

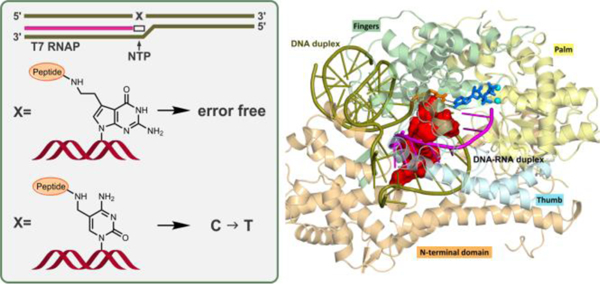
Abstract
DNA-protein cross-links (DPCs) are unusually bulky DNA adducts that block the access of proteins to DNA and interfere with gene expression, replication, and repair. We previously described DPC formation at the N7-guanine position of DNA in human cells treated with antitumor nitrogen mustards and platinum compounds and have shown that DPCs can form endogenously at DNA epigenetic mark 5-formyl-dC. However, insufficient information is available about the effects of these structurally distinct DPCs on transcription. In the present work, we employ a combination of in vitro assays, mass spectrometry, and molecular dynamics simulations to examine the ability of phage T7 RNA polymerase to bypass DPCs conjugated to the C7 position of 7-deaza-dG and the C5 position of dC. These model adducts represent endogenous DPCs induced by exposure to antitumor drugs and formed at epigenetics DNA marks, respectively. Our results reveal that DPCs containing full length proteins significantly inhibit in vitro transcription by T7 RNA polymerase, while short DNA-peptide cross-links (DpCs) are bypassed. DpCs conjugated to the C7 position of 7-deaza-dG are transcribed with high fidelity, while the same polypeptides attached to the C5 position of dC induce transcription errors. Molecular dynamics simulations of DpCs conjugated either to the C5 atom of dC or the C7 position of 7-deaza-dG on the template strand in T7 RNA polymerase explain how the conjugated peptide can be accommodated in the narrow major groove of the DNA-RNA hybrid and how the modified dC can form a stable mismatch with the incoming ATP in the polymerase active site, allowing for transcriptional mutagenesis.
GRAPHICAL ABSTRACT
INTRODUCTION
DNA-protein cross-links (DPCs) are ubiquitous DNA lesions that form when proteins are covalently trapped on genomic DNA upon exposure to endogenous and environmental chemicals.1 DPCs can be induced by formaldehyde, reactive oxygen species, free radicals, ionizing radiation, transition metals, and chemotherapeutic agents such as nitrogen mustards and platinum compounds.1–3 Many nuclear proteins including histones, DNA polymerases, DNA repair proteins, and transcription factors have been shown to participate in DPC formation.4–7 Because of the significant steric hindrance imposed by DPCs, they disrupt DNA structure, interfere with DNA replication, transcription and repair, and are thought to contribute to human disease.1–3, 8, 9
We recently discovered that 5-formylcytosine bases (5fC), which are endogenously present in cells as DNA epigenetic marks, readily form reversible Schiff base conjugates with Lys side chains of histones or other nuclear proteins in vitro and in vivo.10 5-Formylcytosine has been shown to play an important role in epigenetic control of gene expression in vitro and in human cells.11–13 Histone-5fC cross-links were detected in human cells, with global levels reaching 1.2 per 106 dCs.10 Li et al. independently reported the formation of 5fC-histone conjugates in nucleosomal core particles.14 Although Schiff base conjugates are reversible (t1/2 ~1.8 h), they are likely to interfere with DNA replication and transcription if present at replication forks or within actively transcribed regions of the genome.10–13 Our recent DNA polymerase bypass assays performed in vitro and in cells showed that reductively stabilized 5fC-mediated DPCs containing whole proteins (e.g. histone H2A) (Figure 1A) completely block human translesion synthesis (TLS) DNA polymerases η and κ, while corresponding cross-links to short peptides (DpCs) are bypassed by hPol η and κ in an error-prone manner.15, 16
Figure 1.
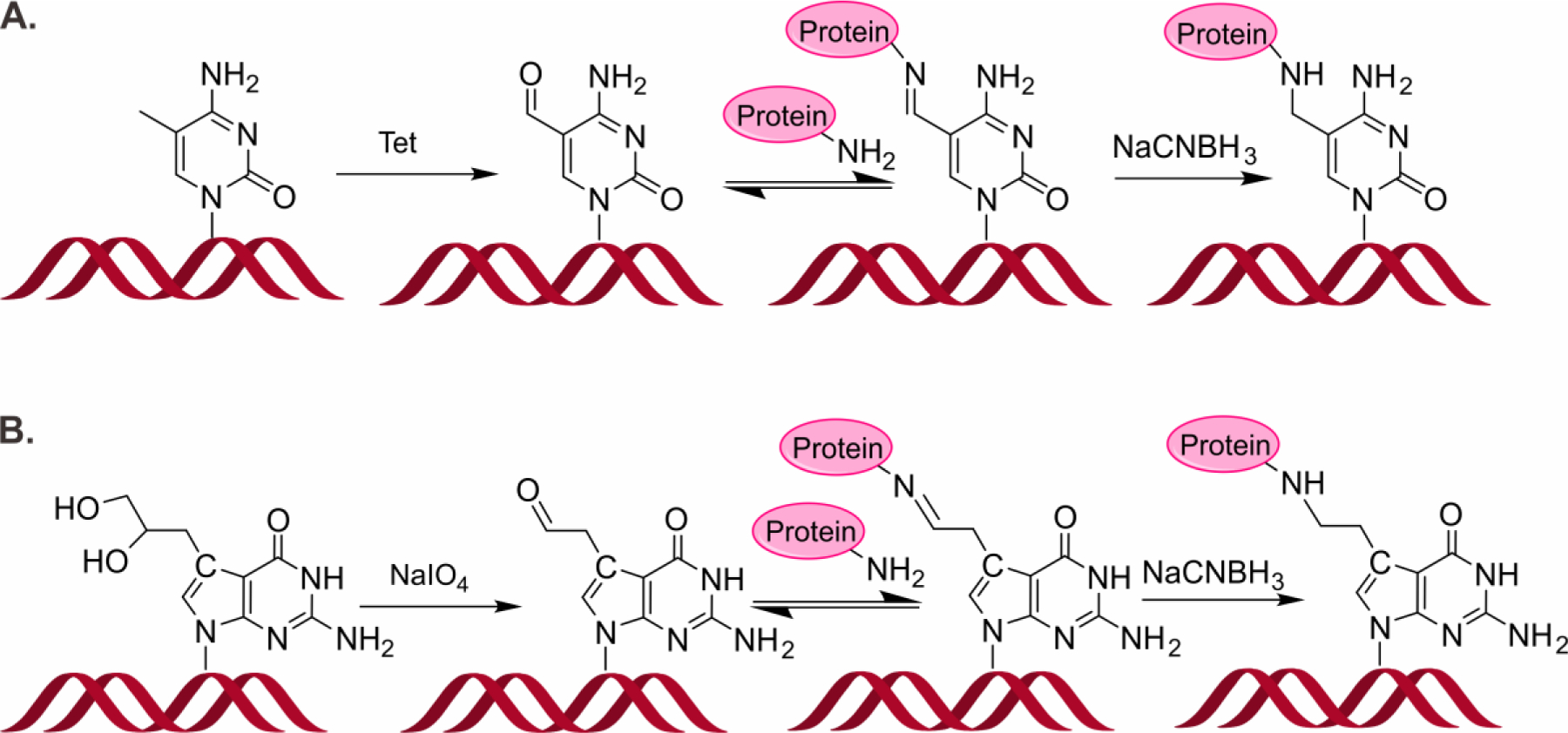
Formation of DNA-protein cross-links conjugated to the C5 position of cytosine (A) and the C7 position of 7-deazaguanine (B).
The most common site of DNA involved in DPC formation following exposure to bis-electrophile treatment (e.g. antitumor nitrogen mustards, 1,2,3,4-diepoxybutane and cis-platinum) is the N7 of guanine.5, 17, 18 Such lesions are inherently unstable due to spontaneous glycosidic bond cleavage under physiological conditions. We have previously synthesized hydrolytically stable DPCs conjugated to the 7 position of 7-deaza-dG, structurally analogous to N7-guanine DPC adducts generated by antitumor nitrogen mustards (Figure 1B) and showed that both TLS hPol η and κ inserted the correct base (C) opposite the short peptide conjugates, although with significantly reduced efficiency.18, 19
In the present work, we examined in vitro transcription in the presence of two types of site-specific, structurally defined DPC conjugated to the C5 position of dC and the C7 position of 7-deaza-dG. These structures resemble DPCs induced endogenously at 5-formyl-C of DNA (Figure 1A) and by antitumor drugs, respectively (Figure 1B). 7-Deaza-dG was used in place of dG in order to prevent spontaneous depurination of the lesions substituted at the 7-G position. A combination of gel electrophoresis and mass spectrometry-based techniques was employed to investigate the effects of DPCs and DpCs on transcription in the presence of T7 RNA polymerase, while the structural aspects of lesion bypass were investigated by molecular dynamics simulations to offer interpretations to the experimental results.
RESULTS AND DISCUSSION
Synthesis and characterization of double-stranded DNA duplexes containing site-specific DNA-protein or DNA-peptide conjugates
Due to their unusually large size and their helix-distorting nature, DPCs are expected to interfere with gene expression and to block the access of proteins required for other necessary biological functions, thereby contributing to cytotoxicity of cross-linking agents.1 In order to investigate the effects of DPCs and DpCs on transcription, we constructed a double-stranded DNA template containing a 17-bp T7 RNAP promoter region. Our double-stranded DNA template is 100-bp long, with DPC lesion located at 49th or 50th nucleotide downstream of the transcription start site (Figures 2 and S1). Two DPC types employed in the present study are proteins/peptides conjugated to the C5 position of cytosine or the C7 position of 7-deazaguanine, respectively. The first type of DPC was synthesized via reductive amination reaction between the aldehyde group of 5-formyl-dC in DNA and Lys side chains of proteins/peptides in the presence of NaCNBH3 10 (Figures 1A and 2A). DNA-protein and DNA-peptide cross-links involving the C7 position of 7-deaza-dG were prepared analogously by reductive amination reactions between Lys/Arg side chains of proteins/peptides and 7-deaza-7-(2-oxoethyl)-dG in DNA (Figures 1B and 2B).20
Figure 2.
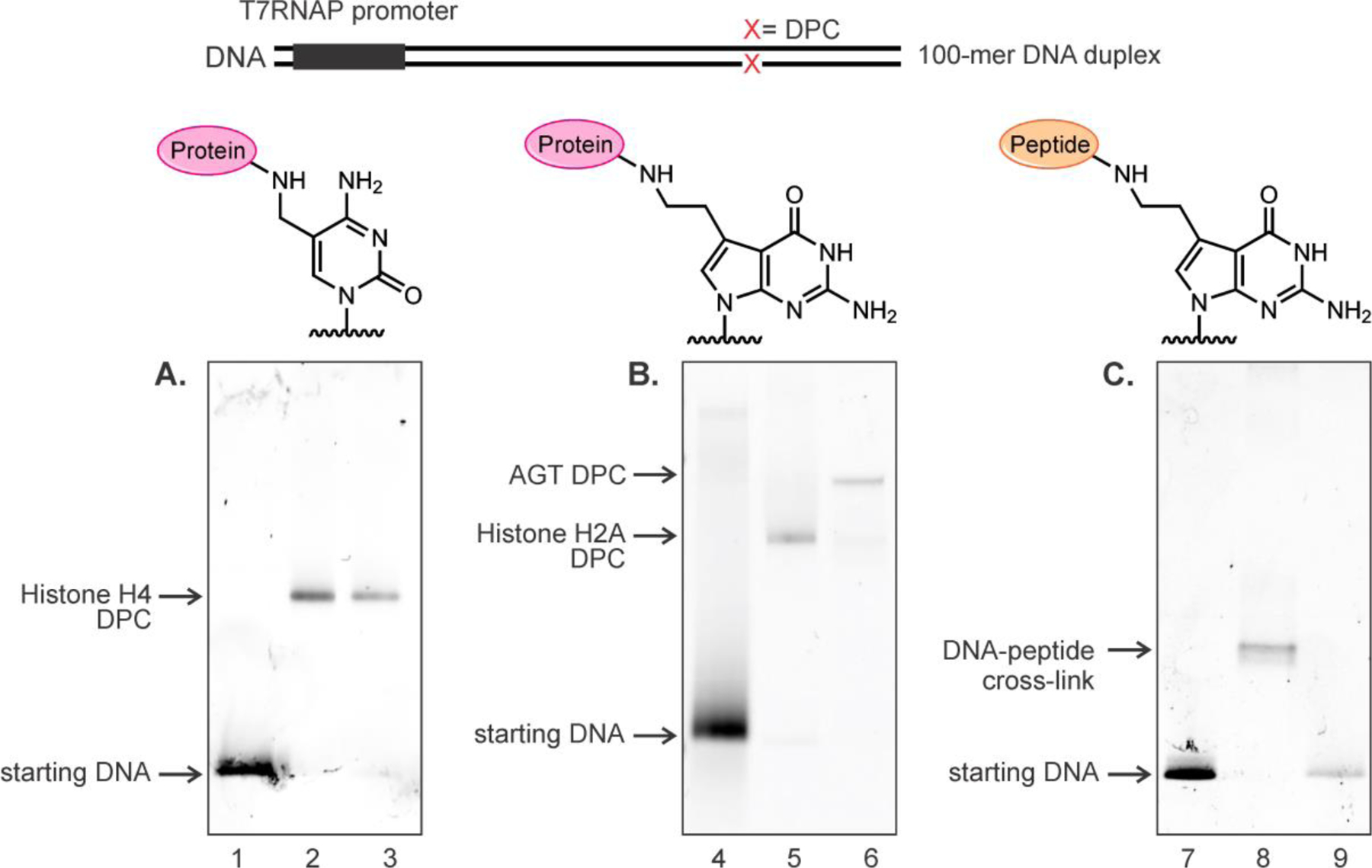
Representative denaturing PAGE images of 100-mer duplexes containing DPCs or DpCs. (A) DNA-protein cross-links conjugated to the C5 of dC. Lanes 1: starting DNA (100-mer duplex containing 5fC); lanes 2 and 3: gel purified 5fC-mediated histone H4 DPCs located on transcribed strands (TS) and non-transcribed strands (NTS). (B) DNA-protein cross-links conjugated to C7 of 7-deazaguanine. Lane 4: starting DNA (100-mer duplex containing 7-deaza-DHP-dG); Lane 5 and 6: gel purified 7-deazaG-mediated histone H2A and AGT DPCs located on transcribed strands (TS). (C) DNA-peptide cross-links at C7 of 7-deazaguanine containing 31-mer peptide. Lane 7: starting DNA (100mer DNA duplex containing 7-deaza-DHP-dG); lane 8: gel purified DpC conjugate located on transcribed strands (TS); lane 9: protease K digested DNA-peptide conjugates.
Protein substrates for DPC synthesis (Histone H4 (11.2 kDa), histone H2A (14.0 kDa), hOGG1 (39 kDa) and AGT (53.1 kDa)) were chosen based on our previous studies reporting their involvement in DNA-protein cross-linking in cells.1, 10 In addition, smaller DNA-peptide conjugates (DpC) were prepared representing putative products of proteolytic cleavage of DPCs by specialized proteases such as Spartan/Wss1.21–23 Four Lys-containing peptides of increasing size (10-mer, 11-mer, 31-mer and 57-mer, see Table 1) were cross-linked to 5-formyl-dC and 7-deaza-7-(2-oxoethyl)-dG in DNA using reductive amination to reveal the effects of DpC size on transcription (Figure 1). The same peptides were used in our earlier studies investigating the formation of DNA-peptide cross-links and their effects on DNA replication.15, 19 DNA-protein and DNA-peptide cross-links were purified by gel electrophoresis and characterized by SDS-PAGE (see representative gel images in Figure 2), and their structures were confirmed by HPLC-ESI-MS/MS as reported previously.10, 20
Table 1.
Amino acid sequences of polypeptides employed to generate model DpCs.
| Peptide | Sequence | Length |
|---|---|---|
| A | EQKLISEEDL | 10-mer |
| B | RPKPQQFFGLM | 11-mer |
| C | YGGFMTSEKSQTPLVTLFKNAIIKNAYKKGE | 31-mer |
| D | YAEGTFISDYSIAMDKIHQQDFVNWLLAQKGKK NDWKHNITQGKKKKKKSKTKCVIM |
57-mer |
Effects of DNA-protein cross-links on transcription catalyzed by T7 RNA polymerases.
T7 RNA polymerase is a single subunit RNA polymerase that requires no ancillary transcription factors and is widely used as a model polymerase in studies of transcriptional bypass and repair of DNA lesions.24–29 For example, T7 RNA Pol was used as a model polymerase to study in vitro transcription in the presence of cisplatin adducts,26 malondialdehyde adducts,28 benzo[a]pyrene lesions,25 and ethyl-dT adducts.24 Although T7 RNA Pol is significantly different from mammalian polymerases, its simplicity and the availability of detailed crystal structures make it possible to conduct detailed modeling and mechanistic studies currently not feasible with human RNA Pol.
The DNA template was created by including a 17-bp T7 RNA Pol promoter region and a modified nucleobase 50 nucleotides downstream from transcription start (Figure 3).30 In the absence of DNA damage, T7 RNA Pol binds to the promoter and generates 72 nt long RNA transcripts (see below).
Figure 3.
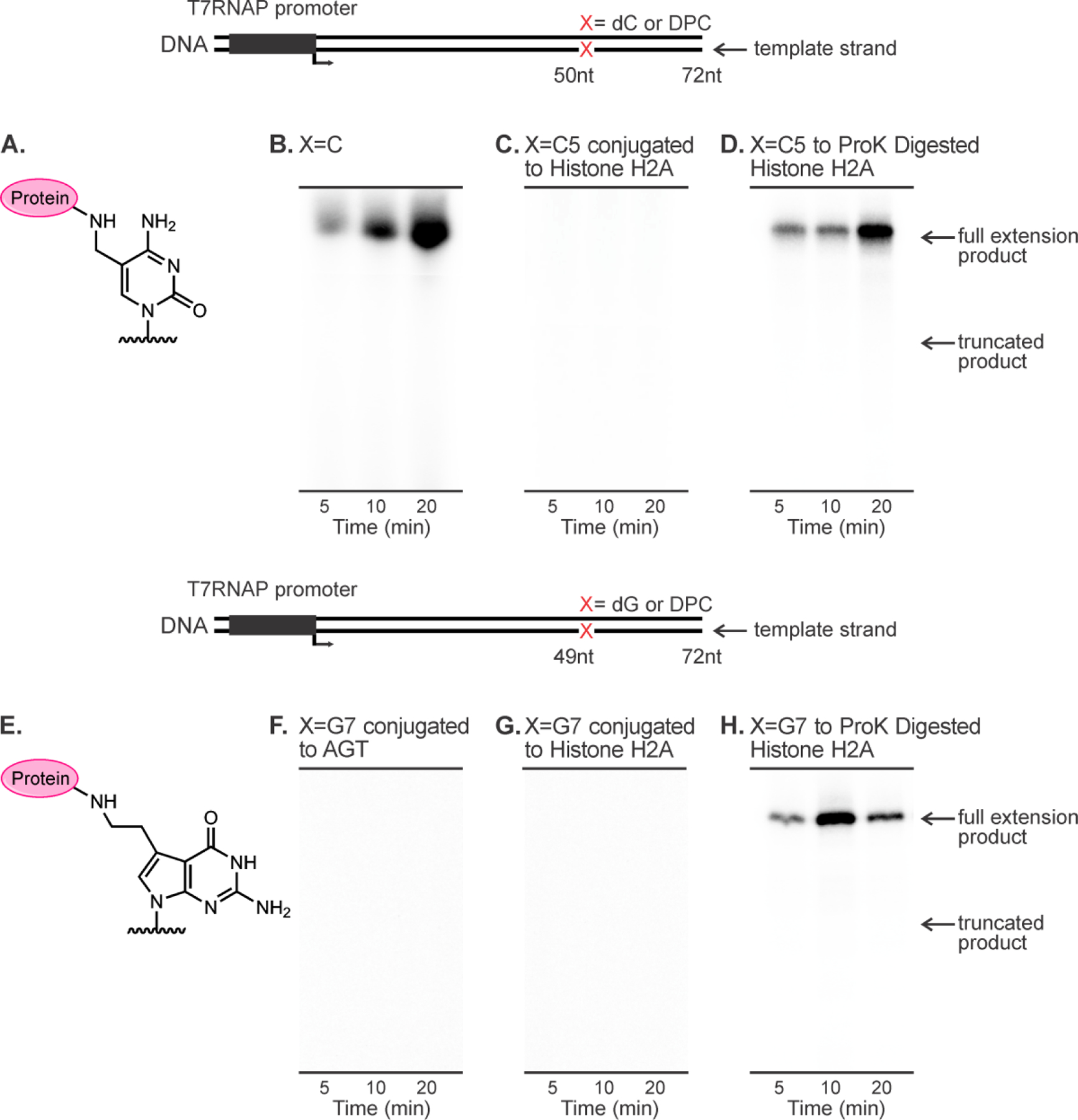
In vitro T7 RNAP bypass assays of DNA-protein cross-links conjugated to C5 of cytosine (top panel) and C7 of 7-deazaguanine (bottom panel) on template strands.
To generate 5fC-mediated DPC substrates for in vitro T7 transcription assays, double-stranded 100-mer DNA duplexes containing site-specific 5fC were incubated with recombinant proteins in sodium phosphate buffer. The resulting Schiff base conjugates were stabilized by reduction in the presence of NaCNBH3 (Figure 1A) and purified by SDS-PAGE as described elsewhere (Figure 2A).15 These lesions structurally resemble 5fC-histone cross-links formed endogenously in human cells.10
7-dG conjugated DPCs were prepared analogously using 100-mer DNA duplexes containing site-specific 7-deaza-7-(2-oxoethyl)-2-deoxyguanosine (7-deaza-DHP-dG), which were formed in situ from 7-deaza-7-(2,3-dihydroxypropan-1-yl)-2'-deoxyguanosine-containing DNA as described in our earlier publication (Figure 1B) and purified by SDS-PAGE (Figure 2B).19 In our model crosslinks, Lys or Arg side chains of proteins and peptides are attached to the 7-deazaguanine residues of DNA via a two-carbon linker. Such conjugates are structurally analogous to DPC lesions induced in cells by antitumor nitrogen mustards and ethylene dibromide.1
To examine the influence of DNA-protein cross-links on transcription, double-stranded 100-mer DNA duplexes containing canonical DNA bases (control) or site-specific DPC lesions to histone H4, histone H2A, and O6-alkylguanine transferase (AGT) proteins at position “X” were subjected to in vitro transcription in the presence of T7 RNA polymerase (Figure 3). Since [α−32P] UTP was added to the transcription reaction, all newly synthesized transcripts contain 32P label and can be detected by phosphorimaging followed by separation by denaturing PAGE (Figure 3). Because no repair proteins were included in this reconstituted transcription system, full-length transcripts indicated bypass of the adducted base rather than removal of the adduct prior to RNAP bypass.
In our control experiments, DNA templates containing only standard DNA bases within the transcribed region yielded the expected full-length transcripts with a length of 72 nucleotides (Figure 3B). In contrast, no full-length transcripts were observed for histone or AGT DPCs conjugated to the C5 position of cytosine or the C7 position of 7-deazaguanine (Figures 3C, F and G). The ability of T7 RNAP to bypass the damaged strand was restored when protein component of DPC was digested with proteinase K treatment (Figures 3D and 3H). When present on coding strand, DPC lesions were less blocking (Figure 4), and full-length transcripts were observed. Collectively, these results indicate that regardless the attachment site in DNA, full-size DNA-protein cross-links on transcribed strand completely inhibit in vitro transcription. However, such transcriptional blockage can be relieved by proteolytic degradation of the protein component of DPC (Figure 3D,H).31 Our results (Figures 3 and 4) differ from an earlier observation by Nakano et al.29 who reported that full size DPCs formed at oxanine residues of DNA could be bypassed by T7 RNAPs when present on transcribed strand of DNA.29 This discrepancy may be due to the different structures of the DPCs lesions employed in the two studies.
Figure 4.
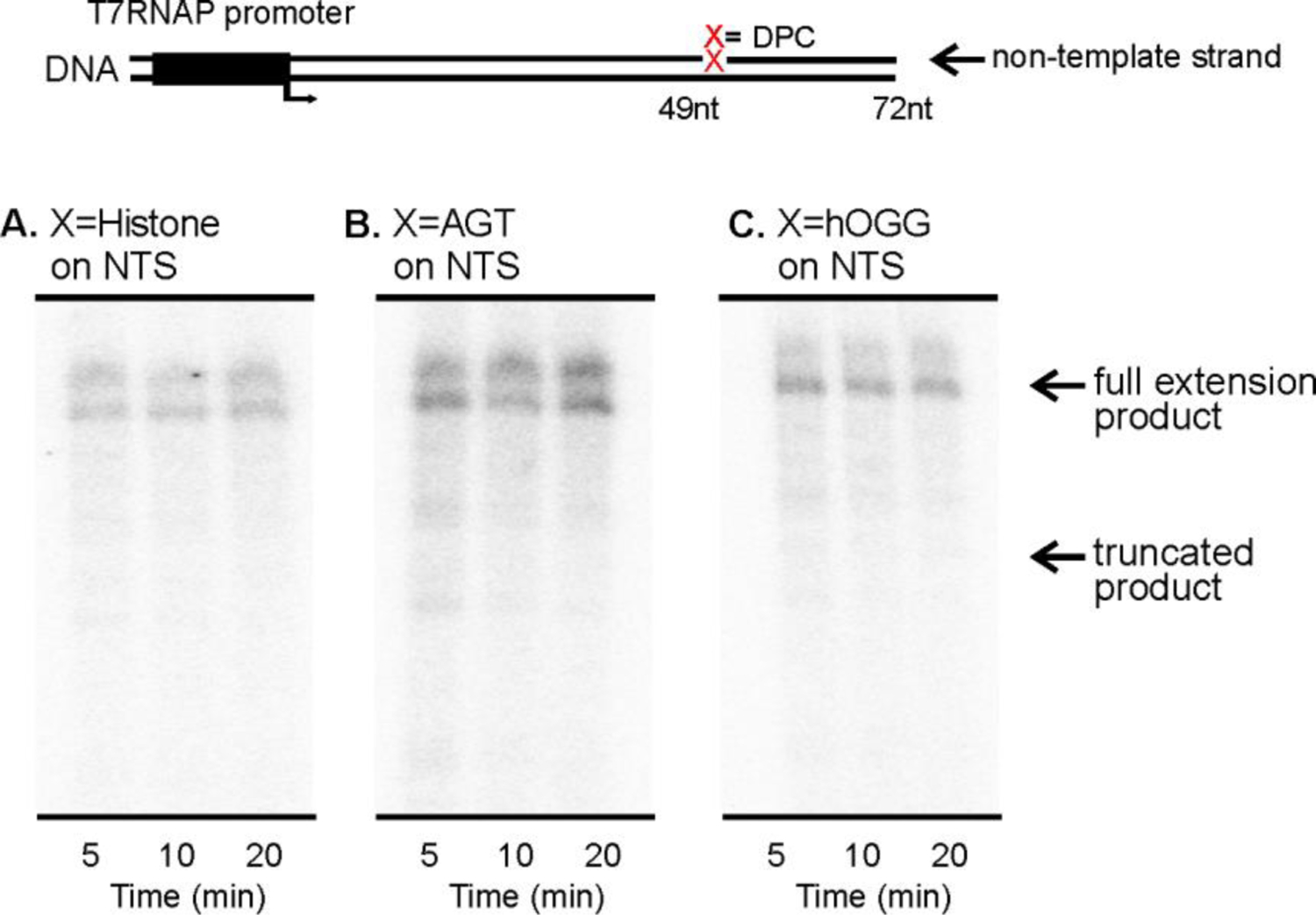
In vitro transcription of DNA containing DNA-protein cross-links present on coding strand of DNA by T7 RNAP. Proteins were conjugated to the C7 of 7-deazaguanine via reductive amination.
The biological responses to stalled RNAPs-DNA complexes require further investigation. For example, RNAPs stalling at DPC lesions may trigger their proteolytic processing and repair via transcription-coupled NER. Chesner et al. recently showed that DPCs present on transcribed strands were repaired 2.5-fold more efficiently than the identical DPC on non-transcribed strands, suggesting the involvement of transcription-coupled nucleotide excision repair (TC-NER) in DPC removal in cells.32
Transcription of DNA template containing DNA-peptide cross-links of varying sizes
In yeast and eukaryotic cells, the protein component of DPCs is proteolytically degraded by specialized proteases to form the corresponding DNA-peptide conjugates (DpCs).21, 23, 31 DpC lesions are expected to be less blocking and may be bypassed by DNA and RNA polymerases. We investigated the effects of DNA-peptide cross-links of varying sizes (10-mer, 11-mer, 31-mer and 57-mer peptides, see Table 1 for amino acid sequences) on T7 RNAP catalyzed transcription. We initially conducted time course experiments using DNA templates containing dG or 7-deaza-DHP-dG located at the 49th nucleotide downstream of the transcription start site. PAGE analysis of transcription products generated upon replication of control templates revealed the presence of only full-length transcripts 72 nts in length (Figures 5A and B). In contrast, in vitro transcription of DNA-peptide cross-links conjugated to C7 position of 7-deaza-dG also generated truncated products (Figure 5C, D and E). The truncated transcripts were about 50 nucleotides in length, suggesting that the stalling sites were at or near the DNA-peptide cross-links located at 49 nts downstream the transcription start site. The T7 RNAP bypass efficiencies were calculated using equation: percent bypass efficiency = (full transcription products)/(full transcription products + truncated transcription products) x 100%. Transcriptional bypass efficiencies gradually decreased as the size of the conjugated peptide increased from 10-mer to 31-mer and to 57-mer peptides (Figure 6).
Figure 5.
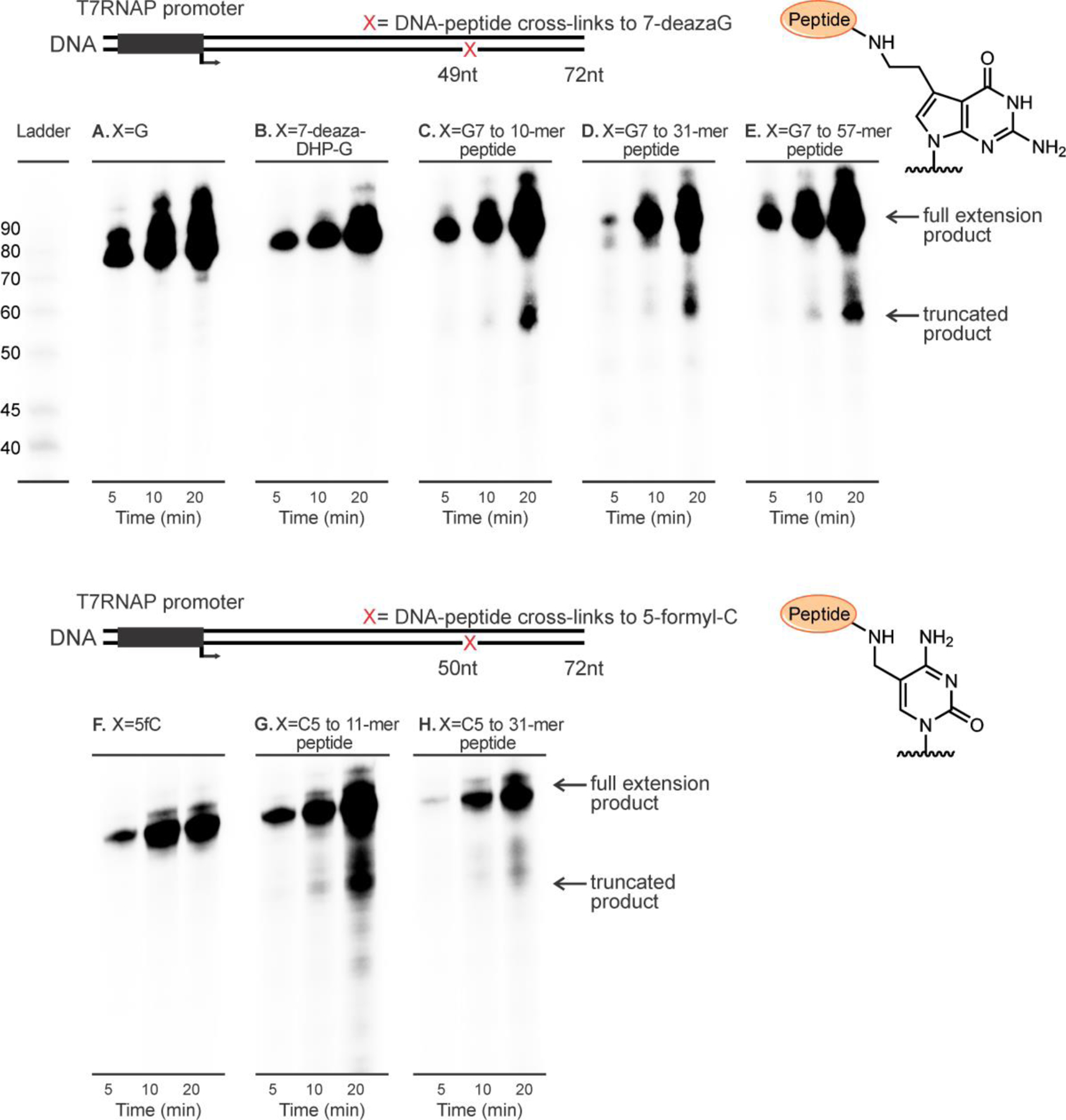
In vitro T7 RNAP bypass assays of DNA-peptide cross-links conjugated to C7 of 7-deazaguanine (top panel) or C5 of cytosine (bottom panel) on template strands.
Figure 6.
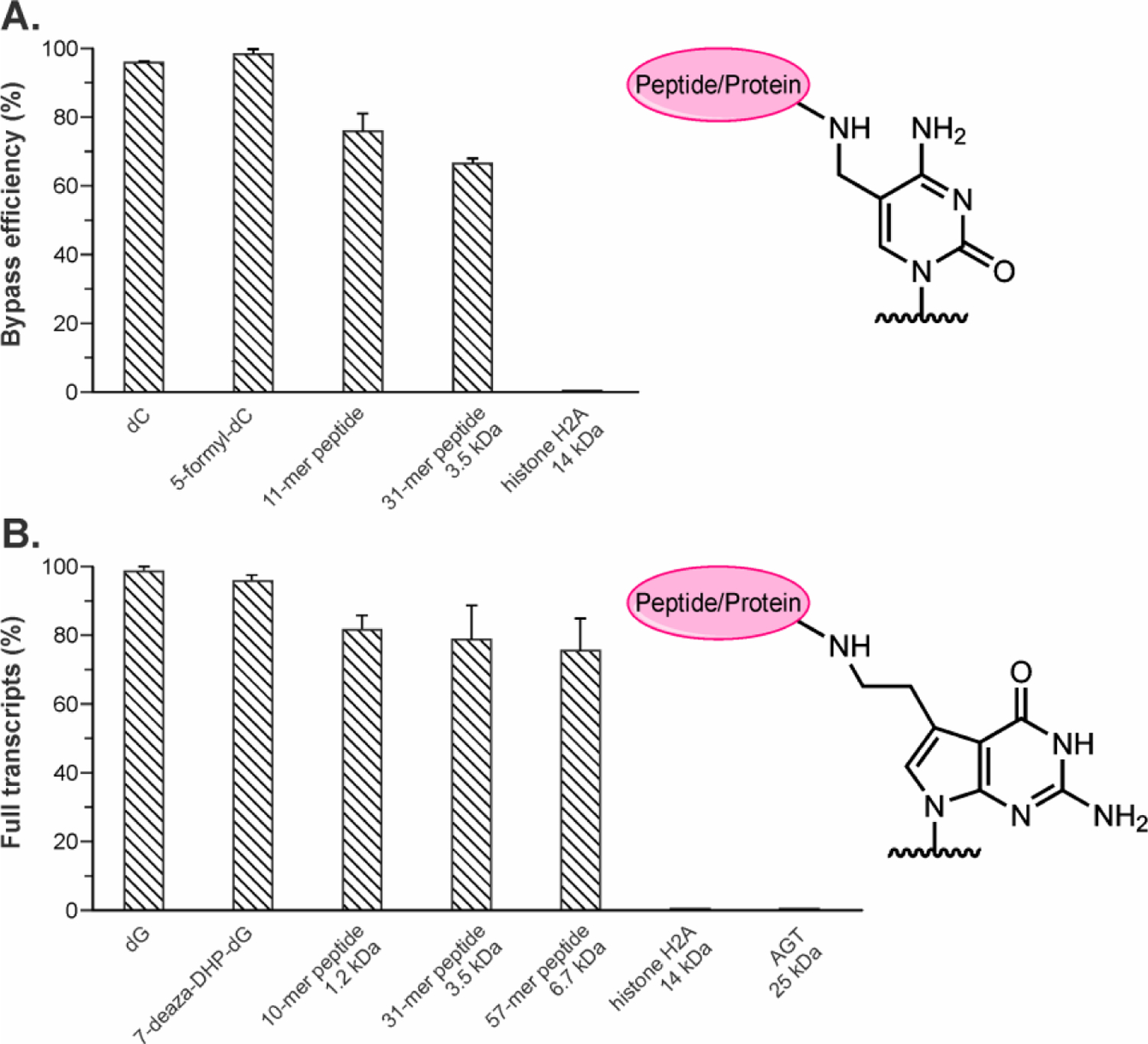
T7 RNAP transcription bypass efficiencies of DNA-protein/peptide cross-links conjugated to C5 of cytosine (A) and C7 of 7-deazaguanine (B) on template strands. Data is a result of 3–5 independent experiments.
Similar results were obtained for DNA-peptide lesions conjugated to C5 of 5fC (bottom panel in Figure 5). As expected, 5-formyl-dC, an endogenous DNA modifications detected in normal cells,33, 34 did not inhibit in vitro T7 RNAP catalyzed transcription (Figure 5F). In contrast, pronounced pausing was observed at about 1–2 nucleotides downstream from 5fC-conjugated DNA-peptide cross-links (Figures 5G and H). These results provide evidence that DNA-peptide cross-links conjugated to either C7 position of 7-deazaG or C5 position of 5fC can be tolerated by T7 RNAPs.
RT-PCR and LC-MS/MS based method to investigate transcriptional mutagenesis.
We next asked whether the presence of DNA-peptide cross-links in DNA interferes with fidelity of transcription by T7 RNAP. For this purpose, RNA transcripts generated upon in vitro transcription of unmodified and DpC containing templates were amplified by RT-PCR and sequenced by LC-MS/MS (Figure 7A). Transcription reactions were performed as described above, but in the presence of non-radioactive NTPs. After complete digestion of the DNA templates using DNAse, the transcripts of interest were amplified by RT-PCR, and the resulting PCR products were digested with two suitable restriction enzymes (Figure S2). The released short oligonucleotides containing the region of interest are readily sequenced by HPLC-ESI-MS/MS. This methodology allows for the detection and quantification of transcriptional mutagenesis at the adduct site (Figure 7A).
Figure 7.
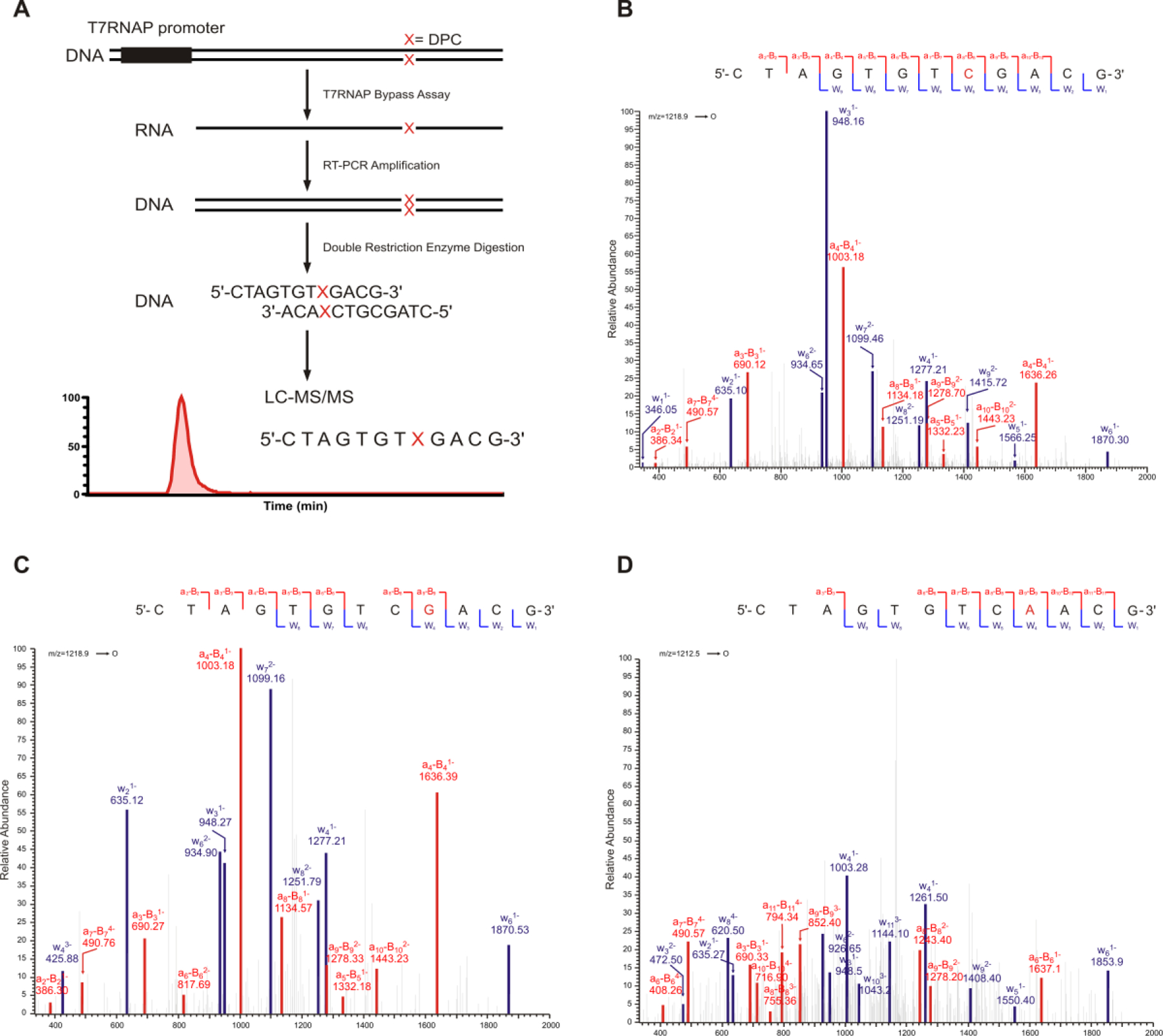
Scheme of RT-PCR and LC-MS/MS based methodology to investigate transcriptional mutagenesis. (A) MS/MS spectrum of the transcription products. (B-D) CID spectra of error free transcription products (5'-CTAGTGTCGACG-3', [M-3H]3−=1218.87) from 7-deaza-dG conjugated peptides. (B) Error free transcription products (5'-CTAGTGTCGACG-3', [M-3H]3−=1218.87) (C) and C to T mutations (5'-CTAGTGTCAACG-3', [M-3H]3−=1213.54) from 5fC conjugated peptides (D).
HPLC-ESI-MS/MS was used to characterize the products of in vitro transcription of templates containing unmodified dG, 7-deaza-DHP-dG, or 7-G conjugated DNA-peptide cross-links of increasing size. HPLC-ESI-FTMS analysis of digested PCR products on Orbitrap Velos allowed for the detection and relative quantification of transcription products. In control experiments with unmodified dG or 7-deaza-DHP-dG, only the error-free transcription products (5'-CTAGTGTCGACG-3', [M-3H]3−=1218.87) and its complementary strands (5'- CTAGCGTCGACA, [M-3H]3−=1208.54) were detected (Figure S3). MS/MS spectrum of this error-free oligonucleotide product was consistent with the predicted spectrum (Figure 7B). Similarly, only error-free transcription products were observed in experiments performed using DNA templates containing polypeptides conjugated to C7 of 7-deazaG (Figure S3 and Table 2). These results demonstrate that the transcription bypass catalyzed by T7 RNAPs opposite the polypeptide conjugated to C7 position of 7-deaza-dG is essentially error-free.
Table 2.
Summary of LC-MS/MS analysis of transcription products generated using DNA templates containing DNA-peptide cross-links at C7 of 7-deazaguanine.
| Template | 5'-CTA GTG TXG ACG-3' 3'-AC AXC TGC GAT C −5' |
|||
|---|---|---|---|---|
| Template | Extension product | Percent product | Base opposite X | Comment |
| dG | GTGTCGACG | 100 | C | error free |
| 7-deaza-DHP-dG | GTGTCGACG | 100 | C | error free |
| 10-mer peptide | GTGTCGACG | 100 | C | error free |
| 31-mer peptide | GTGTCGACG | 100 | C | error free |
| 57-mer peptide | GTGTCGACG | 100 | C | error free |
Similar experiments were conducted using DNA templates containing 11-mer peptides conjugated C-5 position of dC (Figure 7A). Transcription reactions obtained using unmodified dC or 5-formyl-dC (negative controls) contained only the expected error-free products (5'-CTAGCGTCGACA, [M-3H]3−=1208.54). In contrast, transcription reactions conducted using C5-dC DpCs templates revealed the presence of C →T transcriptional mutations in addition to error-free products (Figures 7C and D). In vitro transcription past 5fC-mediated 11-mer DpCs resulted in 21.4% C →T transcription mutations (5'-CTAGTGTCAACG-3', [M-3H]3−=1213.54) and 78.6% error-free products (5'-CTAGTGTCGACG-3', [M-3H]3−=1218.87). In the transcription bypass of 31-mer peptide at 5fC, 6.5% of total products corresponded to C → T transcriptional mutations (5'-CTAGTGTCAACG-3', [M-3H]3−=1213.54) (Figure S4 and Table 3). These results indicate that in contrast to error-free transcription bypass of 7-deaza-dG mediated DNA-peptide cross-links, transcriptional bypass of 5fC-mediated DNA-peptide cross-links by T7 RNAP is highly error-prone, leading to incorporation of A opposite to modified dC and transcriptional errors. Overall, these results indicate that the fidelity of RNA polymerases is highly dependent on the structure of lesion and its attachment site within DNA.
Table 3.
Summary of LC-MS/MS analysis of transcription products generated using DNA templates containing DNA-peptide cross-links at C5 of cytosine.
| Template | 5'-CTA GTG TCX ACG-3' 3'-AC AGX TGC GAT C −5' |
|||
|---|---|---|---|---|
| Template | Extension product | Percent product | Base opposite X | Comment |
| dC | GTGTCGACG | 100 | G | error free |
| 5-formyl-dC | GTGTCGACG | 100 | G | error free |
| 11-mer peptide | GTGTCGACG | 78.6 | G | error free |
| GTGTCAACG | 21.4 | A | C to T | |
| 31-mer peptide | GTGTCGACG | 93.5 | G | error free |
| GTGTCAACG | 6.5 | A | C to T | |
Molecular modeling and molecular dynamics simulations
In order to explore the structural basis for DpC bypass by T7 RNA polymerase and interpret the differences in transcriptional outcomes for lesions conjugated to C-5 of dC and C-7 of 7-deaza-dG, we constructed molecular models of the ternary complex between the DpC containing DNA templates, T7 RNA polymerase, and the incoming nucleotide triphosphates. To create a starting model for our molecular dynamics simulations, we used the high-resolution crystal structure of the T7-RNA polymerase elongation complex with PDB35 ID 1S76.36 The structure that was resolved contains a ternary complex with 21 nucleotides of template DNA and 17 nucleotides of non-template DNA, as well as a 9 base pair DNA–RNA hybrid, and an incoming ribonucleotide triphosphate NTP. The enzyme consists of an N-terminal domain (residues 1−324), thumb (residues 325−411), fingers (residues 566−784), and palm domains (residues 412−565 and 785−883) (Figure 8A). Missing residues 195−199, 233−240, 363−374, and 594−611 in loops of the enzyme were modeled into the T7-RNAP complex using the Modeller37 program. The downstream DNA duplex contains 10 base pairs with 7 nucleotides on the non-template DNA in the open bubble region (Figure 8B). In the crystal structure, the template DNA base is thymine and its partner NTP is ATP.
Figure 8.
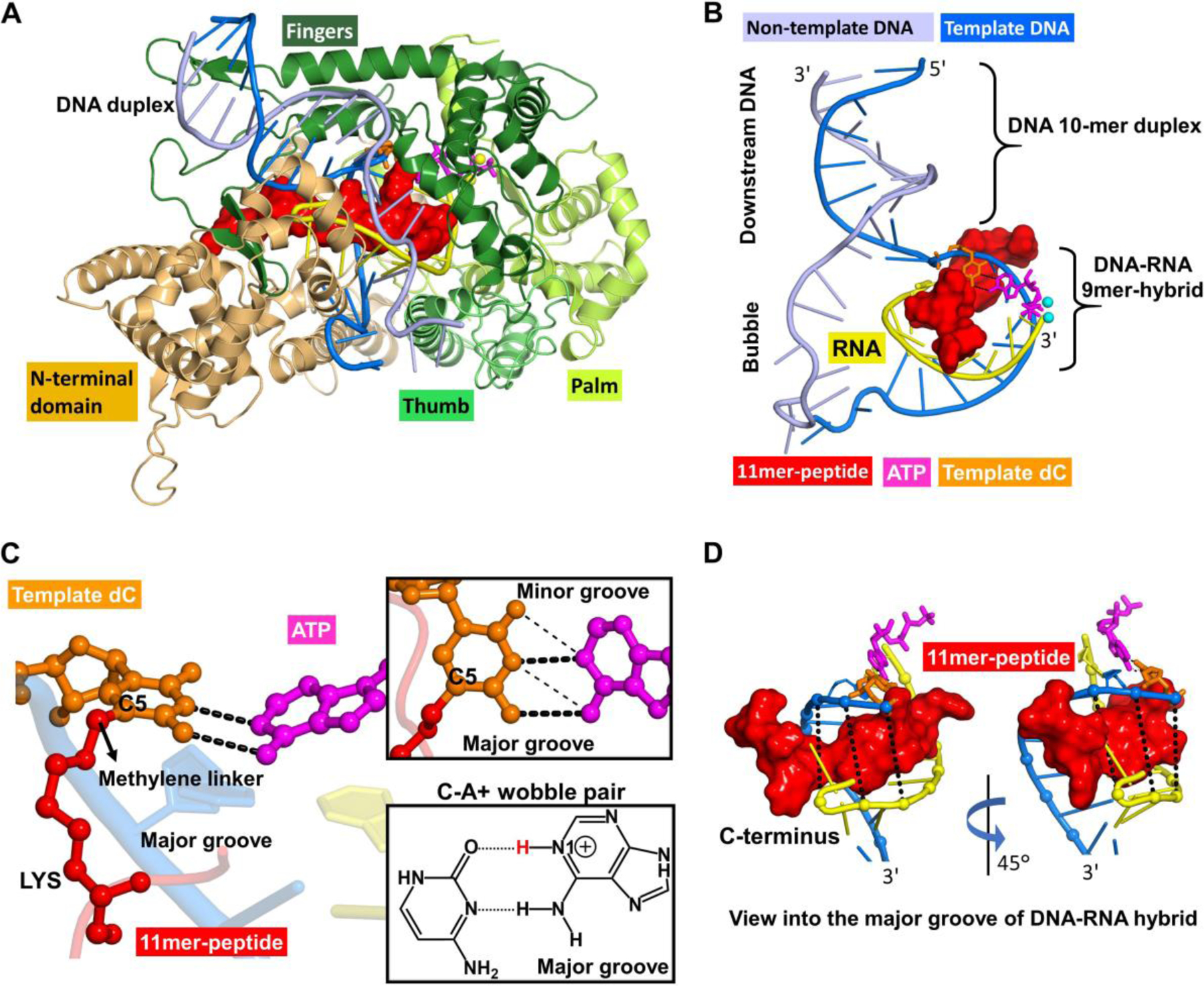
Overall structure of the T7 RNA polymerase complex in the insertion state, showing the 11mer-peptide cross-linked to the templating dC opposite incoming ATP to form a C-A+ mismatch pair. (A) This complex includes protein domains and nucleic acids in cartoon rendering. The enzyme domains consist of the N-terminus (residues 1−325), thumb (residues 326−411), palm (residues 412−553 and 785–883), and fingers (residues 554−784). (B) A view without enzyme. The nucleic acids include template and non-template DNA, RNA and an incoming ATP. The DNA consists of a part with a 10-mer duplex in the downstream region and a noncomplementary segment in the open bubble. A complementary nine nucleotide RNA segment at the 3'-end is hybridized to the template DNA in the bubble region to form the DNA-RNA hybrid 9-mer. (C) A view of the active site from the major groove showing the templating dC paired with incoming ATP via two hydrogen bonds. (inset box, thick dashed lines), which shifted from the standard wobble pairing scheme (thin dashed lines) in our MD simulations. The chemical structure of the C-A+ wobble pair, containing two hydrogen bonds, dC(N3) − A+(N6H2) and dC(O2) − A+(N1H) with glycosidic bonds anti in both dC and A+, is also displayed. The C5 atom of templating dC is conjugated to the side chain of a lysine residue in the 11mer-peptide via a methylene linker. (D) Views of the DNA-RNA hybrid structure highlighting the 11mer-peptide tightly-held in the narrow major groove of the hybrid. The C-terminus of the 11mer-peptide is directed toward the 3'-end of the DNA template strand.
DNA-peptide cross-links studied experimentally in this work (Figure 1) were introduced at the template base. Two types of DNA-protein cross-links were investigated here: a lysine side chain conjugated to the C5 position of dC via a methylene linker and a lysine side chain conjugated to the C7 position of 7-deaza-dG via a two-carbon linker (Figure 1). To construct the 5-dC conjugated DNA-peptide cross-link, the thymine template base was substituted with a C to which a lysine side-chain was conjugated to its C5 atom via a methylene linker (Figure 8C). The 11-mer peptide RPKPQQFFGLM-CONH2 (Table 1) was modeled into the major groove of the DNA-RNA hybrid, via a step-wise build-up procedure, as described previously for the same peptide placed in the duplex DNA major groove in DNA polymerases.15 The peptide has its C-terminus oriented toward the 3'− end of the template strand (Figures 8B,D). To help explain the preferential misinsertion of A opposite the 5fC-peptide lesion, we investigated a mismatch of the modified C with A to form a C-A+ wobble pair. To do so, the NTP was modeled as an ATP that was N1-protonated (termed ATP) (Figure 8C). This model is termed 11mer-dC-ATP. For its Watson-Crick partner, we remodeled the incoming NTP to GTP. The final model was named 11mer-dC-GTP.
A similar procedure was applied to construct the dG conjugated DNA-protein cross-link. The thymine template base was replaced with a G. The N7 position of dG was then substituted with a C7 atom, which was then conjugated to the side chain of the single lysine residue via the two-carbon linker. The 10-mer peptide EQKLISEEDL-CONH2 (Table 1) was then modeled, using step-wise build-up procedure, into the major groove of the DNA-RNA hybrid with its C-terminus oriented toward the 3'− end of the template strand.. For its Watson-Crick partner, we replaced the incoming NTP with CTP; this final model was named 10mer-dG-CTP. In addition, three control models without 11-mer or 10-mer peptide were constructed, termed dC-GTP, dC-ATP, and dG-CTP. Thus six T7 RNA polymerase models were prepared for the MD simulations.
For the unmodified dC-GTP or dG-CTP matched base pair, our molecular dynamics simulations showed three full stable Watson-Crick hydrogen bonds. In the case of the unmodified dC-ATP mismatched base-pair, we found a stable wobble pair with two hydrogen bonds (Figure S5 and Tables S1 and S2). In both DpC containing duplexes, the conjugated peptide fits tightly and penetrates deeply into the narrow and deep A-type major groove of the DNA-RNA hybrid (Figure 8D). Watson-Crick hydrogen bonding is maintained in the presence of the DpC (Table S1), although for the 10mer-dG-CTP, one of the hydrogen bonds is weakened. In the 11mer-dC-ATP, which was initially modeled as a wobble pair, the hydrogen bonding scheme shifted early in the simulation to produce two new hydrogen bonds: dC(N3) − A+(N1H) and dC(N4H) − A+(N6) (Figure 8C). This shift occurred because the modified dC moved toward the minor groove, pushed by the tightly-held peptide in the narrow major groove (Figure S6A). However, the peptide does not similarly affect the position of the template base with normal Watson-Crick pair; likely this is due to the stronger dC-GTP/dG-CTP hydrogen bonds, which inhibit the modified template base from shifting (Figure S6B–C).
We also evaluated the position of the incoming NTP for each of our simulated systems to determine how close the NTP position is to being reaction-ready. For this purpose, we measured the distance between O3' of the primer terminus and the Pα of the NTP (Table S3). We found that the value was ~ 3.5 Å in all cases except for the 11mer-dC-ATP and dG-CTP, whose values were ~ 5 Å, and for the 10mer-dG-CTP, whose average distance was ~ 7.0 Å. Our observation that the largest distance occurs with the 10mer-dG-CTP ternary complex, so that the incoming nucleotide triphosphate is much more displaced than in any of the other systems. In those cases, the distance is much closer to the near reaction-ready state of ~ 3.4 Å observed in a well-organized crystal structure of a ternary complex.38 For the ~ 7.0 Å average distance found for the 10mer-dG-CTP, the incoming CTP is lifted away from the primer terminus and propeller-twisted in the presence of the peptide as well as being more dynamic than the other cases.
Our in vitro transcription experiments described above show that unlike C7-deaza-dG DpC lesions, which did not induce transcriptional mutagenesis, C5-dC conjugated peptides generate large amounts of C → T base substitutions (Table 3). MD simulations were performed to gain insight on the mutagenic bypass via a C-A+ mismatch of the C5-dC conjugated 11mer-peptide in T7 RNA polymerase. Our simulations found a stable dC-ATP mismatch, which is supported by a two-hydrogen bonding scheme that is shifted from the standard wobble pair (Figure 8C). In our simulation, the presence of a peptide tightly held in the major groove of the DNA:RNA heteroduplex caused the modified dC to move toward the minor groove, so that the standard wobble pair is destabilized (Figure S6A). However, the shifted hydrogen bonds are well aligned and stable (Tables S1 and S2, and Figure S7).
Our results for DpC induced C-A+ mismatch in the T7 RNA polymerase active site (current work) were compared to the analogous model generated for DNA polymerase ƞ in our earlier study.15 In our model of DNA polymerase ƞ, the C-A+ mismatch between C5-dC DpC and the incoming dATP is supported by a standard wobble pair. A key difference between the models of the tertiary complexes of DpCs with RNA and DNA polymerase is that RNA polymerase is bound to an A-form helix of DNA-RNA, while in the corresponding complex with DNA polymerase ƞ, the enzyme is bound to B-form DNA-DNA duplex. The A-form duplex has a narrower major groove than the B-form, although they have similar depths (Figure S8). Because the peptide is housed in the major groove in complexes with both polymerases, the difference in the groove width appears to play an important role in defining the structural impact of the peptide in the polymerase active site.
The in vitro transcription data show that the C-A mismatch produces ~ 20 % product in both polymerases (21.4% for T7 RNA polymerase versus 20.1 % for DNA polymerase ƞ). In the DNA polymerase ƞ active site, our simulations predicted the formation of a stable C-A+ wobble pair, explaining our mutagenesis results. In the RNA polymerase, the steric effect of the peptide in the narrower A-form major groove pushes the dC towards the minor groove, which shifts the wobble pair (Figures 8C, S6A and S8). However, two hydrogen bonds are still retained in this shifted hydrogen bonding scheme (Table S1), which explains the similar C-A+ mismatch data.
Furthermore, in the case of the DpC-containing dC-GTP pair in the T7 RNA polymerase, the three hydrogen bonds observed in the simulation are stable and strong; consequently, the dC is prevented from shifting towards the minor groove, which explains the error-free extension product data (78.6%). However, in DNA polymerase ƞ, the structure of the dC-G base-pair is more distorted: the template base dC is greatly propeller-twisted (~ 28°), because the DpC in the wide and spacious major groove draws the dC toward the peptide. As a result, two of the C-G hydrogen bonds are formed only in half of the simulations time (Figure S9), which explains the lower error-free extension product data (67.5 %). By contrast, in T7 RNA polymerase, the much narrower major groove confines the peptide so that the Watson-Crick hydrogen bonds between dC-GTP are well-aligned in the simulations.
An interesting question is why the C7-deaza-dG crosslink to 10mer-peptide is bypassed error-free, while the C5-dC 11mer-peptide manifests C-A+ mismatches to a significant extent in the T7 RNA polymerase. In particular, we wondered why the 10mer-dG did not form a wobble pair with UTP, which is a common mismatch in RNAs.39 Our results that show an unusually long O3' to Pα distance between the RNA primer terminus and CTP provides insight on this phenomenon (Table S3). For the error-free extension, the O3' to Pα distance needs to shorten to achieve a reaction-ready state. The 10mer-peptide held tightly in the narrow major groove, crowds the incoming CTP opposite to template dG, and forces it toward the minor groove, while maintaining distorted Watson-Crick hydrogen bonds. However, to form a G-U mismatched wobble pair, the UTP must be shifted toward the major groove, which is prevented by the bulk and constrained situation of the peptide (Figure S6C).
To our knowledge, this is the first report of the effects of C5-dC conjugated peptides on transcription, whereas we have previously investigated the behavior of DNA polymerases with these peptide lesions.15 The TLS DNA polymerase bypass study of C5-dC peptide conjugates also revealed a significant amount of C to T transitions, while at the same time it generated targeted −1 deletion mutations which were not observed in our in vitro transcription experiments.15 This is quite reasonable, because some of the Y family polymerases are known to slip along the DNA template, resulting in deletion mutations.40
Conclusions
Overall, our results indicate that the presence of DPC adducts on DNA significantly influences the efficiency and fidelity of in vitro transcription catalyzed by T7 RNA Pol. Furthermore, lesion size and the site of cross-linking within DNA are the important factors determining whether transcriptional bypass is accurate or error prone. Specifically, full size protein DPCs can only be bypassed if present at the nontranscribed strand. Intriguingly, C5-dC conjugated DpC lesions such as those formed at endogenously occurring 5-formyl-C residues of DNA induce more transcription errors than N7-G DpC adducts formed by common anticancer agents. However, Schiff base adducts between 5-formyl-dC and histone proteins are reversible, and it is possible that this equilibrium is tightly controlled in vivo in order to minimize transcriptional errors. Future studies are needed to examine the effects of DPCs and DpCs on transcription in living cells. One possibility is that the transcriptional blockage might serve as a signal to recruit DNA repair proteins to the stalled RNAP complex, initiating DPC repair.
METHODS
Synthesis and characterization of DNA oligodeoxynucleotides containing 5fC or 7-deaza-DHP-dG.
5fC-containing 47-mers (ODNs, 5'-CT CGA TAA GGA TCC GAT AGC GTX GAC ACT AGT CTC GCA CCA GGG CGC, where X= 5fC) were prepared using solid phase synthesis on an ABI 394 DNA synthesizer (Applied Biosystems, Foster City, CA). 5-Formyl-dC-III-CE phosphoramidite was obtained from Glen Research (Sterling, VA). Synthetic oligonucleotides were deprotected in 30 % ammonium hydroxide for 17 h at room temperature and then in 80 % acetic acid at 20 °C for 6 h. 47-mer oligonucleotides containing site-specific 7-deaza-DHP-dG (ODNs, 5'-CT CGA TAA GGA TCC GAT AGC GTC XAC ACT AGT CTC GCA CCA GGG CGC, where X= 7-deaza-DHP-dG) were prepared using standard solid phase synthesis on an ABI 394 DNA synthesizer (Applied Biosystems, Foster City, CA) as described previously.20, 41 Synthetic oligonucleotides were purified by 20 % denaturing PAGE, desalted by NAP-10 columns (GE Healthcare, Pittsburgh, PA), and characterized by mass spectrometry. Unmodified DNA oligodeoxynucleotides were purchased from IDT (Coralville, IA). Synthetic 47-mer oligonucleotides containing 5fC or 7-deaza-DHP-dG were phosphorylated and subsequently ligated into double-stranded 100-mer DNA duplexes (see Figure S1 for sequence details).
Synthesis and characterization of DNA oligonucleotides containing DNA-protein and DNA-peptide cross-links.
Site-specific DNA-protein cross-links conjugated to C5 position of dC or C7 position of 7-deaza-dG were prepared by reductive amination strategy developed in our laboratory.10, 19, 20 For C5-dC cross-links, double-stranded 100-mer DNA duplexes containing site-specific 5fC (see figure S1 for sequence, 30 pmol) were incubated with recombinant proteins (histone H2A, histone H4, AGT, or hOGG1, 10-fold molar excess) in 16 μL sodium phosphate buffer (4.5 mM, pH 7.4) for 3 h at 37 °C. To convert transient Schiff base conjugates to stable amine linkages, samples were treated with NaCNBH3 (4 μL of 100 mM solution) at 37 °C overnight (Figure 1A).
To generate DPCs conjugated to C7 of 7-deaza-dG, double-stranded 100-mer DNA duplexes containing 7-deaza-DHP-dG (30 pmol) were oxidized in the presence of 20 mM NaIO4 in sodium phosphate buffer (10 mM, pH 5.4, 40 μL) for 6 h at 4 °C in the dark to convert deaza-DHP-dG to 7-deaza-7-(2-oxoethyl)-dG.20 To generate DNA-protein conjugates, excess NaIO4 was quenched with Na2SO3, and the resulting aldehyde-containing DNA was incubated with the 10-fold molar excess of the proteins of interest in the presence of 10 mM NaCNBH3 at 37 °C overnight (Figure 1B). DPC reaction mixtures were heated at 90 °C for 15 min, cooled down to room temperature, and subsequently purified by 4–12% denaturing PAGE. Gel-purified DNA-protein conjugates were desalted on Micro Bio-spin columns (Bio-Rad, Hercules, CA). DNA duplexes conjugated to 31-mer and 57-mer peptides (Table 1C and D) were prepared analogously, except that 100–200 fold excess of the peptide was used. DNA duplexes conjugated to 10-mer and 11-mer peptides (Table 1A and B) were prepared by cross-linking short peptides to 47-mer oligonucleotides first and subsequently ligated into double-stranded 100-mer DNA duplexes. Briefly, gel-purified 47-mer oligo containing site-specific conjugated 10-mer and 11-mer peptides (75 pmol) were enzymatically ligated with a 53-mer fragment (100 pmol) using a 20-mer fragment (100 pmol) as scaffold. The resulting 100-mer DNA-peptide conjugates were gel purified and then annealed with the complementary stand (Figure S10).
In vitro transcription assays.
In vitro transcription bypass assays using T7RNAP were performed as described below. Briefly, 0.5 mM of ATP, GTP, CTP and 0.01 mM of UTP, 0.625 μM [α- 32P] UTP (800 Ci/mmol), 100 U of T7 RNAP, 40 U of RNAse inhibitor and 0.2 pmol of DNA template were mixed in 1× RNA Pol reaction buffer (40 mM Tris–HCl, pH 7.9, 6 mM MgCl2, 1 mM DTT and 2 mM spermidine) to a total volume of 20 μl. The reaction was initiated by the addition of T7 RNAP, and aliquots of the reaction mixtures (6 μL) were taken at pre-selected time points and quenched with a gel loading buffer (20 mM EDTA in 95 % formamide containing 0.05 % bromophenol blue and xylene cyanol). The quenched aliquots were heated at 90 °C for 10 min, loaded onto 20 % denaturing PAGE containing 7 M urea, run at 80 W for 1.5 h in 1× TBE buffer and visualized using Typhoon FLA 7000 phosphorimager (GH Healthcare, Pittsburgh, PA)
RT-PCR and LC-MS/MS based method to sequence transcription products
In vitro transcription bypass assays were performed for 1 h in the presence of 0.5 mM non-radiolabeled NTP mixtures as described above. The reaction mixtures were treated twice with Ambion DNA-free kit (Thermo Scientific, Watham, MA) to digest the DNA template and subsequently remove DNase and divalent cations, according to the manufacturer’s instructions. cDNA synthesis was performed using SuperScript II Reverse Transcriptase (Thermo Scientific, Watham, MA) with a primer 5'-CTCGATAAGGATCCGCTAGC-3'. A control experiment without the addition of reverse transcriptase was performed, and the products were examined by qPCR using SYBR Green Real-Time PCR Master Kit (Thermo Scientific, Watham, MA) to evaluate potential DNA contamination in isolated RNA samples (Figure S3A). Approximately, 2.5 % of the resulting cDNA were used as templates for RT-PCR amplification with 1.25 U of PfuTurbo DNA polymerase, 0.2 μM primers (5'-CTCGATAAGGATCCGCTAGC-3' and 5'-CGTCATCAACTGCGCCCTGG-3'), and 0.2 mM dNTPs in a total of 50 μL 1× buffer (Agilent Technologies, Inc., Wilmington, CA). PCR amplification started at 95 °C for 8 min, followed with 35 cycles at 95 °C for 15 secs, 62 °C for 15 secs and 72 °C for 1 min. PCR products were purified by QiaQuick Nucleotide Removal Kit (Qiagen, Valencia, CA) and eluted in EB buffer.
To analyze in vitro transcription products, PCR fragments obtained as described above were treated with 150 U of NheI, 150 U of SpeI and 30 U of Shrimp Alkaline Phosphatase in 300 μL of 1× CutSmart buffer (New England Biolabs, Beverly, MA) at 37 °C for 3 h. DNA was extracted with an equal volume of phenol:chloroform:isoamyl alcohol solution (25:24:1), precipitated at −80 °C overnight using 2.5 volumes of ethanol and 0.1 volumes of 3 M sodium acetate at pH 5.2, and dissolved in 12 μL of LCMS grade water. HPLC-ESI-MS/MS sequencing and quantification of transcription products was performed with a Dionex UltiMate 3000 HPLC coupled to a LTQ Orbitrap Velos mass spectrometer (Thermo Scientific, Watham, MA) as described previously.19, 42 Samples were loaded on a Zorbax 300SB-C18 column (150 × 0.5 mm, 5 μm, Agilent Technologies, Inc., Wilmington, CA) eluted at a flow rate of 15.0 μL/min using a gradient of 15 mM ammonium acetate in water (A) and acetonitrile (B). Solvent composition was linearly changed from 2 to 20 % B in 25 min. Orbitrap Velos mass spectrometry analyses were performed at a resolution of 60,000 and a scan range of m/z 300–2000. Relative quantification of transcription products was accomplished using HPLC-ESI-MS peak areas in extracted ion chromatograms. MS/MS fragmentation patterns were consistent with the CID fragments predicted using Mongo Oligo mass calculator version 2.06 (The RNA Institute, College of Arts and Sciences, State University of New York at Albany).
Molecular dynamics simulations
The tleap module of AmberTools1443 was used to construct the molecular topology and coordinate files for the initial models. The Amber ff14SB44 force field with the parmbsc145 nucleic acid parameters was used for modelling the protein and nucleic acids. A rectangular TIP3P46 water box with at least a 10 Å buffer was used to solvate the systems, which were then neutralized with sufficient numbers of Na+ ions. The salt concentration was brought to 0.15 M by the addition of Na+ and Cl− ions. The final systems contained 147 Na+ ions, 100 Cl− ions, and ~ 32,000 water molecules.
All simulations were performed with the CUDA-enabled graphics processing units (GPUs) version of pmemd47, 48 in the AMBER1649 program package. The cut off distances for van der Waals and short-range electrostatic interactions were set to the value of 9 Å. The long-range electrostatic interactions were treated with the particle-mesh Ewald (PME)50 method. The SHAKE algorithm was used to constrain the covalent bonds involving hydrogen atoms, with a time step of 2 fs for the equilibration and production run. Further details for the MD protocols are given in Supplementary Materials and Methods. We performed ~ 1 µs MD simulation for each polymerase system. In each simulation, the polymerase enzyme reached a stable state after ~ 200 ns MD simulation (Figure S7A) and the active site remained stable after ~ 250 ns (Figure S7B). Therefore, structural analyses were carried out based on the MD trajectories with the first 300 ns discarded, yielding a 700 ns production run for ensemble averaging. Within these ensembles, each polymerase domain has an average Ca RMSD of about 1−2 Å, indicating stable conformations throughout the simulations (Table S4).
Discovery Studio software v2.5.5 (Accelrys) was used to model the initial structures. PyMOL (The PyMOL Molecular Graphic System, version 1.3x Schrodinger, LLC) was used to construct molecular images and movies. The CPPTRAJ51 module of AMBER1443 and Curves+52 were used to post-process the simulations.
Supplementary Material
ACKNOWLEDGMENTS
We thank X. Ming and P. W. Villalta (University of Minnesota) for their help with mass spectrometry analysis, and R. Carlson (University of Minnesota) for his help with figure preparation.
FUNDING
National Institute of Environmental Health Sciences [R01 ES-023350 to N.T., R01 ES-025987 to S.B.]; National Cancer Institute [R01 CA-075449 to S.B.]; Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation [MCB060037 to S.B.]; Wayland E. Noland Graduate Student Fellowship and Doctoral Dissertation Fellowship at the University of Minnesota to S.J. (in part).
Footnotes
SUPPORTING INFORMATION AVAILABLE
This material is available free of charge via the Internet. Supplementary Materials and Methods (MD parameters for non-standard residues, structural stability, structural clustering, major groove width); Supplementary Tables; Supplementary Figures; Supplementary Movie.
REFERENCES
- 1.Tretyakova NY, Groehler A 4th, and Ji S (2015) DNA-Protein cross-links: Formation, structural identities, and biological outcomes, Acc. Chem. Res 48, 1631–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barker S, Weinfeld M, and Murray D (2005) DNA-protein crosslinks: their induction, repair, and biological consequences, Mutat. Res 589, 111–135. [DOI] [PubMed] [Google Scholar]
- 3.Ide H, Shoulkamy MI, Nakano T, Miyamoto-Matsubara M, and Salem AM (2011) Repair and biochemical effects of DNA-protein crosslinks, Mutat. Res 711, 113–122. [DOI] [PubMed] [Google Scholar]
- 4.Barker S, Weinfeld M, Zheng J, Li L, and Murray D (2005) Identification of mammalian proteins cross-linked to DNA by ionizing radiation, J. Biol. Chem 280, 33826–33838. [DOI] [PubMed] [Google Scholar]
- 5.Loeber RL, Michaelson-Richie ED, Codreanu SG, Liebler DC, Campbell CR, and Tretyakova NY (2009) Proteomic analysis of DNA-protein cross-linking by antitumor nitrogen mustards, Chem. Res. Toxicol 22, 1151–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Groehler A 4th, Villalta PW, Campbell C, and Tretyakova N (2016) Covalent DNA-protein cross-linking by phosphoramide mustard and nornitrogen mustard in human cells, Chem. Res. Toxicol 29, 190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michaelson-Richie ED, Ming X, Codreanu SG, Loeber RL, Liebler DC, Campbell C, and Tretyakova NY (2011) Mechlorethamine-induced DNA–protein cross-linking in human fibrosarcoma (HT1080) cells, J. Proteome Res 10, 2785–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu F-Y, Lee Y-J, Chen D-R, and Kuo H-W (2002) Association of DNA-protein crosslinks and breast cancer, Mutat. Res 501, 69–78. [DOI] [PubMed] [Google Scholar]
- 9.Izzotti A, Cartiglia C, Taningher M, De Flora S, and Balansky R (1999) Age-related increases of 8-hydroxy-2'-deoxyguanosine and DNA–protein crosslinks in mouse organs, Mutat. Res 446, 215–223. [DOI] [PubMed] [Google Scholar]
- 10.Ji S, Shao H, Han Q, Seiler CL, and Tretyakova NY (2017) Reversible DNA-protein cross-linking at epigenetic DNA marks, Angew. Chem. Int. Ed. Engl 56, 14130–14134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu L, Chen YC, Nakajima S, Chong J, Wang L, Lan L, Zhang C, and Wang D (2014) A Chemical Probe Targets DNA 5-Formylcytosine Sites and Inhibits TDG Excision, Polymerases Bypass, and Gene Expression, Chem. Sci 5, 567–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kellinger MW, Song CX, Chong J, Lu XY, He C, and Wang D (2012) 5-formylcytosine and 5-carboxylcytosine reduce the rate and substrate specificity of RNA polymerase II transcription, Nat. Struct. Mol. Biol 19, 831–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.You C, Ji D, Dai X, and Wang Y (2014) Effects of Tet-mediated oxidation products of 5-methylcytosine on DNA transcription in vitro and in mammalian cells, Sci. Rep 4, 7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li F, Zhang Y, Bai J, Greenberg MM, Xi Z, and Zhou C (2017) 5-Formylcytosine yields DNA–protein cross-links in nucleosome core particles, J. Am. Chem. Soc 139, 10617–10620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ji S, Fu I, Naldiga S, Shao H, Basu AK, Broyde S, and Tretyakova NY (2018) 5-Formylcytosine mediated DNA–protein cross-links block DNA replication and induce mutations in human cells, Nucleic Acids Res 46, 6455–6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naldiga S, Ji S, Thomforde J, Nicolae CM, Lee M, Zhang Z, Moldovan GL, Tretyakova NY, and Basu AK (2019) Error-prone replication of a 5-formylcytosine-mediated DNA-peptide cross-link in human cells, J. Biol. Chem 294, 10619–10627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Michaelson-Richie ED, Loeber RL, Codreanu SG, Ming X, Liebler DC, Campbell C, and Tretyakova NY (2010) DNA−Protein cross-linking by 1,2,3,4-diepoxybutane, J. Proteome Res 9, 4356–4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pande P, Ji S, Mukherjee S, Scharer OD, Tretyakova NY, and Basu AK (2017) Mutagenicity of a model DNA-peptide cross-link in human cells: roles of translesion synthesis DNA polymerases, Chem. Res. Toxicol 30, 669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wickramaratne S, Ji S, Mukherjee S, Su Y, Pence MG, Lior-Hoffmann L, Fu I, Broyde S, Guengerich FP, Distefano M, Scharer OD, Sham YY, and Tretyakova N (2016) Bypass of DNA-protein cross-links conjugated to the 7-deazaguanine position of DNA by translesion synthesis polymerases, J. Biol. Chem 291, 23589–23603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wickramaratne S, Mukherjee S, Villalta PW, Scharer OD, and Tretyakova NY (2013) Synthesis of sequence-specific DNA-protein conjugates via a reductive amination strategy, Bioconjug. Chem 24, 1496–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duxin JP, Dewar JM, Yardimci H, and Walter JC (2014) Repair of a DNA-protein crosslink by replication-coupled proteolysis, Cell 159, 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stingele J, Schwarz MS, Bloemeke N, Wolf PG, and Jentsch S (2014) A DNA-dependent protease involved in DNA-protein crosslink repair, Cell 158, 327–338. [DOI] [PubMed] [Google Scholar]
- 23.Vaz B, Popovic M, Newman JA, Fielden J, Aitkenhead H, Halder S, Singh AN, Vendrell I, Fischer R, Torrecilla I, Drobnitzky N, Freire R, Amor DJ, Lockhart PJ, Kessler BM, McKenna GW, Gileadi O, and Ramadan K (2016) Metalloprotease SPRTN/DVC1 orchestrates replication-coupled DNA-protein crosslink repair, Mol. Cell 64, 704–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.You C, Wang P, Dai X, and Wang Y (2014) Transcriptional bypass of regioisomeric ethylated thymidine lesions by T7 RNA polymerase and human RNA polymerase II, Nucleic Acids Res 42, 13706–13713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Remington KM, Bennett SE, Harris CM, Harris TM, and Bebenek K (1998) Highly mutagenic bypass synthesis by T7 RNA polymerase of site-specific benzo[a]pyrene diol epoxide-adducted template DNA, J. Biol. Chem 273, 13170–13176. [DOI] [PubMed] [Google Scholar]
- 26.Tornaletti S, Patrick SM, Turchi JJ, and Hanawalt PC (2003) Behavior of T7 RNA polymerase and mammalian RNA polymerase II at site-specific cisplatin adducts in the template DNA, J. Biol. Chem 278, 35791–35797. [DOI] [PubMed] [Google Scholar]
- 27.Kalogeraki VS, Tornaletti S, and Hanawalt PC (2003) Transcription arrest at a lesion in the transcribed DNA strand in vitro is not affected by a nearby lesion in the opposite strand, J. Biol. Chem 278, 19558–19564. [DOI] [PubMed] [Google Scholar]
- 28.Cline SD, Riggins JN, Tornaletti S, Marnett LJ, and Hanawalt PC (2004) Malondialdehyde adducts in DNA arrest transcription by T7 RNA polymerase and mammalian RNA polymerase II, Proc. Natl. Acad. Sci. U. S. A 101, 7275–7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakano T, Ouchi R, Kawazoe J, Pack SP, Makino K, and Ide H (2012) T7 RNA polymerases backed up by covalently trapped proteins catalyze highly error prone transcription, J. Biol. Chem 287, 6562–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oakley JL, and Coleman JE (1977) Structure of a promoter for T7 RNA polymerase, Proc. Natl. Acad. Sci. U. S. A 74, 4266–4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaz B, Popovic M, and Ramadan K (2017) DNA–Protein crosslink proteolysis repair, Trends Biochem. Sci 42, 483–495. [DOI] [PubMed] [Google Scholar]
- 32.Chesner LN, and Campbell C (2018) A quantitative PCR-based assay reveals that nucleotide excision repair plays a predominant role in the removal of DNA-protein crosslinks from plasmids transfected into mammalian cells, DNA Repair (Amst) 62, 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, and Zhang Y (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine, Science 333, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pfaffeneder T, Hackner B, Truss M, Munzel M, Muller M, Deiml CA, Hagemeier C, and Carell T (2011) The discovery of 5-formylcytosine in embryonic stem cell DNA, Angew. Chem. Int. Ed. Engl 50, 7008–7012. [DOI] [PubMed] [Google Scholar]
- 35.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, and Bourne PE (2000) The Protein Data Bank, Nucleic Acids Res 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin YW, and Steitz TA (2004) The structural mechanism of translocation and helicase activity in T7 RNA polymerase, Cell 116, 393–404. [DOI] [PubMed] [Google Scholar]
- 37.Fiser A, and Sali A (2003) Modeller: generation and refinement of homology-based protein structure models, Methods Enzymol 374, 461–491. [DOI] [PubMed] [Google Scholar]
- 38.Batra VK, Beard WA, Shock DD, Krahn JM, Pedersen LC, and Wilson SH (2006) Magnesium-induced assembly of a complete DNA polymerase catalytic complex, Structure 14, 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Varani G, and McClain WH (2000) The G x U wobble base pair. A fundamental building block of RNA structure crucial to RNA function in diverse biological systems, EMBO Rep 1, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ling H, Boudsocq F, Woodgate R, and Yang W (2001) Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication, Cell 107, 91–102. [DOI] [PubMed] [Google Scholar]
- 41.Angelov T, Guainazzi A, and Scharer OD (2009) Generation of DNA interstrand cross-links by post-synthetic reductive amination, Org. Lett 11, 661–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wickramaratne S, Boldry EJ, Buehler C, Wang YC, Distefano MD, and Tretyakova NY (2015) Error-prone translesion synthesis past DNA-peptide cross-links conjugated to the major groove of DNA via C5 of thymidine, J. Biol. Chem 290, 775–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Case DA, Darden TA, Cheatham TE 3rd, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Wang B, Hayik S, Roitberg A, Seabra G, Kolossváry I, Wong KF, Paesani F, Vanicek J, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh MJ, Cui G, Roe DR, Mathews DH, Seetin MG, Sagui C, Babin V, Gusarov S, Kovalenko A, and Kollman PA (2014) AMBER 14, University of California, San Francisco. [Google Scholar]
- 44.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, and Simmerling C (2015) ff14SB: Improving the cccuracy of protein side chain and backbone parameters from ff99SB, J. Chem. Theory Comput 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ivani I, Dans PD, Noy A, Perez A, Faustino I, Hospital A, Walther J, Andrio P, Goni R, Balaceanu A, Portella G, Battistini F, Gelpi JL, Gonzalez C, Vendruscolo M, Laughton CA, Harris SA, Case DA, and Orozco M (2016) Parmbsc1: a refined force field for DNA simulations, Nat. Methods 13, 55–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jorgensen WL, Chandreskhar J, Madura JD, Imprey RW, and Klein ML (1983) Comparison of simple potential functions for simulating liquid water, J. Chem. Phys 79, 926–935. [Google Scholar]
- 47.Salomon-Ferrer R, Gotz AW, Poole D, Le Grand S, and Walker RC (2013) Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald, J. Chem. Theory Comput 9, 3878–3888. [DOI] [PubMed] [Google Scholar]
- 48.Gotz AW, Williamson MJ, Xu D, Poole D, Le Grand S, and Walker RC (2012) Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized Born, J. Chem. Theory Comput 8, 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Case DA, R. M. B., Cerutti DS, Cheatham TE III, Darden TA, Duke RE, Giese TJ, Gohlke H,, Goetz AW, N. H., Izadi S, Janowski P, Kaus J, Kovalenko A, Lee TS, LeGrand S, Li P, Lin C, T. L., Luo R, Madej B, Mermelstein D, Merz KM, Monard G, Nguyen H, Nguyen HT, Omelyan I, A. O., Roe DR, Roitberg A, Sagui C, Simmerling CL, Botello-Smith WM, Swails J,, and Walker RC, J. W., Wolf RM, Wu X, Xiao L and Kollman PA (2016) AMBER 2016, University of California, San Francisco. [Google Scholar]
- 50.Darden T, York D, and Pedersen L (1993) Particle mesh Ewald: an N log(N) method for Ewald sums in large systems, J. Chem. Phys 98, 10089–10092. [Google Scholar]
- 51.Roe DR, and Cheatham TE (2013) PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data, J. Chem. Theory Comput 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- 52.Lavery R, Moakher M, Maddocks JH, Petkeviciute D, and Zakrzewska K (2009) Conformational analysis of nucleic acids revisited: Curves+, Nucleic Acids Res 37, 5917–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.