

SUMMARY
Bacteria sense and respond to environmental changes via several broad categories of sensory signal transduction systems. Recently, we described the key features of a previously unrecognized, but widely conserved class of prokaryotic sensory system that we refer to as the LytTR Regulatory System (LRS). Our previous studies suggest that most, if not all, prokaryotic LRS membrane proteins serve as inhibitors of their cognate transcription regulators, but the inhibitory mechanisms employed have thus far remained a mystery. Using the Streptococcus mutans HdrRM LRS as a model, we demonstrate how the LRS membrane protein HdrM inhibits its cognate transcription regulator HdrR by tightly sequestering HdrR in a membrane localized heteromeric HdrR/M complex. Membrane sequestration of HdrR prevents the positive feedback autoregulatory function of HdrR, thereby maintaining a low basal expression of the hdrRM operon. However, this mechanism can be antagonized by ectopically expressing a competitive inhibitor mutant form of HdrR that lacks its DNA binding ability while still retaining its HdrM interaction. Our results indicate that sequestration of HdrR is likely to be the only mechanism required to inhibit its transcription regulator function, suggesting that endogenous activation of the HdrRM LRS is probably achieved through a modulation of the HdrR/M interaction.
Keywords: LytTR Regulatory System, signal transduction, sequestration, Streptococcus mutans, ECF sigma factor, Two-component signal transduction system
INTRODUCTION
The capacity of bacteria to sense and respond to changes in the extracellular environment is fundamental for survival, particularly within highly dynamic and/or competitive niches. The adaptation mechanisms used to respond to such changes can be broadly grouped into several basic categories: one-component systems (Ulrich et al., 2005), phosphosignaling systems (Burnside & Rajagopal, 2012, Cho et al., 2001, Dworkin, 2015, Gao & Stock, 2009, Wright & Ulijasz, 2014), extracytoplasmic function (ECF) sigma factor systems (Mascher, 2013, Staron et al., 2009), and second messenger systems (Corrigan & Grundling, 2013, Hauryliuk et al., 2015, Hengge et al., 2016, Romling et al., 2013). Recently, we identified and described another class of widely distributed prokaryotic sensory system that is distinct from each of these, which we refer to as the LytTR Regulatory System (LRS). LRS are two-protein sensory systems that are encoded within operons with the first gene encoding a transcription regulator from the LytTR Family (Nikolskaya & Galperin, 2002) and the second encoding a transmembrane protein inhibitor of the LRS regulator. Most LRS operons are simply two-gene operons specifically encoding the LRS, but a minority also encode additional proteins, such as ABC transporters (Zou et al., 2018). While nearly all LRS currently remain uncharacterized, we recently identified several features of LRS that are highly conserved, even between distantly related organisms. 1) LRS exist in a basal “off” state. Since LRS membrane proteins serve as inhibitors of their cognate transcription regulators, it is possible to constitutively activate an LRS simply by mutating its inhibitory membrane protein. This is analogous to ECF sigma factor systems, which can be similarly activated by mutating their cognate anti-sigma proteins (Ho & Ellermeier, 2012). 2) LRS inhibitory membrane proteins do not contain obvious enzymatic domains (Zou et al., 2018), such as histidine kinase domains, which are characteristic of two-component signal transduction system (TCSTS) sensor proteins (Gao & Stock, 2009). LRS transcription regulators also lack the signal receiver domains and conserved aspartate residues required for the phosphorylation of TCSTS response regulators (Bourret, 2010, Cho et al., 2001, Zou et al., 2018). Thus, there is currently no evidence to suggest that LRS function through phosphorylation or other common posttranslational modifications. Interestingly, the vast majority of putative LRS membrane proteins that have been identified to date contain either of two Domains of Unknown Function (DUF2154 or DUF3021) (Zou et al., 2018). However, there are no common phenotypes yet associated with these DUF that might implicate a mechanism of LRS membrane protein inhibition toward their cognate transcription regulators. 3) LRS exhibit positive feedback autoregulation via direct repeat sequences located immediately upstream of the LRS operon promoters. These repeats serve as the binding sites for cognate LRS transcription regulators, which function in transcription activation (Zou et al., 2018). In fact, all other characterized regulators from the larger LytTR Family of transcription regulators similarly serve as transcription activators (Nikolskaya & Galperin, 2002). Accordingly, expression of S. mutans LRS operons can be strongly activated by mutating the respective LRS membrane protein-encoding genes and this autoregulation is critically dependent upon the presence of operon direct repeats (Zou et al., 2018).
Despite the broad distribution of putative LRS operons among bacteria and some archaea, characterized LRS are thus far limited to those of S. mutans (Merritt & Qi, 2012, Merritt et al., 2007, Okinaga et al., 2010a, Okinaga et al., 2010b, Xie et al., 2010). Recently, we screened the genome sequences of multiple S. mutans strains and identified five LRS operons that are likely components of the core S. mutans genome (SMU_RS01460/01465, SMU_RS02105/02110, SMU_RS04925/04930, hdrRM, and brsRM) (Zou et al., 2018). Of these, the HdrRM and BrsRM LRS are the most thoroughly characterized and have largely overlapping regulons that include both bacteriocin and natural competence genes. The HdrRM and BrsRM LRS are coregulatory at the transcriptional level as well (Merritt et al., 2007, Okinaga et al., 2010a, Okinaga et al., 2010b, Xie et al., 2010, Zou et al., 2018). In fact, most of the S. mutans LRS exhibit transcriptional regulation of one or more additional LRS operons (Zou et al., 2018). Currently, there is only a limited understanding of LRS activation and it still is unclear which signals or stimuli directly modulate the function of LRS. The hdrRM and brsRM LRS operons are both induced by a rapid switch to high cell density growth conditions (Merritt et al., 2007), while activation of the BrsRM LRS is intimately connected with purine metabolism (Zou et al., 2018). The HdrRM and BrsRM LRS also jointly control a cell death pathway that is mediated by their coregulatory ability (Xie et al., 2010), but the mechanism of lethality is still under active investigation. While we have a detailed knowledge of the regulons and phenotypes controlled by some S. mutans LRS, it is completely unknown how LRS membrane proteins antagonize the function of their cognate LRS transcription regulators. Consequently, in the current study, we employed the HdrRM LRS as a model to decipher the first LRS membrane protein inhibitory mechanism.
RESULTS
HdrR subcellular localization is mediated by HdrM
Our previous studies indicated that HdrM is a critical posttranslational regulator preventing HdrR from engaging the autoregulatory positive feedback loop responsible for hdrRM operon induction (Merritt et al., 2007, Okinaga et al., 2010a, Zou et al., 2018). The lack of any discernable enzymatic domains in HdrM or any other LRS membrane proteins provides no obvious clues to implicate posttranslational modification as a strategy to regulate cognate LRS transcription regulators. However, secondary structure predictions of HdrM suggest that in addition to its three transmembrane segments, the protein also contains a prominent cytoplasmic segment (Zou et al., 2018), implying that protein-protein interactions might somehow control HdrR activity. Thus, we tested three hypothetical scenarios to explain the inhibitory mechanism mediated by HdrM: 1) activation of HdrR proteolysis, 2) sequestration of HdrR, and/or 3) posttranslational modification of HdrR. To test the first scenario, we examined HdrR abundance using the wild-type, hdrM mutant, and a mutant strain separately overexpressing both HdrR and HdrM. HdrM antagonism of HdrR autoregulation was found to be the only mechanism dictating HdrR abundance, as HdrR protein levels were unaffected by HdrM when the expression of both hdrR and hdrM were uncoupled from operon autoregulation (data not shown). Therefore, there was no evidence to suggest regulated proteolysis. For the HdrR sequestration mechanism proposed in the second scenario, we reasoned that such a mechanism would likely require a highly stable interaction between HdrM and HdrR to minimize positive feedback activation of the hdrRM operon. Thus, we repeated the ectopic expression of both HdrR and HdrM and then examined their localization via western blots of both cytoplasmic and membrane fractions. Consistent with expectations, HdrM was found exclusively localized in the membrane fraction (Fig. 1A). For HdrR, nearly all of the detectable protein was confined to the membrane fraction when HdrM was present. However, its localization switched primarily to the cytoplasm in the absence of HdrM (Fig. 1B). Also consistent with our previous results, we found no evidence to suggest that separate ectopic expression of HdrM can trigger the proteolytic degradation of HdrR, as HdrR did not exhibit any obvious reduction in its abundance due to HdrM expression (compare WT vs. ΔM in the unfractionated cell lysate) (Fig. 1B). This further argues against the possibility of an HdrM-mediated proteolytic degradation mechanism targeting HdrR. As an independent confirmation of the HdrRM fractionation results, we created GFP and mCherry fusion proteins of both HdrM and HdrR and then performed in situ imaging to examine their localization. Given the small size of S. mutans cells (≤0.5 μM), it was necessary to perform deconvolution microscopy to discern the subcellular localization of the fluorescent signals. In agreement with the western blot results, it was evident that HdrM fluorescence was primarily localized around the periphery of the cells, as would be expected for a membrane protein (Fig. 1C). Furthermore, HdrM localization switched to the cytoplasm following deletion of its putative transmembrane segments (Fig. 1D). A similar membrane-localized fluorescence pattern could also be observed for HdrR when HdrM was present in the cells (Fig. 1E), whereas HdrR fluorescence was exclusive to the interior of the cells in the absence of HdrM (Fig. 1F). These results all indicated that HdrR membrane localization is critically dependent upon HdrM.
Figure 1. HdrM mediates HdrR membrane localization.
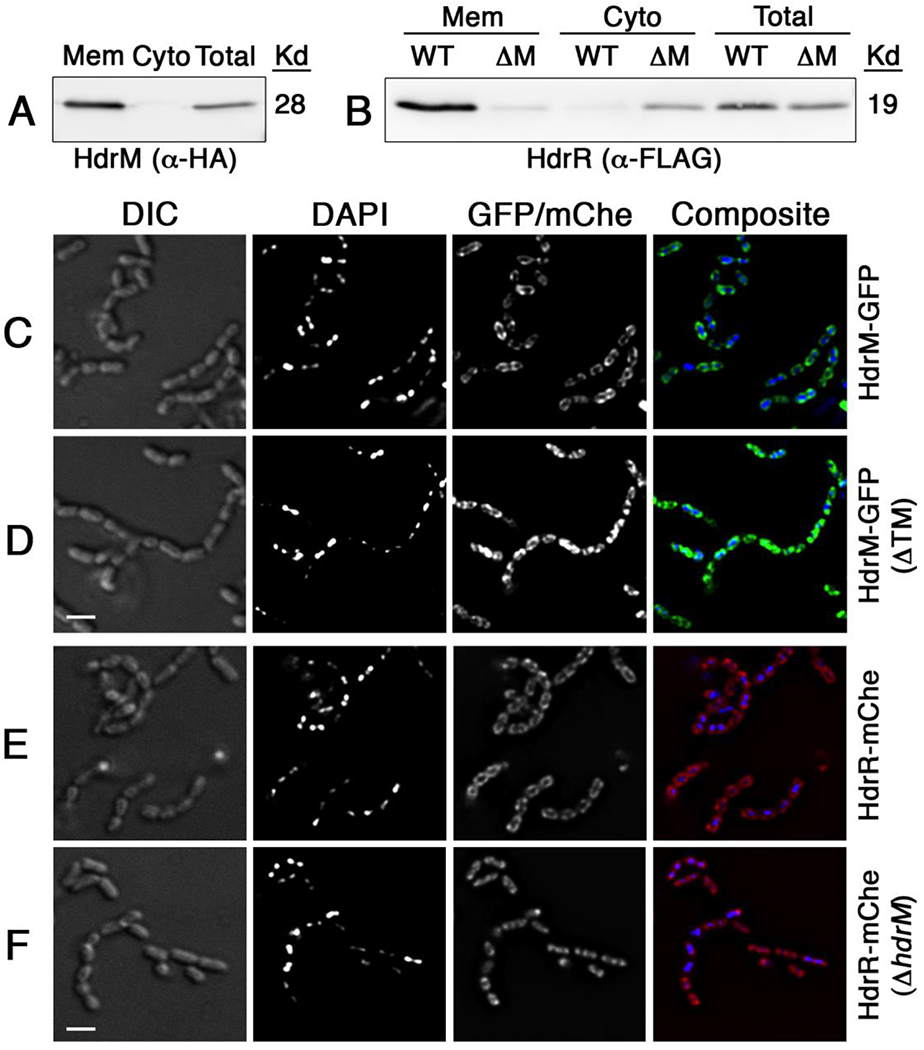
A) The localization of HdrM was detected via α-HA epitope western blots following separate ectopic expression of epitope tagged HdrM-HA and HdrR-FLAG. Several protein fractions were analyzed: membrane (Mem), cytoplasmic (Cyto), and unfractionated (Total). B) The localization of epitope tagged HdrR-FLAG was similarly analyzed via α-FLAG epitope western blots in both the presence of epitope tagged HdrM-HA (WT) and its absence (ΔM). For panels C – F, HdrM and HdrR localization was assessed by creating protein fusions to the green fluorescent protein (GFP) and mCherry (mChe), respectively. Images were acquired using a DeltaVision image restoration microscope with both light microscopy (DIC) and epifluorescence (DAPI and GFP/mChe). Scalebars indicate 1 μM. C) Full length HdrM with a C-terminal fusion to GFP (HdrM-GFP); D) Truncated HdrM lacking its transmembrane segments with a C-terminal fusion to GFP (HdrM-GFP ΔTM); E) HdrR with a C-terminal fusion to mCherry (HdrR-mChe); and F) HdrR with a C-terminal fusion to mCherry in a ΔhdrM background (HdrR-mChe ΔhdrM).
HdrR and HdrM form stable heteromeric protein complexes
We were next interested to determine whether HdrR membrane localization could be attributed to the formation of HdrRM heteromeric protein complexes or via some other indirect mechanism. We performed HdrRM coimmunoprecipitation upon purified membrane fractions, and as shown in Figures 2A and B, HdrR immunoprecipitation could also coimmunoprecipitate HdrM and vice-versa, indicating that the proteins are indeed components of a stable complex. As an independent verification of these results, we also employed yellow fluorescent protein (YFP) bimolecular fluorescence complementation (BiFC) (Hu et al., 2002) to test whether HdrRM complex formation is detectable in situ. In agreement with the coimmunoprecipitation results, yellow fluorescence was only detectable when the YFP fragments were expressed simultaneously as fusions to both HdrR and HdrM (Fig. 2C), which indicates that both proteins stably exist in immediate proximity to each other in situ. Next, we were curious whether the HdrRM complex is likely formed via binary HdrR/M interactions or is dependent upon interactions with an unidentified adaptor protein. To test this, we expressed both hdrR and hdrM in E. coli and performed coimmunoprecipitation. Since E. coli is both distantly related to S. mutans and lacks hdrRM orthologs, we reasoned it would be extremely unlikely to observe HdrRM complex formation in E. coli if an adaptor protein was essential for its assembly. Furthermore, to increase the stringency of coimmunoprecipitation, we performed tandem affinity purification using a dual HA+FLAG epitope tagged HdrR to coimmunoprecipitate an HdrM-GFP fusion protein. As shown in Figure 2D, HdrRM heteromeric complexes were indeed readily detectable in E. coli, suggesting these interactions are both highly avid and likely to form spontaneously without requiring additional components. Based upon the protein localization and interaction data, we conclude that HdrM is highly likely to directly localize HdrR to the cell membrane via the formation of stable heteromeric HdrRM complexes.
Figure 2. HdrR and HdrM form stable heteromeric complexes.
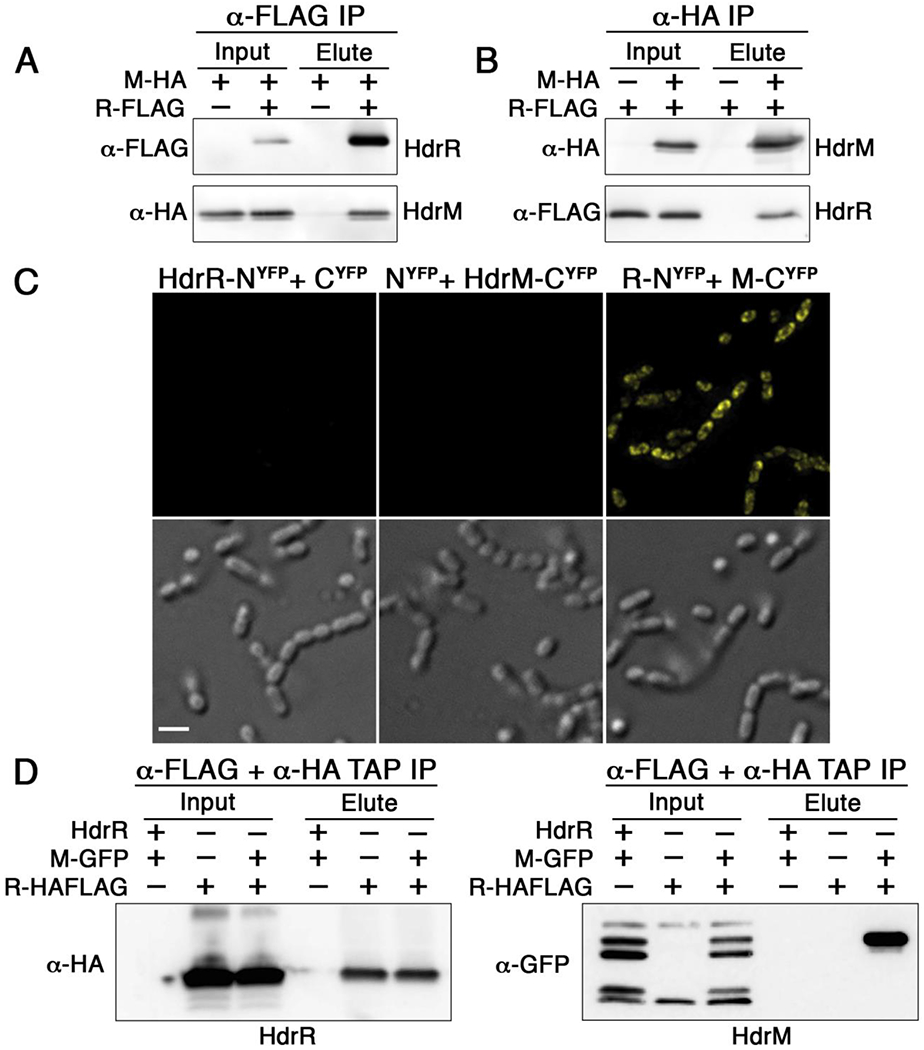
A) Coimmunoprecipitation was used to detect heteromeric complex formation between HA epitope tagged HdrM (M-HA) and FLAG epitope tagged HdrR (R-FLAG). Samples were immunopurified with α-FLAG affinity resin and the results are shown using both α-FLAG and α-HA western blots. Elution samples were loaded in the same order as the input samples. B) The same HdrM-HA and HdrR-FLAG epitope tagged proteins were immunopurified with α-HA affinity resin. Elution samples were loaded in the same order as the input samples. C) Bimolecular fluorescence complementation (BiFC) with the yellow fluorescent protein (YFP) was used to detect the association of HdrR and HdrM in situ. Fluorescent images are shown above their corresponding light microscopy images. In the left panels, the N-terminal fragment of YFP was fused to HdrR (HdrR-NYFP), while the C-terminal YFP fragment was simultaneously expressed as an unfused protein (CYFP). In the middle panels, the N-terminal fragment of YFP was expressed as an unfused protein (NYFP), while the C-terminal YFP fragment was simultaneously expressed as a fusion to HdrM (HdrM-CYFP). In the right panels, the N-terminal fragment of YFP was expressed as fusion to HdrR (R-NYFP), while the C-terminal fragment of YFP was simultaneously expressed as a fusion to HdrM (M-CYFP). Images were captured using identical exposure times and camera settings. Scalebar indicates 1 μM. D) Tandem affinity coimmunoprecipitation in E. coli was used to detect heteromeric complex formation between heterologously expressed HdrR/M. A dual HA+FLAG epitope was fused to the C-terminus of HdrR to facilitate successive tandem affinity immunopurification of E. coli lysates using α-FLAG affinity resin immediately followed by a second immunopurification with α-HA affinity resin. Afterward, tandem affinity purified eluates were analyzed for the presence of HdrR/M via α-HA (HdrR-HA+FLAG) and α-GFP (HdrM-GFP) western blots. E. coli strains heterologously expressed different combinations of the following proteins: unmodified wild-type HdrR (HdrR), HdrM-GFP fusion (M-GFP), and dual epitope tagged HdrR (R-HAFLAG). Elution samples were loaded in the same order as the input samples.
HdrM inhibits HdrR via membrane sequestration
The strict membrane localization of HdrRM heteromeric complexes is consistent with a membrane sequestration inhibitory mechanism. If such a mechanism was truly employed, we hypothesized it should be possible to artificially activate the HdrRM LRS by overexpressing a DNA-binding defective mutant form of HdrR to serve as a competitive inhibitor of the wild-type HdrR/M interaction. To test this, we first performed alanine-scanning mutagenesis to identify HdrR mutants with transcription activator defects. Blocks of six consecutive alanine residues were substituted sequentially throughout the entire length of HdrR for a total of 22 separate HdrR mutants (referred to as Ala1 – Ala22) (Fig. 3A). Each of these mutants was ectopically expressed and tested for its ability to activate a transcription fusion reporter strain harboring a luciferase gene replacement of the hdrRM open reading frames (ORFs) (Fig. 3B). Most of the alanine mutants were found to exhibit major defects in reporter activation with the majority losing their activity entirely (Fig. 3C). Next, each of the activation-defective HdrR mutant proteins was ectopically expressed in a wild-type hdrRM luciferase reporter strain to determine whether any might have retained their HdrM interaction abilities. Of these, expression of the Ala5 and Ala6 mutants was able to trigger >25-fold increased luciferase activity from the reporter, even though both alanine mutants were similarly incapable of directly inducing luciferase activity (Figs. 3C and 4A). This suggested that the HdrR Ala5 and Ala6 mutants both retained their HdrM interaction abilities and were able to displace wild-type HdrR from HdrRM heteromeric complexes, freeing it to activate expression of the reporter. As a final confirmation of the expected Ala5 and Ala6 mutant phenotypes, we directly assayed their DNA binding and HdrM interaction abilities. While both mutants were severely impaired in DNA binding, they still retained their HdrM interaction abilities like that of the wild-type HdrR (Figs. 4B–D). Thus, the results indicate that membrane sequestration of HdrR is sufficient to prevent it from activating gene expression.
Fig. 3. Creation of mutant HdrR proteins with defects in transcription activation.
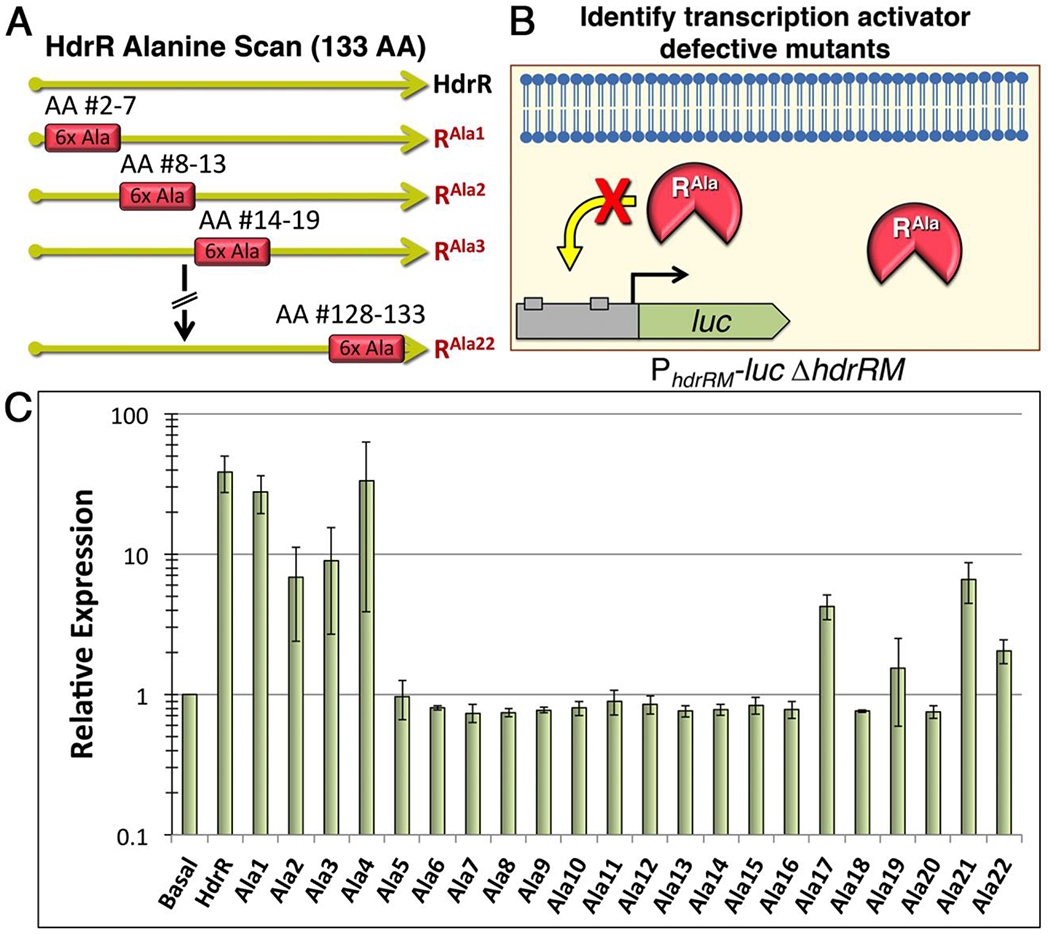
A) Illustration of the alanine scanning mutagenesis scheme. Blocks of 6 consecutive alanine mutations were engineered throughout the entire length of HdrR for a total of 22 separate mutants. The mutants were named sequentially starting with Ala1, which contained alanine substitutions in amino acids #2 – 7. B) Illustration of the luciferase screening procedure to identify mutant proteins defective in hdrRM operon transcription activation. C) Each of the 22 alanine mutant hdrR genes was ectopically expressed to assess its ability to activate gene expression from a reporter strain containing a luciferase ORF replacement of the hdrRM ORFs. Data are presented relative to the parent reporter strain (Basal), which was arbitrarily assigned a value of 1. The results represent the means of three independent experiments ± SD.
Figure 4. HdrM antagonizes HdrR via membrane sequestration.
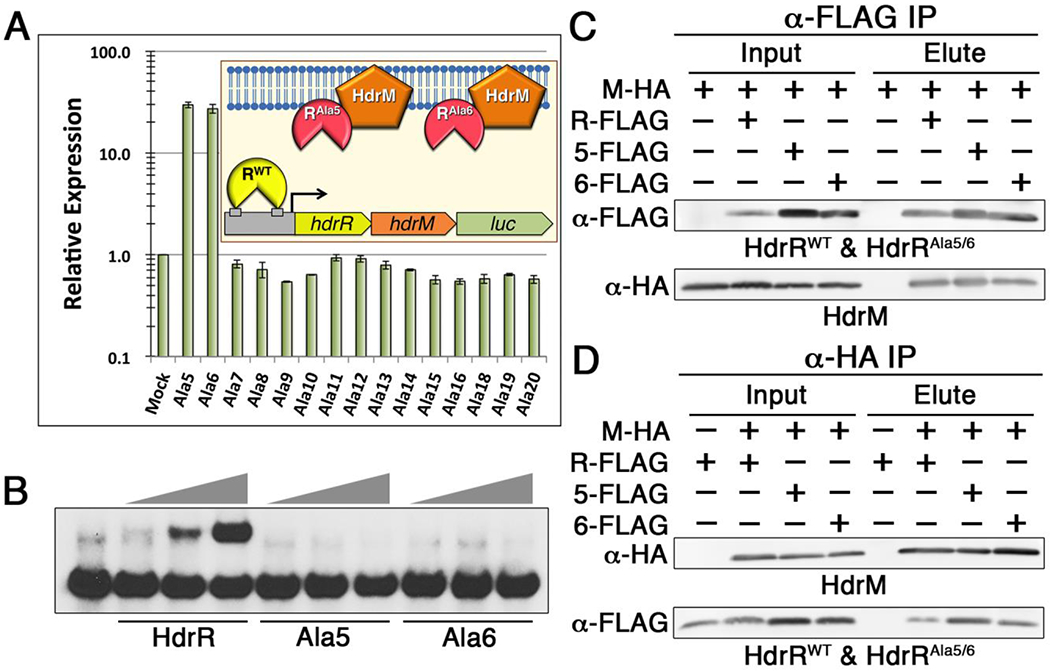
A) A series of DNA binding-defective alanine mutants of HdrR was ectopically expressed to identify mutant proteins capable of activating an hdrRM luciferase reporter. As illustrated in the graph inset, activation of the reporter strain is indicative of competitive inhibition of the wild-type HdrR/M interaction. Based upon the greatly increased luciferase activity triggered by ectopic expression of the Ala5 and Ala6 mutants, both were identified as candidate competitive inhibitors. Data are presented relative to the parent reporter strain (Mock), which was arbitrarily assigned a value of 1. The presented results represent the means of three independent experiments ± SD. B) EMSA of the hdrRM operon promoter region was used to compare the DNA binding affinities of the wild-type HdrR vs. that of the Ala5 and Ala6 HdrR mutants. All EMSA reactions contained a range of 10 – 30 μg of protein incubated with 1 ng of labeled hdrRM promoter region DNA probe. C) Coimmunoprecipitation was used to detect heteromeric complex formation between HA epitope tagged HdrM (M-HA) and FLAG epitope tagged HdrR (R-FLAG), the HdrR Ala5 mutant (5-FLAG), and the HdrR Ala6 mutant (6-FLAG). Samples were immunopurified with α-FLAG affinity resin and the results are shown using both α-FLAG and α-HA western blots. Elution samples were loaded in the same order as the input samples. D) The same epitope tagged proteins were immunopurified with α-HA affinity resin. Elution samples were loaded in the same order as the input samples.
HdrR does not require post-translational modifications for functionality
While the evidence provided strong support for a sequestration mechanism of HdrR antagonism, we were curious whether posttranslational modifications of HdrR might regulate its interaction with HdrM and/or control its DNA binding ability. Therefore, to test the third scenario of HdrR inhibition via posttranslational modification, we purified HdrR directly from S. mutans strains either encoding HdrM (i.e. inactive HdrR) or lacking HdrM (i.e. active HdrR) and compared their molecular masses using mass spectrometry. With hdrM present, the experimentally determined molecular weight of HdrR was 19090.15 Da, while it was 19089.69 Da in the hdrM deletion background (Figs. 5A and B). Since the two proteins exhibited a mass differential of only 0.46 Da between them and both are <4 Da from the predicted HdrR molecular weight (19086.6 Da), it is highly unlikely that posttranslational modifications modulate the ability of HdrR to either activate gene expression in the ΔhdrM background or interact with HdrM during the basal “off” state of the LRS. For example, even a small posttranslational modification like phosphorylation still adds ~80 Da to the molecular weight of a protein. Thus, the mass spectrometry results suggest that sequestration by HdrM is likely to be the sole mechanism required to inhibit HdrR function. However, we cannot presently exclude the possibility that HdrR is posttranslationally modified with a highly labile modification that is lost during protein purification.
Fig. 5. HdrR molecular weight determination ± HdrM.
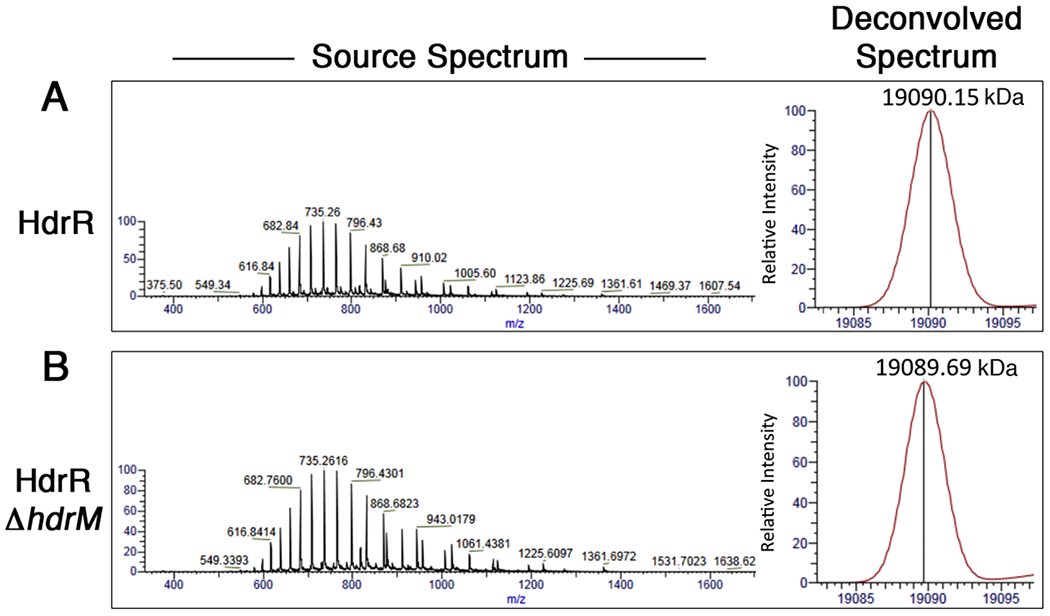
FLAG epitope tagged HdrR was directly purified from S. mutans strains either (A) encoding or (B) lacking hdrM and then subjected to electrospray ionization mass spectrometry as described in Experimental Procedures. Mass spectra of 350 – 2000 m/z were collected and then deconvolved with Protein Deconvolution 4.0 software.
DISCUSSION
The current study provides some of the first insights into one of the outstanding mysteries of LRS: the inhibitory mechanism employed by LRS membrane proteins. Our previous studies found S. mutans LRS membrane proteins to serve exclusively as inhibitors of their cognate LRS regulators, but homology searches and domain analyses of the LRS membrane proteins yielded no leads to suggest an inhibitory mechanism. Thus, we examined several likely mechanisms in the current study. Our results indicate that HdrM antagonism of HdrR is achieved primarily (and perhaps exclusively) through sequestration of HdrR within membrane-localized HdrRM heteromeric complexes (Fig. 6). Based upon the HdrR protein interaction and competitor results (Figs. 2 and 4), this sequestration likely occurs directly between HdrM and HdrR. The protein localization data (Fig. 1) indicate that HdrRM complexes are also likely to be quite stable, as HdrR was almost entirely localized together with HdrM at the cell membrane, despite being a soluble transcription factor. Similarly, HdrRM heteromeric complexes were easily detectable following a stringent tandem affinity purification of heterologously expressed HdrRM in E. coli (Fig. 2D). Such avid binding within the HdrRM complex is presumably required to minimize the basal expression of the hdrRM operon by preventing spurious positive feedback autoregulation in the absence of inducing signals (Fig. 6). In addition, we found no evidence to implicate regulated proteolysis or posttranslational modification of HdrR (Figs. 1B, 5A, and 5B). Thus, we suspect that endogenous activation of the HdrRM LRS is highly likely to involve direct regulation of HdrM function, rather than that of HdrR.
Fig. 6. Membrane Sequestration Model of the HdrRM LRS.
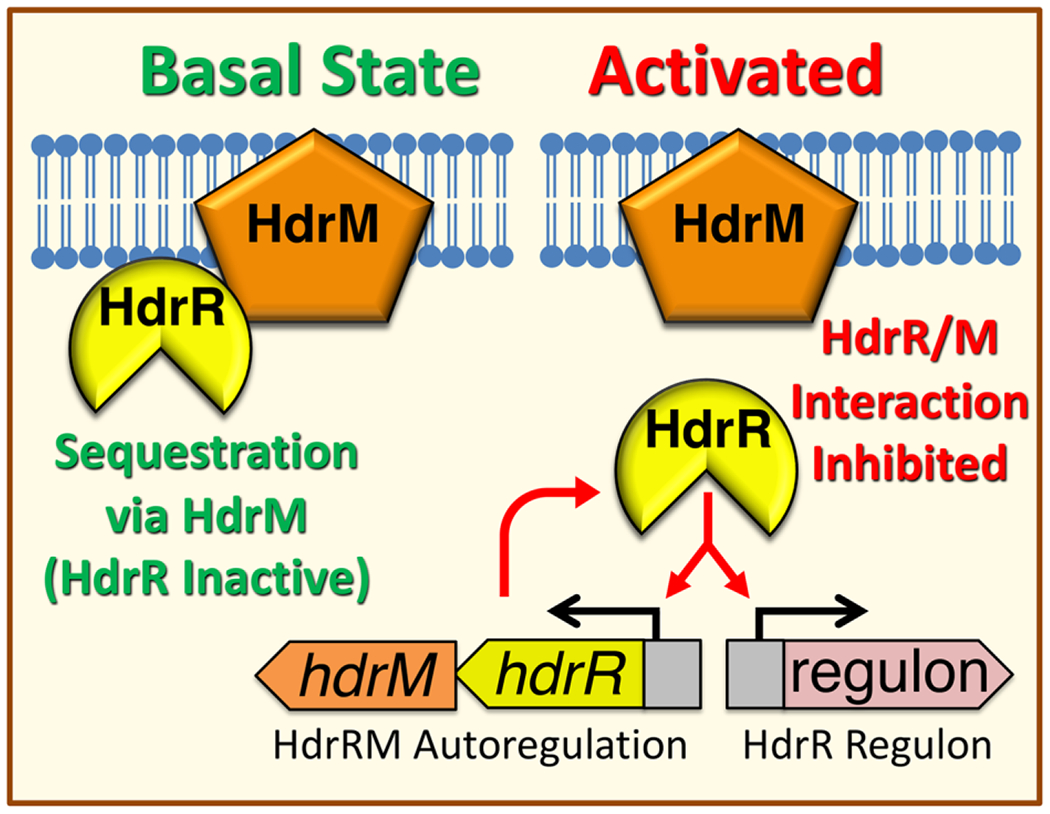
During normal growth conditions, LRS exist in a basal “off” state. For the HdrRM LRS, its spurious activation and subsequent autoregulation are prevented by sequestering HdrR at the cell membrane via stable heteromeric complex formation with the integral membrane inhibitor protein HdrM. Since HdrR is the proximal activator of hdrRM gene expression, both HdrR and HdrM remain as low abundance proteins. Upon activation of the HdrRM LRS due to currently unknown environmental stimuli or through mutagenesis of hdrM, HdrR is released from its membrane sequestration and can then activate gene expression of the hdrRM operon as well as the HdrR regulon. This results in a rapid increase in HdrRM protein abundance due to positive feedback autoregulation, which concurrently triggers a major amplification of the downstream output transcriptional response targeting the HdrR regulon.
Regulator sequestration is a strategy also employed by ECF sigma factor systems. Like LRS, ECF systems are dispensable under normal growth conditions and are maintained in a basal inactive state due to inhibitory sequestration (Luo et al., 2010, Asai et al., 2008). ECF system activation is typically dependent upon positive feedback autoregulation, ultimately triggered by environmental stress (Ho & Ellermeier, 2012, Mascher, 2013, Souza et al., 2014, Helmann, 2016). This combination of regulator sequestration and positive feedback autoregulation may provide additional insights into the biology of LRS. For example, a variety of experimentally verified mathematical models have illustrated how engineering both sequestration and positive feedback into genetic circuits results in ultrasensitive bistable switches with tunable gene expression and functionality over an exceptionally wide concentration of inducing signals (Chen & Arkin, 2012, Shopera et al., 2015). Such systems have sigmoidal response curves that are relatively insensitive to intrinsic noise, while the response characteristics of these bistable switches can be modulated simply by adjusting the concentration of the sequestering molecule (i.e. LRS membrane proteins or anti-σ in ECF systems) (Chen & Arkin, 2012). For LRS, these inhibitor concentration adjustments should be relatively easy to evolve, since it would only require a small number of point mutations to alter the transcript levels and/or translation of genes encoding the sequestering protein. Alternatively, LRS functionality might also be tunable via point mutations directly altering the affinity of the sequestration interaction. The malleability and simplicity of this type of genetic switch mechanism is ideally suited for sensory systems designed to respond to abnormal growth conditions, particularly because they only require minimal resources in a normal growth environment when the system is inactive (i.e. two low abundance proteins), but they can yield a tremendous output response once an activating environmental signal has been detected (Fig. 6). This may explain why LRS and ECF systems are conceptually analogous, as they would appear to be products of convergent evolution. For example, most streptococci encode a very limited number of alternative sigma factors (Souza et al., 2014), and of these, only a small minority is coexpressed with obvious anti-σ genes (unpublished observations). Thus, archetypal ECF σ/anti-σ pairs seem to be quite rare among the streptococci, whereas LRS are commonly encoded by streptococci and many other Firmicutes (Zou et al., 2018). Nevertheless, we cannot exclude the possibility that both LRS and ECF systems are simply derived from a shared ancestral lineage. Furthermore, it is presently unclear whether regulator sequestration is a defining feature of all LRS or if there are multiple inhibitory strategies employed by different LRS. This should become more apparent as additional LRS are characterized.
EXPERIMENTAL PROCEDURES
Bacterial species and culture conditions
All bacterial strains used in this study are listed in Table S1 and were either grown in an anaerobic chamber containing 85% N2, 10% CO2, and 5% H2 at 37 °C or cultured with aeration at 37 °C. All streptococcal strains were cultured using Todd Hewitt medium supplemented with 0.3% wt vol−1 yeast extract (THYE), while E. coli strains were cultured with Lennox LB (LB) medium. For antibiotic selection, cultures were supplemented with the following antibiotics: S. mutans - 10 μg ml−1 erythromycin and 1 mg ml−1 spectinomycin; E. coli - 100 μg ml−1 kanamycin, 50 μg ml−1 chloramphenicol, 250 μg ml−1 erythromycin, and 100 μg ml−1 spectinomycin.
DNA manipulation and strain construction
The strains used in this study are described in Table S1. Phusion DNA polymerase, restriction enzymes, and T4 DNA ligase were all purchased from New England Biolabs. The PCR primers used in this study are listed in Table S2. All mutant strains were sequenced to confirm their expected genotypes.
Creation of hdrRM operon luciferase reporter strains
To create the markerless replacement of the hdrRM ORFs with that of luciferase, we first created an allelic replacement of the hdrRM ORFs with the counterselectable IFDC2 cassette (Xie et al., 2011). Using UA140 genomic DNA as a template, two fragments corresponding to the upstream region and downstream regions of the hdrRM operon were amplified with the primer pairs hdrRupF/hdrRupR-ldh and hdrMdnF-erm/hdrMdnR, respectively. The IFDC2 cassette was amplified using the primer pair ldhF/ermR. The three fragments were mixed and used as template for overlap extension PCR (OE-PCR) with the primer pair hdrRupF/hdrMdnR. The resulting OE-PCR product was transformed into UA140 and selected on medium containing erythromycin to obtain strain RMIFDC2. Next, a DNA fragment containing the hdrR upstream region and luciferase ORF was amplified with the primer pair hdrRupF/lucR-1856 and strain LZ89-luc (Merritt et al., 2007) as a template. Using strain UA140 as a template, a fragment corresponding to the hdrM downstream region was amplified with the primer pair 1856F-luc/hdrMDnR. The two fragments were mixed and assembled with OE-PCR using the primer pair hdrRupF/hdrMdnR. The OE-PCR amplicon was transformed into strain RMIFDC2 and selected on medium containing p-chlorophenylalanine (4-CP) to obtain strain RpLuc. To insert the luciferase ORF downstream of the hdrRM ORFs, a DNA fragment containing the hdrR upstream region and IFDC2 were PCR amplified from strain RMIFDC2 with the primer pair hdrRupF/ermR-lucf. Using the genomic DNA of RpLuc as a template, the luciferase ORF was amplified with the primer pair lucF-erm/lucmR. The two amplicons were assembled using OE-PCR and the primer pair hdrRupF/lucmR. The resulting overlapping PCR products were transformed into RpLuc strain and selected on medium containing erythromycin to obtain the strain RMlucIFDC2. Next, two fragments encompassing the hdrRM locus were amplified from strain UA140 with the primer pair hdrRupF/MterR-luc, while the luciferase ORF was amplified from strain RpLuc with the primer pair lucF-Mter/lucmR. The PCR amplicons were mixed and assembled by OE-PCR using the primer pair hdrRupF/lucmR. The resulting OE-PCR amplicon was transformed into strain RMlucIFDC2 and selected on plates supplemented with 4-CP to obtain the strain hdrRMluc.
Creation of hdrR and hdrM epitope tagged ectopic expression strains
Using plasmid pHdrRoe as a template, the Pldh-hdrR transcription fusion was amplified with the primer pair ldhF-bamHI/hdrR3xFlagOL. The resulting amplicon was subsequently amplified with the primer pair ldhF-bamHI/hdrR3xFlag-hindIII, then digested with BamHI/HindIII, and ligated to the suicide vector pJY4164 to obtain pJYROEflag. To create the plasmid pJYROE, a fragment containing hdrR ORF driven by the ldh promoter was amplified with the primer pair ldhF-bamHI/hdrRR-hindIII from pHdrRoe. The PCR product was digested with BamHI and HindIII, and ligated to pJY4164 to obtain the suicide vector pJYROE. The plasmid pJYROEflag was transformed into strain RpLuc and selected on plates supplemented with erythromycin to obtain the epitope tagged hdrR expression strain ROEflag. To create the hdrM HA epitope tagged expression plasmid pMOEha, an ldh-hdrM transcription fusion was first assembled by creating two PCR amplicons with the primer pairs IdhF-BamHI/IdhR-SpeI and hdrMF-SpeI/hdrMR-EcoRI and UA140 gDNA as a template. The fragments were digested with (BamHI/SpeI) and (SpeI/EcoRI) and ligated to the E. coli-Streptococcus shuttle vector pDL278 after cutting with (BamHI/EcoRI). The resulting plasmid pMOE was then used as a template and amplified with the primer pair IdhF-bamHI/hdrMR-linker2. The resulting PCR amplicon was further amplified with the primer pair IdhF-bamHI/linker2-HAR-EcoRI, then digested with BamHI/EcoRI, and ligated to the shuttle vector pDL278 to create the plasmid pMOEha. The plasmids pMOE and pMOEha were transformed into the strain ROEflag to obtain RMOEflag or the epitope tagged hdrRM expression strain RMOEflagha. pJYROE and pMOEha were both sequentially transformed into strain RpLuc to create the strain RMOEha.
The E. coli strains used for heterologous coimmunoprecipitation studies of HdrRM were created as follows. Using plasmid pJYROEflag as a template both the hdrR ORF and plasmid backbone with ldh promoter were amplified with the primer pairs (ldhp)hdrR-F/(HA3FLAG)hdrR-R and FLAG-F/ldhp-R, respectively. The HA+FLAG dual epitope-encoding fragment was synthesized from IDT DNA (Table S2) and PCR amplified with the primer pair HA3FLAG-F/FLAG-R. The three PCR amplicons were mixed and assembled by the Gibson method (Gibson et al., 2009) to create the plasmid pJYRHAFLAG. The plasmids pJYROE and pMOEgfp were both sequentially transformed into E. coli to create the strain EcRMgfp. The plasmid pJYRHAFLAG was transformed into E. coli to create the strain EcRHAFLAG, while the plasmid pMOEgfp was transformed into strain EcRHAFLAG to create the strain EcRtagMgfp.
Creation of HdrR Ala5 and Ala6 mutant constructs
Using pJYROEflag as a template, two DNA fragments were PCR amplified with the primer pairs JYmF/ala5R and ala5F/JYmR. The amplicons were assembled by POE-PCR to create the suicide vector pJYAla5flag. The suicide vector pJYAla6flag was constructed using the same strategy except the primers ala6R and ala6F are used. For the construction of pJYAla5, two DNA fragments were PCR amplified from pJYROE with the primer pairs JYmF/ala5R and ala5F/JYmR. The amplicons were assembled by POE-PCR to create the suicide vector pJYAla5. The suicide vector pJYAla6 was constructed using the same strategy except the primers ala6R and ala6F were used. The suicide vector pJYAla5flag and the shuttle vector pMOEha were transformed into the strain RpLuc to obtain the strain Ala5MOEflagha. The suicide vector pJYAla6flag and the shuttle vector pMOEha were transformed into the strain RpLuc to obtain the strain Ala6MOEflagha.
Creation of HdrR and HdrM fluorescent protein fusion strains
The HdrR-mCherry and HdrM-GFP protein fusion strains were created as follows. Using pHdrRoe as a template, the ldhP-hdrR transcription fusion was amplified with the primer pair IdhF-bamHI/hdrRR-linker2. The ORF for mCherry was amplified from the plasmid pCWU3 (Wu & Ton-That, 2010) using the primer pair mcherryF-linker2/mcherryR-HindIII. The two amplicons were assembled using OE-PCR, then digested with BamHI/HindIII, and ligated to the suicide vector pJY4164 to create the plasmid pJYROEmChe. The plasmid was then transformed into strain RpLuc to create the strain ROEmChe. Strain ROEmChe was also transformed with the plasmid pMOE to create the strain ROEmCheM. Using pMOEha as a template, the Pldh-hdrM transcription fusion was amplified with the primer pair IdhF-bamHI/linker2R-L1, while the gfp ORF was amplified from the plasmid pldhGFP (unpublished plasmid) with the primer pair GFPF-linker1/GFPR-HindIII. The resulting amplicons were next assembled using OE-PCR with the primer pair IdhF-bamHI/GFPR-HindIII, then digested with BamHI/HindIII, and ligated to the shuttle vector pDL278 to create the plasmid pMOEgfp. This plasmid was transformed into the strain RpLuc to create the strain MOEgfp. Using pMOEgfp as a template, two fragments were amplified with the primer pairs pDLmF/DTMR and pDLmR/DTMF and assembled using prolonged overlap extension PCR (POE-PCR) (Xie et al., 2013) to create the plasmid pMtmOEgfp. This plasmid was transformed into strain RpLuc to create the strain MtmOEgfp.
The HdrR and HdrM BiFC YFP fusion strains were created as follows. Using pJYROE as a template, the ldhP-hdrR transcription fusion was amplified with the primer pair IdhF-bamHI/hdrRR-linker2. The N-terminal fragment of YFP was amplified from the plasmid pEYFP (unpublished plasmid) using the primer pair cYNF-L2/YN155R-HindIII. The two fragments were assembled using OE-PCR, then digested with BamHI/HindIII, and ligated to the suicide vector pJY4164 to create the plasmid pJYRYNOE. This plasmid was then used as a template for amplification with the primer pairs IdhR-L2/JYmF and L2F-ldh/JYmR and assembled by POE-PCR to create the plasmid pJYYNOE. Using the plasmid pMOE as a template, the Pldh-hdrM transcription fusion was amplified with the primer pair IdhF-bamHI/hdrMR-linker2, while the C-terminal YFP fragment was amplified from pEYFP with the primer pair YC155F-L2/YCR-HindIII. The two fragments were assembled by OE-PCR, then digested with BamHI/HindIII, and ligated to the shuttle vector pDL278 to create the plasmid pMYCOE. This plasmid was then used as a template for amplification with the primer pairs IdhR2-L2/pDLmF and L2F-ldh2/pDLmR and assembled by POE-PCR to create the plasmid pYCOE. The plasmids pJYRYNOE and pYCOE were both sequentially transformed into the strain RpLuc to create the strain RYNOE. The plasmids pJYYNOE and pMYCOE were both sequentially transformed into strain RpLuc to create the strain MYCOE. The plasmids pJYRYNOE and pMYCOE were both sequentially transformed into strain RpLuc to create the strain RYNMYCOE.
Creation of HdrR alanine scanning mutants
To perform alanine scanning mutagenesis of HdrR, blocks of six consecutive alanine mutations were introduced into HdrR starting at amino acid #2 and ending at the final amino acid #133 for a total of 22 unique mutant HdrR proteins (see Fig. S3A). Using the plasmid pJYROE as a template, ldhP-hdrR transcription fusion was amplified with the primer pair IdhF-DpDL/HdrRR-DpDL, while the pVA380 streptococcal replicon (LeBlanc et al., 1992) was amplified from the shuttle vector pDL278 (Chen & LeBlanc, 1992) with the primer pair DpDLF-ldh/DpDLR-hdrR. The two amplicons were assembled by POE-PCR to create the plasmid pRwt. This plasmid then served as a template for all subsequent alanine scanning mutagenesis constructs. Plasmid pRwt was transformed into strain RpLuc to create the strain RpLucROE. The first alanine mutant ectopic expression plasmid (pRAla1) was created by amplifying pRwt with the primer pairs pDLmR/Ala1R and pDLmF/Ala1F. The two amplicons were assembled by POE-PCR and transformed into strain RpLuc to create the strain RpLucAla1. Subsequent hdrR mutant ectopic expression plasmids were assembled similarly except the corresponding alanine mutagenesis primers were substituted for Ala1R and Ala1F in the POE-PCR reactions to create the plasmids pRAla2 – pRAla22. Each of the POE-PCR reactions was directly transformed into strain RpLuc to create the plasmid-harboring strains RpLucAla2 – RpLucAla22. The plasmids pRAla5 – pRAla16 and pRAla18 – pRAla20 were extracted and transformed into strain hdrRMluc to create the strains RMlucAla5 – RMlucAla16 and RMlucAla18 – RMlucAla20. The empty vector pVA380 was also transformed into strain hdrRMluc to obtain the negative control strain Rmluc-mock.
Creation of hdrR recombinant expression vectors
The hdrR ORF was amplified from strain UA140 using the primer pair hdrRF-NdeI/HdrRR-Hind. The amplicon was digested with NdeI/HindII and ligated to the expression vector pET29b to create the plasmid pEcROE. Using pEcROE as a template, the primer pairs Ala5F/pETmF and pETmR/Ala5R were used to create two amplicons that were subsequently assembled with POE-PCR to create the vector pEcAla5. The same strategy was used to create the vector pEcAla6, except the primers Ala6F and Ala6R were used for POE-PCR.
Recombinant protein expression and purification
HdrR wild-type, Ala5, and Ala6 mutants were all purified using pET29b and the E. coli BL21(DE3) pLysS expression system. Cultures were grown to OD600 0.6 at 37 °C with aeration before adding 0.1 mM IPTG and culturing for an additional 12 hr. at 20 °C. Cells were harvested by centrifugation (6000 x g, 5 min, 4 °C), washed twice with binding buffer (20 mM Tris, 300 mM NaCl, 5 mM imidazole, 10% glycerol, pH 7.9) and then resuspended in 20 ml of the same buffer. Next, the cells were chilled on ice, lysed by sonication, centrifuged to recover supernatants (20,130 x g, 20 min, 4 °C), and then HdrR-His6, Ala5-His6, and Ala6-His6 were purified using Ni-NTA agarose chromatography (Novagen). Proteins were eluted with 4 ml elution buffer (20 mM Tris, 300 mM NaCl, 500 mM imidazole, 10% glycerol, pH 7.9) and concentrated by ultrafiltration (Millipore membrane, 3 kDa cut-off size). Purified proteins were stored in 10% glycerol at −80 °C.
Cytoplasmic and membrane protein purification
100 ml mid-log cultures of S. mutans were harvested by centrifugation at 3,220 x g at 4 °C for 10 min. Cell pellets were washed with 50 mM Na-phosphate buffer (pH 7.0) and then suspended in 1.2 ml of buffer containing 50 mM Na-phosphate (pH 7.0), 150 mM NaCl, and 100 μM PMSF. The cells were lysed using an Omni Bead Ruptor 24 (Omni International) and then MgCl2 and CaCl2 were added to a final concentration of 1 mM before treating the lysates with DNase I (10 μg ml−1) and RNase (20 μg ml−1) for 1 hr. on ice. The crude cell lysate was clarified by centrifugation at 16,000 x g for 10 min. to remove cell debris and unbroken cells. 1 ml of clarified lysate was further centrifuged at 105,000 x g for 1 hr. at 4 °C to pellet the membranes. After ultracentrifugation, the top 100 μl of supernatant above the membrane pellet was used as the cytoplasmic protein fraction. The membrane protein pellet was washed twice in 1 ml buffer containing 50 mM Na-phosphate and 150 mM NaCl buffer (pH 7.0) and finally resuspended in 50 μl of 50 mM Na-phosphate buffer (pH 7.0). 20 μl of each sample was separated using 12% SDS-PAGE. To determine the purity of isolated membrane fractions, samples were analyzed via western blot to confirm the absence of contaminating lactate dehydrogenase, which is an exceptionally high abundance S. mutans protein that is exclusively localized in the cytoplasm.
Electrophoretic mobility shift assays (EMSAs)
EMSAs were performed similarly as previously described (Zou et al., 2014). Briefly, double-stranded probes were obtained by annealing equal molar concentrations of two oligonucleotides (Table S2) in 50 mM Tris-HCl (pH 8.0), 10 mM MgCl2, 50 mM NaCl and 1 mM EDTA, with the forward primer 5′-end labeled with digoxigenin-11-ddUTP (Roche). The oligonucleotide pair EMSA-hdrRp-F/EMSA-hdrRp-R served as the wild-type probe. 1 ng of DNA probe was incubated individually with various concentrations of HdrR-His6, Ala5-His6, or Ala6-His6 at 25 °C for 20 min in a 20 μl reaction volume. After incubation, the reaction mixtures were separated by electrophoresis and electro-transferred to nylon membranes. Images were detected using chemiluminescence and X-ray films.
Luciferase measurements
Assays of firefly luciferase activity were performed using a previously described methodology (Merritt et al., 2016) with mid-log phase cultures. Reporter data were normalized by dividing luciferase values by their corresponding optical density (OD600) values.
Western blot analysis
Western blots were performed using the WesternDot 625 western blot kit (Invitrogen) as previously described (Liu et al., 2015). Epitope tagged proteins were labeled with primary DYKDDDDK Tag (α-FLAG) and HA Tag (α-HA) monoclonal antibodies (Fisher Scientific) diluted 1:2000.
Deconvolution microscopy
S. mutans strains expressing fluorescent protein fusions were grown overnight in a CO2-enriched environment (candle jar) in THYE medium supplemented with spectinomycin. Stationary phase cultures were diluted 1:20 in fresh medium and grown for 5 hr. and then fixed using 2.6% (wt/vol) paraformaldehyde in 30 mM phosphate buffer pH 7.4 for 15 min. at room temperature followed by 30 min. on ice. Fixed cells were washed with PBS, attached to poly-L-lysine-coated cover glass slips, and mounted in 50% glycerol and 0.1% p-phenylenediamine in PBS pH 8. Cells were imaged using a DeltaVision image restoration microscope (Applied Precision/Olympus), fitted with an Olympus 100×/1.40 NA, UPLS Apo oil immersion objective, and a CoolSNAP QE cooled CCD camera (Photometrics). Z-stack images were taken at 0.15 μm intervals. Image deconvolution and correction of chromatic aberrations were performed using the SoftWoRx software package (Applied Precision/DeltaVision).
Coimmunoprecipitation
Cytoplasmic and membrane protein fractions were prepared using 100 ml of mid-log phase cell culture as described above, except that membrane protein fractions were resuspended in 1 ml buffer containing 50 mM Tris-HCl, 150 mM NaCl (pH 7.4), and 1% Triton X-100. Membrane proteins were solubilized after 1 hr. incubation at 4 °C and then clarified by ultracentrifugation at 105,000 x g for 30 min. at 4 °C. Input protein samples were mixed with 20 μl of ANTI-FLAG M2 Affinity Gel (Sigma) or Anti-HA agarose (Pierce) and rotated for 2 hr. at 4 °C (or overnight). Afterward, the resin was pelleted at 6000 x g for 30 sec., the supernatant was removed, and the resin was washed three times with 0.5 ml TBS buffer (50 mM Tris-HCl and 150 mM NaCl). The immuoprecipitated proteins were eluted by adding 20 μl of SDS buffer (125 mM pH 6.8 Tris-HCl, 4% wt/vol SDS, 20% vol/vol glycerol, 0.004% wt/vol bromophenol blue) to each sample. The sample was boiled for 3 min. and then pelleted by centrifugation at 6000 x g for 30 sec. The resulting supernatant was transferred to a fresh tube and served as the resin eluate. Both input and eluted proteins were detected using α-FLAG and/or α-HA western blots as described. To detect the interaction between HdrR and HdrM in E. coli, heterologously expressed proteins extracted from strains EcRMgfp, EcRHAFLAG, and EcRtagMgfp were mixed with ANTI-FLAG M2 Affinity Gel (Sigma), eluted with FLAG peptide, repurified with Anti-HA agarose (Pierce), and then eluted with 1% (wt/vol) SDS. Both the input and output protein samples were analyzed via western blot using α-HA (Fisher Scientific) and α-GFP (Invitrogen) antibodies diluted 1:2000 to detect the presence of HdrR and HdrM, respectively.
HdrR molecular weight determination via mass spectrometry
Strains RMOEflag and ROEflag were each grown to an optical density of OD600 0.3 – 0.4, collected by centrifugation, washed with TBS buffer (pH 7.4), and then sonicated in the same buffer containing 1 mM PMSF. The cell lysate was collected by centrifugation at 16,000 x g for 30 min. and then the clarified lysates were further was centrifuged at 105,000 x g for 1.5 hr. at 4 °C to pellet the cell membranes. The resulting supernatant was used as the cytoplasmic fractions, while the membrane protein pellets were solubilized with TBS containing 1% Triton X-100. HdrR proteins were immunopurified from cytoplasmic fractions of strain ROEflag and membrane fractions of RMOEflag using ANTI-FLAG M2 Affinity Gel (Sigma-Aldrich) according to the manufacturer’s protocol. Eluted HdrR proteins were separated in duplicate with 15% SDS-PAGE. The resulting gels were negatively stained with the imidazole-zinc procedure (Castellanos-Serra & Hardy, 2006). Protein bands corresponding to HdrR proteins were excised from the gel and directly eluted from the gel slices by passive diffusion. The eluted proteins were concentrated and then injected onto a 1.0 × 250-mm C4 column (214 MS C4; Vydac, Hesperia, CA). Masses were determined by electrospray ionization mass spectrometry on a model LTQ Velos Pro ion trap instrument (ThermoFinnigan, San Jose, CA). The flow rate was 20 μl min.−1 and used a mobile phase A containing water and mobile phase B containing acetonitrile, both with 0.1% formic acid. The sample was loaded at 2% B for 5 min. onto a 1 × 8 mm Opti-Trap protein trap (Optimize Technologies, Oregon City, OR), then separated using a 1 min. gradient to 7.5% B, 30 min. to 60% B, 4 min. to 100% B, 5 min. at 100% B, 1 min to 2% B, and equilibration for 14 min. at 2% B. Mass spectra of 350-2000 m/z were collected in profile mode while averaging 20 μscans, using an automatic gain control of 3 × 104, and a maximum ion time of 200 msec. Mass spectra were deconvolved using Protein Deconvolution 4.0 software (Thermo Scientific).
Statistical analysis
All statistical analyses were performed using GraphPad Prism software version 6.0 to calculate significance via two-tailed Student’s t-tests with Welch’s correction. Statistical significance was assessed using a cutoff value of P < 0.05.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Alison North and the staff of the Rockefeller University BioImaging Resource Center for their assistance with deconvolution microscopy. We also wish to thank Dr. Larry David and the staff of the Oregon Health & Science University Proteomics Shared Resource facility for their assistance with mass spectrometry. Mass spectrometric analysis by the OHSU Proteomics Shared Resource was partially supported by NIH core grants P30EY010572 and P30CA069533 as well as NIH grants S10RR025571 and R01DC002368-15S1. This project was supported by NIH grants DE022083, DE023850, and DE028252 to JM and NIH grants DE021726 and DE029492 to JK.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Asai K, Ishiwata K, Matsuzaki K, and Sadaie Y (2008) A viable Bacillus subtilis strain without functional extracytoplasmic function sigma genes. J Bacteriol 190: 2633–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourret RB (2010) Receiver domain structure and function in response regulator proteins. Curr Opin Microbiol 13: 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnside K, and Rajagopal L (2012) Regulation of prokaryotic gene expression by eukaryotic-like enzymes. Curr Opin Microbiol 15: 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellanos-Serra L, and Hardy E (2006) Negative detection of biomolecules separated in polyacrylamide electrophoresis gels. Nat Protoc 1: 1544–1551. [DOI] [PubMed] [Google Scholar]
- Chen D, and Arkin AP (2012) Sequestration-based bistability enables tuning of the switching boundaries and design of a latch. Mol Syst Biol 8: 620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YY, and LeBlanc DJ (1992) Genetic analysis of scrA and scrB from Streptococcus sobrinus 6715. Infect Immun 60: 3739–3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HS, Pelton JG, Yan D, Kustu S, and Wemmer DE (2001) Phosphoaspartates in bacterial signal transduction. Curr Opin Struct Biol 11: 679–684. [DOI] [PubMed] [Google Scholar]
- Corrigan RM, and Grundling A (2013) Cyclic di-AMP: another second messenger enters the fray. Nat Rev Microbiol 11: 513–524. [DOI] [PubMed] [Google Scholar]
- Dworkin J (2015) Ser/Thr phosphorylation as a regulatory mechanism in bacteria. Curr Opin Microbiol 24: 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, and Stock AM (2009) Biological insights from structures of two-component proteins. Annu Rev Microbiol 63: 133–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, and Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345. [DOI] [PubMed] [Google Scholar]
- Hauryliuk V, Atkinson GC, Murakami KS, Tenson T, and Gerdes K (2015) Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nat Rev Microbiol 13: 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmann JD (2016) Bacillus subtilis extracytoplasmic function (ECF) sigma factors and defense of the cell envelope. Curr Opin Microbiol 30: 122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengge R, Grundling A, Jenal U, Ryan R, and Yildiz F (2016) Bacterial Signal Transduction by Cyclic Di-GMP and Other Nucleotide Second Messengers. J Bacteriol 198: 15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TD, and Ellermeier CD (2012) Extra cytoplasmic function sigma factor activation. Curr Opin Microbiol 15: 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu CD, Chinenov Y, and Kerppola TK (2002) Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol Cell 9: 789–798. [DOI] [PubMed] [Google Scholar]
- LeBlanc DJ, Lee LN, and Abu-Al-Jaibat A (1992) Molecular, genetic, and functional analysis of the basic replicon of pVA380-1, a plasmid of oral streptococcal origin. Plasmid 28: 130–145. [DOI] [PubMed] [Google Scholar]
- Liu N, Niu G, Xie Z, Chen Z, Itzek A, Kreth J, Gillaspy A, Zeng L, Burne R, Qi F, and Merritt J (2015) The Streptococcus mutans irvA gene encodes a trans-acting riboregulatory mRNA. Mol Cell 57: 179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Asai K, Sadaie Y, and Helmann JD (2010) Transcriptomic and phenotypic characterization of a Bacillus subtilis strain without extracytoplasmic function sigma factors. J Bacteriol 192: 5736–5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascher T (2013) Signaling diversity and evolution of extracytoplasmic function (ECF) sigma factors. Curr Opin Microbiol 16: 148–155. [DOI] [PubMed] [Google Scholar]
- Merritt J, and Qi F (2012) The mutacins of Streptococcus mutans: regulation and ecology. Mol Oral Microbiol 27: 57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt J, Senpuku H, and Kreth J (2016) Let there be bioluminescence: development of a biophotonic imaging platform for in situ analyses of oral biofilms in animal models. Environ Microbiol 18: 174–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt J, Zheng L, Shi W, and Qi F (2007) Genetic characterization of the hdrRM operon: a novel high-cell-density-responsive regulator in Streptococcus mutans. Microbiology 153: 2765–2773. [DOI] [PubMed] [Google Scholar]
- Nikolskaya AN, and Galperin MY (2002) A novel type of conserved DNA-binding domain in the transcriptional regulators of the AlgR/AgrA/LytR family. Nucleic Acids Res 30: 2453–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okinaga T, Niu G, Xie Z, Qi F, and Merritt J (2010a) The hdrRM operon of Streptococcus mutans encodes a novel regulatory system for coordinated competence development and bacteriocin production. J Bacteriol 192: 1844–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okinaga T, Xie Z, Niu G, Qi F, and Merritt J (2010b) Examination of the hdrRM regulon yields insight into the competence system of Streptococcus mutans. Mol Oral Microbiol 25: 165–177. [DOI] [PubMed] [Google Scholar]
- Romling U, Galperin MY, and Gomelsky M (2013) Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev 77: 1–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shopera T, Henson WR, Ng A, Lee YJ, Ng K, and Moon TS (2015) Robust, tunable genetic memory from protein sequestration combined with positive feedback. Nucleic Acids Res 43: 9086–9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza BM, Castro TL, Carvalho RD, Seyffert N, Silva A, Miyoshi A, and Azevedo V (2014) sigma(ECF) factors of gram-positive bacteria: a focus on Bacillus subtilis and the CMNR group. Virulence 5: 587–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staron A, Sofia HJ, Dietrich S, Ulrich LE, Liesegang H, and Mascher T (2009) The third pillar of bacterial signal transduction: classification of the extracytoplasmic function (ECF) sigma factor protein family. Mol Microbiol 74: 557–581. [DOI] [PubMed] [Google Scholar]
- Ulrich LE, Koonin EV, and Zhulin IB (2005) One-component systems dominate signal transduction in prokaryotes. Trends Microbiol 13: 52–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright DP, and Ulijasz AT (2014) Regulation of transcription by eukaryotic-like serine-threonine kinases and phosphatases in Gram-positive bacterial pathogens. Virulence 5: 863–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, and Ton-That H (2010) Allelic exchange in Actinomyces oris with mCherry fluorescence counterselection. Appl Environ Microbiol 76: 5987–5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Okinaga T, Niu G, Qi F, and Merritt J (2010) Identification of a novel bacteriocin regulatory system in Streptococcus mutans. Mol Microbiol 78: 1431–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Okinaga T, Qi F, Zhang Z, and Merritt J (2011) Cloning-independent and counterselectable markerless mutagenesis system in Streptococcus mutans. Appl Environ Microbiol 77: 8025–8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Qi F, and Merritt J (2013) Cloning-independent plasmid construction for genetic studies in streptococci. J Microbiol Methods 94: 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Z, Du D, Zhang Y, Zhang J, Niu G, and Tan H (2014) A gamma-butyrolactone-sensing activator/repressor, JadR3, controls a regulatory mini-network for jadomycin biosynthesis. Mol Microbiol 94: 490–505. [DOI] [PubMed] [Google Scholar]
- Zou Z, Qin H, Brenner AE, Raghavan R, Millar JA, Gu Q, Xie Z, Kreth J, and Merritt J (2018) LytTR Regulatory Systems: A potential new class of prokaryotic sensory system. PLoS Genet 14: e1007709. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.