

Abstract
Development of small molecule inhibitors of protein-protein interactions (PPI) is hampered by our poor understanding of the druggability of PPI target sites. Here, we describe the application of alanine-scanning mutagenesis, fragment screening, and FTMap-based computational hot-spot mapping to comprehensively evaluate the druggability of the PPI interface between Kelch like ECH Associated Protein-1 (KEAP1) and Nuclear Factor, Erythroid 2 Like 2 (Nrf2), an important drug target. FTMap identifies four major binding-energy hot spots at the active site, only two of which are exploited by Nrf2, which interacts in a two-pronged manner primarily driven by Nrf2 residues Glu79 and Glu82. We identify fragment hits, and develop a new crystal form of KEAP1 with a binding site free of intermolecular contacts which we use to obtain X-ray complex structures for three of these fragments via crystal soaking. Our results reveal key interactions important for ligand binding to KEAP1, and highlight the role of ligand-induced conformational change of KEAP1 residue Arg415 in allowing access to a deep hole at the bottom of the binding site where FTMap predicts additional binding energy can be gained. Despite the highly charged nature of the DEETGE binding loop on Nrf2, our results suggest that all of these charges may be dispensed with in a synthetic ligand. We additionally discuss how the complementary information provided by alanine scanning, fragment screening and computational hot spot mapping can be integrated to more accurately evaluate PPI druggability.
Graphical Abstract
Introduction
Protein-protein interactions (PPI) mediate a vast number of cellular pathways and processes, making them an important class of targets for drug discovery. However, decades of effort aimed at developing small molecule drugs to modulate PPIs have been rewarded with relatively few successes. The difficulty is thought to arise primarily from the topology of PPI interfaces (1–7), many of which lack any binding pocket or cleft of suitable dimensions to bind a drug-sized small molecule ligand (< 500 Da). For this reason, screening large compound libraries often fails to identify useful hits and leads against PPI targets (2, 4, 8–21). Extensive experience applying fragment-based lead discovery (FBLD) to PPI targets, which enables a more comprehensive and unbiased search of chemical space, has not solved this problem for the majority of cases (22–24), indicating that the difficulty is not simply due to a rarity of compounds that can bind well, or to inappropriate compound libraries. Rather, this outcome indicates that many PPI surface sites are intrinsically undruggable by conventional druglike compounds. There are, however, a number of examples of PPI targets for which high affinity, druglike inhibitors have been obtained (25, 26), indicating that some PPI interfaces, while challenging, are in fact druggable. It is therefore important to have reliable methods for evaluating the druggability of PPI sites, given the considerable time and resources necessary to pursue the discovery of small molecule inhibitors, especially against a challenging target.
A PPI target of high pharmaceutical interest is that of Kelch like ECH Associated Protein-1 (KEAP1) with Nuclear Factor, Erythroid 2 Like 2 (Nrf2). KEAP1 is a 624-residue protein containing a dimerization domain, an intervening domain, and a β-propeller domain comprising six kelch-like repeats (Figure 1). A single Nrf2 molecule binds both β-propeller domains in the KEAP1 dimer, using two distinct motifs in the Neh2 domain of Nrf2 – a high affinity DxETGE motif, and a lower affinity DLG motif (34, 58). Nrf2 is a major activator of the cellular response to oxidative stress (27–29). In the presence of Reactive Oxygen Species (ROS), intracellular levels of Nrf2 increase and Nrf2 migrates to the nucleus where, in complex with Maf, it binds to the Antioxidant Response Element (ARE), an enhancer sequence in the promoter region of many genes that encode antioxidant and Phase II detoxifying enzymes/proteins. The cytoplasmic redox-sensitive protein KEAP1 serves as a repressor of Nrf2 by binding it and mediating its E3 ligase-dependent downregulation (30). Elevated levels of ROS covalently modify cysteine residues on various domains of KEAP1, altering its function so as to suppress Nrf2 ubiquitination (31). Inactivation of KEAP1 thus causes Nrf2 to accumulate, resulting in upregulation of Nrf2 dependent cytoprotective genes (31–34). Up-regulation of Nrf2 has been shown to provide protection against inflammatory damage, and to be beneficial in a range of pathologies including cardiovascular disease and neurodegeneration (27, 35–37). Conversely, elevated Nrf2 levels induced by KEAP1 mutations that abolish Nrf2 binding have been associated with lung, breast, ovarian, and endometrial cancers (38–45).
Figure 1.

Cartoon showing the domain structures of (A) KEAP1 and (B) NrfZ with the fusión constructs of each that were used in this study shown below. The locations of the DLG and DxETGE motifs of Nrf2 are indicated, and the residues selected for mutagenesis are highlighted in red. (C) Crystal structure of the kelch repeat domain of KEAP1 (wheat) in complex with a peptide corresponding to residues 69–84 of Nrf2 (green) encompassing the DxETGE motif (PDB 2FLU), shown in two views.
Given the crucial role of the KEAP1/Nrf2 interaction in regulating cellular defenses against oxidative stress, and its implication in multiple diseases, a therapeutic agent that directly disrupts this PPI is highly desirable (31, 46, 47). Indeed, Tecfidera® (dimethyl fumarate), an approved drug for treating multiple sclerosis, is thought to act, at least in part, by covalently modifying one or more cysteine residues on KEAP1, either directly or as its partially hydrolyzed metabolite, monomethylfumarate, causing elevated levels of Nrf2 and resulting cytoprotective responses (48–51). However, these simple fumarate esters are relatively nonspecific Michael acceptors, and so multiple research groups have tried to identify potent and selective noncovalent inhibitors of the KEAP1/Nrf2 interaction. So far, reported efforts have shown only limited success in identifying potent, druglike inhibitors of KEAP1, with sub-micromolar potency seemingly requiring that the compounds possess one or two unmodified carboxylates (52, 53), which present obstacles for the further development of these chemotypes as oral drugs. Whether or not the apparent difficulty of this target indicates an intrinsic and insurmountable limitation to the druggability of KEAP1 is therefore a pressing question (53–57).
The KEAP1/Nrf2 interface is unusual for a PPI, in that the primary region on Nrf2 that binds KEAP1, a peptide loop with the sequence DEETGEF (34, 58), contains a high density of negatively charged side-chains. The complementary binding site on KEAP1 contains several arginine residues. The highly charged nature of this interaction sharply contrasts with typical PPI interfaces which, while amphipathic, generally involve few aspartate or glutamate residues (59). The behavior of previously characterized PPI targets therefore provides only limited guidance as to the druggability of Nrf2/KEAP1. In particular, it is unclear to what extent the difficulty in identifying potent druglike inhibitors of KEAP1 is due to the unusual charge features of the interface versus other characteristics of the site. A detailed examination of the druggability of Nrf2/KEAP1, using both experimental approaches and physics-based computational methods, can potentially shed light on the overall druggability of this important target, and on particular approaches by which more potent and selective inhibitors might be achieved.
One approach to evaluating the druggability of protein targets, including PPI targets, involves assessing the number, strength and spatial distribution of binding energy hot spots present at the relevant region of the protein’s surface. A well-established experimental method for identifying hot spots at PPI interfaces is alanine scanning mutagenesis (ASM) (60). In this approach, each interface residue in turn is mutated to alanine, and the ensuing effect on the binding affinity of the PPI complex is interpreted in terms of the energetic contribution made by the side-chain atoms of the substituted residue. Experiments of this kind reveal that, in general, binding energy is not distributed uniformly over a PPI interface, but instead is dominated by a small subset of interface residues (60). These residues, and the atoms on the partner protein with which they interact, comprise binding energy hot spots (61–64). Alanine scanning mutagenesis has been widely used to dissect the origins of binding energy at PPI interfaces. However, it has long been realized that interpreting the results in terms of binding energy contributions attributable to particular residues can sometimes be incorrect, for example if a mutation causes a long-range disruption of the structure of the interface (65).
Another experimental method for probing binding energy hot spots is fragment screening (22–24, 66–71). In this approach, the target protein is screened against a library of molecular “fragments” – simple organic compounds of molecular weight less than ~300 Da. – to identify regions on the protein surface that can interact favorably with these small ligands. In an early example of fragment screening, Ringe and coworkers showed that solving the X-ray structure of a protein crystal soaked in organic co-solvents identified particular regions of the protein surface that possessed a general tendency to bind the co-solvent molecules, and established that these binding regions tended to coincide with energetically important subsites within the ligand binding site (72). It is now well established that fragments tend to localize at binding energy hot spots (73, 74).
Binding energy hot spots can also be identified computationally. One well-validated computational method is FTMap, which approaches the problem by placing small virtual probe molecules on a dense grid around the surface of the protein, and then performing a series of energy minimization and clustering steps to identify regions of the protein that have a tendency to interact favorably with the probe. This calculation is repeated for a series of different probe structures, and the results from all probes are superimposed to identify “Consensus Clusters” (CC), which are small surface regions on the protein that display a general tendency to bind small organic compounds (77, 78). FTMap results have been validated by comparison with a large number and variety of experimental binding data, showing that the locations of the FTMap Consensus Clusters closely coincide with binding energy hot spots discovered by alanine scanning mutagenesis (79, 80), multiple solvent crystal structures (81), experimental fragment screening (74), or the binding of larger ligands (82, 104). For example, Zerbe et al. showed that FTMap accurately identified the hot spot residues detected by ASM (80). The agreement between FTMap and ASM was also prospectively validated for the PPI interaction interface between NF-κB Essential Modulator (NEMO) and Inhibitor of kappa-B kinase-β (IKKβ) (79). These studies established that the hot spots identified using FTMap generally coincide closely with those found experimentally by alanine scanning mutagenesis.
The agreement between FTMap hot spots and those revealed by experimental fragment screening has also been studied. For example, it has been shown that the site on a protein where most fragments bind corresponds to the top ranking FTMap hot spot (74). Moreover, conflicting results concerning whether fragments obtained by deconstructing a larger ligand will or will not retain their binding geometry were resolved by showing that the binding mode will be retained if the deconstructed fragment sufficiently complements the top ranked FTMap hot spot (74). The importance of the top ranking hot spot for fragment binding is consistent with the concept of “anchor sites”, introduced by Camacho and coworkers (83), in which one strong hot spot serves to energetically anchor a larger ligand whose binding encompasses that site. A similar hypothesis has been proposed by Blundell et al., who described a new computational method, based on the SuperStar algorithm (84), for predicting fragment binding. They showed that the likelihood of experimentally measured fragment binding was highest at the top-ranked binding hot spots identified by their method (85).
The methods of alanine scanning, fragment screening and computational hot spot mapping each provides information about the locations and strengths of binding energy hot spots at PPI interfaces. However, each method is potentially subject to false positive and or false negative results that complicate interpretation of the data to assess the druggability of a given target. Here, we describe the combined application of alanine scanning mutagenesis, fragment screening, and FTMap to provide a comprehensive evaluation of the druggability of the PPI interface between KEAP1 and Nrf2. We use the results to develop a framework for understanding the overlap and differences in the information provided by these three methods in relation to the structure of the true hot spot ensemble, and discuss how the complementary information these three approaches provide can be used to more accurately evaluate PPI druggability. In applying these methods to KEAP1, we characterized the location and strength of each binding hot spot. Despite the highly charged nature of the binding loop on Nrf2, our results suggest that all of these charges can be dispensed with in a synthetic ligand, and that KEAP1 is likely druggable by conventional druglike ligands. We additionally propose how the binding energy hot spots identified in this study might be exploited to achieve stronger and more selective binding to KEAP1.
Results
Experimental Alanine Scanning Mutagenesis of KEAP1/Nrf2 Binding Interface
Protein Design, Expression and Purification.
A recombinant KEAP1 construct was generated comprising residues 312–624, which encompasses the β-propeller domain that interacts with Nrf2 (58, 86) (Figure 1). Two different Nrf2 Neh2 domain constructs were cloned and expressed; the longer, Nrf21–100, encompasses both the high affinity DxETGE KEAP1-binding motif, which spans residues 74–84, and the low affinity DLG motif spanning residues 24–31 (34, 58), while the shorter, Nrf234–100, lacks the region containing the DLG motif (Figure 1B). All three constructs were subcloned into pET15b vectors, which append an N-terminal His6 tag followed by a tobacco etch virus (TEV) protease cleavage site, expressed in E. coli, and purified by Ni-NTA affinity chromatography. After treatment with TEV protease, the proteins were again passed over a Ni-NTA affinity column to remove the cleaved His6 tag, followed by gel filtration as a final purification step and to remove protein aggregates. The proteins were characterized by SDS-PAGE, MALDI-MS, circular dichroism (CD) spectrometry, and thermal denaturation to establish their identity, purity, homogeneity, and structural integrity, as described in Materials and Methods. Thermal denaturation analysis of KEAP1312–624 showed this construct to have a stable tertiary structure, as expected (Supplementary Figure S1). However, Nrf21–100 and Nrf234–100 showed no cooperative melting transition, and the CD spectra of these constructs indicated structures that are mostly random coil (Supplementary Figure S2), consistent with the Neh2 domain of Nrf2 being largely intrinsically disordered (34, 87, 88).
The binding activities of the recombinant proteins were characterized using a fluorescence anisotropy (FA) competition binding assay. As tracer probe, we used a fluorescein isothiocyanate (FITC) labeled 9-mer synthetic peptide, LDEETGEFL, derived from Nrf2 residues 76–84 which encompass the DxETGE binding motif (57, 89, 90). Titration of recombinant KEAP1312–624 against a fixed concentration of FITC-Nrf2 peptide gave a concentration-dependent increase in anisotropy (Figure 2A), consistent with KEAP1 binding to FITC-Nrf2 (79, 91). Fitting the data to a quadratic binding equation gave an affinity of KD = 11 ± 1 nM (n = 5), consistent with previously reported values for Nrf2 peptides binding to KEAP1 (34, 57, 58, 89). Incubation of various concentrations of unlabeled synthetic Nrf276–84 peptide with fixed, sub-saturating concentrations of KEAP1 (10 nM) and FITC-Nrf2 (5 nM) gave dose-dependent inhibition of the anisotropy signal (Figure 2B). The binding affinity of this unlabeled Nrf2 peptide was determined by fitting the inhibition data to a competitive equilibrium binding model using the curve-fitting software DynaFit (79), as described in Materials and Methods. This approach to data analysis was required because, under our experimental conditions, all three binding components are present at concentrations close to their KD values. Consequently, the approximation that total concentration ≈ free concentration is not valid for any binding component, and so accurate KD values cannot be determined from IC50 values simply by applying a Cheng-Prusoff (92) correction factor (79). Analyzing the binding data as described gave KD = 13 ± 2 nM (n = 3) for the unlabeled 9-mer Nrf2 peptide, similar to that measured for the FITC-labeled peptide.
Figure 2.

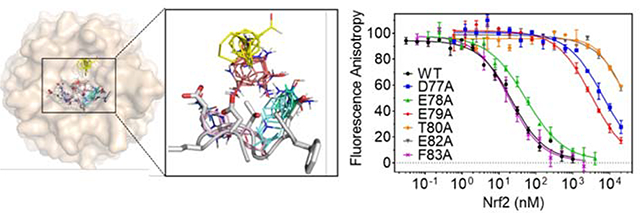
Alanine Scanning Mutagenesis of Nrf2 DxETGE motif. (A) Binding of 5 nM FITC-Nrf276–84 to various concentrations of KEAP1312–624 measured by fluorescence anisotropy. The solid line shows the best fit to a quadratic binding equation. (B) Inhibition of the interaction of FITC-Nrf276–84 (5 nM) to KEAP1312–624 (10 nM) by unlabeled Nrf276–84 peptide. The solid line is the best fit to a competitive inhibition binding model, obtained using DynaFit (see Materials and Methods). (C) Close-up view of the Nrf2 ß-hairpin loop structure with (left) and without (right) KEAP1 protein shown. Residues selected for alanine-scanning mutagenesis are highlighted in pink. (D) Inhibition dose-response curves for wild-type Nrf34.100 and the six alanine variant. Solid lines are best fits of the data to a competitive inhibition binding model, using DynaFit. Error bars show the range of duplicate measurements. Results shown are representative of at least three independent experiments. (E) The decrease in KEAP1/Nrf2 interaction energy for each alanine variant, calculated from AAG = -RTln(KDwt/KDmut).
To validate the binding of the recombinant Nrf2 constructs to the KEAP1 Kelch domain, unlabeled Nrf21–100 or Nrf234–100 were tested as inhibitors in the FA assay (Supplementary Figure S3). Analyzing the inhibition data as described above gave values of KD = 10 ± 2 nM (n = 4) for Nrf234–100, and KD = 10 ± 1 nM (n = 3) for Nrf21–100. The similar KD values seen for the two recombinant Nrf2 constructs, compared to the 9-mer peptide, suggest that the 9-mer comprises the entirety of the high affinity KEAP1 binding motif. Moreover, the insensitivity of the measured affinity to the presence of the DLG motif indicates that, for the longer construct in which both binding motifs are present, binding to KEAP1 is primarily through DxETGE, as expected given its higher affinity (33, 34).
Design and characterization of Nrf2 Alanine Variants.
To probe the molecular origins of the interaction energy that drives KEAP1/Nrf2 binding, the contribution made by each residue in and around the high affinity Nrf2 DxETGE binding motif was systematically assessed using alanine scanning mutagenesis (60, 93–95). The shorter construct, Nrf234–100, was used as the template for the alanine variants. Published X-ray structures of KEAP1 bound by an Nrf2-derived peptide encompassing the DxETGE motif (58, 86) show that Nrf2 residues spanning D77 to F83 (DEETGEF) occupy the binding site on KEAP1. Glycine lacks any side-chain, and therefore G81 was not amenable to alanine scanning mutagenesis. We mutated each of the remaining six residues within the DEETGEF region of Nrf234–100 individually to alanine (Figure 2C). These six variant proteins were cloned, expressed, purified, and characterized as described above for wild-type Nrf234–100. The binding affinity of each variant for KEAP1 was evaluated in the competition FA binding assay (Figure 2D), and the KD values obtained by fitting the inhibition data to the competition binding model, as described above.
The binding affinities measured for the six Nrf2 variants tested in the FA assay are shown in Table 1. Four residues, D77, E79, T80 and E82, when mutated to alanine caused a substantial reduction in binding affinity, giving KDmut/KDWT ratios from 170–2600, corresponding to a reduction in binding energy of ΔΔG = 3.0–4.5 kcal/mol (Figure 2E). In contrast, mutation of either E78 or F83 to alanine resulted in ΔΔG < 0.5 kcal/mol, indicating that these residues do not play a significant role in stabilizing the KEAP1/Nrf2 interaction. Of the four residues found to be important for binding, X-ray crystal structures (PDB entries 2FLU(58) and 4IFL) show that the carboxyl group of E79 engages in a salt-bridge with KEAP1 residues R415 and R483 and forms a hydrogen bond with the hydroxyl group of S508. Similarly, the carboxylate group of E82 forms a salt bridge with KEAP1 residue R380, and hydrogen bonds with the side chains of S363 and N382. In contrast, Nrf2 residues D77 and T80 appear to make relatively little contact with KEAP1 (Supplementary Table 1). These X-ray structures instead show that the carboxyl group of D77 and the hydroxyl group of T80 engage in intramolecular hydrogen-bonding interactions with Nrf2 backbone amide groups and with one another across the hairpin loop of the Nrf2 peptide (Supplementary Figure S4). We investigated the role of T80 further, by mutating it to serine to eliminate the T80 methyl group but preserve its ability to hydrogen bond through its hydroxyl group to D77. Compared to the T80A variant, T80S showed a substantial restoration of the w.t. binding affinity, giving KDT80S/KDWT ~ 30, compared to KDT80A/KDWT ~ 2600 for the T80A variant. This result suggests that the major effect on binding of mutating T80 to alanine arises from loss of hydrogen-bonding interactions involving the T80 hydroxyl group.
Table 1.
Binding Affinities and Binding Energetics for Nrf234–100 Alanine Variants
| Nrf2 | WT | D77A | E78A | E79A | T80A | E82A | F83A |
|---|---|---|---|---|---|---|---|
| KD (nM) | 10±2 | (4.4 ±1.8) ×103 | 25 ± 3 | (1.7 ± 0.2) ×103 | (2.6 ± 1.5) ×104 | (2.2±1.3) ×104 | 18 ± 3 |
| ΔΔG (kcal/mol) | n/a | 3.5 ± 0.3 | 0.5 ± 0.1 | 3.0 ± 0.1 | 4.5 ± 0.4 | 4.4 ± 0.4 | 0.3 ± 0.1 |
To test whether disrupting the D77-T80 interaction changed the binding geometry of the ligand, we obtained a crystal structure of KEAP1 bound by a 9-mer peptide, LDEEAGEFL, containing an alanine at the position corresponding to T80. To do this we developed a new crystal form of human KEAP1 that affords an exposed binding site, devoid of crystal contacts, to facilitate ligand soaking (Supplementary Figure S5A). Previously, crystal contacts have been engineered in KEAP1 for the same purpose, by replacing glutamate residues 540 and 542 with alanine, and simultaneously replacing cysteine residues 319, 613, and 622 with serine to improve protein expression and solubility (102). We introduced these same mutations, and performed crystal screening to identify the crystal form most amenable to soaking. A new crystal form, grown in space group C2 using ammonium sulfate as precipitant, formed different crystal contacts compared to all previously described human KEAP1 structures. The new structure differs from the previously reported structure of unbound KEAP1 developed to be suitable for soaking (102) in that the loops that decorate the periphery of the ligand binding site are involved in fewer contacts, and there is no intermolecular disulfide bond between cysteine 434 and its counterpart on the neighboring chain. Like other KEAP1 structures, crystal packing produces two monomers in the asymmetric unit (chains A and B). In our structure, the blade 2 BC-loop of chain B interacts with the DA loop between blades 1 and 2 of chain A (Supplementary Figure S5A), shifting of chain B away from the chain A ligand binding site, and fully opening the chain A ligand binding site to the solvent channel. The utility of this new crystal form for ligand soaking was validated by soaking crystals with an 8-mer Nrf-2 derived peptide, LDEETGEF. The soaked crystals diffracted to 1.91 Å. The refined structure showed the Nrf2 peptide to have a binding pose that was essentially superimposable with that seen in co-crystal structures of Nrf2 with KEAP1 (58, 86) (Supplementary Figure S5B). Upon soaking into this new crystal form of KEAP1, the T80A variant peptide bound with a geometry very similar to that observed for the unmodified peptide (Supplementary Figure S5C). Interestingly, analysis of the crystallographic B-factors showed that the degree of structural disorder in this bound mutant peptide steadily increased from the middle to each end, whereas no such trend was seen with the soaked wild-type peptide (Supplementary Figure S5D). Taken together, our data suggest that the large contributions that D77 and T80 make to the stability of the complex with KEAP1, as revealed by alanine scanning, do not primarily result from interactions with KEAP1, but rather from stabilization of the bound hairpin conformation of the DxETGE loop of Nrf2.
Evolutionary conservation of Nrf2/KEAP1 Binding Energetics
The DxETGE motif is strictly conserved across 27 Nrf orthologs contained in the National Center for Biotechnology Information (NCBI) Gene database (Supplementary Figure S6A). This motif remains strictly conserved among a larger set of 60 sequences that includes Nrf2 paralogs (Nrf1), while among this larger set the two residues that we identified to be energetically unimportant for binding, E78 and F83, are frequently replaced by Gly and Ser, respectively. On the KEAP1 side of the interface, of the ten residues that contact Nrf2 (58), six of these interact with atoms on Nrf2 that our alanine scanning results show to be involved in energetically important interactions. These residues, S363, R380, N382, R415, R483 and S508, are strictly (S363, R380, R415, S508) or very highly (R483, 97%; N382, 92%) conserved across 37 KEAP1 orthologs, including the highly mobile R415 (Supplementary Figure S6B). The high sequence conservation on both sides of the interface supports the notion that the detailed binding energetics of the Nrf2 DxETGE motif, including occupancy of hot spots B and C by glutamate side-chains, and stabilization of the hairpin loop by polar interactions involving D77 and T80, are highly conserved across species encompassing a wide range of evolutionary complexity. In humans, in addition to Nrf2, several other proteins have been identified to bind KEAP1 through a motif resembling DxETGE (Figure 3). These ligands include Inhibitor of nuclear factor Kappa-B Kinase subunit beta (IKKβ), Partner and Localizer of BRCA2 (PALB2), PGAM Family Member 5, Mitochondrial Serine/Threonine Protein Phosphatase 5 (PGAM5) and Nucleoporin p62 (110–114). In both IKKβ and PGAM5, D77 is replaced by Asn, suggesting that the charged-neutral hydrogen bonds between D77 and T80 and nearby NH groups of Nrf2 can be substituted by neutral-neutral hydrogen bonds. In PGAM5, T80 is replaced by a serine, in keeping with the observation that it is the hydroxyl group of T80, and not the methyl group, that is most important for binding. Both key glutamates on Nrf2, E79 and E82, are strictly conserved among these KEAP1 ligands, with the sole exception of p62 which has a serine in place of E79. However, binding of p62 is regulated by phosphorylation of this serine, which increases the binding affinity for KEAP1 by >30-fold (113, 114). Thus, in p62 the carboxylate of E79 is in effect substituted by the anionic phosphate group. Comparison of the Nrf2 ortholog sequences therefore suggests that these other ligands exploit the hot spot ensemble at the KEAP1 binding sites in very similar ways to achieve binding, despite some minor variations in the sequence of the binding motif.
Figure 3.

Alignment of other human KEAP1 ligands that bind through a motif resembling DxETGE, showing conservation of key binding and structural elements.
Fragment Screening against KEAP1/Nrf2
As a further probe of the locations of binding-energy hot spots at the Nrf2 binding site on KEAP1, the KEAP1 construct was screened against a library of 458 fragment-sized compounds (22, 24, 68, 70, 71), 389 of which were obtained commercially and the remaining 69 from our internal BU-CMD compound collection. To identify fragments binders we employed a ThermoFluor assay, which detects ligands through their ability to increase the melting temperature (Tm) of the protein target (96–99) and, in parallel, the FA competition binding assay described above. For the ThermoFluor screen, fragments were tested individually at a single concentration of 10 mM. Six fragments were found to increase the Tm of KEAP1 by ≥ 3 °C, and an additional seven caused a smaller but still significant stabilization. Compared to the ThermoFluor method, FA assays are typically more sensitive to artifacts resulting from poor compound solubility (100, 101). Screening the fragment collection in the FA assay was therefore performed at a slightly lower compound concentration of 5 mM in a buffer containing 10 % v/v DMSO. FA results for compounds that were not completely soluble under these conditions, defined as compounds giving an absorbance (from light scattering by insoluble aggregates) of >0.1 at 700 nm, were not considered further. The FA screen identified 38 hits (Supplementary Table 2), of which seven were also among the hits found in the ThermoFluor assay (Figure 4A). Attempts to demonstrate dose-dependent inhibition for fragment hits, using the FA inhibition assay, were largely unsuccessful due to the limited solubility of the compounds. However, the chromone hit 4-oxo-4H-1-benzopyran-2-carboxylic acid (ZT0256) showed evidence of dose-dependent inhibition, with an estimated binding affinity for KEAP1, from application of the competition binding model, of KD ~ 1.4 mM (Figure 4B).
Figure 4.

Hits from fragment screening. (A) Structures of the seven fragment hits that showed activity in both the ThermoFluor and FA screening assays. (B) Fragment ZT0256 gave dose-dependent inhibition of KEAP1/Nrf2 binding in the FA competition assay. Analysis of the data using the competition binding model gave an estimated binding affinity for KEAP1 of KD = 1.4 mM.
Characterization of Fragment Hits by X-ray Crystallography.
We attempted to obtain soaked crystal structures with 11 fragment hits (Supplementary Table 2), resulting in three X-ray complex structures. The three fragments bound to the same region of the ligand binding site of KEAP1 (Figure 5A, B). The chromone derivative ZT0256 contains a carboxylate group which engages in salt bridges with the guanidinium groups of R415 and R483, and forms a hydrogen bond with the side chain of S508. The chromone carbonyl group makes a hydrogen bonding interaction with the side chain of S555, while its aromatic moiety engages in π-stacking with Y525 (Figure 5C). A second fragment hit, 4-amino-1,7-dihydro-6H-pyrazolo[3,4-d]pyrimidine-6-thione (ZT0017), also forms a π-stacking interaction with Y525 and hydrogen bonds with S555, and additionally hydrogen bonds with Q530 via its amino group (Figure 5D). The third fragment, 3-(4-hydroxyphenyl) propanoic acid (ZT0633), like the chromone ZT0256, bears a carboxylate group which forms a salt-bridge with R483 and forms a hydrogen bond to S508. The phenyl ring of ZT0633 did not stack with Y525, but instead was bound in an orientation allowed by a different conformation of the side chain of R415. The phenol group of ZT0633 protrudes into the pocket adjacent to S555, and potentially forms a hydrogen bond with the hydroxyl oxygen of this residue (Figure 5E). Thus, among these three fragment hits, both ZT0256 and ZT0633 mimicked the interaction of Nrf2 residue E79 with KEAP1 residues R483 and S508. The π-stacking interactions of ZT0256 and ZT0017 with Y525 were specific to the fragments, however; no similar interaction is seen with Nrf2.
Figure 5.

(A) The three fragment hits for which X-ray crystal structures of complexes were obtained, ZT0256 (green), ZT0017 (magenta) and ZT0633 (blue), overlaid with the Nrf2-derived ETGE peptide (white). (B) Close-up view of overlaid fragments from (A). KEAP1 residues that interact with (C) ZT0256, (D) ZT0017 and (E) ZT0633 (yellow dashed lines indicate polar interactions).
Computational Identification of Hot Spots on KEAP1 by FTMap
To further probe the origins of binding energy at the KEAP1-Nrf2 interface, we characterized the binding site on KEAP1 using the FTMap computational hot spot mapping algorithm (81, 103). In this study, we used a variant of the FTMap method called “focused mapping” (79), in which the initial probe positions are restricted to a defined region of the protein surface. Focused mapping was used so that probes would not be drawn to the large cavity on the opposite surface of the KEAP1 β-propeller domain to that containing the Nrf2 binding site, which is believed not to be involved in ligand binding based on X-ray crystallographic structures obtained by co-crystallization with various ligands, mutagenesis studies, and the locations of disease associated mutations (86). Focused mapping tends to give a more fine-grained picture of the binding potential of the surface site in question, compared to the standard FTMap method. We applied FTMap to both our unliganded and our Nrf2 peptide-bound crystal structures of the Kelch repeat domain of KEAP1. Proteins typically undergo small structural changes upon ligand binding, and we have previously assessed the utility of employing unliganded versus complexed proteins in FTMap analysis. These results show that FTMap analysis of the structure of the unbound protein is most informative about its druggability (82, 104), and for identifying binding sites for compounds discovered through experimental fragment screening (82, 105), while mapping the ligand-bound structure gives the best agreement with experimental alanine scanning data (79, 80).
Mapping the peptide bound KEAP1 structure at the binding site identified four main binding energy hotspots (Figure 6A). The strongest hot spot, which we term hot spot A (CC1, 30 probe clusters), resides deep at the bottom of the binding site, inside the ~6 Å diameter tunnel that passes through the center of the 6-bladed β-propeller structure. This hot spot occupies a hydrophobic interior region of KEAP1, surrounded by L365, A366, I416, V418, V654 and A556, and is not exploited by the natural ligand Nrf2, no atom of which approaches within 6 Å of this hot spot (Figure 6B). The next strongest hot spot, hot spot B (CC2, 16 clusters plus CC6, 4 clusters), lies in the Nrf2 binding groove, at a site defined by KEAP1 residues R415, R483, S508 and Y525. In the KEAP1/Nrf2 complex, hot spot B is partly occupied by the carboxylate side-chain of E79. Another hot spot of comparable strength, hot spot C (CC3, 16 clusters plus CC5, 7 clusters) also lies in the Nrf2 binding groove, close to the polar side chain of KEAP1 residue R380. In the complex hot spot C is occupied by the side chain of Nrf2 residue E82. Finally, a fourth, weaker hot spot, hot spot D, is identified by CC4 (14 clusters), and is located nearby in the shallower part of the binding site adjacent to the Nrf2 binding groove, flanked by residues N414, S421, G433, H436 and I461. Mapping the crystal structure of unliganded KEAP1 gave very similar results, identifying the same four hot spots, A-D (Supplementary Table 3).
Figure 6.

(A) Results of focused mapping of peptide bound KEAP1 using FTMap, showing the consensus clusters (binding energy hot spots) that were identified. FTMap identified hot spots are labeled as A, B, C and D, defined by consensus clusters (shown as thin colored sticks) colored as follows: CC1 (30 probe clusters), red; CC2 (16) pink; CC3 (16), cyan; CC4 (14), yellow; CC5 (7), green; CC6 (4), blue. (B) FTMap results shown with the KEAP1 structure removed, viewed from the side (left image) and top (right image), showing that the side-chains of E79 and E82 overlap with hot spots B and C, respectively. In contrast, the side-chains of D77 and T80 point in towards the center of the hairpin loop, and away from any CCs. Hot spot A defined by CC1 is not approached by Nrf2.
Conformational mobility at the Binding Site on KEAP1
It has been noted previously that KEAP1 residue R415, which is important for the high affinity interaction with Nrf2 (51, 58, 86, 106), adopts distinct conformations in different X-ray crystal structures of the protein (53, 54, 56, 57, 107, 108). However, how the mobility of R415 might affect the shape and properties of the binding site has not previously been described. Examination of the five new KEAP1 crystal structures reported here, together with 17 previously reported crystal structures for human KEAP1, either alone or in complex with various ligands, plus three fragment-bound structures of mouse KEAP1 recently reported by Astex (53) (Supplementary Table 4), shows that the side-chain atoms of R415 can adopt at least three different orientations (Figure 7A). In unbound KEAP1 (Figure 7A, B), the guanidine group of R415 lies at the bottom of the binding site, largely occluding the channel that contains hot spot A. In the soaked structure with ETGE-derived peptides (Figure 7C), the guanidine group is rotated ~60° from this position, allowing it to make a polar interaction with the side-chain of Nrf2 residue E79. Notably, in several structures of KEAP1 bound by small molecule ligands, the side-chain of R415 adopts a third conformation in which the guanidine group points directly out towards solvent (Figure 7D). The positions of other residues at the KEAP1 binding site are essentially identical across the different crystal structures, with the exception of a small displacement of Y525 seen in the structures with some of the Astex fragments and their elaborated derivatives (Supplementary Figure S7).
Figure 7.

Three distinct conformations of Arg415 representative of those seen across different structures of KEAP1. (A) The positions seen for R415 in unbound KEAP1 (magenta, 4IFJ), in KEAP1 in complex with an Nrf2-derived 16-mer peptide encompassing the DxETGE motif (cyan, 4IFL; ligand not shown), and in KEAP1 in complex with a small-molecule ligand (green, 4IQK; ligand not shown). In wheat is the surface of unbound KEAP1 (4IFJ), omitting R415. (B) In unbound KEAP1, R415 (red) largely blocks access to the central channel at the base of the binding site. (C) In the peptide-bound KEAP1, the side-chain of R415 (cyan) is positioned to interact with Nrf2 residue E79. For clarity, the white sticks show only the core ETGE motif from the bound 16-mer peptide. (D) In the presence of small molecule ligands, the R415 side-chain (green) can adopt a position that points out of the binding site, opening the central channel for occupancy by part of the compound (yellow sticks).
Comparing the FTMap results for the unbound and peptide-bound KEAP1 structures indicates that movement of the R415 side-chain from its unliganded position is stabilized by formation of a polar interaction with Nrf2 residue E79. This movement causes the location of hot spot B to shift slightly due to a torsion of the guanidinium group (the position of R415 is unchanged up to Cδ), and also causes a small shift in the location of hot spot C (Supplementary Figure S8). Importantly, this movement of R415 opens access to the deep hole at the bottom of the binding site where FTMap placed the strong hot spot A, which in unbound KEAP1 is almost entirely occluded (Figure 7). In the third, open conformation observed for R415, this deep hole is even more accessible; the diameter of the opening is increased from 6 Å to 9 Å, measured between the α-carbon of G603 and the nearest ε-nitrogen atom of R415. These observations show that the conformation of R415 is affected by ligand binding to KEAP1, and potentially has a significant impact on the accessibility of the deep hole which harbors the putative hot spot A.
Discussion
Alanine scanning mutagenesis, fragment screening, and FTMap are each well-validated as approaches for characterizing the potential of protein surface sites to bind ligands. These methods have intrinsically different sources of false-positive and false-negative results, however, and so their application to a given target often provides complementary rather than redundant information. Here we compare the information obtained when these three methods are applied to understand the binding energetics of the KEAP1/Nrf2 protein-protein interface. We frame the discussion in terms of what information each method provides about the strength and structure of hot spot ensemble at the binding site. The results additionally provide new insight into the energetics of the Nrf2/KEAP1interaction, an important small molecule target of high current interest for drug-discovery.
Comparison of Alanine Scanning with FTMap
Interpreting the results of alanine scanning mutagenesis in terms of binding-energy hot spots assumes that each mutation affects interaction affinity primarily by eliminating stabilizing interactions involving the side-chain atoms of the substituted residue (60, 93–95). It is well recognized, however, that mutating an interface residue to alanine can also have longer range effects, for example by altering the conformation or mobility of neighboring residues (65, 95). Mutation to alanine is likely to have exclusively local effects only if the side-chain of the substituted residue is highly solvent exposed in the unbound protein, with limited interactions with neighboring residues. Comparing alanine scanning results with the locations of binding-energy hot spots determined by FTMap provides insight into which alanine scanning results likely can be interpreted primarily in terms of lost binding interactions, and which are due to other factors (79).
To address this question in the current case we calculated the overlap of the side-chain atoms of the six mutated Nrf2 residues with the FTMap hot spots on KEAP1 (Supplementary Table 1). The results confirm that the side-chain atoms of E79 and E82 overlap with the major hot spots B and C (Figure 6B). This result was robust to whether, for the FTMap analysis, we used unbound KEAP1 or the KEAP1/Nrf2 complex structure with the Nrf2 peptide atoms removed, and is consistent with the large loss in binding energy observed when either E79 or E82 is mutated to alanine. The side-chains of E78 and F83, for which alanine mutations caused negligible loss of binding energy, also reside within the KEAP1 binding site, but make only limited direct contact with KEAP1 (ΔASA < 40 Å2), and neither overlaps with any FTMap hot spot. The alanine scanning results for E79A and E82A can therefore be considered as “true positive” results, and those for E78A and F83A as “true negatives”, with respect to the information they provide about the hot spot ensemble on KEAP1 (Supplementary Table 5).
Interpreting the alanine scanning results for D77 and especially T80 is more complicated. D77 makes little direct contact with KEAP1 (ΔASA = 16 Å2), and has no significant overlap with any FTMap hot spot. Yet its mutation to alanine caused a large reduction in binding affinity, attributable to its stabilization of the Nrf2 hairpin through a network of intramolecular hydrogen bonds. This result can therefore be considered as an alanine scanning “false positive” outcome with respect to hot spot identification, in that the substantial reduction in binding affinity seen upon mutation to alanine is not primarily due the loss of stabilizing contacts between atoms of the D77 side-chain and KEAP1, and therefore does not identify a hot spot. In the case of T80, more than half of the lost affinity for KEAP1 seen upon mutation to Ala was restored when the residue was instead mutated to Ser, confirming the importance of the D77-T80 polar interaction. However, the observation that a substantial amount of binding energy was lost upon mutating T80 to serine, even though the polar interaction with D77 is preserved, suggests that T80 plays a dual role in stabilizing the Nrf2/KEAP1 complex, with a significant contribution from the substantial overlap seen for the T80 side-chain atoms with hot spot C (Supplementary Table 1). The alanine scanning result for T80 is thus a “true positive” result in a qualitative but not a quantitative sense, in that mutation of this residue does identify a hot spot, but its impact is amplified by significant indirect effects. No “false negative” results – that is, residues that interact at hot spots on KEAP1 but do not give a large decrease in binding affinity when mutated to alanine – were found.
In assessing the agreement between alanine scanning and FTMap, it is additionally necessary to consider a fifth class of outcome, in which the existence of a hot spot cannot be tested by alanine mutagenesis because no ligand residue makes contact with the protein receptor at the site in question. Examples of such “null” results are potentially provided by hot spots A and D identified by FTMap. No residue of Nrf2 comes near the locations of either of these putative hot spots (Figure 6B), and thus alanine scanning can provide no information about their existence or strength. This situation could be common, as there is no reason to suppose that a natural ligand must exploit every hot spot available on its binding partner.
The overall picture of Nrf2/KEAP1 binding that results from the above analysis is that, notwithstanding the high density of charged residues that comprise the DEETGE binding motif on Nrf2, the interaction is essentially “two-pronged”, with E79 engaging with hot spot B, and E82 (and to some extent the methyl group of T80) interacting with hot spot C. The function of the remainder of the motif, comprising D77, the hydroxyl group of T80, and presumably G81, appears to be simply to ensure that E79 and E82 assume the correct conformation for optimal interaction with KEAP1. Notably, the conformational change of R415 between unbound and peptide-bound KEAP1 not only brings the R415 side-chain into a position where it can form a salt bridge with E79, but also shifts the locations of hot spots B and C to achieve substantially greater overlap with the side-chain methyl group of T80 (Supplementary Figure S8). Thus, there is a subtle but significant reorganization of the binding site upon Nrf2 binding to KEAP1, mediated by the movement of R415.
Lessons from Fragment Hits
The three fragment hits that were characterized structurally in this study all bound at the strong hot spot B. It was previously shown that the top-ranked hot spot at a protein surface site dominates the free energy of binding, and the vast majority of fragment hits bind at this location (74), though fragment binding at secondary sites can often be observed if very large numbers of fragment hits are characterized crystallographically (109). The binding site on KEAP1 contains two strong hot spots, B and C, of roughly equal energetic importance as judged by the number of FTMap probe clusters they contain. Thus, there is no clear reason why all three of our fragment hits should bind at hot spot B while none bound at hot spot C (the observation that no fragments bound at hot spot A may be due to access to this site being occluded by R415, as discussed below). Three fragment hits against KEAP1 were recently identified by a group from Astex (53), by soaking the compounds into crystals of mouse KEAP1, which is highly homologous to the human protein. Superposition of these Astex fragments onto our FTMap results of shows that two also bind at hot spot B, while the third binds at hot spot C (Figure 8). This result confirms that both of the strong and sterically accessible hot spots present on KEAP1 can serve as binding sites for fragment hits. Although Astex fragment 1 differs from our fragment ZT0633 only by containing a chlorine atom in place of the phenolic hydroxyl, it binds to KEAP1 with a different pose, with its chlorophenyl ring at the bottom of the binding site, and its carboxylate group projecting towards R483 and S508. Astex fragment 2 π-stacks against Y525, an interaction also exploited by our fragments ZT0256 and ZT0017 which likewise contain dinuclear aromatic functions. This interaction closely coincides with the binding mode seen for a prominent cluster of aromatic FTMap probes in hot spot B (Figure 8B). In the cases of ZT0017 and Astex fragment 2, this π-stacking appears to be a main driver of binding. Thus, hot spot B can be occupied in distinct ways by fragments with different structures.
Figure 8.

(A) Overlap of Astex fragment hits with the fragments reported in this study (colored as in Figure 5), superimposed on our structure of unbound KEAP1 (wheat surface). Astex Fragment 1, cyan sticks; Fragment 2, white sticks; Fragment 3, yellow sticks. (B) The three Astex fragment hits overlaid on the FTMap hot spots (thin sticks, colored as in Figure 5).
Integrating Information from Alanine Scanning, FTMap, and Fragment Screening.
When two or all three of the techniques of alanine scanning mutagenesis, FTMap, and experimental fragment screening are applied to a given target, the combination of results expected for different hot spots within the binding site are illustrated in Figure 9. Alanine scanning identifies the subset of hot spots that are exploited by the natural ligand, whether the hot spot is strong or weak. However, alanine scanning is also susceptible to false positive results with regard to hot spot identification in cases where the mutation causes indirect structural effects, and to false negative results in cases of hot spots that are present but not used by the target’s natural protein or peptide ligand. In experimental fragment screening, most fragments bind at the strongest hot spot. Thus, unless X-ray structures are determined for a very large number of bound fragments, this method typically identifies only the strongest one or two hot spots in the ensemble. These hot spots are detected regardless of whether they are exploited by the natural ligand. FTMap detects both strong and weaker hot spots, whether or not they are used in binding the natural ligand. Therefore, among these methods the computational approach is expected to provide the most comprehensive picture of the overall hot spot ensemble, with alanine scanning identifying the subset that is engaged by the natural ligand, and fragment screening identifying the one or two strongest hot spots but providing no information on the existence of other, nearby hot spots that may be important for binding larger ligands (83, 84, 104).
Figure 9.

Diagram illustrating the relationship between the information revealed about the structure of the hot spot ensemble by each of the complementary techniques of FTMap, alanine scanning mutagenesis (ASM), and fragment-based screening (FBS). Alanine scanning identifies the subset of hot spots that are exploited by the natural ligand, whether the hot spot is strong or weak. However, alanine scanning is also susceptible to false positive results with regard to hot spot identification in cases where the mutation causes indirect structural effects, and to false negative results in cases of hot spots that are present but not used by the target’s natural protein or peptide ligand. Experimental fragment screening typically identifies only the strongest one or two hot spots in the ensemble. These hot spots are detected regardless of whether they are exploited by the natural ligand. FTMap detects both strong and weaker hot spots, whether or not they are used in binding the natural ligand.
Computational methods for identifying hot spots, including the FTMap method used here, have their own sources of false positive and false negative results, and these must be considered when evaluating the results achieved when experimental and computational approaches are used in combination to interrogate the hot spot ensemble. In the current study, hot spots A and D are in locations that are not approached by the natural ligand, and therefore are not expected to be identified by alanine scanning. Hot spot D is relatively weak, making it unsurprising that this hot spot was not revealed by experimental fragment screening (74). But why was hot spot A, which FTMap predicts to be strong, not identified by experimental fragment binding? And how confident can we be that either of these two hot spots, which were found only by the computational approach, actually exist?
The accuracy with which FTMap identifies experimentally verifiable hot spots has been tested in many contexts. For example, FTMap was used to analyze the hot spots present at the functional site on 23 well-characterized protein targets, for which a collective 2034 protein-ligand complexes exist in the PDB. For each of these targets, at least 90% of ligands were found to overlap with the top ranked hot spot (average was 98.4%), and every hot spot found by FTMap across all 23 targets was found to be exploited by multiple different ligands (73, 74). This result suggests that the false positive rate for FTMap identification of hot spots relevant for small ligand binding is very low. The reliability of FTMap for identifying hot spots that bind small molecule ligands has been specifically validated for protein-protein interface sites (82). In other work it has been shown that whether an experimental fragment hit retains its binding geometry upon structural elaboration depends strongly on the complementarity of the fragment with the top-ranked hotspot identified by FTMap (74). Additionally, for a set of 121 well-characterized drug targets it was shown that the number, strength, and spatial distribution of FTMap hot spots at the functionally relevant binding site strongly correlated with the experimental druggability of the target – i.e. with whether or not high affinity ligands had been experimentally identified (104). Additionally, the locations and strengths of FTMap hot spots can be used to accurately identify the locations of cryptic binding sites on proteins (116). FTMap’s ability to predict the results of alanine scanning experiments was prospectively validated by application to a protein-protein interface for which experimental alanine scanning data did not previously exist (79), resulting in the experimental confirmation of all predicted hot spots. Moreover, in a larger, retrospective survey, it was shown that FTMap could predict with high accuracy the locations of hot spots at 15 protein-protein interfaces for which both experimental X-ray co-crystal structures and experimental alanine scanning mutagenesis data were available (80). However, in this latter study, although agreement with experiment was excellent overall, in a small number (~10%) of locations where FTMap predicted a hot spot to exist, mutation to alanine of the residue on the partner protein that occupied this site did not lead to a large loss of binding energy, and in ~6% of cases where alanine scanning implied the existence of a hot spot, none was predicted by FTMap. Whether these inconsistencies were due to false results on the part of FTMap or false positive or negative results from alanine scanning is unclear. Thus, although FTMap appears to be highly accurate in its ability to detect binding energy hot spots at protein surfaces, the possibility of a false positive or a false negative result from this method cannot be discounted.
Regarding the existence of hot spot A, access to this hot spot is regulated by the position of the R415 side-chain. In the five available crystal structures of unbound human KEAP1 (including the structure reported here), this residue invariably adopts a conformation in which the guanidine group protrudes into the deep hole at the bottom of the binding site, almost fully blocking access to the region where the putative hot spot A resides (Supplementary Table 4). However, in three of four available structures with a bound Nrf2-derived peptide, R415 adopts a different conformation in which the side-chain is drawn up and out of the bottom of the pocket, to a position in which it is stabilized by a polar interaction with the ligand residue corresponding to Nrf2 E79, opening access to the region harboring hot spot A. There are also a number of ligand-bound structures in which the R415 side-chain adopts a conformation that opens access to the deep hole containing hot spot A, and these too generally involve a charged or highly polar group on the ligand that interacts with the guanidine group of R415 and stabilizes its new position. In the current study, the fragment hits are far too small to both position a charged or polar group to stabilize the R415 side-chain in a position that opens access to the hole containing hot spot A, and at the same time reach into the opened hole to access the hot spot. If accessing hot spot A requires that R415 be stabilized in its alternative position through a polar interaction many Ångstroms distant, this could explain why no fragment was seen to bind at this hot spot. However, we cannot definitively rule out the possibility that hot spot A is a false positive result from FTMap. Experimental evidence that hot spot A is real would require an observation that overlapping with this hotspot in the context of a ligand that also stabilizes the permissive conformation of R415 results in increased binding affinity. To this end, we note that several small molecule inhibitors of KEAP1 that have been reported in the literature derive substantial binding energy by positioning an aromatic moiety in the deep hole that contains hot spot A, establishing that there is binding energy to be had through access with this region of the binding site, though none of these ligands reaches far enough into the hole to achieve full overlap with the hot spot (Supplementary Figure S9).
Druggability of KEAP1.
Of the seven Nrf2 residues that occupy the binding site on KEAP1, four are either Asp or Glu. The unusually high density of charges at the interface makes it difficult to apply conventional criteria when assessing the druggability of KEAP1. Highly charged or polar sites, such as those that bind polynucleic acids, phosphopeptide motifs, or sugars, typically have very low druggability (104, 115). Although a number of low affinity small molecule inhibitors have been reported for KEAP1 (53–57), the only reported compounds with sub-micromolar affinity contain one or two anionic groups (52, 53), typically considered a detriment to druglikeness, leading to a concern that the charged nature of the Nrf2/KEAP1 interface might make it difficult to obtain high affinity, druglike inhibitors. For example, the high affinity KEAP1 inhibitor developed from the Astex fragment hits, which retained a carboxylate group to interact with KEAP1 residue R483, showed only 7% oral bioavailability, and in animal models was administered by i.v. infusion (53).
Our results support the notion that KEAP1 is likely druggable, and potentially provide some guidance as to how a high affinity, druglike ligand might be achieved. Specifically, the alanine scanning results show that E78 makes no contribution to binding, and D77 and the hydroxyl group of T80 contribute only indirectly by stabilizing the loop structure of the motif. In a synthetic ligand, maintaining the shape required to optimally complement the binding site could presumably be achieved by other means, such as macrocyclization. Thus, much of the charged and polar functionality of the Nrf2 binding motif is, in principle, dispensable. Moreover, the results of the fragment screening indicate that it may not be essential to place a carboxylate group in hot spot B to interact with R483, as several fragments can be seen to occupy this site, stabilized instead through a π-stacking interaction with Y525 plus polar contacts with e.g. Q530 and S555. In two cases, ZT0017 and Astex fragment 2, the fragment contains no charged group to interact with R415 or R483. Similarly, the binding pose of Astex fragment 3 shows that the site that in the complex with Nrf2 is occupied by E82 can potentially be satisfied by an uncharged hydrogen bond acceptor such as a sulfonamide, a conclusion that is supported by binding modes observed for published synthetic ligands for KEAP1 (53, 57).
Overall, data from alanine scanning, FTMap, and fragment screening support the possibility that the binding sie on KEAP1 can be well complemented by a suitably designed neutral ligand, despite the highly charged nature of the binding motif on Nrf2. The results suggest that key features of such a compound would be (i) a conformational constraint, such as a macrocyclic structure, that fulfils the role of the D77/T80 interaction in Nrf2 by stabilizing the compound in a conformation that is complementary to the KEAP1 binding pocket; (ii) occupancy of hot spot B with a dinuclear aromatic moiety that engages in a π-stacking interaction with Y525, plus a hydrogen bond acceptor interacting with R415 and R483, and likely additional polar interactions with some or all of S508, S555, and Q530; (iii) a polar interaction that draws the side-chain of R415 into a position that opens up access to the deep hole in the center of the binding site; (iv) penetration of the compound into this hole to position a largely hydrophobic moiety into hot spot A; (v) occupancy of hot spot C by a functionality that contains a neutral hydrogen bond acceptor. Previously reported compounds exploit only a subset of these features, raising the possibility that compounds designed to do so more fully could have improved affinity while dispensing with the pharmaceutically problematic carboxylates that are required in the only sub-micromolar binders so far described. Furthermore, in using structure-based computational techniques to aid inhibitor design, it is clearly crucial to properly consider the orientation of R415, because access to hot spot A is not available if this residue occupies the orientation seen in unbound KEAP1 and in many bound structures.
Material and Methods
Construction of KEAP1 and Nrf2 Bacterial Expression Vectors.
Codon optimized synthetic DNA encoding the human KEAP1 Kelch repeat domain (residues 312–624) was obtained from Genewiz, and was inserted into the pET-15b vector (Novagen) using NdeI and XhoI restriction sites, to obtain a construct with N-terminal six-histidine tag followed by an in-house engineered Tobacco Etch Virus (TEV) cleavage site. A double mutation, E540A/E542A, in a position far from the active binding site that does not affect Nrf2 ligand binding, was introduced to allow crystallization in a form that has the ligand binding site available for soaking experiments (102). Four cysteine-to-serine mutations were also introduced, at positions 319, 613, 622 and 624, to improve protein homogeneity and reduce aggregation (102). To make Nrf21–100 and Nrf234–100, the corresponding DNA was inserted into backbone vector pET-15b using NdeI and BamHI restriction sites. All plasmids were purified using Zyppy Plasmid Miniprep Kit (Zymo research) and confirmed by sequencing. In all cases, the ligated products were then transformed into DH5α competent cells (New England Biolabs) following the manufacturer’s protocol, and selected using Ampicillin (100 μg/mL).
Site-directed mutagenesis.
Site-directed mutagenesis in the bacterial expression plasmids for Nrf2 was performed by following a 2-round PCR procedure. In the first round of reaction, a T7 promoter forward primer and a reverse primer encompassing the mutation were used to create a mega-primer containing the mutation. The first round PCR products were then purified and used as mega-primers for the second round of reaction. After PCR, the products were purified, and 1 μL of DpnI was added and the samples incubated at 37 °C for 1 h. A volume of 2 μL reaction was then transformed into DH5α competent cells. Plasmids containing the desired mutation were selected and confirmed by sequencing as described above.
Expression, Purification and verification of recombinant KEAP1 and Nrf2 protein constructs.
KEAP1 and wild-type and variant Nrf2 constructs were transformed into BL21(DE3) Competent E. coli cells (New England Biolabs) following the manufacturer’s instructions, and colonies were selected on LB plates containing Ampicillin (100 μL/mL). A 50 mL overnight culture in LB with Ampicillin was seeded by a single colony and grown overnight at 37 °C with shaking. New, large scale cultures were inoculated with the overnight cultures by 1:200 dilution and were grown to an OD600 of 0.4 – 0.6. Protein expression was induced by 1 mM isopropyl-beta-D-thiogalactoside (IPTG, GoldBio). KEAP1 protein was expressed at 20 °C with shaking overnight, and all the Nrf2 constructs were expressed at 37 °C for 4 h. Cells were pelleted by centrifugation and stored at −80 °C. Cells were lysed by sonication with addition of Binding Buffer (20 mM sodium phosphate, 500 mM NaCl, 20 mM imidazole, 2.5 mM DTT, pH 7.5) at a concentration of 2 mL/g of cell pellet together with 10% ethylene glycol, 0.1% Pierce Universal Nuclease for Cell Lysis (Thermo Fisher Scientific), and cOmplete ULTRA Tablets, Mini, EDTA-free, EASYpack (1 tablet/2 L of cells, Roshe Life Science). The cell lysate was centrifuged at 27,000 g at 4 °C for 30 min, then the supernatant was collected and filtered through an 0.8 μm syringe filter. The prepared cell lysate was loaded onto a gravity column with 2 mL HisPur Ni-NTA resin that had been pre-equilibrated with 10 resin-bed volumes of binding buffer. Samples were then incubated with the Ni-NTA resin at 4 °C, with rotation to maximize the binding, after which the flow-through was collected. The resin was then washed with 10 column volumes of the binding buffer after which the His-tagged proteins were eluted by 6 column volumes of elution buffer (20 mM sodium phosphate, 500 mM NaCl, 500 mM imidazole, 2.5 mM DTT, pH 7.5). The eluted His-tagged protein was then dialyzed against Dialysis Buffer (20 mM sodium phosphate, 500 mM NaCl, 2.5 mM DTT, pH 7.5) overnight at 4 °C with addition of TEV protease at approximately 1 mg TEV per 50 mg of protein, using TEV protease that was expressed and purified in-house. The reaction mixture was applied to the 2 mL Ni-NTA resin again to separate the untagged protein from the cleaved fragment containing the histidine tag. The cleaved proteins were purified by gel filtration using HiPrep 26/60 Sephacryl S-300 column (GE Healthcare) equilibrated with 25 mM Tris, 2.5 mM DTT for KEAP1, or 20 mM sodium phosphate, 150 mM NaCl, 2.5 mM DTT for Nrf2 constructs, respectively. Purified proteins were concentrated, quantified using absorbance at 280 nm (NanoDrop, Thermo Fisher Scientific) based on calculated extinction coefficients, and aliquots flash frozen and stored at −80 °C. All recombinant proteins were >95% pure as assessed using SDS-PAGE. All protein samples, at 20 μM in 10 μL, were desalted using Zeba Spin Desalting Columns 7K MWCO following the manufacturer’s instructions, and characterized by Waters qTOF Premier LCMS to determine the intact mass.
Circular Dichroism Spectroscopy.
CD spectra were measured using an Applied Photophysics CS/2 Chirascan instrument with a 1 mm path length quartz cuvette at a scan speed of 0.5 nm/s. The CD spectra were collected at 20 °C from 180 to 260 nm, with 10 μM of Nrf2 in 20 mM sodium phosphate, 150 mM NaCl buffer at pH 7.5and 5 μM of KEAP1 in 25 mM Tris, 2.5 mM DTT buffer at pH 8.0, respectively. The buffer background was subtracted. The measured ellipticity in millidegrees (θ) was normalized to mean residue ellipticity (θMRE) using the following equation:
where C is the molar concentration of protein, l is the length path in centimeters, and n is the number of residues. The thermal stability of wild-type Nrf21–100 and Nrf234–100 was measured using samples prepared as described above, by monitoring the increase in elipiticity at 208 nm as the temperature was ramped from 10 °C to 70 °C at a rate of 0.5 °C/min. A faster ramping rate of 1 °C/min was also tested, and no significant difference in results was observed. To determine the reversibility of the thermal denaturation, the CD spectrum was monitored as described above as the temperature was increased to 70 °C, and then the temperature was reduced back to 10 °C at the same rate of 0.5 °C/min, and the spectrum of the protein was measured after cooling to this final temperature.
Fluorescence Anisotropy Assays.
Commercially synthesized FITC-labeled Nrf2 nonamer peptide (LDEETGEFL, residues 76–84) with N-terminal amidation (Genescript) was dissolved in water contaning 0.125% ammonium hydroxide. The concentration of the FITC-Nrf2 peptide was determined from its absorbance at 493 nm, by NanoDrop, using a calculated extinction coefficient. Fluorescence Anisotropy assays were performed in 96-well polypropylene black plates (Corning Costar) in duplicate using a SpectraMax M5 microtiter plate reader (Molecular Devices, Sunnyvale, CA). In binding assays, a total assay volume of 200 μL, made up of 50 μL of water, 50 μL of 4X assay buffer (final concentration 50 mM HEPES, 150 mM NaCl, 0.01% v/v Triton, pH 7.4), 50 μL of FITC-labeled tracer Nrf2 peptide at 20 nM, to give a final constant concentration of 5 nM, and 50 μL of KEAP1 protein at varying concentrations (range 0.003 – 100 KD). The assay mixture was incubated at 25 °C for 30 min, which control experiments showed was sufficient time to reach equilibrium, prior to fluorescence anisotropy reading. Samples were read by excitation at 488 nm and emission at 520 nm, in high sensitivity mode with 100 reads/well. Anisotropy values (r) were calculated by measuring the intensity of the parallel (I∥) and perpendicular (I⊥) components of the fluorescence emission in each well, using the following equation:
Data from direct binding experiments were fitted to quadratic binding equation (1):
| (1) |
where A0 represents the baseline anisotropy signal observed in negative controls omitting KEAP1 protein, and Amax corresponds to the maximal anisotropy signal at saturating KEAP1. [L]T and [R]T are the total concentrations of ligand (FITC-Nrf2 peptide) and protein (KEAP1), respectively, in the final assay, and KD1 is the dissociation constant for the interaction. The term (Amax – A0)/[L]T is the molar response of the assay – i.e. the extent of anisotropy signal change per molar of FITC-labeled ligand.
Competition experiments were carried out in the same assay buffer by keeping FITC-Nrf2 peptide at 5 nM and KEAP1 protein at 10 nM, and varying the concentration of the unlabeled Nrf2 constructs. The unlabeled wild-type and variant Nrf2 proteins were added to each well by two-fold serial dilution. Positive and negative control wells were included in each assay plate to provide a measure of the maximum binding signal corresponding to fully bound complex (Abound) in absence of competitor, and the minimum signal corresponding to free FITC-Nrf2 peptide in solution (Afree). The controls were used to validate the proper functioning of each experiment, and to normalize the binding results obtained for the different competitors across multiple experiments. The competition binding results were fitted using the numerical nonlinear regression software DynaFit 4 (BioKin Ltd.), using the competitive binding model shown below:
In applying this model, the FITC-labeled probe peptide concentration, the KEAP1 concentration and KD1 values were fixed, allowing KD2 and the molar response of the complex to be variable and obtained by fitting. The details of this approach to fitting competition binding data have been reported previously (79).
For fragment screening using the FA competition assay, the compounds were prepared as stock solutions at a concentration of 20 mM in 40% v/v DMSO/water. The initial single dose screening of the fragments against KEAP1/Nrf2 was carried out using KEAP1 at 10 nM, Nrf2 at 5 nM (final concentrations) in the assay buffer, with the compounds added to a final concentration of 5 mM, leading to a final DMSO content of 10%. Compounds that interfered in the assay due to their own fluorescence properties were identified through the resulting elevated raw fluorescence emission signal at 520 nm. A UV-vis spectrum from 280 nm to 750 nm was measured for each compound under assay condition, to identify compounds that absorbed strongly at the excitation or emission wavelengths used in the FA assay, and to identify insoluble compounds that could potentially inhibit through a non-specific mechanism, such as aggregation (100, 117), through their associated light scattering at high wavelengths. Hits from the first round of screening were further verified in dose-response assays, starting from 10 mM compound with 10% DMSO in the assay, using the same competition assay set-up described above.
ThermoFluor Assays.
For fragment screening by ThermoFluor we used a Realplex EP Mastercycler RT PCR instrument (Eppendorf) in a 96-well PCR plate format. The total assay volume of 20 μL contained 10 μM KEAP1, Sypro Orange dye (5X, Thermo Fisher Scientific), and 10 mM of compound in Storage Buffer (25 mM Tris, 2.5 mM DTT, pH 8.0). The plate was sealed with clear seal film to prevent evaporation, and incubated at 20 °C until the temperature equilibrated. Results were collected by monitoring fluorescence at 520 nm as the temperature was ramped up at a rate of 1 °C/min. The Tm values were defined as the temperature corresponding to the peak of the first derivative of the melting curve. Compounds that stabilized KEAP1, resulting in an increased Tm, were considered to be hits.
X-ray Crystallographic Data Collection, Phase Determination and Refinement.
Protein with less than 30% polydispersity at 1 mg/mL as measured by dynamic light scattering was used for crystallization trials. Protein for crystallization was 5.1 mg/mL in 25 mM Tris-HCl pH 8.0 buffer with 10 mM DTT. The initial condition of 1.5 M (NH4)2SO4, 100 mM Bis-Tris, pH 6.5 and 100 mM NaCl was discovered using the Index screen (Hampton Research) using vapor-diffusion with sitting drop geometry in a 96-well format with 2 μl drops (1 μl protein plus 1 μl well). Grid screening around this condition resulted in determination of the final conditions wherein (NH4)2SO4, pH, and PEG concentrations were optimized for each prep: 1.2–1.5 M (NH4)2SO4, 100 mM Bis-Tris, pH 6.0–6.5, and 0 – 0.8% PEG 550 monomethyl ether. For analysis of the unliganded protein, crystals were cryo-protected in mother liquor to which glycerol (30% v/v) was added and frozen in liquid nitrogen. The peptides LDEETGEF and LDEEAGEFL, with N-terminal acetylation and C-terminal amidation, were obtained from GenScript for use in soaking experiments. Stocks of peptide (4 mM) in 25 mM Tris 8.0 and 10 mM DTT were added to the crystallization drop to a final concentration of 1.6 mM peptide and 30–60% glycerol and allowed to equilibrate for 1 hour. Crystals were frozen in liquid nitrogen without further cryo-protection. Fragment soaking was performed by adding a fragment solution to a final concentration of 20 mM in the crystallization drop and incubating from 20 minutes to overnight. Crystals were harvested and transferred to a solution containing the well condition plus 15% glycerol, followed by transfer to a solution containing the well condition plus 30% glycerol, before freezing in liquid nitrogen.
Data for the unliganded structure and the LDEEAGEFL peptide-soaked structure were collected at the Stanford Synchrotron Radiation Lightsource (Menlo Park) on beamline 9–2. Data were scaled and integrated using the HKL2000 suite of programs to a resolution of 1.93 and 2.02 Å, respectively. Data for the 8-mer LDEETGEF peptide soaked structure and the ZT0633 fragment-bound structure were collected at the Advanced Photon Source at Argonne National Laboratory (Lemont, IL) on NE-CAT beamline 24-ID-C. Data were integrated and scaled using the NECAT RAPD online server (http://necat.chem.cornell.edu/) to a resolution of 1.91 and 2.50 Å, respectively. Data for ZT0017 and ZT0256-bound structures were collected at the National Synchrotron Light Source (Brookhaven National Laboratory, Upton, NY) on beamline X-29 using an ADSC Quantum 315r nine quadrant CCD detector and data were integrated and scaled using XDS to resolutions from 2.23 Å. Data collection statistics and PDB accession codes are available in Table S6. Molecular replacement was performed using Phaser (2) with the coordinates from the high resolution (1.85 Å) human native Keap1 structure (PDB 1U6D) followed by model building with Phenix.Autobuild and Coot (4) followed by simulated annealing in Phenix using individual B-factor refinement (occupancy was not refined). Ligands were placed in the corresponding density when the R factor reached ~25%. Ligand placement was confirmed via generation of Polder omit maps. Residues that were visible and modeled in the electron density were as follows: 325 – 614 in unliganded KEAP1; 325 – 613 in LDEETGEFL-bound, LDEEAGEFL-bound, ZT0256-bound, ZT0017-bound, and ZT0633-bound KEAP1. Crystallographic statistics are collected in Supplementary Table 6.
Computational Identification of Hot Spots on KEAP1 by FTMap
Details of the basic FTMap protein mapping program have been previously reported (81, 118). For the analysis of KEAP1 we have used two slightly modified versions of FTMap by adding, respectively, options for masking and focusing. Masking means removing the attractive van der Waals interaction from all atoms in a selected region of the protein. Masking was added because the KEAP1 is 6-bladed β-propeller with a funnel passing through the middle of the protein. The entrance of the funnel is restricted by the side chain of R415 in the unbound protein, and is not accessible. However, FTMap samples the entire space around the protein, including internal cavities, and tends to place a large fraction of probes in the funnel. Masking the inside of the funnel reduces the attraction of the region, and although the strongest hot spot is still in the funnel, more probes are placed in other regions.
For determining the overlap between hot spots and the side chains of the Nrf2 peptide we used a different variant of FTMap to perform focused mapping (118–120). In focused mapping the initial probe positions are restricted to a defined region of the protein surface, and hence the method tends to give a more detailed and fine-grained picture of the binding potential of the surface site in question, compared to standard FTMap. For KEAP1, the mapping was focused to within 3 Angstroms of any atom of the existing ligands in the Nrf2 binding site.
Supplementary Material
References
- 1.Fuller JC, Burgoyne NJ, & Jackson RM (2009) Predicting druggable binding sites at the protein-protein interface. Abstr Pap Am Chem S 238. [DOI] [PubMed] [Google Scholar]
- 2.Whitty A & Kumaravel G (2006) Between a rock and a hard place? Nat Chem Biol 2(3):112–118. [DOI] [PubMed] [Google Scholar]
- 3.Arkin MR & Whitty A (2009) The road less traveled: modulating signal transduction enzymes by inhibiting their protein-protein interactions. Curr Opin Chem Biol 13(3):284–290. [DOI] [PubMed] [Google Scholar]
- 4.Wells JA & McClendon CL (2007) Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 450(7172):1001–1009. [DOI] [PubMed] [Google Scholar]
- 5.Arkin MR & Wells JA (2004) Small-molecule inhibitors of protein-protein interactions: Progressing towards the dream. Nat Rev Drug Discov 3(4):301–317. [DOI] [PubMed] [Google Scholar]
- 6.Arkin MR, Tang YY, & Wells JA (2014) Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing toward the Reality. Chem Biol 21(9):1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hwang H, Vreven T, Janin J, & Weng ZP (2010) Protein-protein docking benchmark version 4.0. Proteins 78(15):3111–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berg T (2008) Small-molecule inhibitors of protein-protein interactions. Curr Opin Drug Disc 11(5):666–674. [PubMed] [Google Scholar]
- 9.Fuentes G, Oyarzabal J, & Rojas AM (2009) Databases of protein-protein interactions and their use in drug discovery. Curr Opin Drug Disc 12(3):358–366. [PubMed] [Google Scholar]
- 10.Cochran AG (2001) Protein-protein interfaces: mimics and inhibitors. Curr Opin Chem Biol 5(6):654–659. [DOI] [PubMed] [Google Scholar]
- 11.Cochran AG (2000) Antagonists of protein-protein interactions. Chem Biol 7(4):R85–R94. [DOI] [PubMed] [Google Scholar]
- 12.Whitty A (2008) Small Molecule Inhibitors of Protein-Protein Interactions: Challenges and Prospects. Gene Family Targeted Molecular Design, ed Lackey K (John Wiley & Sons; ), p 384. [Google Scholar]
- 13.Toogood PL (2002) Inhibition of protein-protein association by small molecules: Approaches and progress. J Med Chem 45(8):1543–1558. [DOI] [PubMed] [Google Scholar]
- 14.Fry DC (2008) Drug-like inhibitors of protein-protein interactions: A structural examination of effective protein mimicry. Curr Protein Pept Sc 9(3):240–247. [DOI] [PubMed] [Google Scholar]
- 15.Fry DC (2006) Protein-protein interactions as targets for small molecule drug discovery. Biopolymers 84(6):535–552. [DOI] [PubMed] [Google Scholar]
- 16.Gerrard JA, Hutton CA, & Perugini MA (2007) Inhibiting Protein-Protein Interactions as an Emerging Paradigm for Drug Discovery. Mini-Rev Med Chem 7(2):151–157. [DOI] [PubMed] [Google Scholar]
- 17.Murray JK & Gellman SH (2007) Targeting protein-protein interactions: Lessons from p53/MDM2. Biopolymers 88(5):657–686. [DOI] [PubMed] [Google Scholar]
- 18.Gadek TR (2003) Strategies and methods in the identification of antagonists of protein-protein interactions. Biotechniques:21–24. [PubMed] [Google Scholar]
- 19.Chene P (2006) Drugs targeting protein-protein interactions. Chemmedchem 1(4):400–411. [DOI] [PubMed] [Google Scholar]
- 20.Sankar U (2005) Protein-protein interactions in drug discovery. Genet Eng News 25(5):30–31. [Google Scholar]
- 21.Scott DE, Bayly AR, Abell C, & Skidmore J (2016) Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat Rev Drug Discov 15(8):533–550. [DOI] [PubMed] [Google Scholar]
- 22.Erlanson DA, Fesik SW, Hubbard RE, Jahnke W, & Jhoti H (2016) Twenty years on: the impact of fragments on drug discovery. Nat Rev Drug Discov 15(9):605–619. [DOI] [PubMed] [Google Scholar]
- 23.Rees DC, Congreve M, Murray CW, & Carr R (2004) Fragment-based lead discovery. Nat Rev Drug Discov 3(8):660–672. [DOI] [PubMed] [Google Scholar]
- 24.Congreve M, Murray CW, Carr R, & Rees DC (2007) Fragment-Based Lead Discovery. Annu Rep Med Chem 42:431–448. [Google Scholar]
- 25.Woodhead AJ, et al. (2010) Discovery of (2,4-Dihydroxy-5-isopropylphenyl)-[5-(4-methylpiperazin-1-ylmethyl)-1,3-dihydroisoindol-2-yl]methanone (AT13387), a Novel Inhibitor of the Molecular Chaperone Hsp90 by Fragment Based Drug Design. J Med Chem 53(16):5956–5969. [DOI] [PubMed] [Google Scholar]
- 26.Oltersdorf T, et al. (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435(7042):677–681. [DOI] [PubMed] [Google Scholar]
- 27.Zhang MJ, et al. (2013) Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog Neurobiol 100:30–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wakabayashi N, et al. (2004) Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the Keap1 sensor modified by inducers. P Natl Acad Sci USA 101(7):2040–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Itoh K, et al. (1997) An Nrf2 small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Bioph Res Co 236(2):313–322. [DOI] [PubMed] [Google Scholar]
- 30.Zhang DD, Lo SC, Cross JV, Templeton DJ, & Hannink M (2004) Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol 24(24):10941–10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kansanen E, Kuosmanen SM, Leinonen H, & Levonen AL (2013) The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol 1(1):45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taguchi K, Motohashi H, & Yamamoto M (2011) Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 16(2):123–140. [DOI] [PubMed] [Google Scholar]
- 33.Tong KI, et al. (2007) Different electrostatic Potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol Cell Biol 27(21):7511–7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tong KI, et al. (2006) Keap1 recruits Neh2 through binding to ETGE and DLG motifs: Characterization of the two-site molecular recognition model. Mol Cell Biol 26(8):2887–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cho HY & Kleeberger SR (2010) Nrf2 protects against airway disorders. Toxicol Appl Pharm 244(1):43–56. [DOI] [PubMed] [Google Scholar]
- 36.Calkins MJ, et al. (2009) The Nrf2/ARE Pathway as a Potential Therapeutic Target in Neurodegenerative Disease. Antioxid Redox Sign 11(3):497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li JQ, Ichikawa T, Janicki JS, & Cui TX (2009) Targeting the Nrf2 pathway against cardiovascular disease. Expert Opin Ther Tar 13(7):785–794. [DOI] [PubMed] [Google Scholar]
- 38.Wong TF, et al. (2011) Association of Keap1 and Nrf2 Genetic Mutations and Polymorphisms With Endometrioid Endometrial Adenocarcinoma Survival. Int J Gynecol Cancer 21(8):1428–1435. [DOI] [PubMed] [Google Scholar]
- 39.Solis LM, et al. (2010) Nrf2 and Keap1 Abnormalities in Non-Small Cell Lung Carcinoma and Association with Clinicopathologic Features. Clin Cancer Res 16(14):3743–3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim YR, et al. (2010) Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J Pathol 220(4):446–451. [DOI] [PubMed] [Google Scholar]
- 41.Jiang T, et al. (2010) High Levels of Nrf2 Determine Chemoresistance in Type II Endometrial Cancer. Cancer Res 70(13):5486–5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang XJ, et al. (2008) Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 29(6):1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shibata T, et al. (2008) Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology 135(4):1358–1368. [DOI] [PubMed] [Google Scholar]
- 44.Singh A, et al. (2006) Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. Plos Med 3(10):1865–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jaramillo MC & Zhang DD (2013) The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Gene Dev 27(20):2179–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen T, Nioi P, & Pickett CB (2009) The Nrf2-Antioxidant Response Element Signaling Pathway and Its Activation by Oxidative Stress. J Biol Chem 284(20):13291–13295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kobayashi M, et al. (2009) The Antioxidant Defense System Keap1-Nrf2 Comprises a Multiple Sensing Mechanism for Responding to a Wide Range of Chemical Compounds. Mol Cell Biol 29(2):493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scannevin RH, et al. (2012) Fumarates Promote Cytoprotection of Central Nervous System Cells against Oxidative Stress via the Nuclear Factor (Erythroid-Derived 2)-Like 2 Pathway. J Pharmacol Exp Ther 341(1):274–284. [DOI] [PubMed] [Google Scholar]
- 49.Brennan MS, et al. (2015) Dimethyl Fumarate and Monoethyl Fumarate Exhibit Differential Effects on KEAP1, NRF2 Activation, and Glutathione Depletion In Vitro. Plos One 10(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Magesh S, Chen Y, & Hu LQ (2012) Small Molecule Modulators of Keap1-Nrf2-ARE Pathway as Potential Preventive and Therapeutic Agents. Med Res Rev 32(4):687–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abed DA, Goldstein M, Albanyan H, Jin HJ, & Hu LQ (2015) Discovery of direct inhibitors of Keap1-Nrf2 protein-protein interaction as potential therapeutic and preventive agents. Acta Pharm Sin B 5(4):285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang ZY, et al. (2015) Structure-Activity and Structure-Property Relationship and Exploratory in Vivo Evaluation of the Nanomolar Keap1-Nrf2 Protein-Protein Interaction Inhibitor. J Med Chem 58(16):6410–6421. [DOI] [PubMed] [Google Scholar]
- 53.Davies TG, et al. (2016) Monoacidic Inhibitors of the Kelch-like ECH-Associated Protein 1: Nuclear Factor Erythroid 2-Related Factor 2 (KEAP1:NRF2) Protein Protein Interaction with High Cell Potency Identified by Fragment-Based Discovery. J Med Chem 59(8):3991–4006. [DOI] [PubMed] [Google Scholar]
- 54.Jnoff E, et al. (2014) Binding Mode and Structure-Activity Relationships around Direct Inhibitors of the Nrf2-Keap1 Complex. Chemmedchem 9(4):699–705. [DOI] [PubMed] [Google Scholar]
- 55.Satoh M, et al. (2015) Multiple binding modes of a small molecule to human Keap1 revealed by X-ray crystallography and molecular dynamics simulation. Febs Open Bio 5:557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jain AD, et al. (2015) Probing the structural requirements of non-electrophilic naphthalene-based Nrf2 activators. Eur J Med Chem 103:252–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marcotte D, et al. (2013) Small molecules inhibit the interaction of Nrf2 and the Keap1 Kelch domain through a non-covalent mechanism. Bioorgan Med Chem 21(14):4011–4019. [DOI] [PubMed] [Google Scholar]
- 58.Lo SC, Li XC, Henzl MT, Beamer LJ, & Hannink M (2006) Structure of the Keap1 : Nrf2 interface provides mechanistic insight into Nrf2 signaling. Embo J 25(15):3605–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lo Conte L, Chothia C, & Janin J (1999) The atomic structure of protein-protein recognition sites. J Mol Biol 285(5):2177–2198. [DOI] [PubMed] [Google Scholar]
- 60.Clackson T & Wells JA (1995) A Hot-Spot of Binding-Energy in a Hormone-Receptor Interface. Science 267(5196):383–386. [DOI] [PubMed] [Google Scholar]
- 61.Thanos CD, DeLano WL, & Wells JA (2006) Hot-spot mimicry of a cytokine receptor by a small molecule. P Natl Acad Sci USA 103(42):15422–15427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moreira IS, Fernandes PA, & Ramos MJ (2007) Hot spots-A review of the protein-protein interface determinant amino-acid residues. Proteins 68(4):803–812. [DOI] [PubMed] [Google Scholar]
- 63.Muller YA, et al. (1997) Vascular endothelial growth factor: Crystal structure and functional mapping of the kinase domain receptor binding site. P Natl Acad Sci USA 94(14):7192–7197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clackson T, Ultsch MH, Wells JA, & de Vos AM (1998) Structural and functional analysis of the 1 : 1 growth hormone : receptor complex reveals the molecular basis for receptor affinity. J Mol Biol 277(5):1111–1128. [DOI] [PubMed] [Google Scholar]
- 65.DeLano WL (2002) Unraveling hot spots in binding interfaces: progress and challenges. Curr Opin Struc Biol 12(1):14–20. [DOI] [PubMed] [Google Scholar]
- 66.Nienaber VL (2011) Fragment-Based Drug Discovery for Diseases of the Central Nervous System. Acs Sym Ser 1076:179–192. [Google Scholar]
- 67.Erlanson DA (2005) Fragment-based drug discovery through tethering. Abstr Pap Am Chem S 230:U2763–U2763. [Google Scholar]
- 68.Hajduk PJ & Greer J (2007) A decade of fragment-based drug design: strategic advances and lessons learned. Nat Rev Drug Discov 6(3):211–219. [DOI] [PubMed] [Google Scholar]
- 69.Winter A, et al. (2012) Biophysical and computational fragment-based approaches to targeting protein-protein interactions: applications in structure-guided drug discovery. Q Rev Biophys 45(4):383–426. [DOI] [PubMed] [Google Scholar]
- 70.Erlanson DA (2006) Fragment-based lead discovery: a chemical update. Curr Opin Biotech 17(6):643–652. [DOI] [PubMed] [Google Scholar]
- 71.Murray CW & Rees DC (2009) The rise of fragment-based drug discovery. Nat Chem 1(3):187–192. [DOI] [PubMed] [Google Scholar]
- 72.Allen KN, et al. (1996) An experimental approach to mapping the binding surfaces of crystalline proteins. J Phys Chem-Us 100(7):2605–2611. [Google Scholar]
- 73.Hall DR, Kozakov D, Whitty A, & Vajda S (2015) Lessons from Hot Spot Analysis for Fragment-Basec Drug Discovery. Trends Pharmacol Sci 36(11):724–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kozakov D, et al. (2015) Ligand deconstruction: Why some fragment binding positions are conserved and others are not (vol 112, pg E2585, 2015). P Natl Acad Sci USA 112(28):E3749–E3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ciulli A, Chirgadze DY, Smith AG, Blundell TL, & Abell C (2007) Crystal structure of Escherichia coli ketopantoate reductase in a ternary complex with NADP(+) and pantoate bound - Substrate recognition, conformational change, and cooperativity. J Biol Chem 282(11):8487–8497. [DOI] [PubMed] [Google Scholar]
- 76.Ciulli A, Williams G, Smith AG, Blundell TL, & Abell C (2006) Probing hot spots at protein-ligand binding sites: A fragment-based approach using biophysical methods. J Med Chem 49(16):4992–5000. [DOI] [PubMed] [Google Scholar]
- 77.Kozakov D, et al. (2015) The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat Protoc 10(5):733–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ngan CH, et al. (2012) FTMAP: extended protein mapping with user-selected probe molecules. Nucleic Acids Res 40(W1):W271–W275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Golden MS, et al. (2013) Comprehensive Experimental and Computational Analysis of Binding Energy Hot Spots at the NF-kappa B Essential Modulator/IKK beta Protein-Protein Interface. J Am Chem Soc 135(16):6242–6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zerbe BS, Hall DR, Vajda S, Whitty A, & Kozakov D (2012) Relationship between Hot Spot Residues and Ligand Binding Hot Spots in Protein-Protein Interfaces. J Chem Inf Model 52(8):2236–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brenke R, et al. (2009) Fragment-based identification of druggable ‘hot spots’ of proteins using Fourier domain correlation techniques. Bioinformatics 25(5):621–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kozakov D, et al. (2011) Structural conservation of druggable hot spots in protein-protein interfaces. P Natl Acad Sci USA 108(33):13528–13533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Meireles LMC, Domling AS, & Camacho CJ (2010) ANCHOR: a web server and database for analysis of protein-protein interaction binding pockets for drug discovery. Nucleic Acids Res 38:W407–W411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Verdonk ML, Cole JC, Watson P, Gillet V, & Willett P (2001) SuperStar: Improved knowledge-based interaction fields for protein binding sites. J Mol Biol 307(3):841–859. [DOI] [PubMed] [Google Scholar]
- 85.Radoux CJ, Olsson TSG, Pitt WR, Groom CR, & Blundell TL (2016) Identifying Interactions that Determine Fragment Binding at Protein Hotspots. J Med Chem 59(9):4314–4325. [DOI] [PubMed] [Google Scholar]
- 86.Li XC, Zhang D, Hannink M, & Beamer LJ (2004) Crystal structure of the Kelch domain of human Keap1. J Biol Chem 279(52):54750–54758. [DOI] [PubMed] [Google Scholar]
- 87.Cino EA, Killoran RC, Karttunen M, & Choy WY (2013) Binding of disordered proteins to a protein hub. Sci Rep-Uk 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Do TN, Choy WY, & Karttunen M (2016) Binding of Disordered Peptides to Kelch: Insights from Enhanced Sampling Simulations. J Chem Theory Comput 12(1):395–404. [DOI] [PubMed] [Google Scholar]
- 89.Inoyama D, et al. (2012) Optimization of Fluorescently Labeled Nrf2 Peptide Probes and the Development of a Fluorescence Polarization Assay for the Discovery of Inhibitors of Keap1-Nrf2 Interaction. J Biomol Screen 17(4):435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen Y, Inoyama D, Kong ANT, Beamer LJ, & Hu LQ (2011) Kinetic Analyses of Keap1-Nrf2 Interaction and Determination of the Minimal Nrf2 Peptide Sequence Required for Keap1 Binding Using Surface Plasmon Resonance. Chem Biol Drug Des 78(6):1014–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Roehrl MHA, Wang JY, & Wagner G (2004) Discovery of small-molecule inhibitors of the NFAT-calcineurin interaction by competitive high-throughput fluorescence polarization screening. Biochemistry-Us 43(51):16067–16075. [DOI] [PubMed] [Google Scholar]
- 92.Cheng Y & Prusoff WH (1973) Relationship between Inhibition Constant (K1) and Concentration of Inhibitor Which Causes 50 Per Cent Inhibition (I50) of an Enzymatic-Reaction. Biochem Pharmacol 22(23):3099–3108. [DOI] [PubMed] [Google Scholar]
- 93.Bogan AA & Thorn KS (1998) Anatomy of hot spots in protein interfaces. J Mol Biol 280(1):1–9. [DOI] [PubMed] [Google Scholar]
- 94.Weiss GA, Watanabe CK, Zhong A, Goddard A, & Sidhu SS (2000) Rapid mapping of protein functional epitopes by combinatorial alanine scanning. P Natl Acad Sci USA 97(16):8950–8954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fersht AR (1987) Dissection of the Structure and Activity of the Tyrosyl-Transfer Rna-Synthetase by Site-Directed Mutagenesis. Biochemistry-Us 26(25):8031–8037. [DOI] [PubMed] [Google Scholar]
- 96.Kranz JK & Schalk-Hihi C (2011) Protein Thermal Shifts to Identify Low Molecular Weight Fragments. Method Enzymol 493:277–298. [DOI] [PubMed] [Google Scholar]
- 97.Lo MC, et al. (2004) Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem 332(1):153–159. [DOI] [PubMed] [Google Scholar]
- 98.Cummings MD, Farnum MA, & Nelen MI (2006) Universal screening methods and applications of ThermoFluor (R). J Biomol Screen 11(7):854–863. [DOI] [PubMed] [Google Scholar]
- 99.Ericsson UB, Hallberg BM, DeTitta GT, Dekker N, & Nordlund P (2006) Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal Biochem 357(2):289–298. [DOI] [PubMed] [Google Scholar]
- 100.Feng BY & Shoichet BK (2006) A detergent-based assay for the detection of promiscuous inhibitors. Nat Protoc 1(2):550–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Feng BY, Shelat A, Doman TN, Guy RK, & Shoichet BK (2005) High-throughput assays for promiscuous inhibitors. Nat Chem Biol 1(3):146–148. [DOI] [PubMed] [Google Scholar]
- 102.Horer S, Reinert D, Ostmann K, Hoevels Y, & Nar H (2013) Crystal-contact engineering to obtain a crystal form of the Kelch domain of human Keap1 suitable for ligand-soaking experiments. Acta Crystallogr F 69:592–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hall DR, Kozakov D, & Vajda S (2012) Analysis of Protein Binding Sites by Computational Solvent Mapping. Methods Mol Biol 819:13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kozakov D, et al. (2015) New Frontiers in Druggability. J Med Chem 58(23):9063–9088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hall DR, Ngan CH, Zerbe BS, Kozakov D, & Vajda S (2012) Hot Spot Analysis for Driving the Development of Hits into Leads in Fragment-Based Drug Discovery. J Chem Inf Model 52(1):199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Khan H, et al. (2015) Molecular effects of cancer-associated somatic mutations on the structural and target recognition properties of Keap1. Biochem J 467:141–151. [DOI] [PubMed] [Google Scholar]
- 107.Saito T, et al. (2016) p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat Commun 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Winkel AF, et al. (2015) Characterization of RA839, a Noncovalent Small Molecule Binder to Keapl and Selective Activator of Nrf2 Signaling. J Biol Chem 290(47):28446–28455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ludlow RF, Verdonk ML, Saini HK, Tickle IJ, & Jhoti H (2015) Detection of secondary binding sites in proteins using fragment screening. P Natl Acad Sci USA 112(52):15910–15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kobayashi A, et al. (2004) Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate for proteasomal degradation of Nrf2. Mol Cell Biol 24(16):7130–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lo SC & Hannink M (2007) PGAM5, a Bcl-XL-interacting protein, is a novel substrate for the redox-regulated Keap1-dependent ubiquitin ligase complex. Faseb J 21(6):A1022–A1022. [DOI] [PubMed] [Google Scholar]
- 112.Lee DF, et al. (2009) KEAP1 E3 Ligase-Mediated Downregulation of NF-kappa B Signaling by Targeting IKK beta. Mol Cell 36(1):131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Komatsu M, et al. (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12(3):213–U217. [DOI] [PubMed] [Google Scholar]
- 114.Ichimura Y, et al. (2013) Phosphorylation of p62 Activates the Keap1-Nrf2 Pathway during Selective Autophagy. Mol Cell 51(5):618–631. [DOI] [PubMed] [Google Scholar]
- 115.Hajduk PJ, Huth JR, & Fesik SW (2005) Druggability indices for protein targets derived from NMR-based screening data. J Med Chem 48(7):2518–2525. [DOI] [PubMed] [Google Scholar]
- 116.Beglov D, et al. (2018) Exploring the Origins of Cryptic Sites on Proteins. Proc. Nat’l Acad. Sci. USA 115:E3416–E3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shoichet BK (2006) Interpreting steep dose-response curves in early inhibitor discovery. J Med Chem 49(25):7274–7277. [DOI] [PubMed] [Google Scholar]
- 118.Kozakov D, et al. (2015) The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat Protoc 10(5):733–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Villar EA, et al. (2014) How proteins bind macrocycles. Nat Chem Biol 10(9):723–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mamonov AB, et al. (2016) Focused grid-based resampling for protein docking and mapping. J Comput Chem 37(11):961–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.