

Abstract
The first structural models of the proposed cis-FeIII(OH)(halide) intermediate in the non-heme iron halogenases were synthesized and examined for their inherent reactivity with tertiary carbon radicals. Selective hydroxylation occurs for these cis-FeIII(OH)(X) (X = Cl, Br) complexes in a radical rebound-like process. In contrast, a cis-FeIII(Cl)2 complex reacts with carbon radicals to give halogenation. These results are discussed in terms of the inherent reactivity of the analogous rebound intermediate in both enzymes and related catalysts.
Non-heme iron enzymes utilize dioxygen to carry out C–H bond transformations, including hydroxylation and halogenation. These enzymes include the αKG-dependent hydroxylase TauD, which carries out the hydroxylation of taurine,1 and αKG-dependent SyrB2,2 CytC3,3 and WelO5,4,5 which carry out the halogenation of natural products.6 The proposed mechanisms of these enzymes are similar, relying on an FeIV(O) oxidant to abstract a hydrogen atom from the C–H substrate. The nascent carbon radical then recombines, or “rebounds”, with either the hydroxyl (FeOH) group in the case of the hydroxylases or the halide (FeX) group in the case of the halogenases.7 A simplifed halogenase mechanism is shown in Scheme 1. Halogenase activity appears to derive from factors that control the selective transfer of Cl· (or X·) to the nearby carbon radical.
Scheme 1.

Proposed Rebound Mechanism
The key factors that control the radical rebound process remain under debate,8–13 in part because of the lack of a direct methods to study the presumably short-lived, unobserved ferric rebound intermediate. A range of different proposals5,14–23 attempting to describe these factors for the halogenases have been put forth. Synthetic iron model complexes also exhibit some halogenation activity24–28 together with hydroxylation activity (Scheme 1). For both enzymes and models, computational studies have indicated that the thermodynamically preferred product should be the alcohol, not the halogenated compound.14,19,22 Thus, halogenation is predicted to arise from a kinetic pathway, in which the reaction barrier for rebound of the halide is significantly lower than that for the hydroxide.14,22 However, addressing the inherent reactivity of FeIII(OH)(X) species experimentally has been out of reach until now.
Herein we report the synthesis of two mononuclear FeIII(OH)(X) (X = Cl, Br) complexes that to our knowledge are the first structural models of the key ferric intermediate proposed for the non-heme iron halogenases. The isolation of these complexes allowed us to examine their inherent reactivity with tertiary carbon radicals. In earlier work, we examined the rebound reactivity of non-heme FeIII(OMe)29 and FeIII(OH)30 complexes, and the latter complex contained pendent H-bond donors to stabilize a terminal OH ligand as well as a labile cis coordination site that has now been exploited to prepare the elusive cis-FeIII(OH)(X) complexes. Reactions with tertiary carbon radicals give exclusively the hydroxylated products, showing a preference for the thermodynamic pathway, while an analogous FeIII(Cl)2 complex is competent to halogenate the radicals.
The tetradentate ligand BNPAPh2O− was used previously to prepare FeIII(BNPAPh2O)(OH)(OTf), a terminal FeIII(OH) complex stabilized by the steric protection and H-bonding of the pendent neopentylamine groups.30 The triflate ligand is cis to the OH−, and we hypothesized that it could be replaced by halide (X−) under suitable conditions. Reaction of BNPAPh2O− with ferrous halide salts gives FeII(BNPAPh2O)(X) (1, X = Cl; 2, X = Br). X-ray diffraction (XRD) revealed the five-coordinate FeII complexes shown in Figure 1. The halide ligands in these approximately trigonal-bipyramidal complexes (τ = 0.626 for 1, τ = 0.635 for 2)31 occupy the axial positions and form H-bonds with the pendent neopentylamine groups: N(H)⋯X(ave) = 3.222 Å in 1 and 3.363 Å in 2 and N–H–X(ave) = 171° in 1 and 166° in 2 (Figure 1). The remaining Fe–N and Fe–O distances are in the typical ranges for high-spin (hs) (S = 2) FeII complexes32–34(Table S1).
Figure 1.
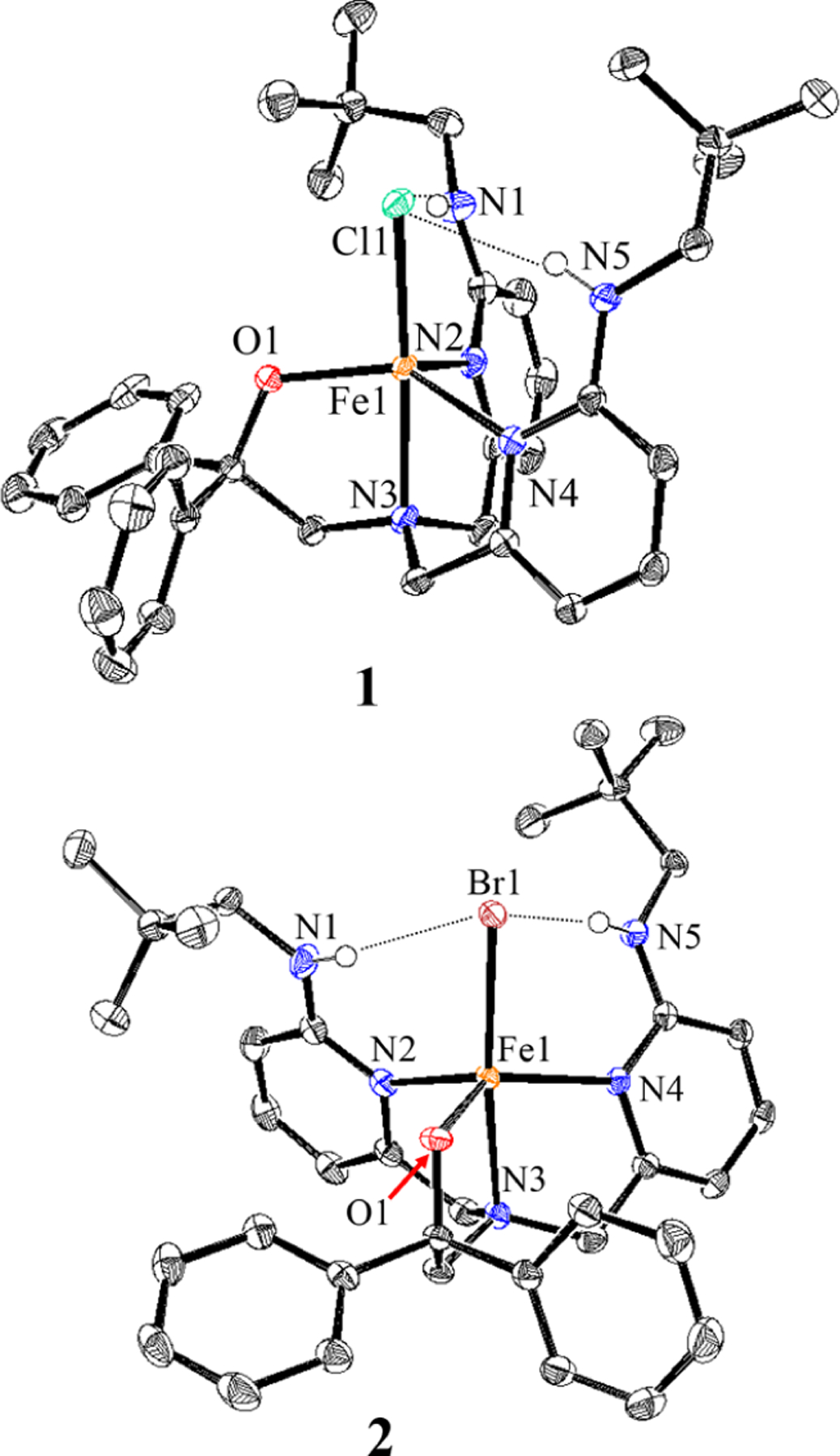
Displacement ellipsoid plots (50% probability level) for 1 and 2 at 110(2) K. Hydrogen atoms (except for N–H) have been omitted for clarity.
The reactions of 1 and 2 with excess O2 in THF at 23 °C lead to a rapid color change from yellow to bright orange. Removal of the volatiles gives dark-orange powders, and recrystallization from THF/n-pentane yields crystals of 3 and 4, respectively, as dark-orange blocks suitable for XRD. The crystal structures of these new species (Figure 2) reveal the six-coordinate complexes FeIII(BNPAPh2O)(OH)(X) (3: X = Cl, 4: X = Br), whose metrical parameters are consistent with hs ferric (S = 5/2) ions.33–37 The OH ligands occupy the sites involved in H-bonding interactions with the pendent amines (3: N(H)⋯O = 2.78 Å, N–H–O = 162°; 4: N(H)···O = 2.77 Å, N–H–O = 159°). These H-bonds are relatively tight, as seen in previously reported FeIII(OH) complexes.32–36,38,39 The halide ligand is bound cis to the OH ligand and exhibits an Fe–X distance that is in the range of those for other hs FeIII(X) compounds.28,40,41 Density functional theory (DFT) calculations at the B3LYP/6–311G* level give optimized geometries for 1–4 that are in good agreement with the crystal structures (Figure S22 and Tables S1 and S2). The formation of 3 and 4 from O2 (Scheme 2) likely follows a mechanism similar to that proposed for FeIII(OH)(OTf)-(BNPAPh2O)30 and related compounds.42,43
Figure 2.
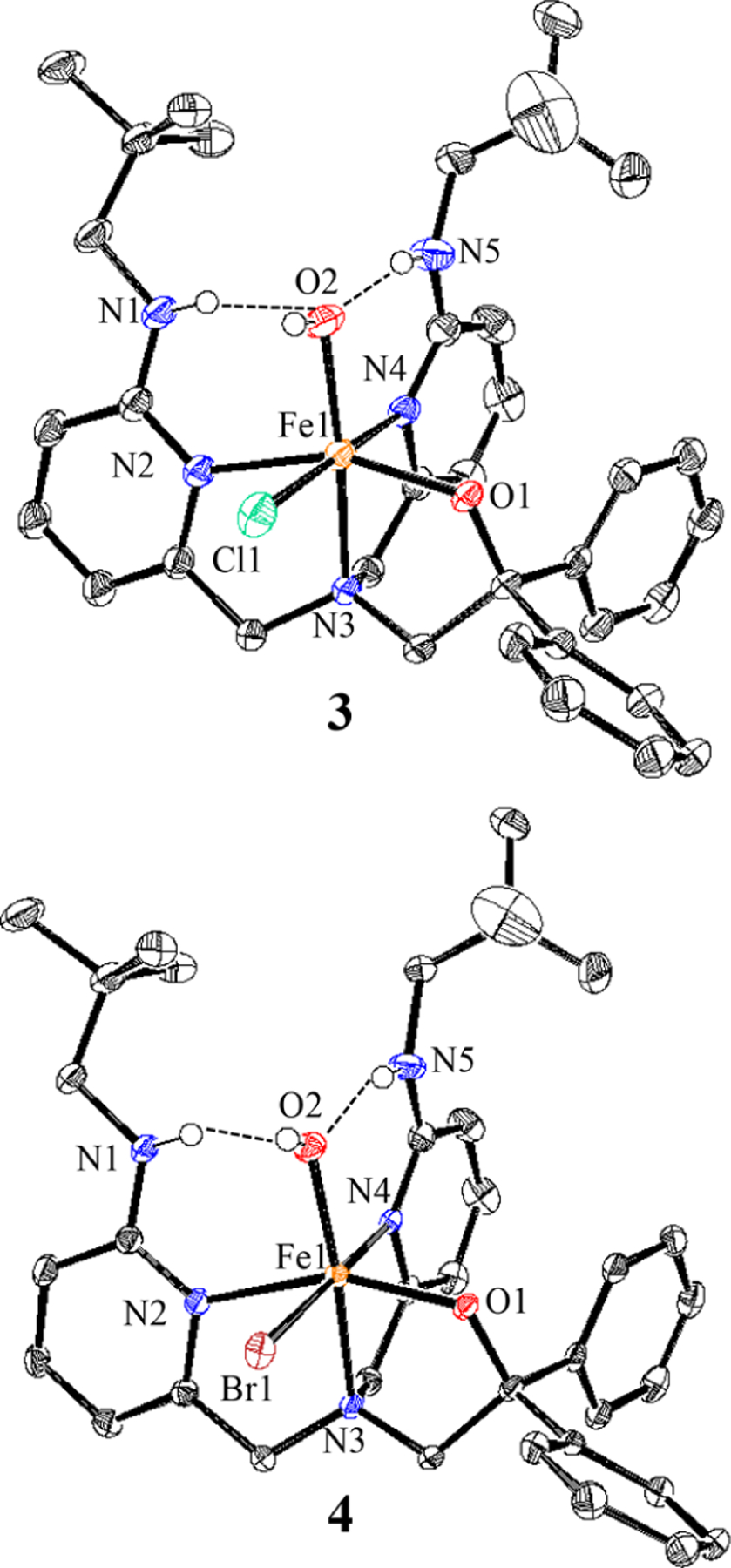
Displacement ellipsoid plots (50% probability level) for 3 and 4 at 110(2) K. Hydrogen atoms (except for N–H and O–H) have been omitted for clarity.
Scheme 2.
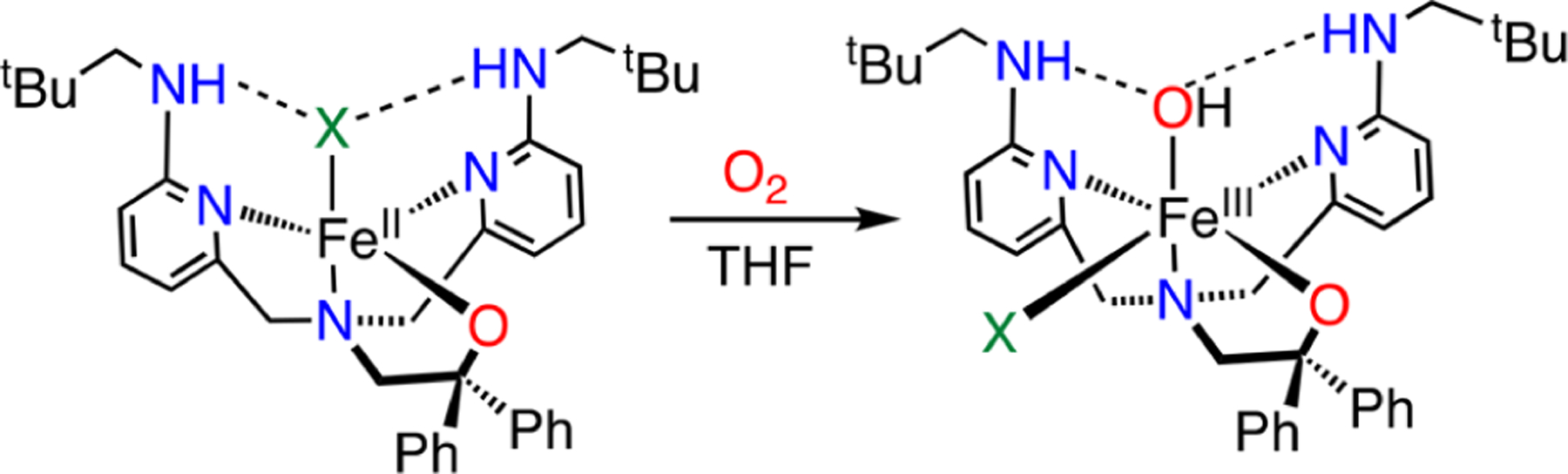
Synthesis of FeIII(OH)(X) Complexes
The UV–vis spectra of 1–4 are relatively featureless in the visible region (Figures S34–S37). The 1H NMR and Mössbauer spectra are more informative. The 1H NMR spectra for complexes 1 and 2 in CD3CN exhibit similar paramagnetically shifted peaks between −12 and +85 ppm (Figure 3). In contrast, the hs FeIII complexes 3 and 4 exhibit fewer and much more broadened peaks (Figures S3 and S4). Enrichment of 1–4 with 57Fe was accomplished by developing an alternative synthetic route involving ligand metathesis of 57FeII(BNPAPh2O)(OTf) or 57FeIII(BNPAPh2O)(OH)(OTf)30 with Bu4NX (X = Cl−, Br−) in 2-MeTHF to give 57Fe-labeled 1–4 in situ. Substitution of X− for OTf− was confirmed by 1H NMR spectroscopy, which gave spectra identical to those seen for independently prepared 3 and 4 (Figures S28 and S29). Mössbauer spectroscopy of 57Fe-labeled 1 and 2 in 2-MeTHF shows a sharp quadrupole doublet for both complexes, with δ = 1.02 mm s−1 and |ΔEQ| = 2.65 mm s−1 for 1 and δ = 1.03 mm s−1 and |ΔEQ| = 2.55 mm s−1 for 2 (Figure 4), indicative of hs FeII (S = 2). As anticipated, the Mössbauer spectra of 3 and 4 show broadened quadrupole doublets with parameters typical of hs FeIII: δ = 0.42 mm s−1 and |ΔEQ| = 1.01 mm s−1 for 3 and δ = 0.41 mm s−1 and |ΔEQ| = 1.10 mm s−1 for 4 (Figure 4). The significant line broadening seen for 3 and 4 can be attributed to population of an intermediate relaxation regime, as reported previously44,45,24 (see the Supporting Information for fitting details).
Figure 3.
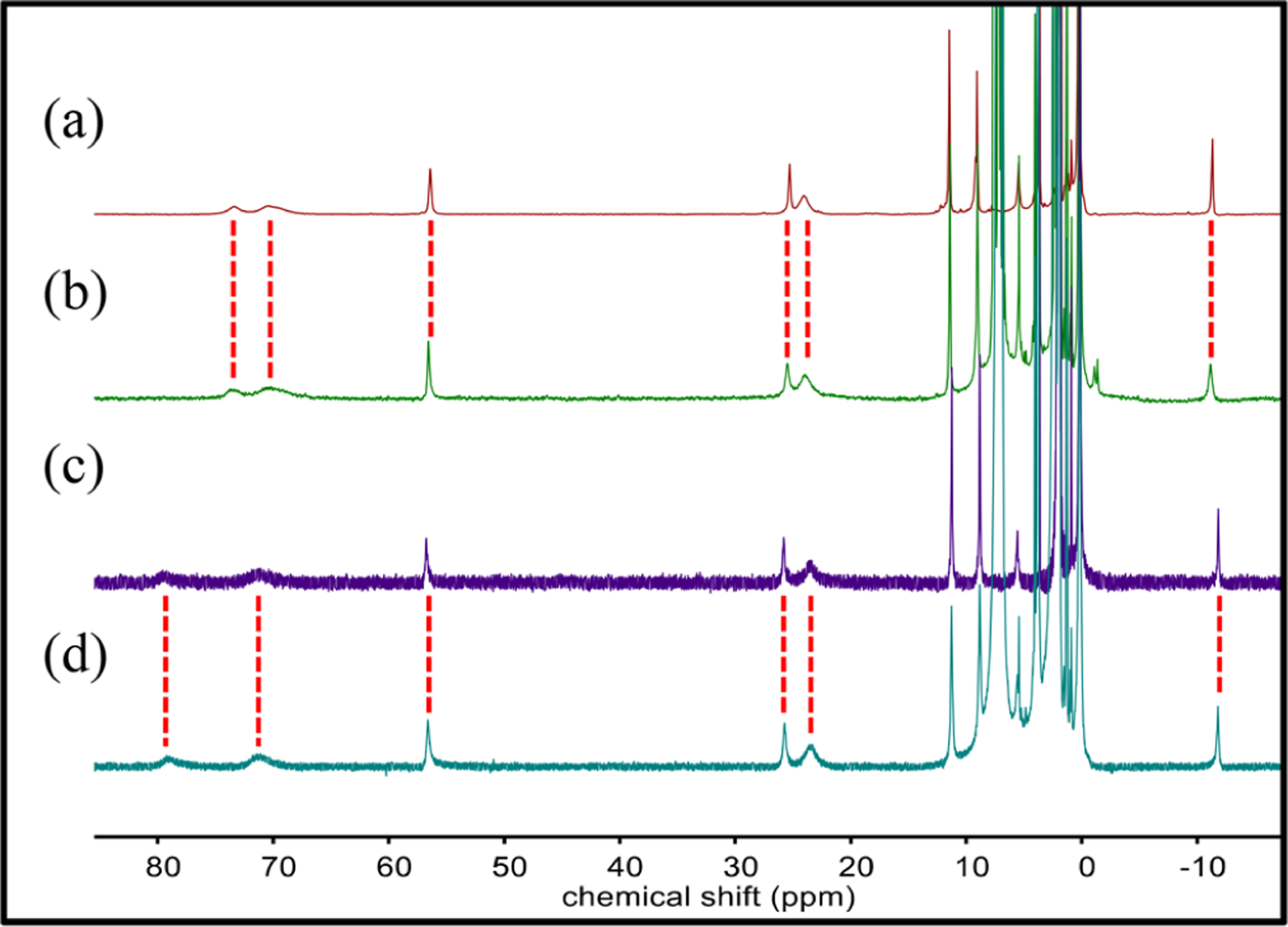
1H NMR spectra in CD3CN of (a) 1, (b) 3 + (p-MeOC6H4)3C·, (c) 2, and (d) 4 + (p-MeOC6H4)3C·.
Figure 4.
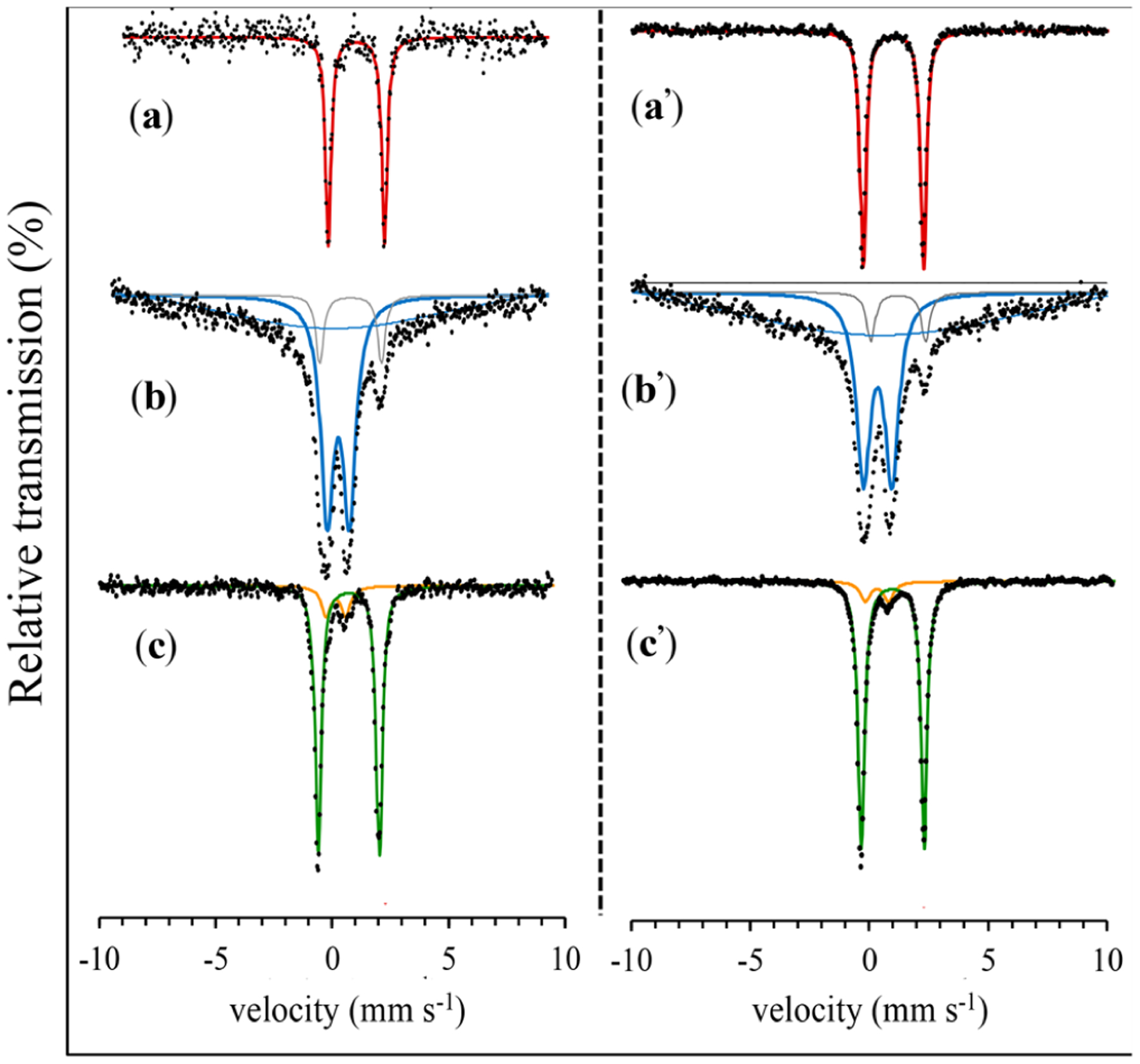
Zero-field 57Fe Mössbauer spectra (80 K) of (a, a′) 1 and 2, (b, b′) 3 and 4, and (c, c′) 3 and 4 + (p-MeOC6H4)3C·.
To our knowledge, complexes 3 and 4 are the first structural models of the FeIII(OH)(X) intermediates proposed for the non-heme iron halogenases and related catalysts, as shown in Figure 1. They also nicely reproduce the H-bonding proposed for the active site in SyrB2.14,19 These complexes are excellent starting points for the study of the inherent rebound reactivity of FeIII(OH)(X) species.
Reactivity studies for 3 and 4 were initiated with the triphenylmethyl radical derivatives (p-YC6H4)3C· (Y = OMe, H, Cl). These tertiary carbon radicals are relatively stable and have been used to show “rebound”-like reactions with Fe and Cu complexes.29,30,39,46–50 Addition of (p-YC6H4)3C· to 3 or 4 was followed by stirring for 1–4 h (depending on the identity of Y) at 23 °C in THF/toluene (5/2 v/v). The volatiles were removed under vacuum, and the reaction mixture was dissolved in CD3CN for analysis by 1H NMR spectroscopy. The disappearance of the peaks for 3 and 4 occurred with the appearance of the spectra for the one-electron-reduced complexes 1 and 2 (Figure 3). These data suggested that the OH group was selectively transferred to the carbon radical.
The reactions with the triphenylmethyl radical derivatives were also monitored by Mössbauer spectroscopy. The final reaction mixture of 57Fe-enriched 3 and excess (p-MeOC6H4)3C·, after removal of the volatiles and dissolution of the resultant solid in 2-MeTHF, showed a sharp quadrupole doublet with δ = 1.03 mm s−1 and |ΔEQ|= 2.69 mm s−1 (94% of the total fit), a close match to that seen for 1 (Figure 4, left panel). Similarly, the ferric complex 4 shows complete conversion to the FeII(Br) complex 2 upon reaction with (p-MeOC6H4)3C· (Figure 4, right panel).
The tertiary alcohols (p-YC6H4)3COH were identified by 1H NMR spectroscopy of the crude reaction mixtures. Peaks arising from the newly formed hydroxyl groups appeared as well-separated singlets, and integration gave a high (>90%) yield of the alcohol in all cases (Figures S13–S18). The formation of Ph3COH (m/z 260.1) and (p-ClC6H4)3COH (m/z 362.0) was confirmed by mass spectrometry. Isotopic substitution of the OH group in 3 via synthesis with 18O2 led to Ph3C18OH (m/z 262.1) with ≥99% isotopic purity. The same reaction with the (p-ClC6H4)3C· and isotopically labeled 4 led to (p-ClC6H4)3C18OH (m/z 364.0) (Figures S30–S33). The data show that selective hydroxylation occurs in a rebound-type process with 3 and 4 (Scheme 3), and the 18O labeling supports a mechanism involving direct transfer of OH·from FeIII to the carbon radical.
Scheme 3.
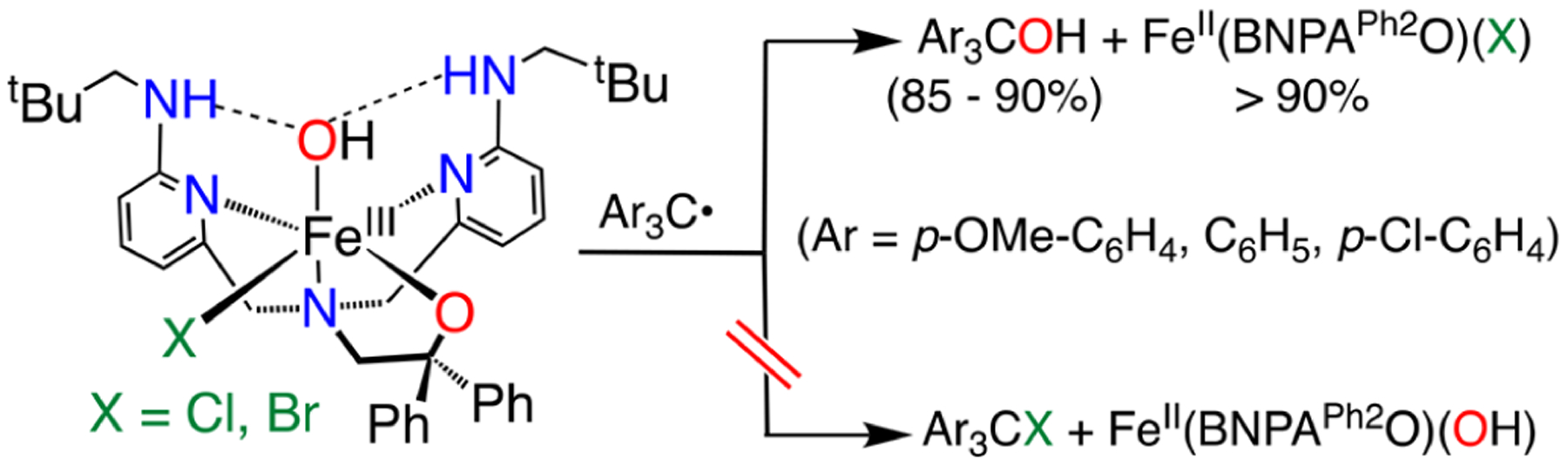
Reaction of 3 and 4 with Triphenylmethyl Radical Derivatives
Metalation of BNPAPh2O− with FeCl3 led to the formation of the dichloro analogue FeIII(BNPAPh2O)(Cl)2 (Figure 5). The structure of 5 shows that the alkoxide ligand is bound trans to the equatorial halide, whereas in complexes 3 and 4 a pyridine arm is bound trans to the equatorial halide. It is not yet known whether the nature of the trans ligand has a significant impact on the reactivity of these complexes. Reaction of 5 with the p-OMe and p-Cl trityl radical derivatives showed complete conversion to 1 by paramagnetic NMR spectroscopy. The formation of the halogenated (p-YC6H4)3CCl was initially monitored by thin-layer chromatography (TLC), and then the products were isolated by silica gel chromatography in good yields (75% for Y = Cl and 84% for Y = OMe). Although it is not possible to determine which of the Cl ligands is transferred, it is clear from these results that halogen transfer occurs easily in the absence of a terminal hydroxide (Scheme 4). This result provides the first example, to our knowledge, of the direct reaction of an isolated FeIII(Cl) complex with a carbon radical to give FeIIand a new C–Cl bond, a process that mimics halogen rebound.
Figure 5.
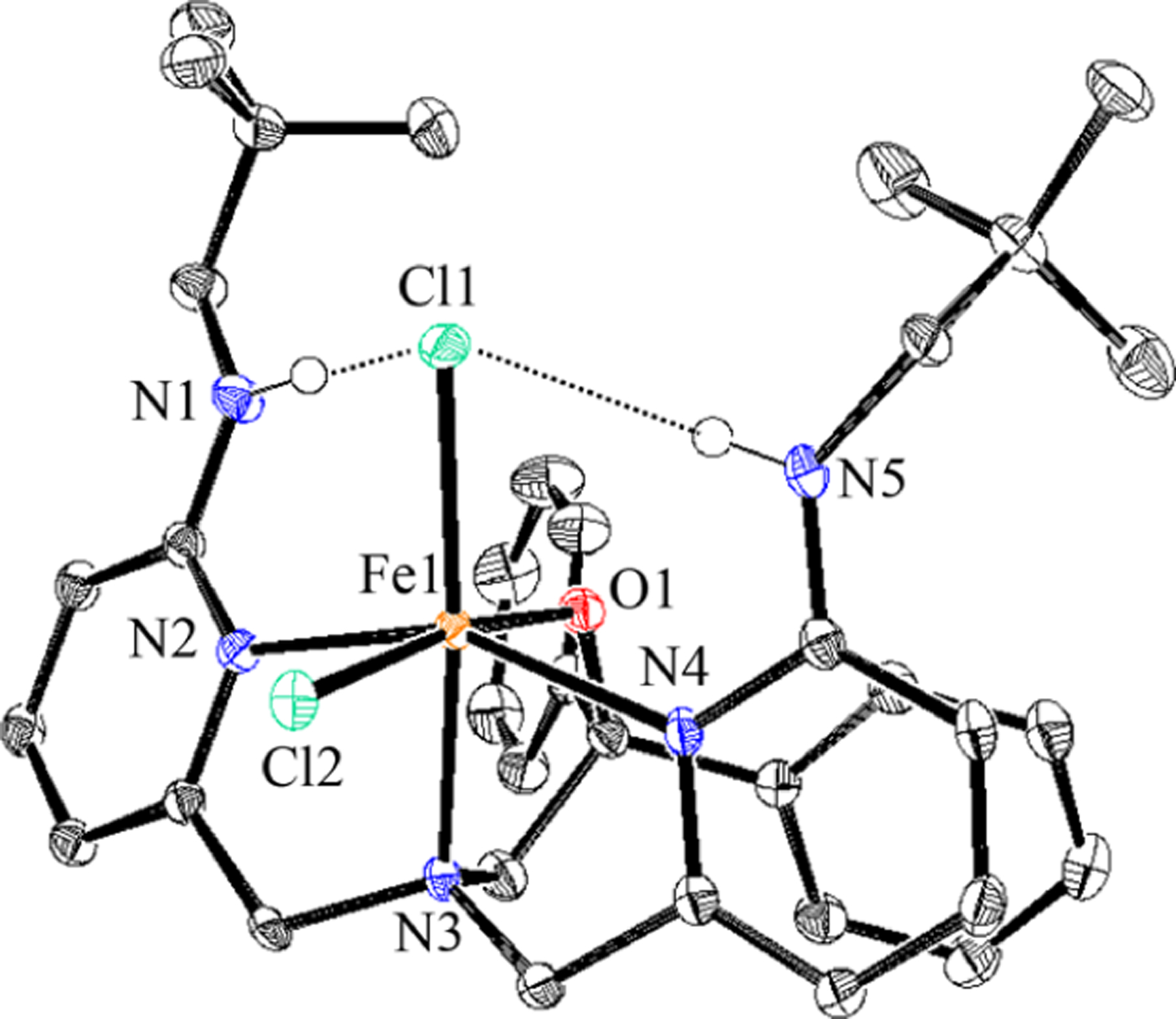
Displacement ellipsoid plot (50% probability level) for 5 at 110(2) K. Hydrogen atoms (except for N–H) have been omitted for clarity.
Scheme 4.
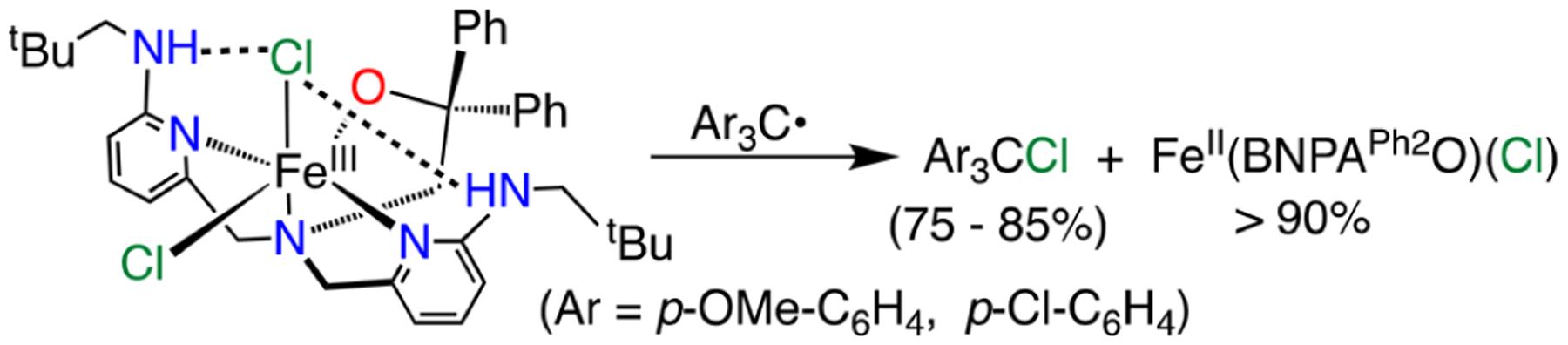
Reaction of 5 with Triphenylmethyl Radical Derivatives
Our findings indicate that the reactions of tertiary carbon radicals with 3 and 4 give hydroxylated products with no evidence of halogenation, although the analogous dichloro complex 5 is competent to transfer the halogen. The lack of halogen transfer for 3 and 4 contrasts with what is seen for both enzymes and halogenase models and thus deserves further comment. The selectivity seen for 3 and 4 is in line with thermodynamic expectations, which are supported by calculations on both enzymes and models that uniformly predict hydroxylation to be thermodynamically favored over halogenation by at least 15 kcal/mol.14,21,22,26 However, the same computational studies also predict that the reaction barriers to halogenation are lower, leading to kinetically favored halogenated products. A range of factors have been offered as contributing to the lower kinetic barriers for halogenation, including substrate orientation,16,23 relocation of the positioning of the OH group on the metal,5,19,20 relative ordering of frontier molecular orbitals,14 relative spin densities on OH versus Cl,15 and steric effects.28 It has also been suggested that H-bonds between the OH group of the putative FeIII(OH)(Cl) intermediate and Arg254/Glu102/water molecules in SyrB2 raise the hydroxylation barrier.14,19
The lack of halogenation seen for 3 and 4 occurs even though OH ligand is held in place and deactivated toward the incoming carbon radical by two tight H-bonds. This inherent reactivity may be a result of the nature of complexes 3 and 4 or may be related to the nature of the radical. It was shown that [FeIV(O)(Cl)(PyTACN)]+ reacts with triphenylmethane to give Ph3 COH,25 suggesting that the tertiary carbon radical may favor hydroxylation. However, [FeIV(O)(Cl)(PyTACN)]+ reacts with other C–H bonds (e.g., cyclohexane), but still no halogenated products were observed. In contrast, some halogenation has been seen for cyclohexane and toluene with a non-heme FeIV(O)(X) (X = Cl, Br) oxidant.24 Reaction of Ph3C· with a low-coordinate FeIII(NHAr)(Cl) complex disfavored halogenation, leading to amination instead.47 The importance of the nature of the carbon radical remains to be determined.
Complexes 3 and 4 are analogous to the key ferric intermediates in non-heme iron halogenases and related catalysts. Future plans include computational studies and other mechanistic investigations of these rebound reactions to understand the key factors that control the rebound selectivity in both enzymes and models.
Supplementary Material
ACKNOWLEDGMENTS
The NIH (GM119374 to D.P.G.) is gratefully acknowledged for financial support. Computer time was provided by the Maryland Advanced Research Computing Center (MARCC).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c00493.
X-ray crystallographic data for 1 (CIF)
X-ray crystallographic data for 2 (CIF)
X-ray crystallographic data for 3 (CIF)
X-ray crystallographic data for 4 (CIF)
X-ray crystallographic data for 5 (CIF)
Synthesis, 1H NMR spectra, 57Fe Mössbauer data, UV–vis spectra, EI-MS spectra, Tables S1–S13, and Figures S1–S41 (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.0c00493
The authors declare no competing financial interest.
REFERENCES
- (1).Martinez S; Hausinger RP Catalytic Mechanisms of Fe(II)- and 2-Oxoglutarate-Dependent Oxygenases. J. Biol. Chem 2015, 290, 20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Vaillancourt FH; Yin J; Walsh CT SyrB2 in syringomycin E biosynthesis is a nonheme FeII α-ketoglutarate- and O2 dependent halogenase. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wong C; Fujimori DG; Walsh CT; Drennan CL Structural Analysis of an Open Active Site Conformation of Nonheme Iron Halogenase CytC3. J. Am. Chem. Soc 2009, 131, 4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hillwig ML; Liu X A new family of iron-dependent halogenases acts on freestanding substrates. Nat. Chem. Biol 2014, 10, 921. [DOI] [PubMed] [Google Scholar]
- (5).Mitchell AJ; Zhu Q; Maggiolo AO; Ananth NR; Hillwig ML; Liu X; Boal AK Structural basis for halogenation by iron- and 2-oxo-glutarate-dependent enzyme WelO5. Nat. Chem. Biol 2016, 12, 636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Latham J; Brandenburger E; Shepherd SA; Menon BRK; Micklefield J Development of Halogenase Enzymes for Use in Synthesis. Chem. Rev 2018, 118, 232. [DOI] [PubMed] [Google Scholar]
- (7).Huang X; Groves JT Beyond ferryl-mediated hydroxylation: 40 years of the rebound mechanism and C–H activation. JBIC, J. Biol. Inorg. Chem 2017, 22, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Cho K-B; Hirao H; Shaik S; Nam W To rebound or dissociate? This is the mechanistic question in C–H hydroxylation by heme and nonheme metal–oxo complexes. Chem. Soc. Rev 2016, 45, 1197. [DOI] [PubMed] [Google Scholar]
- (9).Wu X; Seo MS; Davis KM; Lee Y-M; Chen J; Cho K-B; Pushkar YN; Nam W A Highly Reactive Mononuclear Non-Heme Manganese(IV)–Oxo Complex That Can Activate the Strong C–H Bonds of Alkanes. J. Am. Chem. Soc 2011, 133, 20088. [DOI] [PubMed] [Google Scholar]
- (10).Cho K-B; Shaik S; Nam W Theoretical Investigations into C–H Bond Activation Reaction by Nonheme MnIVO Complexes: Multistate Reactivity with No Oxygen Rebound. J. Phys. Chem. Lett 2012, 3, 2851. [Google Scholar]
- (11).Cho K-B; Wu X; Lee Y-M; Kwon YH; Shaik S; Nam W Evidence for an Alternative to the Oxygen Rebound Mechanism in C–H Bond Activation by Non-Heme FeIVO Complexes. J. Am. Chem. Soc 2012, 134, 20222. [DOI] [PubMed] [Google Scholar]
- (12).Cho K-B; Kang H; Woo J; Park YJ; Seo MS; Cho J; Nam W Mechanistic Insights into the C–H Bond Activation of Hydrocarbons by Chromium(IV) Oxo and Chromium(III) Superoxo Complexes. Inorg. Chem 2014, 53, 645. [DOI] [PubMed] [Google Scholar]
- (13).Dhuri SN; Cho K-B; Lee Y-M; Shin SY; Kim JH; Mandal D; Shaik S; Nam W Interplay of Experiment and Theory in Elucidating Mechanisms of Oxidation Reactions by a Nonheme RuIVO Complex. J. Am. Chem. Soc 2015, 137, 8623. [DOI] [PubMed] [Google Scholar]
- (14).Srnec M; Solomon EI Frontier Molecular Orbital Contributions to Chlorination versus Hydroxylation Selectivity in the Non-Heme Iron Halogenase SyrB2. J. Am. Chem. Soc 2017, 139, 2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Timmins A; Fowler NJ; Warwicker J; Straganz GD; de Visser SP Does Substrate Positioning Affect the Selectivity and Reactivity in the Hectochlorin Biosynthesis Halogenase? Front. Chem 2018, 6, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Matthews ML; Neumann CS; Miles LA; Grove TL; Booker SJ; Krebs C; Walsh CT; Bollinger JM Jr. Substrate positioning controls the partition between halogenation and hydroxylation in the aliphatic halogenase, SyrB2. Proc. Natl. Acad Sci. U. S. A 2009, 106, 17723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Pandian S; Vincent MA; Hillier IH; Burton NA Why does the enzyme SyrB2 chlorinate, but does not hydroxylate, saturated hydrocarbons? A density functional theory (DFT) study. Dalton Trans 2009, 6201. [DOI] [PubMed] [Google Scholar]
- (18).de Visser SP; Latifi R Carbon Dioxide: A Waste Product in the Catalytic Cycle of α-Ketoglutarate Dependent Halogenases Prevents the Formation of Hydroxylated By-Products. J. Phys. Chem. B 2009, 113, 12. [DOI] [PubMed] [Google Scholar]
- (19).Huang J; Li C; Wang B; Sharon DA; Wu W; Shaik S Selective Chlorination of Substrates by the Halogenase SyrB2 Is Controlled by the Protein According to a Combined Quantum Mechanics/Molecular Mechanics and Molecular Dynamics Study. ACS Catal. 2016, 6, 2694. [Google Scholar]
- (20).Borowski T; Noack H; Radoń M; Zych K; Siegbahn PEM Mechanism of Selective Halogenation by SyrB2: A Computational Study. J. Am. Chem. Soc 2010, 132, 12887. [DOI] [PubMed] [Google Scholar]
- (21).Noack H; Siegbahn PEM Theoretical investigation on the oxidative chlorination performed by a biomimetic non-heme iron catalyst. JBIC, J. Biol. Inorg. Chem 2007, 12, 1151. [DOI] [PubMed] [Google Scholar]
- (22).Quesne MG; de Visser SP Regioselectivity of substrate hydroxylation versus halogenation by a nonheme iron(IV)–oxo complex: possibility of rearrangement pathways. JBIC, J. Biol. Inorg. Chem 2012, 17, 841. [DOI] [PubMed] [Google Scholar]
- (23).Martinie RJ; Livada J; Chang W.-c.; Green MT; Krebs C; Bollinger JM; Silakov A Experimental Correlation of Substrate Position with Reaction Outcome in the Aliphatic Halogenase, SyrB2. J. Am. Chem. Soc 2015, 137, 6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Puri M; Biswas AN; Fan R; Guo Y; Que L Jr. Modeling Non-Heme Iron Halogenases: High-Spin Oxoiron(IV)–Halide Complexes That Halogenate C–H Bonds. J. Am. Chem. Soc 2016, 138, 2484. [DOI] [PubMed] [Google Scholar]
- (25).Planas O; Clémancey M; Latour J-M; Company A; Costas M Structural modeling of iron halogenases: synthesis and reactivity of halide-iron(iv)-oxo compounds. Chem. Commun 2014, 50, 10887. [DOI] [PubMed] [Google Scholar]
- (26).Comba P; Wunderlich S Iron-Catalyzed Halogenation of Alkanes: Modeling of Nonheme Halogenases by Experiment and DFT Calculations. Chem. - Eur. J 2010, 16, 7293. [DOI] [PubMed] [Google Scholar]
- (27).Chatterjee S; Paine TK Hydroxylation versus Halogenation of Aliphatic C–H Bonds by a Dioxygen-Derived Iron–Oxygen Oxidant: Functional Mimicking of Iron Halogenases. Angew. Chem., Int. Ed 2016, 55, 7717. [DOI] [PubMed] [Google Scholar]
- (28).Rana S; Biswas JP; Sen A; Clémancey M; Blondin G; Latour J-M; Rajaraman G; Maiti D Selective C–H halogenation over hydroxylation by non-heme iron(iv)-oxo. Chem. Sci 2018, 9, 7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Pangia TM; Davies CG; Prendergast JR; Gordon JB; Siegler MA; Jameson GNL; Goldberg DP Observation of Radical Rebound in a Mononuclear Nonheme Iron Model Complex. J. Am. Chem. Soc 2018, 140, 4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Yadav V; Gordon JB; Siegler MA; Goldberg DP Dioxygen-Derived Nonheme Mononuclear FeIII(OH) Complex and Its Reactivity with Carbon Radicals. J. Am. Chem. Soc 2019, 141, 10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Addison AW; Rao TN; Reedijk J; van Rijn J; Verschoor GC Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc., Dalton Trans 1984, 1349. [Google Scholar]
- (32).Gordon Z; Miller TJ; Leahy CA; Matson EM; Burgess M; Drummond MJ; Popescu CV; Smith CM; Lord RL; Rodríguez-López J; Fout AR Characterization of Terminal Iron(III)–Oxo and Iron(III)–Hydroxo Complexes Derived from O2 Activation. Inorg. Chem 2019, 58, 15801. [DOI] [PubMed] [Google Scholar]
- (33).Cook SA; Ziller JW; Borovik AS Iron(II) Complexes Supported by Sulfonamido Tripodal Ligands: Endogenous versus Exogenous Substrate Oxidation. Inorg. Chem 2014, 53, 11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).MacBeth CE; Gupta R; Mitchell-Koch KR; Young VG; Lushington GH; Thompson WH; Hendrich MP; Borovik AS Utilization of Hydrogen Bonds To Stabilize M–O(H) Units: Synthesis and Properties of Monomeric Iron and Manganese Complexes with Terminal Oxo and Hydroxo Ligands. J. Am. Chem. Soc 2004, 126, 2556. [DOI] [PubMed] [Google Scholar]
- (35).Mukherjee J; Lucas RL; Zart MK; Powell DR; Day VW; Borovik AS Synthesis, Structure, and Physical Properties for a Series of Monomeric Iron(III) Hydroxo Complexes with Varying Hydrogen-Bond Networks. Inorg. Chem 2008, 47, 5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Soo HS; Komor AC; Iavarone AT; Chang CJ A Hydrogen-Bond Facilitated Cycle for Oxygen Reduction by an Acid- and Base-Compatible Iron Platform. Inorg. Chem 2009, 48, 10024. [DOI] [PubMed] [Google Scholar]
- (37).Ogo S; Yamahara R; Roach M; Suenobu T; Aki M; Ogura T; Kitagawa T; Masuda H; Fukuzumi S; Watanabe Y Structural and Spectroscopic Features of a cis (Hydroxo)-FeIII- (Carboxylato) Configuration as an Active Site Model for Lipoxygenases. Inorg. Chem 2002, 41, 5513. [DOI] [PubMed] [Google Scholar]
- (38).Ogo S; Wada S; Watanabe Y; Iwase M; Wada A; Harata M; Jitsukawa K; Masuda H; Einaga H Synthesis, Structure, and Spectroscopic Properties of [Feiii(tnpa)(OH)(PhCOO)]ClO4: A Model Complex for an Active Form of Soybean Lipoxygenase-1. Angew. Chem., Int. Ed 1998, 37, 2102. [DOI] [PubMed] [Google Scholar]
- (39).Drummond MJ; Ford CL; Gray DL; Popescu CV; Fout AR Radical Rebound Hydroxylation Versus H-Atom Transfer in Non-Heme Iron(III)-Hydroxo Complexes: Reactivity and Structural Differentiation. J. Am. Chem. Soc 2019, 141, 6639–6650. [DOI] [PubMed] [Google Scholar]
- (40).Cheruzel LE; Wang J; Mashuta MS; Buchanan RM Structure and properties of an Fe(iii) complex containing a novel amide functionalized polyimidazole ligand. Chem. Commun 2002, 2166. [DOI] [PubMed] [Google Scholar]
- (41).Çelenligil-Çetin R; Paraskevopoulou P; Dinda R; Staples RJ; Sinn E; Rath NP; Stavropoulos P Synthesis, Characterization, and Reactivity of Iron Trisamidoamine Complexes That Undergo Both Metal- and Ligand-Centered Oxidative Transformations. Inorg. Chem 2008, 47, 1165. [DOI] [PubMed] [Google Scholar]
- (42).MacBeth CE; Golombek AP; Young VG; Yang C; Kuczera K; Hendrich MP; Borovik AS O2 Activation by Nonheme Iron Complexes: A Monomeric Fe(III)-Oxo Complex Derived From O2. Science 2000, 289, 938. [DOI] [PubMed] [Google Scholar]
- (43).Goldsmith CR; Stack TDP Hydrogen Atom Abstraction by a Mononuclear Ferric Hydroxide Complex: Insights into the Reactivity of Lipoxygenase. Inorg. Chem 2006, 45, 6048. [DOI] [PubMed] [Google Scholar]
- (44).Gupta R; Lacy DC; Bominaar EL; Borovik AS; Hendrich MP Electron Paramagnetic Resonance and Mössbauer Spectroscopy and Density Functional Theory Analysis of a High-Spin FeIV–Oxo Complex. J. Am. Chem. Soc 2012, 134, 9775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Mørup S Magnetic Relaxation Phenomena. In Mössbauer Spectroscopy and Transition Metal Chemistry: Fundamentals and Applications; Gütlich P, Bill E, Trautwein AX, Eds.; Springer: Berlin, 2011; pp 201–234. [Google Scholar]
- (46).Zaragoza JPT; Yosca TH; Siegler MA; Moenne-Loccoz P; Green MT; Goldberg DP Direct Observation of Oxygen Rebound with an Iron-Hydroxide Complex. J. Am. Chem. Soc 2017, 139, 13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Iovan DA; Betley TA Characterization of Iron-Imido Species Relevant for N-Group Transfer Chemistry. J. Am. Chem. Soc 2016, 138, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Lu X; Li X-X; Seo MS; Lee Y-M; Clémancey M; Maldivi P; Latour J-M; Sarangi R; Fukuzumi S; Nam W A Mononuclear Nonheme Iron(IV)–Amido Complex Relevant for the Compound II Chemistry of Cytochrome P450. J. Am. Chem. Soc 2019, 141, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Jang ES; McMullin CL; Käß M; Meyer K; Cundari TR; Warren TH Copper(II) Anilides in sp3 C-H Amination. J. Am. Chem. Soc 2014, 136, 10930. [DOI] [PubMed] [Google Scholar]
- (50).Pangia TM; Yadav V; Gérard EF; Lin Y-T; de Visser SP; Jameson GNL; Goldberg DP Mechanistic Investigation of Oxygen Rebound in a Mononuclear Nonheme Iron Complex. Inorg. Chem 2019, 58, 9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.