

CONSPECTUS:
Immune checkpoint blockade (ICB) therapy elicits antitumor response by inhibiting immune suppressor components, including programmed cell death protein 1 and its ligand (PD-1/PD-L1) and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4). Despite improved therapeutic efficacy, the clinical response rate is still unsatisfactory as revealed by the fact that only a minority of patients experience durable benefits. Additionally, “off-target” effects after systemic administration remain challenging for ICB treatment. To this end, the local and targeted delivery of ICB agents instead could be a potential solution to maximize the therapeutic outcomes while minimizing the side effects.
In this Account, our recent studies directed at the development of different strategies for the local and targeted delivery of ICB agents are discussed. For example, transdermal microneedle patches loaded with anti-programmed death-1 antibody (aPD1) and anti-CTLA4 were developed to facilitate sustained release of ICB agents at the diseased sites. Triggered release could also be achieved by various stimuli within the tumor microenvironment, including low pH and abnormally expressed enzymes. Recently, the combination of an anti-programmed death-ligand 1 antibody (aPD-L1) loaded hollow-structured microneedle patch with cold atmospheric plasma (CAP) therapy was also reported. Microneedles provided microchannels to facilitate the transdermal transport of CAP and further induce immunogenic tumor cell death, which could be synergized by the local release of aPD-L1. In addition, in situ formed injectable or sprayable hydrogels were tailored to deliver immunomodulatory antibodies to the surgical bed to inhibit tumor recurrence after primary tumor resection. In paralell, inspired by the unique targeting ability of platelets toward the inflammatory sites, we engineered natural platelets decorated with aPD-L1 for targeted delivery after tumor resection to inhibit tumor recurrence. We further constructed a cell–cell combination delivery platform based on conjugates of platelets and hematopoietic stem cells (HSCs) for leukemia treatment. With the homing ability of HSCs to the bone marrow, the HSC–platelet–aPD1 assembly could effectively deliver aPD1 in an acute myeloid leukemia mouse model. Besides living cells, we also leveraged HEK293T-derived vesicles with PD1 receptors on their surfaces to disrupt the PD-1/PD-L1 immune inhibitory pathway. Moreover, the inner space of the vesicles allowed the packaging of an indoleamine 2,3-dioxygenase inhibitor, further reinforcing the therapeutic efficacy. A similar approach has also been demonstrated by genetically engineering platelets overexpressing PD1 receptor for postsurgical treatment. We hope the local and targeted ICB agent delivery methods introduced in this collection would further inspire the development of advanced drug delivery strategies to improve the efficiency of cancer treatment while alleviating side effects.
Graphical Abstract
1. INTRODUCTION
The ongoing gained insights from the different pathways that tumors leverage to evade human immune surveillance have led to a paradigm shift for oncology treatment.4 A critical mechanism underlying the immunosuppression is the immune checkpoint, which is responsible for the maintenance of self-tolerance to protect the host from autoimmunity.5 However, in carcinogenesis, the dysregulated expression of immune checkpoint proteins results in immune resistance against antigen-specific T cells.6 Two important negatively regulated immune checkpoint pathways, PD-1/PD-L1 and CTLA-4/B7, are prime targets for blockade and have been well studied clinically in the past decades. PD-1 and its ligands primarily function to attenuate T cell response and thereby induce peripheral tolerance.7–9 Interaction of PD-1/PD-L1 attenuates the T-cell receptor (TCR) downstream signal and decreases T cell activation via tyrosine phosphorylation of the PD-1 cytoplasmic domain and recruitment of Src homology region 2 domain-containing phosphatase-2 (SHP-2).10 CTLA-4 is an activation-induced molecule expressed on the surface of late-stage T cells that competitively binds to B7 molecules (CD80 and CD86) to suspend the positive co-stimulation through the CD28/B7 pathway.6 CTLA-4 is constitutively overexpressed on regulatory T cells (Tregs), which inhibits the initiation of T cell response against tumors.11,12 It has also been reported that CTLA4 is expressed in a variety of cells including activated B cells, placental fibroblasts, and monocytes, as well as several human tumor cells, expanding its possible role in immune regulation related to other cells.13,14 Other checkpoints, including CD47, lymphocyte activation gene 3, T cell immunoglobulin and mucin domain 3, diacylglycerol kinase α, and V-set immunoglobulin domain suppressor of T cell activation, have also been recognized for their regulatory roles in tumor immunity.15 With these in mind, blocking the interaction between ligands and receptors using monoclonal antibodies has been proven to be an impressive breakthrough in the clinic.16 To date, several immune checkpoint blockade (ICB) agents, such as ipilimumab, pembrolizumab, and nivolumab, have been approved by the U.S. FDA for treating a variety of tumors, including melanoma, advanced renal cell carcinoma, metastatic non-small-cell lung cancer, and metastatic bladder cancer.17–19
Although the therapeutic efficacy of ICB for many advanced tumors has been demonstrated to be superior to that of other treatment options, satisfactory treatment outcome was achieved only in a minority of patients.20,21 Meanwhile, these ICB therapies may generate immune-related adverse events (irAEs) of different grades, which could become life-threatening.22,23 The mechanism underlying the irAEs is possibly linked to the disruption of immunologic homeostasis.23 Dose-limiting auto-immune pathologies were observed in the systemic administration of ICB agents, reflecting the overactivation of the immune system.24 According to a meta-analysis of up to 1265 patients receiving ipilimumab in 2015, dose-dependent risk of irAEs was discovered, with incident of all-grade irAEs for patients receiving 3 mg/kg ipilimumab being 61% and that for patients receiving 10 mg/kg rising to 79%.22 Alternatively, the local and targeted delivery of immunomodulatory antibodies has been developed in recent years as promising ways to overcome these issues.25,26 Through various drug delivery systems, the pharmacokinetics and pharmacodynamics of ICB agents can be tuned to maximize treatment efficacy and reduce systemic side effects. Additionally, other immunomodulatory therapeutics can be readily co-delivered to turn the tumor microenvironment from cold to hot for synergistic treatment response.16 In this Account, we have summarized our recent studies in developing techniques for local and targeted delivery of ICBs, mainly focusing on synthetic material-based and cellular engineering-based strategies (Figure 1).
Figure 1.
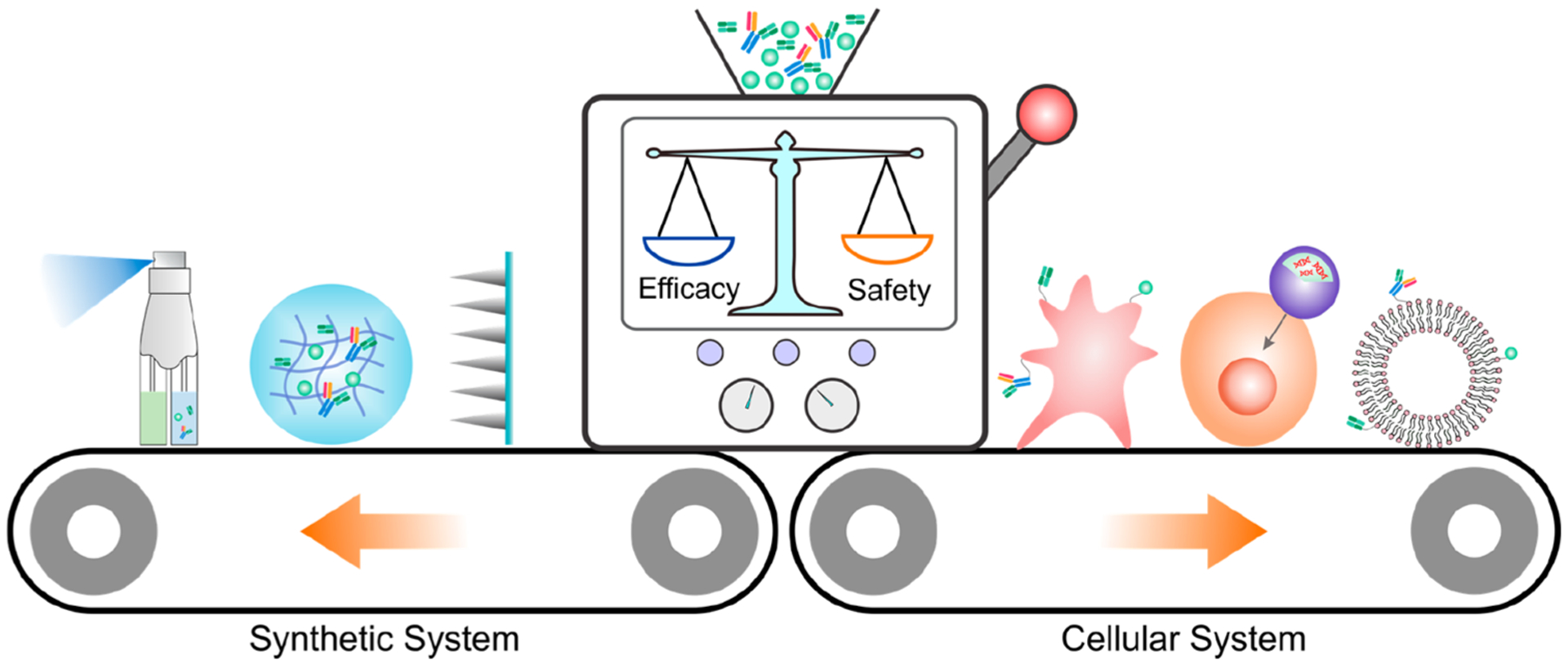
Schematic illustration of different strategies for the local and targeted delivery of ICB agents, with a focus on the synthetic system and cellular system.
2. LOCAL DELIVERY WITH SYNTHETIC SYSTEMS
With tunable drug loading capacity, controlled release ability, and unique hierarchical structures, synthetic drug delivery systems, such as microneedles and hydrogel depots, have been proposed for local delivery applications.27 In this context, a variety of synthetic systems for local delivery of ICB agents have been developed with goal of maximized maintenance of the immunotherapeutic window as well as reducing systemic toxicity.
2.1. Microneedle Patch
Microneedle (MN) patches, as representative painless transdermal drug delivery systems, have been utilized for many different biomedical applications. By adjustment of the formulations, MNs could facilitate the transdermal local delivery of distinct cargoes, ranging from proteins to small molecules, to achieve controlled and sustained cargo release.28,29 Our group designed biodegradable microneedle (MN) patches for the minimally invasive localized delivery of anti-programmed death-1 antibody (aPD1) for melanoma treatment (Figure 2).30 In this design, the MN was integrated with pH-sensitive dextran nanoparticles loaded with glucose oxidase (GOx) and aPD1 antibody (MN-GOx-aPD1). The decrease in pH caused by generation of gluconic acid from glucose oxidation promoted self-degradation of the nanoparticles within the MN, which enabled sustained release of aPD1 within the tumor environment (Figure 2A–C). Furthermore, testing in the in vivo mouse melanoma model proved the superior tumor inhibition ability of a single MN administration compared to intratumoral injection with the same dosage (1 mg/kg) (Figure 2D). As revealed by immunofluorescence staining, the CD8+ and CD4+ T cell infiltration of the tumor treated with MN-GOx-aPD1 was significantly higher than that in the untreated tumors (Figure 2E). In addition, the flexibility of MN fabrication easily enabled co-delivery of two or more ICB agents to achieve a synergistic therapeutic effect. Such design provides a MN-based GOx/CAT enzymatic system to facilitate sustained release of ICB therapeutics to modulate the immunosuppressive microenvironment.
Figure 2.
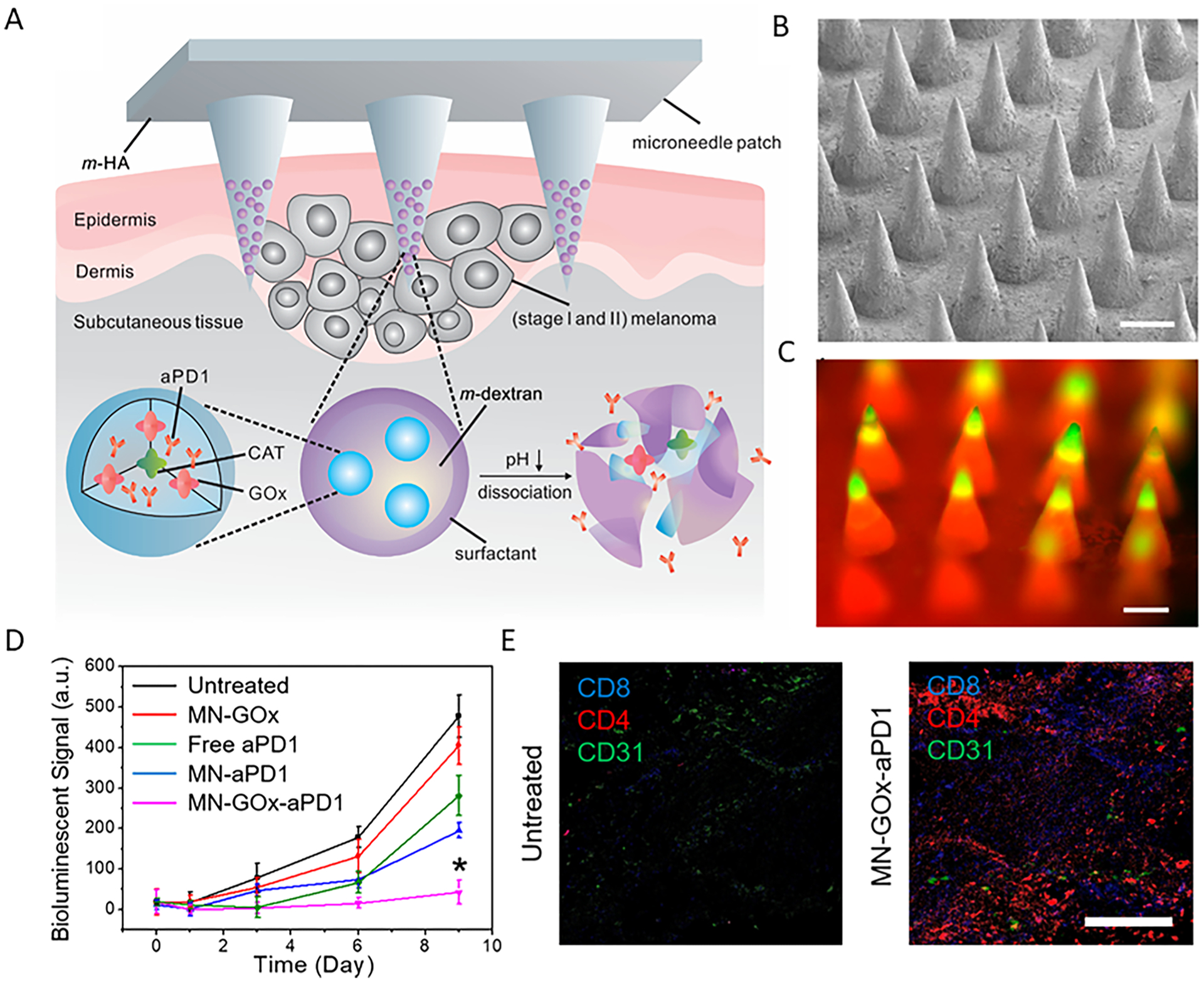
(A) Schematic summary of MN patch-based aPD1 delivery platform. m-HA, matrix hyaluronic acid; CAT, catalase. (B) Representative MN patch under scanning electron microscopy (scale bar 200 μm). (C) Fluorescence microscopy image of the FITC-labeled antibody containing MN patch (scale bar 200 μm). (D) Quantified tumor bioluminescence intensity of B16F10 tumors after different treatments, *P < 0.05. (E) Immunofluorescence of tumor sections to demonstrate the infiltration of CD4+ T cells and CD8+ T cells (scale bar 100 μm). Reprinted and modified with permission from 30. Copyright © 2016, American Chemical Society.
In another study, we tested a MN-based delivery strategy for blockade of PD1 and indoleamine 2,3-dioxygenase (IDO).31 IDO is an immunosuppressive molecule that catalyzes tryptophan degradation along the kynurenine pathway, which creates an immunosuppressive milieu by directly limiting T cell function and recruiting regulatory T cells to the tumor. PD1 antibody was encapsulated in the hyaluronic acid nanoparticles modified by 1-methyl-DL-tryptophan (1-MT), which is an inhibitor of IDO and could be further released in response to hyaluronidase at the tumor sites. The formulation was based on the drug delivery strategy of “drug A in carriers formed by incorporation of drug B”. The platform exhibited prolonged retention of drugs at the tumor sites, which could limit the side effects caused by off-target effects. By utilizing a B16F10 mouse melanoma tumor model, the combination treatment significantly delayed tumor growth and prolonged mouse survival, indicating that synergistic effects of IDO and PD-1 blockade could elicit robust immune response against melanoma.
Recently, we further exploited a combination therapy of cold atmospheric plasma (CAP) and ICB therapy based on a hollow structured MN platform (Figure 3A,B).3 CAP, an ionized gas at near room temperature, has shown promising potential in biomedical applications, especially for cancer treatment.32 The main factor underlying the anticancer ability of CAP is the ratio of reactive oxygen species (ROS) and reactive nitrogen species (RNS).33 Hollow-structured microneedles (hMNs) were implemented as microchannels to facilitate the transdermal penetration of CAP to the tumor tissues and further induce immunogenic death of tumor cells (Figure 3C–F). Afterward, the released tumor-associated antigens (TAAs) could in turn serve as “danger signals” to promote the maturation of dendritic cells (DCs), which then induce T cell-mediated antitumor immunity. The generated immune response was further augmented by the aPD-L1 loaded inside the hMNs. In contrast, CAP alone and CAP/solid-structured MNs (sMNs) did not delay the growth of tumor, probably due to the low penetration of CAP (Figure 3G). The T cell infiltration inside the tumor corresponds with the therapeutic outcome in mice with different treatments, as both CD4+ and CD8+ T cell infiltration greatly increased after combination treatment with CAP and aPD-L1–hMNs (Figure 3H,I). To check the induction of tumor-specific systemic immune responses, we studied a dual-tumor model with only one side treated with CAP/aPD-L1-hMN, where the growth of a distant tumor in mice treated with combination therapy of CAP and ICB was inhibited as well (Figure 3J). These results suggest that the flexibility in microneedle design allows the augmentation of therapeutic efficacy of local delivery of an ICB inhibitor by combination with other therapeutic moduli such as transdermal CAP treatment.
Figure 3.
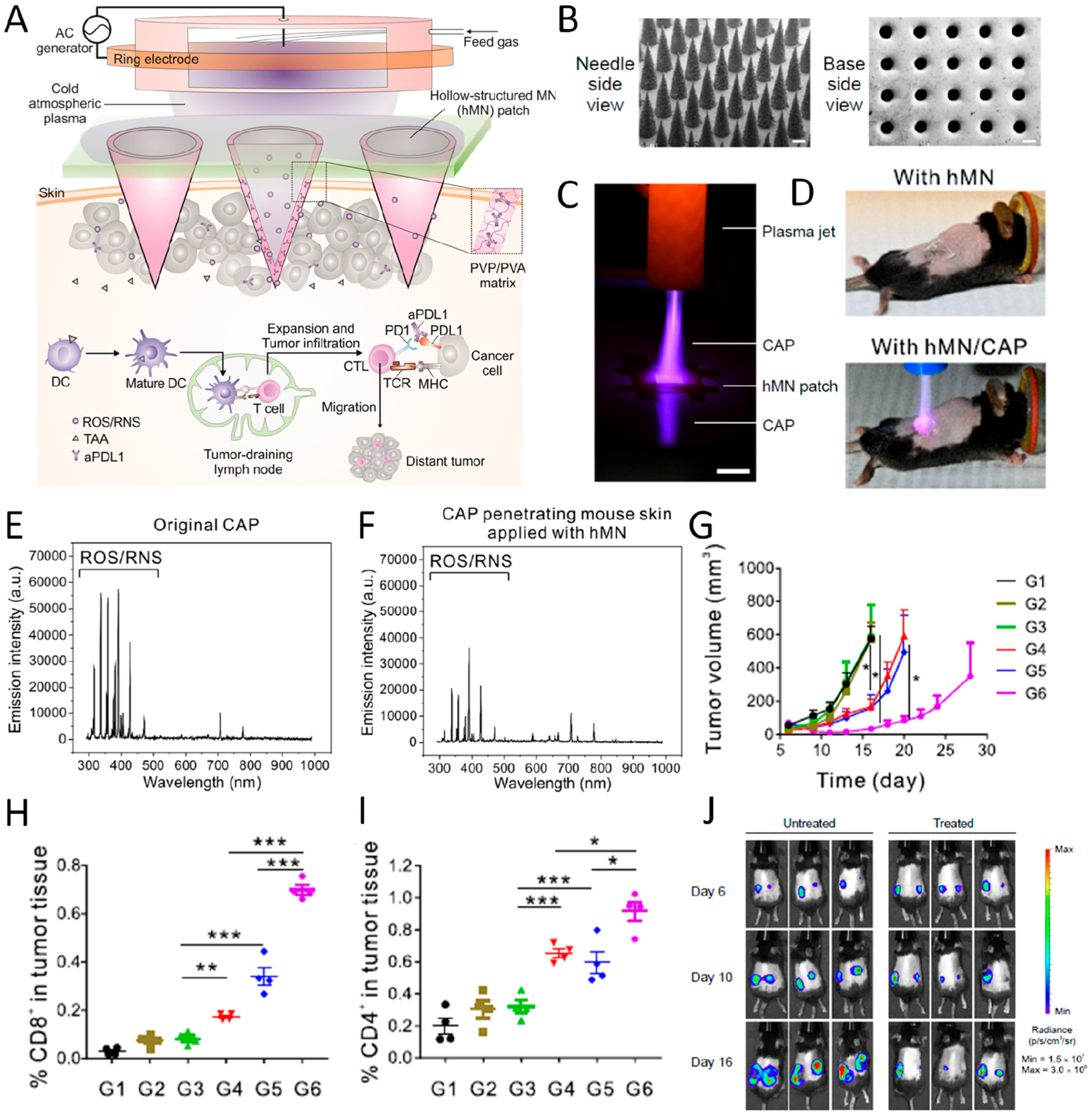
(A) Illustration of hollow-structured microneedle (hMN)-mediated cold atmospheric plasma (CAP) and ICB therapy. TAA, tumor-associated antigen; PVP, poly(vinyl pyrrolidone); PVA, poly(vinyl alcohol). (B) SEM images of MNs from top and bottom side (scale bar 200 μm). (C) Photograph of CAP penetrating through hMN (scale bar 1 cm). (D) Images of mice treated with hMN and hMN combined with CAP therapy. (E, F) Optical emission spectrum of original CAP and CAP penetrating mouse skin applied with hMN. (G) Tumor growth curve of mice with different treatments including untreated (G1) and treated with CAP (G2), CAP with solid-structured microneedle (sMN) (G3), CAP with hMN (G4), hMN loaded with aPD-L1 (G5), and CAP with hMN loaded with aPD-L1 (G6). (H, I) Percentage of CD8+ and CD4+ T cells in tumor 3 days after treatment. Data are presented as mean ± SEM. (J) Bioluminescence image of untreated mice and mice treated with CAP and hMN-aPD-L1 in a distant tumor model. Data are presented as mean ± SEM *P < 0.05; **P < 0.01; ***P < 0.001. Reprinted and modified with permission from ref 3. Copyright 2020 National Academy of Sciences.
2.2. Hydrogel
Hydrogels are typically a three-dimensional cross-linked porous network of polymers.34 Owing to their unique properties such as high biocompatibility, diverse components, and tunable mechanical properties, hydrogels have sparked particular interest in the application of drug delivery, tissue engineering, and hygiene products.27
For ICB therapy, our group developed a ROS-degradable platform for local co-delivery of ICB antibodies and chemotherapeutic agents. The hydrogel was formed in situ quickly after mixing N1-(4-boronobenzyl)-N3-(4-boronophenyl)- N1,N1,N3,N3-tetramethylpropane-1,3-diaminium (TSPBA) with poly(vinyl alcohol) (PVA) (Figure 4).1 Gemcitabine (GEM), a chemotherapeutic agent that can induce tumor cell immunogenic death upon activation of the immune system, was coloaded with aPD-L1 inside the hydrogel (Figure 4A–C). According to the in vitro result, GEM and antibody were released at a higher rate when incubated in H2O2 solution compared to that in PBS, due to the oxidization and hydrolysis of the TSPBA part. Notably, more than 90% of GEM was released within 10 h, while the antibody exhibited a slower and sustained release, with approximate 30% of antibody released within 10 h and 70% antibody released within 3 days (Figure 4D,E). The hydrogel could serve not only as a reservoir for the therapeutic agents but also as a sponge for ROS scavenging, which could further reinforce the therapeutic efficacy by mediating the immunosuppressive tumor microenvironment. B16F10 melanoma-bearing mice peritumorally implanted with aPD-L1-GEM@ Gel showed significant tumor inhibition, with 50% survival after 60 days; while for other control groups, none of the mice survived over the same time (Figure 4F). Much higher infiltration of CD4+ and CD8+ T cells was detected in the tumor residue compared with untreated tumors, highlighting the reverse in the immunosuppressive tumor microenvironment (TME) elicited by local delivery (Figure 4G). Afterward, we further evaluated the systemic immune response using a dual-tumor model with the left side treated with aPD-L1-GEM@Gel. The growth of both hydrogel-implanted and untreated tumor was significantly inhibited compared to the blank group, suggesting that the platform could not only promote tumor regression but also elicit systemic immune response and T cell memory (Figure 4H).
Figure 4.
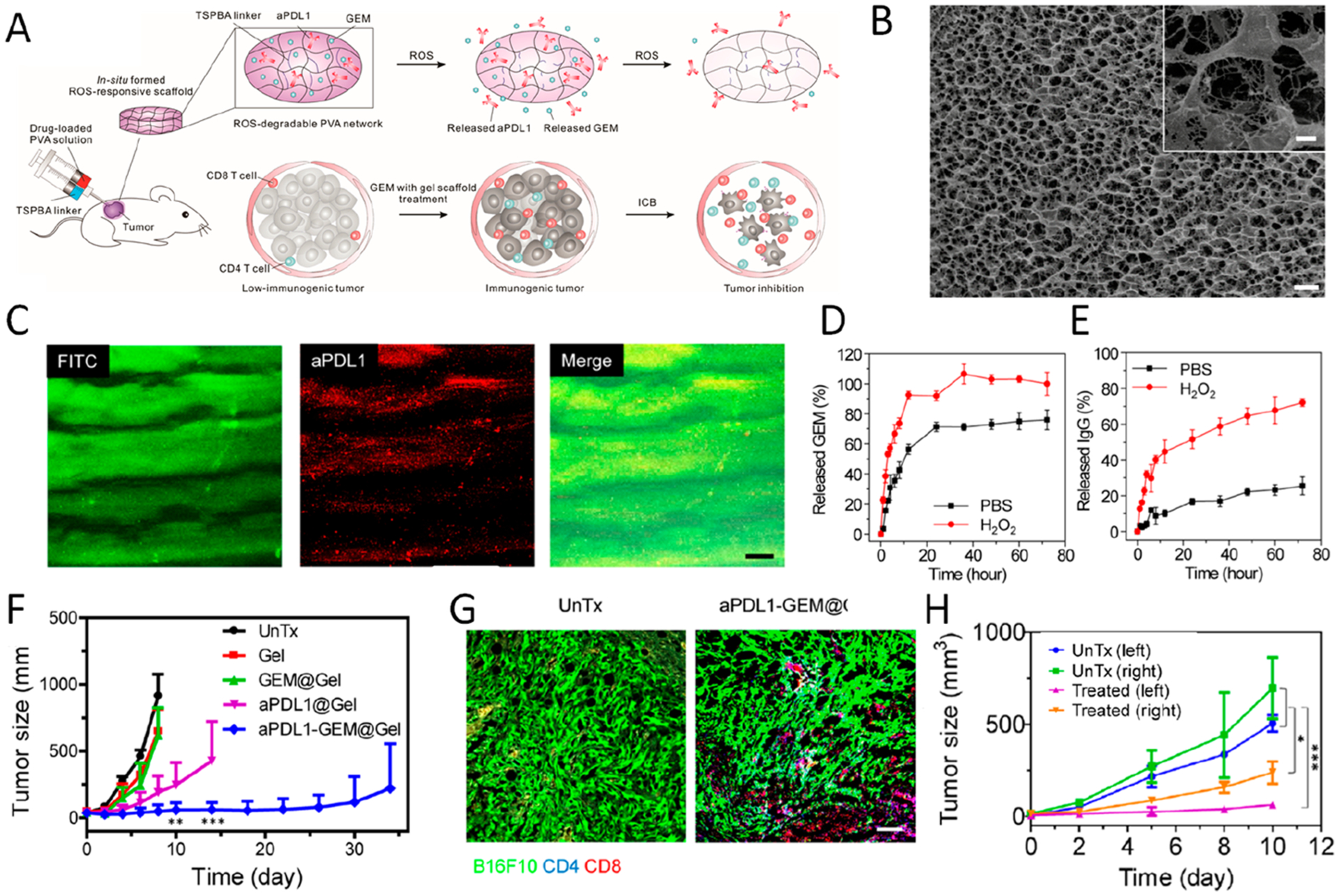
(A) Schematic summary of local co-delivery of GEM and aPD-L1 into the tumor microenvironment using a ROS-degradable hydrogel scaffold. (B) Cryo-scanning electron microscope (Cryo-SEM) image of gel scaffold loaded with GEM and aPD-L1 (scale bar 0.5 μm). Inset, magnified image of the scaffold (scale bar 0.1 μm). (C) ROS-degradable hydrogel under fluorescent microscopy; green, FITC-GEM; red, Cy5.5–aPD-L1 (scale bar 25 μm). (D, E) Release profiles of GEM and aPD-L1 from hydrogels in the presence or absence of H2O2 (1 mM). Data are mean ± SEM. (F) Average tumor growth curves of mice with different treatments. Growth kinetics represent mean ± SEM. (G) Immunofluorescence staining of tumor sections showing infiltration of CD4+ and CD8+ T cells (scale bar 100 μm). (H) Growth curve of left and right tumors. Control mice were untreated, whereas in treated group, hydrogels were implanted only on the left tumor. *P < 0.05; **P < 0.01; ***P < 0.005. Reprinted and modified with permission from ref 1. Copyright 2018 American Association for the Advancement of Science.
In another study, we designed an in situ formed dual-responsive hydrogel depot to co-deliver aPD1 and Zebularine (Zeb), a DNA methyltransferase inhibitor. Zeb and CaCO3 nanoparticles loaded with aPD1 were coencapsulated into a hydrogel formed by cross-linking PVA with TSPBA. The dual-responsive payload release could be triggered in the ROS-rich and acidic TME. Meanwhile, the local release of Zeb could induce TAA release, which further augmented the antitumor immunity in combination with PD-1 blockade.35
We also harnessed polypeptide-based hydrogels for controlled and sustained payload release.36 A triblock copolymer containing a central poly(ethylene glycol) and two flanked polypeptide blocks was synthesized, P(Me-D-1MT)-PEG-P(Me-D-1MT), where Me and D-1MT represent l-methionine and dextro-1-methyl tryptophan, respectively. The copolymer solution underwent sol-to-gel transition in response to higher temperature. The sulfoether group within the Me-containing polypeptide could be oxidized to sulfoxide or sulfone in response to H2O2, thus triggering the transition to polymeric hydrophilicity (Figure 5A–C). Such structural change enabled the intratumoral release of D-1MT, as well as aPD-L1. After intratumoral injection, the in situ formed hydrogel enabled the prolonged retention of aPD-L1 in the tumor site compared with free aPD-L1 (Figure 5D). Moreover, the therapeutic efficacy of aPD-L1-encapsulating hydrogel was further confirmed in a mouse melanoma model. Only limited tumor inhibition was observed in the mice treated with blank hydrogel or free aPD-L1 and D-1MT. In contrast, the tumor growth in mice intratumorally injected with aPD-L1 loaded hydrogel was distinctly delayed. The improved therapeutic outcome was also associated with increased infiltration of T cells inside the tumor (Figure 5E–G). When the cytotoxicity was assessed, some pathologic damage was observed in the spleen of free D-1MT and aPD-L1 treated mice, probably due to the toxicity associated with high administered dosage. Meanwhile, the ex vivo H&E staining of major organs of aPD-L1-loaded gel treated mice did not exhibit significant damage and inflammation as compared with control groups, further demonstrating that the systemic side effects were reduced by the gel system.
Figure 5.
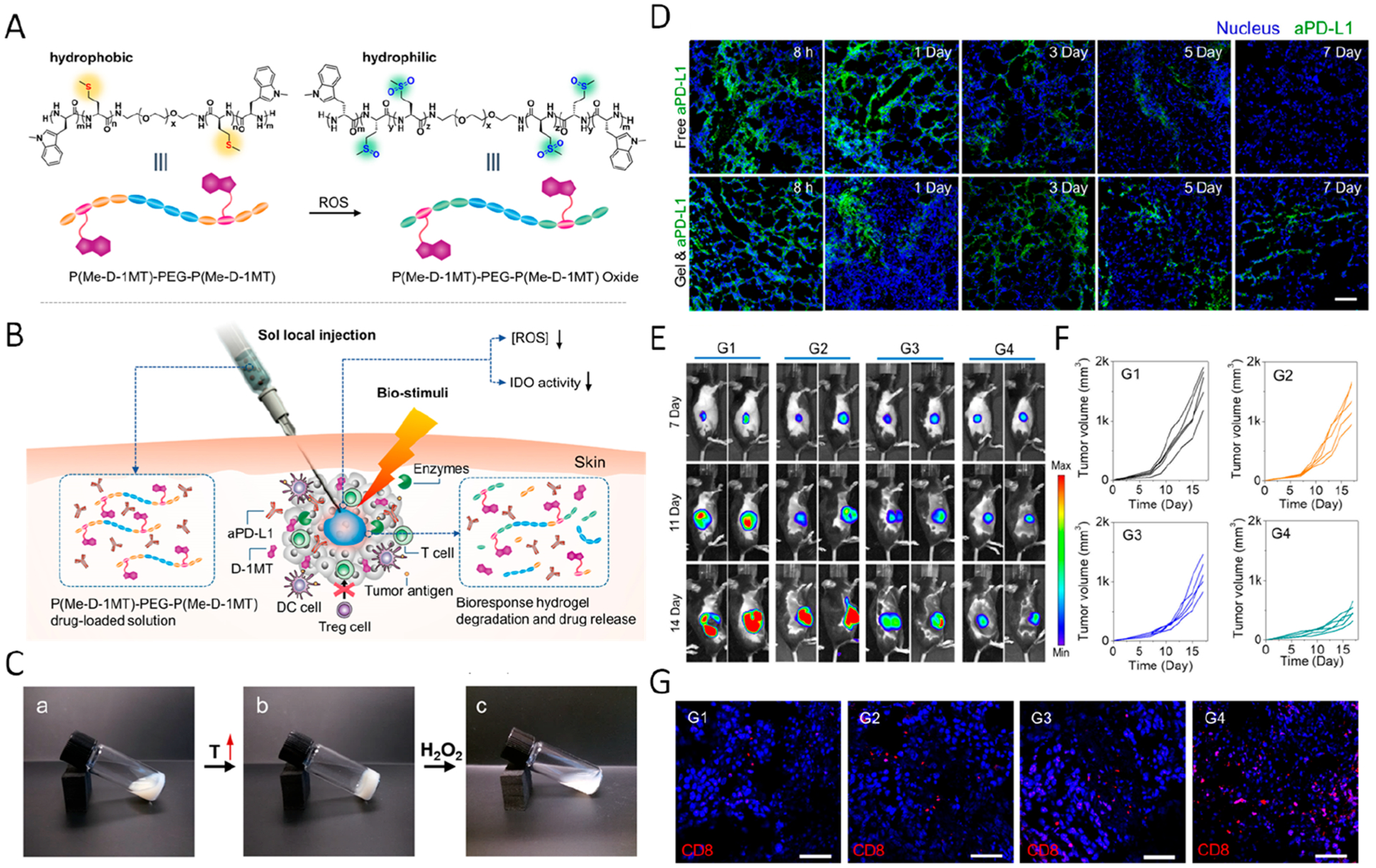
(A) Polymeric hydrophobicity transformation of P(Me-D-1MT)–PEG–P(Me-D-1MT) triggered by ROS. (B) Schematic summary of in situ formed polypeptide hydrogels for controlled release of the cargo. (C) Phase change of the gel in response to increasing temperature and H2O2. (D) Fluorescent imaging showing the local retention of aPD-L1 under different formations. (E) Bioluminence imaging showing tumor growth in mice with different treatments (G1, PBS; G2, blank P(Me-D-1MT)–PEG–P(Me-D-1MT) hydrogel; G3, free aPD-L1 and D-1MT; G4, a-PD-L1 loaded P(Me-D-1MT)–PEG–P(Me-D-1MT) hydrogel) (F) Tumor growth curve in different groups. (G) Immunofluorescence imaging demonstrating CD8+ T cell infiltration. Reprinted and modified with permission from ref 36. Copyright 2018 John Wiley & Sons, Inc.
In order to achieve a simpler in situ gelation method, we also engineered a sprayed fibrin gel incorporating anti-CD47-containing CaCO3 nanoparticles (aCD47@CaCO3@fibrin) as a convenient administration method for postsurgical treatment (Figure 6).37 CD47 is a transmembrane protein that inhibits phagocytosis by binding to signal regulatory protein α (SIRPα) expressed on macrophages, dendritic cells, and neutrophils. As abundant CD47 expression was observed in various types of tumors, inhibiting the CD47–SIRPα pathway would be promising to promote the phagocytosis of tumor cells by macrophages.38 In this design, upon mixing fibrinogen and thrombin solutions by using a dual-channel sprayer, an immunotherapeutic gel could be formed in situ instantly at the tumor resection sites (Figure 6A). CaCO3 nanoparticles could serve as reservoirs for tuning the release of aCD47 and scavenging protons to modulate the acidity of the tumor sites as well (Figure 6B–E). By virtue of an incomplete tumor resection mouse model, the recurrence of tumor residues sprayed with aCD47@CaCO3@fibrin were significantly inhibited, and 50% of the mice in this group had no detectable recurrence and survived after two months (Figure 6F). By analyzing the cells in the tumor residues treated with sprayed hydrogel, we discovered that the infiltration of both M1- and M2-type macrophages was increased after CD47 inhibition. Notably, the proportion of both CD8+ and CD4+ T cells in the tumor residue increased compared with the control groups. In order to evaluate the systemic immune response by aCD47@ CaCO3@fibrin, a dual-tumor model was used, with only one side of the tumor resected incompletely (Figure 6G). Local recurrence as well as the growth of the distant tumor was inhibited after treatment of aCD47@CaCO3@fibrin (Figure 6H). Compared with untreated control, no significant toxic effect was observed in mice at 1, 7, and 14 days after aCD47@CaCO3@fibrin treatment, as evidenced by normal levels of blood cell counts and hemoglobin, as well as various serum biochemistry data. Consistently, the infiltration of M1-type macrophage and CD8+ T cells was increased after treatment compared with those in untreated groups, indicating that CD47 blockade also triggers systemic immune response by enhanced presentation of tumor-specific antigens on antigen presenting cells.
Figure 6.
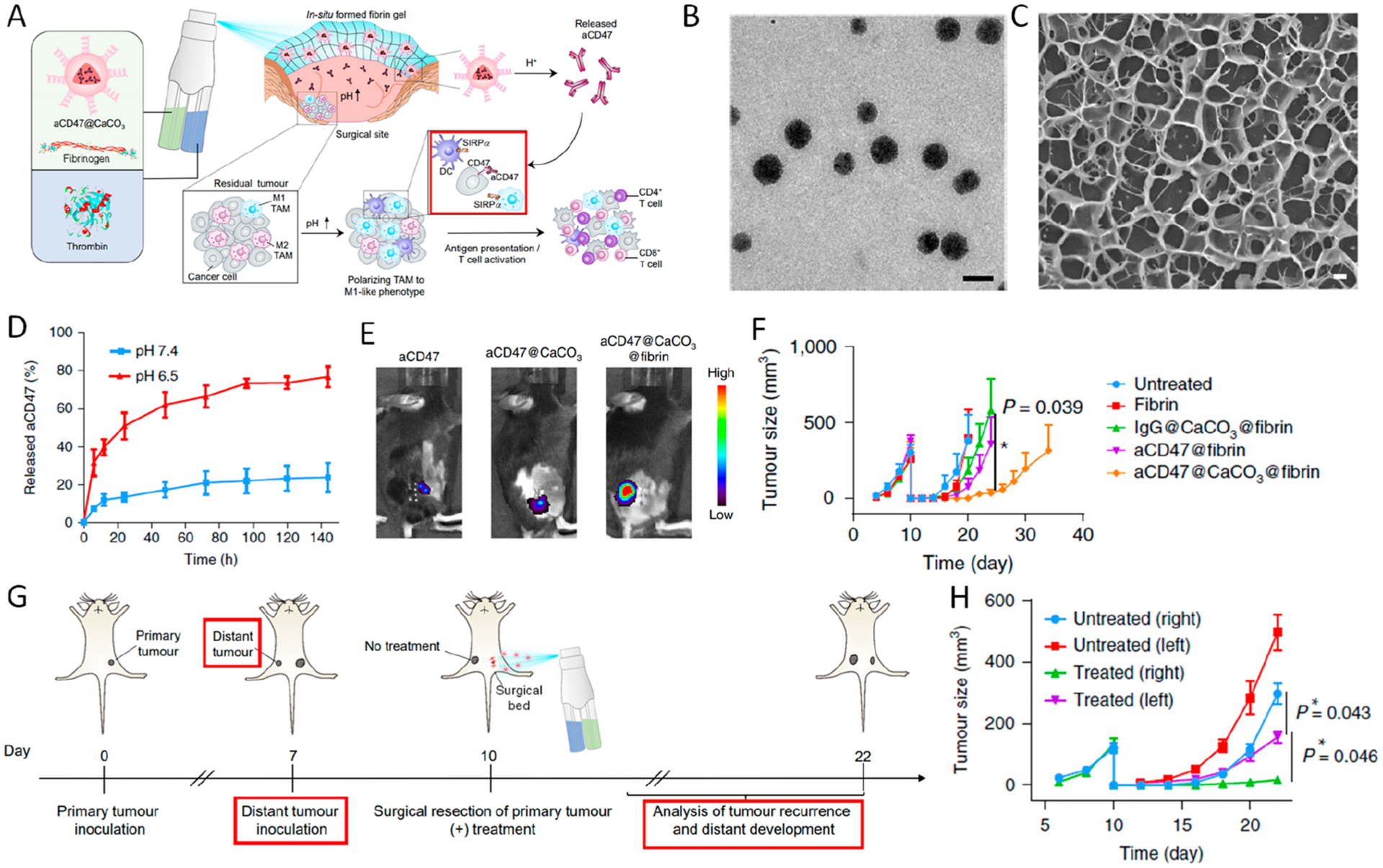
(A) Schematic summary of in situ spraying of aCD47@CaCO3 nanoparticle-containing fibrin gel in the postsurgery tumor bed. (B) Transmission electron microscopy (TEM) image of aCD47@CaCO3 nanoparticles (scale bar 100 nm). (C) Cryo-SEM images of fibrin gel loaded with aCD47@CaCO3 nanoparticles (scale bar 1 μm). (D) Cumulative release profiles of aCD47 from fibrin at different pH. Data are presented as mean ± SEM. (E) In vivo retention of Cy5.5-labeled aCD47 administered in different formulations after 6 days. (F) Average tumor growth curves in different groups. (G) Schematic illustration of aCD47@CaCO3@fibrin therapy in a mouse model of incomplete tumor resection and distant tumor. (H) Growth curves for left and right tumors in untreated and treated mice. Data are presented as mean ± SEM, *P < 0.05. Reprinted and modified with permission from ref 37. Copyright 2019 Springer Nature Limited.
Together, these results highlight that the versatility in hydrogel design using different blocks enables co-delivery of immunotherapeutic agents and other drugs to allow control-lable, sustainable, and local elicitation of antitumor immunity.
3. TARGETED DELIVERY WITH CELLULAR SYSTEMS
In addition to synthetic formulations and devices, drug delivery systems based on natural cellular particulates, which takes advantage of unique host attributes such as specific targeting ability, evasion of the immune system, and cargo release behavior, can also be leveraged for transporting ICB agents.39–41
Platelets, best known for their function in thrombosis, are non-nucleated and terminally differentiated cells released by megakaryocytes (MKs).42,43 Recently, studies have revealed new associations between platelets and inflammatory responses, making platelets the most abundant circulating “immune cells” in the bloodstream.44 The lifespan of platelets circulating in bloodstream is 8–9 days, which means that platelets could be applied as carriers to greatly enhance the pharmacokinetics and distribution of drugs. Moreover, growing evidence has shown that platelets can bind to circulating tumor cells, contributing to the evasion of the immune system and promoting tumor metastasis.45 As the monitor of vascular damage, platelets can migrate to the surgical wound for hemostasis. Inspired by these intrinsic properties of platelets, we conjugated aPD-L1 to the surface of platelets via a chemical linkage for the treatment of postsurgical tumor recurrence (Figure 7A–C).46 The aPD-L1-conjugated platelets (P-aPD-L1) could be recruited to tumor resection sites based on their natural wound-homing capacity (Figure 7D).40 Upon activation, platelet-derived microparticles (PMPs) with aPDL-1 presenting on the surface were released, enriching the available concentration at the tumor site and increasing the bioavailability of aPD-L1 to tumor cells. Compared to mice treated with free aPD-L1, those in the P-aPD-L1 group exhibited a lower signal in the liver, probably due to the targeting ability of platelets to the wound site. Mice bearing triple-negative mammary carcinomas or melanoma were intravenously injected with P-aPD-L1 after tumor resection. It was observed that P-aPD-L1 could facilitate the accumulation of aPD-L1 to the resected side, recruit T cells to the remaining tumor residue, and finally inhibit tumor recurrence (Figure 7E,F). Additionally, according to another B16F10 lung metastasis model, P-aPD-L1 treatment could also reduce lung metastasis (Figure 7G). Although traceable PD-L1 has been reported to be marginally expressed on platelets under normal conditions,47 the amount did not contribute to the therapeutic outcomes. Beyond that, taking advantage of the inflammation targeting ability of platelets also inspired other treatments of different diseases, such as acute myocardial infarction.48,49 Moreover, a combinational therapeutic strategy of platelets with other inflammation-inducing strategies, such as thermal ablation, photodynamic therapy, and radiotherapy, can also be conceived.50
Figure 7.
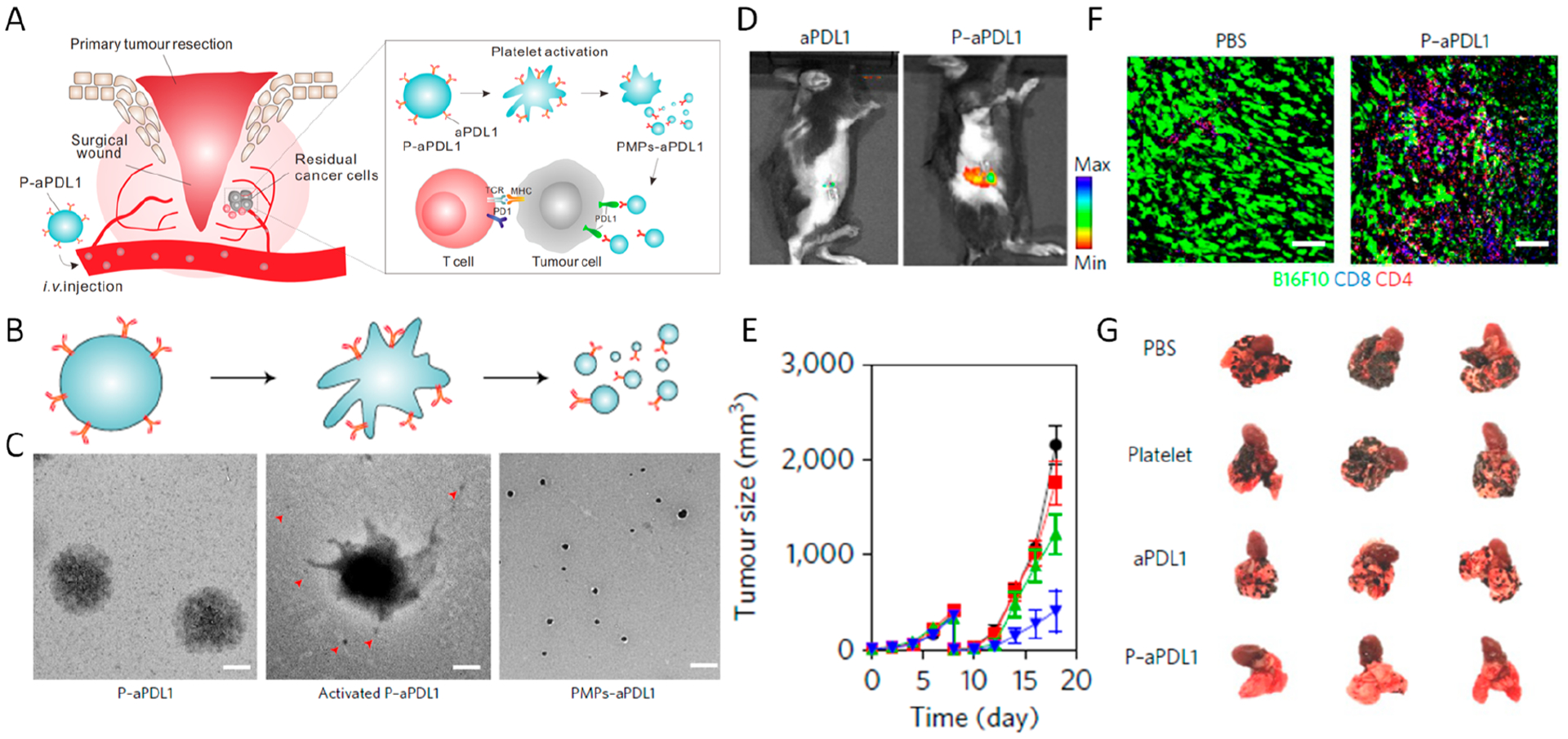
(A) Schematic summary of targeted delivery of aPD-L1 to the tumor resection site using aPD-L1-conjugated platelets. TCR, T cell receptor; MHC, major histocompatibility complex. (B, C) TEM images of P-aPD-L1 before (left) and after (middle and right) activation. The red arrowheads indicate the PMPs released from the platelet (scale bar 0.5 μm). (D) Fluorescence images (aPD-L1–Cy5.5) of mice 2 h after intravenous injection of P-aPD-L1 or an equivalent dose of free aPD-L1. (E) Sizes of recurrent B16F10 tumors with different postsurgical treatments. Data are mean ± SEM. (F) Immunofluorescence staining of residual tumors showing infiltration of CD4+ and CD8+ T cells (scale bar 50 μm). (G) Representative lung photographs in different treatment groups in a melanoma metastasis model. Reprinted and modified with permission from ref 46. Copyright 2017 Springer Nature Limited.
Blood-originated platelets are terminally differentiated anucleate cells; however, the source and flexibility of modification are the major limitations in platelet applications. Toward this issue, in vitro production from MKs can provide a large source of platelets, which can also be genetically modified. In this regard, we further investigated the usage of genetically modified platelets produced by MKs in vitro to express transgenes. In our study, mouse MK progenitor cells L8057 were genetically modified to stably express PD-1, which can further produce PD-1-presenting platelets under stimulation by phorbol 12-myristate 13-acetate (PMA) (Figure 8A,B).51 While blank platelets exhibited limited binding affinity to B16 melanoma cells, PD-1-presenting platelets could strongly bind to B16 cells through PD-1/PD-L1 interactions. Similarly, using an incomplete tumor resection model, we proved that PD-1-presenting platelets could effectively bind to the tumor resection site through thrombosis and PD-1/PD-L1 interactions (Figure 8C). Intravenous infusion of PD-1-presenting platelets greatly reverted the immune-tolerant microenvironment and inhibited the tumor recurrence through a T cell-based mechanism (Figure 8D). As an extension, PD-1 expressing platelets were further loaded with cyclophosphamide, which could deplete Tregs when loaded at a low concentration. Cyclophosphamide-loaded PD-1 expressing platelets could release the cargo upon activation of platelets, further promoting the therapeutic potency by blocking the PD-L1 pathway and depleting Tregs (Figure 8E,F).
Figure 8.
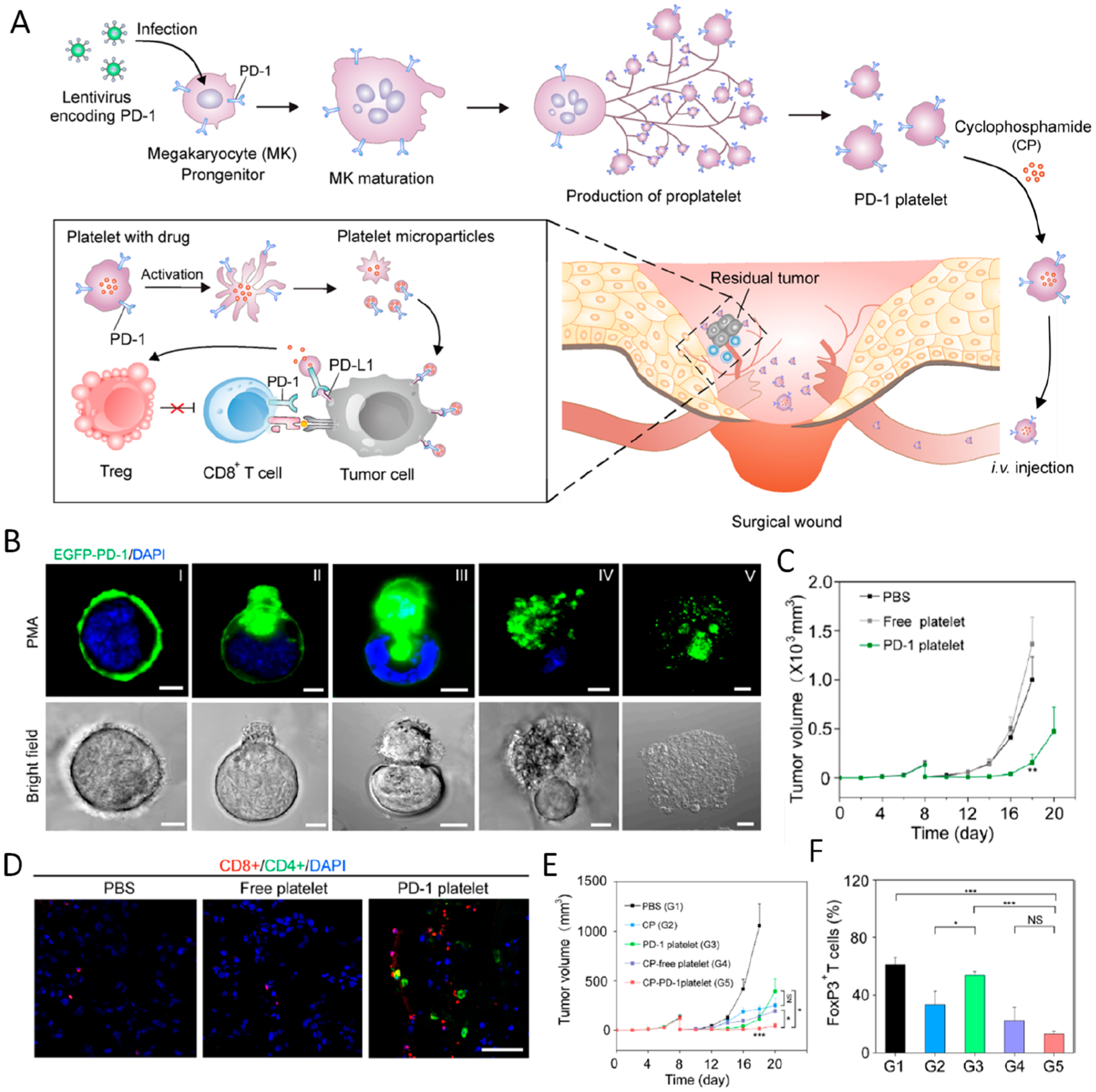
(A) Schematic summary of the production of PD-1-expressing platelets. (B) Evolution process of PD-1-expressing proplatelets from MKs (scale bar 10 μm). (C) Average tumor volumes of mice with different treatments. Data are shown as the mean ± SEM. (D) Immunofluorescence of tumor sections verifying the infiltration of CD4+ and CD8+ T cells (scale bar 100 μm). (E) Average tumor volumes of mice with different treatment groups as indicated. Data are shown as the mean ± SEM. (F) Quantification of FoxP3 expression in CD4+ T cells within the tumors analyzed by flow cytometry (gated on CD4+ T cells). *P < 0.05; **P < 0.01; ***P < 0.005. Reprinted and modified with permission from ref 51. Copyright 2018 American Chemical Society.
Recently, we extended this strategy to generate PD-L1-overexpressing platelets and leveraged them for type-1 diabetes treatment. When administered in vivo, these platelets could accumulate inside pancreas and inhibit autoreactive T cells, thus protecting the β-cells from damage. As a result, the PD-L1-bearing platelets could induce the newly hyperglycemic nonobese diabetic mice to maintain normoglycemia.52
Acute myeloid leukemia (AML) is characterized by a clonal disorder of hematopoietic progenitor cells in the bone marrow.53 Preclinical results suggest that the therapeutic outcome in AML could be optimized by PD-1 blockade.54 Inspired by the homing ability of hematopoietic stem cells (HSCs) to the bone marrow, we developed an HSC–platelet combination cell delivery paradigm to facilitate the delivery of aPD1 to the bone marrow in AML mice (Figure 9).2 In our design, platelets decorated with aPD1, which serve as reservoirs for the antibody, were conjugated to the surface of HSCs through a click chemical linker (Figure 9A,B). After intravenous administration, both the HSC–platelet–aPD1 assembly (S-P-aPD1) and HSC–aPD1 (S-aPD1) exhibited 25-fold greater aPD1 signal in the bone marrow compared with that of other groups at equivalent aPD1 dosage in C57BL/6J mice (Figure 9C). To investigate the in vivo treatment efficacy of S-P-aPD1 against AML, C57BL/6J mice were intravenously injected with C1498 cells and then treated with three doses of saline, free aPD1, S-aPD1, P-aPD1, S + aPD1, and S-P-aPD1. The leukemia bioluminescence signals significantly decreased in mice treated with S-P-aPD1 after 2 weeks. In marked contrast, only minor tumor inhibition was observed in mice treated with aPD1. The limited therapeutic efficacy of P-aPD1 alone was attributed to insufficient bone marrow accumulation. S-aPD1, which exhibited bone marrow homing ability, did not elicit efficient immune response probably due to various reasons, including steric hindrance of cell–cell interaction, internalization of aPD1 by HSCs, and insufficient aPD1 release (Figure 9D). After treatments, both CD3+ and CD8+ T cell levels in the peripheral blood from S-P-aPD1 groups showed significant increase compared to those of groups treated with aPD1, suggesting boost of T-cell immune response by S-P-aPD1 (Figure 9E,F). Of note, when rechallenged with C1498 cells, mice treated with S-P-aPD1 had no detectable tumor growth, while no tumor inhibition was observed in naive mice, indicating the formation of the immune memory. (Figure 9G)
Figure 9.
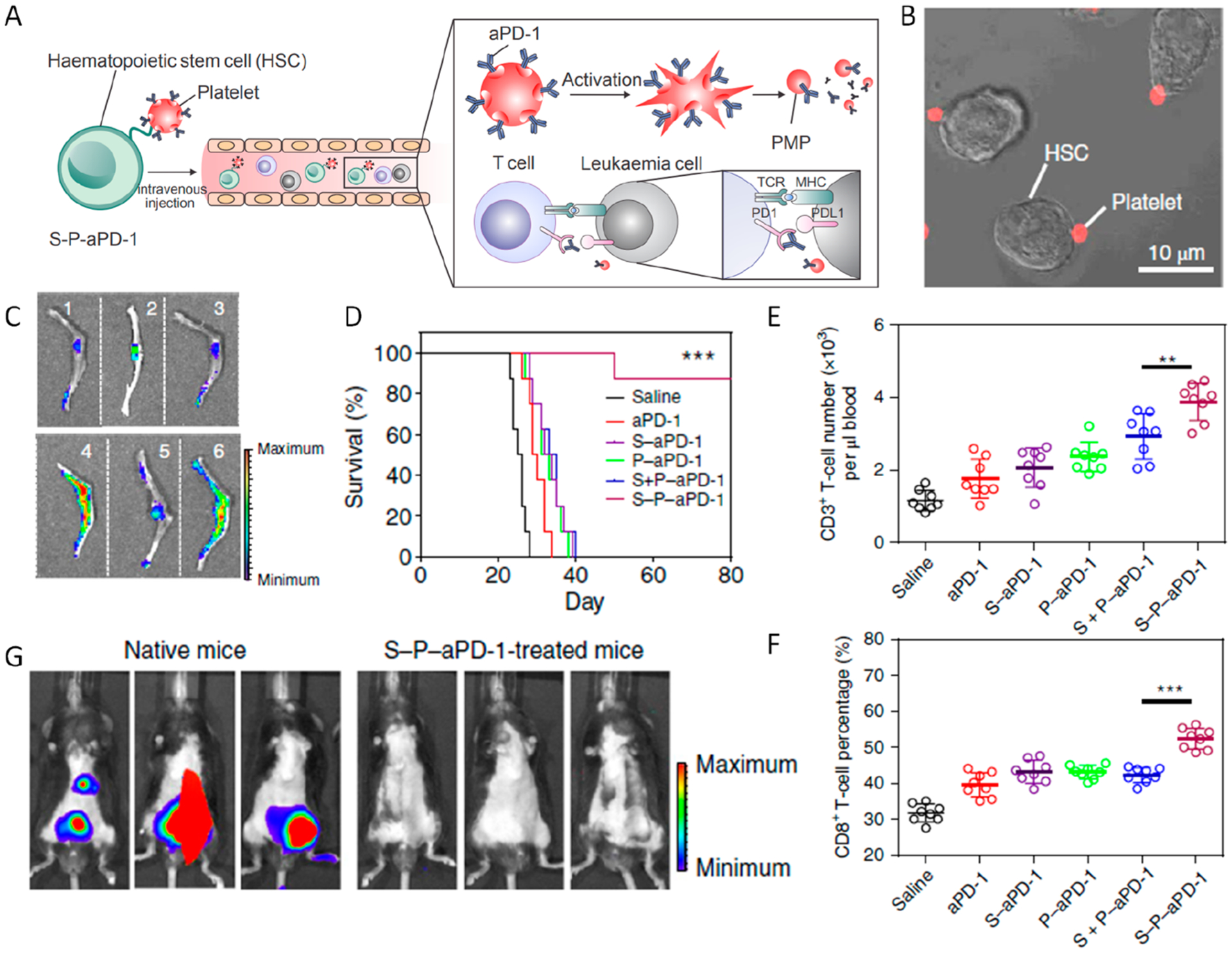
(A) Schematic summary of HSC–platelet dual cell assembly assisted aPD1 delivery. (B) Confocal microscopy image of S-P-aPD1 conjugates. (C) Fluorescence images of bone tissues from mice treated with saline (1), Cy5.5-labeled free aPD1 (2), P-aPD1 (3), S-aPD1 (4), S + P-aPD1 (5), and S-P-aPD1 (6). (D) Survival curves for mice with different groups indicated. (E, F) Quantitative analysis of the percentage of CD3+ and CD8+ T cells. Data are presented as mean ± SD. **P < 0.01; ***P < 0.005. (G) Bioluminescence of mice rechallenged with C1498 cells after 3 weeks. Reprinted and modified with permission from ref 2. Copyright 2018 Springer Nature Limited.
Besides living cells, natural cell-derived vesicles have recently received considerable interest due to their unique properties, including targeting and drug loading abilities.55 For ICB therapy, we developed HEK293T-derived extracellular vesicles as a drug delivery platform and checkpoint inhibitor carriers. HEK293T cells were transfected to stably express PD-1 receptor on the surface.56 PD-1-expressing HEK293T cells were lysed and extruded through a 0.8 μm pore-sized polycarbonate membrane filter to obtain PD-1 bearing nanovesicles (NVs). Next, 1-MT, a small molecule IDO inhibitor, was loaded inside the NVs. When administered via tail vein, PD-1 NVs mainly accumulated in kidney, liver, and tumor sites. The dual blockade of IDO and PD-L1 significantly suppressed the growth of the tumor in a B16F10-luc model.
Therefore, by rationally interfacing the inherent favorable interactions between natural particulates or the living systems and the advances in bio-nanotechnology, genetic engineering, as well as bioconjugation chemistry, advanced ICB delivery systems with targeting capacity or other properties could be engineered depending on the clinical indications.
4. SUMMARY AND OUTLOOK
In this Account, we have introduced our recent efforts in the development of anticancer therapeutic platforms for achieving the local and targeted delivery of ICB agents to tumors while enhancing potency and reducing side effects. A MN-based transcutaneous delivery platform could facilitate the controlled release and prolong the retention time of the ICB agent as well as other synergistic therapeutics. Hydrogels formed in situ, either by directly injecting cross-linking agents or by spraying gel-containing solution, serve as an effective reservoir of ICB agents for passive release or triggered active release, further reinforcing the inhibition of tumor recurrence after surgery. Meanwhile, the excellent biocompatibility of natural cellular particulates, together with the advances in bioconjugate chemistry and genetic engineering techniques, allows targeted delivery of immunomodulatory antibodies. The local and targeted delivery of ICB agents could be a promising therapeutic strategy to boost the immune response by remodulation of the tumor microenvironment, which could enhance tumor infiltration by CD8+ T cells.57,58
Although considerable progress has been made in the field of local and targeted delivery of ICB agents, several challenges still need to be addressed for acceleration of clinical translation. First, for the synthetic delivery system, the loading efficiency, responsive drug release, and degradation kinetics should be fully optimized to maximize therapeutic outcomes. Although the availability of ICB agents for such delivery systems is relatively high, their implementation could be limited by the administration sites. For tumors in deep tissues, further improvement in administration of such delivery strategies is needed. The stability of the loaded ICB proteins should also be carefully assessed before clinical translation, as the potential deformation of the protein during fabrication and storage could affect the therapeutic potency of ICB therapeutics. Regarding cell-based delivery systems, the cell source is often limited, especially for clinical applications. Therefore, stem cell-mediated production offers a promising approach with merits of personalized medication, but quality control of cellular products poses a challenge.59 Generally, cell-based delivery systems have to be quickly administered after formation, which limits the development of “off-the-shelf” ICB delivery platforms of such formulations. Also, for acceleration of translation, immunogenicity and the good manufacturing practice (GMP) of both natural and genetically engineered cell-based ICB delivery system need to be thoroughly evaluated, thereby avoiding potential side effects associated with adopted cells and high liver uptake. Furthermore, with burgeoning applications of ICB to treat various types of cancer, patients must face different choices of ICB agents with unknown outcomes. As mentioned above, the clinical response rate for ICB treatments is usually low. Identification of biomarkers to predict the therapeutic efficacy of ICB treatments could thus be valuable. Lastly, irAEs should be thoroughly elucidated based on comparisons between systemic and local, targeted treatments in order to augment the merits of reducing side effects.
KEY REFERENCES.
Wang, C.; Wang, J.; Zhang, X.; Yu, S.; Wen, D.; Hu, Q.; Ye, Y.; Bomba, H.; Hu, X.; Liu, Z. In situ formed reactive oxygen species-responsive scaffold with gemcitabine and checkpoint inhibitor for combination therapy. Sci. Transl. Med. 2018, 10, eaan3682.1 Chemoimmunotherapy based on the in situ formed hydrogel scaffold boosted the immune response against tumors.
Hu, Q.; Sun, W.; Wang, J.; Ruan, H.; Zhang, X.; Ye, Y.; Shen, S.; Wang, C.; Lu, W.; Cheng, K. Conjugation of haematopoietic stem cells and platelets decorated with anti-PD-1 antibodies augments anti-leukaemia efficacy. Nat. Biomed. Eng. 2018, 2, 831–840.2 A “cell-combination” strategy for delivery of PD-1 antibodies to the bone marrow in the leukemia model.
Chen, G.; Chen, Z.; Wen, D.; Wang, Z.; Li, H.; Zeng, Y.; Dotti, G.; Wirz, R. E.; Gu, Z. Transdermal cold atmospheric plasma-mediated immune checkpoint blockade therapy. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 3687–3692.3 Hollow microneedles loaded with anti- programmed death-ligand 1 antibody (aPD-L1) facilitate the transdermal penetration of cold atmosphere plasma to the tumor tissues and further induce immunogenic death of tumor cells.
ACKNOWLEDGMENTS
The work described in this Account was supported by grants from the start-up packages of University of California, Los Angeles (UCLA), NIH (R01CA234343-01A1), and Jonsson Comprehensive Cancer Center at UCLA.
Biographies
Xiao Han received his B.S. and M.S. degrees at Soochow University in 2016 and 2019, respectively. In 2019, he joined Prof. Zhen Gu’s group at University of California, Los Angeles, to continue his Ph.D. studies. His research interests include drug delivery and immunotherapy.
Hongjun Li received his B.S. degree in chemistry in 2012, and Ph.D. in Biology in 2017 at University of Science and Technology of China. From 2017 to 2020, he worked as a postdoctoral researcher sequentially in the South China University of Technology and University of California, Los Angeles. His research interests focus on design and synthesis of biomaterials for cancer treatment.
Daojia Zhou is an undergraduate student at the University of California, Los Angeles, receiving her B.S. degree in bioengineering in 2021. Her research interests include biomimetics and medical devices.
Zhaowei Chen is a Professor in the College of Chemistry at Fuzhou University. Dr. Chen received his B.S. degree at Northwestern Polytechnical University in 2010 and Ph.D. in Inorganic Chemistry and Chemical Biology at Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, in 2016. From 2016 to 2020, he worked as a postdoctoral researcher sequentially in the Joint Department of Biomedical Engineering at the UNC and NCSU, Argonne National Laboratory, and University of California, Los Angeles. Then, he joined Fuzhou University as a Minjiang Scholar Professor. His research interests focus on design and synthesis of bioinspired and biomimetic materials for theranostic, biocatalytic, and energy conversion applications.
Zhen Gu is a Qiushi Chair Professor and Dean of College of Pharmaceutical Sciences at Zhejiang University. Dr. Gu received his B.S. degree in Chemistry and M.S. degree in Polymer Chemistry and Physics from Nanjing University. In 2010, he obtained his Ph.D. from the Department of Chemical and Biomolecular Engineering at the University of California, Los Angeles (UCLA). He was a Postdoctoral Associate at MIT and Harvard Medical School during 2010 to 2012. Before he moved to Zhejiang University in 2020, he was a Full Professor in the Department of Bioengineering and Director of the NIH Biotechnology Training in Biomedical Sciences and Engineering Program at UCLA. From 2012 to 2018, he was working in the Joint Department of Biomedical Engineering at the University of North Carolina at Chapel Hill and North Carolina State University, where he was appointed as a Jackson Family Distinguished Professor. His group studies controlled drug delivery, biomaterials and cell therapy, especially for cancer and diabetes treatment.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.accounts.0c00339
The authors declare the following competing financial interest(s): Z.G. is a scientific co-founder of ZenCapsule Inc., Zhenle Inc., Zenomics Inc., Lizen Inc. and wskin Inc.
REFERENCES
- (1).Wang C; Wang J; Zhang X; Yu S; Wen D; Hu Q; Ye Y; Bomba H; Hu X; Liu Z; et al. In situ formed reactive oxygen species–responsive scaffold with gemcitabine and checkpoint inhibitor for combination therapy. Sci. Transl. Med 2018, 10, No. eaan3682. [DOI] [PubMed] [Google Scholar]
- (2).Hu Q; Sun W; Wang J; Ruan H; Zhang X; Ye Y; Shen S; Wang C; Lu W; Cheng K Conjugation of haematopoietic stem cells and platelets decorated with anti-PD-1 antibodies augments anti-leukaemia efficacy. Nat. Biomed. Eng 2018, 2, 831–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Chen G; Chen Z; Wen D; Wang Z; Li H; Zeng Y; Dotti G; Wirz RE; Gu Z Transdermal cold atmospheric plasma-mediated immune checkpoint blockade therapy. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 3687–3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Patel SA; Minn AJ Combination cancer therapy with immune checkpoint blockade: mechanisms and strategies. Immunity 2018, 48, 417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wei SC; Duffy CR; Allison JP Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discovery 2018, 8, 1069–1086. [DOI] [PubMed] [Google Scholar]
- (6).Pardoll DM The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Keir ME; Butte MJ; Freeman GJ; Sharpe AH PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol 2008, 26, 677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Topalian SL; Hodi FS; Brahmer JR; Gettinger SN; Smith DC; McDermott DF; Powderly JD; Carvajal RD; Sosman JA; Atkins MB; et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med 2012, 366, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Freeman GJ; Long AJ; Iwai Y; Bourque K; Chernova T; Nishimura H; Fitz LJ; Malenkovich N; Okazaki T; Byrne MC; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med 2000, 192, 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Baumeister SH; Freeman GJ; Dranoff G; Sharpe AH Coinhibitory pathways in immunotherapy for cancer. Annu. Rev. Immunol 2016, 34, 539–573. [DOI] [PubMed] [Google Scholar]
- (11).Grosso JF; Jure-Kunkel MN CTLA-4 blockade in tumor models: an overview of preclinical and translational research. Cancer Immun. Res 2013, 13, 5. [PMC free article] [PubMed] [Google Scholar]
- (12).Egen JG; Kuhns MS; Allison JP CTLA-4: new insights into its biological function and use in tumor immunotherapy. Nat. Immunol 2002, 3, 611–618. [DOI] [PubMed] [Google Scholar]
- (13).Contardi E; Palmisano GL; Tazzari PL; Martelli AM; Fala F; Fabbi M; Kato T; Lucarelli E; Donati D; Polito L; et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int. J. Cancer 2005, 117, 538–550. [DOI] [PubMed] [Google Scholar]
- (14).Zhang H; Dutta P; Liu J; Sabri N; Song Y; Li WX; Li J Tumor cell-intrinsic CTLA 4 regulates PD-L1 expression in non-small cell lung cancer. J. Cell. Mol. Med 2019, 23, 535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Sadreddini S; Baradaran B; Aghebati-Maleki A; Sadreddini S; Shanehbandi D; Fotouhi A; Aghebati-Maleki L Immune checkpoint blockade opens a new way to cancer immunotherapy. J. Cell. Physiol 2019, 234, 8541–8549. [DOI] [PubMed] [Google Scholar]
- (16).Sanmamed MF; Chen L A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell 2018, 175, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).US FDA. FDA labeling information – Yervoy (ipilimumab) http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/125377s0000lbl.pdf (2011).
- (18).US FDA. FDA labeling information – Keytruda (pembrolizumab) https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125514s016lbl.pdf (2014).
- (19).US FDA. FDA labeling information - Opdivo (nivolumab) https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125554s055lbl.pdf, (2014).
- (20).Brahmer JR; Tykodi SS; Chow LQ; Hwu W-J; Topalian SL; Hwu P; Drake CG; Camacho LH; Kauh J; Odunsi K; et al. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med 2012, 366, 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Larkin J; Chiarion-Sileni V; Gonzalez R; Grob JJ; Cowey CL; Lao CD; Schadendorf D; Dummer R; Smylie M; Rutkowski P; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med 2015, 373, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Bertrand A; Kostine M; Barnetche T; Truchetet M-E; Schaeverbeke T Immune related adverse events associated with anti-CTLA-4 antibodies: systematic review and meta-analysis. BMC Med. 2015, 13, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Postow MA; Sidlow R; Hellmann MD Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med 2018, 378, 158–168. [DOI] [PubMed] [Google Scholar]
- (24).Khedri M; Rafatpanah H; Abnous K; Ramezani P; Ramezani M Cancer immunotherapy via nucleic acid aptamers. Int. Immuno-pharmacol 2015, 29, 926–936. [DOI] [PubMed] [Google Scholar]
- (25).Chen Q; Wang C; Chen G; Hu Q; Gu Z Delivery strategies for immune checkpoint blockade. Adv. Healthcare Mater 2018, 7, 1800424. [DOI] [PubMed] [Google Scholar]
- (26).Riley RS; June CH; Langer R; Mitchell MJ Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discovery 2019, 18, 175–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hoare TR; Kohane DS Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar]
- (28).Ye Y; Yu J; Wen D; Kahkoska AR; Gu Z Polymeric microneedles for transdermal protein delivery. Adv. Drug Delivery Rev 2018, 127, 106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lee K; Goudie MJ; Tebon P; Sun W; Luo Z; Lee J; Zhang S; Fetah K; Kim H-J; Xue Y Non-transdermal microneedles for advanced drug delivery. Adv. Drug Delivery Rev 2019, DOI: 10.1016/j.addr.2019.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wang C; Ye Y; Hochu GM; Sadeghifar H; Gu Z Enhanced cancer immunotherapy by microneedle patch-assisted delivery of anti-sPD1 antibody. Nano Lett. 2016, 16, 2334–2340. [DOI] [PubMed] [Google Scholar]
- (31).Ye Y; Wang J; Hu Q; Hochu GM; Xin H; Wang C; Gu Z Synergistic transcutaneous immunotherapy enhances antitumor immune responses through delivery of checkpoint inhibitors. ACS Nano 2016, 10, 8956–8963. [DOI] [PubMed] [Google Scholar]
- (32).Yan D; Sherman JH; Keidar M Cold atmospheric plasma, a novel promising anti-cancer treatment modality. Oncotarget 2017, 8, 15977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Chernets N; Kurpad DS; Alexeev V; Rodrigues DB; Freeman TA Reaction chemistry generated by nanosecond pulsed dielectric barrier discharge treatment is responsible for the tumor eradication in the B16 melanoma mouse model. Plasma Processes Polym. 2015, 12, 1400–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Li J; Mooney DJ Designing hydrogels for controlled drug delivery. Nat. Rev. Mater 2016, 1, 16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ruan H; Hu Q; Wen D; Chen Q; Chen G; Lu Y; Wang J; Cheng H; Lu W; Gu Z A dual-bioresponsive drug-delivery depot for combination of epigenetic modulation and immune checkpoint blockade. Adv. Mater 2019, 31, 1806957. [DOI] [PubMed] [Google Scholar]
- (36).Yu S; Wang C; Yu J; Wang J; Lu Y; Zhang Y; Zhang X; Hu Q; Sun W; He C; et al. Injectable bioresponsive gel depot for enhanced immune checkpoint blockade. Adv. Mater 2018, 30, 1801527. [DOI] [PubMed] [Google Scholar]
- (37).Chen Q; Wang C; Zhang X; Chen G; Hu Q; Li H; Wang J; Wen D; Zhang Y; Lu Y; et al. In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment. Nat. Nanotechnol 2019, 14, 89–97. [DOI] [PubMed] [Google Scholar]
- (38).Willingham SB; Volkmer J-P; Gentles AJ; Sahoo D; Dalerba P; Mitra SS; Wang J; Contreras-Trujillo H; Martin R; Cohen JD; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 6662–6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Chen Z; Wang Z; Gu Z Bioinspired and Biomimetic Nanomedicines. Acc. Chem. Res 2019, 52, 1255–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Chen Z; Wen D; Gu Z Cargo-encapsulated cells for drug delivery. Sci. China: Life Sci 2020, 63, 599–601. [DOI] [PubMed] [Google Scholar]
- (41).Chen Z; Hu Q; Gu Z Leveraging Engineering of Cells for Drug Delivery. Acc. Chem. Res 2018, 51, 668–677. [DOI] [PubMed] [Google Scholar]
- (42).Lu Y; Hu Q; Jiang C; Gu Z Platelet for drug delivery. Curr. Opin. Biotechnol 2019, 58, 81–91. [DOI] [PubMed] [Google Scholar]
- (43).Andrews RK; Berndt MC Platelet physiology and thrombosis. Thromb. Res 2004, 114, 447–453. [DOI] [PubMed] [Google Scholar]
- (44).Morrell CN; Aggrey AA; Chapman LM; Modjeski KL Emerging roles for platelets as immune and inflammatory cells. Blood 2014, 123, 2759–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Gay LJ; Felding-Habermann B Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Wang C; Sun W; Ye Y; Hu Q; Bomba HN; Gu Z In situ activation of platelets with checkpoint inhibitors for post-surgical cancer immunotherapy. Nat. Biomed. Eng 2017, 1, 0011. [Google Scholar]
- (47).Rolfes V; Idel C; Pries R; Plötze-Martin K; Habermann J; Gemoll T; Bohnet S; Latz E; Ribbat-Idel J; Franklin BS; Wollenberg B PD-L1 is expressed on human platelets and is affected by immune checkpoint therapy. Oncotarget 2018, 9, 27460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Li Z; Hu S; Huang K; Su T; Cores J; Cheng K Targeted anti-IL-1β platelet microparticles for cardiac detoxing and repair. Sci. Adv 2020, 6, No. eaay0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Ma Q; Fan Q; Xu J; Bai J; Han X; Dong Z; Zhou X; Liu Z; Gu Z; Wang C Calming Cytokine Storm in Pneumonia by Targeted Delivery of TPCA-1 Using Platelet-derived Extracellular Vesicles. Matter 2020, 3, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Han X; Chen J; Chu J; Liang C; Ma Q; Fan Q; Liu Z; Wang C Platelets as platforms for inhibition of tumor recurrence post-physical therapy by delivery of anti-PD-L1 checkpoint antibody. J. Controlled Release 2019, 304, 233–241. [DOI] [PubMed] [Google Scholar]
- (51).Zhang X; Wang J; Chen Z; Hu Q; Wang C; Yan J; Dotti G; Huang P; Gu Z Engineering PD-1-Presenting Platelets for Cancer Immunotherapy. Nano Lett. 2018, 18, 5716–5725. [DOI] [PubMed] [Google Scholar]
- (52).Zhang X; Kang Y; Wang J; Yan J; Chen Q; Cheng H; Huang P; Gu Z Engineered PD-L1-Expressing Platelets Reverse New-Onset Type 1 Diabetes. Adv. Mater 2020, 32, 1907692. [DOI] [PubMed] [Google Scholar]
- (53).Bonnet D; Dick JE Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med 1997, 3, 730–737. [DOI] [PubMed] [Google Scholar]
- (54).McClanahan F; Hanna B; Miller S; Clear AJ; Lichter P; Gribben JG; Seiffert M PD-L1 checkpoint blockade prevents immune dysfunction and leukemia development in a mouse model of chronic lymphocytic leukemia. Blood 2015, 126, 203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Toledano Furman NE; Lupu-Haber Y; Bronshtein T; Kaneti L; Letko N; Weinstein E; Baruch L; Machluf M Reconstructed stem cell nanoghosts: a natural tumor targeting platform. Nano Lett. 2013, 13, 3248–3255. [DOI] [PubMed] [Google Scholar]
- (56).Zhang X; Wang C; Wang J; Hu Q; Langworthy B; Ye Y; Sun W; Lin J; Wang T; Fine J; et al. PD-1 Blockade Cellular Vesicles for Cancer Immunotherapy. Adv. Mater 2018, 30, 1707112. [DOI] [PubMed] [Google Scholar]
- (57).Wei SC; Levine JH; Cogdill AP; Zhao Y; Anang N-AA; Andrews MC; Sharma P; Wang J; Wargo JA; Pe’er D; Allison JP Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell 2017, 170, 1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Blackburn SD; Shin H; Freeman GJ; Wherry EJ Selective expansion of a subset of exhausted CD8 T cells by αPD-L1 blockade. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 15016–15021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Andrews PW; Cavagnaro J; Deans R; Feigal E; Horowitz E; Keating A; Rao M; Turner M; Wilmut I; Yamanaka S Harmonizing standards for producing clinical-grade therapies from pluripotent stem cells. Nat. Biotechnol 2014, 32, 724–726. [DOI] [PubMed] [Google Scholar]