

Abstract
Catalytic C–H oxyfunctionalization reactions have garnered significant attention in recent years with their ability to streamline synthetic routes toward complex molecules. Consequently, there have been significant strides in the design and development of catalysts that enable diversification through C–H functionalization reactions. Enzymatic C–H oxygenation reactions are often complementary to small molecule based synthetic approaches, providing a powerful tool when deployable on preparative-scale. This review highlights key advances in scalable biocatalytic C–H oxyfunctionalization reactions developed within the past decade.
Graphical Abstract
1. Introduction
Catalytic C–H oxidation reactions are of immense interest to the synthetic community, largely due to their potential to unlock novel synthetic strategies and maximize step efficiency through late-stage functionalization of complex molecules.1–3 Although C–H oxidation reactions were once glamorous exceptions to the rules that define retrosynthetic logic, these transformations are now routinely incorporated into synthetic strategies.1–4 Initial research in this field focused on using transition metal complexes to guide oxidation events based on either the inherent differences between C–H bonds in a given compound, or using the existing moieties within a substrate to direct the selectivity outcome of the reaction.5,6
In parallel to advances in small molecule based tools, biocatalytic methods for selective C–H oxidation reactions are rapidly emerging. These advances are fuelled by DNA sequencing and bioinformatic technologies, enabling the identification of powerful enzymes with untapped potential,7 and with protein engineering strategies allowing for manipulation of these catalysts.8–11 Additionally, enzymes offer unprecedented selectivity profiles in C–H oxyfunctionalization transformations, that are often complementary to those obtained through small molecule organometallic catalysts.12 Furthermore, the safety, sustainability, and procedural simplicity of enzymatic reactions are attractive features that drive the development of biocatalytic methods.13, 14
Consequently, biocatalytic methods are becoming increasingly embraced by mainstream organic chemists.15–19 The advantages of leveraging enzymes in synthesis are exemplified by the recent work from Merck and Codexis in the development of a three-step biocatalytic cascade for the manufacture of the investigational HIV treatment drug islatravir.20 This incredible accomplishment is just one example of biocatalytic reactions on industrial-scale.19–21
As the organic synthesis community chooses which emerging biocatalytic methods to adopt, demonstrating the scalability of these reactions is of paramount importance.22, 23 However, the feasibility of preparative-scale biocatalytic oxygenation is not uniformly demonstrated as many reports detail reactions only on analytical-scale, leaving questions on enzyme expression-levels, stability, amenability to scaling, and overall robustness.19, 22–25 Despite these challenges, there are numerous biocatalytic oxyfunctionalization reactions of C–H bonds that are reliably scalable.12
In this review, we highlight key advances in scalable biocatalytic C–H oxyfunctionalization reactions from 2010 onwards. Since this review focuses exclusively on the addition of oxygen to substrates, we chose to use the term “oxyfunctionalization” as opposed to “oxidation”, as the latter term encompasses a broader meaning in organic chemistry.12, 26–28 The contents of this review are broadly categorized based on the type of C–H bond undergoing biocatalytic oxyfunctionalization. In most examples presented in this review, the biocatalysts are well characterized and belong to the broader classes of cytochromes P450,29 α-oxo acid and non-heme Fe(II)-dependent (NHI) oxygenases,30 and unspecific peroxygenases (UPOs).31, 32 However, in a subset of examples presented, whole-cell biotransformations are carried out,33 in which the exact enzyme responsible for the transformation is not identified.
The examples presented in this review were chosen from research articles in which preparative-scale reactions were carried out. Although the meaning of “preparative-scale” varies, it is broadly understood as a laboratory-scale process in which the product is isolated in sufficient quantities to allow for purification and/or full characterization. This scale often ranges from a few milligrams to multiple grams of the starting material. Most examples presented in this review were reported on milligram-scale. A select few examples, however, were reported on gram- and kilogram-scale, as noted.
1. Benzylic functionalizations
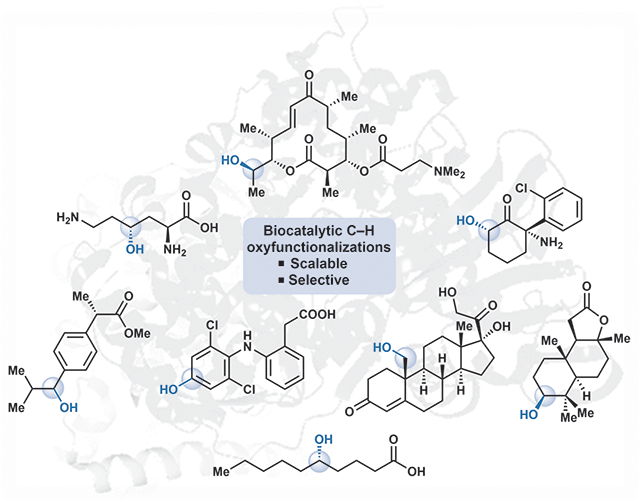
Benzylic oxidation reactions are amongst the most common type of C–H activation reactions carried out using a variety of small molecule oxidants.34 In the past 10 years, a number of biocatalytic benzylic C–H oxidations have been reported, with many enzymes tuned to have increased activity or desired selectivity using directed evolution techniques.8, 12 Most commonly, cytochrome P450 variants have been employed for selective benzylic hydroxylations on a wide variety of aromatic substrates.35 The prevalence of P450s in the biocatalytic oxygenation literature is in part based on decades of successful protein engineering on this class of enzymes.36–40 Pietruszka and coworkers improved the hydroxylation efficiency of cytochrome P450BM3 (B. megaterium) towards aromatic scaffolds containing protected alcohol and ester functionalities.38 They accomplished this by coupling a well-known active site mutation (F87A) to increase the space above the heme cofactor, along with site-saturation mutagenesis of three important amino acids (at positions 47, 51, and 188) that are involved in substrate recognition and selectivity.38 Profiling the activity of this enzyme library, they found a variant with two substitutions, F87A and L188C, that showed a 535-fold improvement in catalytic efficiency compared to wild-type P450BM3 on methyl 2-methoxy-3-methyl benzoate.38 A preparative-scale biotransformation using the F87A/L188C variant provided full conversion to the benzylic alcohol product 1 in 73% isolated yield (Fig. 1).38
Figure. 1.
P450-catalyzed benzylic C–H hydroxylations
Research by Reetz and coworkers led to the development of the P450BM3 A328F variant capable of benzylic hydroxylation on a variety of indane and tetralin substrates with high levels of site-selectivity and moderate-to-high enantioselectivity in the generation of the corresponding benzyl alcohol products (see 2, Fig. 1).41 In addition to the P450BM3 variants developed by Reetz, Yu and coworkers identified P450LaMO (L. aggregata) variants are efficient biocatalysts for benzylic hydroxylations of tetralin-like alkylbenzenes.42 Reetz and coworkers also engineered a P450BM3 variant (F87V) specifically for the oxidation of 6-iodotetralone. The benzylic hydroxylation product 3 was used as a coupling partner in several different palladium-catalyzed cross-coupling reactions, to rapidly access further functionalized products.39 The benzylic hydroxylation product 3 was obtained through milligram-scale biotransformation in 37% isolated yield and 99.9% ee.39
Directed evolution has been used to develop variants with stereo-complementarity allowing for selective access to either enantiomer of a chiral lactone through benzylic hydroxylation.43 Pietruszka generated two P450BM3 variants (R47Y and F87V/L188Q) capable of affecting benzylic hydroxylation of methyl 2-ethyl benzoate (4, Fig. 1), the product of which underwent subsequent cyclization to provide the lactone product 5 in high conversion, and with moderate yield and enantioselectivity.43 Zhou and Wong created a library of P450BM3 variants to enable biocatalytic hydroxylation of pharmaceutical drugs.44 Upon screening a library of 65 P450BM3 variants against the commonly administered tricyclic antidepressant amitriptyline, more than twenty variants displayed activity resulting in N-demethylation, whereas a select few performed benzylic hydroxylation to form (Z)-6 (Fig. 1).44, 45
Fasan and coworkers took a unique approach to protein engineering by incorporating four unnatural aromatic amino acids into 11 active-site positions of P450BM3 variant V78A/A184V by amber stop codon suppression.46 Out of the resulting 44 variants, 36 variants (dubbed “uP450s”) could be expressed in sufficient quantities to study the practical utility of incorporating aromatic unnatural amino acids within the active site of a cytochrome P450.46 The activity and selectivity of the uP450 library was explored through cell lysate reactions with several model substrates including (S)-ibuprofen methyl ester. Using the unmodified P450BM3 variant V78A/A184V, reaction with (S)-ibuprofen methyl ester afforded a 62:38 ratio of benzylic to homobenzylic hydroxylation products.46 Among the uP450s tested, variants with improved selectivity for either position were found. In particular, the site-selectivity for benzylic hydroxylation (forming product 7, Fig. 1) could be raised to 88% by the incorporation of 4-acetyl phenylalanine for the alanine at position 78 of the P450BM3 enzyme.46
Protein engineering has also been demonstrated to improve the activity of several other cytochrome P450 enzymes in addition to P450BM3.47 Bell and coworkers reported a CYP101B1 with a single substitution, H85F, that was capable of affecting benzylic hydroxylation on several toluene and naphthalene substrates, forming products 8-10 (Fig. 1) in high site- and stereoselectivity.47 Yu described several P450LaMO variants capable of oxidizing simple alkylbenzenes.42 Using both random and site-saturation mutagenesis, Yu and coworkers developed the RJ33 variant (T121P/Y385F/M329L) which provided improved selectivity for the benzylic hydroxylation of several substrates including ethylbenzene and propyl benzene, compared to the wild type enzymes.42
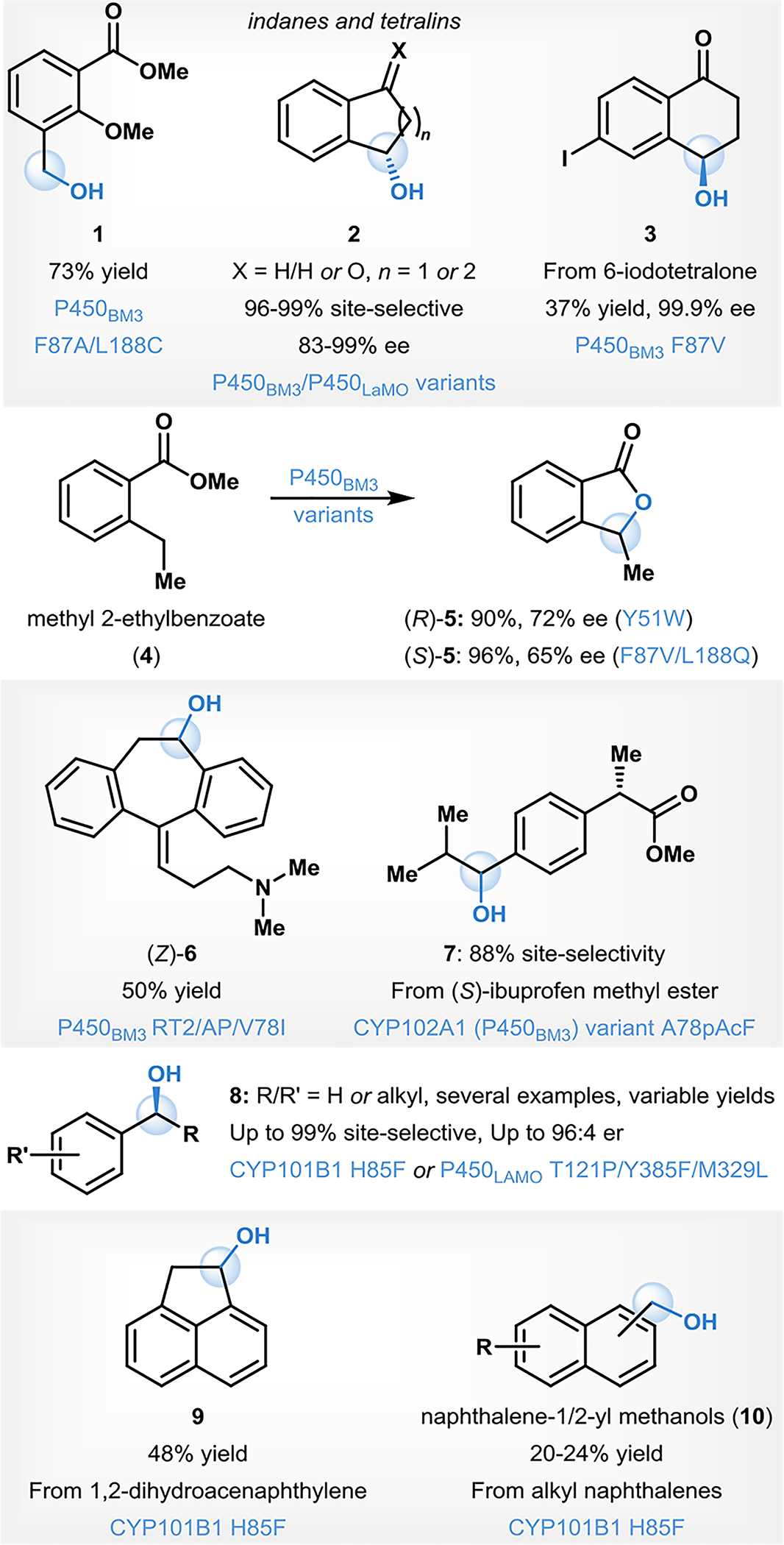
In addition to cytochromes P450, which have been the most well-studied family of enzymes for biocatalytic benzylic hydroxylation reactions, several other enzyme types have also been reported to possess this function.48–53 For example, Chen and coworkers identified two strains of the soil fungus P. plecoglossicidas (ZMU-T02 and ZMU-T06) for the whole-cell biocatalytic benzylic hydroxylation of tetrahydroquinoline and the seven-membered benzoazepine-like homolog to form the corresponding products 11 and 12 in high yield and enantioselectivity (Fig. 2A).48 Van Berkel and coworkers employed the flavin-dependent enzyme vanillyl alcohol oxidase (VAO) to affect benzylic hydroxylation of 4-ethylphenol on 10 gram-scale in 94% conversion and with a 36% isolated yield and 97% enantiomeric excess (ee) of the hydroxylated product 13 (Fig. 2A).49 The preparative-scale reaction was conducted with a cell-free extract containing VAO, and the authors attributed the overall low isolated yield in comparison to the high conversion to challenges in effectively extracting the alcohol product from the potassium phosphate reaction buffer.49
Figure. 2.
Biocatalytic benzylic C–H oxyfunctionalizations using non-450 enzymes
One advantage of using other classes of enzymes for scalable benzylic hydroxylation lies in the fact that unlike cytochromes P450, many other oxidases do not require an external reductase partner or NADPH recycling system.54 For example, our group has recently explored the use of α-ketoglutarate dependent NHI-enzymes for benzylic hydroxylation.50 This family of enzymes is well known for its scalability and ease of use.30, 55 We demonstrated the ability of two NHI enzymes, CitB and ClaD, to effectively perform benzylic hydroxylation on a wide range of phenols and resorcinols (14), using both whole-cell and crude-lysate formats to routinely conduct preparative-scale reactions with isolated yields ranging from 7–82% (Fig. 2B).50 The ortho-phenolic benzylic alcohol products (15) of these reactions are facile precursors to reactive ortho-quinone methide (o-QM; 16) intermediates for subsequent diversification.50, 56 The ease and scalability of performing benzylic hydroxylation using NHI enzymes allowed for a simple platform for direct hydroxylation and o-QM diversification in a single reaction vessel to isolate benzylic C–H functionalized products (17).50
In the same vein, Hollman and coworkers have developed several cascade strategies to achieve scalable benzylic hydroxylation using a nonspecific peroxygenase from A. aegerita (rAaeUPO).51–53 Peroxygenases are a class of enzyme capable of oxidizing a variety of C–H bonds including benzylic C–H bonds; however, the scalability of these reactions is hindered by their dependence on H2O2 as the stoichiometric oxidant, which is both difficult to use on a large scale and detrimental to enzyme activity at high concentrations.32, 57 Hollman and coworkers have been able to overcome these limitations by coupling peroxygenase activity to either photocatalysis or multi-enzyme cascades.51, 52 Initially, they employed an inorganic photocatalyst (Au-TiO2) for the reduction of oxygen in the presence of methanol to catalytically generate H2O2 for use by the peroxygenase to carry out benzylic hydroxylation of propyl benzene (18, Fig. 2C).51 This process was then further streamlined by their use of photocatalytic water oxidation catalysts (WOCs) to directly oxidize water to H2O2, thereby avoiding the need for methanol in the reaction mixture.52 More recently, Hollmann and coworkers have collaboratively developed a multi-enzyme cascade which combines rAaeUPO with a formic acid oxidase from A. oryzae (AoFOx) to generate the needed H2O2 in situ from formic acid without the need for a photocatalyst or UV light.53 This method improved upon previously designed multi-enzyme cascades by using a single enzyme for H2O2 generation to carry out benzylic hydroxylation of ethylbenzene (20). Ultimately, these strategies allowed them to achieve scalable, selective oxidation of several different types C–H bonds using peroxygenase rAaeUPO, including the benzylic hydroxylation of 20 to form 21 in 31% yield and 99% ee (Fig. 2C).
2. Allylic C–H oxyfunctionalizations
Several inorganic and organometallic reagents have been developed for the oxidation of allylic C–H moieties in organic compounds.34 Allylic and benzylic C–H bonds are considered activated, as intermediates leading to oxidations of such systems are resonance stabilized.58 Tremendous progress has been made in the scalable biocatalytic hydroxylation of allylic C–H bonds in cases of simple hydrocarbon substrates (covered in this section) and in complex natural product scaffolds (covered in Section 5).
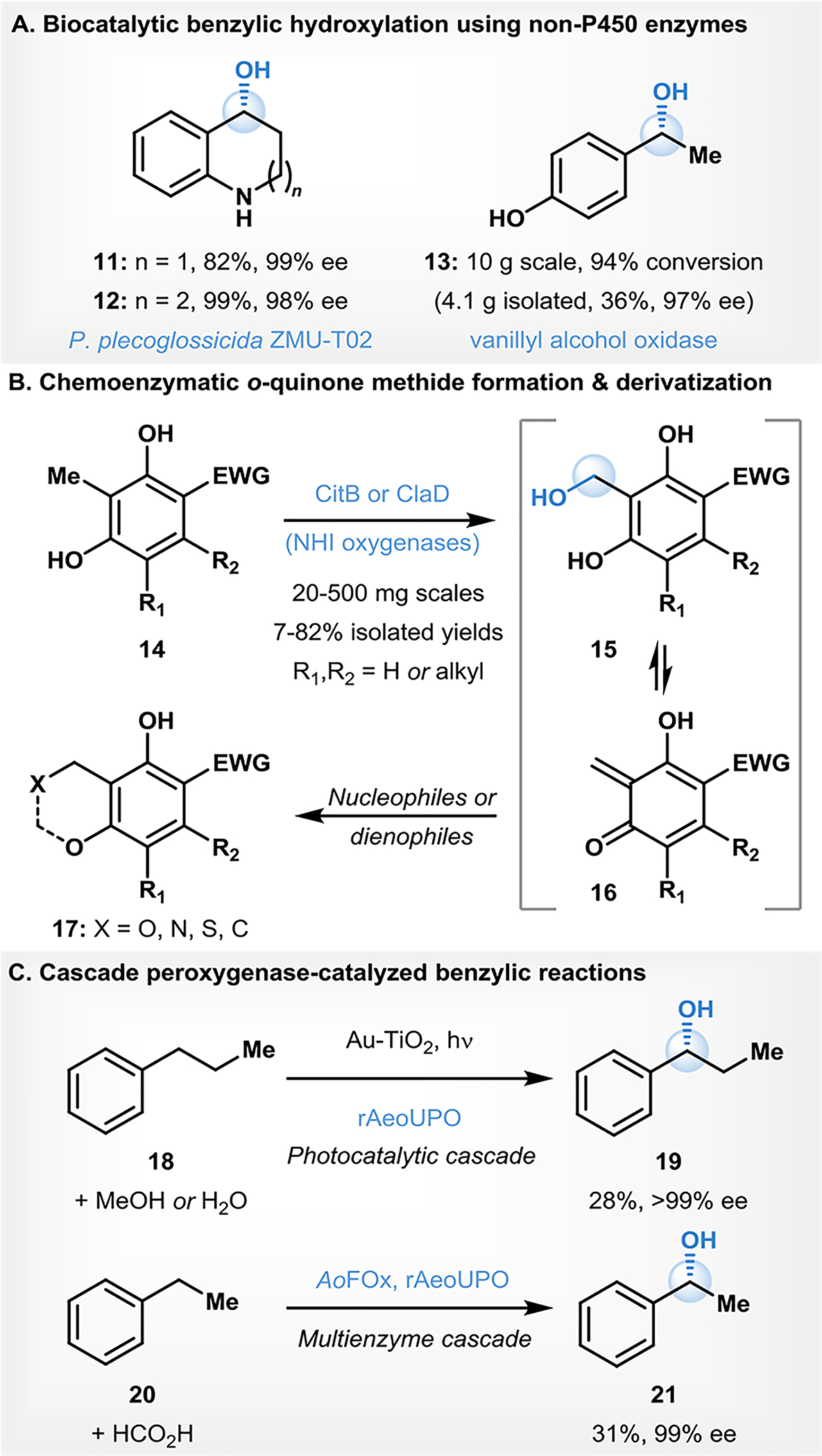
Zorn and Maison described the allylic oxidations of simple terpenoids and a number of related cycloalkenes with a cell-free lysate of the edible fungus Pleurotus sapidus (PSA) to form the corresponding alcohol or enone products (e.g., 22-27, Fig. 3) with up to 58% isolated yields.59 Maison and coworkers subsequently expanded on this work by investigating this oxidation with additional substrates.60 Using PSA lysate, a number of simple isoprenoid substrates were successfully converted to enone products (e.g., 28, 29) in preparative-scale reactions with yields ranging from 11–45%.59, 60
Figure. 3.
Biocatalytic allylic C–H oxidations
Reetz and coworkers demonstrated site- and enantioselective P450BM3-catalyzed allylic C–H hydroxylation of functionalized cycloalkenes.61 In this work, P450BM3 is engineered to provide variants suitable for preparative-scale allylic hydroxylations with cyclohexene-1-carboxylic acid methyl ester.61 Both enantiomers of the chiral product 30 (Fig. 3) could be isolated in up to 88% yield using the appropriate variant (F87V/A328N for R-selective hydroxylation, and I263G/A328S for S-selective oxidation). Reetz further expanded on this work in demonstrating that P450BM3 F87I/A328V variant could catalyze the allylic oxidation of 1-methyl cyclohexene.61 A similar transformation was reported by Wińska and coworkers in the case of a bicyclic cyclohexene-lactone scaffold using A. cylindrospora AM336 to affect allylic hydroxylation forming product 31 in 7% isolated yield (Fig. 3).62
Brenna and coworkers reported a biocatalytic asymmetric synthesis of cyclic γ-oxo esters via sequential C–H hydroxylation, oxidation of the resulting alcohol, and, finally, alkene reduction.63 In this work, the C–H hydroxylation of cyclohexene and cyclopentene-1-carboxylic acid ester was carried out using R. oryzae immobilized on polymer beads, providing the corresponding allylic hydroxylation products 32 in 65–88% yield (Fig. 3).63 Bell described selective aliphatic C–H bond functionalization of substrates containing a protected hydroxyl group by cytochrome P450 enzymes.64 When benzoic acid cyclohex-2-enyl ester was used as the substrate, the P450cam variant F87A/Y96F catalyzed the allylic oxidation of this compound with high levels of selectively affording the product trans-33 in 24% yield.64 Pietruszka and coworkers reported a P450BM3 catalyzed enantioselective allylic hydroxylation of ω-alkenoic acids and esters.37 Three ester substrates and one acid substrate were oxidized with P450BM3 variant A74G/L88Q, providing the corresponding allylic hydroxylation products 34 in 20–80% yields and with ee’s ranging from 92% to >97% (Fig. 3).37
Kaluzna and group described a kilogram-scale, selective and sustainable P450-based biocatalytic allylic hydroxylation of α-isophorone for the production of 4‑hydroxy-α-isophorone (35, Fig. 3).65 Two consecutive 100-liter scale reactions were carried out within 10 hours to afford 35 in excellent ee (>99%) and high purity (>98% HPLC, GC, NMR) with isolated yields of 51% and 61%, for batches 1 and 2, respectively.65 Overall, the two batches delivered a total of 1 kilogram of 4-hydroxy-α-isophorone.65 A subsequent report from Mattevi and Turner described a biocatalytic one-pot iterative double oxidation of α-isophorone to ketoisophorone (36).66 A milligram-scale oxidation was accomplished in a cascade with P450WAL (variant of the self-sufficient chimeric P450cam-RhFRed; used for the initial hydroxylation), and alcohol dehydrogenase Cm-ADH10 (used for the alcohol oxidation to the ketone), affording ketoisophorone (36) in 56% yield (Fig. 3).
3. Aromatic C–H hydroxylations
Hydroxylated aromatic compounds (phenols) are widely distributed in Nature, serve as components of important natural products, and constitute important feedstocks in the chemical industry.67 Therefore, extensive efforts have been devoted to the synthesis of phenols through different methods.68 The direct C–H hydroxylation of arenes is the most streamlined route towards the synthesis of phenols, yet it continues to remain elusive in synthetic organic chemistry.69 Biocatalytic aromatic C–H hydroxylation reactions have emerged as a powerful tool to enable the synthesis of hydroxylated arene derivatives.70
The development of synthetic methods to access drug metabolites and study their biophysical and metabolic properties is an important endeavor executed by both the pharmaceutical industry and the food and drug administration (FDA).71 Methodology development for accessing hydroxylated metabolites of the commonly consumed non-steroidal anti-inflammatory drug (NSAID) diclofenac (37) has received considerable attention from the biocatalytic community.44, 72–77 Biocatalytic hydroxylation of diclofenac has been carried out using a variety of enzymes and whole-cell microorganisms ranging in milligram- to gram-scales, to afford aromatic C–H hydroxylation products such as 4’- or 5- hydroxydiclofenac (38 and 39, Fig. 4).45, 72, 74–78 Biocatalytic hydroxylation of a deuterated diclofenac (37-d7) was reported by Atzeodt and coworkers.73 Using Cunninghamella echinulate (var elegans) ATCC9245, hydroxylation of a d7-diclofenac substrate afforded the 4’-hydroxylated product (38-d6) in 65% isolated yield.73 Scalable biocatalytic aromatic C–H hydroxylation has also been demonstrated in the case of other drug molecules such as chlorzoxazone and mefenamic acid (Fig. 4).44, 79 Chlorzoxazone was selectively oxidized to afford milligram quantities of 6-hydroxy chlorzoxazone (40) in 98% yield using a P450BM3 variant.44 Commandeur and coworkers reported P450BM3 variants that affect preparative-scale aromatic C–H hydroxylation of mefenamic acid and other structurally related NSAIDs.79 Using the P450BM3 variant M11 V87F, mefenamic acid was oxidized to afford 4’-hydroxymefenamic acid (41) in 53% isolated yield.79
Figure 4.
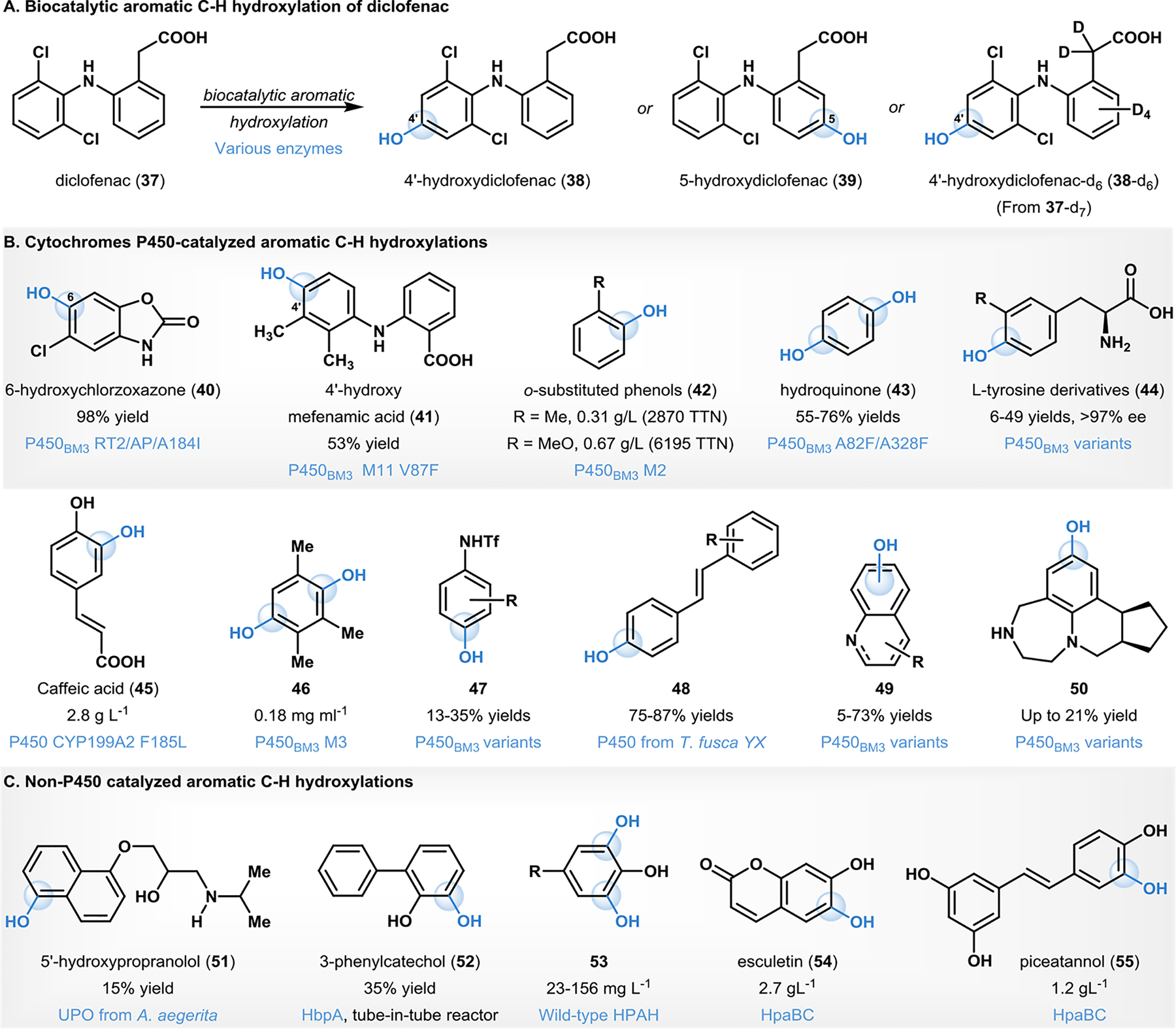
Biocatalytic aromatic C–H oxyfunctionalizations
Efficient biocatalytic hydroxylations of highly hydrophobic aromatic substrates such as benzene and its alkylated derivatives have been reported (Fig. 4).80–83 For example, Schwaneberg and coworkers reported a regioselective ortho-hydroxylation of monosubstituted benzenes by P450BM3 variants.80 On preparative scales, both anisole and toluene were oxidized to the respective ortho-hydroxylated products (42) at levels 0.67gL−1 (6195 TTN) and 0.31 gL−1 (2870 TTN), respectively.80 Li and Reetz employed the P450BM3 A82F/A328F variants to form hydroquinone through either dihydroxylation of benzene (54% yield), or monohydroxylation of phenol (76% yield).81
Amino acids constitute important building blocks in the pharmaceutical industry, and enabling access to canonical amino-acid derivatives has attracted considerable attention from the synthetic community.84 Kroutil and Faber reported the synthesis of L-tyrosine derivatives (44, Fig. 4) using a biocatalytic one-pot, two-step cascade from substituted benzenes, pyruvate, and ammonia as starting materials.85 P450BM3 variants were used for ortho-selective hydroxylation of monosubstituted arenes in the first step of the transformation. Subsequent biocatalysis using tyrosine-phenol lyase enabled C–C coupling and simultaneous asymmetric amination with pyruvate and ammonia forms L-3,4-dihydroxyphenylalanine (DOPA) derivatives with isolated yields ranging from 6–49% and >97% ee in milligram scales.85 Section 4 of this review covers alkyl C–H hydroxylations in amino acid scaffolds (vide infra).
Biocatalytic C–H hydroxylations have been reported on a variety of other functionalized arenes using cytochrome P450 enzymes (Fig. 4).86–90 For example, Furuya and coworkers used CYP199A2 variant F185L in whole-cell biotransformation to catalyze the oxidation of p-coumaric acid to form caffeic acid (45; product yield 2.8 g L−1 in 24 h).86 Dennig et al., demonstrated that the aromatic ring in pseudocumene could be selectively oxidized using an engineered P450BM3 variant to deliver 0.18 mg mL−1 (0.9% isolated yield) of hydroquinone derivative 46.88 Aromatic hydroxylations on functionalized anilide scaffolds was reported by Wong and Robertson using P450BM3 variants to form products (47) in 13–35% isolated yields.89 Engineered cytochromes P450 from Thermobifida fusca were employed by Urlacher and coworkers in carrying out para-selective aromatic hydroxylations of stilbenes, to form the corresponding products (48) in 75–87% isolated yields.90 Wong reported P450BM3 variants for aromatic C–H hydroxylations of quinolines and tetrahydroquinolines to form the corresponding products (49) in 5–73% isolated yields in synthetically relevant scales of 1.5 gL−1day−1.45
Late-stage functionalization of complex molecular scaffolds is a challenging synthetic endeavor and is increasingly employed in lead optimization of drug candidates.91 In this regard, Lange and coworkers reported the regioselective aromatic hydroxylation of protected vabicaserin substrates using P450BM3 variants.92 This biocatalytic method was also shown to be highly chemoselective for aromatic hydroxylation, even in the presence of more reactive tertiary benzylic sites within the substrate.92 Following biocatalytic aromatic hydroxylation and N-aryl sulfonamide deprotection, the products (e.g., 50, Fig. 4) were obtained in up to 21% isolated yield.92
In addition to cytochromes P450, other enzymes and whole-cell biotransformations are reported to affect aromatic C–H hydroxylations (Fig. 4).93–97 For example, Alcade and coworkers reported an aromatic C–H hydroxylation catalyzed by an engineered unspecific peroxygenase (UPO) from Agrocybe aegerita, to enable access to the human drug metabolite 5’-hydroxypropranolol (51) in 15% isolated yield.93 Buehler and coworkers reported a synthesis of 3-phenylcatechol (52) on gram-scale using a continuous segmented flow tube-in-tube reactor (TiTR) employing 2-hydroxybiphenyl-3-monooxygenase (HbpA) for catalyzing the aromatic hydroxylation of 2-hydroxybiphenyl.94 The TiTR hydroxylation of 2-hydroxybiphenyl was carried out to obtain 3-phenylcatechol (52) in 35% isolated yield.94 Chaiyen and coworkers reported the efficacy of p-hydroxyphenylacetate-3-hydroxylase (HPAH) as an efficient biocatalyst for the synthesis of trihydroxyphenolic acids (53).95, 96 In their work, p-coumaric acid and caffeic acid (45) were used directly from an extract of Palm oil mill effluent.96 Yan and Lin reported the use of the non-P450 native E. coli hydroxylase HpaBC in enabling efficient ortho hydroxylation of plant phenylpropanoids such as the coumarin umbelliferone and stilbene-derivative resveratrol, yielding corresponding products esculetin (54) and piceatannol (55) in 2.7 g L−1 and 1.2 g L−1 (in almost quantitative yields), respectively.97
4. Functionalizations of amino acids
Amino acids are an important class of organic molecules that serve as important building blocks in the asymmetric synthesis of pharmaceutical agents and natural products.84 However, several challenges continue to exist in the synthetic manipulation of native amino acid scaffolds. For instance, direct C–H functionalization approaches using transition metal complexes typically functionalize the β-position of α-amino acids,98 and there are limited reports on functionalization at the γ-,99 and δ-positions.100,101 Additionally, synthetic C–H functionalizations of amino acid scaffolds require several protecting group manipulations, adding extra steps and cost to a synthetic campaign.99–101 In stark contrast to the existing chemical methods, Nature’s catalysts mediate these transformations with a broader array of site-selectivities, without requiring protecting group manipulations (vide infra). This section reviews some of the major advances made in the past decade towards scalable biocatalytic C–H oxyfunctionalizations of amino-acids.
Several biocatalytic amino-acid oxygenations are carried out as part of chemoenzymatic total synthesis campaigns towards bioactive molecules.84, 102–104 For example, Shimizu and coworkers developed a stereospecific C4 biocatalytic hydroxylation of L-isoleucine (56, Fig. 5) using L-isoleucine dioxygenase (IDO), to produce (4S)-hydroxy-isoleucine (4S-(OH)-56).102 The latter is a natural nonproteinogenic amino acid that exhibits insulinotropic biological activity and is used for the treatment of diabetes.105 Hüttel and coworkers developed a facile method for the production of cis-3-, cis-4-, and trans-4-proline hydroxylase (α-ketoglutarate (α-KG) dependent (NHI) monooxygenases), and the application of this approach for the site- and stereoselective hydroxylation of L-proline (57) and it’s six-membered ring homologue L-pipecolic acid (58).106, 107 The Renata group developed a scalable hydroxylation reaction with L-pipecolic acid 58 to form cis-3-hydroxy-pipecolic acid (3R-(OH)-58) using the α-KG dependent NHI oxygenase GetF. This transformation was leveraged in the total synthesis of the peptide complex GE81112 B1.103
Figure 5.
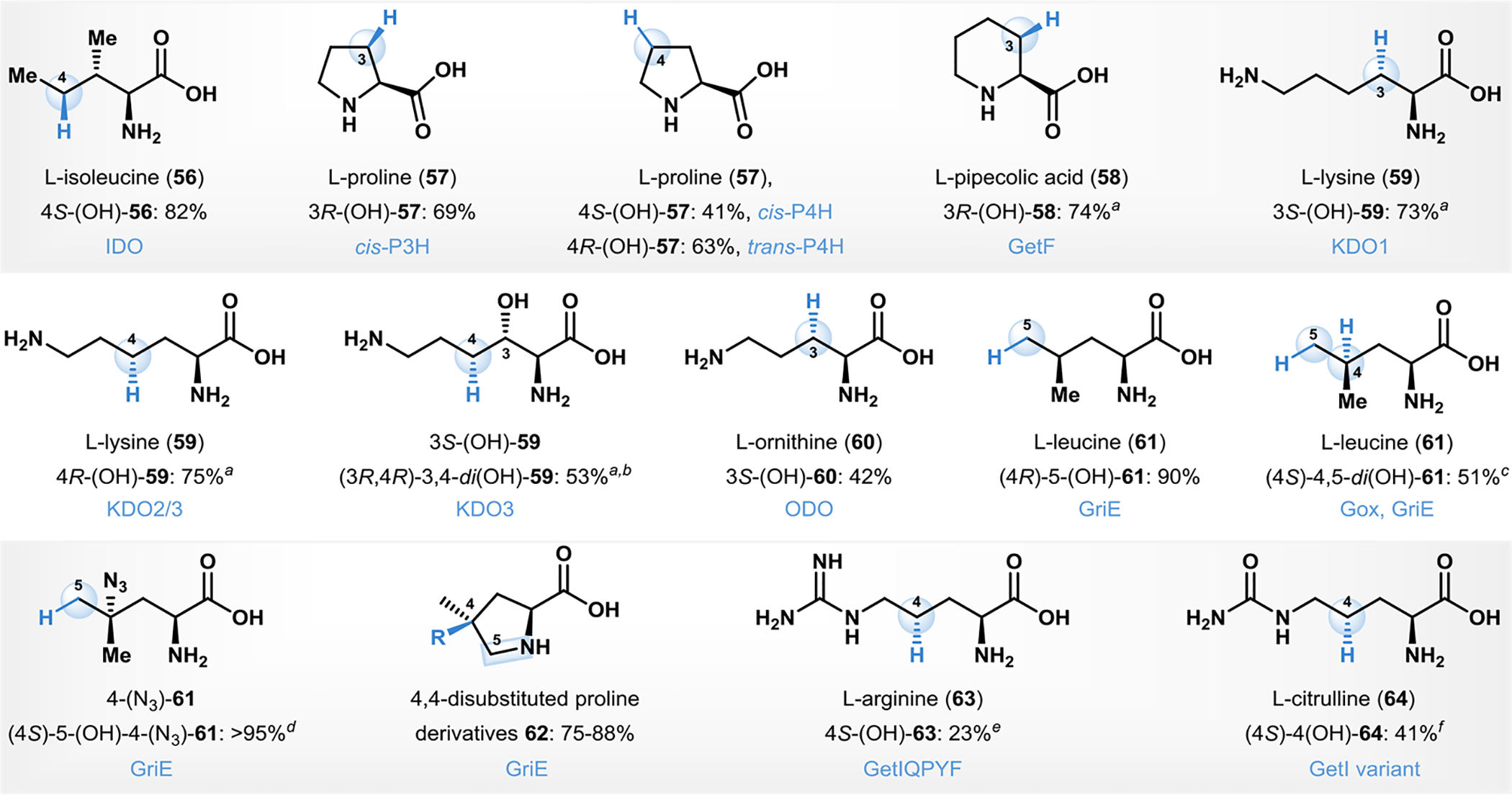
Biocatalytic C–H hydroxylation of amino acids. Note: aYields based on the boc protected derivative(s), bOverall yield starting from L-lysine, cYield obtained after Fmoc protection and cyclization to the lactone derivative. dPercent conversion provided.eNMR yield. fMulti-step cascade is carried out prior to isolation.
Zaparucha and coworkers developed a genome-mining approach, which led to the discovery of α-KG dependent NHI oxygenases hydroxylating the side chains of basic amino acids lysine (59) and ornithine (60).108,109 Two of the enzymes, KDO2, and KDO3 (lysine dixoygenases) demonstrated hydroxylation activity at the C4 position of these basic amino acids.108 The biocatalysts developed were regioselective, and the reactions were carried out on milligram preparative scales.108 The C3-selective hydroxylation of L-lysine developed by Zaparucha was employed in a multigram scale by Renata and coworkers in their total synthesis of tambromycin.108, 110 The Renata group subsequently identified a lysine-4-hydroxylase GlbB from the glidobactin biosynthetic pathway and evaluated its synthetic potential. The α-KG dependent NHI oxygenase GlbB catalyzes the C4-selective C–H hydroxylation of L-lysine (59) and L-leucine (61), and affects monooxygenation of the sulfur in L-methionine.111 GlbB mediated C4-selective C–H hydroxylation of L-lysine was subsequently carried out on multigram scale in the chemoenzymatic total synthesis of cepafungin I.104
Renata and coworkers subsequently developed a general strategy to hydroxylate the δ-position of proteinogenic amino acids using an α-KG dependent non-heme oxygenase GriE.84,55 The enzyme GriE was shown to hydroxylate L-leucine (61, Fig. 5) to afford (4R)-5-hydroxy-leucine ((4R)-5-(OH)-61) in 90% yield and with complete diastereoselectivity.84 This facile chemistry was demonstrated on several other amino acids bearing structural similarity to L-leucine with isolated product yields ranging from 18–90%.84 The group also developed a one-pot dihydroxylation cascade employing the α-KG dependent NHI oxygenase Gox to hydroxylate the 4-position of L-leucine (61).84, 112 Gox hydroxylation followed by treatment with GriE afford diol (4S)-4,5-di(OH)-61.84 This chemistry was extended to leucine derivatives, for example, hydroxylation of 4-azido-L-leucine (4-(N3)-61) afforded the C5 hydroxylated product in >95% conversion and in 58% isolated yield (obtained after boc protection).55, 84 Hydroxylated leucine derivatives functionalized at C4 (e.g. (4S)-5-(OH)-4-(N3)-61) are synthetic precursors to functionalized proline derivatives. At sufficiently high concentrations of GriE, iterative oxidation was carried out with the formation of an intermediate imine, followed by reduction with NH3.BH3 to afford 4,4’-difunctionalized proline derivatives 62 in excellent yields and with complete stereocontrol.84
Renata and coworkers carried out the characterization of a citrulline 4-hydroxylase from NRP GE81112 biosynthesis, and establish the role of an α-KG dependent NHI enzyme GetI in the production of GE81112.113 Homology modeling and multiple sequence alignments facilitated the rational engineering of this enzyme to deliver a hydroxylase for the C4-selective hydroxylation of L-arginine (63, Fig. 5). Sequence similarity analysis predicted the substrate recognition of GetI and facilitated its rational engineering to become a specific 4-arginine hydroxylase with four mutations. The GetI-mediated C4-selective hydroxylation was also employed by the Renata group for L-citrulline (64) towards the concise total synthesis of the peptide complex GE81112 B1.103
5. Functionalization of complex molecules
5.1. Biocatalytic C–H hydroxylation of steroid scaffolds
The steroid skeleton is present in a wide variety of natural products and synthetic molecules with diverse bioactivities.114 Recent developments in biocatalytic C–H oxyfunctionalization reactions have enabled access to hydroxylated steroid derivatives that are not easily accessible using traditional synthetic methods.114–117 Scalable biocatalytic C–H hydroxylation of testosterone has been carried out with selective access to hydroxylated derivatives at the 2, 14, 15 and 16 positions (see Fig. 6A), often in a site- and stereoselective manner.118–120 The Reetz group developed P450BM3 (F87A) based variants for the scalable site- and stereoselective hydroxylation of testosterone.118 Selective formation of 2β-hydroxytestosterone (65) was possible with the variant containing two substitutions (A330W/F87A) to deliver 65 in a 97:3 regioisomeric ratio (rr) of 65:67 in 79% total conversion.118 Hydroxylation to afford 15β-hydroxytestosterone (67) was achieved using the variant harboring three substitutions (V78L/A82F/F87A), providing product in 86% conversion and a 3:91 ratio of 65:67. Using whole-cell biotransformation, Thulasiram and coworkers used the fungus Mucor hiemalis to obtain 14α-hydroxylated derivative (66) in 11% isolated yield.119 In addition to the 14α-monohydroxylated product 66, the authors observed the formation of 7α,14α- and 6β,14α- dihydroxylated products in 32% and 12.5% isolated yields, respectively.119 The exact biocatalyst was not identified from Mucor hiemalis; however, the authors carried out additional experiments reporting that the 14α-hydroxylase activity is NADPH dependent, and concluded that this reactivity likely originates from a cytochrome P450 family of enzymes.119 A subsequent report from Scheibner and coworkers demonstrated the efficacy of an unspecific peroxygenase (UPO) Cg/UPO, isolated from the fungus Chaetomium globosum in the selective hydroxylation of testosterone.120 Whereas the major product formed was the 4,5-β-epoxide, a minor product was formed from the selective C–H hydroxylation at C16 resulting in 16-α-hydroxytestosterone (68) in 7% isolated yield.120
Figure 6.
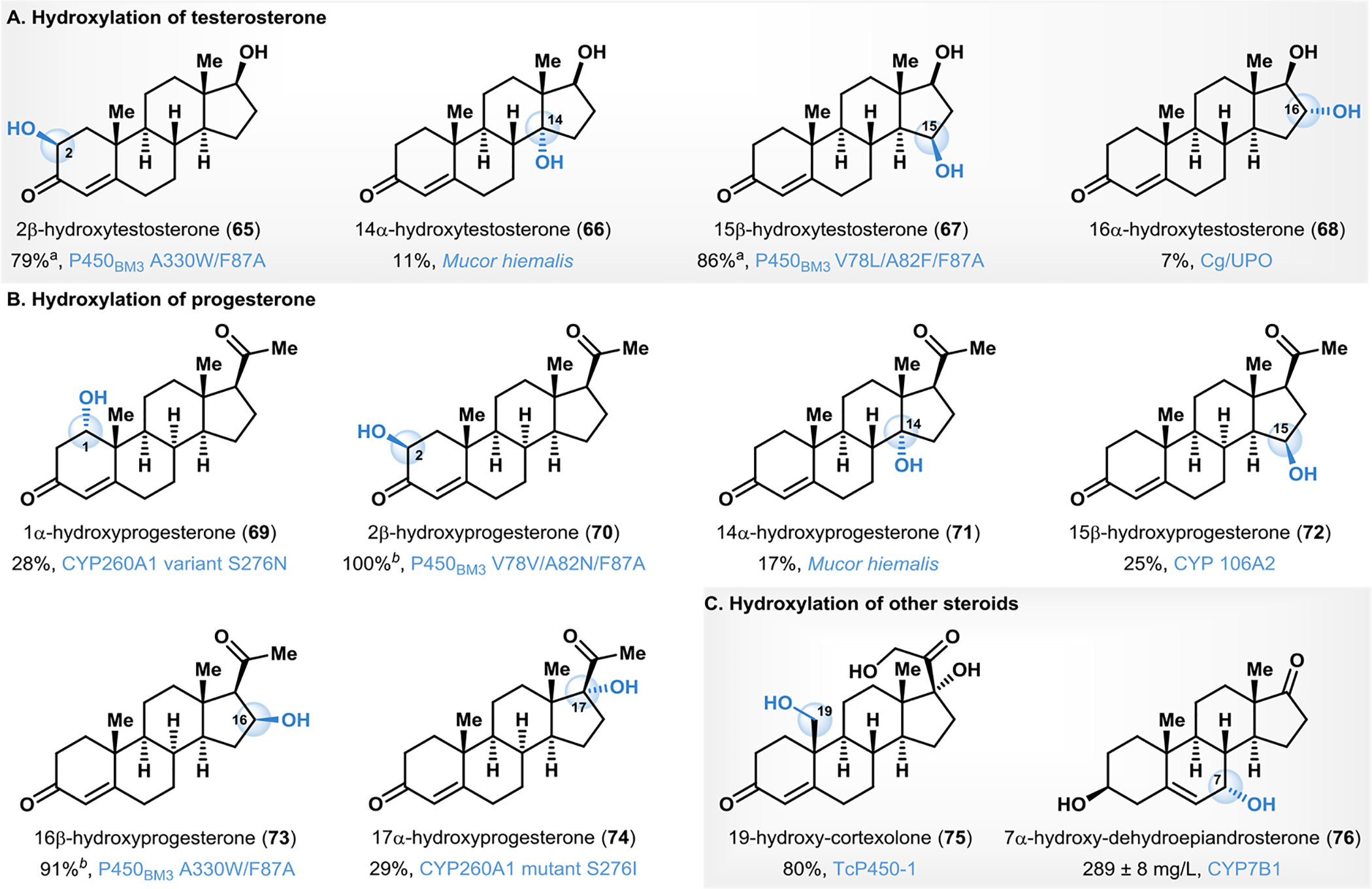
Biocatalytic C–H monohydroxylation of steroidal frameworks. aPercent conversion.bPercent site-selectivity.
Several groups have developed biocatalytic C–H hydroxylation reactions with the steroidal hormone progesterone to enable access to progesterone derivatives monohydroxylated at positions 1,2,14, 15, 16 and 17 (Fig. 6B).118, 119, 121, 122 Bernhardt has reported variants of CYP260A1 isolated from S. cellulosum that are effective mediators of progesterone hydroxylation with the complementary site- and stereoselectivity.121 The S276N variant enabled access predominantly to 1α-hydroxyprogesterone (69, 28%), whereas the S276I variant resulted in 17α-hydroxyprogesterone (74, 29%).121 Engineered P450BM3 variants developed by Reetz for the selective hydroxylation of testosterone were also screened for activity with progesterone.118 The variants V78V/A82N/F87A and A330W/F87A resulted in site-selective formation of 2β-hydroxyprogesterone (70) as the sole product, and 16β-hydroxyprogesterone (73; 73:70 = 91:9), respectively.118 Thulasiram and coworkers used Mucor hiemalis to obtain 14α-hydroxylated progesterone (71) in 17% isolated yield.119 Similar to the product distribution obtained in case of testosterone, progesterone hydroxylation with Mucor hiemalis resulted in a mixture of other dihydroxylated products accounting for the remainder of the mass balance.119 Lütz and coworkers demonstrated the efficacy of CYP106A2 from Bacillus megaterium in the selective hydroxylation of progesterone resulting in 15β-hydroxyprogesterone (72, 25% yield).122 Dihydroxy derivatives of progesterone have also been selectively accessed, for example, Arnold reported a P450BM3 variant for hydroxylation of 11α-hydroxyprogesterone to generate 2α,11α-dihydroxy progesterone in 20% isolated yield.123
Additional scaffolds have been explored in the context of biocatalytic C–H hydroxylation in pursuit of new steroid-based drugs (Fig. 6C).124–126 Hydroxylation at the C19-position of steroidal frameworks is extremely challenging using synthetic chemical methods,127–129 yet functionalization of this position proves pivotal to accessing bioactive C19-hydroxylated steroids. Zhou overcame this synthetic bottleneck by optimizing the production of 19-hydroxycortexolone (75) in multigram scales through hydroxylation of 17-acetyl-cortexolone with TcP450–1, a cytochrome P450 enzyme.125 The product 75 could be transformed into 19-hydroxy-deoxycorticosterone or 19-hydroxy-androstenedione, which are valuable precursors to other 19-functionalized bioactive steroids.125 Song and coworkers employed P450 CYP7B1 in a bioelectrocatalytic 7α-hydroxylation of dehydroepiandosterone (DHEA).126 This method employed an electricity-driven NADPH regeneration with a concomitant electron shuttle to accomplish cofactor regeneration. Using this setup, production of 7α-hydroxy-DHEA (76) was achieved in 289 ± 8 mg L−1 (67–71% yield; Fig. 6C).126
5.2. Biocatalytic C–H hydroxylation in terpene scaffolds
Biocatalytic late-stage C–H hydroxylation is a powerful tool for achieving diversification of complex terpenoid natural products. Fasan and coworkers developed a method to map the reactivity patterns of engineered P450 variants in high-throughput using a set of chromogenic probes, to obtain an indirect map of the size and geometry of their active sites.130 Through analysis of their resulting fingerprints, reliable predictions were made as to which C–H hydroxylation events could be possible on complex terpenoid natural products (e.g., 77-79, Fig. 7A).130 This fingerprinting approach is a powerful tool for expediting the discovery of P450 catalysts for the functionalization of several natural product scaffolds.
Figure 7.
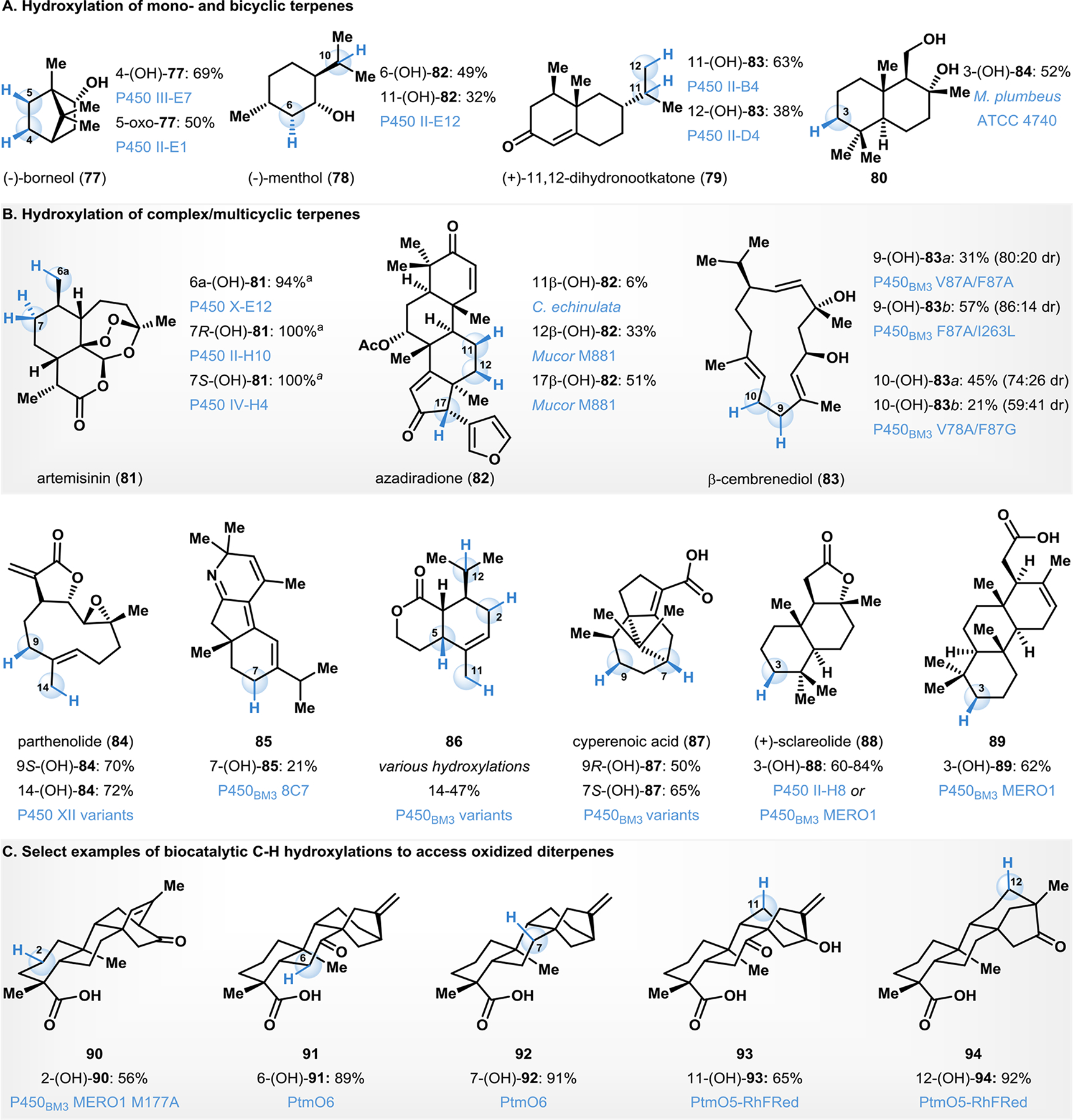
Biocatalytic C–H hydroxylation in diverse terpene scaffolds.
Terpene C–H functionalization has been significantly explored in the context of chemoenzymatic total synthesis of bioactive natural products.131–134 For example, a key transformation executed by Barrero en-route to (+)-myrrhanol C involved C3 hydroxylation of diol 80 (Fig. 7A).131 This transformation was carried out as biotransformation with M. plumbeus ATCC 4740. The C3-hydroxylated product was subsequently transformed to myrrhanol C through subsequent chemical steps.131
The structurally complex natural product artemisinin (81) has been long used for the effective treatment of malaria,135 and is a promising lead natural product for various drug developments.136 This has resulted in a surge of method developments to functionalize 81 to obtain derivatives bearing enhanced therapeutic activity.137, 138 Using 81 as a model substrate, Fasan demonstrated the high throughput P450 fingerprinting approach to discover three efficient biocatalysts for the selective hydroxylation of a primary and secondary C–H bonds. Whereas the parent P450 formed a 83:10:7 ratio of C7(S) : C7(R) : C6a hydroxylated products from 81, they evolved P450s selectively deliver each of these products with IV-H4 provided (7S)-hydroxylation, II-H10 affording (7R)-hydroxylation and X-E12 delivering the C6a-hydroxy product all in excellent yields (Fig. 7B).139
Several groups have reported biocatalytic methods to carry out site-selective C–H hydroxylations in structurally complex terpenoid scaffolds.140–148 For example, Thulasiram developed a whole cell biocatalytic platform for the selective C–H hydroxylations of azadiradione (82).140, 141 The 11 β-selective hydroxylation of 82 was carried out using the fungal species, C. echinulate, in 6% isolated yield.140 Upon incubation of substrate 82 to the fungus Mucor M881, site-isomeric C12 and C17 β-hydroxylated products formed in 33% and 51% isolated yields, respectively (Fig. 7B).141 Urlacher and coworkers developed a P450- catalyzed chemo-, site- and stereoselective oxidation of the tobacco cembranoid natural product β-cembrenediol (83).142 This macrocyclic terpenoid diol possesses antitumor and neuroprotective biological activities.142 Several P450BM3 variants were engineered to selectively hydroxylate 83 at C9 (with variant F87A/I263L) and C10 positions (with the variant L75A/V87A/F87G) in moderate site- and diastereoselectivity (Fig. 7B). Subsequent follow-up work by Urlacher demonstrated selective hydroxylation of synthetic macrocyclic scaffolds structurally similar to 83 using engineered P450BM3 variants.149
Parthenolide (84) is a sesquiterpene lactone with promising antitumor properties against stem-cell cancers, including leukemia.143, 150 Fasan and coworkers developed biocatalysts for the late-stage C–H hydroxylation of two aliphatic sites (C9 and C14) of 84 (Fig. 7B).143 Engineered P450 XII-F12 was selective for C9-hydroxylation of 84, affording the hydroxylated product in 70% isolated yield. Alternatively, P450 VII-H11 was a productive catalyst for C14-hydroxylation of 84, providing the desired product in 72% isolated yield.143 Arnold and Stoltz employed a biocatalytic late-stage C–H hydroxylation to access the norditerpenoid alkaloid nigelladine A.132 In their synthetic plan, an engineered P450BM3 variant was employed to affect allylic C–H oxidation of 85, followed by oxidation with DMP to afford the natural product in 21% yield over two steps.132 The appropriate enzyme variant was readily identified from a focused library of three enzymes, allowing for completion of the synthesis without the need for extensive screening.132
Robertson and coworkers engineered a panel of P450BM3 variants for the oxidation of terpenoid scaffolds (e.g., 86, Fig. 7B) relevant to the synthesis of hydroxylated eleutherobin analogs.144 Eleutherobin is a diterpene isolated from the soft coral Eleutherobia species and is cytotoxic towards a variety of human cancer cell lines with potency and mechanism of action comparable to that of taxol.151 In the course of their research, the authors subjected the lactone 86 to P450BM3 variants to achieve selective hydroxylation at four separate C–H sites.144 A related lactol substrate was also tested to assess the influence of the carbonyl group on reactivity. In case of the lactol substrate, however, the same P450BM3 variants were less selective.144
The sesquiterpenoid natural product cyperenoic acid (87) isolated from C. crassifolius possesses an antiangiogenic promoting effect.152 You and coworkers developed a P450BM3 variant to hydroxylate the C7- and C9a- positions of 87 (Fig. 7B).145 These hydroxylated derivatives of 87 can significantly inhibit the release of vascular endothelial growth factor.152 You evolved P450BM3 through one or two generations of mutations, and the resulting variants selectively delivered hydroxylation products, with the F87A/A330W/F331L variant providing C7 hydroxylation and L75V/F87A/T88F/A330W variant providing C9 hydroxylation in excellent yields and site- and stereoselectivities.
The C3-selective hydroxylation of the sesquiterpene lactone sclareolide (88) was developed by Fasan and coworkers using a P450BM3 variant II-H8 (15 mutations from the wild type) to affect the desired transformation on a milligram scale in 84% isolated yield (Fig. 7B).130 In a chemoenzymatic campaign towards meroterpene natural products, Renata and coworkers, further engineered this variant and developed a protocol for scaling the C3 oxidation of 88 up to gram-scale.133 They identified a variant 1857 V328A (BM3 MERO1) as an efficient biocatalyst affording >95% conversion to 3-hydroxy sclareolide. This protocol was conducted on the multigram scale to deliver 60–70% isolated yields of the 3-(OH)-88.133 This biocatalyst was also applied to the C3 hydroxylation of terpenic acid 89 en-route to chemoenzymatic syntheses of other meroterpenoids.133
Renata and coworkers recently disclosed a chemoenzymatic strategy to access complex ent-kauranes, ent-atisanes, and ent-trachylobanes (Fig. 7C).134 Owing to the structural complexities of these terpenoids, the total synthesis of these molecules presents a tremendous challenge, and semisynthetic access is limited by the lack of chemical tools for modification of such complex scaffolds.134 NHI α-ketoglutarate-dependent dioxygenase from platensimycin biosynthesis were involved in this campaign, for example, the enzyme PtmO6 was shown to exhibit exquisite selectivity for C7 position, and a capability to oxidize C6 in case of C7 oxidized to a carbonyl functionality. Select examples (90-94) are shown in Fig. 7C.
5.3. Biocatalytic C–H hydroxylation in macrolide scaffolds
Sherman and Podust characterized a P450 monooxygenase PikCD50N-RhFRED from the pikromycin biosynthetic pathway from Streptomyces venezuelae.153 Their studies revealed structural and biochemical evidence for a salt bridge between the desosamine N,N-dimethylamino-functionality of the natural substrate (95, Fig. 8), and carboxylate residues within the active site and they proposed that this interaction controls the efficiency and selectivity of the C–H hydroxylation event. Building on this work, Sherman and coworkers developed a substrate engineering approach that involved replacing desosamine with various synthetic N,N-dimethylamino anchoring groups (Fig. 8).154 While the natural anchoring group on desosamine affords a 1:1 ratio of C-10 to C-12 hydroxylated products, synthetic anchors could be tuned to afford selective hydroxylation of either C-10 (96: 74% yield, C-10:C-12 rr = >20:1) or C-12 (97: 73% yield, C-10:C-12 rr = 1:4) hydroxylated products, demonstrating the utility of substrate engineering as an orthogonal approach to protein engineering in modulating regioselective C–H functionalization in biocatalysis.154
Figure 8.
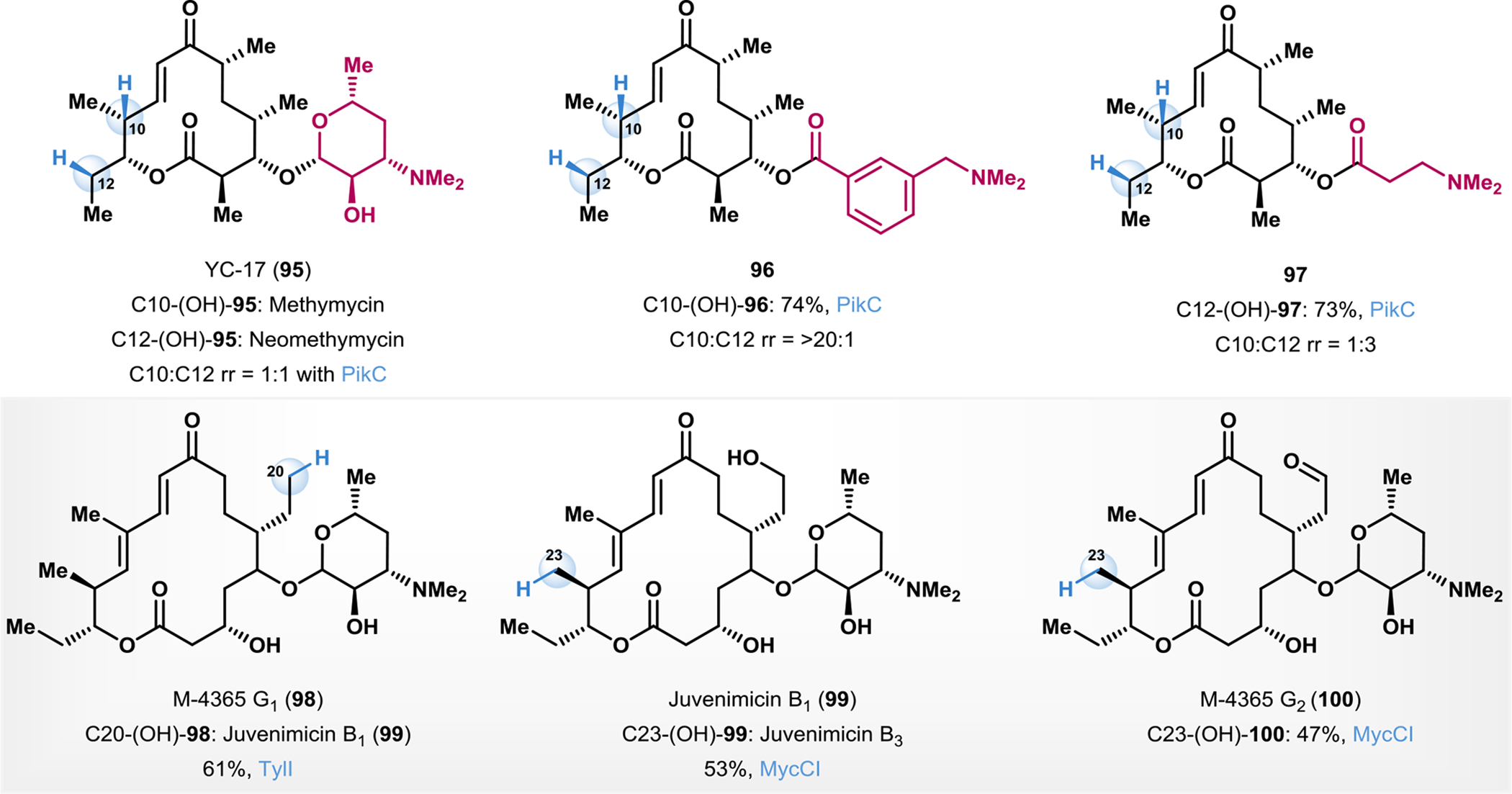
C–H hydroxylation in complex macrolide scaffolds.
Sherman and coworkers carried out scalable late-stage structural diversification of the macrolide natural product M-4365 G1 (98, Fig. 8) using P450-mediated oxidation reactions.155 The P450 TylI obtained from the tylosin biosynthetic pathway was effective in selective C–H hydroxylation of 98 at C20 to yield juvenimicin B1 (M-4365 G3, 99) in 61% isolated yield.155 The Sherman group subsequently employed the P450 MyCCl from the mycinamicin biosynthetic pathway of Micromonospora griseorubida to selectively affect the C–H hydroxylation at position 23 to afford juvenimicin B3 (99) in 53% isolated yield.155 The P450 MycCI was also effective in carrying out the C23 hydroxylation of an aldehyde variant of juvenimicin B1 (M-4365 G2, 100) to yield the corresponding alcohol in 47% yield.155
6. Oxygenation in other systems
6.1. Carbonyl α-oxygenations
Compounds containing an α-hydroxy carbonyl moiety can be useful synthetic intermediates with a variety of applications in C–C and C–X bond forming reactions.156 In particular, α-hydroxy ketones and α’-hydroxy enones have emerged as useful templates for metal-catalyzed and organocatalyzed asymmetric bond-forming reactions.157 Consequently biocatalytic methods to affect C–H hydroxylation at α-positions of carbonyl compounds are on the rise.156–160 Reetz and coworkers developed P450BM3 variants through directed evolution to obtain monooxygenases selective for α-oxidative hydroxylation of aryl ketones.158 In the case of alkyl-aryl ketones (e.g., 101; Fig. 9A), P450BM3 variants were developed for either R- or S- selective hydroxylation reactions to afford a variety of products in 4–24% isolated yields (102). In the case of alkyl-benzyl ketones (e.g., 103, Fig. 9B), oxygenation was accomplished site-selectively at the benzylic position affording either enantiomer of the products 104 in 4–56% isolated yields.158 Hall and Faber described an oxidative biocatalytic cascade for the conversion of fatty acids to α-keto acids using an internal H2O2 recycling protocol (Fig. 9C).159 Specifically, octanoic acid (105) was converted to 2-hydroxyoctanoic acid (106) by P450SPα (α-hydroxylase from S. paucimobili) in >99% ee, and this product was subsequently transformed to the corresponding ketone (107) using an α-hydroxy-acid oxidase (α-HAO) in 91% isolated yield.159 This approach could successfully hydroxylate saturated fatty acids of various chain lengths (C6 to C10). Urlacher and coworkers have developed a one-step P450-monooxygenase mediated N-demethylation and consecutive carbonyl α-hydroxylation on the anesthetic (S)-ketamine (108) to form (2S, 6S)-hydroxynorketamine (109, Fig. 9D).160 The latter compound is also a biological metabolite of 108, and has been found to have a similar antidepressant effect as 108 but with fewer undesirable side effects.161 This reaction could be carried out in milligram preparative scales to provide 109 in good yields and excellent diastereoselectivity (Fig. 9D).160
Figure 9.
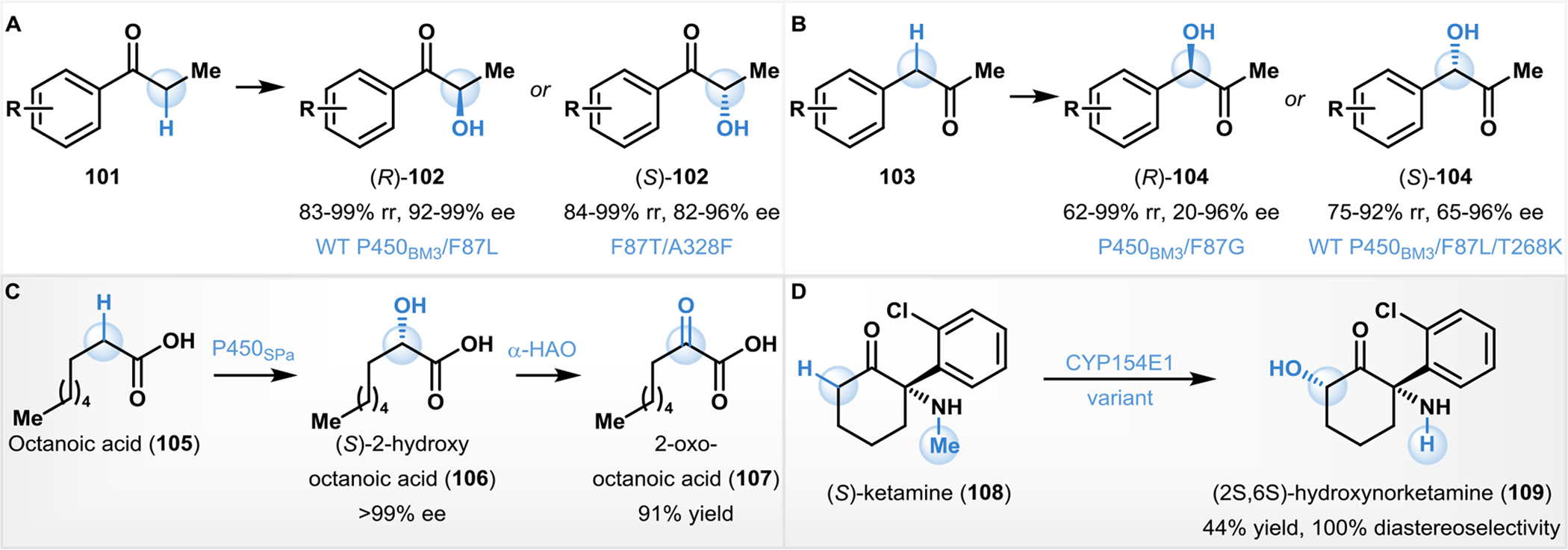
Biocatalytic carbonyl α-C–H oxygenation reactions.
6.2. Hydroxylations of unactivated C–H bonds
Hydroxylation of unactivated C–H bonds is a challenging endeavor in synthetic chemistry, particularly in cases of substrates lacking a directing group or significant electronic bias within the C–H bonds of the substrate.162 Scalable, biocatalytic C–H hydroxylation has been demonstrated in a variety of such unactivated and other systems.163–166 For example, Flitsch and coworkers explored the ability of R. rhodochrous (NCIMB 9703) to catalyze the site- and stereoselective hydroxylation of a range of benzyloxy tetrahydro- pyrans and furans.163 Hydroxylation of a tetrahydrofuran substrate occurred site-selectively at C4 to afford product 110 in 26% yield and 93% ee (Fig. 10).163 Structurally related tetrahydropyran derivatives, however, yielded mixtures of diastereomers in this transformation.163 In a chemoenzymatic cascade to a bis-(2-substituted benzofuran) product, Schwaneberg and coworkers developed a P450BM3 based variant for the selective C–H oxidation of an N-methyl group to form hemiaminal species such as 111 (Fig. 10).164 The intermediate subsequently undergoes further elimination reactions and dimerizes with the formation of the final product in their cascade.164
Figure 10.
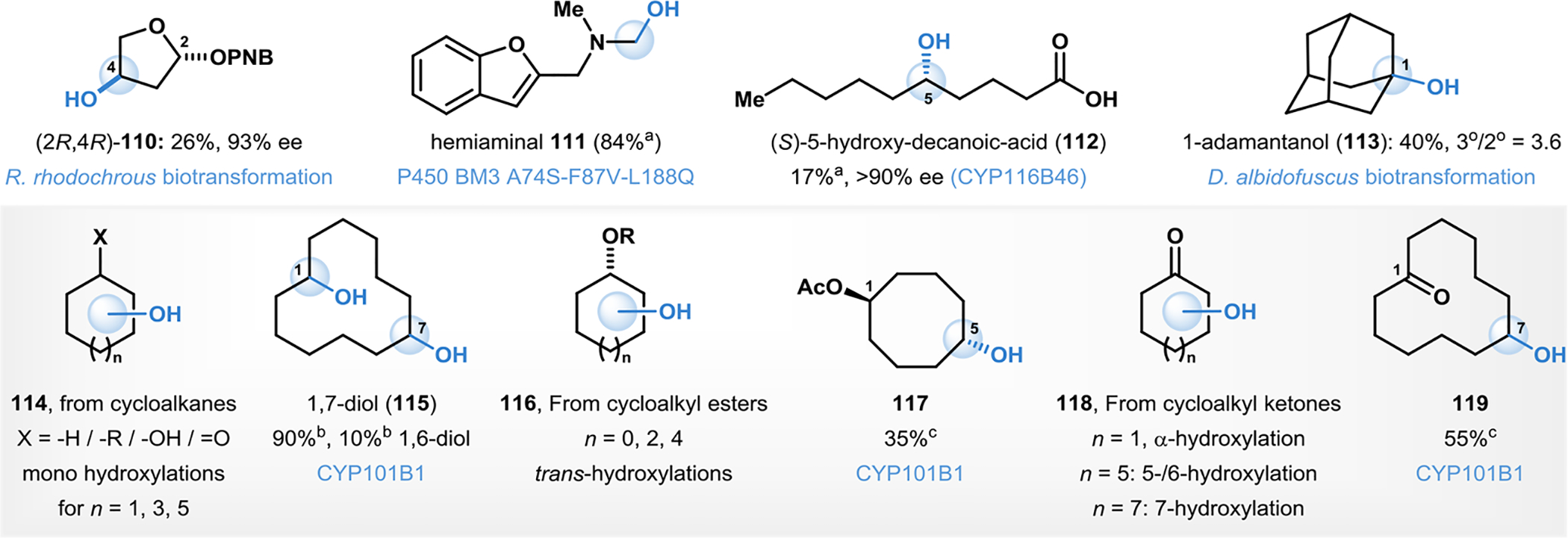
C–H oxygenation in unactivated systems. aProduct isolated after a cascade of reaction(s). bPercent site-selectivity. cPercent conversion.
The high levels of site-selectivity in enzymatic reactions are often perfectly exemplified in selective oxyfunctionalizations of long-chain alkane substrates, bearing several identical methylene C–H bonds.165 For example, Flitsch and coworkers demonstrated the efficacy of a P450 monooxygenase CYP116B46 from Tepidiphilus thermophilus in carrying out the site- and enantioselective C5-hydroxylation of decanoic acid to form (5S)-hydroxydecanoic acid (112, Fig. 10).165 Following cyclization to the corresponding lactone, the product was isolated in approximately 17% yield and >99% ee.165
Preparative aerobic oxidations with basidiomycetous enzymes for selective C–H hydroxylations were demonstrated by Zhuk in the oxidation of adamantane to 1-adamantanol (113) delivering the alcohol in 40% isolated yield, and with a site-selectivity ratio for hydroxylation of the tertiary positions over secondary C–H bonds was found to be 3.6 (Fig. 10).167 A kinetic isotope effect (kH/kD = 2.25) was measured, providing indirect evidence for the participation of fungal metalloenzymes in the C–H activation step.167 A variety of simple mono-, di- and non-substituted cycloalkanes have been successfully enzymatically hydroxylated.52, 64, 168–170 Bell and coworkers used P450 cam variant F87A-Y96F to carry out C4-hydroxylation of cyclohexyl benzoate to form 4-benzyloxy-cyclohexanol in 48% isolated yield and in >95% site-selectivity (Fig. 10).64 The corresponding benzyl ether substrate resulted in the same site- and stereochemical outcome.64 Reetz and coworkers developed a site- and stereoselective hydroxylation of monosubstituted cyclohexanes to form cyclohexanols (114) using P450BM3 variants.168 Using achiral substrates, the method developed by Reetz affects stereoselective hydroxylation with the simultaneous generation of additional chiral centers in a single C–H functionalization event.168 Reetz subsequently developed whole-cell cascade reactions in which E. coli cells harboring P450BM3 in combination with the appropriate alcohol dehydrogenase was employed in up to four-step cascade reactions starting with cyclohexane, cyclohexanol or cyclohexanone to form either enantiomer or the meso form of cyclohexane 1,2-diol.169 Hollman and coworkers developed a visible-light driven catalytic water oxidation to render a peroxygenase platform catalytically active in the hydroxylation of C6–C8 cycloalkanes (forming products 114, Fig. 10).52 Bell and coworkers developed hydroxylation of several non-activated methylene C–H bonds in cycloalkanes, cycloalkyl ketones and cycloalkyl esters (forming products 114-119, Fig. 10) with the cytochrome P450 monooxygenase CYP101B1 from Novosphin gobium aromaticivorans.170 Cycloalkanes were oxidized to the corresponding mono-alcohol or diol products (in the case of cycloheptane; 115, Fig. 10).170 Cycloalkyl ketones and esters were oxidized selectively on the opposite side of the ring relative to the carbonyl substituent (118, 119, Fig. 10).170
Conclusions
The diversity of complex molecules available for use as chemical probes, pharmaceutical agents, reagents for sequencing polymeric biomolecules, or as smart materials, at any given point in time depends on our ability to access them. Methods for C–H functionalization have transformed what constitutes an accessible compound. Nature’s catalysts perform chemical transformations with exquisite control over selectivity and stand to be valuable tools in cases where these transformations are readily scalable.
The use of biocatalytic oxyfunctionalization reactions has increased tremendously over the past decade, transforming structurally complex scaffolds, including the steroids, terpenoids, and macrolide antibiotics, among several others, presented in this review. The potential for outstanding levels of site-selectivity on such complex scaffolds motivates research in this area and has driven progress in recent years. Preparative-scale biocatalytic reactions are becoming increasingly prevalent in organic synthesis, and industrial adoption of these methods are on the rise.19–21
Traditional challenges associated with applying biocatalytic transformations in synthesis have included enzyme availability and limited information on, or limited, substrate promiscuity, thus, contributing to the slow adoption of biocatalytic methods by the mainstream synthetic community. In recent years, however, the repertoire of biocatalytic methods has increased tremendously, and chemoenzymatic methods of synthesis are taking up speed. To accelerate this trend, more broad commercialization of enzymes to increase access is needed.
Cytochrome P450s are extensively utilized in oxidative biocatalysis due to the breadth of transformations that these enzymes are capable of, however, their redox requirement often significantly hinders their utility on preparative scales.29 In this regard, α-oxo acid-dependent non-heme iron(II) (NHI) monooxygenases have demonstrated great promise for preparative- and industrial-scale applications.30 Alternatively, peroxygenases are a relatively under-explored class of enzymes for their potential in preparative-scale oxyfunctionalization reactions.31 Peroxygenases possess the specificity of P450s without the need for a cofactor recycling system.32 Currently, both classes of enzymes present challenges with regard to selectivity and total substrate turnover, however, advances in enzyme evolution approaches are expected to render them the optimal choice of biocatalysts in the future.31,32
Whereas the majority of examples presented in this review report excellent yields of isolated products, this is not uniformly true of all biocatalytic oxyfunctionalization reactions. Several factors can contribute to low yields of product in biocatalytic reactions. Enzyme instability is often a critical factor. This is, however, often circumvented with the use of biotransformations involving whole cells as opposed to using purified enzymes. Substrate solubility is often an issue with highly hydrophobic substrates. Cosolvents are often employed in some cases to improve product yields. For several examples presented in this review, the substrates were added to the biocatalytic mixtures as solutions in water-miscible organic solvents such as DMSO, DMF, ethanol, methanol, and acetone. These solvents often comprised about 1–5% v/v of the reaction mixtures, and are primarily added to improve substrate solubility. Low enzyme activity for a given transformation can also lead to a decrease in the yield of product formation. In addition, protein engineering strategies can also provide a fruitful path to improved catalytic activity. Several metrics are reflected in the isolated yields of preparative-scale reactions, allowing chemists to compare biocatalytic options with other approaches in synthetic planning.17 Although there are several examples reviewed here with excellent yields of the oxyfunctionalized products, this is not uniform for all enzymatic reactions. However, with the advances in protein engineering strategies, improvement of reaction yields is possible, and this is becoming increasingly routine.81, 145, 169, 171
Complementary selectivity profiles are often observed in enzymatic reactions compared to small molecule-mediated reactions on the same substrate.12, 132, 134 For synthetic chemists driven to devise and implement the most efficient syntheses, strategically combining the best of both small- and large-molecule catalysts is the key. Several examples presented in this review embrace chemoenzymatic strategies, wherein biocatalytic C–H oxyfunctionalization strategies were used along with powerful small molecule-mediated methods in a single sequence.84, 103, 104, 113, 133, 134, 164 Chemoenzymatic routes toward a diversity of molecules are possible with existing tools, and it stands to change the way we design and make molecules.
Acknowledgements
We are grateful to the University of Michigan’s Life Sciences Institute, University of Michigan’s Department of Chemistry, and the National Institutes of Health R35 GM124880 for support. We are grateful to Dr. Christopher Perry, Lara E. Zetzsche, Tyler J. Doyon, Di Yang, and Attabey Rodríguez Benítez for helpful discussions and proofreading this manuscript.
Biographies

Suman Chakrabarty graduated with a B.Sc. degree in 2011 from St. Joseph’s College, and a M.Sc. degree in Chemistry in 2013 from Christ University, both based in Bangalore, India. He moved to USA in 2013 and joined Prof. James Takacs’ lab at the University of Nebraska-Lincoln, developing rhodium-catalyzed asymmetric hydroboration reactions to form chiral tertiary boronic esters. After obtaining his Ph.D. in 2019, he joined Prof. Alison Narayan’s lab as a postdoctoral fellow at the University of Michigan, where he is currently developing biocatalytic routes towards complex molecule synthesis.

Ye Wang graduated from Sichuan University, China in 2014 with a B.Sc. degree in chemistry. Then he joined Prof. Shu-Li You’s lab at Shanghai Institute of Organic Chemistry (SIOC), Chinese Academy of Sciences doing research on iridium-catalyzed asymmetric allylic substitution and desymmetric dearomatization reactions. After receiving his Ph.D. in 2019, he moved to the University of Michigan as a postdoctoral fellow in Prof. Alison Narayan’s lab, developing methods for biocatalytic complex molecule synthesis and diversification of amino acids.

Jonathan Perkins graduated from Roanoke College in 2014 with a B.S. degree in Chemistry and Physics, before moving on to obtain his M.S. in Chemistry from the College of William and Mary in 2017, conducting research in the lab of Prof. Jonathan R. Scheerer. He is currently a third-year graduate student at the University of Michigan working on his Ph.D. in the lab of Prof. Alison Narayan. His research interests include biocatalysis, synthesis of complex natural products, chemoenzymatic method development, and bioinformatics.

Alison Narayan is a native Michigander with a B.S. in chemistry from the University of Michigan. She holds a Ph.D. in organic chemistry from the University of California, Berkeley, where she worked with Prof. Richmond Sarpong. She then worked with Prof. David Sherman as a Life Sciences Research Foundation Postdoctoral Fellow. She joined the Department of Chemistry and the Life Sciences Institute at the University of Michigan as an assistant professor in 2015. Her research group develops biocatalytic tools for complex molecule synthesis.
Footnotes
Footnotes relating to the title and/or authors should appear here.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
References
- 1.White MC and Zhao J, J. Am. Chem. Soc, 2018, 140, 13988–14009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saint-Denis TG, Zhu R-Y, Chen G, Wu Q-F and Yu J-Q, Science, 2018, 359, eaao4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crabtree RH and Lei A, Chem. Rev, 2017, 117, 8481–8482. [DOI] [PubMed] [Google Scholar]
- 4.Sterckx H, Morel B and Maes BUW, Angew. Chem. Int. Ed 2019, 58, 7946–7970. [DOI] [PubMed] [Google Scholar]
- 5.Neufeldt SR and Sanford MS, Acc. Chem. Res, 2012, 45, 936–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.White MC, Synlett, 2012, 23, 2746–2748. [Google Scholar]
- 7.Keller NP, Nat. Rev. Microbiol, 2019, 17, 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arnold FH, Angew. Chem. Int. Ed, 2019, 58, 14420–14426. [DOI] [PubMed] [Google Scholar]
- 9.Fasan R, Jennifer Kan SB and Zhao H, ACS Catalysis, 2019, 9, 9775–9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arnold FH, Angew. Chem. Int. Ed, 2018, 57, 4143–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen K and Arnold FH, Nat. Catal, 2020, 3, 203–213. [Google Scholar]
- 12.Dong JJ, Fernández‐Fueyo E, Hollmann F, Paul CE, Pesic M, Schmidt S, Wang Y, Younes S and Zhang WY, Angew. Chem. Int. Ed, 2018, 57, 9238–9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abdelraheem EMM, Busch H, Hanefeld U and Tonin F, React. Chem. Eng, 2019, 4, 1878–1894. [Google Scholar]
- 14.Sheldon RA and Brady D, ChemSusChem, 2019, 12, 2859–2881. [DOI] [PubMed] [Google Scholar]
- 15.Turner NJ and O’Reilly E, Nat. Chem. Biol, 2013, 9, 285–288. [DOI] [PubMed] [Google Scholar]
- 16.de Souza ROMA, Miranda LSM and Bornscheuer UT, Chem. Eur. J, 2017, 23, 12040–12063. [DOI] [PubMed] [Google Scholar]
- 17.Clouthier CM and Pelletier JN, Chem. Soc. Rev, 2012, 41, 1585–1605. [DOI] [PubMed] [Google Scholar]
- 18.Sheldon RA, Brady D and Bode ML, Chem. Sci, 2020, 11, 2587–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adams JP, Brown MJB, Diaz-Rodriguez A, Lloyd RC and Roiban G-D, Adv. Synth. Catal, 2019, 361, 2421–2432. [Google Scholar]
- 20.Huffman MA, Fryszkowska A, Alvizo O, Borra-Garske M, Campos KR, Canada KA, Devine PN, Duan D, Forstater JH, Grosser ST, Halsey HM, Hughes GJ, Jo J, Joyce LA, Kolev JN, Liang J, Maloney KM, Mann BF, Marshall NM, McLaughlin M, Moore JC, Murphy GS, Nawrat CC, Nazor J, Novick S, Patel NR, Rodriguez-Granillo A, Robaire SA, Sherer EC, Truppo MD, Whittaker AM, Verma D, Xiao L, Xu Y and Yang H, Science, 2019, 366, 1255–1259. [DOI] [PubMed] [Google Scholar]
- 21.Hughes G and Lewis JC, Chem. Rev, 2018, 118, 1–3. [DOI] [PubMed] [Google Scholar]
- 22.Devine PN, Howard RM, Kumar R, Thompson MP, Truppo MD and Turner NJ, Nat. Rev. Chem, 2018, 2, 409–421. [Google Scholar]
- 23.Qu G, Li A, Acevedo-Rocha CG, Sun Z and Reetz MT, Angew. Chem. Int. Ed, 2020, 59, 2–30. [DOI] [PubMed] [Google Scholar]
- 24.Goodwin NC, Morrison JP, Fuerst DE and Hadi T, ACS Med. Chem. Lett, 2019, 10, 1363–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Truppo MD, ACS Med. Chem. Lett, 2017, 8, 476–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang Y, Wei J, Qiu X and Jiao N, Chem. Rev, 2018, 118, 4912–4945. [DOI] [PubMed] [Google Scholar]
- 27.Hollmann F, Arends IWCE, Buehler K, Schallmey A and Bühler B, Green Chem, 2011, 13, 226–265. [Google Scholar]
- 28.Holtmann D, Fraaije MW, Arends IWCE, Opperman DJ and Hollmann F, Chem. Commun, 2014, 50, 13180–13200. [DOI] [PubMed] [Google Scholar]
- 29.Ortiz de Montellano PR, Chem. Rev, 2010, 110, 932–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zwick CR and Renata H, Nat. Prod. Rep, 2020, Advance Article, DOI: 10.1039/C9NP00075E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Lan D, Durrani R and Hollmann F, Curr. Opin. Chem. Biol, 2017, 37, 1–9. [DOI] [PubMed] [Google Scholar]
- 32.Bormann S, Baraibar AG, Ni Y, Holtmann D and Hollmann F, Catal. Sci. Technol, 2015, 5, 2038–2052. [Google Scholar]
- 33.Garzón-Posse F, Becerra-Figueroa L, Hernández-Arias J and Gamba-Sánchez D, Molecules, 2018, 23, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andrus MB, Sci. Synth, 2011, DOI: 10.1055/sos-SD-203-00294. [DOI] [Google Scholar]
- 35.Burton SG, Trends Biotechnol, 2003, 21, 543–549. [DOI] [PubMed] [Google Scholar]
- 36.Jung ST, Lauchli R and Arnold FH, Curr. Opin. Biotechnol, 2011, 22, 809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neufeld K, Henßen B and Pietruszka J, Angew. Chem. Int. Ed, 2014, 53, 13253–13257. [DOI] [PubMed] [Google Scholar]
- 38.Neufeld K, Marienhagen J, Schwaneberg U and Pietruszka J, Green Chem, 2013, 15, 2408–2421. [Google Scholar]
- 39.Ilie A, Harms K and Reetz MT, J. Org. Chem, 2018, 83, 7504–7508. [DOI] [PubMed] [Google Scholar]
- 40.Fasan R, ACS Catal, 2012, 2, 647–666. [Google Scholar]
- 41.Roiban GD, Agudo R, Ilie A, Lonsdale R and Reetz MT, Chem. Commun, 2014, 50, 14310–14313. [DOI] [PubMed] [Google Scholar]
- 42.Li R-J, Li A, Zhao J, Chen Q, Li N, Yu H-L and Xu J-H, Catal. Sci. Technol, 2018, 8, 4638–4644. [Google Scholar]
- 43.Holec C, Hartrampf U, Neufeld K and Pietruszka J, ChemBioChem, 2017, 18, 676–684. [DOI] [PubMed] [Google Scholar]
- 44.Ren XK, Yorke JA, Taylor E, Zhang T, Zhou WH and Wong LL, Chem. Eur. J, 2015, 21, 15039–15047. [DOI] [PubMed] [Google Scholar]
- 45.Li Y and Wong LL, Angew. Chem. Int. Ed, 2019, 58, 9551–9555. [DOI] [PubMed] [Google Scholar]
- 46.Kolev JN, Zaengle JM, Ravikumar R and Fasan R, ChemBioChem, 2014, 15, 1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sarkar MR, Lee JHZ and Bell SG, ChemBioChem, 2017, 18, 2119–2128. [DOI] [PubMed] [Google Scholar]
- 48.Zheng D, Yang M, Zhuo JR, Li K, Zhang H, Yang J, Cui B and Chen Y, J. Mol. Catal. B-Enzym, 2014, 110, 87–91. [Google Scholar]
- 49.Ewing TA, Kühn J, Segarra S, Tortajada M, Zuhse R and van Berkel WJH, Adv. Synth. Catal, 2018, 360, 2370–2376. [Google Scholar]
- 50.Doyon TJ, Perkins JC, Dockrey SAB, Romero EO, Skinner KC, Zimmerman PM and Narayan ARH, J. Am. Chem. Soc, 2019, 141, 20269–20277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang W, Burek BO, Fernández‐Fueyo E, Alcalde M, Bloh JZ and Hollmann F, Angew. Chem. Int. Ed, 2017, 56, 15451–15455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang W, Fernández-Fueyo E, Ni Y, van Schie M, Gacs J, Renirie R, Wever R, Mutti FG, Rother D, Alcalde M and Hollmann F, Nat. Catal, 2018, 1, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tieves F, Willot SJP, van Schie M, Rauch MCR, Younes SHH, Zhang W, Dong J, de Santos PG, Robbins JM, Bommarius B, Alcalde M, Bommarius AS and Hollmann F, Angew. Chem.-Int. Edit, 2019, 58, 7873–7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Reilly E, Köhler V, Flitsch SL and Turner NJ, Chem. Commun, 2011, 47, 2490–2501. [DOI] [PubMed] [Google Scholar]
- 55.Zwick CR and Renata H, J. Org. Chem, 2018, 83, 7407–7415. [DOI] [PubMed] [Google Scholar]
- 56.Singh MS, Nagaraju A, Anand N and Chowdhury S, RSC Adv, 2014, 4, 55924–55959. [Google Scholar]
- 57.Wang Y, Lan D, Durrani R and Hollmann F, Curr. Opin. Chem. Biol, 2017, 37, 1–9. [DOI] [PubMed] [Google Scholar]
- 58.Nakamura A and Nakada M, Synthesis, 2013, 45, 1421–1451. [Google Scholar]
- 59.Rickert A, Krombach V, Hamers O, Zorn H and Maison W, Green Chem, 2012, 14, 639–644. [Google Scholar]
- 60.Weidmann V, Kliewer S, Sicka M, Bycinskij S, Kleczka M, Rehbein J, Griesbeck AG, Zorn H and Maison W, J. Mol. Catal. B-Enzym, 2015, 121, 15–21. [Google Scholar]
- 61.Agudo R, Roiban G-D and Reetz MT, Chembiochem, 2012, 13, 1465–1473. [DOI] [PubMed] [Google Scholar]
- 62.Winska K, Grabarczyk M, Maczka W, Zarowska B, Maciejewska G and Aniol M, Z. Naturforsch. C. J. Biosci, 2017, 72, 209–217. [DOI] [PubMed] [Google Scholar]
- 63.Brenna E, Crotti M, Gatti FG, Monti D, Parmeggiani F, Pugliese A and Tentori F, Green Chem, 2017, 19, 5122–5130. [Google Scholar]
- 64.Bell SG, Spence JTJ, Liu S, George JH and Wong L-L, Org. Biomol. Chem, 2014, 12, 2479–2488. [DOI] [PubMed] [Google Scholar]
- 65.Kaluzna I, Schmitges T, Straatman H, van Tegelen D, Müller M, Schürmann M and Mink D, Org. Process Res. Dev, 2016, 20, 814–819. [Google Scholar]
- 66.Tavanti M, Parmeggiani F, Castellanos JRG, Mattevi A and Turner NJ, ChemCatChem, 2017, 9, 3338–3348. [Google Scholar]
- 67.Shahidi F and Ambigaipalan P, J. Funct. Food, 2015, 18, 820–897. [Google Scholar]
- 68.Hao L, Ding G, Deming DA and Zhang Q, Eur. J. Org. Chem, 2019, 2019, 7307–7321. [Google Scholar]
- 69.Wedi P and van Gemmeren M, Angew. Chem. Int. Ed, 2018, 57, 13016–13027. [DOI] [PubMed] [Google Scholar]
- 70.Gröger H, Angew. Chem. Int. Ed, 2014, 53, 3067–3069. [DOI] [PubMed] [Google Scholar]
- 71.Thompson TN, in Annual Reports in Medicinal Chemistry, Vol 44, ed. Macor JE, Elsevier Academic Press Inc, San Diego, 2009, vol. 44, pp. 459–474. [Google Scholar]
- 72.Dragan CA, Peters FT, Bour P, Schwaninger AE, Schaan SM, Neunzig I, Widjaja M, Zapp J, Kraemer T, Maurer HH and Bureik M, Appl. Biochem. Biotechnol, 2011, 163, 965–980. [DOI] [PubMed] [Google Scholar]
- 73.Atzrodt J, Blankenstein J, Brasseur D, Calvo-Vicente S, Denoux M, Derdau V, Lavisse M, Perard S, Roy S, Sandvoss M, Schofield J and Zimmermann J, Bioorg. Med. Chem, 2012, 20, 5658–5667. [DOI] [PubMed] [Google Scholar]
- 74.Quinn L, Dempsey R, Casey E, Kane A and Murphy CD, J. Ind. Microbiol. Biotechnol, 2015, 42, 799–806. [DOI] [PubMed] [Google Scholar]
- 75.Klenk JM, Nebel BA, Porter JL, Kulig JK, Hussain SA, Richter SM, Tavanti M, Turner NJ, Hayes MA, Hauer B and Flitsch SL, Biotechnol. J, 2017, 12: 1600520. [DOI] [PubMed] [Google Scholar]
- 76.Tavanti M, Porter JL, Sabatini S, Turner NJ and Flitsch SL, ChemCatChem, 2018, 10, 1042–1051. [Google Scholar]
- 77.Hata T, Kawai S, Okamura H and Nishida T, Biodegradation, 2010, 21, 681–689. [DOI] [PubMed] [Google Scholar]
- 78.Klenk JM, Kontny LH, Escobedo-Hinojosa W, Nebel BA and Hauer B, J. Appl. Microbiol, 2019, 127, 724–738. [DOI] [PubMed] [Google Scholar]
- 79.Venkataraman H, Verkade-Vreeker MCA, Capoferri L, Geerke DP, Vermeulen NPE and Commandeur JNM, Bioorg. Med. Chem, 2014, 22, 5613–5620. [DOI] [PubMed] [Google Scholar]
- 80.Dennig A, Lulsdorf N, Liu H and Schwaneberg U, Angew. Chem. Int. Ed, 2013, 52, 8459–8462. [DOI] [PubMed] [Google Scholar]
- 81.Zhou H, Wang B, Wang F, Yu X, Ma L, Li A and Reetz MT, Angew. Chem. Int. Ed, 2019, 58, 764–768. [DOI] [PubMed] [Google Scholar]
- 82.Munday SD, Dezvarei S, Lau IC-K and Bell SG, ChemCatChem, 2017, 9, 2512–2522. [Google Scholar]
- 83.Rousseau O, Ebert M, Quaglia D, Fendri A, Parisien AH, Besna JN, Iyathurai S and Pelletier JN, ChemCatChem, 2020, 12, 837–845. [Google Scholar]
- 84.Zwick CR and Renata H, J. Am. Chem. Soc, 2018, 140, 1165–1169. [DOI] [PubMed] [Google Scholar]
- 85.Dennig A, Busto E, Kroutil W and Faber K, ACS Catal, 2015, 5, 7503–7506. [Google Scholar]
- 86.Furuya T, Arai Y and Kino K, Appl. Environ. Microbiol, 2012, 78, 6087–6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dhammaraj T, Pinthong C, Visitsatthawong S, Tongsook C, Surawatanawong P and ChaiyenT P, ACS Chem. Biol, 2016, 11, 2889–2896. [DOI] [PubMed] [Google Scholar]
- 88.Dennig A, Weingartner AM, Kardashliev T, Müller CA, Tassano E, Schürmann M, Ruff AJ and Schwaneberg U, Chem. Eur. J, 2017, 23, 17981–17991. [DOI] [PubMed] [Google Scholar]
- 89.O’Hanlon JA, Ren X, Morris M, Wong LL and Robertson J, Org. Biomol. Chem, 2017, 15, 8780–8787. [DOI] [PubMed] [Google Scholar]
- 90.Ruhlmann A, Antovic D, Muller TJJ and Urlacher VB, Adv. Synth. Catal, 2017, 359, 984–994. [Google Scholar]
- 91.Moir M, Danon JJ, Reekie TA and Kassiou M, Expert. Opin. Drug Discov, 2019, 14, 1137–1149. [DOI] [PubMed] [Google Scholar]
- 92.Vickers C, Backfisch G, Oellien F, Piel I and Lange UEW, Chem. Eur. J, 2018, 24, 17936–17947. [DOI] [PubMed] [Google Scholar]
- 93.de Santos PG, Canellas M, Tieves F, Younes SHH, Molina-Espeja P, Hofrichter M, Hollmann F, Guallar V and Alcalde M, ACS Catal, 2018, 8, 4789–4799. [Google Scholar]
- 94.Tomaszewski B, Schmid A and Buehler K, Org. Process Res. Dev, 2014, 18, 1516–1526. [Google Scholar]
- 95.Dhammaraj T, Phintha A, Pinthong C, Medhanavyn D, Tinikul R, Chenprakhon P, Sucharitakul J, Vardhanabhuti N, Jiarpinitnun C and Chaiyen P, ACS Catal, 2015, 5, 4492–4502. [Google Scholar]
- 96.Pinthong C, Phoopraintra P, Chantiwas R, Pongtharangkul T, Chenprakhon P and Chaiyen P, Process Biochem, 2017, 63, 122–129. [Google Scholar]
- 97.Lin Y and Yan Y, Biotechnol. Bioeng, 2014, 111, 1895–1899. [DOI] [PubMed] [Google Scholar]
- 98.He J, Li SH, Deng YQ, Fu HY, Laforteza BN, Spangler JE, Homs A and Yu JQ, Science, 2014, 343, 1216–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Morcillo SP, Dauncey EM, Kim JH, Douglas JJ, Sheikh NS and Leonori D, Angew. Chem. Int. Ed, 2018, 57, 12945–12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu T, Myers MC and Yu J-Q, Angew. Chem. Int. Ed, 2017, 56, 306–309. [DOI] [PubMed] [Google Scholar]
- 101.Quinn RK, Könst ZA, Michalak SE, Schmidt Y, Szklarski AR, Flores AR, Nam S, Horne DA, Vanderwal CD and Alexanian EJ, J. Am. Chem. Soc, 2016, 138, 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Smirnov SV, Kodera T, Samsonova NN, Kotlyarova VA, Rushkevich NY, Kivero AD, Sokolov PM, Hibi M, Ogawa J and Shimizu S, Appl. Microbiol. Biotechnol, 2010, 88, 719–726. [DOI] [PubMed] [Google Scholar]
- 103.Zwick C, Sosa M and Renata H (2019): Concise Chemoenzymatic Total Synthesis of GE81112 B1. ChemRxiv. Preprint. 10.26434/chemrxiv.9684554.v1 [DOI] [Google Scholar]
- 104.Amatuni A, Shuster A, Adibekian A and Renata H (2020): Concise Chemoenzymatic Total Synthesis and Identification of Cellular Targets of Cepafungin I. ChemRxiv. Preprint. 10.26434/chemrxiv.11852496.v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Broca C, Breil V, Cruciani-Guglielmacci C, Manteghetti M, Rouault C, Derouet M, Rizkalla S, Pau B, Petit P, Ribes G, Ktorza A, Gross R, Reach G and Taouis M, Am. J. Physiol.-Endocrinol. Metab, 2004, 287, E463–E471. [DOI] [PubMed] [Google Scholar]
- 106.Mattay J and Huttel W, ChemBioChem, 2017, 18, 1523–1528. [DOI] [PubMed] [Google Scholar]
- 107.Klein C and Hüttel W, Adv. Synth. Catal, 2011, 353, 1375–1383. [Google Scholar]
- 108.Baud D, Saaidi P-L, Monfleur A, Harari M, Cuccaro J, Fossey A, Besnard M, Debard A, Mariage A, Pellouin V, Petit J-L, Salanoubat M, Weissenbach J, de Berardinis V and Zaparucha A, Chemcatchem, 2014, 6, 3012–3017. [Google Scholar]
- 109.Baud D, Peruch O, Saaidi PL, Fossey A, Mariage A, Petit J-L, Salanoubat M, Vergne-Vaxelaire C, de Berardinis V and Zaparucha A, Adv. Synth. Catal, 2017, 359, 1563–1569. [Google Scholar]
- 110.Zhang X, King-Smith E and Renata H, Angew.Chem. Int.Ed, 2018, 57, 5037–5041. [DOI] [PubMed] [Google Scholar]
- 111.Amatuni A and Renata H, Org. Biomol. Chem, 2019, 17, 1736–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Smirnov SV, Sokolov PM, Kodera T, Sugiyama M, Hibi M, Shimizu S, Yokozeki K and Ogawa J, FEMS Microbiol. Lett, 2012, 331, 97–104. [DOI] [PubMed] [Google Scholar]
- 113.Zwick CR, Sosa MB and Renata H, Angew. Chem. Int. Ed, 2019, 58, 18854–18858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Donova MV and Egorova OV, Appl. Microbiol. Biotechnol, 2012, 94, 1423–1447. [DOI] [PubMed] [Google Scholar]
- 115.Khatri Y, Ringle M., Lisurek M, von Kries JP, Zapp J and Bernhardt R, ChemBioChem, 2016, 17, 90–101. [DOI] [PubMed] [Google Scholar]
- 116.Lu W, Feng J, Chen X, Bao Y-J, Wang Y, Wu Q, Ma Y and Zhu D, Appl. Environ. Microbiol, 2019, 85, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lu W, Chen X, Feng JH, Bao YJ, Wang Y, Wu QQ and Zhu DM, Appl. Environ. Microbiol, 2018, 84, e00503–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kille S, Zilly FE, Acevedo JP and Reetz MT, Nat. Chem, 2011, 3, 738–743. [DOI] [PubMed] [Google Scholar]
- 119.Kolet SP, Haldar S, Niloferjahan S and Thulasiram HV, Steroids, 2014, 85, 6–12. [DOI] [PubMed] [Google Scholar]
- 120.Kiebist J, Schmidtke KU, Zimmermann J, Kellner H, Jehmlich N, Ullrich R, Zänder D, Hofrichter M and Scheibner K, Chembiochem, 2017, 18, 563–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Khatri Y, Jozwik IK, Ringle M, Ionescu IA, Litzenburger M, Hutter MC, Thunnissen A and Bernhardt R, ACS Chem. Biol, 2018, 13, 1021–1028. [DOI] [PubMed] [Google Scholar]
- 122.Zehentgruber D, Hannemann F, Bleif S, Bernhardt R and Lutz S, Chembiochem, 2010, 11, 713–721. [DOI] [PubMed] [Google Scholar]
- 123.Lewis JC, Mantovani SM, Fu Y, Snow CD, Komor RS, Wong CH and Arnold FH, ChemBioChem, 2010, 11, 2502–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang S-K, Dai C-F and Duh C-Y, J. Nat. Prod, 2006, 69, 103–106. [DOI] [PubMed] [Google Scholar]
- 125.Wang J, Zhang Y, Liu H, Shang Y, Zhou L, Wei P, Yin W-B, Deng Z, Qu X and Zhou Q, Nat. Commun, 2019, 10, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang Z, Li F, Cao Y, Tian Y, Li J, Zong Y and Song H, Catal. Sci. Technol, 2019, 9, 4877–4887. [Google Scholar]
- 127.Renata H, Zhou QH and Baran PS, Science, 2013, 339, 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Renata H, Zhou Q, Dunstl G, Felding J, Merchant RR, Yeh C-H and Baran PS, J. Am. Chem. Soc, 2015, 137, 1330–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang Y, Ju W, Tian H, Tian W and Gui JH, J. Am. Chem. Soc, 2018, 140, 9413–9416. [DOI] [PubMed] [Google Scholar]
- 130.Zhang KD, Damaty SE and Fasan R, J. Am. Chem. Soc, 2011, 133, 3242–3245. [DOI] [PubMed] [Google Scholar]
- 131.Domingo V, Lorenzo L, del Moral JFQ and Barrero AF, Org. Biomol. Chem, 2013, 11, 559–562. [DOI] [PubMed] [Google Scholar]
- 132.Loskot SA, Romney DK, Arnold FH and Stoltz BM, J. Am. Chem. Soc, 2017, 139, 10196–10199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li J, Li F, King-Smith E and Renata H, Nat. Chem, 2020, 12, 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhang X, King-Smith E, Dong L-B, Yang L-C, Rudolf JD, Shen B, Renata H (2019): Divergent Synthesis of Complex ent-Kauranes, ent-Atisanes and ent-Trachylobanes via a Hybrid Oxidative Approach. ChemRxiv. Preprint. 10.26434/chemrxiv.7663112.v2 [DOI] [Google Scholar]
- 135.Balint GA, Pharmacol. Ther, 2001, 90, 261–265. [DOI] [PubMed] [Google Scholar]
- 136.Lee S, Mini-Rev. Med. Chem, 2007, 7, 411–422. [DOI] [PubMed] [Google Scholar]
- 137.Pinheiro LCS, Feitosa LM, Da Silveira FF and Boechat N, An. Acad. Bras. Cienc, 2018, 90, 1251–1271. [DOI] [PubMed] [Google Scholar]
- 138.Ponnapalli MG, Sura MB, Sudhakar R, Govindarajalu G and Sijwali PS, J. Agric. Food Chem 2018, 66, 10490–10495. [DOI] [PubMed] [Google Scholar]
- 139.Zhang K, Shafer BM, Demars MD, Stern HA and Fasan R, J. Am. Chem. Soc, 2012, 134, 18695–18704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Haldar S, Mulani FA, Aarthy T and Thulasiram HV, J. Org.Chem, 2015, 80, 6490–6495. [DOI] [PubMed] [Google Scholar]
- 141.Haldar S, Kolet SP and Thulasiram HV, Green Chem, 2013, 15, 1311–1317. [Google Scholar]
- 142.Le-Huu P, Heidt T, Claasen B, Laschat S and Urlacher VB, ACS Catal, 2015, 5, 1772–1780. [Google Scholar]
- 143.Kolev JN, O’Dwyer KM, Jordan CT and Fasan R, ACS Chem. Biol, 2014, 9, 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Syntrivanis L-D, Wong LL and Robertson J, Eur. J. Org. Chem, 2018, 2018, 6369–6378. [Google Scholar]
- 145.Li Y, Qin B, Li X, Tang J, Chen Y, Zhou L and You S, Chemcatchem, 2018, 10, 559–565. [Google Scholar]
- 146.Meguro A, Tomita T, Nishiyama M and Kuzuyama T, ChemBioChem, 2013, 14, 316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Narayan ARH, Jiménez-Osés G, Liu P, Negretti S, Zhao W, Gilbert MM, Ramabhadran RO, Yang Y-F, Furan LR, Li Z, Podust LM, Montgomery J, Houk KN and Sherman DH, Nat. Chem, 2015, 7, 653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hall EA, Sarkar MR, Lee JHZ, Munday SD and Bell SG, ACS Catal, 2016, 6, 6306–6317. [Google Scholar]
- 149.Le-Huu P, Rekow D, Krüger C, Bokel A, Heidt T, Schaubach S, Claasen B, Hölzel S, Frey W, Laschat S and Urlacher VB, Chem. Eur. J, 2018, 24, 12010–12021. [DOI] [PubMed] [Google Scholar]
- 150.Alwaseem H, Frisch BJ and Fasan R, Bioorg. Med. Chem, 2018, 26, 1365–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Long BH, Carboni JM, Wasserman AJ, Cornell LA, Casazza AM, Jensen PR, Lindel T, Fenical W and Fairchild CR, Cancer Res, 1998, 58, 1111–1115. [PubMed] [Google Scholar]
- 152.Huang W, Wang J, Liang Y, Ge W, Wang G, Li Y and Chung HY, J. Ethnopharmacol, 2015, 175, 185–191. [DOI] [PubMed] [Google Scholar]
- 153.Li S, Ouellet H, Sherman DH and Podust LM, J. Biol. Chem, 2009, 284, 5723–5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Negretti S, Narayan ARH, Chiou KC, Kells PM, Stachowski JL, Hansen DA, Podust LM, Montgomery J and Sherman DH, J. Am. Chem. Soc, 2014, 136, 4901–4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lowell AN, DeMars MD, Slocum ST, Yu F, Anand K, Chemler JA, Korakavi N, Priessnitz JK, Park SR, Koch AA, Schultz PJ and Sherman DH, J. Am. Chem. Soc, 2017, 139, 7913–7920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hoyos P, Sinisterra J-V, Molinari F, Alcántara AR and Domínguez de María P, Acc. Chem. Res, 2010, 43, 288–299. [DOI] [PubMed] [Google Scholar]
- 157.Palomo C, Oiarbide M and García JM, Chem. Soc. Rev, 2012, 41, 4150–4164. [DOI] [PubMed] [Google Scholar]
- 158.Agudo R, Roiban GD, Lonsdale R, Ilie A and Reetz MT, J. Org. Chem, 2015, 80, 950–956. [DOI] [PubMed] [Google Scholar]
- 159.Gandomkar S, Dennig A, Dordic A, Hammerer L, Pickl M, Haas T, Hall M and Faber K, Angew. Chem. Int. Ed, 2018, 57, 427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bokel A, Rühlmann A, Hutter MC and Urlacher VB, ACS Catalysis, 2020, 10, 4151–4159. [Google Scholar]
- 161.Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, Alkondon M, Yuan P, Pribut HJ, Singh NS, Dossou KSS, Fang Y, Huang X-P, Mayo CL, Wainer IW, Albuquerque EX, Thompson SM, Thomas CJ, Zarate CA Jr and Gould TD, Nature, 2016, 533, 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tang X, Jia X and Huang Z, Chem. Sci, 2018, 9, 288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.O’Reilly E, Aitken SJ, Grogan G, Kelly PP, Turner NJ and Flitsch SL, Beilstein J. Org. Chem, 2012, 8, 496–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Mertens MAS, Thomas F, Noth M, Moegling J, El-Awaad I, Sauer DF, Dhoke GV, Xu WJ, Pich A, Herres-Pawlis S and Schwaneberg U, Eur. J. Org. Chem, 2019, 2019, 6341–6346. [Google Scholar]
- 165.Manning J, Tavanti M, Porter JL, Kress N, De Visser SP, Turner NJ and Flitsch SL, Angew. Chem. Int. Ed, 2019, 58, 5668–5671. [DOI] [PubMed] [Google Scholar]
- 166.Lukowski AL, Ellinwood DC, Hinze ME, DeLuca RJ, Du Bois J, Hall S and Narayan ARH, J. Am. Chem. Soc, 2018, 140, 11863–11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zhuk TS, Goldmann M, Hofmann J, Pohl JCS and Zorn H, J. Mol. Catal. B-Enzym, 2015, 122, 87–92. [Google Scholar]
- 168.Roiban GD, Agudo R and Reetz MT, Angew. Chem. Int. Ed, 2014, 53, 8659–8663. [DOI] [PubMed] [Google Scholar]
- 169.Li AT, Ilie A, Sun ZT, Lonsdale R, Xu JH and Reetz MT, Angew. Chem. Int. Ed, 2016, 55, 12026–12029. [DOI] [PubMed] [Google Scholar]
- 170.Sarkar MR, Dasgupta S, Pyke SM and Bell SG, Chem. Commun, 2019, 55, 5029–5032. [DOI] [PubMed] [Google Scholar]
- 171.Renata H, Wang ZJ and Arnold FH, Angew. Chem. Int. Ed, 2015, 54, 3351–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]