

Abstract
Macrophages are key effectors of host defense and metabolism, making them promising targets for transient genetic therapy. Gene editing through delivery of the Cas9-ribonucleoprotein (RNP) provides multiple advantages over gene delivery-based strategies for introducing CRISPR machinery to the cell. There are, however, significant physiological, cellular, and intracellular barriers to the effective delivery of the Cas9 protein and guide RNA (sgRNA) that have to date, restricted in vivo Cas9 protein-based approaches to local/topical delivery applications. Herein we describe a new nanoassembled platform featuring co-engineered nanoparticles and Cas9 protein that has been developed to provide efficient Cas9-sgRNA delivery and concomitant CRISPR editing through systemic tail-vein injection into mice, achieving >8% gene editing efficiency in macrophages of the liver and spleen.
Keywords: protein delivery, CRISPR-Cas9 ribonucleoprotein delivery, in vivo delivery, gene editing, phagocyte targeting
Graphical Abstract
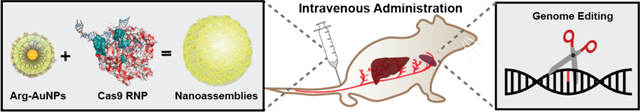
CRISPR-Cas9-ribonucleoprotein (RNP) delivery provides multiple advantages over gene delivery-based strategies. However, cellular barriers to nuclear delivery of Cas9 RNP restrict in vivo Cas9 protein-based approaches to systemic delivery applications. The nanoassemblies provide efficient CRISPR editing through systemic tail-vein injection into mice, achieving >8% gene editing efficiency in macrophages of the liver and spleen.
1. Introduction
Macrophage cells have become a key target for gene editing due to their central immunological role.[1] As secondary immune cells, macrophages are a critical part of innate immunity for host defense against infectious agents.[2] Unlike many other cells of the immune system, macrophages are resident in all tissues, have tremendous plasticity, and play key roles in the regulation of metabolic cellular processes.[3] Conversely, malfunctioning macrophages have been known to precipitate abnormal metabolism[4] and contribute to disorders such as lysosomal storage diseases,[5] atherosclerosis,[6] and Alzheimer’s disease,[7] thus making them attractive therapeutic targets.
Reprogramming the genome of secondary immune cells is a strategy that prolongs the therapeutic effect for the duration of the cell’s lifetime.[8,9,10] This strategy has recently been explored for the therapeutic correction or alteration of the macrophage genome, using the CRISPR-Cas9 gene editing system.[11] Specific, targeted delivery to macrophage cells is advantageous as it avoids unwanted effects in non-disease cells, and minimizes required dosage.[12] Administration of the CRISPR-Cas9 system specifically to macrophages is a promising yet underutilized approach to the treatment of disease, mostly due to poor specificity and barriers to nuclear access, such as endosomal entrapment.[13]
CRISPR-Cas9-based systems have been widely utilized for the treatment of genetic diseases both in vitro[14, 15] and in animal models using ex vivo and in vivo strategies.[16,17] In the majority of such approaches, genes or mRNA encoding Cas9 elements are packed into viral or non-viral vectors and injected into animal models through either local or systemic injections.[14,18] Adeno-associated viruses (AAVs) have been widely utilized for gene delivery of CRISPR systems in vivo.[16,19] Genes delivered by these methods are continuously expressed, and consequentially persist longer in host cells, posing concerns for the host including high immunogenicity, unwanted gene editing, and insertional mutagenesis.[20,21] Furthermore, AAV vectors can package genes only up to the size of ~4.7 kbp, thus requiring the use of a smaller CRISPR-Cas9 system, such as SaCas9 (3.16kb)[22] or CjCas9 (2.95kb),[23] to be capable of being packaged with its sgRNA into a single AAV vector for in vivo genome editing.
Non-viral delivery of Cas9 mRNA has been used as an option to bypass many of the challenges associated with viral delivery.[24,25,26,27] However, these systems still face potential issues arising from continuous expression of the Cas9 protein and lower transfection efficiency compared to other methods, limiting the usage of these platforms.[29]
Direct delivery of the Cas9 protein and its associated single guide RNA (sgRNA, together comprising the CRISPR-Cas9-RNP) complex offers an attractive alternative approach to CRISPR-based genome editing. Protein-mediated strategies are transient,[28] and therefore limit the exposure of the genome to the editing machinery, which can result in unwanted off-target editing.[29] Transient protein delivery also decreases the risk of immunogenicity in the host arising from persistent expression of active Cas9. There have been recent reports describing delivery of the CRISPR-Cas9-RNP into animals for gene editing using gold nanoparticles[30] or DNA nanoclews.[24] Notably, these vehicles enter cells through endocytic pathways, making them vulnerable to entrapment in the endo/lysosome, which is a major barrier to efficacy. In practice, these CRISPR-Cas9-RNP vectors were injected locally into the target organs, resulting in localized genomic editing. While such methods have potential for treating certain locally-affected diseases,[14,31,32] passive, cell-specific targeting using systemic administration has remained a challenge.[33]
Systemic administration of the CRISPR-Cas9-RNP accompanied by macrophage-targeted delivery would provide a less invasive approach to macrophage gene editing, with better translatability in the clinic.[34] To our knowledge, however, there has been no reported approach to achieve effective gene editing by systemic administration using the CRISPR-Cas9-RNP,[35] in large part due to inefficient endosomal escape (and hence, editing) by current systems.[29]
In previous work, we developed strategies for protein-nanoparticle assembly that use protein engineering to append an oligo(glutamic acid) tag to either the C- or N-terminus of the protein (Figure 1). These E-tagged proteins form hierarchical nanocomposites with nanoparticles featuring arginine head-groups (ArgNPs) through carboxylate-guanidinium interaction, a phenomenon our previous studies demonstrate does not occur when using proteins lacking an E-tag.[28] These nanocomposites have multiple layers of hierarchy that are highly responsive to stimuli from the microenvironment.[36] Significantly, these nanocomposites were found to provide highly efficient delivery of a range of E-tagged proteins, including Cas9, directly to the cytosol[37] in a range of laboratory cell lines.[28,38]
Figure 1.
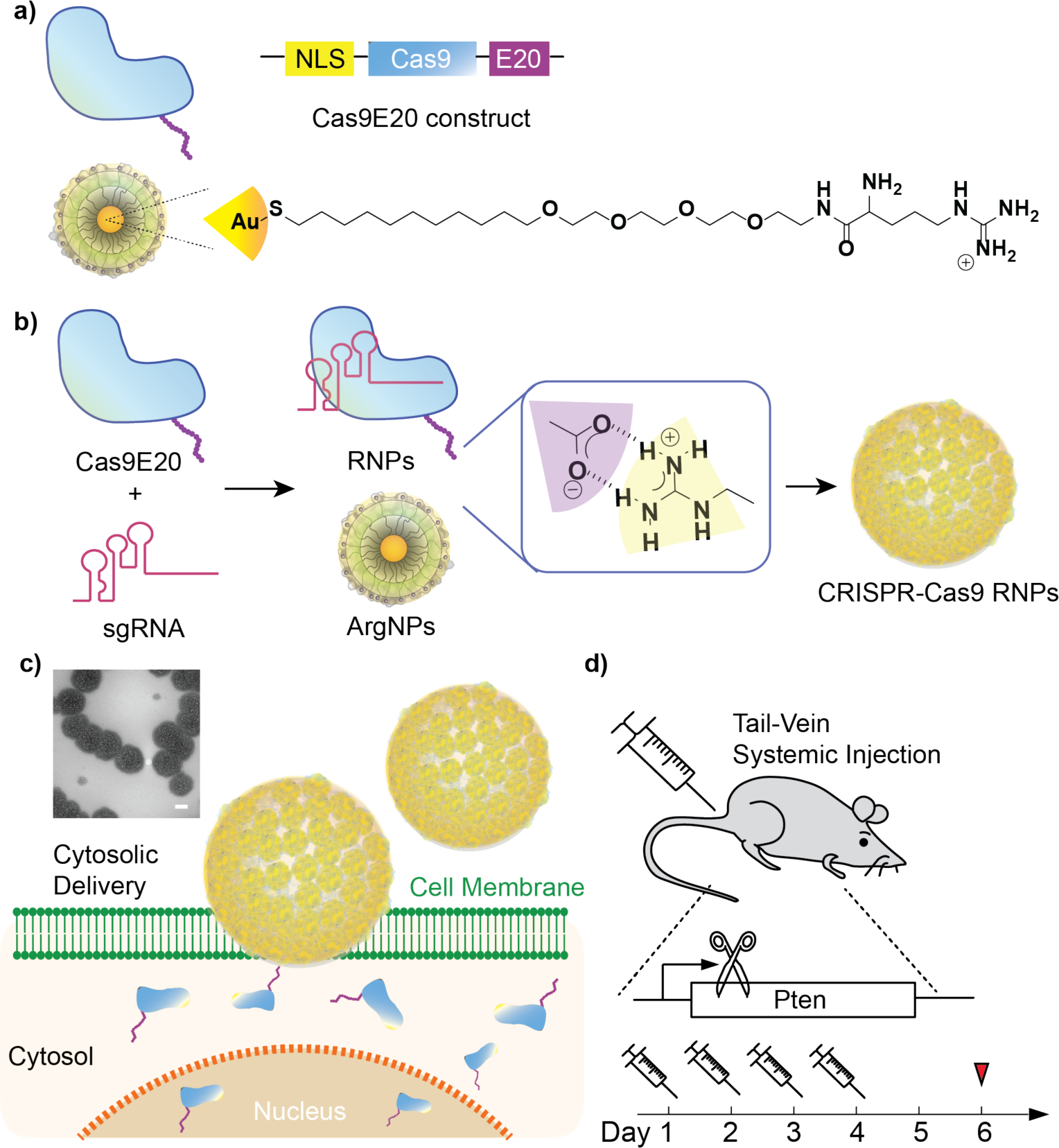
Schematic depiction of systemic delivery of CRISPR-Cas9-RNP in vivo for gene editing. (a) The generalized construct of Cas9E20 protein and chemical structure of nanoparticle ligand. (b) Co-engineered Cas9E20 and arginine-functionalized gold nanoparticles were fabricated to form nanocomposites via electrostatic interaction. (c) These nanocomposites were then delivered through a membrane fusion mechanism and accumulated in the nucleus for genome editing. Inset shows TEM imaging of multiple composites, scale bar: 100 nm. (d) Injections of CRISPR-Cas9 RNP into BALB/c mice (n=5) through tail-vein administration, with dosing at days 1,2,3,4 and sacrifice/editing analysis on day 6.
We hypothesized that the ability of the co-engineered particle-protein assemblies to directly access the cytosol, coupled by their ability to protect their protein cargo from degradation, would make them effective vectors for systemic delivery. We additionally postulated that the size of these nanocomposites would provide selective passive targeting of macrophages in major clearance organs.[39] We report herein macrophage-specific delivery of the CRISPR-Cas9-RNP into mice through systemic tail-vein injection. Carrier nanocomposites were fabricated through co-engineering of ArgNPs with Cas9 protein engineered to include a 20-glutamic acid tag (Cas9E20) (Figure 1a). When ArgNPs and Cas9E20 along with the associated sgRNAs were complexed, a single carrier nanocomposite was formed (Figure 1b). Systemic injection of this complex provided efficient (>8% and >4%) in vivo gene editing of the PTEN gene, specifically in macrophages of the liver and spleen, respectively. This platform thus shows potential as a selective immunotherapeutic for the treatment of macrophage-related diseases.[38,40,41,42]
2. Results and discussion
2.1. Characterization of co-engineered Cas9 protein and gold nanoparticles.
The Cas9E20:sgRNA complex was prepared by incubating the two components at room temperature for 30 min. ArgNPs were subsequently added to the preassembled Cas9E20:sgRNA complex to form the final nanocomposites (see Methods section).[28] The mean size of these nanocomposites was 285 nm (± 23 nm), as determined by dynamic light scattering (DLS) (Figure S1). Further characterization of these assemblies may be found in our previous report.[36] The stability of the CRISPR-Cas9-RNP in serum was also evaluated, and these nanocomposites were found to be stable over a relevant 24-hour period in serum (Figure S2). We then investigated the gene editing capability of these CRISPR-Cas9-RNP nanocomposites by systemically administering them to mice (Figure 1c–d). To maximize genome editing efficiency, the nanocomposites were administered via tail-vein injection into BALB/c mice (n = 5) for four days (one dose per day), at a dose of Cas9E20 (0.9 nmol), sgRNA (0.9 nmol) and ArgNPs (1.8 nmol), for each injection. The stock concentrations of Cas9E20 and ArgNPs were 4.68μM and 31.8μM, respectively. The concentration of engineered protein was determined by bicinchoninic acid assay (BCA assay, with BSA used as a standard) and the concentration of arginine nanoparticle was determined by UV-visible spectrometry. Mice were sacrificed 2 days after final injection, and the care given during handling adhered to all standards set forth by the IACUC.
2.2. Biodistribution of gold as detected by LA-ICP-MS.
To assess Cas9-RNP delivery and in vivo distribution of nanocomposites, inductively coupled plasma mass spectrometry (ICP-MS) was used to determine the content of gold in different organs. These biodistribution analyses provide valuable information for understanding the biological fate of the CRISPR-Cas9-RNP delivery vehicle when in circulation. Quantification of gold in organs indicated that compared to controls thast exhibited baseline gold content (Figure S3), in experimental animals, 55% and 40% of total detected gold accumulated in the spleen and liver, respectively (Figure 2a). This predominant accumulation of nanocomposites in these organs is generally as expected, due to their roles as the major detoxifying and filtering organs in the mononuclear phagocyte system (MPS, also known as the reticuloendothelial system, or RES). This distinction also makes them key targets for the treatment of organ-specific diseases related to the immune system.[43] Less than 5% of nanocomposites were distributed in other organs, including the kidney, lung, heart, and small intestine.
Figure 2.
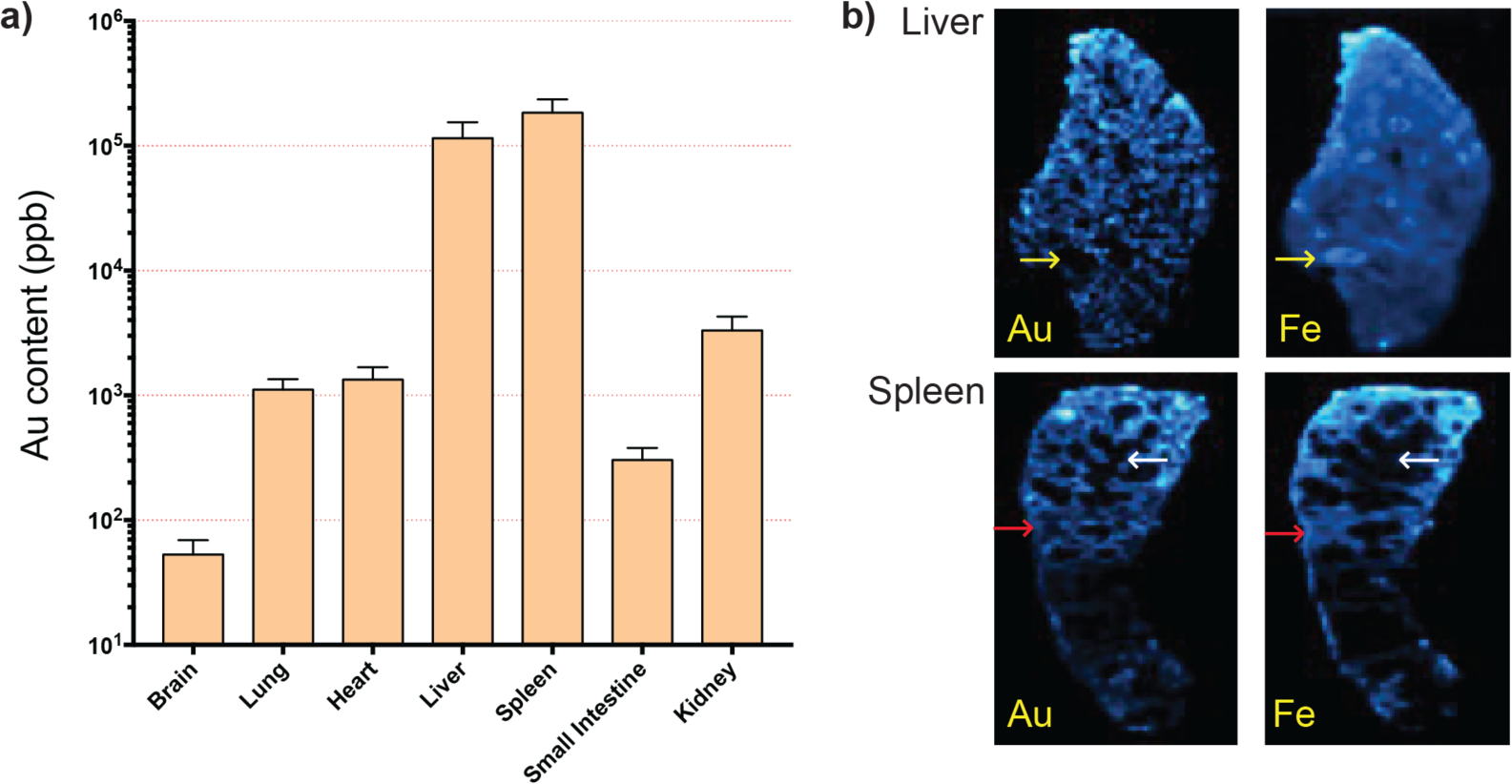
Tissue localization of gold after systemic administration. (a) Biodistribution of gold in mouse organs after 4 doses of intravenously injected CRISPR-Cas9-RNP, quantitatively analyzed by ICP-MS. Gold concentration in tissue was calculated by dividing the gold amount (ng) per tissue by organ weight (g) in each organ (n = 3, mean ± SD) (b) LA-ICP-MS mapping image of liver and spleen mouse organs of mice injected with nanocomposites. Yellow arrows denote a vein; red arrows denote areas of the red pulp; white arrows denote areas of the white pulp.
From each mouse, laser ablation (LA)-ICP-MS was used for imaging the sub-organ distribution of the species 197Au, 57Fe, and 66Zn in liver and spleen. Each organ was sliced at 20 μm, all individual pixels were calibrated to render the images with a size of 50 μm × 50 μm by summing 5-second increments of data collected at a scan rate of 10 μm s−1, and images were analyzed using a resolution of 100 μm. LA-ICP-MS provided the metal content (Au, Fe, and Zn) as a function of position, showing the sub-organ distribution of each of the metals in the organs, with Zn used as an evenly distributed control. (Figure S4) The Fe map of the liver also showed an even distribution throughout; a logical result considering the liver’s responsibilities in blood filtration (Figure 2b). Notably, the Au and Au/Fe image analysis suggested clustered behavior of Au in the liver tissue (Figure S5). Previous reports on NPs with different surface charges show that positively-charged NPs interact preferentially with cells over negatively or neutrally-charged NPs.[44,45] In accordance with these studies, and based on the role of MPS-associated macrophages in phagocytosis of foreign species, we hypothesized that this accumulation was potentially due to interaction between the nanocomposites and these phagocytes.[45,46]
The Fe map of the spleen (Figure 2b) allows us to distinguish between the red pulp (red arrows) and white pulp (white arrows), characteristic of the sub-organ distribution of the spleen. The similar intensities in the gold and iron signals suggests that the gold nanoparticles are located predominantly in the red pulp, which is a reasonable result due to the filtration responsibilities of the region.[47] The Au/Fe matrix enables the visualization of areas in which the Au content is higher in comparison to the Fe signal. (Figure S5) The red pulp of the spleen is home to numerous monocytes, subcellular species which are circulated through the blood and ultimately differentiate into macrophage cells.[48] It is notable that the AuNPs are highly present in the marginal zone between the red and white pulp, which is associated with the initial stages of degradation and clearance by spleen macrophage cells. (Figure 2b) These cells are components of the immune system responsible for the targeted destruction of foreign bodies, and therefore interesting therapeutic targets.[49]
2.3. Editing efficiency of the PTEN gene in vivo.
Encouraged by the biodistribution as determined by ICP-MS, we next investigated the specificity and efficacy of gene knockout by targeting the PTEN gene following systemic injection, using a previously validated vector.[28] The dosage amount and schedule were determined by a series of optimization experiments, and it was determined that at least 4 injections would provide statistically significant editing events. As such, our studies utilized a series of 4 injections for each experimental group. Cells were first isolated, and genomic DNA was extracted following standard protocol.[50] In the spleen and liver, insertion/deletion (indel) frequencies were observed as analyzed by T7 Endonuclease 1 (T7E1) assay. These results are consistent with ICP-MS biodistribution analyses and reveal a co-localization between AuNPs and CRISPR-Cas9-RNP in the liver and spleen. By contrast, neither control mice injected with Cas9E20-sgRNA lacking gold nanoparticles nor those injected with Cas9E20-AuNPs without sgRNA, exhibited genome editing in the 7 major organs examined (Figure S6). No significant bodyweight change was noted for mice injected with nanocomposites as compared to PBS only or Cas9E20 only injection (Figure S7). Notably, this gene editing efficiency is comparable with other gene delivery-based approaches.[14,19,24]
2.4. Macrophage-specific treatment with CRISPR-Cas9-RNP.
Encouraged by the biodistribution and selective genome editing results, we carried out further in vivo experiments to investigate which type of cells internalized the Cas9-RNP. In a follow-up study, we first labeled different immune cells, including T cells, B cells, and macrophages using respective antibodies (specific antibodies can be found in the Methods section). After lysing red blood cells, immune cells in the spleen and liver were sorted by FACS to a high level of purity (Figure S8), and the gold content in different cell types was analyzed by ICP-MS. As shown in Figure 3, in the spleen and liver, the distribution of gold was much higher in macrophages than in T cells and B cells. These results suggest that the CRISPR-Cas9-RNP effectively targeted macrophages through systemic injection. The gene editing capability of this platform was evaluated on sorted cell populations using the T7E1 assay as shown in Figure 4a and Figure S9, and demonstrated editing in the PTEN gene of splenic and hepatic macrophages. These results were confirmed using Interference for CRISPR Edits (ICE) analysis (Figure 4b and Figure S10). [51]
Figure 3.
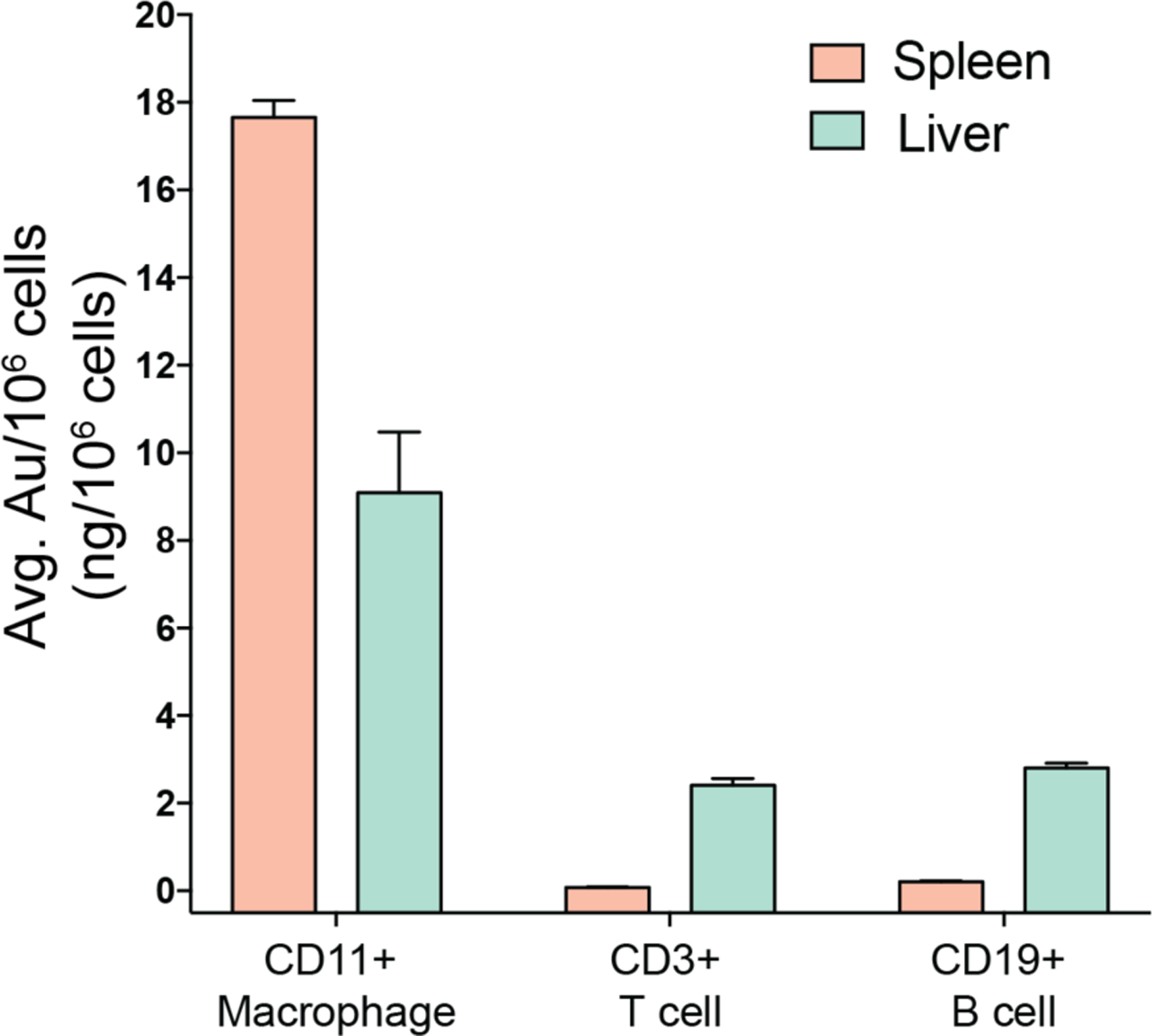
Average gold content in sorted cell populations of CD11+ macrophages, CD3+ T cells, and CD19+ B cells of the liver and spleen demonstrates notable macrophage specificity, as identified by ICP-MS.
Figure 4.
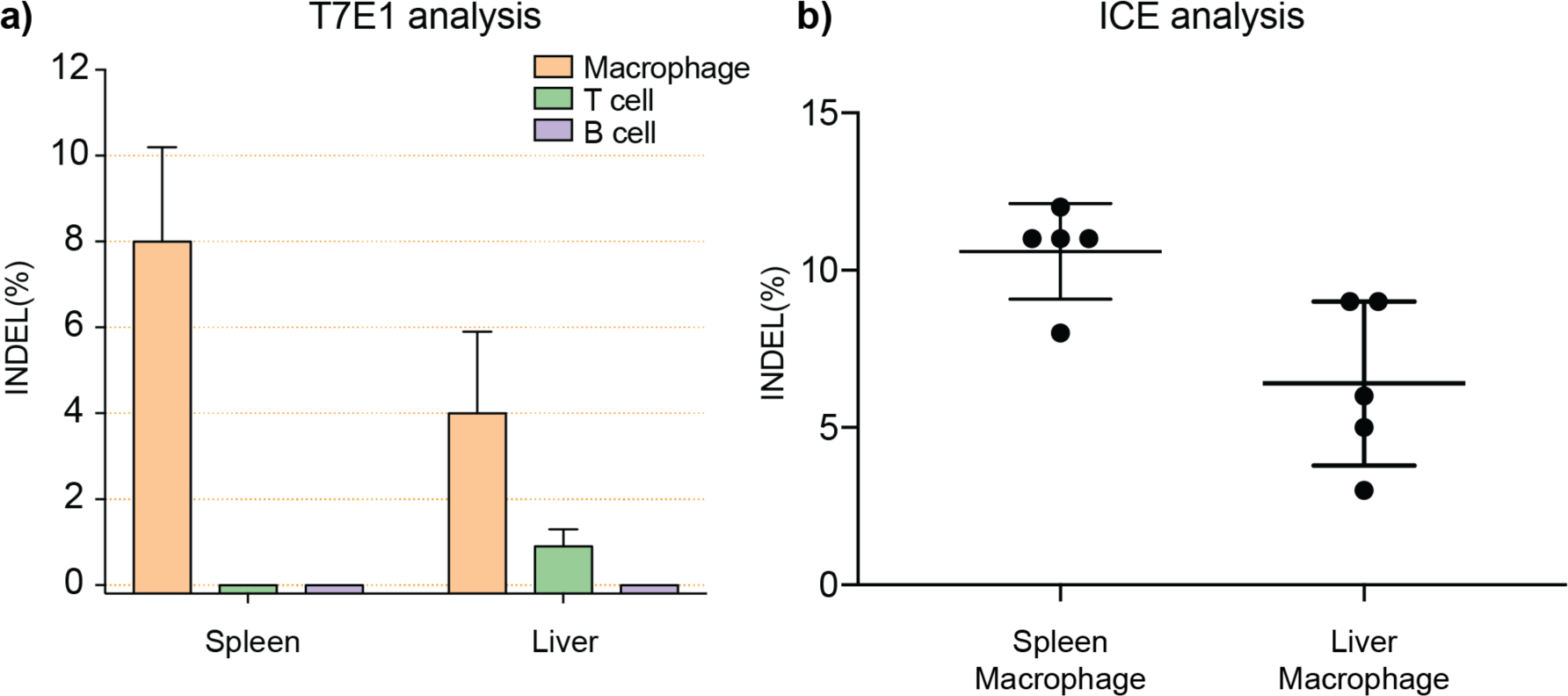
Efficient gene editing as analyzed by (a) T7E1 assay and (b) Interference for CRISPR Edits (ICE) analysis.
2.5. Cytokine secretion, liver panel, and off-target effect evaluation.
Direct delivery of the recombinant Cas9 protein boasts a notoriously low immune response compared to other approaches. However, macrophages are critical regulators of the immune system, thus making an assessment of the immune response prudent. We assessed cytokine secretion and liver function after systemic injection of our nanocomposites to evaluate adverse immunological effects following treatment. The level of the macrophage-associated inflammatory cytokine TNF-α was measured by ELISA. TNF-alpha serves as a general, broad response cytokine for macrophage activation, triggered by non-self-stimulus, and as such can act as a gauge for the presence of an immune response in the host. As shown in Figure 5a, the levels in experimental groups were comparable to those of the PBS-treated control group, suggesting a lack of substantial immune response after 4 single-dose injections of CRISPR-Cas9-RNP nanocomposites. In addition, it is well-known that the immune response associated with certain other delivery methods, such as adenoviral vectors, can lead to liver damage.[52] We thus examined liver function by serum ALT and Bilirubin assays. Blood was collected two days after all injections, and serum was separated using a Microtainer tube with serum separator. As shown in Figure 5b, alanine aminotransferase and Bilirubin levels were assayed in triplicate and elicited similar results to PBS treatment.
Figure 5.
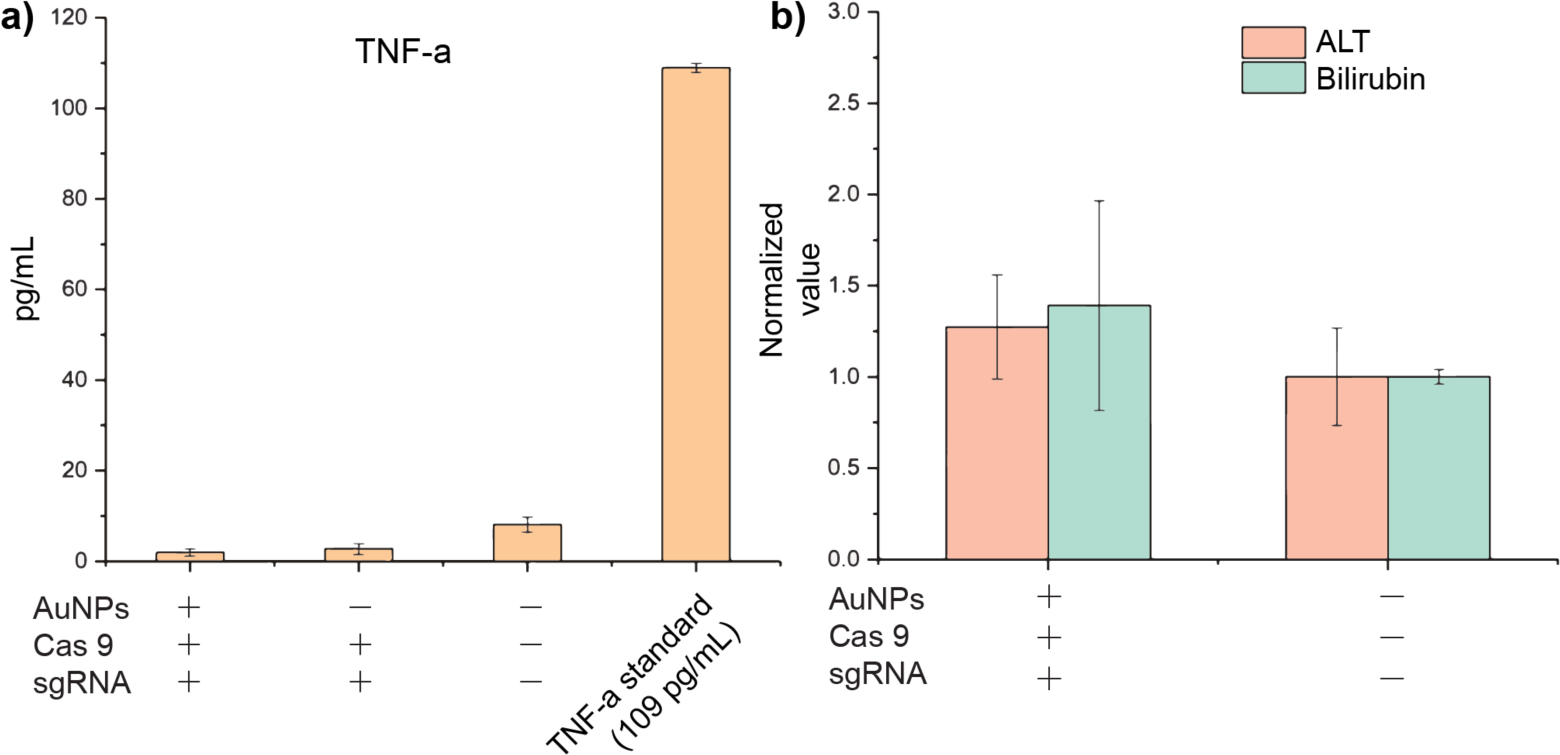
Serum levels of (a) TNF-α cytokine and (b) alanine aminotransferase (ALT) and Bilirubin.
Together, these data suggest the absence of an immune response following the systemic introduction of our nanocomposites. This low immune response is unsurprising due to the transient nature of CRISPR-Cas9-RNP delivery.
Finally, to demonstrate the on-target activity of CRISPR-Cas9-RNP genome editing, four potential off-target sites of PTEN (Table S1) predicted by the online service, Benchling,[53] were evaluated. No indel events were detected at these four sites of interest (Figure S11). Compared to other methods, the low frequency of off-target events using the CRISPR-Cas9-RNP is well-documented[29] and is therefore expected.
3. Conclusion
In summary, we have demonstrated an in vivo application of the CRISPR-Cas9 gene editing system using a transient, immunologically silent delivery method with marked specificity for macrophages of the liver and spleen. Our nanocomposite system maintains the capacity for systemic circulation while simultaneously displaying effective CRISPR-Cas9-RNP delivery and efficient genomic editing in secondary immune cells. These advances promise to not only alleviate many of the issues associated with endocytic entrapment for in vivo therapeutics, but also provide a promising platform for targeted delivery to macrophages. Furthermore, the concurrent intracellular delivery and macrophage targeting of these systems allows the capability to engineer phagocytes ex vivo and in vivo, and thus could be an effective and very appealing approach to the development of immunotherapeutics. In short, our approach presents a promising opportunity for not only the treatment of diseases of the liver and spleen, but also for transient modulation of immunological host defense.
4. Experimental Section
Expression, purification and characterization of engineering E-tagged Cas9E20:
Glutamic acid tag (E-tag) was inserted to N-terminus of SpCas9 through site directed mutagenesis (SDM).[54] Recombinant proteins (Cas9E20) were expressed in E. coli BL21 Rosetta strain using standard protein expression protocol. Briefly, protein expression was carried out in 2xYT media with an induction condition of 0.5 mM IPTG at 18°C for 16 hours. At this point, the cells were harvested, and the pellets were lysed using 1% Triton-X-100/DNase-I treatment. Proteins were purified using HisPur cobalt columns. Proteins were finally preserved in PBS buffer containing 300 mM salt. The purity of native proteins was determined using 8% SDS-PAGE gel. The concentration of Cas9 protein was determined using a NanoDrop 2000 (Thermo Fisher Scientific) from absorbance at 280 nm.
sgRNA synthesis:
sgRNAs were in vitro transcribed from dsDNA template (containing the protospacer and the tracrRNA sequence) using AmpliScribe-T7-Flash Transcription kit according to manufacturer’s protocol. In vitro transcribed sgRNAs were purified using an RNA purification kit (Zymo research RNA Clean & Concentrator). The concentration of sgRNA was determined using a NanoDrop 2000 (Thermo Fisher Scientific) and the final sgRNA products were stored at −80°C for the subsequent experiments.
Nanoparticle synthesis and characterization:
Arginine-functionalized gold nanoparticles (ArgNPs) were prepared according to our previous methods.[55] The arginine-functionalized thiol ligand was synthesized first. Following this, ArgNPs were prepared by conventional place-exchange reaction of 2-nm sized 1-pentanethiol-protected gold nanoparticles (Au-C5) with HS-C11-TEG-NH-Arginine. The resultant ArgNPs were dissolved in distilled water, purified by dialysis, and the characterizations of the ArgNPs were performed as in our previous report.
Nanocomposite fabrication:
Cas9E20-RNP:ArgNPs nanocomposites were prepared through a simple mixing procedure. Cas9E20 (0.9 nmol per dose) and sgRNAs (0.9 nmol per dose) were assembled in 1×PBS for 30 min at room temperature first, then ArgNPs (50 μM stock in 5 mM PB, pH 7.4) were added to 100 μL of 1×PBS in another vial, followed by adding the preassembled Cas9E20-RNP at appropriate molar ratio [usually at 2:1 ratio (ArgNPs, 1.8 nmol per dose)/(Cas9E20-RNP, 0.9 nmol per dose)].[56] The nanocomposites were incubated at room temperature for another 10 min. DMEM was added to the nanocomposites to make the final volume up to 200 μL for one injection.
In vivo administration of nanocomposites:
Female BALB/c mice were purchased from Jackson Laboratory. All animal studies were performed following authorized protocol (IACUC Protocol ID 2016–0051) and the policies of UMass Institutional Animal Care and Use Committee. BALB/c mice (n=5) received four intravenous injections of nanocomposites through tail vein and were euthanized 2 days after the last injection.
MS ICP-MS biodistribution:
From each mouse, organs were extracted for detection of gold content. Tissue or sorted cells were digested with 2 mL of a 3:1 v/v mixture of HNO3:H2O2 and left for 24h for complete digestion. After 24 hours, the solution was diluted to 10 mL using MQ water. Solutions were analyzed by ICP-MS using a Perkin Elmer NexION 300X (Waltham, MA). A daily performance test and a set of freshly prepared Au calibration solutions (0 ppb to 20 ppb) were used as standards to perform quantitative measurements.
LA-ICP-MS Imaging:
Each organ was flash-frozen using liquid nitrogen and then sliced to 20 μm using a LEICA CM1850 cryostat microtome. Tissue slices were attached to regular glass slides and used for LA-ICP-MS imaging and E&H staining. A CETAC LSX-213 G2 laser ablation system (Photon Machines, Omaha, NE) was attached to the Perkin Elmer NexION 300X (Waltham, MA). The Laser Ablation LA Parameters correspond to: laser spot size, 100 μm; line distance, 10 μm; scan speed, 10 μm/s; energy, 3.34 J. The data was analyzed by a python script and images were generated in ImageJ.
Immunofluorescence staining for flow cytometry sorting analyses:
After mice were sacrificed, the desired organs were ground and processed to form a single cell suspension in cell staining buffer. Red blood cell lysis buffer was added to lyse the red blood cell before Fc-receptor blocking. Purified rat anti-mouse CD16/CD32 (BD Pharmingen) was used to block non-specific staining of antibodies. After blocking, FITC anti-mouse/human CD11b antibody (BioLegend), brilliant violet 421 anti-mouse CD19 antibody (BioLegend), and PE anti-mouse CD3 antibody (BioLegend) were used to labeled macrophages, B cell, and T cells, and the pool of cells were analyzed and sorted by FACS (BD FACSAriz II).
Determining allele modification frequencies via T7E1 assay:
After mice were sacrificed, all organs were harvested, ground, and different types of immune cells were taken for immunofluorescence staining. The sorted cells were then subjected to genomic DNA extraction using QuickExtract genomic DNA isolation kit (Epicentre Biotechnologies). PCR was performed to generate amplicons surrounding sgRNA target sites. The sequences of on-targeting and off-targeting primer pairs are shown in Supplementary Table 1. The purified PCR products were denatured, re-annealed and digested with T7 Endonuclease I (New England BioLabs). Digested samples were resolved by electrophoresis in a 4%−20% gradient TBE gel (Invitrogen), stained with SYBR gold (ThermoFisher), marker used was O’GeneRuler 100bp Plus (Thermo Scientific), visualized by gel imager (BioRad) and indel frequencies were determined based on standard protocol.[50]
Serum TNF-α, ALT, and Bilirubin assays:
Blood was collected after sacrificing the mice, and serum was separated using a Microtainer tube with serum separator (BD) at 5,000 r.p.m. for 10 min, and the supernatant serum was carefully collected for TNF-α by ELISA (R&D Systems, MN, USA)
Serum total ALT and Bilirubin levels were assayed in triplicate using the ALT Color Endpoint Reagent Kit and Total Bilirubin Reagent Set (TECO Diagnostics), respectively, according to the manufacturer’s instructions.
Statistical analysis:
All data are shown as means ± standard deviations (SD). Statistical analysis was performed by one-way ANOVA.
Supplementary Material
Acknowledgements
This research was supported by the NIH (GM077173 and EB022641), National Research Service Award T32 GM008515 from the NIH, the NSF (CHE-18081991) and a UMass OTCV grant. Yi-Wei Lee and Rubul Mout contributed equally to this work.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
References
- [1].Naldini L, Nature 2015, 526, 351–360. [DOI] [PubMed] [Google Scholar]
- [2].Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M, J. Pathol 2013, 229, 176–185. [DOI] [PubMed] [Google Scholar]
- [3].Ganz T, Blood, 2003, 102, 783–788. [DOI] [PubMed] [Google Scholar]
- [4].Hotamisligil GS, Nature, 2006, 444, 860–867. [DOI] [PubMed] [Google Scholar]
- [5].Sands MS, Davidson BL, Mol Ther. 2006, 13, 839–849. [DOI] [PubMed] [Google Scholar]
- [6].Moore KJ, Tabas I, Cell, 2011, 145, 341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fiala M, Lin J, Ringman J, Kermani-Arab V, Tsao G, Patel A, Lossinsky AS, Graves MC, Gustavson A, Sayre J, Sofroni E, Suarez T, Chiappelli F, Bernard G, J. Alzheimers Dis 2005, 7, 221–232. [DOI] [PubMed] [Google Scholar]
- [8].Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier EA, Science 2012, 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM, Science 2013, 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cho SW, Kim S, Kim JM, Kim JS, Nat. Biotechnol 2013, 31, 230–232. [DOI] [PubMed] [Google Scholar]
- [11].Luo YL, Xu CF, Li HJ, Cao ZT, Liu J, Wang JL, Du XJ, Yang XZ, Gu Z, Wang J, ACS Nano, 2018, 12, 994–1005. [DOI] [PubMed] [Google Scholar]
- [12].Sander JD; Joung JK CRISPR-Cas Systems for Editing, Regulating and Targeting Genomes. Nat. Biotechnol 2014, 32, 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pei Y, Yeo Y, J. Control. Release, 2016, 240, 202–211. [DOI] [PubMed] [Google Scholar]
- [14].Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, Maeder ML, Joung JK, Chen ZY, Liu DR, Nat. Biotechnol 2014, 33, 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang HX, Song Z, Lao YH, Xu X, Gong J, Cheng D, Chakraborty S, Park JS, Li M, Huang D, Yin L, Cheng J, Leong KW, Proc. Natl. Acad. Sci 2018, 115, 4903–4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Swiech L, Heidenreich M, Banerjee A, Habib N, Li Y, Trombetta J, Sur M, Zhang F, Nat. Biotechnol 2014, 33, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kim SM, Shin SC, Kim EE, Kim SH, Park K, Oh SJ, Jang M, ACS Nano 2018, 12, 7750–7760. [DOI] [PubMed] [Google Scholar]
- [18].Yin H, Song CQ, Suresh S, Wu Q, Walsh S, Rhym LH, Mintzer E, Bolukbasi MF, Zhu LJ, Kauffman K, Nat. Biotechnol 2017, 35, 1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, Bhattacharyya S, Shelton JM, Bassel-Duby R, Olson EN, Science 2016, 351, 400–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chew WL, Tabebordbar M, Cheng JKW, Wu EY, Ng AHM, Zhu K, Wagers AJ, Church GM, Nat. Methods 2016, 13, 868–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang D, Mou H, Li S, Li Y, Hough S, Tran K, Li J, Yin H, Anderson DG, Sontheimer EJ, Weng Z, Gao G, Xue W, Hum. Gene Ther 2015, 26, 432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA, Zhang F, Nature 2015, 520, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kim E, Koo T, Park SW, Kim D, Kim K, Cho H-Y, Song D, Lee K, Jung M, Kim S, Kim J, Kim J, Kim J-S, Nat. Comm 2017, 8, 14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yin H, Song CQ, Dorkin JR, Zhu LJ, Li Y, Wu Q, Park A, Yang J, Suresh S, Bizhanova A, Gupta A, Bolukbasi MF, Walsh S, Bogorad RL, Gao G, Weng Z, Dong Y, Koteliansky V, Wolfe SA, Langer R, Xue W, Anderson DG, Nat. biotech 2016, 34, 328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sun W, Ji W, Hall JM, Hu Q, Wang C, Beisel CL, Gu Z, Angew. Chem. Int. Edit 2015, 54, 12029–12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kim S, Kim D, Cho SW, Kim J, Kim J-S, Genome Res. 2014, 24, 1012–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yu X, Liang X, Xie H, Kumar S, Ravinder N, Potter J, Jeu X, Chesnut JD, Biotechnol. Lett 2016, 38, 919–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mout R, Ray M, Yesilbag Tonga G, Lee Y-W, Tay T, Sasaki K, Rotello VM, ACS Nano 2017, 11, 2452–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mout R, Ray M, Lee Y-W, Scaletti F, Rotello VM, Bioconjugate Chem. 2017, 28, 880–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lee K, Conboy M, Park HM, Jiang F, Kim HJ, Dewitt MA, Mackley VA, Chang K, Rao A, Skinner C, et al. Nat. Biomed. Eng 2017, 1, 889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wang M, Zuris JA, Meng F, Rees H, Sun S, Deng P, Han Y, Gao X, Pouli D, Wu Q, Georgakoudi I, Liu DR, Xu Q, Proc. Natl. Acad. Sci 2016, 113, 2868–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ha JS, Lee JS, Jeong J, Kim H, Byun J, Kim SA, Lee HJ, Chung HS, Lee JB, Ahn D-R, J. Control. Release 2017, 250, 27–35. [DOI] [PubMed] [Google Scholar]
- [33].Lammers T, Kiessling F, Hennink WE, Storm G, J. Control. Release, 2012, 161, 175–183. [DOI] [PubMed] [Google Scholar]
- [34].Xue HY, Liu S, Wong HL, Nanomedicine, 2014, 9, 295–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang M, Glass ZA, Xu Q, Gene Ther. 2016, 24, 144–150. [DOI] [PubMed] [Google Scholar]
- [36].Mout R, Tonga GY, Wang L-S, Ray M, Roy T, Rotello VM, ACS Nano 2017, 11, 3456–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mout R, Ray M, Tay T, Sasaki K, Tonga GY, Rotello VM, ACS Nano 2017, 11, 6416–6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ray M, Lee YW, Hardie J, Mout R, Yesilbag Tonga G, Farkas ME, Rotello VM, Bioconjug. Chem 2018, 29, 445–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Walkey CD, Olsen JB, Guo H, Emili A, Chan WC, J. Am. Chem. Soc 2012, 134, 2139–2147. [DOI] [PubMed] [Google Scholar]
- [40].Moyes KW, Lieberman NAP, Kreuser SA, Chinn H, Winter C, Deutsch G, Hoglund V, Watson R, Crane CA, Hum. Gene Ther 2017, 28, 200–215. [DOI] [PubMed] [Google Scholar]
- [41].Kalos M & June CH, Immunity 2013, 39, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Cantore A, Milani M, Annoni A, Liu T, Bartolaccini S, Biffi M, Russo F, Peters R, Lombardo A, Nichols TC, Ayuso E, Naldini L, Blood 2017. 130, 605. [Google Scholar]
- [43].Wang Y, Huang LA, Nat. biotechnol, 2013, 31, 611–612. [DOI] [PubMed] [Google Scholar]
- [44].Kim ST, Saha K, Kim C, Rotello VM, Accounts Chem. Res 2013, 46, 681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Elci SG, Yan B, Kim ST, Saha K, Jiang Y, Klemmer GA, Moyano DF, Tonga GY, Rotello VM, Vachet RW, Analyst 2016, 141, 2418–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Willekens FLA, Werre JM, Kruijt JK, Roerdinkholder-Stoelwinder B, Groenen-Döpp YAM, van den Bos AG, Bosman GJCGM, van Berkel TJC, Blood 2005, 5, 2141–2145. [DOI] [PubMed] [Google Scholar]
- [47].Elci SG, Jiang Y, Yan B, Kim ST, Saha K, Moyano DF, Yesilbag Tonga G, Jackson LC, Rotello VM, Vachet RW, ACS Nano 2016, 10, 5536–5542. [DOI] [PubMed] [Google Scholar]
- [48].Bayik D, Tross D, Haile LA, Verthelyi D, Klinman DM, Blood Advances 2017, 1, 2510–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hirayama D, Iida T, Nakase H, Int. J. Mol. Sci 2018, 19, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ran FA, Hsu PD, Wright J, Agawala V, Scott DA, Zhang F, Nat. Protoc 2013, 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Synthego Performance Analysis ICE Analysis. 2019. v1.1 Retrieved from https://ice.synthego.com/#/ (accessed 1 July 2019).
- [52].Appledorn DM, McBride A, Seregin S, Scott JM, Schuldt N, Kiang A, Godbehere S, Amalfitano A, Gene Ther, 2008, 15, 1606–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Benchling [Biology Software] (2019). Retrieved from www.benchling.com (accessed 1 July 2019).
- [54].Gagnon JA, Valen E, Thyme SB, Huang P, Ahkmetova L, Pauli A, Montague TG, Zimmerman S, Richter C, Schier AF, PLoS One, 2014, 9, e98186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Yang XC, Samanta B, Agasti SS, Jeong Y, Zhu ZJ, Rana S, Miranda OR, Rotello VM, Angew. Chem., Int. Ed 2011, 50, 477–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Mout R, Rotello VM, Bio-protocol, 2017, 7, e2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.