

Abstract
Transcription and metabolism both influence cell function, but dedicated transcriptional control of metabolic pathways that regulate cell fate has rarely been defined. We discovered, using a chemical suppressor screen, that inhibition of the pyrimidine biosynthesis enzyme dihydroorotate dehydrogenase (DHODH) rescues erythroid differentiation in bloodless zebrafish moonshine (mon) mutant embryos defective for transcriptional intermediary factor 1 gamma (tif1γ). This rescue depends on the functional link of DHODH to mitochondrial respiration. The transcription elongation factor TIF1γ directly controls coenzyme Q (CoQ) synthesis gene expression. Upon tif1γ loss, CoQ levels are reduced, and a high succinate/α-ketoglutarate ratio leads to increased histone methylation. A CoQ analog rescues mon’s bloodless phenotype. These results demonstrate that mitochondrial metabolism is a key output of a lineage transcription factor that drives cell fate decisions in the early blood lineage.
Vertebrate embryos produce circulating red blood cells (RBCs) that are required for oxygen and carbon dioxide transport (1). During embryonic development, three overlapping hematopoietic waves can be distinguished that all produce RBCs (2). In mammals, primitive erythroblasts that emerge in the blood islands within the extraembryonic yolk sac give rise to primitive erythrocytes of the first transient wave, and a second wave generates definitive erythroid-myeloid progenitors in the hemogenic endothelium of the yolk sac. Definitive erythrocytes of the third wave arise from multipotent hematopoietic stem cells that are born in the aortic endothelium of the aorta-gonad-mesonephros region and sustain hematopoiesis throughout the lifetime of the animal. Primitive erythrocytes supply the embryo with the oxygen needed for its rapid proliferation. Failure to initiate the first wave of erythropoiesis leads to embryonic lethality (3). Erythroid lineage differentiation is regulated by key transcription factors (4), but the cellular mechanisms that allow the generation and differentiation of the first erythroid progenitors remain largely unknown. Stem and descendent progenitor cells differ by their metabolic profiles (5), but there is little in vivo evidence for a link between transcriptional regulation and metabolic changes during cell fate decisions.
DHODH inhibitors rescue defective tif1γ-dependent erythropoiesis
TIF1γ is essential for erythropoiesis from zebrafish to humans (6–10). Zebrafish moonshine (mon) mutant embryos defective for tif1γ do not make RBCs because of a transcription elongation block characterized by aberrantly paused RNA polymerase II (Pol II) (9–11). To uncover other factors required for tif1γ-dependent erythropoiesis, we performed a high-content chemical suppressor screen of 3120 compounds with known bioactivities to rescue erythropoiesis in the mon mutant. We incubated embryos with compounds from 5.3 hours post fertilization (hpf) until 22 hpf, the time of gastrulation and primitive erythropoiesis, and assayed embryonic βe3 globin expression using whole-mount in situ hybridization (WISH; Fig. 1A). This strategy identified the drug leflunomide as an inhibitor of dihydroorotate dehydrogenase (DHODH), the enzyme catalyzing the fourth step in the de novo pyrimidine biosynthesis pathway (12). Although mon embryos treated with leflunomide or its active metabolite, A771726 (13), showed increased βe3 globin expression, their wild-type and heterozygous siblings were blocked in erythroid differentiation (Fig. 1B and fig. S1, A and B). The fact that both leflunomide and A771726 rescued erythropoiesis in mon mutants ruled out an involvement of the aryl hydrocarbon receptor in this process, of which leflunomide has been described as an agonist (14). The structurally divergent DHODH inhibitor brequinar (15) similarly rescued the blood defect in tif1γ-depleted embryos, both at the level of βe3 globin expression (fig. S1C) and hemoglobin synthesis (Fig. 1C and fig. S1D), indicative of terminal erythropoiesis. In control embryos, DHODH inhibition led to a reduction in the number of hemoglobinized differentiated RBCs (Fig. 1C and fig. S1D). Modeling predicted the structures of zebrafish and human DHODH, including the binding pockets for A771726 and brequinar, to be highly conserved (fig. S2, A to C). These results demonstrate that inhibition of DHODH activity is required to rescue defective tif1γ-dependent erythropoiesis.
Fig. 1. Chemical suppressor screen for the mon zebrafish mutant identifies DHODH inhibitors to rescue primitive erythropoiesis.
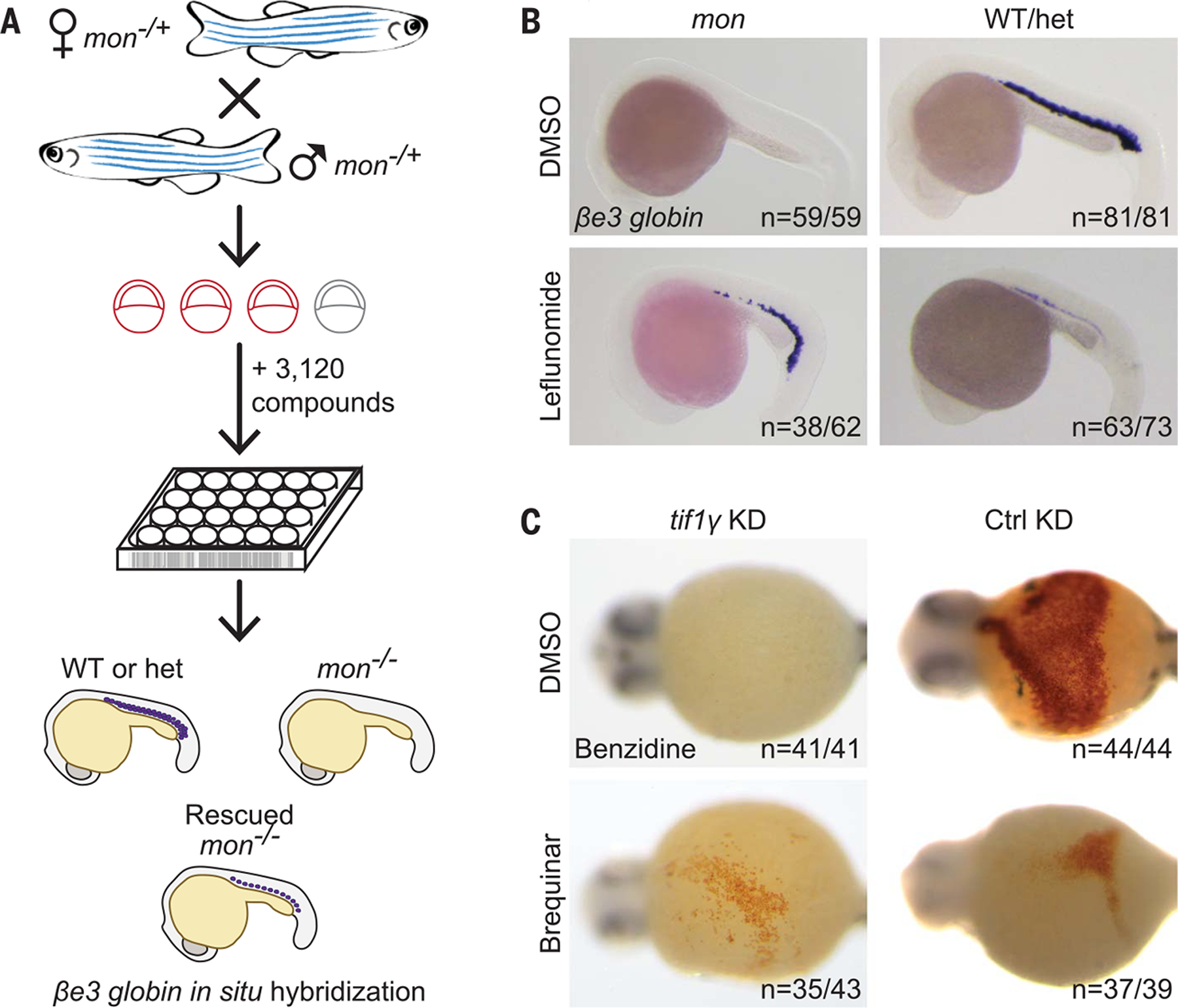
(A) Cartoon depicting chemical screening strategy. WT, wild type; het, mon heterozygous. (B) WISH for βe3 globin in mon and WT or het zebrafish embryos at 22 hpf treated with 7 μM leflunomide or DMSO. N (number of biological replicates) = 4. Cumulative results are shown. (C) Benzidine staining of embryos at 44 hpf after morpholino-mediated knockdown of tif1γ (tif1γ KD) or standard control knockdown (Ctrl KD) and treatment with 3.5 μM brequinar or DMSO from 5.3 hpf until 11.7 hpf. N = 3. Data from one representative experiment are shown. n = number of embryos with depicted phenotype/number of all embryos tested (see also fig. S1).
We performed time course studies to establish the developmental window during which DHODH is active in mon embryos. Treating embryos with leflunomide during gastrulation, but not later, was sufficient to rescue erythropoiesis in mon embryos (fig. S3). These results assign a critical early function for tif1γ, when specification of the erythroid lineage is initiated within mesoderm at the end of gastrulation. Blastula transplantation experiments showed a non-cell-autonomous role for tif1γ (fig. S4, A and B) in addition to its cell-autonomous role in erythropoiesis (fig. S4, C and D). During its development, mesoderm lies above the yolk sac, a metabolically active tissue that is a source of nutrients for the embryo proper and is analogous to the mammalian yolk sac and placenta (16, 17). A non-cell-autonomous role of tif1γ could suggest a more direct metabolic function in the early embryo.
tif1γ loss leads to TCA cycle changes, DNA and histone hypermethylation
Because DHODH is the only enzyme of the pyrimidine de novo synthesis pathway located in mitochondria (18), we investigated whether mitochondrial metabolism was altered in mon mutant embryos. RNA-sequencing (RNA-seq) experiments for tif1γ-depleted embryos at 10 hpf (fig. S5 and data S1) revealed that 77% (182 genes) of genes encoding proteins with mitochondrial function (19) [95% (20 genes) when considering genes that encode for subunits of the respiratory chain] were transcriptionally down-regulated upon tif1γ loss of function (Fig. 2A, middle and right). By contrast, only 55% (or 1986 genes) of nonmitochondrial genes were transcriptionally down-regulated (Fig. 2A, left). Furthermore, mitochondrial genes were more significantly down-regulated compared with nonmitochondrial genes (fig. S6). These results indicate that mitochondrial gene expression specifically depends on tif1γ. DHODH inhibition did not alter the expression of p53 target genes (20), cell cycle genes, or genes involved in mitochondrial fission or fusion in tif1γ-depleted embryos (fig. S7, A and B; fig. S8; and data S2) but did promote differentiation processes including hematopoiesis specifically upon tif1γ loss, as shown by Metascape pathway analysis (fig. S9, A to D). Untargeted metabolomics analysis of mon drl:EGFP embryos at 22 hpf (Fig. 2B and fig. S10, A and B) or tif1γ-depleted embryos at 11 or 22 hpf (fig. S10, C and D) revealed reduced levels of several tricarboxylic acid (TCA) cycle metabolites, including citrate, its isomerization intermediate cis-aconitate, isocitrate, oxaloacetate, α-ketoglutarate (α-KG, also called 2-oxoglutarate), malate, and the TCA-cycle-related metabolite aspartate, suggesting a more general mitochondrial defect.
Fig. 2. tif1γ loss affects the TCA cycle and histone methylation.
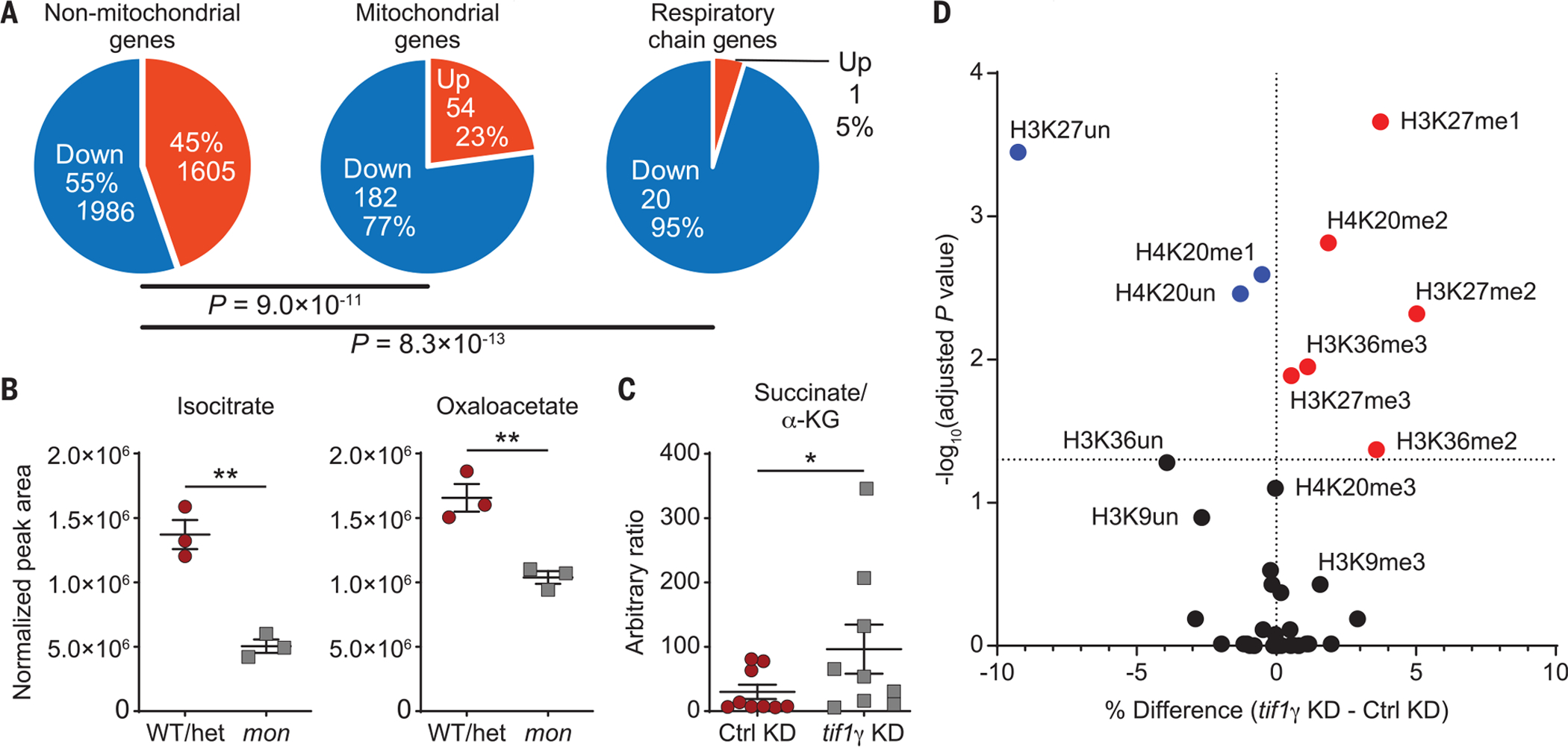
(A) RNA-seq data at 10 hpf showing all up- or down-regulated [FPKM (fragments per kilobase of transcript per million mapped reads) >1, q < 0.05] nonmitochondrial (left), mitochondrial (middle), and respiratory chain genes (right) in tif1γ KD or Ctrl KD embryos treated with DMSO from 5.3 hpf until 10 hpf. P values were determined by Pearson’s chi-squared test with default parameters. (B) Levels of the indicated TCA cycle metabolites in mon;Tg(drl:EGFP) or sibling WT or het embryos at 22 hpf as measured by untargeted metabolomics. N = 3. Normalization by median correction. Data are shown as means ± SEM by two-tailed t test. (C) Succinate/α-KG ratio in tif1γ KD or Ctrl KD embryos at 22 hpf as measured by untargeted metabolomics. Data are shown as means ± SEM by two-tailed t test. (D) Volcano plot of percentage occupancy data of histone posttranslational modifications (PTMs) in tif1γ KD compared with Ctrl KD embryos at 22 hpf. Each dot represents a single modification at the amino acid level. x-axis shows the percentage difference between the means of tif1γ KD and Ctrl KD samples. Adjusted P values were determined by multiple t tests between all five replicates across the two conditions. Significantly enriched histone PTMs in tif1γ KD and Ctrl KD samples are shown in red and blue, respectively. **P < 0.01; *P < 0.05; ns, not significant (see also figs. S5 to S12).
The activity of 2-oxoglutarate-dependent dioxygenases (OGGDs), which include DNA and histone demethylases, can be inhibited by an increased ratio of succinate, L-2-hydroxyglutarate (2HG), or fumarate to α-KG (21). Increased DNA and histone methylation levels have been observed after conditional knockout of mitochondrial complex III subunit Rieske iron-sulfur protein (encoded by UQCRFS1) in the hematopoietic system (22, 23), and a biallelic variant in UQCRFS1 in humans has been recently described to cause anemia (24). In-depth TCA cycle metabolic analyses revealed that the succinate/α-KG ratio was significantly increased in tif1γ-depleted embryos (Fig. 2C), and the 2HG/α-KG and fumarate/α-KG ratios were elevated (fig. S10E). To address whether histone and DNA methylation levels were altered upon tif1γ loss, we profiled global chromatin modifications in histones extracted from tif1γ-depleted embryos at 22 hpf using quantitative targeted mass spectrometry (fig. S11, A and B). The absence of tif1γ led to significantly increased levels of the methylated histone species H3K27me1, H3K27me2, and H3K27me3, H3K36me2 and H3K36me3, as well as H4K20me2 (Fig. 2D and fig. S12, A and B), modifications that are removed by OGGDs (25). The levels of histone acetylation, phosphorylation, and ubiquitination remained unchanged (fig. S12A). The expression of histone demethylases in tif1γ knockdown and mon does not account for the changes in methylation (data S3). We next evaluated DNA methylation, which can be reversed by OGGDs. Reduced representation bisulfite sequencing (RRBS) experiments revealed significant hypermethylation of background (genome excluding CpG islands) CpG dinucleotides (CpGs; fig. S13, A and B, and data S4), which represented 94% of all CpGs covered by RRBS with at least 10 reads (fig. S13C). Our results indicate that the altered mitochondrial TCA cycle activity in the absence of tif1γ leads to an increased succinate/α-KG ratio that in turn results in changes in the epigenome with higher histone and DNA methylation levels.
Mitochondrial function is impaired upon tif1γ loss
To investigate whether tif1γ loss compromises mitochondrial function, we performed a high-throughput single-embryo analysis of the oxygen consumption rate (OCR). Embryos depleted for tif1γ exhibited a functional decrease in oxidative aerobic metabolism at 14 hpf compared with control embryos, as demonstrated by a significant decrease in the OCR at baseline (Fig. 3A and fig. S14A). Although DHODH inhibition by leflunomide treatment of wild-type embryos led to a reduced OCR, tif1γ depletion did not result in a further reduction. To estimate the reserve respiratory capacity, we treated embryos with the uncoupling agent carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP), which abolishes the proton gradient and thus leads to an increase in oxygen consumption. Upon addition of FCCP, tif1γ depletion rescued the reduced OCR of DHODH-inhibited embryos (Fig. 3A and fig. S14B), showing that the function of DHODH in mitochondrial respiration is intimately linked to TIF1γ. In agreement with decreased baseline oxygen consumption of tif1γ-depleted embryos, we found that the ratio of oxidized to reduced nicotinamide adenine dinucleotides (NAD+/NADH), which directly correlates with the OCR, was lowered in mon embryos (Fig. 3B). By comparison, embryos of the same age that were depleted for gata1a, the master transcription factor of the erythroid lineage and thus for RBCs (fig. S14C), showed an increased OCR over control-depleted embryos (fig. S14, D to F). Gata1− mouse embryonic stem cells give rise to an abnormal and highly proliferative blast population (26) that might exhibit high levels of mitochondrial respiration (22). These data show that mitochondrial respiration is required for blood formation and that tif1γ is required for normal mitochondrial function.
Fig. 3. tif1γ loss leads to a functional mitochondrial defect.
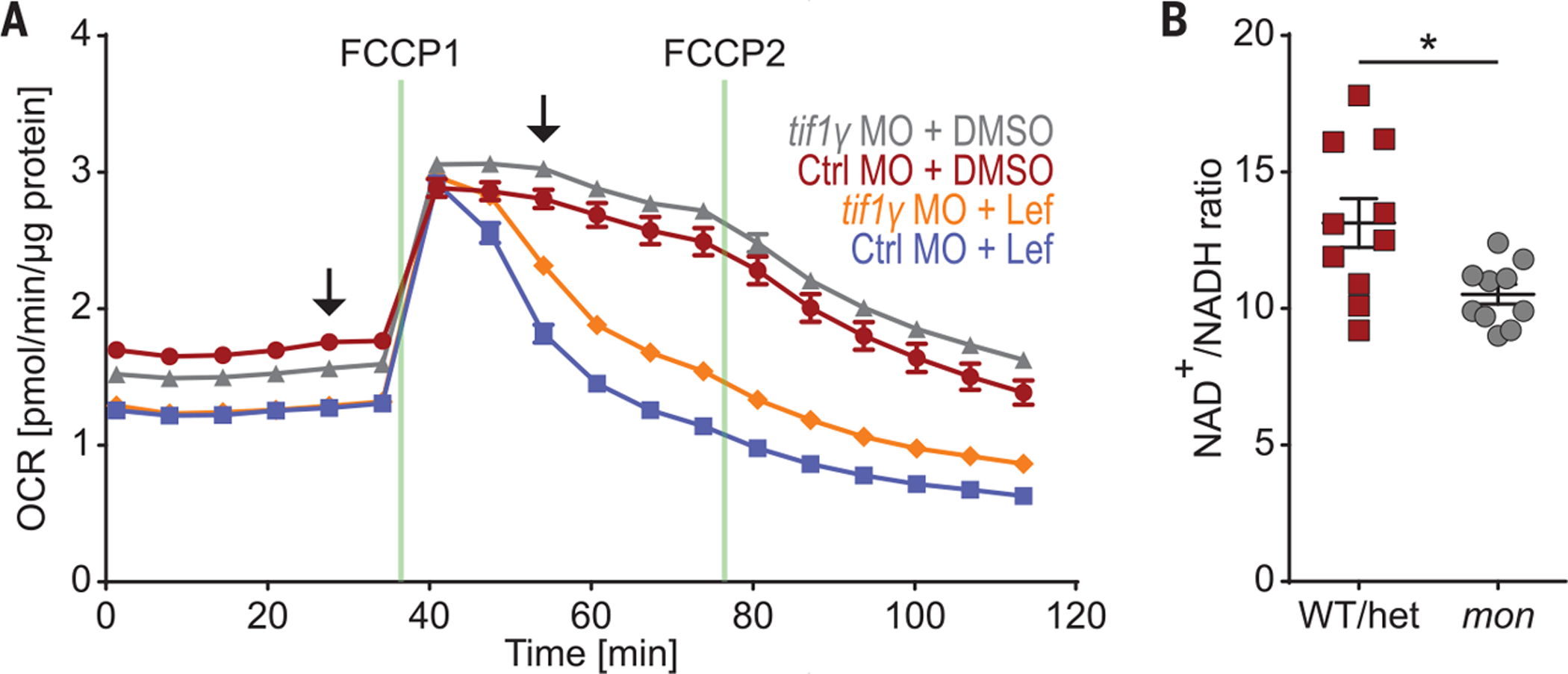
(A) OCR as measured by Seahorse analysis of single tif1γ KD or Ctrl KD embryos at 16 hpf treated with 7 μM leflunomide or DMSO from 5.3 until 11 hpf. N = 3. Data from one representative experiment are shown. Data are shown as means ± SEM. Arrows indicate time points for which statistical analysis is shown in fig. S14, A and B. (B) Analysis of NAD+/NADH ratio in mon and sibling WT or het embryos at 48 hpf using the NAD/NADH-Glo Assay. N = 3. Cumulative results are shown. One data point indicates one embryo. *P < 0.05, two-tailed t test.
tif1γ drives erythropoiesis by affecting the electron transport chain
DHODH catalyzes the oxidation of dihydroorotate into orotate by transferring electrons to CoQ in the electron transport chain (ETC) (27). Thus, DHODH functionally links the production of pyrimidines with the mitochondrial respiratory chain (Fig. 4A). Uridine, which is converted through the salvage pathway into uridine monophosphate, can bypass the inhibition of de novo pyrimidine synthesis (fig. S15A). Although the erythroid differentiation block caused by DHODH inhibition in wild-type and heterozygous embryos could be partially reversed by the addition of uridine (fig. S15, B and C), the rescue of the mon blood defect by DHODH inhibition was not affected by uridine (Fig. 4B and fig. S15B), which could be caused by a “rewired metabolism” in mon mutants that may allow for more salvage pathway activity. To test whether the rescue of the mon blood defect by DHODH inhibition depends on DHODH’s impact on mitochondrial electron transport, we cotreated mon embryos with leflunomide and rotenone, an inhibitor of complex I of the ETC. Indeed, rotenone reversed the rescue of mon’s blood defect by leflunomide, whereas on its own it did not rescue blood in mon (Fig. 4C). The antioxidant N-acetyl-L-cysteine did not rescue the mon blood defect (fig. S16). These results demonstrate that DHODH inhibition rescues the blood defect in mon embryos through its link to the ETC.
Fig. 4. Function of tif1γ in erythropoiesis is linked to the ETC.
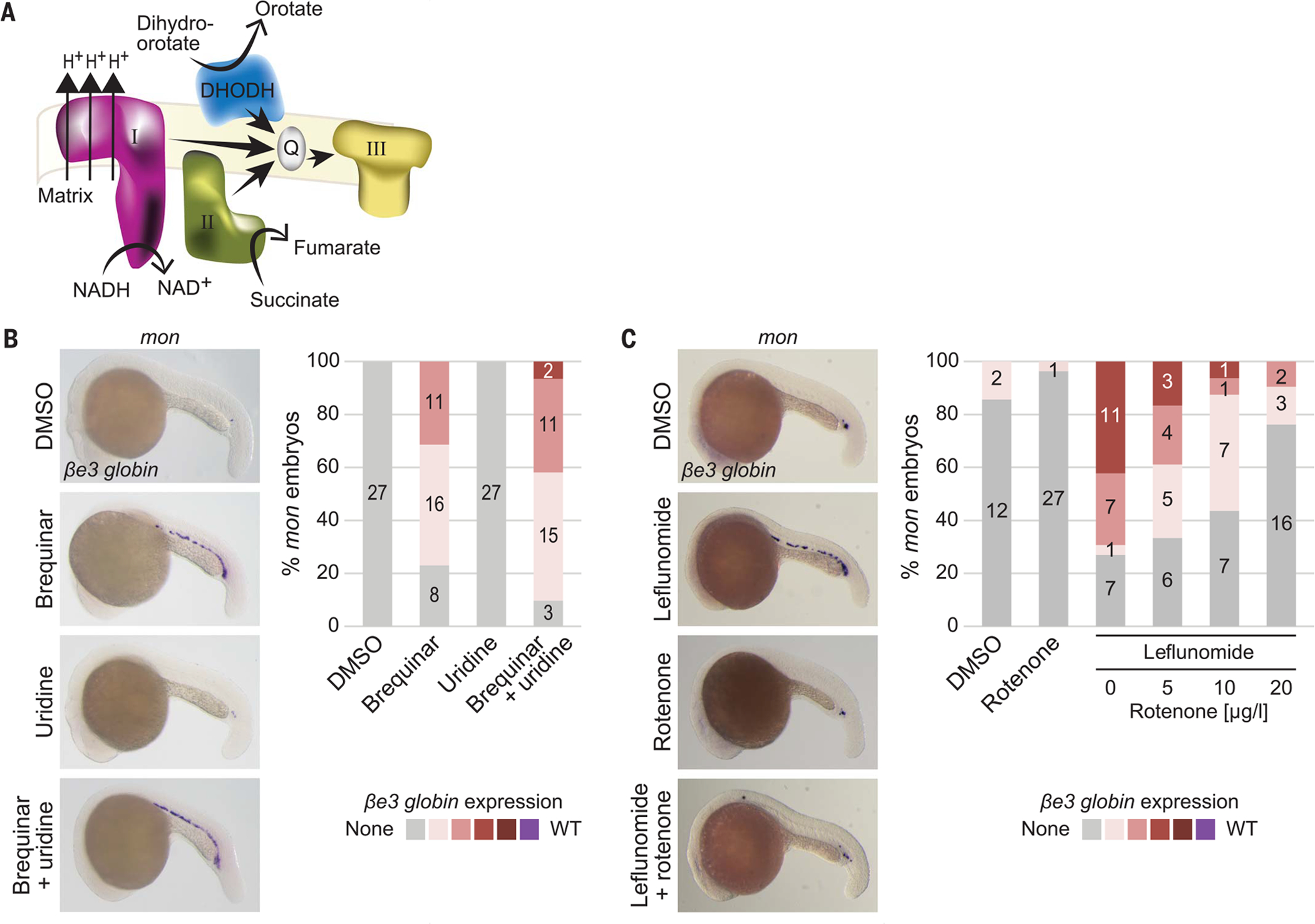
(A) Cartoon depicting relationship of DHODH with some components of the mitochondrial ETC. (B and C) Representative βe3 globin WISH images (left) and quantification (right) of mon embryos at 22 hpf treated with DMSO or 3.5 μM brequinar alone or in combination with 100 mM uridine [(B); N = 4] or treated with DMSO or 7 μM leflunomide alone or in combination with 5, 10, or 20 μg/L rotenone [(C); N = 3]. Cumulative results are shown. Numbers in columns in (B) and (C) refer to the number of embryos with the color-coded degree of βe3 globin staining (see also fig. S15).
TIF1γ directly regulates CoQ levels in erythropoiesis
In the inner mitochondrial membrane, electrons are transferred from complex I or II to complex III in a manner dependent on CoQ, an electron carrier that cycles between three redox states (28). DHODH competes with complexes I and II of the ETC for CoQ as an electron acceptor (Fig. 4A). CoQ consists of a benzoquinone ring head group joined to a polyisoprenoid side chain (29), which is synthesized by an enzyme encoded by PDSS1 and PDSS2 in humans and zebrafish. The head group subsequently is modified by several COQ enzymes (fig. S17). COQ2 deficiency in humans leads to pancytopenia and anemia in infancy, and quinone deficiencies can be rescued by an exogeneous ubiquinone analog (30). To evaluate the role of TIF1γ in the CoQ biosynthesis pathway, we generated stable human HepG2 cell lines that expressed one of two different TIF1γ shRNAs (shTIF1γ) or control shRNAs (shCtrl) under doxycycline control. We chose HepG2 cells as metabolically active cells akin to the yolk sac in the early zebrafish embryo. In these clonal cell lines, shTIF1γ-1 and shTIF1γ-2 induction specifically reduced TIF1γ expression by 98% and 50%, respectively, after 48 hours (Fig. 5A and fig. S18A), which correlated with reduced TIF1γ protein levels (fig. S18B). At this time point, we performed RNA-seq analysis, comparing both shTIF1γ with both shCtrl clones, in combination with chromatin immunoprecipitation-sequencing (ChIP-seq) analysis for TIF1γ chromatin occupancy, to identify differentially expressed genes that are also direct TIF1γ targets. Direct TIF1γ target genes frequently belonged to metabolic pathways (fig. S18C). In particular, several genes encoding CoQ pathway enzymes were significantly down-regulated upon shTIF1γ-1 and shTIF1γ-2 knockdown, including PDSS1 by 88% and 35% and COQ8A by 42% and 69%, respectively, as well as PDSS2 by 57% and COQ2 by 84% in the more potent shTIF1γ-1 condition. Expression of the actin gene ACTA2 remained unaffected by TIF1γ knockdown (fig. S18A). At the protein level, PDSS1 was down-regulated twofold upon TIF1γ knockdown (Fig. 5B). ChIP-seq analysis revealed that TIF1γ bound to the loci of these CoQ pathway genes and was often accompanied by the histone mark H3K27Ac, decorating active promoters and enhancers (Fig. 5C and fig. S19). These data establish that CoQ biosynthesis genes are direct targets of TIF1γ.
Fig. 5. CoQ is a functional target of tif1γ in primitive erythropoiesis.
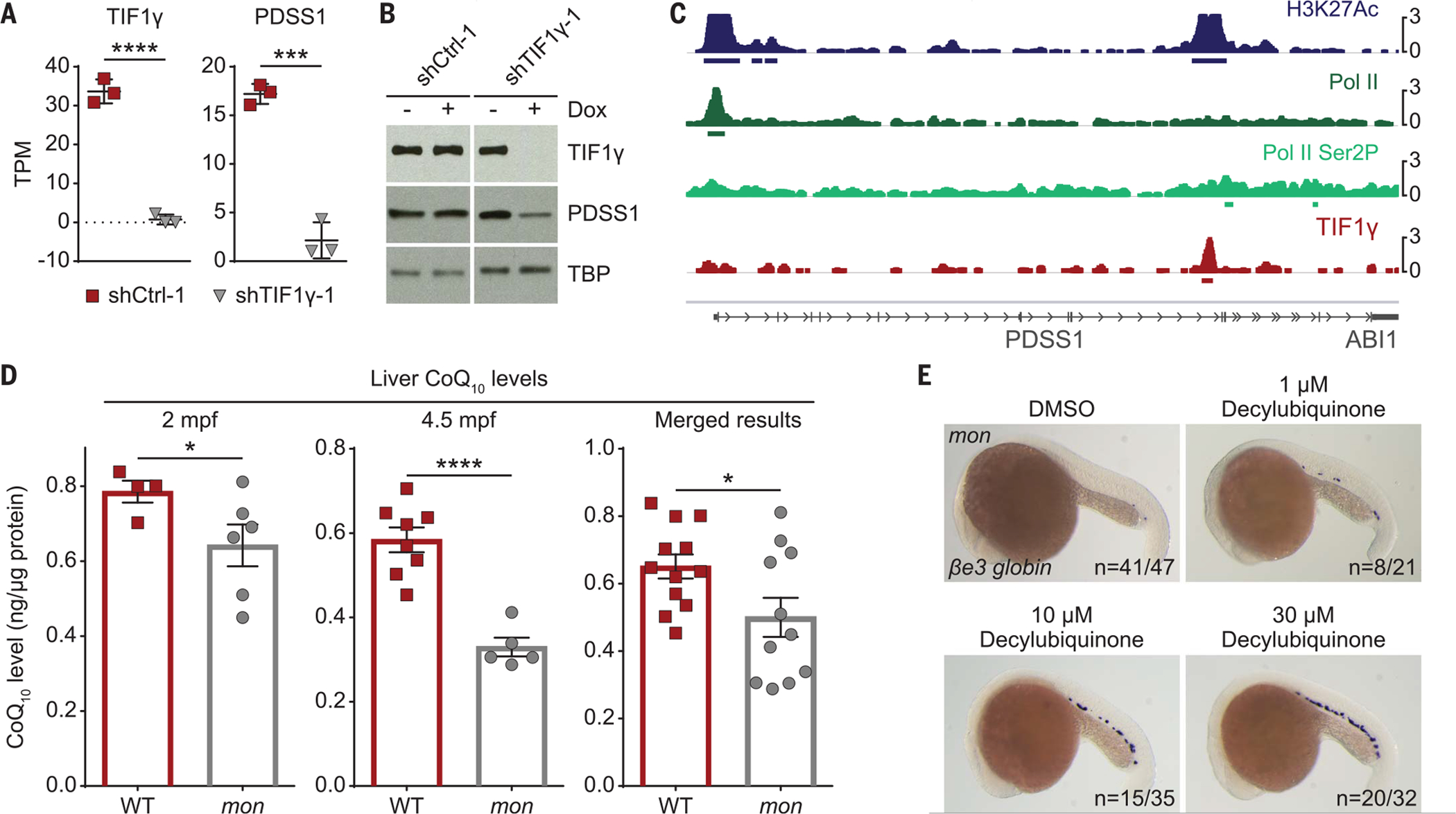
(A) RNA-seq data showing transcripts per kilobase million (TPM) of TIF1γ and PDSS1 in two stable HepG2 clones carrying either an inducible shRNA targeting TIF1γ (shTIF1γ-1) or a control shRNA (shCtrl-1) 48 hours after induction with doxycycline. N = 3. Results were analyzed with two-tailed t test. (B) Western blot of TIF1γ and PDSS1 for the experiment described in (A). TBP was used as the loading control. (C) IGV output of a 52-kb genomic region around PDSS1 on chromosome 10 showing the chromatin occupancy by ChIP-seq for H3K27Ac, Pol II, Pol II phosphorylated at serine 2 (Pol II Ser2P), and TIF1γ in HepG2 cells. (D) CoQ10 quantification of adult mon and WT livers at indicated time points by HPLC. mpf, months postfertilization. Merged results are shown on the right. Each dot represents five pooled livers (except for one sample per genotype at 4.5 mpf, which only contained three livers). Normalization to total protein amount per sample. (E) Representative βe3 globin WISH images of embryos at 22 hpf treated with DMSO or 1, 10, 20, or 30 μM decylubiquinone. N = 3. Cumulative results are shown. ****P < 0.0001; ***P < 0.001; *P < 0.05 (see also figs. S18 to S21).
We hypothesized that down-regulation of CoQ pathway genes upon TIF1γ depletion and exhausted CoQ levels could be functionally relevant to the role of tif1γ in embryonic erythropoiesis. To test this idea, we measured CoQ levels in the zebrafish. We first determined that, as in humans but unlike in mice (28), the zebrafish CoQ isoprenoid side chain consists of 10 subunits (fig. S20A). Although the mon mutant is embryonically lethal, in rare cases, homozygous mutant mon fish survive into adulthood despite producing only very few mature RBCs (9). In other animals, livers have been described to contain normal levels of CoQ (31). In a large-scale effort, we generated sufficient numbers of mon fish, dissected their livers at 2 and 4.5 months, and found that CoQ10 levels were significantly reduced (Fig. 5D) and CoQ biosynthesis gene expression was down-regulated (fig. S20B and supplementary text). Treatment of mon embryos from the onset of gastrulation until 22 hpf with the CoQ analog decylubiquinone rescued βe3 globin expression in 26% (33/126) of mon embryos (Fig. 5E and fig. S21). We propose that in the absence of tif1γ, mitochondrial respiratory capacity is impaired because of insufficient electron transfer from complexes I and II to complex III of the ETC, thus shifting the balance in the hemogenic mesoderm to the undifferentiated cell state. Supplying mon embryos with CoQ boosts mitochondrial respiration and promotes erythroid differentiation.
Discussion
Tissue differentiation can be regulated by metabolic activities. It was previously unclear how lineage transcription factors induce distinct changes during cell fate specification and lineage differentiation. Our work demonstrates that the metabolic state of the tissue required for early erythroid lineage differentiation is under the direct transcriptional control of TIF1γ. CoQ is a critical downstream effector of TIF1γ transcriptional activity, regulating the balance between nucleotide synthesis and ETC activity in embryonic erythropoiesis. This highlights a previously unappreciated role of mitochondrial respiration in driving the early commitment of the erythroid lineage. It has been proposed that transcriptional and metabolic processes influence each other (32, 33). We demonstrate that this is the case in early erythropoiesis, where exogenous CoQ can drive erythroid differentiation in the mon mutant, including the expression of embryonic globin as a late lineage marker. This study thus offers the possibility for metabolic therapeutics that could be used to target otherwise difficult-to-treat deficiencies in transcriptional programs.
Supplementary Material
ACKNOWLEDGMENTS
We thank R. Mori and M. C. Nonato for generating the DHODH alignment and structural model; L. Krug for excellent care of our fish; C.-Y. Chiang, E. Lee, S. Kemet, W. Chaudhry, T. Schlaeger, D. Gupta, L. Rubin, T. Arvanites, K. Nybakken, and V. Oza for technical assistance with the chemical suppressor screen; S. Freyer, J. Weiss, and H. Moreau for other technical assistance; M. Prasad for help with RNA-seq data mapping; members of the Zon laboratory for discussions; and A. B. Cantor and B. Boettner for critical reading of the manuscript.
Funding:
This work was supported by the National Heart, Lung, and Blood Institute (grants 4R01HL048801, 5P01HL032262, 5U01HL134812, and 1P01HL131477 to L.I.Z.); the National Institute of Diabetes and Digestive and Kidney Diseases (grants 1U54DK110805 and 3R24DK092760 to L.I.Z.); Harvard Catalyst (L.I.Z.); the Canadian Institutes of Health Research (foundation grant FDN0159916 to S.H. and Y.W.); the National Cancer Institute (grant 5R01CA213062 to M.C.H.); the National Institute of General Medical Sciences (grant R35GM127045 to S.L.S.); the National Human Genome Research Institute (grant U54-HG008097 to M.P.); the Cancer Research Institute (I.E.); and the American Lebanese Syrian Associated Charities (B.J.A.).
Footnotes
Competing interests: B.J.A. is a shareholder in Syros Pharmaceuticals. C.A.C. is a founder and Chief Scientific Officer of Sana Biotechnology. S.L.S. serves on the Board of Directors of the Genomics Institute of the Novartis Research Foundation (GNF), is a shareholder in and serves on the Board of Directors of Jnana Therapeutics, is a shareholder in Forma Therapeutics, is a shareholder in and advises Decibel Therapeutics and Eikonizo Therapeutics, serves on the Scientific Advisory Boards of Eisai Co., Ltd., Ono Pharma Foundation, Exo Therapeutics and F-Prime Capital Partners, and is a Novartis Faculty Scholar. R.A.Y. is a founder of and shareholder in Syros Pharmaceuticals, Camp4 Therapeutics, Omega Therapeutics and Dewpoint Therapeutics. M.C.H. serves on the Scientific Advisory Board of Pori Therapeutics. S.A.C. is a member of the scientific advisory boards of Kymera, PTM BioLabs and Seer, and is a scientific adviser to Pfizer and Biogen. L.I.Z. is a founder of and holds stock in Fate Therapeutics, Camp4 Therapeutics and Scholar Rock, and is a consultant to Celularity. All other authors declare no competing interests.
Data and materials availability: The RNA-seq, ChIP-seq, and RRBS data of this study are deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE136456.
SUPPLEMENTARY MATERIALS
science.sciencemag.org/content/372/6543/716/suppl/DC1
Materials and Methods
Supplementary Text
Figs. S1 to S21
Table S1
References (34–75)
Data S1 to S4
MDAR Reproducibility Checklist
REFERENCES AND NOTES
- 1.Baron MH, Stem Cells 31, 849–856 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palis J, Front. Physiol 5, 3 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lensch MW, Daley GQ, Curr. Top. Dev. Biol 60, 127–196 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Orkin SH, Zon LI, Cell 132, 631–644 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cabezas-Wallscheid N et al. , Cell 169, 807–823. e19 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Aucagne R et al. , J. Clin. Invest 121, 2361–2370 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bai X et al. , Dev. Biol 373, 422–430 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He W et al. , Cell 125, 929–941 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Ransom DG et al. , PLOS Biol. 2, E237 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ransom DG et al. , Development 123, 311–319 (1996). [DOI] [PubMed] [Google Scholar]
- 11.Bai X et al. , Cell 142, 133–143 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lieberman I, Kornberg A, Biochim. Biophys. Acta 12, 223–234 (1953). [DOI] [PubMed] [Google Scholar]
- 13.Williamson RA et al. , J. Biol. Chem 270, 22467–22472 (1995). [DOI] [PubMed] [Google Scholar]
- 14.O’Donnell EF et al. , PLOS ONE 5, e13128 (2010).20957046 [Google Scholar]
- 15.Liu S, Neidhardt EA, Grossman TH, Ocain T, Clardy J, Structure 8, 25–33 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Carvalho L, Heisenberg CP, Trends Cell Biol. 20, 586–592 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Donovan A et al. , Nature 403, 776–781 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Chen JJ, Jones ME, Arch. Biochem. Biophys 176, 82–90 (1976). [DOI] [PubMed] [Google Scholar]
- 19.Pagliarini DJ et al. , Cell 134, 112–123 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer M, Oncogene 36, 3943–3956 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martínez-Reyes I, Chandel NS, Nat. Commun 11, 102 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ansó E et al. , Nat. Cell Biol 19, 614–625 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weinberg SE et al. , Nature 565, 495–499 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gusic M et al. , Am. J. Hum. Genet 106, 102–111 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Islam MS, Leissing TM, Chowdhury R, Hopkinson RJ, Schofield CJ, Annu. Rev. Biochem 87, 585–620 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Stachura DL, Chou ST, Weiss MJ, Blood 107, 87–97 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madak JT, Bankhead A 3rd, Cuthbertson CR, Showalter HD, Neamati N, Pharmacol. Ther 195, 111–131 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Hekimi S, Trends Cell Biol. 26, 367–378 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Stefely JA, Pagliarini DJ, Trends Biochem. Sci 42, 824–843 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mollet J et al. , J. Clin. Invest 117, 765–772 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Casey AC, Bliznakov EG, Cancer Res. 33, 1183–1186 (1973). [PubMed] [Google Scholar]
- 32.Donati S, Sander T, Link H, Wiley Interdiscip. Rev. Syst. Biol. Med 10, e1396 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Guijas C, Montenegro-Burke JR, Warth B, Spilker ME, Siuzdak G, Nat. Biotechnol 36, 316–320 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.