq datasets (as in a–c). e, Browser track of 50 cell uliCUT&RUN for Sox2, Nanog, Oct4 and a corresponding no-antibody control over the Nanog locus and accompanying upstream and downstream genomic regions.
Fig. 6 |. Expected results for CTCF blastocyst or single-cell uliCUT&RUN.
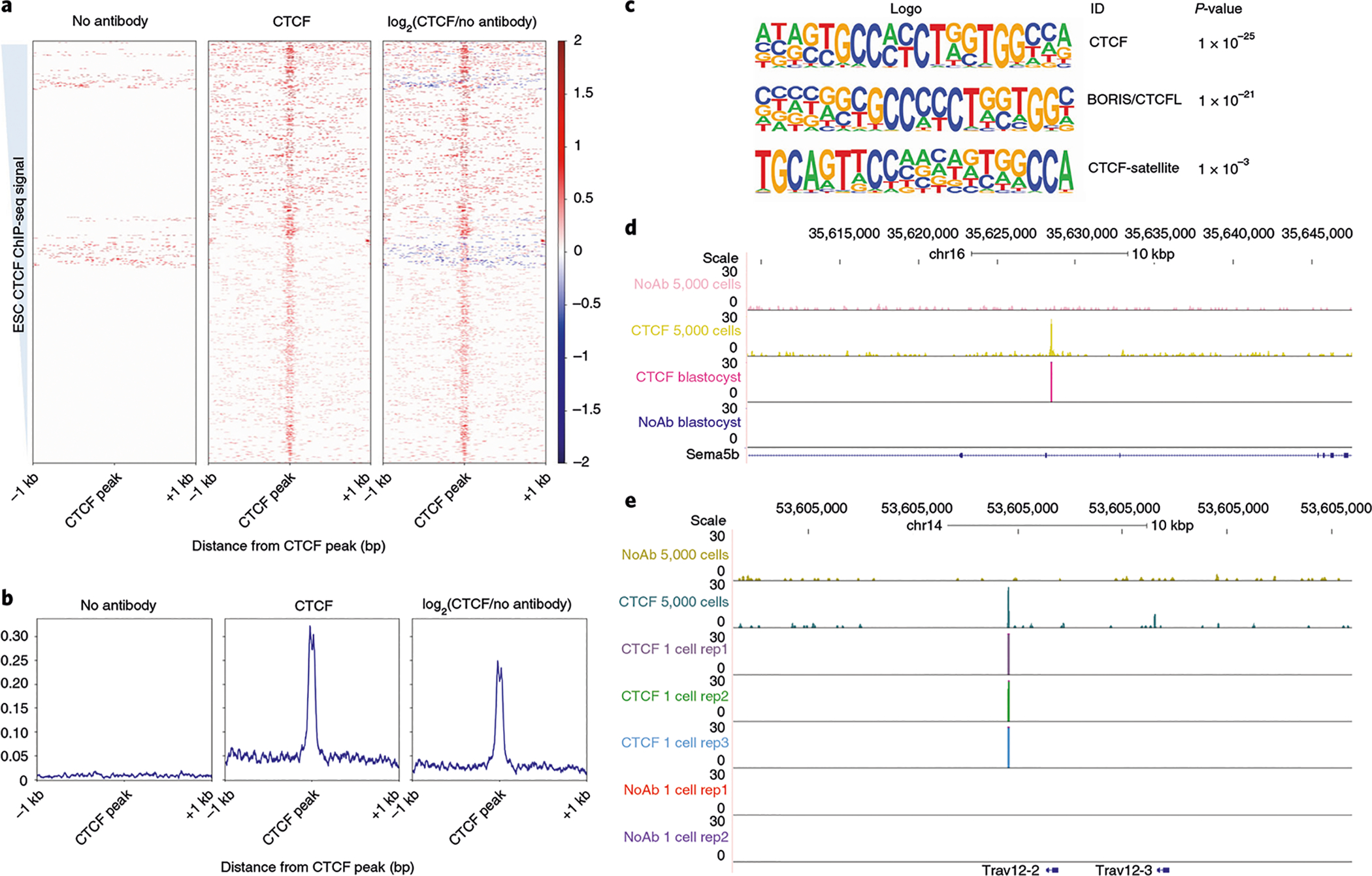
a, Heatmaps generated with deepTools for individual blastocysts subjected to uliCUT&RUN. Left panel contains signal for no-antibody control, the middle panel contains signal for CTCF and the right panel shows signal for the log2 enrichment of CTCF/no antibody. Data are shown over previously published CTCF ChIP-seq signal (GSE11431 (ref.84)), sorted from strongest CTCF peak to weakest CTCF peak within the ChIP-seq data. b, Metaplots generated with deepTools, over previously published CTCF ChIP-seq peaks. Panels as in a. c, Motif search performed with HOMER after peak calling from single blastocyst relative to no antibody demonstrating enrichment of CTCF motif within single blastocyst uliCUT&RUN data. d, Browser track of individual blastocyst CTCF and control uliCUT&RUN with CUT&RUN in high cell number (50,000) shown for comparison. e, Example browser tracks of single-cell CTCF and control uliCUT&RUN.
Beyond what we have outlined, uliCUT&RUN data are amenable to additional avenues of analysis. Bioinformatic analysis tools specific to CUT&RUN, including SEACR56 and CUT&RUNTools57, are valuable resources for blastocyst and bulk uliCUT&RUN-based genomic research. Single-cell uliCUT&RUN data should be amenable to analyses for single-cell epigenomics research, similar to those described for scATAC-seq data analysis58–62. In principle, after quality filtering, individual cell read data would be compiled into a matrix of counts per cell at a set of provided genomic features (e.g., previously identified binding sites or cis-regulatory elements). Next, data transformation would be needed to account for the scarcity and binary nature of single cell uliCUT&RUN data (as described in ref.58). Finally, dimensional reduction analysis and clustering to identify subgroups of genomic loci with heterogeneous factor of interest occupancies between individual cells would be performed (as described in ref.58). We conceptualize these data could be used to determine the dynamic range of target protein occupancy at genomic loci within and between cell types in complex tissue specimens. When combined with scATAC-seq and scRNA-seq datasets, differences in factor of interest occupancies could reveal connections between differences in chromatin accessibility and transcriptomic output between healthy patients’ and diseased patients’ biopsies to study the evolution of cancerous cells in situ. Bioinformatic tools used to perform data transformation, dimensional reduction and clustering with single-cell epigenomic datasets that we propose for use include the R packages scABC61 and Destin59. However, many additional tools exist for the study of single-cell epigenomic datasets, and so we envision that additional novel avenues of investigation show great potential.
Materials
Biological materials
Mice from line of interest (FVB/NJ, Jackson Laboratories, stock no. 001800; RRID: JAX:001800) ! CAUTION Experiments utilizing animals should conform to all governmental and institutional standards and regulation. Dispose of all biological waste in accordance with government and institutional standards.
Cell line of interest (ES-E14TG2a, ATCC, cat. no. CRL-1821; RRID: CVCL_9108) ! CAUTION Cell lines utilized should be checked routinely to ensure no mycoplasma contamination.
Reagents
Antibody to protein of interest
(Optional) Positive control antibody (CTCF Millipore, cat. no. 07-729; RRID: AB_441965)
(Optional) Negative control antibody (guinea pig anti-rabbit IgG, Antibodies-Online, cat. no. ABIN101961; RRID: AB_10775589)
100 % (vol/vol) Triton X-100 (Sigma Aldrich, cat. no. 9002-93-1) ! CAUTION Triton X-100 is hazardous; use a lab coat, gloves and goggles when handling.
Potassium chloride (KCl; Sigma Aldrich, cat. no. P3911)
Sodium chloride (NaCl; Sigma Aldrich, cat. no. S5150-1L)
HEPES (Fisher Scientific, cat. no. BP310-500)
BSA (Sigma Aldrich, cat. no. A1933)
Glycerol, 100% (vol/vol) (Fisher Scientific, cat. no. BP229-1)
Spermidine (Sigma Aldrich, cat. no. S2626)
Manganese chloride (MnCl2; Sigma Aldrich, cat. no. 244589)
EGTA (Sigma Aldrich, cat. no. E3889)
Nuclease-free water (New England Biolabs, cat. no. B1500S)
PBS (Thermo Fisher Scientific, cat. no. 10010023)
RNase A (New England Biolabs, cat. no. T3010)
EDTA (0.5 M; Fisher Scientific, cat. no. BP2482100)
Glycogen (20 mg/mL; Thermo Fisher Scientific, cat. no. 10814010)
Protease inhibitors (Thermo Fisher Scientific, cat. no. 78430)
BioMag Plus Concanavalin A (ConA) beads (Polysciences, cat. no. 86057-10)
Recombinant pA-MNase, purified from pK19pA-MN (Addgene, cat. no. 86973; RRID: Addgene_86973) ▲ CRITICAL See Supplementary Methods for pA-MNase purification.
(Optional) pAG-MNase (Epicypher, cat. no. 15-1016) ▲ CRITICAL See Supplementary Methods for pAG-MNase information.
Calcium chloride (CaCl2; Fisher Scientific, cat. no. AAJ62905AP)
Sodium dodecyl sulfate (SDS; Thermo Fisher Scientific, cat. no. BP166-500) ! CAUTION SDS is poisonous if inhaled; handle with care in well-ventilated spaces using gloves, eye protection and an N95-grade respirator when handling.
Proteinase K (New England Biolabs, cat. no. P8107S)
Phenol/chloroform/isoamyl alcohol (PCI; Thermo Fisher Scientific, cat. no. 15593049) ! CAUTION Phenol is harmful if swallowed or upon skin contact; handle in a chemical fume hood using gloves, a lab coat and goggles.
Chloroform (Thermo Fisher Scientific, cat. no. C298-500) ! CAUTION Chloroform is a skin irritant and harmful if swallowed; handle in a chemical fume hood using gloves, a lab coat and goggles.
Ethanol (EtOH; Fisher Scientific, cat. no. 22032601) ! CAUTION 100% (vol/vol) EtOH is highly flammable; handle in a chemical fume hood using gloves, a lab coat and goggles.
Sodium hydroxide (NaOH; Fisher Scientific, cat. no. S318-1) ! CAUTION NaOH is an eye/skin irritant as a solid and corrosive in solution. Handle in a chemical fume hood using gloves, a lab coat and goggles.
Potassium hydroxide (KOH; Fisher Scientific, cat. no. P250-1) ! CAUTION KOH is an eye/skin irritant as a solid and corrosive in solution. Handle in a chemical fume hood using gloves, a lab coat and goggles.
1× Tris-EDTA (TE) buffer (Thermo Fisher Scientific, cat. no. 12090015)
Buffer QG (Qiagen, cat. no. 19055)
Buffer PE (Qiagen, cat. no. 19065)
10× T4 DNA Ligase buffer (New England Biolabs, cat. no. B0202S)
T4 DNA polymerase (New England Biolabs, cat. no. M0203S)
Taq DNA polymerase (New England Biolabs, cat. no. M0273S)
dNTP set (100 mM; New England Biolabs, cat. no. N0446S)
ATP (100 mM; Thermo Fisher Scientific, cat. no. R0441)
PEG 4000 (VWR, cat. no. A16151)
T4 PNK (New England Biolabs, cat. no. M0201S)
AMPure XP beads (Beckman Coulter, cat. no. A63881) ! CAUTION Because of potential variability between AMPure XP bead lots, it is recommended that your AMPure bead solution be calibrated. See manufacturer’s instructions or Supplementary Methods.
Quick Ligase with 2× Quick Ligase buffer (New England Biolabs, cat. no. M2200S)
NEBNext Multiplex Oligos for Illumina kit (includes NEB index primers, NEB adapter, and USER enzyme; New England Biolabs, cat. nos. E7335S/L, E7500S/L, E7710S/L, E7730S/L)
KAPA HotStart HiFi DNA polymerase with 5× KAPA HiFi buffer (Roche, cat. no. 07958889001)
Sodium acetate (NaOAc; Thermo Fisher Scientific, cat. no. BP333-500)
Agarose, molecular biology grade (Thermo Fisher Scientific, cat. no. BP160-100)
Ethidium bromide (Thermo Fisher Scientific, cat. no. 15585011)
Tris base (Tris; Fischer, cat. no. BP152-5)
Hydrochloric acid (Fisher Chemical, cat. no. A144-212) ! CAUTION Hydrochloric acid is very corrosive; handle in a chemical fume hood using gloves, a lab coat and goggles.
Glacial acetic acid (Sigma, cat. no. A6283) ! CAUTION Hydrochloric acid is very corrosive; handle in a chemical fume hood using gloves, a lab coat and goggles.
DNA ladder (100 bp; New England Biolabs, cat. no. N3231S/L)
-
Heterologous S. cerevisiae DNA spike-in
Prepared from crosslinked, MNase-digested and agarose gel extracted genomic DNA purified of protein/RNA and diluted to 10 ng/mL. We recommend yeast genomic DNA, but other organisms can be used if needed. Please refer to Supplementary Methods and Supplementary Fig. 2 for production protocol.
TBE buffer (Thermo Fisher Scientific, B52)
For embryo uliCUT&RUN experiments
Pregnant mare serum gonadotropin (PMSG; IDT Biologika)
Human chorionic gonadotropin (hCG; MSD Animal Health, cat. no. 140-927)
Bovine pancreatic trypsin inhibitor (BPTI; Sigma Aldrich, cat. no. A6106)
Hyaluronidase (Sigma Aldrich, cat. no. H4272)
Trypsin, 0.5% (vol/vol)/EDTA, 0.2% (vol/vol) (Thermo Fisher Scientific, 25300054)
KSOM medium (Millipore, cat. no. MR-020P-5F)
Mineral oil (Fisher, cat. no. 0121-1)
M2 medium (Sigma Aldrich, cat. no. M7167)
Acid Tyrode’s solution (Sigma Aldrich, cat. no. MR-004-D)
(Optional) Drug or microinjection reagents
For cell-based uliCUT&RUN experiments
Cell-specific medium for cell culture (DMEM and associated components for ES E14TG2a cells)
For alternative library build protocol
Isopropanol (Sigma Aldrich, cat. no. I9516) ! CAUTION 100% isopropanol is highly flammable; handle in a chemical fume hood using gloves, a lab coat and goggles.
For heterologous DNA spike-in production
37% (wt/vol) Formaldehyde (Thermo Fisher Scientific, cat. no. AC119690010) ! CAUTION Formaldehyde is flammable and can be toxic if in contact with skin. Handle in a chemical fume hood using gloves, a lab coat and goggles.
Glycine (Thermo Fisher Scientific, cat. no. G46-500)
NP-40, 10% (wt/vol) (Thermo Fisher Scientific, cat. no. 28324)
RNase A (Thermo Fisher Scientific, cat. no. EN0531)
Magnesium chloride (MgCl2; Thermo Fisher Scientific, cat. no. M33-500)
Equipment
Pipette aid (Drummond Scientific, cat. no. 4-000-100)
Standard laboratory materials such as serological pipettes and pipette tips
Laminar flow hood (Bakery Company, cat. no. SG404)
Incubator with temperature and atmosphere control (Thermo Fisher Scientific, cat. no. 51030284)
Sterile tissue culture plates (10 cm; Thermo Fisher Scientific, cat. no. 150464)
Sterile tissue culture plates (6 cm; Thermo Fisher Scientific, cat. no. 150462)
Micropipette set (Rainin, cat. no. 30386597)
Waterbath and/or thermomixer (Thermo Fisher Scientific, cat. no. FSGPD02 or Eppendorf cat. no. 5384000020)
Clear microfuge tubes (1.5 mL; Thermo Fisher Scientific, cat. no. 90410)
Phase lock tubes (Qiagen, cat. no. 129046)
DNA spin columns (Epoch Life Sciences, cat. no. 1920-250)
Tabletop vortexer (Thermo Fisher Scientific, cat. no. 02215414)
Minifuge (Benchmark Scientific, cat. no. C1012)
Tube magnetic rack (1.5 mL; Thermo Fisher Scientific, cat. no. 12321D)
Tube rotator (VWR, cat. no. 10136084)
Tube-compatible cold centrifuge (1.5 mL; Eppendorf, cat. no. 5404000537)
Thermocycler (Eppendorf, cat. no. 2231000666)
PCR tubes (200 μL; Eppendorf, cat. no. 951010022)
Centrifuge tubes, plastic (15 mL; Eppendorf, cat. no. 022362304)
Qubit fluorometer (Thermo Fisher Scientific, cat. no. Q33238)
Qubit 1× dsDNA HS Assay Kit (Thermo Fisher Scientific, cat. no. Q33230)
Qubit Assay tubes (Thermo Fisher Scientific, cat. no. Q32856)
Ice bucket
Electrophoresis equipment
Standard mouse dissection equipment, including a mouth pipette to manipulate embryos ! CAUTION Mouth pipetting is potentially hazardous and a potential source of contamination; perform mouth pipetting manipulations according to manufacturer’s safety instruction.
Cell sorter and associated training (BD FACSAria II cell sorter)
Computer with 64-bit processer and access to a supercomputing cluster
Software
bcl2fastq2, v2.20.0 (https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html; RRID: SCR_015058)
FastQC, v0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/; RRID: SCR_014583)
SAMtools63, v1.9 (http://www.htslib.org/download/; RRID: SCR_002105)
Bowtie 2 (ref.64), v2.3.5.1 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml; RRID: SCR_005476)
Picard, v2.18.12 (https://broadinstitute.github.io/picard/; RRID: SCR_006525)
deepTools65, v3.3.0 (https://deeptools.readthedocs.io/en/develop/index.html; RRID: SCR_016366)
HOMER66, v4.10.3 (http://homer.ucsd.edu/homer/; RRID: SCR_010881)
Reagent setup
Stock solutions
▲ CRITICAL We recommend preparing stock solutions prior to buffer preparation. All solutions should be prepared with nuclease free water at room temperature (RT, 22–25 °C) and filter sterilized or autoclaved after preparation as specified, unless otherwise stated. We also provide recommendations on their storage conditions.
1 M HEPES-KOH (pH 7.5 or 7.9; 100 mL). Add 23.8 g HEPES to 80 mL water. Add KOH to reach a pH of 7.5 or 7.9 (prepare each separately). Make up to a final volume of 100 mL with water, and filter sterilize. Can be stored at RT for up to a year.
1 M KCl (100 mL). Dissolve 7.455 g KCl into water, and make up to a final volume of 100 mL. Autoclave. Can be stored at RT up to a year.
10% (vol/vol) Triton X-100 (10 mL). Combine 1 mL 100% (vol/vol) Triton X-100 with 9 mL water. Do not filter sterilize. Can be stored at RT up to a year.
80% (vol/vol) Glycerol (100 mL). Combine 80 mL 100% (vol/vol) glycerol with 20 mL water. Can be stored at RT up to a year.
1 M MnCl (100 mL). Dissolve 12.58 g MnCl in water, and make up to a final volume of 20 mL. Filter sterilize and store at RT for up to a year protected from light.
30% (wt/vol) BSA (50 mL). Combine 15 g BSA with 40 mL water, and let shake at 4 °C overnight to dissolve. The next day, make up to a final volume of 50 mL, and filter sterilize. Store this solution at RT for up to a year.
0.2 M EGTA (100 mL). Dissolve 7.61 g EGTA in 80 mL water, and add NaOH to adjust pH to 8.0. Make up to a final volume of 100 mL. Filter sterilize and store at RT for up to a year.
10% (wt/vol) SDS (100 mL). Dissolve 10 g SDS in 80 mL water, and make up to a final volume of 100 mL. Filter sterilize and store at RT for up to a year.
2 M Spermidine (20 mL). Dissolve 5.8 g spermidine to 16 mL water, and make up to a final volume of 20 mL. Filter sterilize. Store at 4 °C for up to a year protected from light.
100% (vol/vol) EtOH (100 mL). Prepare aliquot of 50 mL 100% (vol/vol) EtOH in an airtight vessel. Can be stored at RT with a tight lid up to a month.
80% (vol/vol) EtOH (20 mL; prepare this fresh on the day of the assay). Add 16 mL 100% (vol/vol) to 4 mL water, and mix well. Can be stored at RT for the day.
0.5× TBE buffer (1 L). Mix 500 mL 1× TBE buffer with 500 mL water. Can be stored at RT for up to a year.
40% (wt/vol) PEG 4000 (20 mL). Dissolve 8 g PEG 4000 to 16 mL water, and make up to final volume of 20 mL. Filter sterilize. Can be stored at RT for up to a year.
3 M NaOAc (pH 5.2; 100 mL). Dissolve 24.61 g NaOAc into 80 mL water. Add glacial acetic acid to adjust pH to 5.2. Make up to a final volume of 100 mL, and filter sterilize. Can be stored at RT for up to a year.
0.01 M Tris-HCl (pH 8.0; 100 mL). Dissolve 0.12g Tris in 80 mL water. Use concentrated HCl to pH to 8.0. Make up to a final volume of 100 mL and filter sterilize. Can be stored at RT for up to a year.
Stock solutions for heterologous DNA spike-in generation
2.5 M Glycine (100 mL). Dissolve 18.75 g glycine in 80 mL water, and make up to a final volume of 100 mL. Filter sterilize and store at RT for up to a year.
1 M Tris-HCl pH 7.5 (100 mL). Dissolve 12.1 g Tris in 80 mL water. Use concentrated HCl to adjust pH to 7.5. Make up to a final volume of 100 mL and filter sterilize. Can be stored at RT for up to a year.
0.5 M EDTA (100 mL). Dissolve 18.61 g EDTA in 80 mL water. Add NaOH to adjust pH to 8.0. Make up to a final volume of 100 mL and filter sterilize. Can be stored at RT for up to a year.
1 M CaCl2 (100 mL). Dissolve 14.70 g CaCl2 in 80 mL water, and make up to a final volume of 100 mL. Filter sterilize the solution and store up at RT for up to a year.
1 M MgCl2 (100 mL). Dissolve 20.33 g MgCl2 in 80 mL water, and make up to a final volume of 100 mL. Filter sterilize the solution and store at RT for up to a year.
Buffers
Nuclear extraction (NE) buffer (50 mL). Combine 1 mL of 1 M HEPES-KOH (pH 7.9), 500 μL of 1 M KCl, 12.5 μL of 2 M spermidine, 500 μL of 10% (vol/vol) Triton X-100, 12.5 mL of 80% (vol/vol) glycerol and 35.49 mL of nuclease-free water. This buffer can be stored up to a year at 4 °C.
Binding buffer (20 mL). Combine 400 μL of 1 M HEPES-KOH, pH 7.9, 200 μL of 1 M KCl, 20 μL of 1 M CaCl2, 20 μL of 1 M MnCl2 and 19.36 mL of nuclease-free water. This buffer can be stored up to a year at 4 °C.
Wash buffer (100 mL). Combine 2 mL of 1 M HEPES-KOH (pH 7.5), 3 mL of 5 M NaCl, 25 μL of 2 M spermidine, 333 μL of 30% (wt/vol) BSA and 94.64 mL of nuclease-free water. This buffer can be stored up to a year at 4 °C.
2XRSTOP+ buffer (5 mL). Combine 200 μL of 5 M NaCl, 200 μL of 0.5 M EDTA, 100 μL of 0.2 M EGTA and 4.46 mL of nuclease-free water. This buffer can be stored up to a year at RT.
Procedure
Obtaining blastocysts or single cells
-
1On the day of experiments, prepare these buffers prior to beginning as follows:
- Prepare aliquot of 500 μL NE buffer per blastocyst or 200 μL NE buffer per single cell in a vessel (such as a 15 mL or 50 mL conical tube, depending on the number of samples). Add 1× protease inhibitors and mix well by inverting. Keep on ice. This buffer is used during sample harvest and nuclear extraction.
- Make fresh blocking buffer by adding 20 μL 0.5 M EDTA and 1× protease inhibitors to 5 mL wash buffer. Mix well by inverting. Keep on ice. Make 1 mL per blastocyst or single cell. This buffer is used during nuclear extraction.
- Prepare aliquot of 5 mL wash buffer per blastocyst or single cell in a vessel (such as a 50 mL conical tube). Add 1× protease inhibitors and mix well by inverting. Keep on ice. This buffer is used from nuclear extraction to chromatin digestion.
- Prepare a fresh dilution of 80% (vol/vol) EtOH from 100% (vol/vol) EtOH. This is used during sample purification using AMPure beads.
-
2We outline several options for obtaining starting material for uliCUT&RUN. Fresh blastocysts can be either harvested directly from mice (option A) or collected as zygotes and cultured to the blastocyst stage (option B). To obtain single cells from cells in culture, follow option C, and to obtain single cells from tissue, follow D.
- Harvest fresh blastocyst from mice ● Timing ~6 d
-
Superovulate the female mice. Briefly, inject 5–10 IU PMSG into the females; 46–48 h later, inject 5 IU hCG (or the recommended dosage for your strain) and allow the mice to copulate overnight. Confirm the copulation the next morning by the presence of a mating plug (this morning timepoint is Day 0.5).▲ CRITICAL STEP Use the recommended amount of hCG dosage for your mouse strain. See Luo et al. and Jackson Laboratories (https://www.jax.org/news-and-insights/1998/july/superovulation-technique#) for information on estimating hCG dosage if needed67.
- Isolate the impregnated females in a separate enclosure for ~72 h.
- At 84 h postfertilization (Day 3.5), sacrifice the females, then dissect and place the uterus into a 6 cm plate.
- Insert a blunt syringe filled with PBS into the uterine horn, and inject enough 1× PBS to fully expand it.
- While the uterine horn is expanded, make an excision at the section of the uterine horn close to the oviduct. The pressure from the uterine horn expansion will flush the blastocyst out into the plate with 1× PBS.
- Repeat this process with the remaining uterine horn. This process should yield 20–25 blastocysts per female mouse (pending mouse line; for example, in an FVB mouse line you would obtain this number if mice have been superovulated).
-
Transfer harvested blastocysts via mouth pipetting to a new dish for blastocyst preparation (Step 2).▲ CRITICAL STEP Harvest an additional blastocyst for use as the control sample (can be either no antibody or IgG).
-
- Harvest zygotes and culture to the blastocyst stage (recommended if microinjections or drug treatment are being used) ● Timing ~6 d
- Superovulate the females as above, and allow the mice to copulate overnight. Confirm the copulation the next morning by the presence of a mating plug. (This morning timepoint is Day 0.5.)
-
In a 6 cm plate, place several droplets of KSOM medium into linear groupings of 4+ droplets (~15 μL of total droplet volume; the number of droplets depends on the number of zygotes harvested). The first three droplets will be used for pre-equilibrated KSOM medium washes, while the fourth (and more) will be used to incubate the zygotes to the blastocyst stage. Place a drop of mineral oil (suitable for embryo culture) over each KSOM medium droplet, and place the 6 cm plate into a 37 °C 5% (vol/vol) CO2 incubator for at least 1 h (or up to overnight) to pre-equilibrate KSOM medium.▲ CRITICAL STEP If zygotes are to be incubated with a drug, compounds can be solubilized in DMSO or water and diluted to working concentration in pre-equilibrated KSOM medium to be used at this stage.
- On a separate 6 cm plate, make a row of three droplets of M2 medium, one droplet of hyaluronidase (1 mg/mL) and three additional droplets of M2 medium. Each droplet should contain ~15 μL of liquid.
- Sacrifice the females, dissect the oviduct from the uterus and place oviduct into a 6 cm plate.
- Submerge the oviduct in ~20–50 μL prewarmed (to 37 °C) M2 medium.
- (vi) Carefully open the oviduct. Zygotes will be released into M2 medium.
- Using a mouth pipette, carefully transfer the zygotes to the first M2 medium droplet to perform the first wash. This will remove any oviduct debris. Zygotes can be transferred in groups of 1–15.
- Carefully transfer the zygotes to the second, then third M2 medium droplet to perform two additional washes.
- Transfer the zygotes to the hyaluronidase (1 mg/mL) droplet, and incubate for 1–2 min at RT. During this incubation, the cumulus cells that surround the zygote are removed.
-
After incubation, transfer the zygotes through the next three M2 medium droplets to perform three additional washes, to remove cumulus cell debris.▲ CRITICAL STEP If zygotes are to be microinjected, perform microinjections in M2 medium directly following the washes to remove cumulus cell debris. Incubate zygotes in M2 medium for 5 min, then proceed as detailed below.
- Retrieve KSOM medium-containing plate from incubator.
- To a single pre-equilibrated KSOM droplet, add 5–15 zygotes to perform a KSOM wash. By batch mouth pipetting, transfer zygotes through the two additional KSOM droplets to perform additional KSOM medium washes.
- Transfer the batch of zygotes to the final KSOM medium droplet. When possible, let zygotes develop together to increase the likelihood of zygotes advancing to the blastocyst stage.
- Place the plate back into the 37 °C 5% (vol/vol) CO2 incubator, and wait for zygotes to develop to the blastocyst stage, ~72 h.
- At Day 3.5, visually confirm the blastocyst development under a microscope. Note that some zygote treatments might result in delayed development.
- On a new 6 cm plate, place three droplets of M2 medium, ~15 μL each.
- Using standard embryo mouth pipetting, transfer the blastocysts to the first M2 medium droplet, then through the remaining two droplets to perform a total of three M2 medium washes.
-
Proceed directly to the blastocyst preparation stage of this protocol (Step 2).▲ CRITICAL STEP Harvest an additional blastocyst for use in the control sample (can be either no antibody or IgG).
- Obtain single cells from cultured cells ● Timing ranges based on sorting conditions; ~30 min to 2 h
- Grow a suspension culture or adherent cells to the cell-type-specific optimal density in the user’s preferred growth conditions. Any experimental manipulation should be performed prior to cell harvest.
- For adherent cells in a plate, aspirate away the medium and use a disassociation agent (such as trypsin) to detach cells. Transfer the dissociated cells (or cell suspension, for cells grown in suspension) to a 15 mL tube and centrifuge at 600g for 5 min at 4 °C.
- Gently resuspend the cells in the recommended volume PBS plus 1% (vol/vol) FBS for your cell type.
-
Using a FACS instrument with the recommended settings for your cell type, sort the single cells into individual wells of the 96-well plate containing 100 μL NE buffer.▲ CRITICAL STEP Harvest additional single cells for use as the control sample (either no antibody or IgG).
- Proceed immediately to the nuclear extraction at Step 10.
- Obtain single cells from a tissue sample ● Timing ranges based on sorting conditions; ~30 min to 2 h
- Obtain a single-cell suspension in the recommended volume of PBS plus 1% (vol/vol) FBS according to the tissue extraction protocol of the user’s preference (e.g., mechanical or proteolytic extraction).
- Add 100 μL NE buffer to each well of a 96-well plate.
-
Using a FACS instrument with the recommended settings for your cell type, sort the single cells into individual wells of the 96-well plate containing 100 μL NE buffer.▲ CRITICAL STEP Harvest additional single cells for use as the control sample (either no antibody or IgG).
- Proceed immediately to the nuclear extraction at Step 10.
Blastocyst preparation ● Timing 15 min
-
3
Acquire the desired number of appropriately staged blastocysts (obtained by harvest, Step 2A or culture, Step 2B).
-
4
In a 6 cm plate, make a row of two droplets of M2 medium, one droplet of acid Tyrode’s solution and two additional droplets of M2 medium. Each droplet should contain ~15 μL of liquid. Make a row of droplets per group of blastocysts to be processed (keeping differentially treated blastocysts separate).
-
5
In groups of up to five blastocysts, mouth pipette the blastocysts into the first M2 medium droplet to wash. Transfer the blastocysts to the second M2 medium droplet for a second wash.
-
6
Transfer washed blastocysts to the acid Tyrode’s solution droplet, and incubate for 1 min. During this incubation, the zona pellucida is digested.
-
7
Transfer the blastocysts to the next M2 medium droplet, and then the two M2 medium droplets for two additional M2 medium washes.
-
8
From the final M2 medium droplet, carefully mouth pipette a single blastocyst into a 1.5 mL microfuge tube containing 300 μL NE buffer.
-
9
Proceed to the nuclear extraction immediately. Alternatively, refer to Box 1 in the case of a limited number of blastocysts or if multiple assays are to be conducted on the same blastocyst.
? TROUBLESHOOTING
Box 1 |. Procedure for disaggregating blastocysts.
Procedure
After acid Tyrode’s solution incubation, transfer an individual blastocyst into 3 μL 0.5% (vol/vol) trypsin/0.2% (vol/vol) EDTA.
Incubate the blastocyst in 0.5% (vol/vol) trypsin/0.2% (vol/vol) EDTA for 3 min, with gentle but frequent mouth pipetting to disaggregate the cells.
Add 3 μL 1 mg/mL BPTI to stop the reaction.
Gently subdivide the cell suspension into a desired number of equal parts into separate 1.5 mL microfuge tubes containing 300 μL NE buffer.
Proceed to nuclear extraction immediately.
Nuclear extraction ● Timing 45 min
-
10
Centrifuge the 1.5 mL tubes containing individual blastocysts (Step 2A or B) or the 96-well plate containing single cells (Step 2C or D) at 600g for 2 min at 4 °C.
-
11
Incubate on ice for 10 min.
? TROUBLESHOOTING
-
12
Prepare the ConA bead mixture during the 10 min incubation. To a 1.5 mL microfuge tube, add 20 μL ConA bead slurry per blastocyst (for blastocyst samples obtained from Step 1A or B) or add 15 μL ConA bead slurry per single cell (for single cells from Step 1C or D) to batch prepare for multiple samples in a single microfuge tube.
-
13
Add 850 μL binding buffer, and mix well by pipetting up and down several times with a P1000. Be careful to avoid bubbles.
-
14
Place the microfuge tube into the magnetic stand, and let sit until solution becomes clear because of magnetic beads adhering to the side of the vessel (~2 min).
-
15
Once cleared, while leaving the microfuge tube in the magnetic stand, remove supernatant with a P1000.
-
16
Remove the microfuge tube from the magnetic stand, and resuspend the ConA beads in 1 mL binding buffer.
-
17
Briefly spin the microfuge tube in a MyFuge mini centrifuge at RT (~3 s), and place the tube back into the magnetic stand and allow the solution to clear (~2 min).
-
18
Perform one additional bead wash with 1 mL binding buffer. Resuspend the ConA beads in 150 μL binding buffer per blastocyst or 50 μL binding buffer per single cell to complete the ConA bead mixture.
▲ CRITICAL STEP Be careful to remove all of the supernatant without disturbing beads to reduce bead loss.
? TROUBLESHOOTING
-
19Add the ConA beads as described in Option A for blastocyst samples or (B) for single-cell samples:
- For blastocyst samples
- While gently vortexing (setting 3 out of 10 on a vortexer), slowly add 150 μL ConA bead mixture to each sample and incubate at RT for 10 min.
- For single-cell samples
-
Add 50 μL ConA bead mixture to each sample well, mixing thoroughly but gently via pipetting, and transfer the samples to individual 1.5 mL microfuge tubes and incubate them at RT for 10 min.▲ CRITICAL STEP Mixing gently and effectively is important to ensure efficient nucleus binding. We also recommend pipetting up and down upon ConA bead addition.? TROUBLESHOOTING
-
-
20
Place the samples into the magnetic rack, allow them to clear (~5 min) and remove the supernatant with a pipette.
▲ CRITICAL STEP Now that nuclei are bound to beads, complete binding of magnetic beads to the magnet is absolutely essential to reduce DNA loss.
-
21
Remove the samples from the magnetic stand, and gently resuspend the beads in 1 mL blocking buffer.
▲ CRITICAL STEP Complete and gentle mixing of nuclei with blocking buffer is critical to protect nuclei from preemptive pA-MNase digestion.
? TROUBLESHOOTING
-
22
Incubate the samples for 5 min at RT.
-
23
Place the samples onto the magnetic stand, and let the solution clear on the magnetic rack (~5 min). Then perform one wash with 1 mL wash buffer.
-
24
Remove the supernatant and gently resuspend the beads 250 μL wash buffer for blastocysts or 125 μL wash buffer for single cells, so as not to generate bubbles.
Antibody incubation ● Timing 2 h 10 min
-
25
To prepare the antibody solution, in a separate microfuge tube, add the optimized amount of antibody to 250 μL wash buffer per blastocyst or to 125 μL wash buffer per single cell. We generally recommend starting with 1:100 final dilution (5 μL of antibody for blastocysts and 2.5 μL of antibody for single cells). However, it is important to note that some antibodies could benefit from higher or lower concentrations, so a titration series experiment might be necessary.
▲ CRITICAL STEP For multiple experiments using the same antibody, these antibody solutions can be batch prepared.
▲ CRITICAL STEP For the no-antibody control sample, a no-antibody solution should be prepared. If using an IgG antibody control, prepare IgG antibody solution for use as described above.
? TROUBLESHOOTING
-
26
Mix the antibody solution well by pipetting up and down carefully, so as not to generate bubbles.
-
27
While gently vortexing, add 250 μL of the antibody solution to each blastocyst or 125 μL of antibody solution to each single cell.
▲ CRITICAL STEP Thoroughly but gently mix by pipetting into the tube while vortexing on low setting. Draw and expel liquid into the pipette tip slowly for additional mixing.
▲ CRITICAL STEP For no-antibody control, add either 250 μL of wash buffer for blastocyst controls or 125 μL wash buffer for single-cell controls.
? TROUBLESHOOTING
-
28
Place the samples onto the tube rotator and rotate at 4 °C for 2 h.
? TROUBLESHOOTING
-
29
After the incubation, place the samples on the magnetic rack, let the solution clear (~5 min) and perform one wash with 1 mL wash buffer.
-
30
Resuspend the samples in 250 μL wash buffer for blastocyst samples or 125 μLwash buffer for single-cell samples.
pA-MNase or pAG-MNase targeting and chromatin digestion ● Timing 1 h 15 min
-
31
To prepare the pA-MNase mixture, add the optimized amount of pA-MNase or pAG-MNase to 250 μL wash buffer per blastocyst or 125 μL wash buffer per single cell in a microfuge tube. The final dilution of pA-MNase/pAG-MNase will depend on the enzymatic activity of user-generated recombinant pA-MNase/pAG-MNase or according to manufacturer’s instructions from commercially sourced pA-MNase/pAG-MNase.
-
32
Mix the pA-MNase or pAG-MNase solution well by pipetting up and down.
▲ CRITICAL STEP If purified pA-MNase is used, the enzymatic activity needs to first be assayed. See Supplementary Methods for details; we typically find 1:200 or 1:400 dilution works appropriately. If commercial pAG-MNase, the amount to be added will be specified by manufacturers.
▲ CRITICAL STEP Thoroughly but gently mix by pipetting into the tube while vortexing on low setting. Draw and expel liquid into the pipette tip slowly for additional mixing.
-
33
While vortexing gently, add 250 μL pA-MNase mixture to each blastocyst or 125 μL pA-MNase mixture to each single cell.
? TROUBLESHOOTING
-
34
Place the samples in a tube rotator, and rotate for 2 h at 4 °C.
-
35
Place the samples onto the magnetic rack, allow the solutions to clear (~5 min) and perform one wash with 1 mL wash buffer.
-
36
Resuspend each sample in 150 μL wash buffer.
-
37
Place the samples on ice/water slurry to equilibrate to 0 °C for 5–10 min.
▲ CRITICAL STEP Slurry is a mixture of ice and water with enough ice to keep tubes stable but enough water so that tubes are entirely submerged and covered in ice-cold liquid.
-
38
Quickly remove the samples from the ice/water slurry, add 3 μL 100 mM CaCl2 while gently vortexing, then flick the tubes two to three times and place the samples back into the ice bath for 30 min.
▲ CRITICAL STEP Mixing well at this step is critical for efficient and uniform digestion. When adding CaCl2 to samples, take care to hold firmly directly below the lip of the microfuge tube so as to not contaminate but not warm the sample with body heat.
? TROUBLESHOOTING
-
39
During the 30 min incubation, make 2XRSTOP buffer. To 5 mL 2XRSTOP buffer, add 25 μL RNase A (10 mg/mL) and 10 μL glycogen (20 mg/mL). For uliCUT&RUN with blastocysts, add 2 μL heterologous gDNA spike-in (10 ng/μL). For uliCUT&RUN with single cells, add 0.5 μL heterologous gDNA spike-in (10 ng/μL). Mix well by vortexing.
-
40
At 30 min exactly (and in the order of CaCl2 addition), add 150 μL of 2XSTOP+ buffer to each sample.
DNA isolation ● Timing 1 h 30 min
-
41
Incubate the samples at 37 °C for 20 min.
-
42
Centrifuge the samples at 16,000g for 5 min at 4 °C. Transfer the supernatant to a new microfuge tube.
-
43
Add 3 μL 10% (vol/vol) SDS and 2.5 μL proteinase K, and mix the samples well by inversion. Quickly spin each sample with a MyFuge at RT to collect all liquid to the bottom of the tube, and incubate at 70 °C for 10 min.
-
44
Add 300 μL PCI to each sample, and vortex well for 10–15 s.
-
45
Transfer the samples to phase lock tubes, and centrifuge at 16,000g for 5 min at 4 °C.
-
46
Add 300 μL chloroform directly to the phase lock tube where the supernatant containing soluble DNA is above the matrix, and invert well for 10–15 s. Centrifuge at 16,000g for 5 min at 4 °C.
-
47
Carefully transfer the aqueous layer from each sample to a new 1.5 mL microfuge tube.
-
48
To each sample, add 5 μL glycogen (20 mg/ml), 30 μl 3 M NaOAc pH 5.2 and 750 μL 100% (vol/vol) EtOH, and mix well by vortexing for 10–15 s.
-
49
Incubate at −20 °C for at least 20 min, or overnight if convenient.
■ PAUSE POINT Samples at this point can be safely stored at −20 °C for up to a year if needed.
-
50
Spin the samples at 16,000g for 30 min at 4 °C.
-
51
Carefully remove the supernatant without disturbing the sample pellet.
-
52
Add 1 mL 80% (vol/vol) EtOH, vortex briefly and spin the samples at 16,000g for 5 min at 4 °C.
-
53
Carefully remove the supernatant without disturbing the pellet.
-
54
Allow the samples to air dry with tube caps open for ~5 min.
-
55
Resuspend the DNA pellet in 36.5 μL 0.1× TE.
■ PAUSE POINT Samples at this point can be safely stored at −20 °C for up to a year if needed.
? TROUBLESHOOTING
Library build
▲ CRITICAL For the entirety of the library build, keep the samples on ice when not in use/in an incubation step, and assemble all reactions on ice unless otherwise stated.
End repair ● Timing 1 h
-
56
Allow 40% (vol/vol) PEG 4000 to come to RT before use.
-
57
Transfer each sample to a 200 μL PCR tube.
-
58
Dilute T4 DNA polymerase 1:20 (0.5 U/μL final) in 17 μL nuclease-free water, 2 μL 10× T4 DNA Ligase buffer and 1 μL T4 DNA polymerase.
-
59Prepare the end repair/3’ master mix per sample as follows:
Reagent Volume (μL) Final concentration 10× T4 DNA Ligase buffer 5 1× dNTPs (10 mM each) 2.5 0.5 mM each ATP (10 mM) 1.25 0.25 mM 40% (vol/vol) PEG 4000 3.13 2.5% (vol/vol) T4 PNK (10 U/μL) 0.63 0.125 U/μL Diluted T4 DNA polymerase (0.5 U/μL) 0.5 0.0025 U/μL Taq DNA polymerase 0.5 — Total volume 13.5 — -
60
Add 13.5 μL end repair/3’A master mix to each sample for a final volume of 50 μL, and mix well by pipetting.
-
61
Spin the samples briefly in a MyFuge at RT to collect any mix from the sides of the tubes.
-
62
On a thermocycler, with a heated lid set to >20 °C, incubate the samples at 12 °C for 15 min, then 37 °C for 15 min and finally 72 °C for 20 min.
-
63
Place the samples on ice, and proceed immediately with the adapter ligation.
Adapter ligation ● Timing 35 min
-
64
Allow 2× Quick Ligase buffer to come to RT before use. Mix well by vortexing.
-
65
Dilute the NEBNext adapter to 1.5 μM by diluting 1:10 in adapter dilution mix (included in NEBNext kit, cat. no. E7103).
? TROUBLESHOOTING
-
66Assemble the adapter ligation master mix per sample as follows:
Reagent Volume (μL) 2× Quick Ligase buffer 55 Quick Ligase 5 1.5 μM NEB adapter 5 Master mix volume 65 -
67
Add 65 μL adapter ligation master mix to each sample for a total volume of 115 μL, and mix well by pipetting.
-
68
Incubate the samples at 20 °C (without a heated lid) for 15 min.
-
69
Place the samples directly on ice.
-
70
To each sample, add 3 μL USER enzyme. Mix each sample well by pipetting up and down.
-
71
Incubate the samples at 37 °C for 15 min with a heated lid set at ≥45 °C.
-
72
Proceed immediately into the AMPure XP bead purification.
AMPure XP bead purification ● Timing 30 min
▲ CRITICAL Owing to potential variability between AMPure XP bead lots, it is recommended that your AMPure bead solution be calibrated according to the manufacturer’s instructions. The fragment range being selected for is 100–750 bp, so your beads should be able to select within this range.
▲ CRITICAL Allow AMPure XP beads to equilibrate at RT for ~30 min to decrease viscosity of PEG in solution, and vortex well prior to use.
-
73
Transfer the samples into 1.5 mL microfuge tubes.
-
74
To each sample, add 39 μL (0.33×) AMPure XP beads.
▲ CRITICAL STEP Adding the specified amount of AMPure XP beads at this step is critical to sample purification, as DNA fragment size selection is dependent upon the ratio of AMPure XP bead to sample volume. After adding the AMPure XP beads, the final amount of PEG in solution is ~11.18% when considering previously added PEG.
? TROUBLESHOOTING
-
75
Thoroughly mix the beads with the samples by pipetting up and down and vortexing briefly.
▲ CRITICAL STEP Mixing thoroughly is critical to ensure efficient DNA fragment binding.
-
76
Perform a brief spin in a MyFuge at RT (do not pellet the beads), and incubate the samples at RT for 15 min to allow DNA to bind to the beads.
-
77
Place the samples in the magnetic rack, and allow the solution to clear (~5–10 min).
-
78
Leaving the samples in the magnetic rack, carefully remove the supernatant without disturbing the beads.
-
79
Add 200 μL 80% (vol/vol) EtOH, and incubate the samples on the magnetic rack until the solution clears (~2 min).
-
80
Carefully remove and discard the supernatant without disturbing the beads.
-
81
Perform an additional wash with 200 μL 80% (vol/vol) EtOH while leaving the samples in the magnetic rack.
-
82
Briefly spin the samples in a MyFuge at RT to collect residual EtOH from the sides of the vessel.
-
83
Place the samples onto the magnetic rack, let the solution clear (~2 min) and remove any residual EtOH without disturbing the beads.
-
84
Let the samples sit in the magnetic rack with the lids open for ~2 min to air dry.
▲ CRITICAL STEP Removing all residual EtOH is critical to promote sufficient DNA recovery from beads. However, do not overdry beads, as this decreases DNA recovery efficiency. Beads will have a ‘matte’ appearance when they are ready for elution.
-
85
Remove the samples from the magnetic rack, and add 29 μL 10 mM Tris-HCl, pH 8.0.
-
86
Thoroughly resuspend the beads by pipetting up and down.
▲ CRITICAL STEP Complete resuspension of beads is essential for complete elution of DNA from beads.
-
87
Incubate the samples at RT for 5 min to elute the DNA from the AMPure XP beads.
-
88
Place the samples onto the magnetic rack, and allow the solution to clear (~5 min).
-
89
Once cleared, transfer 27.5 μL of the supernatant to a new 200 μL PCR tube, while being careful to not collect the beads.
■ PAUSE POINT Samples at this point can be safely stored at −20 °C for up to a year if needed.
Library enrichment ● Timing 25 min
▲ CRITICAL Sample index assignment occurs at this step. Each sample should be carefully assigned a unique NEB i7 index.
-
90
Place the samples on ice (or thaw on ice if necessary).
-
91
Prepare fresh 1:10 dilutions for the universal i5 index and all i7 index primers to be used by diluting each primer with nuclease-free water. Prepare 5 μL of universal i5 index dilution for each sample, and at least 5 μL of i7 each index dilution to be used.
? TROUBLESHOOTING
-
92
Add 5 μL of the appropriate i7 index primer dilution to the assigned sample.
▲ CRITICAL STEP Take care to add the correctly assigned i7 index primer dilution to each sample. This is critical to ensure the proper assignment of sequencing reads to the appropriate sample during data analysis.
-
93Prepare the library enrichment master mix per sample as follows:
Reagent Volume (μL) Final concentration 5× KAPA HiFi buffer 10 1× dNTPs (10 mM total) 1.5 0.3 mM Universal i5 index dilution 5 1.5 mM KAPA HotStart HiFi DNA polymerase (1 U/μL) 1 0.2 U/μL Master mix final volume 17.5 — -
94
To each sample, add 17.5 μL library enrichment master mix to a total volume of 50 μL. Mix the samples well by pipetting up and down, and briefly spin with a MyFuge at RT.
-
95Run the following program on a thermocycler:
Number of cycles Denature Anneal Extend 1 98 °C, 45 s — — 18 for blastocyst or 22 for single cell 98 °C, 15 s 60 °C, 10 s — 1 — — 72 °C, 60 s — — — 4 °C, hold ? TROUBLESHOOTING
-
96
Proceed directly to library purification.
Library purification and quality check ● Timing 2 h
-
97
Prepare a 15 cm, 1.5% (wt/vol) agarose (TBE) gel with the desired intercalating agent. We use ethidium bromide as an intercalating agent and 0.5× TBE buffer for our gels and as running buffer, but other intercalating agents and 1× Tris acetate EDTA can be used as alternatives if desired.
-
98
Transfer the samples to fresh 1.5 mL microfuge tubes.
▲ CRITICAL STEP Allow the AMPure XP beads to incubate at RT for at least 30 min prior to use.
-
99
Add 60 μL (1.2×) of AMPure XP beads to each sample. Mix the samples well by pipetting up and down, and perform a brief spin with a MyFuge at RT to collect the entire sample from tubes.
? TROUBLESHOOTING
-
100
Incubate the samples for 15 min at RT. Place the samples into the magnetic stand, and incubate at RT until each sample clears (~5 min).
-
101
Carefully remove the supernatant without disturbing the beads, and perform a wash with 200 μL of 80% (vol/vol) EtOH without disturbing the beads.
-
102
Perform two additional 80% (vol/vol) EtOH washes.
-
103
Briefly spin the samples with a MyFuge at RT, and place the samples back onto the magnetic rack.
Allow the beads to readhere to the sides of the tubes, and carefully remove residual EtOH.
▲ CRITICAL STEP Removing all residual EtOH is critical to ensure sufficient DNA recovery.
-
104
On the magnetic rack, allow the samples to air dry for ~2 min.
-
105
Carefully resuspend each sample in 27.5 μL nuclease-free water by pipetting up and down.
-
106
Incubate the samples for ~5 min at RT to elute the DNA library from the beads.
-
107
Place the samples back into the magnetic rack, and incubate until the solution clears (~2 min).
-
108
Transfer 25 μL of each sample to a fresh 1.5 mL tube, and place these tubes on ice.
■ PAUSE POINT Samples at this stage can be stored at −20 °C for up to a year until needed.
-
109
Assemble and ready the gel electrophoresis apparatus.
-
110
For each sample, mix 5 μL of sample with DNA loading dye and load samples onto the agarose gel along with 3 μL NEB 100 bp DNA ladder.
-
111
Run the gel at 100–120 V for ~60 min. Under UV light, visualize the gel to confirm the absence of adapter dimer or primer dimer in the library build for each sample. See Fig. 4a for an example of a well-constructed blastocyst library.
? TROUBLESHOOTING
Sample preparation and sequencing ● Timing 1–2 d
-
112
Quantify the sample libraries with the Qubit 1× HS dsDNA quantification kit, and pool them for multiplexing according to standard Illumina protocols. For a more detailed description of library pooling and multiplexing, refer to https://www.illumina.com/documents/products/datasheets/datasheet_sequencing_multiplex.pdf.
-
113
If using an Illumina NextSeq, sequence libraries using the 75 mid- or high-output cartridge, doing 2 × 42 paired-end sequencing. We have found that a maximum depth of 1,000,000 (uniquely) mapped (deduplicated) reads is sufficient to determine TF occupancies in blastocysts, and 100,000 (uniquely) mapped (deduplicated) reads are sufficient for single cells; however, sequencing depth might need to be optimized for your protein of interest.
Data analysis ● Timing 1 d to 1 week
-
114
The output of an Illumina sequencing run is a BCL file, containing all sequenced reads and associated index assignments. Using this BCL file as input, use bcl2fastq to demultiplex, trim adapter sequences and create paired fastq files (R1 and R2). This step might not be necessary in most contexts as it is often automated using the Illumina BaseSpace processing.
-
115
Once demultiplexed, use FastQC to assess the quality of each fastq file. See the developer’s website for examples of high-quality and low-quality data reports (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). We recommend not to proceed with low-quality fastq files, as these largely limit the applications in the downstream analyses because of the small number of sequencing reads per sample.
-
116
Next, use bowtie 2 to align paired fastq files for each sample with the sample reference genome (such as mm10 for mouse) and the spike-in control reference genome (such as sacCer3 for S. cerevisiae). Refer to the script below as an example.
Example bowtie 2 script
$ bowtie2 -q -N 1 -X 1000 -x /path/to/samplereferencegenome -1 /path/to/sample1_R1.fastq -2 /path/to/sample2_R2.fastq -S sample1_sample.sam $ bowtie2 -q -N 1 -X 1000 -x /path/to/spike_in_referencegenome -1 /path/to/sample1_R1.fastq -2 /path/to/sample2_R2.fastq -S sample1_spikein.sam
? TROUBLESHOOTING
-
117
Using Picard, SAMtools and a custom script, remove PCR duplicates and low-quality reads (MAPQ<10) from each sample, and then generate coordinate-sorted alignment files and fragment size distribution files. Refer to script below as an example.
Example Picard/SAMtools/custom script
$ java -Xmx4g -jar /path/to/picard.jar SortSam INPUT=sample1_sample. sam OUTPUT=sample1_sample_Picard.bam VALIDATION_STRINGENCY=LENIENT TMP_DIR=/tmp SORT_ORDER=coordinate $ java -Xmx4g -jar /path/to/picard.jar MarkDuplicates INPUT=sample1_sample_Picard.bam OUTPUT=sample1_sample_Picard2.bam VALIDATION_STRINGENCY=LENIENT TMP_DIR=/tmp METRICS_FILE=dup.txt REMOVE_DUPLICATES=true $ samtools view -h -o sample1_sample_Picard2.sam sample1_sample_Picard2.bam $ samtools view -Sq 10 sample1_sample_Picard2.sam > sample1_sample_Picard2_filtered.sam $ perl -e ‘ $col=8; while (<>) { s/\r?\n//; @F = split /\t/, $_; $val = $F [$col]; if (! exists $count{$val}) { push @order, $val } $count{$val}++; } foreach $val (@order) { print “$val\t$count{$val}\n” } warn “\nPrinted number of occurrences for “, scalar(@order), “ values in $. lines.\n\n”; sample1_sample_Picard2_filtered.sam > sample1_sample_Picard2_ filtered_unique_reads.txt $ perl -e ‘ $col=8; while (<>) { s/\r?\n//; @F = split /\t/, $_; $val = $F [$col]; if (! exists $count{$val}) { push @order, $val } $count{$val}++; } foreach $val (@order) { print “$val\t$count{$val}\n” } warn “\nPrinted number of occurrences for “, scalar(@order), “ values in $. lines.\n\n”; ‘ sample1_spikein.sam > sample1_spikein_unique_reads.txt -
118
At this stage, the analysis will have generated SAM and coordinate-sorted BAM alignment files filtered of PCR duplicates and low-quality mapped reads (e.g., sample1_sample_Picard2_filtered.sam), and a text file containing the sizes of all fragments obtained from the sample or spike-in (e.g., sample1_spikein_unique_reads.txt) aligned to the respective reference genome. As a quality control, we recommend inputting these text files into a spread sheet program such as Microsoft Excel or R to quickly check the fragment size distribution is as expected. See Fig. 4b,c for an example.
-
119
With the SAM alignment files, use the following lines of custom code to create size class-filtered SAM files. This step aids to enrich for DNA fragment lengths of the expected size bound by the protein of interest and to decrease background noise. For TFs and histone modifications, we recommend sorting between 1–120 bp and 150–500 bp, respectively. This might need to be optimized for your protein of interest. Refer to the following code as an example of how to sort for fragments 1–120 bp.
Example size class generation script
$ awk ‘ $9 <= 120 && $9 >= 1 || $9 >= -120 && $9 <= -1 ‘ sample1_sample_filtered.sam > sample1_sample_1_120.sam $ cp bowtie2.header sample1_sample_1_120.header $ cat sample1_sample_1_120.sam >> sample1_sample_1_120.header $ rm sample1_sample_1_120.sam $ mv sample1_sample_1_120.header sample1_sample_1_120.sam $ samtools view -S -t /path/to/chrom.sizes -b -o sample1_sample_1_120.bam sample1_sample_1_120.sam $ rm 10000_ sample1_sample_1_120.sam
-
120
The size class-sorted BAMs are used as inputs for downstream programs that should be selected with the user’s goal in mind. If data visualizations of factor occupancies over annotated genomic loci are needed, we recommend deepTools. Data visualizations are frequently used in our lab to efficiently analyze uliCUT&RUN data. We utilize deepTools for spike-in normalization as well. If other data visualization software is used, we recommend still utilizing deepTools to generate spike-in normalized bigwig files, although other options are available. Another common analysis performed once a protein’s localization has been defined is motif calling, which we conduct with HOMER. Other analysis programs can be utilized as desired but might need to be optimized for uliCUT&RUN analysis. As examples, we have provided two analyses. The first shows how to normalize samples to spike-in read counts and generate data visualizations of factor occupancy over annotated TSSs with deepTools. The second demonstrates how to perform motif calling analysis with HOMER.
Factor occupancy visualization over TSSs with deepTools
$ icpm_sample1=1(sum(sample1_spikein_unique_reads.txt$2)/1000000)) $ bamCoverage --bam sample1_sample_1_120.bam -scaleFactors $icpm_sample1 -o sample1_sample_1_120.bw $ bamCoverage --bam sample1_sample_1_120.bam -o sample1_sample_1_120.bw $ computeMatrix reference-point -R /path/to/TSSs.bed -S $ -o sample1_sample_1_120.mat \ --referencePoint TSS -b 2000 -a 2000 bs 20 $ plotHeatmap -m sample1_sample_1_120.mat --zMin -3 --zMax 3 --colorMap ‘seismic’ \ --whatToShow ‘plot, heatmap and colorbar’ --outFileName sample1_ sample_1_120.png
Motif analysis over defined regions of target occupancy with HOMER
$ makeTagDirectory sample1_1_120/ sample1_sample1_1_120.bam $ makeUCSCfile sample1_1_120 -o auto $ annotatePeaks.pl /path/to/regions_of_interests.bed. /path/to/reference_genome -size 4000 -hist 20 -d /path/to/tag_directory/sample_1_1_120 > sample1 1_120_regions_of_interests.txt $ findMotifsGenome.pl /path/to/sample1_1_120_regions_of_interest. xtx_file.txt /path/to/ref_genome /output/directory/sample1_1_120_regions_of_interests_peaks.txt -size 4000
? TROUBLESHOOTING
Troubleshooting
Troubleshooting advice can be found in Table 3.
Table 3 |.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 9 | Limited number of embryos | Blastocyst loss during harvest or preparation. Alternatively, different assays are desired for the same blastocyst | Use an alternative method described in Box 1 if disaggregating the blastocyst is desired (useful if parallel assays on cells from same blastocyst are needed) |
| 19 | ConA bead clumping after nuclei addition | Incorrect ratio of ConA beads to sample in Step 18 | Increase the amount of ConA beads added per sample (1.2–1.5×) to help decrease bead clumping and reduce sample loss |
| 55 | Low DNA recovery | Incomplete membrane lysis at Step 11 | Increase NE buffer incubation time to 15–30 min on ice. Alternatively, small amounts of additional detergent (such as digitonin61) can be added. This is not necessary for E14 ES cells and other cell types we have tested |
| Inefficient nucleus binding at Step 19 | Ensure thorough but gentle mixing of ConA beads and nuclei by pipetting into tube while vortexing on low setting. Mixing for longer can help | ||
| Not enough antibody used at Step 25 | We recommend starting with a 1:100 dilution of antibody, but because not all antibodies have similar affinities and/or background binding, a dilution series might need to be used to find the optimal dilution | ||
| Inefficient antibody mixing at Step 27 | Thoroughly but gently mix by pipetting into tube while vortexing on low setting. Draw and expel liquid into the pipette tip slowly for additional mixing | ||
| Incubation length not optimized with selected antibody at Step 28 | Perform an antibody incubation titration series from 30 min to 2 h to optimize antibody incubation | ||
| Not enough pA-MNase added at Step 30 | Perform a pA-MNase titration series to determine catalytic activity of purified pA-MNase (Supplementary Methods) | ||
| Insufficient compatibility between pA-MNase and mouse antibody. Alternatively, purified pA-MNase is not of sufficient quality for uliCUT&RUN | Use commercially available pAG-MNase (see Supplementary Methods for guidelines for use with uliCUT&RUN) | ||
| Target protein-DNA interactions cannot be captured with a native approach | Utilize crosslinking with uliCUT&RUN, with the optimizations as described in ref.58 | ||
| 111 | High background signal in all samples | Improper nucleus blocking at Step 21 | Ensure thorough but gentle mixing by pipetting into tube. Additional mixing might be necessary |
| Too much pA-MNase or pAG-MNase used at Step 33 | Perform a pA-MNase titration series to determine catalytic activity of purified pA-MNase | ||
| Inefficient temperature control at Step 38 | When adding CaCl2 to samples, take care to hold firmly directly below the lip of the microfuge tube so as to avoid contaminating the sample or warming it with body heat | ||
| Inefficient CaCl2 mixing upon addition at Step 38 | Proper mixing can be achieved by additional inversions and flicking after CaCl2 addition | ||
| Overdigestion with pA-MNase at Step 38 | Perform a shorter digestion time period of MNase digestion after calcium addition (15–20 min) | ||
| Primer or adapter dimer present | Inefficient library purification at Steps 99–108 | Prepare another 15 cm, 1.5% (wt/vol) agarose gel, and perform a gel extraction of each sample as outlined in the alternative library build protocol found in Supplementary Methods | |
| Improper adapter dilution used at Step 65 | Confirm that the proper adapter dilution was used. It might be necessary to perform an adapter dilution series to determine optimal dilution | ||
| Improper primer dilution used at Step 91 | Confirm that the proper primer dilution was used. Alternatively, it might be necessary to perform a primer dilution series to determine optimal dilution | ||
| Low library recovery | Incorrect AMPure XP handling used at Step 99 | Incubate beads at RT for at least 15–30 min prior to use. Ensure the proper amount of beads are added to the sample and mixed thoroughly. After incubation, allowing the beads to magnetize and the solution to clear for 10 min on the stand can reduce sample loss | |
| Library underamplified at Step 95 | Confirm use of 1:10 index dilutions for PCR. If still underamplified, we recommend performing a library cycle amplification series to optimize number of PCR cycles as the PCR cycles listed may or may not be sufficient for all antibodies or target proteins. If issue persists, see Supplementary Methods for a qPCR-based approach for cycle optimization | ||
| No-antibody sample contains more free pA-MNase than antibody-containing samples, which can contribute to higher background digestion | As an alternative to the no-antibody control, use an IgG antibody control. We have previously confirmed this as a suitable alternative if high background is an issue13 | ||
| Sample is incompatible with the outlined library build protocol | See Supplementary Methods for an alternative library build with inline adapters or TruSeq adapters (see also Supplementary Fig. 1). Use of NEBNext kit (with NEBNext stem-loop adapters) rather than user-sourced materials is also an option | ||
| High background in no-antibody control | Incorrect amounts of AMPure XP beads added at Step 74 or 99 | Ensure that the correct amount of beads was added to each sample. If incorrect size selection persists, you might have to titrate AMPure beads to make sure size selection is performed properly. See manufacturer’s instructions | |
| 120 | Components of suggested bioinformatics pipeline are unavailable or alternatives are desired | An alternative aligner at Step 116 | BWA69 or STAR70 aligners could be used as alternatives to bowtie 2. See developers’ instructions |
| An alternative data visualization package at Step 120 | HOMER80 and SeqPlots81 can be used in place of deepTools to generate heatmaps for visualization. See developers’ instructions | ||
| An alternative software for motif analysis at Step 120 | MACS82 and MEME SUITE83 can serve as options for motif analysis. See developers’ instructions |
Timing
The times indicated are for ~20 samples or fewer.
Steps 1–2A, harvesting fresh blastocysts: ~6 d, dependent on user experience
Steps 1–2B, harvesting cultured blastocysts: ~6 d, dependent on user experience
Steps 1–2C or 1–2D, preparing single cells by FACS: ~1 h, dependent on sorting conditions and user experience
Steps 3–9, blastocyst preparation: 15 min
Steps 10–24, nuclear extraction and bead binding: 45 min
Steps 25–30, antibody targeting: 2 h 10 min
Steps 31–40, pA-MNase targeting and chromatin digestion: 1 h 15 min
Steps 41–55, DNA isolation: 1 h 30 min
Steps 56–63, end repair: 1 h
Steps 64–72, adapter ligation: 35 min
Steps 73–89 AMPure XP bead purification: 30 min
Steps 90–96, library enrichment: 25 min
Steps 97–111, library purification and quality check: 2 h
Steps 112–113, sample preparation and sequencing: 1 d to 2 weeks, dependent on sequencing facility
Steps 114–120, data analysis: 1 d to 1 week, dependent on user experience
Anticipated results
In this protocol, we describe an approach to profile protein localization on chromatin from single embryos or single mammalian cells. Relative to traditional ChIP-seq experiments, CUT&RUN and associated variants result in lower background signal, allowing more reliable identification of peaks and binding locations of proteins on chromatin41. Our modifications have produced robust data from extremely low cell numbers, especially when assessing TF occupancy13. Of note, CUT&Tag provides reliable results for histone modifications from low numbers of cells41. Importantly, as all these experiments are antibody based, selection of an appropriate and target-specific antibody is of utmost importance.
A typical uliCUT&RUN analysis against a TF in 50 cells or blastocysts will yield results as shown in Figs. 4–6 (data from GSE111121)13. This will include spike-in-normalized occupancy data visualized as heatmaps for the target protein at genomic locations of interest (e.g., transcription start sites or enhancers), often in comparison with the accompanying no-antibody control (or IgG control if applicable; Fig. 5a). The sample and control profiles can be used to generate differential heatmaps, which can further highlight differences in the occupancy profiles between sample and control (Fig. 6a, right panel). The occupancy data of the target protein can be averaged at all regions of interest (genome wide or in subsets) and visualized in graphs referred to as metaplots to reveal site-specific changes in occupancy (Fig. 5d). Finally, peaks can be called and assessed independently. Using peak information, underlying DNA sequence motifs can be determined (Fig. 6c). Finally, the occupancy profile of a sample can be viewed in a locus-specific manner with browser tracks (Fig. 5e). Because of the binary nature of single-cell data, heatmaps and metaplots may not be as informative as browser tracks (Fig. 6e)13. However, as we discussed previously, we imagine that single-cell uliCUT&RUN data are amenable to dimensional reduction analysis (as described in ref.58) and other novel avenues of bioinformatic analyses. Taken together, these data visualizations show that localization of the TF CTCF can be profiled effectively in single blastocysts and single cells with uliCUT&RUN. Traditional CUT&RUN and other enzyme tethering assays have improved our ability to assess and understand chromatin-associated proteins16, but uliCUT&RUN now extends the horizons of this potential research, most critically into the clinical and biomedical setting where extremely low cell numbers and rare samples can be assayed.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The GEO accession number for the sequencing data reported in this paper is GSE111121, originally generated for ref.13.
Code availability
Code for sequencing data analysis is available at https://github.com/bjp86/Patty_uliCUTandRUNAnalysis.
Supplementary Material
Acknowledgements
We thank the Henikoff group for the original development of CUT&RUN and discussion regarding application. We thank A. Boskovic and T. Fazzio for assistance in development and application of uliCUT&RUN. We thank members of the Hainer lab for helpful comments on the manuscript. This work was supported by the National Institutes of Health grant R35GM133732 to S.J.H.
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41596-021-00516-2.
References
- 1.Spitz F & Furlong EEM Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet 13, 613–626 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Villar D, Flicek P & Odom DT Evolution of transcription factor binding in metazoans—mechanisms and functional implications. Nat. Rev. Genet 15, 221–233 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mitsis T et al. Transcription factors and evolution: an integral part of gene expression (Review). World Acad. Sci. J 3–8 (2020) [Google Scholar]
- 4.Andersson R & Sandelin A Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet 21, 71–87 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Whyte WA et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lambert SA et al. The human transcription factors. Cell 172, 650–665 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Zaret KS & Mango SE Pioneer transcription factors, chromatin dynamics, and cell fate control. Curr. Opin. Genet. Dev 37, 76–81 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwafuchi-Doi M & Zaret KS Cell fate control by pioneer transcription factors. Dev 143, 1833–1837 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu P, Zhang W, Xin H & Deng G Single cell isolation and analysis. Front. Cell Dev. Biol 4, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pijuan-Sala B et al. A single-cell molecular map of mouse gastrulation and early organogenesis. Nature 566, 490–495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng S et al. Single-cell RNA-seq reveals cellular heterogeneity of pluripotency transition and X chromosome dynamics during early mouse development. Cell Rep 26, 2593–2607.e3 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Petropoulos S et al. Single-cell RNA-seq reveals lineage and X chromosome dynamics in human pre-implantation embryos. Cell 165, 1012–1026 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hainer SJ, Bošković A, McCannell KN, Rando OJ & Fazzio TG Profiling of pluripotency factors in single cells and early embryos. Cell 177, 1319–1329.e11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hainer SJ & Fazzio TG High-resolution chromatin profiling using CUT&RUN. Curr. Protoc. Mol. Biol 126, 1–22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skene PJ & Henikoff S A simple method for generating high resolution maps of genome-wide protein binding. eLife 4, 1–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu J et al. Super-resolution imaging reveals the evolution of higher-order chromatin folding in early carcinogenesis. Nat. Commun 11, 1–17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmid M, Durussel T & Laemmli UK ChIC and ChEC: genomic mapping of chromatin proteins. Mol. Cell 16, 147–157 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Solomon MJ & Varshavsky A Formaldehyde-mediated DNA-protein crosslinking: a probe for in vivo chromatin structures. Proc. Natl Acad. Sci. USA 82, 6470–6474 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilmour DS & Lis JT In vivo interactions of RNA polymerase II with genes of Drosophila melanogaster. Mol. Cell. Biol 5, 2009–2018 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilmour DS & Lis JT Detecting protein-DNA interactions in vivo: distribution of RNA polymerase on specific bacterial genes. Proc. Natl Acad. Sci. USA 81, 4275–4279 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Irvine RA, Lin IG & Hsieh C-L DNA methylation has a local effect on transcription and histone acetylation. Mol. Cell. Biol 22, 6689–6696 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Albert I et al. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature 446, 572–576 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Dingwall C, Lomonossoff GP & Laskey RA High sequence specificity of micrococcal nuclease. Nucleic Acids Res 9, 2659–2674 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hörz W & Altenburger W Sequence specific cleavage of DNA by micrococcal nuclease. Nucleic Acids Res 9, 2643–2658 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klein DC & Hainer SJ Genomic methods in profiling DNA accessibility and factor localization. Chromosom. Res 28, 69–85 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fosslie M et al. Going low to reach high: small-scale ChIP-seq maps new terrain. Wiley Interdiscip. Rev. Syst. Biol. Med 12, 1–24 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buenrostro JD, Wu B, Chang HY & Greenleaf WJ ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 2015, 21.29.1–21.29.9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buenrostro JD et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmidl C, Rendeiro AF, Sheffield NC & Bock C ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nat. Methods 12, 963–965 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao Z, Chen C, He B, Tan K & Lu C A microfluidic device for epigenomic profiling using 100 cells. Nat. Methods 12, 959–962 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shankaranarayanan P et al. Single-tube linear DNA amplification (LinDA) for robust ChIP-seq. Nat. Methods 8, 565–568 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Rotem A et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol 33, 1165–1172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grosselin K et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat. Genet 51, 1060–1066 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Rhee HS & Pugh BF Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 147, 1408–1419 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He Q, Johnston J & Zeitlinger J ChIP-nexus enables improved detection of in vivo transcription factor binding footprints. Nat. Biotechnol 33, 395–401 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keller CA et al. Effects of sheared chromatin length on ChIP-seq quality and sensitivity. G3 (Bethesda) 10.1093/g3journal/jkab101 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steensel BV & Henikoff S Identification of in vivo DNA targets of chromatin proteins using tethered Dam methyltransferase. Nat. Biotechnol 18, 424–428 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Kind J et al. Genome-wide maps of nuclear lamina interactions in single human. cells. Cell 163, 134–147 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zentner GE, Kasinathan S, Xin B, Rohs R & Henikoff S ChEC-seq kinetics discriminates transcription factor binding sites by DNA sequence and shape in vivo. Nat. Commun 6, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaya-Okur HS et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun 10, 1–10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carter B et al. Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq). Nat. Commun 10, 1–5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu B et al. The landscape of RNA Pol II binding reveals a stepwise transition during ZGA. Nature 587, 139–144 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Ku WL et al. Single-cell chromatin immunocleavage sequencing (scChIC-seq) to profile histone modification. Nat. Methods 16, 323–325 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skene PJ & Henikoff S An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 6, 1–35 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skene PJ, Henikoff JG & Henikoff S Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc 13, 1006–1019 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Kaya-Okur HS, Janssens DH, Henikoff JG, Ahmad K & Henikoff S Efficient low-cost chromatin profiling with CUT&Tag. Nat. Protoc 15, 3264–3283 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skene PJ & Henikoff S A simple method for generating high resolution maps of genome-wide protein binding. eLife 4, 1–9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meers MP, Bryson TD, Henikoff JG & Henikoff S Improved cut&run chromatin profiling tools. eLife 8, 1–16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nagano T et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shah RN et al. Examining the roles of H3K4 methylation states with systematically characterized antibodies. Mol. Cell 72, 162–177.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng XY & Gehring M Low-input chromatin profiling in Arabidopsis endosperm using CUT&RUN. Plant Reprod 32, 63–75 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Chereji RV, Bryson TD & Henikoff S Quantitative MNase-seq accurately maps nucleosome occupancy levels. Genome Biol 20, 198 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oomen ME, Hansen AS, Liu Y, Darzacq X & Dekker J CTCF sites display cell cycle-dependent dynamics in factor binding and nucleosome positioning. Genome Res 29, 236–249 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Janssens DH et al. Automated in situ chromatin profiling efficiently resolves cell types and gene regulatory programs. Epigenetics Chromatin 11, 1–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meers MP, Tenenbaum D & Henikoff S Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling. Epigenetics Chromatin 12, 1–11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu Q, Liu N, Orkin SH & Yuan GC CUT and RUNTools: a flexible pipeline for CUT and RUN processing and footprint analysis. Genome Biol 20, 1–12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baek S & Lee I Single-cell ATAC sequencing analysis: from data preprocessing to hypothesis generation. Comput. Struct. Biotechnol. J 18, 1429–1439 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Urrutia E, Chen L, Zhou H & Jiang Y Destin: toolkit for single-cell analysis of chromatin accessibility. Bioinformatics 35, 3818–3820 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cai S, Georgakilas GK, Johnson JL & Vahedi G A cosine similarity-based method to infer variability of chromatin accessibility at the single-cell level. Front. Genet 9, 1–10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zamanighomi M et al. Unsupervised clustering and epigenetic classification of single cells. Nat. Commun 9, 1–8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baker SM, Rogerson C, Hayes A, Sharrocks AD & Rattray M Classifying cells with Scasat, a single-cell ATAC-seq analysis tool. Nucleic Acids Res 47, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li H et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ramírez F, Dündar F, Diehl S, Grüning BA & Manke T DeepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luo C et al. Superovulation strategies for 6 commonly used mouse strains. J. Am. Assoc. Lab. Anim. Sci 50, 471–478 (2011). [PMC free article] [PubMed] [Google Scholar]
- 68.Adli M & Bernstein BE Whole-genome chromatin profiling from limited numbers of cells using nano-ChIP-seq. Nat. Protoc 6, 1656–1668 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zwart W et al. A carrier-assisted ChIP-seq method for estrogen receptor-chromatin interactions from breast cancer core needle biopsy samples. BMC Genomics 14, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Valensisi C, Liao JL, Andrus C, Battle SL & Hawkins RD cChIP-seq: a robust small-scale method for investigation of histone modifications. BMC Genomics 16, 1–11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ng JH et al. In vivo epigenomic profiling of germ cells reveals germ cell molecular signatures. Dev. Cell 24, 324–333 (2013). [DOI] [PubMed] [Google Scholar]
- 72.Liu X et al. Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos. Nature 537, 558–562 (2016). [DOI] [PubMed] [Google Scholar]
- 73.Lara-Astiaso D et al. Immunogenetics. Chromatin state dynamics during blood formation. Science 345, 943–949 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van Galen P et al. a multiplexed system for quantitative comparisons of chromatin landscapes. Mol. Cell 61, 170–180 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zheng X et al. Low-cell-number epigenome profiling aids the study of lens aging and hematopoiesis. Cell Rep 13, 1505–1518 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dahl JA et al. Broad histone H3K4me3 domains in mouse oocytes modulate maternal-to-zygotic transition. Nature 537, 548–552 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang B et al. Allelic reprogramming of the histone modification H3K4me3 in early mammalian development. Nature 537, 553–557 (2016). [DOI] [PubMed] [Google Scholar]
- 78.Zarnegar MA, Reinitz F, Newman AM & Clarke MF Targeted chromatin ligation, a robust epigenetic profiling technique for small cell numbers. Nucleic Acids Res 45, 1–9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Harada A et al. A chromatin integration labelling method enables epigenomic profiling with lower input. Nat. Cell Biol 21, 287–296 (2019). [DOI] [PubMed] [Google Scholar]
- 80.Akalin A, Franke V, Vlahoviček K, Mason CE & Schübeler D Genomation: a toolkit to summarize, annotate and visualize genomic intervals. Bioinformatics 31, 1127–1129 (2015). [DOI] [PubMed] [Google Scholar]
- 81.Stempor P & Ahringer J SeqPlots—interactive software for exploratory data analyses, pattern discovery and visualization in genomics. Wellcome Open Res 1, 14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bailey TL et al. MEME Suite: tools for motif discovery and searching. Nucleic Acids Res 37, 202–208 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen X et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133, 1106–1117 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The GEO accession number for the sequencing data reported in this paper is GSE111121, originally generated for ref.13.