

Autism-associated CHD8/CHD7/kismet deficiency in blood-brain barrier glia causes developmental hyperserotonemia and sleep defects.
Abstract
Sleep disturbances in autism and neurodevelopmental disorders are common and adversely affect patient’s quality of life, yet the underlying mechanisms are understudied. We found that individuals with mutations in CHD8, among the highest-confidence autism risk genes, or CHD7 suffer from disturbed sleep maintenance. These defects are recapitulated in Drosophila mutants affecting kismet, the sole CHD8/CHD7 ortholog. We show that Kismet is required in glia for early developmental and adult sleep architecture. This role localizes to subperineurial glia constituting the blood-brain barrier. We demonstrate that Kismet-related sleep disturbances are caused by high serotonin during development, paralleling a well-established but genetically unsolved autism endophenotype. Despite their developmental origin, Kismet’s sleep architecture defects can be reversed in adulthood by a behavioral regime resembling human sleep restriction therapy. Our findings provide fundamental insights into glial regulation of sleep and propose a causal mechanistic link between the CHD8/CHD7/Kismet family, developmental hyperserotonemia, and autism-associated sleep disturbances.
INTRODUCTION
Sleep is essential for health and cognition. In neurodevelopmental disorders, sleep disturbances are among the most common co-occurring features, observed with an incidence ranging between 50 and 80% (compared to ~20% in typically developing children) (1, 2). Sleep problems adversely affect quality of life in patients and their families, to an extent that can exceed the burden of other physical and cognitive deficits (1, 3). Despite evidence that sleep interventions can improve behavioral and cognitive outcomes (4), the mechanisms underlying sleep disturbances in the context of these disorders are unclear and understudied.
We propose that monogenic (Mendelian) neurodevelopmental syndromes that are characterized by highly prevalent and specific sleep disturbances offer unique opportunities to investigate disease pathologies and mechanisms, including those underlying sleep difficulties. At the same time, this work holds the potential to provide novel fundamental insights into the biology of sleep and its mechanistic links to cognitive disorders.
Pathogenic mutations in the CHD8 (chromodomain helicase DNA binding protein 8) gene are a leading risk factor for autism spectrum disorders (ASDs) and among the most frequent findings revealed by large-scale exome sequencing efforts (5–7). They define an ASD subtype with distinctive clinical features [Online Mendelian Inheritance in Man (OMIM) 615032], including sleep complaints in most individuals with CHD8 mutations, while other clinical comorbidities such as intellectual disability (ID) remain variable (5, 8). However, the nature of CHD8-associated sleep problems (i.e., which parameters of sleep are affected) is unclear, and whether CHD8 mutations increase the risk of sleep complaints compared to other genetic forms of ASD is controversial (9). Disruptive de novo mutations in the CHD8 paralog CHD7 lead to CHARGE syndrome (OMIM 214800), a rare and phenotypically variable neurodevelopmental disorder characterized predominantly by severe ID (10, 11). Although initially less attention has been paid to behavioral problems, it is now well established that children with CHARGE syndrome also frequently exhibit autistic features and sleep disturbances (12, 13). In these patients, sleep problems are significantly associated with anxiety and autistic-like behaviors, greatly influencing their quality of life (12). CHD8 and CHD7 interact with each other and are part of a large multisubunit complex that organizes chromatin assembly (14). Their molecular interaction suggests that the overlapping clinical features of both disorders have a common mechanistic basis.
Work in Drosophila melanogaster, the fruit fly, has made seminal contributions to our understanding of sleep and neurodevelopmental disorders (15, 16). In Drosophila, kismet (kis) is the sole ortholog of CHD8 and CHD7 (17). Neuronal Kismet regulates morphology and synaptic physiology of the larval neuromuscular junction, axonal morphology and pruning of Kenyon cells, memory, and motor function (17–19). Moreover, Kismet is essential in a subset of circadian neurons for light-dependent circadian responses (20).
Here, we analyzed the role of the CHD8/CHD7/Kismet family in sleep regulation across species. First, we characterized the nature of sleep complaints in individuals with CHD8 and CHD7 mutations. We found specific defects in sleep initiation and maintenance that are highly consistent with each other and closely recapitulated in Drosophila. Leveraging the genetic tractability of Drosophila, we investigated the underlying spatiotemporal mechanisms of these sleep disturbances. Our data demonstrate that Kismet is required in glia, not in neurons, for sleep integrity at both larval and adult stages. We further localize sleep disturbances to a glial subtype constituting the blood-brain barrier (BBB) and to high serotonin levels during development, linking several previously unconnected dots in autism pathology. Last, despite the developmental origin of Kismet sleep defects, we provide a translatable behavioral strategy to ameliorate adult sleep abnormalities.
RESULTS
Individuals with mutations in CHD8 and CHD7 suffer from sleep initiation and maintenance defects
Previous studies suggest a high prevalence of sleep complaints in individuals with mutations in CHD8 (5, 9). A more detailed examination of sleep disturbances in this population has not been described. We extracted detailed sleep data from 23 individuals with disruptive CHD8 variants from the Simons Simplex Collection (SSC) and National Institute of Mental Health-funded TIGER study, two large studies of neurodevelopmental disorder cohorts characterized by rare, ASD-associated single gene etiologies. This cohort showed significantly higher rates of difficulties falling asleep and maintaining asleep (i.e., frequent or prolonged nighttime awakenings) compared to a large sample of SSC individuals with idiopathic ASD (Fig. 1A and table S1). In addition, we observed a higher frequency of daytime complaints; however, this tendency did not withstand correction for multiple testing. There was no significant difference in rates of other sleep difficulties between groups.
Fig. 1. Mutations in the conserved CHD8/7/kismet family leads to sleep disturbances.
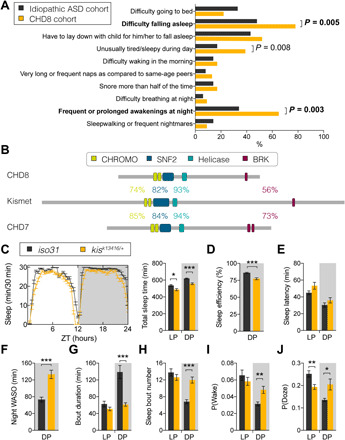
(A) Individuals with disruptive CHD8 mutations present with higher rates of difficulties falling asleep and frequent/prolonged night awakenings compared to a SSC idiopathic ASD cohort. Groups were compared using Fisher’s exact tests. Bonferroni correction for multiple comparisons was applied to the significance level (P < 0.005). Values in bold withstand correction for multiple testing. (B) High conservation of protein domains between human CHD8 and CHD7 and Drosophila Kismet. Percentages indicate similarity of amino acid sequence between Kismet and CHD8 (top) or CHD7 (bottom). CHROMO, chromatin organization modifier. (C) Sleep profile and quantification of TST of kisk13416/+ flies (n = 55) and iso31 genetic background controls (n = 61). kisk13416/+ mutants show decreased TST during the light period [LP; Zeitgeber time (ZT) 0 to ZT12, P = 0.016, t = 3.244, df = 96.44] and dark period (DP; ZT12 to ZT24, P < 0.0001, t = 5.201, df = 87.4). (D) SE is reduced in kisk13416/+ (P < 0.0001, t = 5.201, df = 87.4). (E) Latency to the first sleep bout after lights-on or lights-off is unaffected (P > 0.8). (F) WASO is increased at night (P < 0.0001, t = 5.2, df = 88.14). (G) Average bout duration in kisk13416/+ flies is strongly reduced specifically during the DP (P < 0.0001, t = 4.781, df = 68.78), while (H) the number of bouts is increased (P < 0.0001, t = 1.039, df = 111.1). (I) P(Wake) is significantly increased during the DP (P = 0.0048, t = 3.442, df = 48.07), while (J) P(Doze) is decreased in LP (P = 0.0075, t = 3.255, df = 59.37) but increased in DP (P = 0.042, t = 2.692, df = 36.62) in kisk13416/+ mutants compared to iso31. Data are represented as means ± SEM. Two-tailed unpaired Welch’s t test with Bonferroni correction for multiple testing. P values are indicated as follows: *P < 0.05, **P < 0.01, and ***P < 0.001.
Next, we further pursued in-depth sleep phenotyping in a small cohort consisting of one individual with a CHD8 (individual 01) and three with CHD7 mutations (individuals 02 to 04). All four individuals presented with sleep initiation and maintenance problems as indicated by increased sleep onset latency (SOL), increased wake after sleep onset (WASO), decreased sleep efficiency (SE; proportion of time spent asleep while lying in bed), and shorter total sleep time (TST) compared to reference values. Hence, all individuals exhibited chronic insomnia disorder as defined by the ICSD (International Classification of Sleep Disorders)–3 criteria (Table 1). Notably, except in one individual with a sleep-related rhythmic movement disorder, a common finding in children, no other sleep disorders [e.g., obstructive sleep apnea, as previously reported in some individuals with CHARGE syndrome (21)] were present. Objective measures of sleep, polysomnography (PSG) or actigraphy, were collected in individuals 02 and 04, respectively (fig. S1), validating the results of the sleep diaries. Our results are also consistent with previously reported sleep initiation and maintenance problems in CHARGE syndrome based on questionnaires, albeit these were collected, in part, for individuals lacking a genetic diagnosis (3, 13). Together, our data strongly suggest a specific role for both CHD8 and CHD7 in sleep initiation and maintenance.
Table 1. One individual with a CHD8 (01) and three individuals with CHD7 (02 to 04) pathogenic mutations, seen at a sleep clinic, also reported difficulties falling asleep and night awakenings.
Quantitative sleep parameters from parent-reported sleep-wake diaries confirmed these complaints: SOL and WASO were increased, and SE was decreased. Values in bold are deviating from established healthy reference values (68, 79, 80). See also Fig. 1, fig. S1, and tables S1 and S2.
| Individuals | 01 | 02 | 03 | 04 |
| Gender | Male | Female | Female | Female |
| Age of examination (years) | 4 | 3 | 8 | 19 |
| Molecular diagnosis | CHD8 | CHD7 | CHD7 | CHD7 |
| c.4062 + 5G > C | c.4493_4506del | c.2643 T > A | c.5181 > G | |
| p.(Ser1350fs) | p.(lle1498fs) | p.(Tyr881*) | p.(Tyr1727*) | |
| Main sleep complaints | Difficulty falling asleep | Difficulty falling asleep | Difficulty falling asleep | Difficulty falling asleep |
| Night awakenings | Night awakenings | Night awakenings | Night awakenings | |
| ICSD-3 diagnosis | Chronic insomnia | Chronic insomnia | Chronic insomnia | Chronic insomnia; Sleep-related rhythmic movement disorder |
| Sleep-wake diaries characteristics | ||||
| Days measured | 14 | 14 | 7 | 16 |
| TIB (min) | 715.7 | 677.1 | 762.9 | 647.8 |
| Total sleep time (min) | 474.6 | 559.3 | 514.3 | 400.3 |
| SOL (min) | 0 | 20.4 | 160.7 | 197.8 |
| WASO (min) | 241.1 | 97.5 | 87.9 | 49.7 |
| SE (%) | 66.7 | 82.5 | 67.6 | 61.7 |
Kismet mutants recapitulate human sleep maintenance defects
To address the potential role of the Drosophila CHD8/CHD7 ortholog Kismet (Fig. 1B) in sleep, we first set out to characterize sleep in kisk13416 mutants. While homozygous kisk13416 flies are not viable, kisk13416/+ flies showed a mild but significant decrease in TST both during the light and dark periods (Fig. 1C). To allow for comparison with human sleep measures, we assessed orthologous sleep metrics (22) and found SE to be decreased (Fig. 1D). Whereas sleep latency was unaffected (Fig. 1E), mutants spent more time awake at night after their first sleep episode (WASO) (Fig. 1F). The elevated WASO in these animals was due to fragmented nighttime sleep, with a strong reduction in average sleep bout duration (from 140 min in controls to 61 min in kisk13416/+) and an increase in the number sleep bouts (Fig. 1, G and H), revealing altered sleep architecture. We assessed circadian rhythms of locomotor activity in kismet mutants by means of fast Fourier transform (FFT). They showed normal rhythmicity (fig. S2).
We further determined two recently defined conditional probabilities to measure Drosophila sleep depth and pressure (23); P(Wake) can be regarded as a behavioral correlate of sleep depth or arousal, whereas P(Doze) reflects sleep pressure. kisk13416/+ mutants showed elevated P(Wake) and P(Doze) at night, suggesting that sleep-wake instability drives nighttime sleep fragmentation (Fig. 1, I and J). P(Doze) was also decreased during the day, indicating reduced sleep drive during this time. Together, kismet mutant flies show reduced sleep duration with altered sleep architecture, indicating defects in sleep maintenance. These sleep deficits resemble the identified sleep disturbances in individuals with CHD8/7 mutations.
Kismet is required in glia for sleep integrity
To collect independent evidence for the role of Kismet in sleep integrity and to identify the cellular substrates through which it exerts its function, we next knocked down its expression in various cell types using the UAS-Gal4 system and inducible RNA interference (RNAi). Unexpectedly, kismet knockdown with the pan-neuronal elav-Gal4 did not recapitulate kisk13416/+ sleep defects (fig. S3, A to C). In contrast, pan-glial expression of two nonoverlapping RNAi constructs, UAS-kisRNAi-1 and UAS-kisRNAi-2, both gave rise to a significant reduction in TST at night (Fig. 2, A and B). This loss was due to a markedly decreased mean sleep bout duration (Fig. 2, Ai and Bi), partially compensated by an increase in the number of sleep bouts (Fig. 2, Aii and Bii). Thus, pan-glial knockdown recapitulated the reduced and fragmented nighttime sleep, causing even more severe defects than shown by the heterozygous kismet mutants. This indicates that Kismet is required in glia for sleep integrity.
Fig. 2. Pan-glial kismet knockdown leads to decreased and fragmented nighttime sleep, a phenotype of developmental origin.
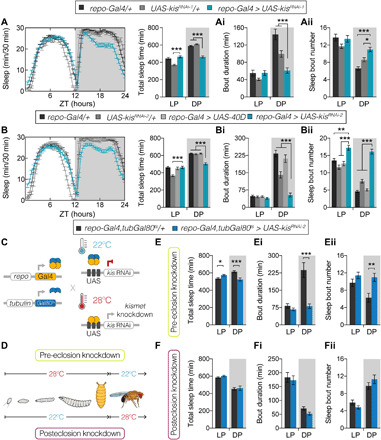
(A, B, E, and F) Sleep profiles and/or quantification. (Ai, Bi, Ei, and Fi) Average duration and (Aii, Bii, Eii, and Fii) number of sleep bouts during the LP (ZT0 to ZT12) and DP (ZT12 to ZT24). (A, Ai, and Aii) Pan-glial kismet knockdown flies (repo-Gal4,UAS-Dcr2 > UAS-kisRNAi-1, in turquois, n = 65) compared to background controls (repo-Gal4,UAS-Dcr2/+ in black, n = 74; UAS-kisRNAi-1/+ in gray, n = 61) show reduced sleep time (P < 0.0001) and sleep fragmentation, characterized by reduced bout length (P < 0.0001), accompanied by increased bout number (P < 0.0001 and P = 0.011, respectively) exclusively during DP. (B, Bi, and Bii) Pan-glial kismet knockdown (repo-Gal4,UAS-Dcr2 > UAS-kisRNAi-2, in turquois, n = 77) compared to background (repo-Gal4,UAS-Dcr2/+ in black, n = 90; UAS-kisRNAi-2/+ in dark gray, n = 65) and UAS-40D controls (repo-Gal4,UAS-Dcr2 > UAS-40D, in light gray, n = 67), causes reduced sleep time (P < 0.0001) and reduced bout length (P < 0.0001), accompanied by increased bout number (P = 0.0027 and P < 0.0001, respectively), during DP. (C) Schematic representation of the TARGET system and (D) performed temperature shifts to achieve pre- and posteclosion knockdown. (E, Ei, and Eii) Pre-eclosion pan-glial kismet knockdown (repo-Gal4,tubGal80ts > UAS-kisRNAi-2, in blue, n = 44) show decreased and fragmented sleep (P = 0.0006, P = 0.0003, and P = 0.006, respectively) during DP compared to controls (repo-Gal4,tubGal80ts/+, in black, n = 31). (F, Fi, and Fii) Posteclosion pan-glial kismet knockdown (repo-Gal4,tubGal80ts > UAS-kisRNAi-2, in blue, n = 51) does not alter sleep compared to controls (repo-Gal4,tubGal80ts/+, in black, n = 34). Data are represented as means ± SEM. (A, Ai, Aii, B, Bi, and Bii): Kruskal-Wallis test with Dunn’s correction. (E, Ei, Eii, F, Fi, and Fii): Two-tailed unpaired Welch’s t test. Bonferroni correction for multiple testing. P values are indicated as follows: *P < 0.05, **P < 0.01, and ***P < 0.001. See also fig. S4.
Adult sleep integrity relies on Kismet during development
To investigate whether Kismet is required in glia for sleep integrity during development or acutely in adulthood, we used temporal and regional gene expression targeting (TARGET) to restrict kismet knockdown to either before or after eclosion (Fig. 2, C and D). Flies carrying the repo-Gal4 driver and ubiquitous GAL4 repressor tubGal80ts kept at restrictive temperature (22°C, knockdown OFF) throughout their life span showed comparable TST and architecture to background controls (fig. S4, A, Ai, and Aii). In contrast, when kept at the permissive temperature (28°C, knockdown ON), flies exhibited decreased and fragmented nighttime sleep (fig. S4, B, Bi, and Bii). Pan-glial kismet knockdown confined to pre-eclosion stages with either UAS-kisRNAi line resulted in decreased and fragmented nighttime sleep in the adult, recapitulating the defects in kismet knockdown throughout life span (Fig. 2, E, Ei, and Eii, and fig. S4, C, Ci, and Cii). In contrast, kismet knockdown exclusively in adult glia did neither alter sleep quantity nor architecture (Fig. 2, F, Fi, and Fii, and fig. S4, D, Di, and Dii). Together, these results demonstrate that Kismet is required during pre-eclosion developmental stages in glia for adult sleep integrity.
Kismet is required in subperineurial glia of the BBB for sleep integrity
We next asked whether Kismet acts in neuropil-associated glia [astrocyte-like glia (ALG) and ensheathing glia (EG)], cortex glia (CG), or surface glia [perineurial glia (PG) and subperineurial glia (SPG)]. While previous work has implicated almost all glia subtypes in sleep regulation (24–26), sleep loss and fragmentation have exclusively been linked to ALG (27, 28). Using specific Gal4 drivers (29, 30), we noted that kismet knockdown in ALG (NP3233-Gal4) showed mild sleep fragmentation during both day and night (fig. S5, A, Ai, and Aii). Knockdown in EG (NP6520-Gal4) or CG (NP2222-Gal4) did not cause nighttime sleep fragmentation (fig. S5, B, Bi, Bii, C, Ci, and Cii).
Notably, kismet knockdown using the well-characterized R54C07-Gal4 driver targeting SPG presented with the combination of decreased nighttime sleep time and reduction of average sleep bouts observed in kisk13416/+ and pan-glial knockdown, with both UAS-kisRNAi lines (Fig. 3, A, Ai, Aii, B, Bi, and Bii). Furthermore, kismet knockdown with an independent SPG driver line (NP2276-Gal4) resulted in decreased total sleep and bout duration and increased bout number (fig. S5, E, Ei, and Eii). SPG are a group of about 300 cells that, together with the PG, form the Drosophila BBB (29). The inner SPG layer forms the paracellular barrier that separates the brain from the hemolymph, whereas PG provide physical and metabolic support and harbor a peripheral molecular clock that modulates circadian changes in BBB permeability (31). However, we found that kismet knockdown in PG (R85G01-Gal4) did not alter nighttime sleep (Fig. 3, C, Ci, and Cii). It has also been shown that inhibition of endocytosis of the surface glia increases baseline sleep and induces resistance to mechanical sleep deprivation (26). In contrast, SPG kismet knockdown did not affect homeostatic sleep rebound after mechanical deprivation (fig. S6, A and B). Together, these findings demonstrate that loss of kismet in SPG underlies reduced sleep and severe sleep fragmentation.
Fig. 3. Kismet function in sleep integrity maps to SPG of the BBB.
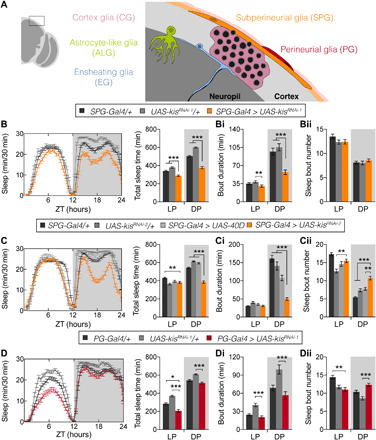
(A) Schematic representation of glia subtypes in the adult Drosophila brain. (B to D) Sleep profiles and quantification. (Bi, Ci, and Di) Average duration and (Bi, Ci, and Di) number of sleep bouts in the LP (ZT0 to ZT12) and DP (ZT12 to ZT24). (B, Bi, and Bii) Kismet knockdown in SPG with the R54C07-Gal4 driver (SPG-Gal4 > UAS-kisRNAi-1, in orange, n = 104) compared to background controls (SPG-Gal4/+, in black, n = 134; UAS-kisRNAi-1/+ in gray, n = 85) shows reduced sleep time during both LP (P = 0.0006) and DP (P < 0.0001), due to shorter sleep bouts (P < 0.0001). (C, Ci, and Cii) Kismet knockdown in SPG with the R54C07-Gal4 driver (SPG-Gal4 > UAS-kisRNAi-2, in orange, n = 108) compared to background controls (SPG-Gal4/+, in black, n = 150; UAS-kisRNAi-2/+ in dark gray, n = 86) and UAS-40D control (SPG-Gal4 > UAS-40D, in light gray, n = 66) shows reduced sleep time during DP (P < 0.0001). Knockdown flies also display shorter nighttime bouts (P < 0.0001) accompanied by an increase in their number (P < 0.0001 and P = 0.0024, respectively). (D, Di, and Dii) Kismet knockdown in PG with the R85G01-Gal4 driver (PG-Gal4 > UAS-kisRNAi-1, in red, n = 80) compared to background controls (PG-Gal4/+ controls in black, n = 84; UAS-kisRNAi-1/+ in gray, n = 62) shows reduced sleep time at LP (P = 0.013 and P < 0.0001, respectively). Nighttime sleep is unaffected. Data are represented as means ± SEM. Kruskal-Wallis test with Dunn’s correction. Bonferroni correction for multiple testing. P values are indicated as follows: *P < 0.05, **P < 0.01, and ***P < 0.001. See also figs. S5 and S6.
Loss of Kismet in SPG disrupts sleep architecture in developing larvae
Given that kismet knockdown in glia before eclosion is sufficient to alter sleep architecture in the adult fly, we wondered whether sleep problems may already manifest during development. Second instar Drosophila larvae have recently been shown to exhibit quiescent periods that meet the behavioral criteria of sleep (32). Kismet knockdown in SPG did not alter the total amount of larval sleep (Fig. 4A) but disrupted sleep architecture, resulting in decreased bout length (Fig. 4Ai) accompanied by an increase in bout number (Fig. 4Aii). As observed for adult sleep, larval sleep remained unaffected by kismet knockdown in PG (Fig. 4, B, Bi, and Bii). Together, Kismet is required in second instar larvae for proper sleep architecture, and this role too localizes to SPG.
Fig. 4. Loss of Kismet in SPG leads to fragmented sleep in the larval stage.
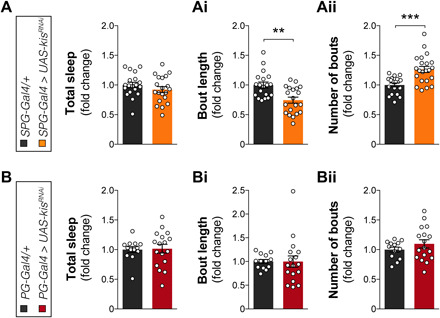
Kismet knockdown in SPG with the R54C07-Gal4 driver (SPG-Gal4 > UAS-kisRNAi-2, in orange, n = 20) compared to background controls (SPG-Gal4/+, in black, n = 19) does not alter the (A) total amount of larval (second instar) sleep (quiescence bouts) (P = 0.7, t = 1.149, df = 36.78) but causes (Ai) decreased duration (P = 0.003, t = 3.585, df = 36.98) and (Aii) increased number (P = 0.0003, t = 4.32, df = 35.14) of sleep bouts. (B) Kismet knockdown in PG with the R85G01-Gal4 driver (PG-Gal4 > UAS-kisRNAi-2, in red, n = 17) compared to background controls (PG-Gal4/+, in black, n = 13) does not alter the total amount of larval (second instar) sleep (P > 0.9, t = 0.178, df = 26.72), (Bi) average sleep bout duration (P > 0.9, t = 0.0186, df = 20.04) and (Bii) number (P = 0.7, t = 1.183, df = 25.44). Data are represented as means ± SEM. Two-tailed unpaired Welch’s t test with Bonferroni correction for multiple testing. P values are indicated as follows: **P < 0.01 and ***P < 0.001.
Glial Kismet loss leads to hyperserotonemia
To shed light onto the mechanisms by which loss of Kismet affects sleep, we first considered whether established sleep regulatory mechanisms may mediate Kismet function. Extensive research has highlighted the crucial role of dopamine in sleep and arousal and how dysfunctions of the dopaminergic system can lead to reduced and fragmented sleep (33). Therefore, we performed high-performance liquid chromatography (HPLC) to measure dopamine, using a column that simultaneously allowed us to assess serotonin (5-hydroxytryptamine) levels. Fly heads from pan-glial kismet knockdown and controls were collected at Zeitgeber time 6 (ZT6) and 18 (ZT18), without disturbing the light:dark (LD) cycle. Dopamine levels at ZT6 were unaltered, whereas we observed a significant, although mild, decrease of total dopamine levels at nighttime (ZT18) (Fig. 5A). In contrast, serotonin showed a notable 2-fold (ZT6) and 2.4-fold (ZT18) increase (Fig. 5B).
Fig. 5. Sleep defects upon glial kismet loss are caused by developmental hyperserotonemia.
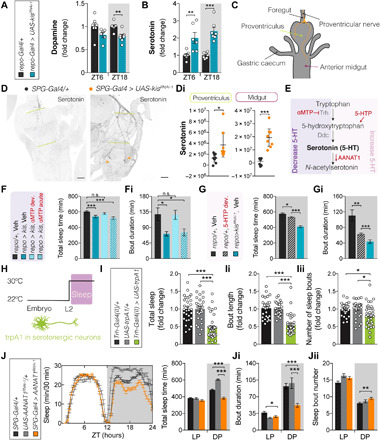
(A and B) repo-Gal4,UAS-Dcr2 > UAS-kisRNAi-1 (blue, n = 6 to 7) exhibit decreased dopamine at ZT18 (P = 0.0028) and ≥2-fold increased serotonin at ZT6 (P = 0.0077) and ZT18 (P = 0.0004) compared to controls (black, n = 7). (C) Scheme of the larval digestive tract and serotonergic innervations. (D) Representative images and (Di) quantification of α-serotonin immunolabeling. SPG kismet knockdown (R54C07-Gal4 > UAS-kisRNAi-1, orange, n = 8) causes increased serotonin in distal innervations (arrowheads) and diffuses signal in the proventriculus (P = 0.037) and anterior midgut (P = 0.0002) compared to controls (black, n = 8). Scale bars, 50 μm. (E) Serotonin metabolism in Drosophila and used approaches to manipulating it. (F, G, and J) Sleep profiles and/or quantification. (Fi, Gi, and Ji) Average duration and (Jii) number of sleep bouts during LP (ZT0 to ZT12) and DP (ZT12 to ZT24). (F and Fi) Pan-glial knockdown (repo-Gal4,UAS-Dcr2 > UAS-kisRNAi-1, dark blue, n = 32) fed with αMTP during development (light blue, n = 49), but not adulthood (light blue dotted, n = 30), shows restored TST and sleep bout duration to vehicle-reared controls (black, n = 41). (G and Gi) Increasing serotonin in wild type (hatched black, n = 72) by developmental 5-hydroxytryptophan (5-HTP) feeding leads to decreased sleep due to significantly reduced sleep bout length compared to vehicle-fed animals (black, n = 70), phenocopying kismet knockdown (blue, n = 62). (H) Temperature program for thermogenetically controlled larval sleep recordings: (I to Iii) Thermogenetic activation of serotonergic neurons [Trh-Gal4(II) > UAS-dTrpA1, green, n = 37] leads to decreased sleep amount (P < 0.0001), bout length (P < 0.0001), and number (P = 0.035 and P = 0.02, respectively) compared to parental controls (Trh-Gal4(II)/+, n = 29; UAS-dTripA1/+, n = 28). (J to Jii) SPG AANAT1 knockdown (SPG-Gal4 > UAS-AANAT1RNAi-1, n = 97) leads to decreased total sleep (P < 0.0001) and bout length (P < 0.0001) compared to controls (SPG-Gal4/+, n = 125; UAS-AANAT1RNAi-1/+, n = 37). Data are represented as means ± SEM, except (Di) represented as means ± 95% confidence interval. (A, B, and Di): Two-tailed unpaired Welch’s t test. (F, Fi, G, Gi, H, I, Ii, and Iii): Kruskal-Wallis test with Dunn’s correction. (J, Ji, and Jii): One-way analysis of variance (ANOVA). Bonferroni correction for multiple testing. P values: *P < 0.05, **P < 0.01, and ***P < 0.001. n.s., not significant. See fig. S7.
Following up on this finding, we investigated serotonin levels during development. We performed antiserotonin fluorescence immunohistochemistry and quantitative imaging (via photon counting, see Materials and Methods), in pan-glial and SPG-specific kismet knockdown wandering third instar larvae. We focused on the projections of serotonergic neurons of the subesophagal ganglion that innervate the proventriculus and anterior midgut of the larval digestive tract (Fig. 5C) (34). In controls, serotonin was visible in the proventricular nerve and at the level of proventricular innervations but absent or hard to discern in the distal projections that innervate the anterior midgut. In contrast, loss of glial Kismet led to elevated serotonin signal (Fig. 5D and fig. S7A), with serotonergic midgut innervations strongly labeled. As the innervated tissues appeared to show slightly higher levels of staining too, we quantified total signal intensities of α-serotonin signal across the proventriculus and in the anterior midgut, containing both the serotonergic terminals and the innervated tissues. We observed significantly increased serotonin at the midgut region of pan-glial kismet knockdown larvae (fig. S7, B, Bi, Bii, C, Ci, and Cii). Similarly, serotonin was significantly increased at the level of the proventriculus and midgut of SPG-specific kismet knockdown larvae (Fig. 5Di and fig. S7, E and Ei). Together, these results demonstrate that Kismet loss in glia leads to increased serotonin during development. The finding that glial loss of Kismet leads to increased serotonin is of considerable interest, as elevated serotonin (hyperserotonemia) is one of the most replicated, yet genetically unsolved, biomarkers in ASD (35, 36).
Developmental hyperserotonemia underlies Kismet’s glia-dependent sleep defects
To investigate whether restoring dopamine signaling can reverse the sleep defects of pan-glial kismet knockdown, we supplemented the food with methylphenidate (MPH; 1 mg/ml a dopamine transporter blocker that increases extracellular dopamine levels by blocking its reuptake), a dose that we and others has previously been shown to rescue sleep and other behavioral deficits of dopamine-related mutant conditions (33, 37). However, MPH did not rescue sleep duration or architecture in the setting of kismet knockdown (fig. S8, A, Ai, and Aii).
Next, we addressed the hypothesis that the observed high serotonin levels underlie the reduced and fragmented sleep in the setting of glial kismet knockdown, by testing the effect of pharmacological serotonin depletion. Adult administration of 20 mM α-methyl-dl-tryptophan (αMTP), a specific Trh (CG9122) inhibitor (Fig. 5E) that has been show to induce a ~2-fold decrease in serotonin levels in fly heads (38), failed to improve sleep deficits of kismet pan-glial knockdown (Fig. 5, F and Fi). Administration of the same dose throughout development led to high lethality in control flies but notably allowed pan-glial kismet knockdown flies to survive. In this population, αMTP supplementation fully averted the adult sleep fragmentation (Fig. 5, F and Fi). Thus, elevated serotonin levels during development underlie, at least partially, the sleep fragmentation observed in adult kismet pan-glial knockdown flies.
Next, we sought to determine whether increased serotonin levels are sufficient to induce decreased and fragmented sleep. First, we attempted to pharmacologically increase serotonin levels by feeding its precursor 5-hydroxytryptophan (5-HTP; Fig. 5E). Administration of 5 mM 5-HTP specifically during development led to decreased sleep time and shorter sleep bouts in adults (Fig. 5, G and Gi). Second, recent work has shown that acute opto- or thermogenetic activation of serotonergic neurons in Drosophila adults leads to a decrease in sleep bout length and increase in sleep bout number, indicating that increased release of serotonin can fragment sleep (39). As Kismet’s sleep phenotype is due to increased serotonin during development, we evaluated the effect of activating serotonergic neurons at this time. We expressed the heat-activated channel dTrpA1 using the Trh-Gal4 driver and activated these neurons by raising the temperature while monitoring sleep in second instar larvae (Fig. 5H). Activation of serotonergic neurons with two different drivers led to a large reduction in sleep bout length and total sleep amount (Fig. 5, I, Ii, and Iii, and fig. S8, B, Bi, and Bii).
Last, we increased serotonin by reducing its metabolization. Serotonin acetylation, catalyzed by the arylalkylamine N-acetyltransferase 1 (AANAT1) enzyme (Fig. 5E), is the main mechanism of neurotransmitter inactivation in Drosophila, which stands in contrast to mammals where oxidation plays a central role (40). We therefore knocked down AANAT1 in all glia and in SPG. Whereas pan-glial AANAT1RNAi-1 knockdown was lethal, AANAT1 knockdown with a second RNAi line (AANAT1RNAi-2) resulted in fragmented nighttime sleep (fig. S8, C, Ci, and Cii). Moreover, AANAT1RNAi-1 induction in SPG was viable and phenocopied the reduced and fragmented nighttime sleep (Fig. 5, J, Ji, and Jii). The latter finding was replicated with a third independent RNAi line targeting AANAT1 (UAS-AANAT1RNAi-3) (fig. S8, D, Di, and Dii). Together, our data provide strong evidence for loss of glial Kismet causing sleep loss and fragmentation through increased serotonin levels during development.
Sleep restriction therapy restores sleep architecture in glial Kismet models
We aimed to identify approaches to improving sleep quality in our preclinical Kismet models. Approaches suitable for direct translation into the clinic should (i) require only postdevelopmental intervention and (ii) should be noninvasive and nonhazardous. Human sleep restriction therapy (SRT) is a key component of cognitive behavioral therapy for insomnia (CBT-I), the first-line intervention to treat chronic insomnia in humans (41). By restricting the sleep opportunity window, SRT enhances sleep drive leading to a more efficient and consolidated sleep. We recently established a model for SRT in flies and demonstrated that SRT in flies improves SE and continuity in short-sleeping Drosophila mutants and a fly model for Alzheimer’s disease (22). Whether SRT can reverse sleep defects in neurodevelopmental disorders models is unknown.
We first restricted sleep opportunity in pan-glial kismet knockdown flies by reducing dark period duration. An SRT regime of 14:10 LD did not increase TST compared with pan-glial or SPG kismet knockdown flies in 12:12 LD (fig. S9, A and Ai); however, SE was fully rescued to control levels (Fig. 6, A, Ai, B, and Bi). Furthermore, SRT rescued sleep bout duration (Fig. 6, C and Ci) and bout number (Fig. 6, D and Di). Most notably, both pan-glial and SPG kismet knockdown flies showed a significant decrease in WASO, back to control at 12:12 LD levels (Fig. 6, E and Ei). SRT further reduced latency to sleep at night compared to flies in a 12:12 LD regime (fig. S9, B and Bi). Together, our data show that a behavioral regime can rescue sleep defects of Kismet models despite their developmental origin, including disturbed sleep architecture, decreased efficiency and increased WASO, The critical sleep metrics affected in individuals with CHD8 and CHD7 mutations.
Fig. 6. Sleep restriction therapy rescues sleep architecture in Drosophila glia Kismet models.
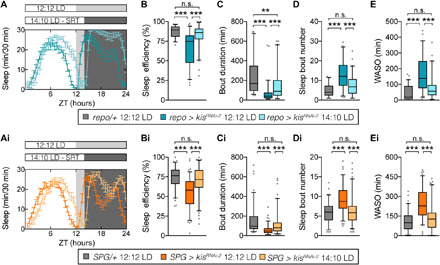
(A) Representative sleep profiles of pan-glial kismet knockdown flies entrained either at 12:12 LD (repo-Gal4,UAS-Dcr2 > UAS-kisRNAi-2, in dark blue, n = 13) or at 14:10 LD (repo-Gal4,UAS-Dcr2 > UAS-kisRNAi-2, in light blue, shortened DP ZT14 to ZT24, n = 15). (Ai) Representative sleep profiles of SPG kismet knockdown flies entrain either at 12:12 LD (SPG-Gal4 > UAS-kisRNAi-2, in dark orange, n = 16) or at 14:10 LD (SPG-Gal4 > UAS-kisRNAi-2, in light orange, n = 16). (B to E) Quantification of sleep parameters of control flies entrained at 12:12 LD (repo-Gal4,UAS-Dcr2/+, in gray, n = 61), and pan-glial kismet knockdown flies entrain either at 12:12 LD (repo-Gal4,UAS-Dcr2 > UAS-kisRNAi-2, in dark blue, n = 62) or at 14:10 LD (repo-Gal4,UAS-Dcr2 > UAS-kisRNAi-2, in light blue, n = 63). (Bi, Ci, Di, and Ei) Quantification of sleep parameters of control flies entrained at 12:12 LD (SPG-Gal4/+, in gray, n = 85), and SPG kismet knockdown flies entrain either at 12:12 LD (SPG-Gal4 > UAS-kisRNAi-2, in dark orange, n = 97) or at 14:10 LD (SPG-Gal4 > UAS-kisRNAi-2, in light orange, n = 100). (B and Bi) SE, (C and Ci) average duration, and (D and Di) number of sleep bouts in the DP. (E and Ei) WASO; minutes spent awake after first sleep episode during DP. SRT successfully improves SE and architecture in pan-glial and SPG kismet knockdown flies. Data are represented as a boxplot that extends from the 25th to the 75th percentiles with the median indicated. Whiskers indicate the 5th and 95th percentiles. Kruskal-Wallis test with Dunn’s correction. Bonferroni correction for multiple testing. P values are indicated as follows: **P < 0.01 and ***P < 0.001. See also fig. S9.
Human CHD8 and CHD7 are expressed in glia and components of the BBB
Last, to address whether a conserved role in glia or other cells forming the BBB can underlie the sleep disturbances shared by Drosophila Kismet mutants and individuals with mutations in CHD8 or CHD7, we revisited recent high-resolution single-cell RNA sequencing (RNA-seq) datasets of human cortex at prenatal and postnatal stages. We found CHD8 and CHD7 to be modestly to highly expressed in all cells forming the human BBB, both during development (radial glia, astrocytes, endothelial, and mural cells) and in adulthood (astrocytes, endothelial cells, and pericytes) (Fig. 7, A, Ai, and Aii). At adult stage, CHD7 shows a notable enrichment in glia compared to neuronal populations, whereas CHD8 is widely expressed across the brain (Fig. 7, B, Bi, and Bii). In conclusion, cutting edge single-cell data support a role for both kismet orthologs in glia and other cell types constituting the human BBB, both during development and in adulthood.
Fig. 7. Human CHD8 and CHD7 are highly expressed in glia and components of the BBB.

(A and B) (Ai and Bi) CHD8 and (Aii and Bii) CHD7 expression in single-cell RNA-seq data of (A, Ai, and Aii) prenatal and (B to Bii) adult human cortex support a role for both kismet orthologs in glia throughout life span. Scatterplots after principal components analysis and t-stochastic neighbor embedding from left to right: colored by cluster, CHD8, and CHD7 expression.
DISCUSSION
Sleep problems in ASD-associated neurodevelopmental syndromes and idiopathic ASD are a common complaint. The negative impact of sleep loss on these vulnerable patient groups and their families is particularly high, and therapeutic options are urgently needed. Whether sleep is under direct control of genes implicated in ASD and related neurodevelopmental disorders is unknown for the vast majority of disorders. Motivated by reports on frequent and severe sleep disturbances in individuals with mutations in the chromatin remodeler CHD8 and its paralog CHD7, we here characterized in-depth their sleep deficits and dissected the underlying mechanisms using Drosophila as a model. Our study provides previously unknown fundamental insights into the regulation of sleep, linking autism-related CHD8 and CHD7 genes, BBB glia, and developmental hyperserotonemia with sleep maintenance defects, and identifies a safe, validated, and translatable behavioral strategy to reduce sleep challenges for patients.
Specific and highly conserved sleep deficits caused by mutations in the CHD8/CHD7/Kismet chromatin remodeling family
By extracting sleep data from two large and comprehensive ASD cohorts, combined with more detailed sleep phenotyping in a smaller cohort, we found that individuals with disruptive CHD8 and CHD7 mutations show difficulties falling asleep and frequent and/or prolonged night awakenings, identifying specific problems in sleep maintenance. Despite sleep disturbances being one of the most prevalent complaints reported in CHD8 individuals (5, 8), it has been questioned whether the frequency of sleep defects is elevated over the already high prevalence associated with ASD (42) and thus whether CHD8 does play a specific role in sleep (9). Resolving various aspects of sleep disorders, we found sleep initiation and maintenance to be specifically enriched compared to idiopathic ASD, highlighting the importance of examining sleep defects with the greatest possible resolution. Moreover, using objective sleep assessment shows that the same complaints—short TST, low SE, and increased SOL and WASO—are present in individuals with mutations in its paralog CHD7, in accordance with previous CHARGE syndrome literature based on sleep questionnaires (3, 12, 13). These results must be interpreted in the context of the limited cohort size that was available to us. Because of this limitation, it also remains to be determined whether sleep disturbance severity in individuals with CHD7 compared to CHD8 mutations correlate with the overall more severe and broader clinical manifestation of CHARGE syndrome. However, the shared sleep characteristics of both disorders made them highly suitable for sleep studies in Drosophila with its single orthologous gene.
Similar to humans, we identified decreased and fragmented nighttime sleep in Drosophila kisk13416/+ mutants, denotating disturbed sleep maintenance. Although flies also sleep during the day, longer sleep episodes preferentially occurring during the night are thought to reflect a crucial, deep sleep state (39). In kismet mutants, this disruption in sleep architecture results in decreased SE and increased night WASO, paralleling the sleep defects identified in individuals with mutations in CHD8/CHD7. Although sleep differences in the kismet mutant primarily rely on glial Kismet function, the absence of sleep phenotypes in pan-neuronal knockdown flies does not exclude that Kismet may also modulate sleep through executing similar functions in neuronal entities regulating sleep. Considerable evidence suggests key roles for sleep in synaptic development and plasticity (43). Kismet has previously been shown to regulate synapse assembly and synaptic transmission (18), and our findings establish a role for Kismet in sleep. Recent work in flies has likewise found that insomniac controls synaptic homeostasis in addition to its role in adult sleep (44). These results set the stage for investigating how sleep and synaptic functions are coupled at a genetic level. Our study reveals specific, highly conserved, and consistent defects in sleep maintenance caused by mutations in the CHD8/CHD7/Kismet chromatin remodeling gene family.
Kismet and BBB glia play a role in sleep integrity, potential implications for individuals with CHD8/CHD7 mutations
Despite Kismet’s predominant expression in neurons (17, 18), we mapped its role in sleep to glia and more specifically to SPG, a key component of the Drosophila BBB. Thus, our data raise the intriguing possibility that glia and the BBB are involved in the pathophysiology of the disorder. Despite only representing 2% of the glia population (29), kismet knockdown in SPG fully recapitulated pan-glial manipulations. Notably, this does not rule out that Kismet also operates in other glial subtypes to affect sleep. We observed mild but significant sleep fragmentation upon kismet knockdown in ALG and a decrease in sleep time upon EG manipulation. These additional roles of Kismet likely contribute to minor differences in phenotype between mutant, pan-glial, and SPG-specific phenotypes. We focused on the most marked and innovative phenotype and revealed that SPG dysregulation can give rise to reduced and fragmented sleep.
Inhibition of endocytosis in SPG and PG has recently been found to increase and consolidate sleep and to confer resistance to sleep deprivation (26). Our finding that sleep rebound remains intact upon kismet knockdown and that its function is confined to SPG, not PG, suggests that SPG regulate different sleep processes through multiple mechanisms. Apart from sleep, it has been shown that the permeability of the Drosophila BBB is rhythmically controlled by a molecular clock residing in PG (31). Whether Kismet loss also leads to changes in BBB permeability and how this relates to loss of CHD8 and/or CHD7 are exciting questions for future research, as emerging data suggest a potential connection of BBB changes to other dysregulated states, including epilepsy (45).
Notably, Kismet loss in SPG already disrupts sleep architecture in Drosophila larvae, resulting in shorter and more frequent bouts. Thus, our work reveals the importance of glia cells, specifically of SPG, in larval sleep and shows that Kismet’s function in sleep persists from early development to adulthood. In contrast, classic short-sleeping mutants do not show larval sleep defects, and even a key regulator of arousal such as dopamine does not show the same waking effects in larvae as in adults (32). It is conceivable that Kismet’s function in glia and its persistent role in sleep throughout life span are directly related. SPG are specified during embryogenesis and maintain numbers and function from early larval specification to adulthood (30), while neurons undergo extensive remodeling during metamorphosis (46). Hence, SPG-encoded regulation of sleep may provide a mechanism for persistent sleep control across the life span.
Could dysregulation of the BBB contribute to CHD8- and CHD7-associated pathologies? Our single-cell expression analyses revealed that both genes are expressed in glia in prenatal and adult human cortex. Although the mature mammalian BBB is not exclusively constituted by glia cells, it shares many commonalities with the Drosophila BBB, including structural features, function, and regulatory mechanisms (45). However, in mammalian embryogenesis, the first cellular barrier separating the nervous system from the vascular elements is constituted by radial glia (47). Most relevantly, both CHD8 and CHD7 are expressed in prenatal endothelial and mural cells and in radial glia. Both genes are also highly expressed in the components of the mature BBB (astrocytes, endothelial cells, and pericytes). They are also highly expressed in oligodendrocyte precursor cells (OPCs) and oligodendrocytes, together supporting a sustained role of CHD8/7 in glia and previous findings in mouse that have revealed a key role for both genes in OPC survival and oligodendrocyte differentiation, myelination, and postinjury remyelination (48–50). On the basis of their expression and in light of the highly specific and conserved aspects of sleep that are affected in both species, it appears plausible that glia and/or other BBB-contributing cells, and a function of CHD8 and CHD7 therein, are relevant to the pathophysiology of these neurodevelopmental disorders.
Origin and consequences of hyperserotonemia caused by glial loss of Kismet
We found that glial loss of Kismet leads to increased levels of serotonin, both in adults and in third instar larvae. Multiple studies have demonstrated elevated platelet serotonin in idiopathic ASD cohorts; this finding, referred to as hyperserotonemia, has proven to be one of the most replicated and consistent biomarkers in ASD (35, 36). However, despite ~60 years since the initial study reporting hyperserotonemia, its origin and role in ASD-associated clinical features have remained largely unsolved. Our study provides several important steps forward in this field. First, the genetic basis of hyperserotonemia in ASD has remained largely unknown (51). Our findings link hyperserotonemia to the evolutionary conserved CHD8/CHD7/Kismet family, highly implicated in ASD (6, 7). Second, the etiology of sleep problems associated with autism is unclear; they have classically been regarded as secondary to other behavioral deficits (52). Moreover, whereas the involvement of serotonin in sleep regulation has been known for many years, it has remained unclear whether serotonin plays a sleep- or a wake-promoting effect (34, 53). Recent work has shown that acute activation of serotonergic neurons in Drosophila adults leads to sleep fragmentation without changing sleep duration (39), whereas we observed that serotonergic neuronal activation during development (in second instar larvae) leads to a decrease in sleep amount and shorter sleep bouts. Together, these data suggest that the serotonergic regulation of sleep is neuronally mediated but context dependent (i.e., dependent on developmental stage and/or duration of serotonin dysregulation), which contributes to the complexity of serotonin’s role in sleep. Moreover, not unexpectedly, it appears to depend on the cell type of origin. Whereas we did not (yet) observe a change in total sleep amount in kismet knockdown L2 larvae, activation of serotonergic neurons at the same stage diminished total sleep. It is conceivable that this difference is due to a stronger activation of serotonergic neurons in the latter case. Despite this complexity, our multiple lines of evidence obtained through genetic, thermogenetic, and pharmacological manipulations demonstrate that high serotonin levels during development cause sleep disturbances later in life and suggest hyperserotonemia as a sleep-relevant mechanism in individuals with mutations in CHD8/7.
Last, increased serotonin has been associated with various ASD-relevant behaviors, including social behavior, in both patients and animal models (54, 55). Thus, our findings may account for other ASD-associated clinical features than just the sleep disturbances in individuals with CHD8/7 mutations. As we observed increased serotonin levels in the projections to the proventriculum and anterior midgut, serotonin imbalance in these organs could contribute to other physiological and/or behavioral problems [e.g., gastrointestinal problems reported in individuals with CHD8 mutations (5)]. In conclusion, our study establishes a causal relationship between BBB glia, hyperserotonemia, sleep, and one of the most frequent risk factors of ASD, providing substantial progress in autism research.
How does Kismet loss lead to hyperserotonemia? Although direct target genes of Kismet in glia remain to be determined, a recent targeted DNA adenine methyltransferase identification (DamID) analysis in intestinal stem cells (56) identified four genes associated with the serotonergic system: the receptors 5-HT2A, 5-HT2B, and 5-HT7 and AANAT1. Since we showed that increasing serotonin levels via AANAT1 knockdown in glia and SPG phenocopied the fragmented nighttime sleep observed in kismet knockdown, glial AANAT1 deregulation may account for or contribute to the observed sleep defects in the Drosophila models and participate in CHD8/CHD7 pathophysiology. Human AANAT (homolog of AANAT1) is required for melatonin synthesis, and its activity is reduced in ASD cohorts (51), which additionally present with low plasma and urine melatonin levels (57). Previous work has shown that AANAT1 mutants exhibit increased rebound upon sleep deprivation (58), and that astrocytic AANAT1 is crucial for proper homeostatic rebound, while its loss in neurons leads to a more consolidated sleep (59). Our work shows yet another previously unidentified and cell-specific role of AANAT1 in sleep regulation and is consistent with evidence for serotonergic regulation of sleep in Drosophila. Further work is needed to understand which dysregulated Kismet target genes in SPG lead to hyperserotonemia and how these findings may relate to human CHD8 and CHD7 pathology.
How does hyperserotonemia arising from Kismet dysfunction lead to sleep disturbances? Our data show that pharmacologically reducing serotonin levels in pan-glial kismet knockdown flies restores adult sleep integrity when applied during development. The fact that αMTP treatment causes lethality in control animals with normal serotonin levels, but is not lethal in kismet knockdown larvae, further supports a link between Kismet and developmentally increased serotonin. Reversely, pharmacologically increasing serotonin levels with 5-HTP in the same critical window lead to a decreased and fragmented sleep in wild-type flies. The effect of 5-HTP may not be as notable as kismet knockdown due to the light sensitivity of this compound as treated animals were kept at 12:12 LD. Moreover, it has been reported that during development, serotonin restricts the growth of serotonergic neurons via a negative feedback loop (60). Thus, increased serotonin levels upon Kismet loss during critical periods for circuit formation and maturation may lead to loss of serotonergic terminals and miswiring of neural networks, in analogy to existing prominent hypotheses how hyperserontonemia can cause ASD-associated clinical features (55).
Applicability of SRT in CHD8/7 syndromes and other neurodevelopmental disorders
The developmental trajectories of sleep disturbances in young children with neurodevelopmental disorders suggest that we should instigate intervention as early as possible, yet there is an urgent need to remediate sleep problems in older children and adults as well. We show here that SRT improved sleep quality in both pan-glial and SPG kismet knockdown flies. With this behavioral approach, we effectively rescued shortened sleep bout duration and increased bout number, restored SE and WASO to control levels, and significantly improved sleep latency. Notably, these aberrancies in sleep metrics in Drosophila reflect those perturbed in CHD8 and CHD7 individuals. It is remarkable that sleep opportunity restriction, applied only in adulthood, can override sleep defects that are, as we demonstrated, of developmental origin. Hence, we propose that SRT provides a viable strategy to improve sleep in patients with CHD8/CHD7 mutations and perhaps other forms of autism and related neurodevelopmental disorders. Although not quantitatively assessed, components of CBT-I including sleep opportunity restriction (i.e., shortening bedtimes) were advised to CHD8/7 individuals 01 to 04 seen in our sleep center and showed positive effects on the insomnia, as documented in the patient files (table S2).
In this study, we shed light onto mechanisms and cellular substrates regulating sleep, linking the evolutionary CHD8/CHD7/Kismet family to BBB glia, hyperserotonemia, and ASD-associated sleep defects. With our work, we also aim to contribute to a paradigm shift away from the commonly held view that sleep deficits are coupled inevitably to the neurodevelopmental disorders in which they occur and are therefore refractory to therapy.
MATERIALS AND METHODS
Study design: Sleep assessment of CHD8 and CHD7 individuals
In neurodevelopmental disorder cohorts, the prevalence of CHD8 and CHD7 loss-of-function mutations lies in the range of 0.15 to 0.40% and 0.28%, respectively (5, 61, 62). We assessed sleep characteristics in a cohort of individuals with CHD8 mutations using structured questionnaires. Probands included 23 individuals with disruptive mutations to CHD8. Six individuals were drawn from the SSC (63, 64), and 17 probands were drawn from an ongoing study of individuals with mutations to ASD-associated genes (NIMH TIGER) (9). Pathogenic or likely pathogenic mutations to CHD8 were confirmed through review of clinical or research genetic testing results; individuals with other pathogenic mutations to neurodevelopmental disorder and/or sleep related genes were excluded from analyses. To establish a comparison group of individuals with ASD but no known genetic etiology (“idiopathic” ASD), 2305 probands without pathogenic ASD-associated single-gene variants or deleterious copy number variants as confirmed by whole-exome sequencing were drawn from the SSC (64, 65). Age and sex for the CHD8 group and the idiopathic comparison group are presented in table S3. Written consent was obtained from participants, and all procedures were approved by the University of Washington Institutional Review Board.
A comprehensive, structured medical history interview initially designed for the large-scale SSC was completed with primary caregivers of all participants in SSC and NIMH TIGER, which included questions about history of sleep problems. Caregivers were asked whether their child had ever experienced the sleep problems listed in table S1, which included bedtime problems, excessive daytime sleepiness, sleep-disordered breathing, and nighttime awakenings. The same sleep history questions were used in both SSC and TIGER. Fisher’s exact tests were used to determine whether rates of specific sleep problems in individuals with disruptive CHD8 mutations differed from a large, idiopathic ASD group obtained through the SSC. Bonferroni correction for multiple comparisons was applied to the significance level (P < 0.005). Groups differed by gender [χ2(1, n = 2311 = 9.84, P = 0.002)], with the CHD8 sample containing a significantly larger proportion of females than the SSC group. Groups did not differ by age [t(22.19) = −1.52, P > 0.05]. Analyses were completed in SPSS version 26 (IBM Corp., 2019).
Second, to further address the clinical significance of CHD7/8-associated sleep problems, we searched for individuals with CHD7 or CHD8 pathogenic mutations that have been seen in a tertiary sleep medicine center upon referral due to sleep complaints. Therefore, we screened all referrals between 2012 and 2019 for individuals with mutations in those genes, identifying one additional individual with a pathogenic CHD8 mutation and three patients with CHD7 mutations. All four patients underwent a basic clinical workup including a full history taking, physical examination, and a consensus sleep diary (66), based on which time in bed (TIB), TST, SOL, WASO, and SE were calculated per individual with a 15-min bin resolution provided by the parent-reported sleep-wake calendars. SE was calculated as TST divided by TIB and multiplied by 100. SOL was calculated as the time between going to bed and sleep onset. WASO was calculated as the total time awake between sleep onset until the end of TIB. When appropriate, wrist actigraphy (MotionWatch 8, CamNtech, Cambridgeshire, UK) or video-PSG was performed. A sleep disorder diagnosis was made according to the ICSD-3 criteria (66, 67). Mean sleep duration was compared to values typically characterizing a pediatric population (68). Effective and ineffective treatments (pharmacological and behavioral) either received in the past or applied after consultation in the sleep clinic are reported in table S2. Informed consent was according to local ethical procedures.
Drosophila strains and husbandry
Fly stocks were reared on standard medium containing cornmeal, yeast, sugar, and agar at 25°C and 60% humidity in a 12:12 LD cycle. The following fly stocks were used for this study: iso31, kisk13416 mutants [(Bloomington Drosophila Stock Center (BDSC) #10442)] backcrossed 6× to iso31, UAS-kisRNAi1 (VDRC #46685), UAS-kisRNAi2 (VDRC #109414), UAS-AANAT1RNAi1 (VDRC #47906), UAS-AANAT1RNAi2 (VDRC #105064), UAS-AANAT1RNAi3 (BDSC #36726), background control GD library [Vienna Drosophila Resource Center (VDRC#60000 )], background control KK library (VDRC #60.100), background control Transgenic RNAi Project (TRiP) library attP40 (BDSC #36304), repo-Gal4 (UAS-Dcr2/CyO; repo-Gal4/TM3, Sb), repo-2-Gal4 (repo-Gal4/TM6b, Tb, Sb), repo-Gal4,tubGal80ts (repo-Gal4,tubGal80ts/TM3, Sb), elav-Gal4 (UAS-Dcr2; elav-Gal4), SPG-Gal4 (R54C07-Gal4, BDSC #50472), PG-Gal4 (R85G01-Gal4, BDSC #40436), ALG-Gal4 [NP3233-Gal4, [Drosophila Genomics Resource Center (DGRC) #113173], EG-Gal4 (NP6520-Gal4, DGRC #105240), CG-Gal4 (NP2222-Gal4, DGRC #112830), SPG-2-Gal4 (NP2276-Gal4, DGRC #112853), Trh-Gal4(II) (BDSC #38388), Trh-Gal4(III) (BDSC #38389), and UAS-dTrpA1 backcrossed 6× to iso31. The UAS-kisRNAi1 line has previously been shown to induce a strong knockdown (≈90%) (17, 20). The pKC43 insertion sites of the KK RNAi lines used in this study were verified by diagnostic polymerase chain reaction according to the findings of Green and colleagues (69). The hairpin construct of the UAS-kisRNAi-2 line (VDRC #109414) was found to be inserted in both 40D and 30B landing sites, in agreement with information meanwhile available at VDRC. Thus, an additional control for the effect of the UAS insertion into the 40D locus was used (VDRC #60101), referred to as UAS-40D control.
Adult sleep analysis
Locomotor activity and sleep were recorded with the Drosophila Activity Monitor (DAM2) system from TriKinetics (Waltham, MA, USA). Briefly, 3- to 5-day-old male flies were individually transferred without CO2 anesthesia to glass tubes (65 mm by 5 mm) containing standard food, loaded into the DAM systems, allowed to acclimatize to activity monitors and food for at least 12 hours, and monitored for 4 days at 25°C in a 12:12 LD cycle. For SRT experiments, the acclimation and monitoring were performed simultaneously in two different incubators at 12:12 LD and 14:10 LD. Motion was detected via the monitors’ infrared light beams, and activity and sleep (defined in Drosophila as 5 or more minutes of inactivity) (58, 70) parameters were extracted from the beam breaks using the publicly available Sleep and Circadian Analysis MATLAB Program (71) for MATLAB. P(Wake) and P(Doze) were calculated from the two first days of activity monitoring recordings with the MATLAB scripts deposited in GitHub (23, 39). SE in Drosophila is depicted as time spent asleep divided by the opportunity window (ZT12 to ZT24 for flies entrained at 12:12 LD or ZT14 to ZT24 for flies entrained at 14:10 LD). WASO was calculated by subtracting the latency to the first sleep episode from the total time spent awake during the dark period (ZT12 to ZT24 for flies entrained at 12:12 LD or ZT14 to ZT24 for flies entrained at 14:10 LD). All sleep data are the average over the 4 days of data acquisition, except for SRT where data are the average of days 3 and 4 to allow entrainment to the new light regime.
Circadian analysis
Three- to five-day-old male flies were loaded into the DAM system as described above and entrained to a 12:12 LD cycle for 3 days before being transferred to constant darkness (DD). Locomotor activity during days 2 to 7 in DD was analyzed in ClockLab software (Actimetrics, Wilmette, IL). FFT (72) was performed, and the maximum amplitude of the FFT was calculated and compared across genotypes. According to their FFT, flies were categorized as strongly rhythmic (FFT ≥ 0.05), moderately rhythmic (0.05 > FFT ≥ 0.03), weakly rhythmic (0.03 > FFT ≥ 0.01), or arrhythmic (FFT < 0.01).
TARGET system experiments
The temperature-dependent TARGET system (73) was used for temporal mapping experiments. Flies were reared at either 22°C (restrictive temperature) or 28°C (permissive temperature) and, after eclosion (day 0), were either kept at the same temperature or swapped as indicated to confine gene knockdown to pre- or posteclosion periods. Genetic background controls were reared in parallel with the experimental flies. All experiments were conducted in a 12:12 LD cycle.
Drug supplementation experiments
MPH (Brocacef, Maarssen) was supplemented in fly food at a final concentration of 1 mg/ml (33, 37) only during sleep monitoring. αMTP (Sigma-Aldrich, M8377) was diluted in fly food to achieve a final concentration of 20 mM (38). αMTP was administered only during sleep monitoring for acute treatment, or only throughout development in 5 ml of food containing the drug (until DAM on vehicle food). 5-HTP (Sigma-Aldrich, H9772) was diluted in fly food to achieve a final concentration of 5 mM and administered only during development in 5 ml of food containing the drug, and adults after eclosion were reared and monitored on regular food. All drugs were dissolved in Milli-Q water.
Sleep deprivation
Flies in DAM2 monitors were placed on a TriKinetics vortexer mounting plate (Waltham, MA, USA), and their activity was recorded for three consecutive days. On the second day, they were shaken for 2 s randomly within every 20-s window for 12 hours during the dark period (ZT12 to ZT24). The capability of each tested genotype to rebound was assessed by comparing the total sleep during ZT0 to ZT3 in the preceding day under unperturbed conditions (baseline) and the same time period following sleep deprivation (recovery).
Larval rearing and sleep assay
Experiments were carried out as previously described (32, 74). Briefly, to collect developmentally synchronized second instar larvae, adult flies were placed in an embryo collection chamber, kept at 12:12 LD and 25°C, and allowed to lay eggs on a petri dish containing 3% agar, 2% sucrose, and 2.5% apple juice with yeast paste on top for 24 hours. Animals developed on this medium for 48 hours, after which late first instar larvae near ecdysis (based on the presence of double mouth hooks and double vertical plates) were collected. After they finished molting into second instars, single larvae were carefully transferred into LarvaLodge wells containing 100 ml of 3% agar and 2% sucrose medium covered with a thin layer of yeast paste. The LarvaLodge was covered with a transparent acrylic sheet and placed into a behavior room at 25°C and DD for imaging via infrared light. Notably, larval sleep is neither light- nor circadian-regulated (32). Time-lapse images were acquired every 6 s with the software IC Capture (The Imaging Source). Images were analyzed using custom-written MATLAB software (74). The first hour of data acquisition was discarded to allow acclimation. All data shown are the average of the 4 hours following acclimation. For dTrpA1 activation experiments, embryo collection chambers were kept instead at 22°C and 12:12 LD, and temperature was raised to 30°C during sleep recordings.
High-performance liquid chromatography
HPLC was used to quantify dopamine and serotonin levels in fly head extracts. Three- to 5-day-old male flies entrained in a 12:12 LD cycle were frozen by immersion in liquid nitrogen at ZT6 and ZT18 after at least 24-hour recovery from CO2. Samples were acquired without disturbing the LD cycle. Fly heads were detached from their bodies by gentle vortexing. Per sample, three heads were transferred to an Eppendorf tube containing 100 μl of ice-chilled 0.1 M perchloric acid (pH 3.0) and homogenized using a hand tissue grinder. Homogenates were kept on ice for 10 min to denaturalize the proteins, followed by centrifugation at 14,000 rpm for 15 min at 4°C. The supernatant was transferred to an Ultrafree-MC filter with a pore size of 0.45 μm (Merck Millipore), filtered by centrifugation for 1 hour at 14,000 rpm at 4°C, and stored at −80°C until HPLC analysis. Ten microliters of the filtrate was analyzed using HPLC with electrochemical detection (EICOMPAK SC-30DS, EICOM) using a mobile phase consisting of 80% citrate-acetate buffer (0.1 M) (pH 3.5), methanol (20%) with sodium octane sulfonate (220 mg/liter) and EDTA-2Na (5 mg/liter), pumped at a rate of 360 μl/min through a 3.0 mm by 100 mm column (EICOMPAK SC-30DS, EICOM), as described by the manufacturer.
Immunohistochemistry, imaging, and quantification
Proventriculus and anterior midgut of male from wandering third instar larvae were dissected in phosphate-buffered saline (PBS) and immediately fixed in 4% paraformaldehyde dissolved in PBS for 30 min at room temperature. Tissues were washed for 30 min with PBS with 0.6% Triton X-100 (PBST) and blocked for 2 hours at room temperature with 3% goat serum in PBST. Tissues were incubated overnight at 4°C with primary antibody against serotonin (1:500; Sigma-Aldrich, S5545). Secondary donkey anti-rabbit antibody conjugated with Alexa Fluor 488 (1:1000; Thermo Fisher Scientific, A-21206) was incubated overnight at 4°C. Samples were then washed again five times in PBST and mounted in ProLong Gold Antifade Mountant (Thermo Fisher Scientific, P36930).
Samples were imaged using a Leica SP8 laser scanning confocal microscope with Hybrid Detector technology for quantitative imaging through single-photon counting. Images were analyzed using ImageJ to calculate the corrected total cell fluorescence (CTCF): CTCF = integrated density [corresponding to the sum of all pixel values the region of interest (ROI)] – (area ROI × mean fluorescence of background readings), with ROIs being per image (i) the entire proventriculus and (ii) region of the anterior midgut. Two regions per image in the proventriculus not containing the proventricular ganglion nor serotonergic innervation were selected, averaged, and used to determine the mean fluorescence of background readings.
CHD8 and CHD7 expression in human cortex
The t-stochastic neighbor embedding projections along with the cluster annotations were retained from the original body of work for both the developing (75) and the adult cortex (76). The single-cell RNA-seq counts per million for both datasets were processed into Unique molecular identifier (UMI) counts using quantile normalization (77) with the shape parameter set to “2.3.” SCTransform (78) was applied on the post quantile normalized count matrix. The plots for expression are the log-normalized expression corrected through SCTransform.
Statistical analysis
Adult and larval Drosophila sleep data statistical analysis was carried out in GraphPad Prism version 7 for Macbook (GraphPad Software, San Diego, CA, USA). Datasets of only two groups that followed a Gaussian distribution but showed differences in their standard deviation with the controls were analyzed with a two-tailed unpaired Welch’s t test. For groups of more than two genotypes, Kruskal-Wallis test with Dunn’s multiple comparisons test was performed to assess significance between the tested groups. Post hoc Bonferroni correction for multiple testing was further applied for the number of tests performed on a dataset per genotype to determine the corrected two-sided significance level. Only P values that pass the corrected significance level are indicated in the figures. All data shown are from at least three independent experiments (N = 3) unless specified otherwise. For sleep deprivation experiments, statistical significance was determined with a two-tailed paired t test between baseline and recovery day per each genotype. Post hoc Bonferroni correction for multiple testing was further applied to correct for the number of tests performed on a dataset per genotype. Only P values that withstand multiple testing correction are indicated in the figures. At least three independent biological replicates were performed per genotype. Statistical significance of dopamine and serotonin levels was determined with a two-tailed unpaired t test between the tested groups of three independent biological replicates.
Acknowledgments
We thank the individuals with CHD8 and CHD7 mutations and their families for participating in our research. We are grateful to the Griffith laboratory for support with sleep probability analysis. We thank J. IntHoud for statistics support; and S. Schirmeier, A. Seghal, J. M. Kramer, the VDRC, the Kyoto DGRC, and the Bloomington Drosophila Stock Center for reagents. We thank E. Winkler, N. Mora-Garcia, C. Souglakos, T. Schillemans, I. Janssen, and Chrono@work for experimental support. We thank P. Lasko and members of the Schenck and Kayser labs for helpful discussions. R.A.B. is currently employed by Apple Inc. Funding: This work was, in part, supported by a Radboudumc personal PhD fellowship and a traveling fellowship by The Company of Biologists (DMMTF1908278) to M.C.-T., an NIH grant (F30AG058409) to S.J.B., a Simons Foundation grant (SFARI 491371) to T.J.N., National Institute of Mental Health grants nos. MH100047 (“ZEBRA” study) to R.A.B. and MH101221 (“TIGER” study) to E.E.E., and by National Institute of Child Health and Human Development grant no. U54 HD083091 to the University of Washington’s Center on Human Development and Disability. E.E.E. is an investigator of the Howard Hughes Medical Institute. The work was further supported by NIH grants K08 NS090461 and DP2 NS111996, a Burroughs Wellcome Career Awards for Medical Scientists, a March of Dimes Basil O’Connor Scholar Award, and a Sloan Research Fellowship to M.S.K., by funding from the Australian National Health and Medical Research Council to the Centre for Research Excellence in Neurocognitive Disorders (CRE-NCD, grant no. APP1117394) to A.S., a Radboudumc junior researcher fellowship to T.K. and A.S., and by grants from the Netherlands Organisation for Scientific Research (NWO) no. 91718310 (ZonMw Vidi to T.K.) and no. 09150181910022 (ZonMw Vici to A.S.). Author contributions: Conceptualization: M.C.-T. and A.S. Methodology: M.C.-T., S.J.B., M.S., I.E., M.M.M.V., and M.S.K. Software: N.N.G. and M.S. Formal analysis: M.C.-T., N.N.G., S.J.B., L.V.v.R., E.C.K.-N., B.v.R., J.D., and C.N.K. Investigation: M.C.-T., N.N.G., L.V.v.R., E.C.K.-N., B.v.R., I.T., and J.D. Resources: M.M.M.V., C.M.H., R.A.B., S.P., R.K.E., E.E.E., T.K., and M.S.K. Data curation: M.C.-T., C.M.H., R.A.B., R.K.E., E.E.E., and A.S. Writing—original draft: M.C.-T. and A.S. Writing—review and editing: M.C.-T., N.N.G., S.J.B., L.V.v.R., E.C.K.-N., I.E., C.M.H., R.A.B., S.P., R.K.E., E.E.E., T.K., M.S.K., and A.S. Visualization: M.C.-T. Supervision: M.C.-T., T.J.N., R.A.B., R.K.E., E.E.E., M.S.K., and A.S. Project administration: M.C.-T., M.S.K., and A.S. Funding acquisition: M.C.-T., R.A.B., S.P., E.E.E., T.K., M.S.K., and A.S. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/23/eabe2626/DC1
REFERENCES AND NOTES
- 1.Richdale A. L., Schreck K. A., Sleep problems in autism spectrum disorders: Prevalence, nature, & possible biopsychosocial aetiologies. Sleep Med. Rev. 13, 403–411 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Esbensen A. J., Schwichtenberg A. J., Sleep in neurodevelopmental disorders. Int. Rev. Res. Dev. Disabil. 51, 153–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hartshorne T. S., Heussler H. S., Dailor A. N., Williams G. L., Papadopoulos D., Brandt K. K., Sleep disturbances in CHARGE syndrome: Types and relationships with behavior and caregiver well-being. Dev. Med. Child Neurol. 51, 143–150 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Phillips N. L., Moore T., Teng A., Brookes N., Palermo T. M., Lah S., Behavioral interventions for sleep disturbances in children with neurological and neurodevelopmental disorders: A systematic review and meta-analysis of randomized controlled trials. Sleep 43, zsaa040 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Bernier R., Golzio C., Xiong B., Stessman H. A., Coe B. P., Penn O., Witherspoon K., Gerdts J., Baker C., Vulto-van Silfhout A. T., Schuurs-Hoeijmakers J. H., Fichera M., Bosco P., Buono S., Alberti A., Failla P., Peeters H., Steyaert J., Vissers L. E. L. M., Francescatto L., Mefford H. C., Rosenfeld J. A., Bakken T., O’Roak B. J., Pawlus M., Moon R., Shendure J., Amaral D. G., Lein E., Rankin J., Romano C., de Vries B. B. A., Katsanis N., Eichler E. E., Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158, 263–276 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Satterstrom F. K., Kosmicki J. A., Wang J., Breen M. S., De Rubeis S., An J.-Y., Peng M., Collins R., Grove J., Klei L., Stevens C., Reichert J., Mulhern M. S., Artomov M., Gerges S., Sheppard B., Xu X., Bhaduri A., Norman U., Brand H., Schwartz G., Nguyen R., Guerrero E. E., Dias C.; Autism Sequencing Consortium; iPSYCH-Broad Consortium, Betancur C., Cook E. H., Gallagher L., Gill M., Sutcliffe J. S., Thurm A., Zwick M. E., Børglum A. D., State M. W., Cicek A. E., Talkowski M. E., Cutler D. J., Devlin B., Sanders S. J., Roeder K., Daly M. J., Buxbaum J. D., Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584.e23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stessman H. A. F., Xiong B., Coe B. P., Wang T., Hoekzema K., Fenckova M., Kvarnung M., Gerdts J., Trinh S., Cosemans N., Vives L., Lin J., Turner T. N., Santen G., Ruivenkamp C., Kriek M., van Haeringen A., Aten E., Friend K., Liebelt J., Barnett C., Haan E., Shaw M., Gecz J., Anderlid B.-M., Nordgren A., Lindstrand A., Schwartz C., Kooy R. F., Vandeweyer G., Helsmoortel C., Romano C., Alberti A., Vinci M., Avola E., Giusto S., Courchesne E., Pramparo T., Pierce K., Nalabolu S., Amaral D. G., Scheffer I. E., Delatycki M. B., Lockhart P. J., Hormozdiari F., Harich B., Castells-Nobau A., Xia K., Peeters H., Nordenskjöld M., Schenck A., Bernier R. A., Eichler E. E., Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 49, 515–526 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cotney J., Muhle R. A., Sanders S. J., Liu L., Willsey A. J., Niu W., Liu W., Klei L., Lei J., Yin J., Reilly S. K., Tebbenkamp A. T., Bichsel C., Pletikos M., Sestan N., Roeder K., State M. W., Devlin B., Noonan J. P., The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat. Commun. 6, 6404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beighley J. S., Hudac C. M., Arnett A. B., Peterson J. L., Gerdts J., Wallace A. S., Mefford H. C., Hoekzema K., Turner T. N., O’Roak B. J., Eichler E. E., Bernier R. A., Clinical phenotypes of carriers of mutations in CHD8 or its conserved target genes. Biol. Psychiatry 87, 123–131 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vissers L. E. L. M., van Ravenswaaij C. M. A., Admiraal R., Hurst J. A., de Vries B. B. A., Janssen I. M., van der Vliet W. A., Huys E. H. L. P. G., de Jong P. J., Hamel B. C. J., Schoenmakers E. F. P. M., Brunner H. G., Veltman J. A., van Kessel A. G., Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 36, 955–957 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Zentner G. E., Layman W. S., Martin D. M., Scacheri P. C., Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am. J. Med. Genet. A 152A, 674–686 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartshorne N., Hudson A., MacCuspie J., Kennert B., Nacarato T., Hartshorne T., Blake K., Quality of life in adolescents and adults with CHARGE syndrome. Am. J. Med. Genet. A 170, 2012–2021 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Kennert B. A., Harshorne T. S., Kanouse S., Johnson C., Parent survey of sleep problems among children with CHARGE syndrome. Res. Dev. Disabil. 101, 103614 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Batsukh T., Pieper L., Koszucka A. M., von Velsen N., Hoyer-Fender S., Elbracht M., Bergman J. E. H., Hoefsloot L. H., Pauli S., CHD8 interacts with CHD7, a protein which is mutated in CHARGE syndrome. Hum. Mol. Genet. 19, 2858–2866 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Dubowy C., Sehgal A., Circadian rhythms and sleep in Drosophila melanogaster. Genetics 205, 1373–1397 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coll-Tané M., Krebbers A., Castells-Nobau A., Zweier C., Schenck A., Intellectual disability and autism spectrum disorders ‘on the fly’: Insights from Drosophila. Dis. Model. Mech. 12, dmm039180 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Melicharek D. J., Ramirez L. C., Singh S., Thompson R., Marenda D. R., Kismet/CHD7 regulates axon morphology, memory and locomotion in a Drosophila model of CHARGE syndrome. Hum. Mol. Genet. 19, 4253–4264 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghosh R., Vegesna S., Safi R., Bao H., Zhang B., Marenda D. R., Liebl F. L. W., Kismet positively regulates glutamate receptor localization and synaptic transmission at the Drosophila neuromuscular junction. PLOS ONE 9, e113494 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Latcheva N. K., Viveiros J. M., Marenda D. R., The Drosophila chromodomain protein kismet activates steroid hormone receptor transcription to govern axon pruning and memory In Vivo. iScience 16, 79–93 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dubruille R., Murad A., Rosbash M., Emery P., A constant light-genetic screen identifies KISMET as a regulator of circadian photoresponses. PLOS Genet. 5, e1000787 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trider C.-L., Corsten G., Morrison D., Hefner M., Davenport S., Blake K., Understanding obstructive sleep apnea in children with CHARGE syndrome. Int. J. Pediatr. Otorhinolaryngol. 76, 947–953 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Belfer S. J., Bashaw A. G., Perlis M. L., Kayser M. S., A Drosophila model of sleep restriction therapy for insomnia. Mol. Psychiatry 26, 492–507 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiggin T. D., Goodwin P. R., Donelson N. C., Liu C., Trinh K., Sanyal S., Griffith L. C., Covert sleep-related biological processes are revealed by probabilistic analysis in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 117, 10024–10034 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Estes P. S., Daniel S. G., McCallum A. P., Boehringer A. V., Sukhina A. S., Zwick R. A., Zarnescu D. C., Motor neurons and glia exhibit specific individualized responses to TDP-43 expression in a Drosophila model of amyotrophic lateral sclerosis. Dis. Model. Mech. 6, 721–733 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stahl B. A., Peco E., Davla S., Murakami K., Caicedo Moreno N. A., van Meyel D. J., Keene A. C., The taurine transporter Eaat2 functions in ensheathing glia to modulate sleep and metabolic rate. Curr. Biol. 28, 3700–3708.e4 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Artiushin G., Zhang S. L., Tricoire H., Sehgal A., Endocytosis at the Drosophila blood-brain barrier as a function for sleep. eLife 7, e43326 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerstner J. R., Perron I. J., Riedy S. M., Yoshikawa T., Kadotani H., Owada Y., Van Dongen H. P. A., Galante R. J., Dickinson K., Yin J. C. P., Pack A. I., Frank M. G., Normal sleep requires the astrocyte brain-type fatty acid binding protein FABP7. Sci. Adv. 3, e1602663 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vanderheyden W. M., Goodman A. G., Taylor R. H., Frank M. G., Van Dongen H. P. A., Gerstner J. R., Astrocyte expression of the Drosophila TNF-alpha homologue, Eiger, regulates sleep in flies. PLOS Genet. 14, e1007724 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kremer M. C., Jung C., Batelli S., Rubin G. M., Gaul U., The glia of the adult Drosophila nervous system. Glia 65, 606–638 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yildirim K., Petri J., Kottmeier R., Klämbt C., Drosophila glia: Few cell types and many conserved functions. Glia 67, 5–26 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Zhang S. L., Yue Z., Arnold D. M., Artiushin G., Sehgal A., A circadian clock in the blood-brain barrier regulates xenobiotic efflux. Cell 173, 130–139.e10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szuperak M., Churgin M. A., Borja A. J., Raizen D. M., Fang-Yen C., Kayser M. S., A sleep state in Drosophila larvae required for neural stem cell proliferation. eLife 7, e33220 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van der Voet M., Harich B., Franke B., Schenck A., ADHD-associated dopamine transporter, latrophilin and neurofibromin share a dopamine-related locomotor signature in Drosophila. Mol. Psychiatry 21, 565–573 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schoofs A., Hückersfeld S., Surendran S., Pankratz M. J., Serotonergic pathways in the Drosophilia larval enteric nervous system. J. Insect Physiol. 69, 118–125 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Gabriele S., Sacco R., Persico A. M., Blood serotonin levels in autism spectrum disorder: A systematic review and meta-analysis. Eur. Neuropsychopharmacol. 24, 919–929 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Pagan C., Delorme R., Callebert J., Goubran-Botros H., Amsellem F., Drouot X., Boudebesse C., Le Dudal K., Ngo-Nguyen N., Laouamri H., Gillberg C., Leboyer M., Bourgeron T., Launay J.-M., The serotonin-N-acetylserotonin-melatonin pathway as a biomarker for autism spectrum disorders. Transl. Psychiatry 4, e479 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Swinderen B., Brembs B., Attention-like deficit and hyperactivity in a Drosophila memory mutant. J. Neurosci. 30, 1003–1014 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dierick H. A., Greenspan R. J., Serotonin and neuropeptide F have opposite modulatory effects on fly aggression. Nat. Genet. 39, 678–682 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Liu C., Meng Z., Wiggin T. D., Yu J., Reed M. L., Guo F., Zhang Y., Rosbash M., Griffith L. C., A serotonin-modulated circuit controls sleep architecture to regulate cognitive function independent of total sleep in Drosophila. Curr. Biol. 29, 3635–3646.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin C. A., Krantz D. E., Drosophila melanogaster as a genetic model system to study neurotransmitter transporters. Neurochem. Int. 73, 71–88 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller C. B., Espie C. A., Epstein D. R., Friedman L., Morin C. M., Pigeon W. R., Spielman A. J., Kyle S. D., The evidence base of sleep restriction therapy for treating insomnia disorder. Sleep Med. Rev. 18, 415–424 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Gregory A. M., Sadeh A., Sleep, emotional and behavioral difficulties in children and adolescents. Sleep Med. Rev. 16, 129–136 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Tononi G., Cirelli C., Sleep and the price of plasticity: From synaptic and cellular homeostasis to memory consolidation and integration. Neuron 81, 12–34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kikuma K., Li X., Perry S., Li Q., Goel P., Chen C., Kim D., Stavropoulos N., Dickman D., Cul3 and insomniac are required for rapid ubiquitination of postsynaptic targets and retrograde homeostatic signaling. Nat. Commun. 10, 2998 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cuddapah V. A., Zhang S. L., Sehgal A., Regulation of the blood-brain barrier by circadian rhythms and sleep. Trends Neurosci. 42, 500–510 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramaekers A., Magnenat E., Marin E. C., Gendre N., Jefferis G. S. X. E., Luo L., Stocker R. F., Glomerular maps without cellular redundancy at successive levels of the Drosophila larval olfactory circuit. Curr. Biol. 15, 982–992 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Saunders N. R., Habgood M. D., Dziegielewska K. M., Barrier mechanisms in the brain, II. Immature brain. Clin. Exp. Pharmacol. Physiol. 26, 85–91 (1999). [DOI] [PubMed] [Google Scholar]
- 48.He D., Marie C., Zhao C., Kim B., Wang J., Deng Y., Clavairoly A., Frah M., Wang H., He X., Hmidan H., Jones B. V., Witte D., Zalc B., Zhou X., Choo D. I., Martin D. M., Parras C., Lu Q. R., Chd7 cooperates with Sox10 and regulates the onset of CNS myelination and remyelination. Nat. Neurosci. 19, 678–689 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao C., Dong C., Frah M., Deng Y., Marie C., Zhang F., Xu L., Ma Z., Dong X., Lin Y., Koenig S., Nait-Oumesmar B., Martin D. M., Wu L. N., Xin M., Zhou W., Parras C., Lu Q. R., Dual requirement of CHD8 for chromatin landscape establishment and histone methyltransferase recruitment to promote CNS myelination and repair. Dev. Cell 45, 753–768.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marie C., Clavairoly A., Frah M., Hmidan H., Yan J., Zhao C., Van Steenwinckel J., Daveau R., Zalc B., Hassan B., Thomas J.-L., Gressens P., Ravassard P., Moszer I., Martin D. M., Lu Q. R., Parras C., Oligodendrocyte precursor survival and differentiation requires chromatin remodeling by Chd7 and Chd8. Proc. Natl. Acad. Sci. U.S.A. 115, E8246–E8255 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benabou M., Rolland T., Leblond C. S., Millot G. A., Huguet G., Delorme R., Leboyer M., Pagan C., Callebert J., Maronde E., Bourgeron T., Heritability of the melatonin synthesis variability in autism spectrum disorders. Sci. Rep. 7, 17746 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Missig G., McDougle C. J., Carlezon W. A. Jr., Sleep as a translationally-relevant endpoint in studies of autism spectrum disorder (ASD). Neuropsychopharmacology 45, 90–103 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oikonomou G., Altermatt M., Zhang R. W., Coughlin G. M., Montz C., Gradinaru V., Prober D. A., The serotonergic raphe promote sleep in zebrafish and mice. Neuron 103, 686–701.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tanaka M., Sato A., Kasai S., Hagino Y., Kotajima-Murakami H., Kashii H., Takamatsu Y., Nishito Y., Inagaki M., Mizuguchi M., Hall F. S., Uhl G. R., Murphy D., Sora I., Ikeda K., Brain hyperserotonemia causes autism-relevant social deficits in mice. Mol. Autism. 9, 60 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Muller C. L., Anacker A. M. J., Veenstra-VanderWeele J., The serotonin system in autism spectrum disorder: From biomarker to animal models. Neuroscience 321, 24–41 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gervais L., van den Beek M., Josserand M., Sallé J., Stefanutti M., Perdigoto C. N., Skorski P., Mazouni K., Marshall O. J., Brand A. H., Schweisguth F., Bardin A. J., Stem cell proliferation is kept in check by the chromatin regulators kismet/CHD7/CHD8 and Trr/MLL3/4. Dev. Cell 49, 556–573.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Melke J., Botros H. G., Chaste P., Betancur C., Nygren G., Anckarsäter H., Rastam M., Ståhlberg O., Gillberg I. C., Delorme R., Chabane N., Mouren-Simeoni M.-C., Fauchereau F., Durand C. M., Chevalier F., Drouot X., Collet C., Launay J.-M., Leboyer M., Gillberg C., Bourgeron T.; PARIS study , Abnormal melatonin synthesis in autism spectrum disorders. Mol. Psychiatry 13, 90–98 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shaw P. J., Cirelli C., Greenspan R. J., Tononi G., Correlates of sleep and waking in Drosophila melanogaster. Science 287, 1834–1837 (2000). [DOI] [PubMed] [Google Scholar]
- 59.Davla S., Artiushin G., Li Y., Chitsaz D., Li S., Sehgal A., van Meyel D. J., AANAT1 functions in astrocytes to regulate sleep homeostasis. eLife 9, e53994 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gaspar P., Cases O., Maroteaux L., The developmental role of serotonin: News from mouse molecular genetics. Nat. Rev. Neurosci. 4, 1002–1012 (2003). [DOI] [PubMed] [Google Scholar]
- 61.O’Roak B. J., Vives L., Fu W., Egertson J. D., Stanaway I. B., Phelps I. G., Carvill G., Kumar A., Lee C., Ankenman K., Munson J., Hiatt J. B., Turner E. H., Levy R., O’Day D. R., Krumm N., Coe B. P., Martin B. K., Borenstein E., Nickerson D. A., Mefford H. C., Doherty D., Akey J. M., Bernier R., Eichler E. E., Shendure J., Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaplanis J., Samocha K. E., Wiel L., Zhang Z., Arvai K. J., Eberhardt R. Y., Gallone G., Lelieveld S. H., Martin H. C., McRae J. F., Short P. J., Torene R. I., de Boer E., Danecek P., Gardner E. J., Huang N., Lord J., Martincorena I., Pfundt R., Reijnders M. R. F., Yeung A., Yntema H. G.; Deciphering Developmental Disorders Study, Vissers L. E. L. M., Juusola J., Wright C. F., Brunner H. G., Firth H. V., Fitz Patrick D. R., Barrett J. C., Hurles M. E., Gilissen C., Retterer K., Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature 586, 757–762 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fischbach G. D., Lord C., The Simons Simplex Collection: A resource for identification of autism genetic risk factors. Neuron 68, 192–195 (2010). [DOI] [PubMed] [Google Scholar]
- 64.Sanders S. J., He X., Willsey A. J., Ercan-Sencicek A. G., Samocha K. E., Cicek A. E., Murtha M. T., Bal V. H., Bishop S. L., Dong S., Goldberg A. P., Jinlu C., Keaney J. F. III, Klei L., Mandell J. D., Moreno-De-Luca D., Poultney C. S., Robinson E. B., Smith L., Solli-Nowlan T., Su M. Y., Teran N. A., Walker M. F., Werling D. M., Beaudet A. L., Cantor R. M., Fombonne E., Geschwind D. H., Grice D. E., Lord C., Lowe J. K., Mane S. M., Martin D. M., Morrow E. M., Talkowski M. E., Sutcliffe J. S., Walsh C. A., Yu T. W.; Autism Sequencing Consortium, Ledbetter D. H., Martin C. L., Cook E. H., Buxbaum J. D., Daly M. J., Devlin B., Roeder K., State M. W., Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Iossifov I., O’Roak B. J., Sanders S. J., Ronemus M., Krumm N., Levy D., Stessman H. A., Witherspoon K. T., Vives L., Patterson K. E., Smith J. D., Paeper B., Nickerson D. A., Dea J., Dong S., Gonzalez L. E., Mandell J. D., Mane S. M., Murtha M. T., Sullivan C. A., Walker M. F., Waqar Z., Wei L., Willsey A. J., Yamrom B., Lee Y.-h., Grabowska E., Dalkic E., Wang Z., Marks S., Andrews P., Leotta A., Kendall J., Hakker I., Rosenbaum J., Ma B., Rodgers L., Troge J., Narzisi G., Yoon S., Schatz M. C., Ye K., McCombie W. R., Shendure J., Eichler E. E., State M. W., Wigler M., The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Carney C. E., Buysse D. J., Ancoli-Israel S., Edinger J. D., Krystal A. D., Lichstein K. L., Morin C. M., The consensus sleep diary: Standardizing prospective sleep self-monitoring. Sleep 35, 287–302 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sateia M. J., International classification of sleep disorders-third edition: Highlights and modifications. Chest 146, 1387–1394 (2014). [DOI] [PubMed] [Google Scholar]
- 68.Paruthi S., Brooks L. J., D’Ambrosio C., Hall W. A., Kotagal S., Lloyd R. M., Malow B. A., Maski K., Nichols C., Quan S. F., Rosen C. L., Troester M. M., Wise M. S., Recommended amount of sleep for pediatric populations: A consensus statement of the american academy of sleep medicine. J. Clin. Sleep Med. 12, 785–786 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Green E. W., Fedele G., Giorgini F., Kyriacou C. P., A Drosophila RNAi collection is subject to dominant phenotypic effects. Nat. Methods 11, 222–223 (2014). [DOI] [PubMed] [Google Scholar]
- 70.Hendricks J. C., Finn S. M., Panckeri K. A., Chavkin J., Williams J. A., Sehgal A., Pack A. I., Rest in Drosophila is a sleep-like state. Neuron 25, 129–138 (2000). [DOI] [PubMed] [Google Scholar]
- 71.Donelson N., Kim E. Z., Slawson J. B., Vecsey C. G., Huber R., Griffith L. C., High-resolution positional tracking for long-term analysis of Drosophila sleep and locomotion using the “Tracker” program. PLOS ONE 7, e37250 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cavanaugh D. J., Geratowski J. D., Wooltorton J. R. A., Spaethling J. M., Hector C. E., Zheng X., Johnson E. C., Eberwine J. H., Sehgal A., Identification of a circadian output circuit for rest:activity rhythms in Drosophila. Cell 157, 689–701 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McGuire S. E., Roman G., Davis R. L., Gene expression systems in Drosophila: A synthesis of time and space. Trends Genet. 20, 384–391 (2004). [DOI] [PubMed] [Google Scholar]
- 74.Churgin M. A., Szuperak M., Davis K. C., Raizen D. M., Fang-Yen C., Kayser M. S., Quantitative imaging of sleep behavior in Caenorhabditis elegans and larval Drosophila melanogaster. Nat. Protoc. 14, 1455–1488 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nowakowski T. J., Bhaduri A., Pollen A. A., Alvarado B., Mostajo-Radji M. A., Di Lullo E., Haeussler M., Sandoval-Espinosa C., Liu S. J., Velmeshev D., Ounadjela J. R., Shuga J., Wang X., Lim D. A., West J. A., Leyrat A. A., Kent W. J., Kriegstein A. R., Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hodge R. D., Bakken T. E., Miller J. A., Smith K. A., Barkan E. R., Graybuck L. T., Close J. L., Long B., Johansen N., Penn O., Yao Z., Eggermont J., Höllt T., Levi B. P., Shehata S. I., Aevermann B., Beller A., Bertagnolli D., Brouner K., Casper T., Cobbs C., Dalley R., Dee N., Ding S.-L., Ellenbogen R. G., Fong O., Garren E., Goldy J., Gwinn R. P., Hirschstein D., Keene C. D., Keshk M., Ko A. L., Lathia K., Mahfouz A., Maltzer Z., McGraw M., Nguyen T. N., Nyhus J., Ojemann J. G., Oldre A., Parry S., Reynolds S., Rimorin C., Shapovalova N. V., Somasundaram S., Szafer A., Thomsen E. R., Tieu M., Quon G., Scheuermann R. H., Yuste R., Sunkin S. M., Lelieveldt B., Feng D., Ng L., Bernard A., Hawrylycz M., Phillips J. W., Tasic B., Zeng H., Jones A. R., Koch C., Lein E. S., Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Townes F. W., Irizarry R. A., Quantile normalization of single-cell RNA-seq read counts without unique molecular identifiers. Genome Biol. 21, 160 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hafemeister C., Satija R., Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Consensus Conference Panel, Watson N. F., Badr M. S., Belenky G., Bliwise D. L., Buxton O. M., Buysse D., Dinges D. F., Gangwisch J., Grandner M. A., Kushida C., Malhotra R. K., Martin J. L., Patel S. R., Quan S. F., Tasali E., Joint Consensus Statement of the American Academy of Sleep Medicine and Sleep Research Society on the recommended amount of sleep for a healthy adult: Methodology and discussion. Sleep 38, 1161–1183 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mollayeva T., Thurairajah P., Burton K., Mollayeva S., Shapiro C. M., Colantonio A., The Pittsburgh sleep quality index as a screening tool for sleep dysfunction in clinical and non-clinical samples: A systematic review and meta-analysis. Sleep Med. Rev. 25, 52–73 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/23/eabe2626/DC1