

Abstract
A lack of expression of miR-143 and miR-145 has been demonstrated to be a frequent feature of colorectal tumors. Activating KRAS mutations have been reported in 30–60% of colorectal cancers and an inverse correlation between Kras and miR-143/145 expression has been observed. Previously, we have demonstrated that oncogenic Kras leads to repression of the miR-143/145 cluster in pancreatic cancer and is dependent on the Ras responsive element (RRE) binding protein (RREB1), which negatively regulates miR-143/145 expression. In the present study, we have found that RREB1 is overexpressed in colorectal adenocarcinoma tumors and cell lines, and the expression of the miR-143/145 primary transcript is inversely related to RREB1 expression. In colorectal cancer cell lines, the miR-143/145 cluster is repressed by RREB1 downstream of constitutively active KRAS. RREB1 is activated by the MAPK pathway and negatively represses the miR-143/145 promoter through interaction with two RREs. In addition, overexpression of miR-143 or miR-145 in HCT116 cells abrogates signaling through the MAPK, PI3K and JNK pathways by downregulation of both KRAS and RREB1 in addition to downregulation of a cohort of genes in the MAPK signaling cascade. These results establish a complex network of regulation through which the miR-143/145 cluster is able to modulate KRAS signaling in colorectal cancer.
Keywords: miRNA, miR-143, miR-145, KRAS, RREB1, colorectal
INTRODUCTION
MicroRNAs (miRNAs) are 18–24 nucleotide single-stranded RNAs that cause accelerated turnover and reduced translation of imperfectly complementary target messenger RNAs. During the last decade, over 700 human miRNAs have been identified and implicated in the regulation of a wide range of cellular processes including differentiation, proliferation and apoptosis.1,2 Accordingly, a large body of evidence has established an important role for miRNAs in cancer pathogenesis. Specific miRNAs have been shown to act as critical components of key oncogenic and tumor suppressor pathways highlighting the importance of identifying miRNAs that function in the primary signaling pathways that regulate tumorigenesis. For example, the miR-17-92 cluster is directly induced by the Myc oncogenic transcription factor and is able to promote cellular proliferation, survival and tumor angiogenesis.3 miR-34 family members are directly regulated by p53 and activate cell-cycle checkpoints and apoptosis.4,5
Since miRNAs likely regulate the majority of protein coding transcripts and mutations of KRAS have been implicated in a broad range of human cancers accentuating the widespread role of Ras signaling in tumor development and progression,6,7 growing attention has been applied toward understanding how miRNA-mediated regulation is integrated into RAS signaling pathways. The earliest link between miRNAs and RAS came from the Slack group who demonstrated the 3′UTRs of human RAS contain multiple sites for let-7 miRNA, allowing let-7 to regulate RAS expression.8 Recently, miR-21 has been found to be upregulated by oncogenic RAS and the genetic deletion of miR-21 suppressed Kras-driven tumors.9 miR-21 is induced by AP-1 downstream of the MAPK pathway and may involve a feed-forward loop since among its direct targets are multiple negative regulators of the Ras/MEK/ERK (extracellular signal-regulated kinase) pathway including PDCD4 (an inhibitor of AP-1).
Previously, we have demonstrated that expression of oncogenic Kras in a transformed pancreatic ductal epithelial cell line (HPNE) leads to repression of both miR-143 and miR-145.10 These two miRNAs are found in a polycistronic cluster on chromosome 5q32; miR-143 and miR-145 are co-transcribed from a single promoter resulting in a primary transcript that contains both miRNAs. The KRAS repression of the miR-143/145 cluster is dependent on the Ras responsive element (RRE) binding protein 1 (RREB1), which negatively regulates the miR-143/145 promoter. RREB1 has been implicated as a potential human oncogene and is found to be overexpressed in thyroid cancer where it confers increased transactivation of the calcitonin gene in response to Ras signal transduction.11-16 Furthermore, RREB1 represses expression of the p16INK promoter and the development of pristane-induced plasma cell tumors in BALB/c mice is attributable to a polymorphism in the RREB1 binding site.17 Both KRAS and RREB1 are experimentally determined targets of miR-143 and miR-145 demonstrating the existence of a feed-back circuit that potentiates Kras-mediated tumorigenesis through repression of the anti-tumorigenic miR-143/145 locus.10,18
Interestingly, a lack of miR-143 and miR-145 expression has been demonstrated to be a frequent feature of colorectal tumors.19,20 Activating KRAS mutations have been reported in 30–60% of colorectal cancers21,22 and activation of the Ras/Raf/MEK/ERK cascade is a common feature of this malignancy and promotes cell proliferation in the colonic epithelium.23 Multiple studies have demonstrated the suppression of miR-143 and miR-145 in colon cancer and the adenomatous and cancerous stages of colorectal neoplasia highlighting the importance of these two miRNAs in influencing colorectal tumorigenesis.19,20,24 In colon cancer tissues, there is an inverse correlation between oncogenic Kras and miR-143/145 expression, suggesting that Kras negatively regulates the miR-143/145 cluster in colorectal cancer.18,25 However, the molecular mechanism of miR-143/145 repression in colorectal cancer has not been determined.
In the present study, we have found that RREB1 is overexpressed in colorectal adenocarcinoma tumors and colon cancer cell lines harboring activating KRAS mutations. In normal colon, RREB1 is expressed at the base of the colonic crypts where miR-143 and miR-145 are not expressed. The colorectal cancer cell lines have negligible levels of miR-143/145 expression whereas RREB1 is highly expressed. In colorectal cancer cell lines, the miR-143/145 cluster is repressed by KRAS signaling downstream of the MAPK pathway. miR-143/145 expression is repressed by RREB1 directly binding to two RREs in the miR-143/145 promoter. In addition, overexpression of miR-143 or miR-145 in HCT116 cells abrogates signaling through the MAPK, PI3K and c-Jun NH2-terminal kinase (JNK) pathways by downregulation of both KRAS and RREB1 in addition to downregulation of a cohort of genes in the MAPK signaling cascade.
RESULTS
RREB1 is expressed in colorectal cancer and negatively regulates miR-143 and miR-145 downstream of oncogenic Kras
Using the Oncomine database, we found three data sets using mRNA gene expression arrays that identified RREB1 as significantly dysregulated in colon cancer.26 In the only study representing adenocarcinoma, RREB1 was significantly upregulated 1.5-fold versus normal colon (Figure 1a).27 The two additional studies on the Oncomine database demonstrated that RREB1 was significantly upregulated 1.5-fold in colorectal adenoma versus intestinal mucosa and 2.0-fold in colon adenoma versus ascending colon.28,29 We confirmed upregulation of RREB1 in colon cancer by performing immunohistochemistry (IHC) staining of RREB1 in several anonymous colon cancer tissues. RREB1 was readily detectable in these primary cancers (Figure 1b; Supplementary Figure 1A). Furthermore, we isolated total RNA from four tumors and matched normal adjacent tissues and performed qRT–PCR analysis of RREB1 mRNA. We found a statistically significant upregulation of RREB1 in all four tumors compared with normal adjacent tissue, which correlated with RREB1 protein expression as determined by IHC from the same tumors (Supplementary Figures 1A and B).
Figure 1.

RREB1 expression in colorectal cancer. (a) RREB1 expression in normal colon versus colon adenocarcinoma. The P-value indicated fold change 1.5-fold.27 (b) RREB1 IHC in anonymous colon cancer (T) samples. Positive RREB1 staining is brown. Original magnification × 160. (c) RREB1 IHC in normal colon samples. For orientation, the muscularis mucosa, representing the base of the glandular colonic tissue, is present at the bottom of each panel and the colonic lumen is present at the top. (d) Quantification of positive RREB1 staining at the top and bottom of colonic crypts. Analysis using Aperio Score parameter generated for 14 separate clusters of 10+ neighboring crypt segments. The P-value calculated using a paired t-test for means. (e) Log2 normalized qRT–PCR expression analysis of RREB1 mRNA and miR-143/145 primary transcript expression in a panel of colorectal carcinoma cell lines and three normal colon tissue samples (Colon-1, 2, 3). The activating KRAS mutation found in each cell line is indicated. *Activating BRAF mutation.
In normal colon, a recent study found miR-143 and miR-145 were predominantly expressed in epithelial cells on the colonic surface and maturation zone of the crypts by locked nucleic acid-in situ hybridization.30 We used IHC to determine the expression of RREB1 in normal colonic crypts. RREB1 was found predominantly in the cytoplasm with some nuclear localization detected. RREB1 staining was quantified using the Aperio stain intensity score (range 100–300) on 14 separate measures of 10+ neighboring base or surface crypt segments. We found that RREB1 is highly expressed at the bottom of the crypts where miR-143/145 are not appreciated by locked nucleic acid-in situ hybridization (Figure 1c, P=0.0001; Figure 1d).30
To determine RREB1 expression levels in a tractable system, we performed a qRT–PCR analysis of RREB1 mRNA and the miR-143/145 primary transcript RNA, representing expression of the miR-143/145 cluster, from a panel of colorectal cancer cell lines and normal colon tissues. The panel of colorectal cancer cell lines all harbor activating KRAS mutations except for Colo205, which contains an activating BRAF mutation (known to be mutually exclusive to KRAS mutations in this cancer type). RREB1 mRNA was readily detectible in RNA isolated from the colorectal cancer cell lines and exhibited expression levels 3- to 20-fold higher than RREB1 in normal colon tissues (Figure 1e, P=0.001). Conversely, the miR-143/145 primary transcript was expressed at very low levels in colorectal cancer cell lines compared with normal colon tissues where the primary transcript was readily detectable (P=2.8 E–07). Some of the miR-143/145 signal in normal colon tissue may derive from non-epithelial cells such as smooth muscle cells.31 RREB1 demonstrated an inverse expression relationship to the miR-143/145 primary transcript (with a Pearson correlation coefficient of −0.8). Western blot analysis of protein from colorectal cancer cell lines revealed readily detectible levels of RREB1 (protein isoforms α/β/δ)32 compared with HPNE which we previously determined to have low RREB1 expression and HPNE-KrasG12D which expresses RREB1 (Supplementary Figure 1C). Colorectal cancer cell lines expressed the mature miR-143 and miR-145 at greatly reduced levels compared with HPNE cells by northern blot (Supplementary Figure 1D). The absence of miR-143/145 expression was not due to loss of heterozygosity of 5q32 (containing miR-143 and miR-145) since 5q32 was diploid in the colorectal cancer cell lines studied (Supplementary Figure 1E).
To demonstrate RREB1-dependent repression of miR-143/145 in colon cancer, we used siRNA to transiently knockdown KRAS or RREB1 in colon carcinoma cell lines DLD1 and HCT116 to look for corresponding changes in pri-miR-143/145 expression. Acute knockdown of KRAS using siRNA partially reverses repression of pri-miR-143/145 in both DLD1 and HCT116 cell lines compared with siRNA-negative control (Figure 2a). Similarly, acute knockdown of RREB1 with siRNA in both DLD1 and HCT116 also reversed the transcriptional repression of pri-miR-143/145 compared with siRNA-negative control (Figure 2a). In addition, to determine if the regulation of miR-143/145 was unique to KRAS we also performed the transient knockdown of HRAS and NRAS. Acute knockdown of HRAS or NRAS using siRNA had no significant effect on de-repression of pri-miR-143/145 (Supplementary Figure 2A) or expression of RREB1 (Supplementary Figure 2B) in either DLD1 or HCT116 cell lines compared with siRNA-negative control.
Figure 2.

Kras regulation of RREB1 leads to repression of miR-143/145. (a) qRT–PCR analysis of KRAS, RREB1 and pri-miR-143/145 expression in DLD1 and HCT116 cell lines transfected with negative control or KRAS-targeting or RREB1-targeting siRNAs. Error bars for this and subsequent panels represent standard deviations from three independent measurements. (b) qRT–PCR analysis of RREB1 and pri-miR-143/145 expression in DLD1 and HCT116 cell lines with (KRAS) or without (Null) oncogenic KRAS. (c) TaqMan analysis of miR-143 and miR-145 expression in DLD1 and HCT116 cell lines with (KRAS) or without (Null) oncogenic KRAS. For all panels, the P-value calculated using a paired t-test (n.s. not significant was P>0.05).
To further demonstrate the link between KRAS and miR-143/145 repression, we used qRT–PCR to analyze RREB1 and pri-miR-143/145 expression in DLD1 and HCT116 cell lines where activated KRAS was disrupted by homologous recombination.33 We observed a decrease in RREB1 mRNA in cell lines, where activated KRAS was disrupted (null) compared with cells that retained constitutively active KRAS (Figure 2b). Both DLD1 and HCT116 cell lines with knocked out activated KRAS had a significant increase in both the level of pri-miR-143/145 transcript (Figure 2b) and mature miR-143 and miR-145 (Figure 2c) consistent with de-repression of the miR-143/145 cluster associated with decreased RREB1. Therefore, the data suggest that RREB1 negatively regulates miR-143/145 cluster expression in a KRAS-dependent manner in colorectal cancer.
RREB1 regulation of miR-143 and miR-145 is downstream of the MAPK pathway
Previously, two independent lines of evidence suggest that RREB1 is activated downstream of the MAPK pathway: first, RREB1 was found to regulate the calcitonin gene promoter through both activated Ras signaling and activated Raf signaling and second, the p16INK promoter can be downregulated by RREB1 in a Ras- or MEK-dependent manner.13,16 To support the hypothesis that RREB1 is activated by RAS signaling downstream of the MAPK pathway, we looked at miR-143 and miR-145 expression and RREB1 expression in HPNE-KrasG12D after blocking signaling through the two main KRAS effector pathways MAPK and PI3K. Following serum deprivation, cells were treated with small molecule inhibitors of MEK (U0126) or AKT (LY294002) for 24 h and RNA isolated for analysis of corresponding changes in RREB1 mRNA and miR-143/145 primary transcript expression. Blocking MEK activity in HPNE-KrasG12D cells with U0126 resulted in a greater than twofold increase in mature miR-143 and miR-145 and a similar increase in expression of the primary transcript of these miRNAs (Figure 3a; Supplementary Figure 3A). Following U0126 treatment, we saw a significant decrease in expression of RREB1 mRNA (Figure 3b). These results agree with previous findings that RREB1 may be activated downstream of the MAPK pathway.
Figure 3.
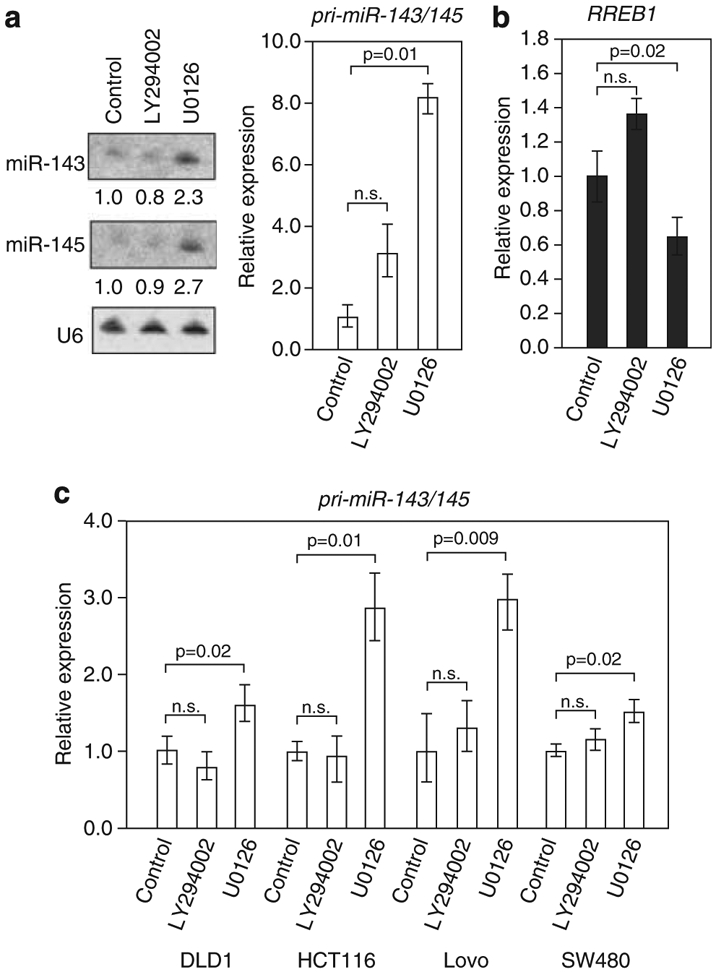
Small molecule inhibition of the MAPK pathway derepresses the miR-143/145 cluster. (a) Northern blot analysis of miR-143 and miR-145 (left) and qRT–PCR analysis of pri-miR-143/145 expression (right) in HPNE-KrasG12D cells treated with DMSO (control), or LY294002 or U0126. (b) qRT–PCR analysis of RREB1 expression in HPNE-KrasG12D cells treated with DMSO (control), LY294002 or U0126. (c) qRT–PCR analysis of pri-miR-143/145 expression in DLD1, HCT116, Lovo and SW480 cell lines treated with the indicated small molecule inhibitors. For all panels, the P-value calculated using a paired t-test (n.s. not significant was P>0.05).
To demonstrate this mechanism of activation of RREB1 in colorectal cancer, we treated DLD1, HCT116, Lovo and SW480 colorectal cancer cell lines with small molecule inhibitors LY294002 and U0126 for 24 h. RNA was isolated for analysis of corresponding changes in RREB1 mRNA and miR-143/145 primary transcript expression. In all four cell lines, we did not observe any statistically significant changes in expression of RREB1 looking at mRNA expression levels or protein expression with any of the conditions tested (Supplementary Figures 3B and C). However, expression of the pri-miR-143/145 transcript was consistently and significantly increased in cells treated with U0126 inhibitors of MAPK signaling pathway but not LY294002 or DMSO control (Figure 3c). These results suggest that RREB1 activation may require phosphorylation downstream of the MAPK pathway. Indeed, the Scansite2.0 algorithm predicted a short RREB1 protein sequence motif that may be a substrate for ERK1.
The miR-143/145 promoter contains two RREs regulated by RREB1 Previously, we mapped the miR-143/145 primary transcript and determined an ~1-kb region proximal to the first exon of the primary transcript contained the miR-143/145 promoter.10 Close inspection of the miR-143/145 promoter region using the UCSC genome browser conserved transcription factor binding site track revealed the presence of a high scoring motif matching the recognition site for RREB1 called an RAS responsive element (RRE-1; Figure 4a). In addition, manual scanning of the miR-143/145 promoter sequence allowed us to detect a second RRE, which we call RRE-2 that closely resembled both RRE-1 and the RREB1 consensus sequence (Figure 4a).13 We cloned the 1-kb human genomic segment encompassing the highly conserved transcription start site (TSS) (corresponding to the region shown in Figure 4a) into a promoterless luciferase reporter vector. The miR-143/145 promoter construct was repressed when introduced into DLD1 or HCT116 cells that retain oncogenic KRAS relative to DLD1 and HCT116 cells where oncogenic KRAS was knocked out by homologous recombination (Figure 4b).
Figure 4.

miR-143/145 promoter contains two RREs regulated by RREB1. (a) Magnified view of the 5′end of the human miR-143/145 pri-miRNA showing evolutionary conservation of the proximal promoter region that contains two RREs (RRE-1 and RRE-2). Sequences of RRE-1 and RRE-2 are compared with the consensus RREB1 binding site.13 (b) Activity of the miR-143/145 promoter reporter construct in DLD1 and HCT116 cell lines with (KRAS) or without (Null) oncogenic KRAS. The P-value calculated using a paired t-test. (c) Activity of the wild-type and mutant miR-143/145 promoter reporter constructs in NIH3T3 and NIH3T3-Kras cells. Values represent firefly luciferase activity produced from each plasmid normalized to renilla luciferase activity produced from a co-transfected control vector. Error bars represent standard deviations from three independent transfections, each measured in triplicate. (d) qPCR analysis of FLAG-RREB1 chromatin immunoprecipitates in NIH3T3 cells. Signal obtained from the miR-143/145 promoter amplicons containing the RREB1 binding sites (RRE-1 and RRE-2), an amplicon 300 bp downstream (Dn) and a negative control amplicon 5 kb upstream (5 kb) are shown. Error bars represent standard deviations derived from three independent measurements and the P-value calculated using a paired t-test.
Point mutations were then introduced into critical nucleotides within the RREB1 binding sites, RRE-1 and/or RRE-2, to directly assess the contribution of each RRE to promoter activity (Figure 4c). While these mutations had little effect on promoter function in NIH3T3 cells, mutation of either RRE sequence potently de-repressed the promoter in NIH3T3-Kras cells (Figure 4c). Mutation of the RRE-2 sequence enhanced promoter activity more in both NIH3T3 and NIH3T3-Kras cells, suggesting that RREB1 has a stronger affinity for RRE-2 than for RRE-1 or another transcription factor was able to repress the promoter through this sequence. In addition, the RRE-1 mutant promoter was de-repressed relative to the wild-type promoter in DLD1, Lovo and SW480 cell lines demonstrating colon cancer cell lines regulate the miR-143/145 promoter through an RREB1-dependent mechanism (Supplementary Figure 4).
Finally, we performed chromatin immunoprecipitation to document direct interaction of RREB1 with the endogenous miR-143/145 promoter. Following transfection of FLAG-tagged human RREB1 into NIH3T3 cells (which normally express RREB1 at low levels), enrichment of the miR-143/145 promoter elements RRE-1 and RRE-2 was evident in FLAG immunoprecipitates compared with transfection with empty vector or immunoprecipitation with control antibody (Figure 4d). These experiments demonstrate that RREB1 directly represses the activity of the miR-143/145 promoter in cells expressing activated Kras in colorectal cancer.
miR-143/145 modulate KRAS signaling through downregulation of a cohort of targets
Both KRAS and RREB1 have been experimentally confirmed as miR-143 and miR-145 targets, respectively.10,18 The ability of miR-143 and miR-145 to negatively regulate KRAS and RREB1 expression establishes the existence of a feedback circuit through which repression of these miRNAs in response to Kras activity further potentiates signaling through the Kras effector pathways. To demonstrate this circuit was active in colorectal cancer cell lines, we looked at total Ras, Kras and RREB1 expression in HCT116 cells with enforced miR-143 or miR-145 expression. We found that total Ras, Kras and RREB1 protein levels were all significantly decreased in HCT116 cells transfected with either miR-143 or miR-145 mimics compared with control mimic or mock transfection (Figure 5a). We wanted to confirm that the reduction in total Ras was due to decreased Kras expression since the pan-Ras antibody detects H-, K- and N-Ras isoforms. Previously, N- and H-Ras have been reported to be abundant in HCT116 cells;34 therefore, we looked at the contribution of each Ras isoform to the pool of total Ras expression by qRT–PCR. Although there was no change in H-Ras mRNA in HCT116 cells with enforced miR143/145 expression, interestingly both N-Ras and Kras mRNAs were reduced mirroring the total reduction in Ras observed by western blot (Figure 5a; Supplementary Figure 5A). It has been shown that constitutive Kras activity promotes elevated levels of N-Ras;34 therefore, the decrease in N-Ras expression we observed is likely an indirect effect of Kras downregulation by miR-143/145.
Figure 5.

Several miR-143/145 targets are components in Kras signaling pathways. (a–c) Western blot analysis of indicated protein expression (a, c) and qRT–PCR analysis of pri-miR-143/145 expression (b) in HCT116 cells treated with control mimic, miR-143 or miR-145 mimic, or untransfected. (d, e) Luciferase activity derived from the indicated 3′UTR reporter constructs following transfection into MiaPaCa2 cells with control mimic (−) or miR-143 mimics (d, +) or miR-145 mimics (e, +). All values were normalized to renilla luciferase activity produced from a co-transfected control plasmid. For each transfection condition, activity produced from the wild-type construct was normalized to the activity produced by the mutant construct. Error bars represent standard deviations from three independent transfections, each measured in triplicate. P-values for significant experiments indicated (two-tailed t-test) *Double miR-145 mutant. Colored bars represent statistically significantly results. (f, g) Western blot confirmation of miR-143 and miR-145 targets (f) or total JNK (t-JNK) or phospho-JNK (p-JNK) expression (g) in HCT116 cells treated with control mimic, miR-143 or miR-145 mimic or untransfected.
In addition, treatment of HCT116 cells with miR-143 or miR-145 mimic also resulted in a significant increase in the miR-143/145 primary transcript (Figure 5b) demonstrating the existence of a feedback circuit that is fully functional in HCT116 cells. As a further test of the feedback circuit in colorectal cancer, we also examined the activity of the MAPK and the PI3K pathways in HCT116 cells transfected with miR-143 or miR-145 mimics (Figure 5c). Expression of these miRNAs dramatically suppressed ERK1/2 and AKT phosphorylation, key effectors in the MAPK and PI3K signaling cascades compared with control mimic or mock transfection.
To gain further insight into the mechanistic contribution of miR-143/145 repression to KRAS signaling, we used the Diana-miR-Path computational tool to identify predicted targets of miR-143 and miR-145 found in KRAS signaling pathways.35 Since miR-143 and miR-145 expression had such a potent effect on inhibition of ERK1/2 activation, we chose to validate predicted targets of miR-143/145 that enhance signaling through the MAPK pathway rather than inhibitors of the pathway. The Diana-miR-Path analysis predicted six miR-143 and six miR-145 targets that are found in the MAPK pathway that met this criteria (listed in Figures 5d and e). All of the 3′UTRs contained one miR-143 or miR-145 binding site, except for MEKK11 which contained two miR-145 sites. To demonstrate that miR-143 or miR-145 can regulate these targets, a reporter plasmid was constructed with the putative miR-143 or miR-145 binding site containing UTRs cloned into the 3′end of luciferase.
Using luciferase assays, we were able to experimentally validate two of the predicted miR-143 targets (Figure 5d colored bars); however, the other four miR-143 targets were negative in the luciferase assays (Figure 5d, gray bars). Significant repression of luciferase expression was observed in cells co-transfected with miR-143 mimic and 3′UTR reporter for the ERK1 and ERK5. In addition, we were able to experimentally validate four of the predicted miR-145 targets (Figure 5e, colored bars) but the other two predicted miR-145 targets were negative in the luciferase assays (Figure 5e, gray bars). Significant repression of luciferase expression was observed in cells co-transfected with miR-145 mimic and 3′UTR reporter for MEK kinase kinase-4 (HGK), JNKK (JNK activating kinase-1), MEKK (MEK kinase-1) and fibroblast growth factor-5 (FGF5). Repression of luciferase activity was relieved by introducing mutations into the appropriate miRNA binding site within each 3′UTR (Figures 5d and e). To validate the luciferase assays, we used western blot to examine protein expression in HCT116 and SW480 cells treated with miR-143 or miR-145 mimics. Expression of miR-143 or miR-145 resulted in a 50–90% decrease of endogenous protein levels of targets compared with control mimic or mock transfection (Figure 5f; Supplementary Figure 5B). We were unable to validate FGF5 protein levels due to lack of an appropriate antibody.
Since two of the validated miR-145 targets HGK and JNKK are direct effectors of the JNK signaling cascade, we looked at the effect of miR-143/145 expression on activation of JNK signaling. HCT116 cells treated with miR-143 or miR-145 mimics or two pancreatic cancer cell lines, MiaPaCa2 and Panc-2.03, with stable expression of miR-143 and/or miR-145 were used to examine JNK phosphorylation as an indicator of activated JNK (Figure 5g; Supplementary Figure 5C). Expression of miR-145 dramatically suppressed JNK phosphorylation in all three cell lines. Interestingly, in HCT116 and Panc-2.03 cells, miR-143 expression also had a potent effect on JNK pathway inhibition perhaps through cross-talk between the Ras and JNK pathways (Figure 5f; Supplementary Figure 5C).
DISCUSSION
In the present study, we have demonstrated that RREB1 negatively regulates miR-143/145 expression downstream of oncogenic Kras in colorectal cancer. RREB1 is a zinc-finger transcription factor that both activates and represses gene expression in response to RAS signaling. RREB1 has emerged from multiple screens as a likely human oncogene and was identified in a genetic screen as 1 of 15 genes most likely to be a driver of colorectal cancer.15,36 Our previous studies have demonstrated that RREB1 negatively regulates the miR-143/145 cluster downstream of constitutively active Kras in pancreatic cancer. Activating KRAS mutations occur in approximately half of colorectal cancers,37 which correlates with a mechanism of RREB1 transcriptional regulation of the miR-143/145 cluster in colorectal cancer where miR-143/145 are frequently dysregulated. Indeed, miR-143 and miR-145 have been demonstrated to consistently display reduced steady-state levels of the mature miRNA at the adenomatous and cancer stages of colorectal neoplasia19 and have been documented to show reduced expression in colorectal cancer cell lines and tissues isolated from patients;20,38 however until now, the mechanism of miR-143/145 repression has remained elusive.
Our data demonstrate that RREB1 is expressed in colon cancer as well as in normal colon tissue. Previously and in the present study, RREB1 mRNAs have been shown to be upregulated in human colorectal cancers versus matched normal colon.36 In colon cancer, there was a dynamic range of RREB1 expression relative to adjacent normal crypts possibly reflecting the level of activation of Kras in these tumors. In normal colon, RREB1 is expressed at the base of the colonic crypts where miR-143/145 have been shown to be not expressed.30 As the crypt cells mature and proliferate toward the colonic surface, RREB1 expression is significantly reduced (Figures 1b and c). Our data suggest that if the stem cell of origin for colon cancer is derived from an undifferentiated epithelial cell at the base of the colonic crypts39 that miR-143 and miR-145 expression is not lost in colon cancer but rather maintained in the repressed state through a mechanism likely involving RREB1-mediated repression.
Previously, we have demonstrated that restoring miR-143/145 expression in pancreatic cancer cells blocks anchorage-independent growth in vitro and abrogates tumorigenesis in vivo.10 These phenotypes are consistent with enforced expression of miR-143/145 in colon cancer cell lines. For example, miR-143 overexpression reduces colony-forming ability of SW480 cells in soft agar and prevents HCT116 xenograft growth in nude mice.40,41 Overexpression of miR-145 in HCT116 cells has been shown to diminish colony formation and prevent tumor formation in nude mice.42 These phenotypes implicate the same mechanism of the Kras/RREB1 axis repression of the miR-143/145 cluster that we have demonstrated in pancreatic cancer cells is operational in colon adenocarcinoma. Further support of this mechanism can be inferred by the observation that both DLD1 and HCT116 with knocked out oncogenic KRAS have been reported to lose the ability for anchorage-independent growth in vitro and tumor formation in vivo compared with their counterparts that retain activated KRAS.33 In the current study, we have used these cell lines to demonstrate that the miR-143/145 cluster is regulated by RREB1 downstream of oncogenic KRAS. The anti-tumorigenic effect of the miR-143/145 cluster is likely the result of suppressed signaling through the Ras pathway by directly targeting KRAS and RREB1 causing shutdown of downstream RAS effector pathways.
Although activating KRAS mutations occur in 30–60% of colorectal cancers,21,22 wild-type Ras can be constitutively activated by upstream signals. In colorectal cancer, tumors with wild-type Ras were identified with p21 activation ratios above that of control colonocytes attributed in part to increased expression of c-erbB-2 receptor.43 Thus, stimulation of upstream tyrosine kinase receptors can activate wild-type Ras resulting in significant correlations between phospho-ERK1/2 and Ras activation.43 In addition, epidermal growth factor receptor can also suppress miR-143 and miR-145 in colonic tumorigenesis30 and also activates wild-type Kras in colon cancer cells. These results provide a compelling explanation for the lack of expression of the miR-143/145 cluster observed in colorectal cancer19 and suggest that cancer cells can downregulate these tumor suppressive miRNAs even in the genetic background of wild-type RAS.
The Ras-Raf-MEK-ERK cascade activates a number of growth promoting genes and confers anchorage independence and loss of contact inhibition to RAS transformed cells. The potent anti-tumorigenic effects of miR-143/145 on tumorigenic phenotypes are likely the result of modulation of this signaling cascade. Indeed, signaling through the ERK1/2, p38 and JNK MAPK cascades has been shown to regulate proliferation in Caco-2 cells.44 Multiple studies have implicated dysregulated MAPK signaling as a contributor to tumor pathogenesis. The miR-143 targeting of KRAS and ERK1 and the miR-145 targeting of MEKK (which directly activates MEK) clearly demonstrates how these two miRNAs are capable of negatively modulating signaling through this pathway. Previously, another study has demonstrated ERK5 to be a miR-143 target and downregulation of ERK5 may contribute to the miR-143 induced arrest of cell proliferation and the abrogation of tumor growth in mice in prostate cancer.45-47 Activated ERK5 modulates cell survival, differentiation and proliferation and transient overexpression of miR-143 in HCT116, SW480 and DLD1 colon cancer cell lines has been shown to downregulate ERK5 steady-state levels.41 Knockdown of HGK by RNAi has been found to inhibit hepatocellular carcinoma growth and invasion.48,49 In a screen of single cell-derived progenies from SW480 cells, HGK was identified as a potential predictor of metastasis and survival in colorectal cancer.50 Interestingly, ERK1, ERK5 and MEKK protein expression was decreased when HCT116 cells overexpressed either miR-143 or miR-145. This observation is consistent with several of our prior observations that these miRNAs act synergistically to modulate KRAS signaling and likely implicate ERK1, ERK5 and MEKK proteins in the feedback circuit described previously.
We have observed considerable ablation of JNK signaling in HCT116 cells overexpressing miR-143 and miR-145. The JNK is a member of the MAPK group and downstream targets include the AP-1 family of transcription factors. The JNK signaling transduction pathway has been demonstrated to increase cell proliferation and contribute to cancer. Furthermore, activation of JNK is essential for cell transformation and proliferation in response to oncogenic Ras signaling.51 JNK has been demonstrated to be critically required for RAS-induced transformation of mouse embryonic fibroblast in vitro and lung tumor formation in vivo.52 In addition, increased JNK activity accelerated tumorigenesis in a mouse model of colorectal carcinogenesis presumably through activation of the AP-1 transcription factor.53 Oncogenic Ras has been shown to interact with and therefore activate JNK leading to activation of Jun thus enabling the activated AP-1 complex.54 Furthermore, activated JNK can phosphorylate Raf, and MEK can phosphorylate JNK leading to a strong positive feedback loop that potentiates continuous mitogenic signaling.54 These data are consistent with our results that show decreased signaling through both the JNK and MAPK pathways in HCT116 cells with overexpression of miR-143 and miR-145, which is determinant on downregulation of the experimentally validated targets described in our study.
In conclusion, repression of miR-143/145 is necessary to sustain Ras pathway activity even in the setting of activating KRAS mutations. The ability of miR-143/145 expression to completely abrogate Ras signaling that occurs when these miRNAs are expressed, as evidenced by suppression of the MAPK, PI3K and JNK pathways, undoubtedly contributes significantly to their tumor suppressor activity. This aspect of the miR-143/145 target network is likely especially important in colorectal cancer and pancreatic cancer where activating KRAS mutations are common. Delivery of these miRNAs, or analogs that elicit similar downstream effects, represents a promising therapeutic approach for tumor types with activating KRAS mutations.
MATERIALS AND METHODS
Cell lines
Colorectal cancer cell lines Lovo, SW480, SW620, SW837 and SW948 were cultured in DMEM (4.5 mg/ml glucose) supplemented with 10% fetal bovine serum and antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin). Cell lines Colo205, DLD1 and SKCO-1 were cultured in RPMI supplemented with 10% FBS and antibiotics. HCT116 was cultured in McCoys-5A media supplemented with 10% FBS and antibiotics. HPNE, HPNE-KrasG12D, PDAC, NIH3T3 and NIH3T3-Kras cell lines were cultured as described.55-57 Cells were incubated at 37 °C in a humidified atmosphere of 5% CO2 in air.
Immunohistochemistry
Anonymous normal colon and colon cancer samples (n=8) were obtained from the surgical pathology archives at The Johns Hopkins Hospital with approval of the local Investigational Review Board. Slides were incubated with RREB1 (Abcam, Cambridge, MA, USA) for 30 min at room temperature followed by a rabbit HRP polymer conjugate (SuperPicTure; Invitrogen, Camarillo, CA, USA) for 10 min. Slides were stained with Impact DAB (Vector Labs, Burlingame, CA, USA) and counterstained with hematoxylin (Richard-Allen, Kalamazoo, MI, USA). A normal colon slide was digitized (Aperio, Vista, CA, USA) and analyzed using the Aperio Tool Kit for color deconvolution using the default settings. The base of the crypt was determined based on its proximity to the muscularis mucosa, which defines the base of the lamina propria. The tops of the crypt were either directly adjacent to the intestinal lumen or closer to the lumen than the crypt segments identified to be the base of the crypt. All images were interpreted by a board certified anatomic pathologist.
RNA northern blot analysis
Total RNA was isolated from cells with TRIZOL (Invitrogen) according to manufacturer’s protocol. Northern blots were performed as described previously.57 Quantification of miRNA loading was normalized to U6 snRNA.
qRT–PCR
Total RNA was digested with DNase I. Reverse transcription was performed using the SuperScript First-strand Synthesis System (Invitrogen) with random hexamers. Quantitative PCR was performed using an ABI 7900 Sequence Detection System with the SYBR Green PCR core reagent kit (Applied Biosystems, Carlsbad, CA, USA). Transcript abundance was normalized to β-actin expression. Primer sequences are provided in Supplementary Table 1. For measuring mature miR-143 and miR-145 levels, predesigned Taqman primers and probes (Applied Biosystems) were used according to manufacturer’s instructions.
MiRNA mimics and siRNA gene knockdown
MicroRNA mimics (Dharmacon, Lafayette, CO, USA) or siRNAs (siGENOME smart pool) were suspended in 1× universal buffer (60mm KCl, 6mm HEPES-KOH pH 7.5, 0.2mm MgCl2), heated to 90 °C for 3 min and cooled on ice. Mimics (100 nm final) or siRNAs (10 nm final) were complexed with oligofectamine (mimics; Invitrogen) or DharmaFECT2 (siRNAs) in OPTIMEM serum-free media at room temperature for 20 min and then added to cells for 72 h.
MAPK small molecule inhibitors
In all, 2.0×105 cells were plated in a six-well plate and grown for 24 h followed by an additional 24 h in serum-free media. Small molecule inhibitors were added to complete media at final concentrations of 10 μm (LY294002), or 5 μm (U0126) and added to cells for 24 h. RNA isolation and cDNA synthesis were described above.
Luciferase reporter assays
In all, 0.8×105 cells were plated in triplicate wells of a 24-well plate and transfected 16 h later with 50 ng of pGL3-IRES-promoter reporter construct and 4 ng of phRL-SV40 (Promega, Madison, WI, USA) or 100 ng of the wild-type or mutant 3′UTR reporter construct and 0.5 ng of phRL-SV40 using Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions. Twenty-four hours after transfection, cells were lysed and assayed for firefly and renilla luciferase activity using the Dual-Luciferase Reporter Assay System (Promega). Where indicated control mimic, miR-143 or miR-145 mimics (Dharmacon) were co-transfected at 15 nm final concentration.
Cloning full-length human RREB1
A partial RREB1 cDNA (Open Biosystems, Lafayette, CO, USA; clone ID: BC032062) was obtained that contains the 5′end of the RREB1 transcript but not a functional stop codon. We RT–PCR amplified the 3′end of the RREB1 transcript from total RNA isolated from HPNE-Kras cells. This sequence was designed to take advantage of a unique BglII site at the 5′end of the PCR product to allow cloning into the human RREB1-pCMV-SPORT6.1 vector. The 3.4-kB PCR product was cloned into BglII and XbaI sites to generate full-length human RREB1-pCMV-SPORT6.1. An N-terminal FLAG tag was introduced using PCR of full-length RREB1 and back cloning into pCMV-SPORT6.1 using AgeI and XbaI sites. Primer sequences are provided in Supplementary Table 1.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed in NIH3T3 cells as described previously,10 except that human pCMV-SPORT-FLAG-RREB1 was used. N-terminal FLAG-RREB1 expression was confirmed by western blot (not shown). Immunoprecipitations were performed with 2 μg anti-FLAG-M2 antibody (Sigma-Aldrich, St Louis, MO, USA) or 2 μg mouse IgG1 control antibody (Dako, Carpinteria, CA, USA). Real-time PCR was performed using the SYBR Green PCR master mix (Applied Biosystems) to assess binding to the miR-143/145 promoter. Sequences of primers used to amplify chromatin immunoprecipitation samples are provided in Supplementary Table 1.
Western blotting
All antibodies were obtained from Cell Signaling Technologies, Beverly, MA, USA, except Kras antibody from Santa Cruz Biotechnology, Santa Cruz, CA, USA (K-Ras (F234): sc-30) and RREB1 antibody was from Abcam.
Statistical analysis
Statistical analysis was done using Student’s t-test, assuming equal variance, and P-values were calculated based on two-tailed test. A P-value of <0.05 was considered statistically significant.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Dr Joshua Mendell and Dr Anirban Maitra for helpful discussions and feedback during preparation of the manuscript. We thank Dr James Eshleman for kindly providing cell lines and Norman Barker for photography assistance. We would like to thank Dr Dan Durocher and members of the Durocher laboratory at the Samuel Lunenfeld Research Institute at Mount Sinai Hospital Toronto for kindly providing laboratory space and reagents and helpful discussions during the review process of the manuscript. This work was supported in part through the NIH (R01CA120185) and by a Clinician Scientist Award to MKH from the Doris Duke Foundation Grant #: 2009040. OAK is a Life Sciences Research Foundation Fellow.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
REFERENCES
- 1.Ambros V The functions of animal microRNAs. Nature 2004; 431: 350–355. [DOI] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116: 281–297. [DOI] [PubMed] [Google Scholar]
- 3.Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell 2008; 133: 217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 2007; 26: 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y et al. A microRNA component of the p53 tumour suppressor network. Nature 2007; 447: 1130–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barbacid M Ras genes. Annu Rev Biochem 1987; 56: 779–827. [DOI] [PubMed] [Google Scholar]
- 7.Hingorani SR, Tuveson DA. Ras redux: rethinking how and where Ras acts. Curr Opin Genet Dev 2003; 13: 6–13. [DOI] [PubMed] [Google Scholar]
- 8.Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A et al. RAS is regulated by the let-7 microRNA family. Cell 2005; 120: 635–647. [DOI] [PubMed] [Google Scholar]
- 9.Hatley ME, Patrick DM, Garcia MR, Richardson JA, Bassel-Duby R, van Rooij E et al. Modulation of K-Ras-dependent lung tumorigenesis by MicroRNA-21. Cancer Cell 2010; 18: 282–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kent OA, Chivukula RR, Mullendore M, Wentzel EA, Feldmann G, Lee KH et al. Repression of the miR-143/145 cluster by oncogenic Ras initiates a tumor-promoting feed-forward pathway. Genes Dev 2010; 24: 2754–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Date S, Nibu Y, Yanai K, Hirata J, Yagami K, Fukamizu A. Finb, a multiple zinc finger protein, represses transcription of the human angiotensinogen gene. Int J Mol Med 2004; 13: 637–642. [PubMed] [Google Scholar]
- 12.Mukhopadhyay NK, Cinar B, Mukhopadhyay L, Lutchman M, Ferdinand AS, Kim J et al. The zinc finger protein ras-responsive element binding protein-1 is a coregulator of the androgen receptor: implications for the role of the Ras pathway in enhancing androgenic signaling in prostate cancer. Mol Endocrinol 2007; 21: 2056–2070. [DOI] [PubMed] [Google Scholar]
- 13.Thiagalingam A, De Bustros A, Borges M, Jasti R, Compton D, Diamond L et al. RREB-1, a novel zinc finger protein, is involved in the differentiation response to Ras in human medullary thyroid carcinomas. Mol Cell Biol 1996; 16: 5335–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thiagalingam A, Lengauer C, Baylin SB, Nelkin BD. RREB1, a ras responsive element binding protein, maps to human chromosome 6p25. Genomics 1997; 45: 630–632. [DOI] [PubMed] [Google Scholar]
- 15.Uren AG, Kool J, Matentzoglu K, de Ridder J, Mattison J, van Uitert M et al. Large-scale mutagenesis in p19(ARF)- and p53-deficient mice identifies cancer genes and their collaborative networks. Cell 2008; 133: 727–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang S, Qian X, Redman C, Bliskovski V, Ramsay ES, Lowy DR et al. p16 INK4a gene promoter variation and differential binding of a repressor, the ras-responsive zinc-finger transcription factor, RREB. Oncogene 2003; 22: 2285–2295. [DOI] [PubMed] [Google Scholar]
- 17.Zhang L, Zhao J, Edenberg HJ. A human Raf-responsive zinc-finger protein that binds to divergent sequences. Nucleic Acids Res 1999; 27: 2947–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen X, Guo X, Zhang H, Xiang Y, Chen J, Yin Y et al. Role of miR-143 targeting KRAS in colorectal tumorigenesis. Oncogene 2009; 28: 1385–1392. [DOI] [PubMed] [Google Scholar]
- 19.Michael MZ, O’Connor SM, van Holst PNG, Young GP, James RJ. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res 2003; 1: 882–891. [PubMed] [Google Scholar]
- 20.Akao Y, Nakagawa Y, Naoe T. MicroRNAs 143 and 145 are possible common onco-microRNAs in human cancers. Oncol Rep 2006; 16: 845–850. [PubMed] [Google Scholar]
- 21.Kressner U, Glimelius B, Bergström R, Påhlman L, Larsson A, Lindmark G. Increased serum p53 antibody levels indicate poor prognosis in patients with colorectal cancer. Br J Cancer 1998; 11: 1848–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brink M, de Goeij AF, Weijenberg MP, Roemen GM, Lentjes MH, Pachen MM et al. K-ras oncogene mutations in sporadic colorectal cancer in The Netherlands Cohort Study. Carcinogenesis 2003; 24: 703–710. [DOI] [PubMed] [Google Scholar]
- 23.Calcagno SR, Li S, Colon M, Kreinest PA, Thompson EA, Fields AP et al. Oncogenic K-ras promotes early carcinogenesis in the mouse proximal colon. Int J Cancer 2008; 122: 2462–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Slaby O, Svoboda M, Fabian P, Smerdova T, Knoflickova D, Bednarikova M et al. Altered expression of miR-21, miR-31, miR-143 and miR-145 is related to clinicopathologic features of colorectal cancer. Oncology 2007; 72: 397–402. [DOI] [PubMed] [Google Scholar]
- 25.Mosakhani N, Sarhadi VK, Borze I, Karjalainen-Lindsberg ML, Sundström J, Ristamäki R et al. MicroRNA profiling differentiates colorectal cancer according to KRAS status. Genes Chromosomes Cancer 2012; 51: 1–9. [DOI] [PubMed] [Google Scholar]
- 26.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D et al. ONCO-MINE: a cancer microarray database and integrated data-mining platform. Neoplasia 2004; 6: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaiser S, Park YK, Franklin JL, Halberg RB, Yu M, Jessen WJ et al. Transcriptional recapitulation and subversion of embryonic colon development by mouse colon tumor models and human colon cancer. Genome Biol 2007; 8: R131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaspar C, Cardoso J, Franken P, Molenaar L, Morreau H, Möslein G et al. Cross-species comparison of human and mouse intestinal polyps reveals conserved mechanisms in adenomatous polyposis coli (APC)-driven tumorigenesis. Am J Pathol 2008; 172: 1363–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sabates-Bellver J, Van der Flier LG, de Palo M, Cattaneo E, Maake C, Rehrauer H et al. Transcriptome profile of human colorectal adenomas. Mol Cancer Res 2007; 5: 1263–1275. [DOI] [PubMed] [Google Scholar]
- 30.Zhu H, Dougherty U, Robinson V, Mustafi R, Pekow J, Kupfer S et al. EGFR signals downregulate tumor suppressors miR-143 and miR-145 in Western diet-promoted murine colon cancer: role of G1 regulators. Mol Cancer Res 2011; 9: 960–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN et al. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 2009; 460: 705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nitz MD, Harding MA, Smith SC, Thomas S, Theodorescu D. RREB1 transcription factor splice variants in urologic cancer. Am J Pathol 2011; 179: 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shirasawa S, Furuse M, Yokoyama N, Sasazuki T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science 1993; 260: 85–88. [DOI] [PubMed] [Google Scholar]
- 34.Keller JW, Haigis KM, Franklin JL, Whitehead RH, Jacks T, Coffey RJ. Oncogenic K-RAS subverts the antiapoptotic role of N-RAS and alters modulation of the N-RAS: gelsolin complex. Oncogene 2007; 26: 3051–3059. [DOI] [PubMed] [Google Scholar]
- 35.Papadopoulos GL, Alexiou P, Maragkakis M, Reczko M, Hatzigeorgiou AG. DIANA-mirPath: integrating human and mouse microRNAs in pathways. Bioinformatics 2009; 25: 1991–1993. [DOI] [PubMed] [Google Scholar]
- 36.Starr TK, Allaei R, Silverstein KA, Staggs RA, Sarver AL, Bergemann TL et al. A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science 2009; 323: 1747–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M et al. Genetic alterations during colorectal-tumor development. N Engl J Med 1988; 319: 525–532. [DOI] [PubMed] [Google Scholar]
- 38.Bandres E, Cubedo E, Agirre X, Malumbres R, Zarate R, Ramirez N et al. Identification by Real-time PCR of 13 mature microRNAs differentially expressed in colorectal cancer and non-tumoral tissues. Mol Cancer 2006; 5: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barker N, Ridgway RA, van Es JH, van de Wetering H, Begthel H, van den Born M et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009; 457: 608–611. [DOI] [PubMed] [Google Scholar]
- 40.Ng EK, Tsang WP, Ng SS, Jin HC, Yu J, Li JJ et al. MicroRNA-143 targets DNA methyltransferases 3A in colorectal cancer. Br J Cancer 2009; 101: 699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borralho PM, Simões AE, Gomes SE, Lima RT, Carvalho T, Ferreira DM et al. miR-143 overexpression impairs growth of human colon carcinoma xenografts in mice with induction of apoptosis and inhibition of proliferation. PLoS One 2011; 6: e23787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sachdeva M, Zhu S, Wu F, Wu H, Walia V, Kumar S et al. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci USA 2009; 106: 3207–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bissonnette M, Khare S, von Lintig FC, Wali RK, Nguyen L, Zhang Y et al. Mutational and nonmutational activation of p21ras in rat colonic azoxymethane-induced tumors: effects on mitogen-activated protein kinase, cyclooxygenase-2, and cyclin D1. Cancer Res 2000; 60: 4602–4609. [PubMed] [Google Scholar]
- 44.Buzzi N, Colicheo A, Boland R, de Boland AR. MAP kinases in proliferating human colon cancer Caco-2 cells. Mol Cell Biochem 2009; 328: 201–208. [DOI] [PubMed] [Google Scholar]
- 45.Esau C, Kang X, Peralta E, Hanson E, Marcusson EG, Ravichandran LV et al. MicroRNA-143 regulates adipocyte differentiation. J Biol Chem 2004; 279: 52361–52365. [DOI] [PubMed] [Google Scholar]
- 46.Clape C, Fritz V, Henriquet C, Apparailly F, Fernandez PL, Iborra F et al. miR-143 interferes with ERK5 signaling, and abrogates prostate cancer progression in mice. PLoS One 2009; 4: e7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borralho PM, Kren BT, Castro RE, da Silva IB, Steer CJ, Rodrigues CM. MicroRNA-143 reduces viability and increases sensitivity to 5-fluorouracil in HCT116 human colorectal cancer cells. FEBS J 2009; 276: 6689–6700. [DOI] [PubMed] [Google Scholar]
- 48.Han SX, Zhu Q, Ma JL, Zhao J, Huang C, Jia X et al. Lowered HGK expression inhibits cell invasion and adhesion in hepatocellular carcinoma cell line HepG2. World J Gastroenterol 2010; 16: 4541–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu AW, Cai J, Zhao XL, Jiang TH, He TF, Fu HQ et al. ShRNA-targeted MAP4K4 inhibits hepatocellular carcinoma growth. Clin Cancer Res 2011; 17: 710–720. [DOI] [PubMed] [Google Scholar]
- 50.Hao JM, Chen JZ, Sui HM, Si-Ma XQ, Li GQ, Liu C et al. A five-gene signature as a potential predictor of metastasis and survival in colorectal cancer. J Pathol 2010; 220: 475–489. [DOI] [PubMed] [Google Scholar]
- 51.Dérijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T et al. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 1994; 76: 1025–1037. [DOI] [PubMed] [Google Scholar]
- 52.Cellurale C, Sabio G, Kennedy NJ, Das M, Barlow M, Sandy P et al. Requirement of c-Jun NH(2)-terminal kinase for Ras-initiated tumor formation. Mol Cell Biol 2011; 31: 1565–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sancho R, Nateri AS, de Vinuesa AG, Aguilera C, Nye E, Spencer-Dene B et al. JNK signalling modulates intestinal homeostasis and tumourigenesis in mice. EMBO J 2009; 28: 1843–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adler V, Qu Y, Smith SJ, Izotova L, Pestka S, Kung HF et al. Functional interactions of Raf and MEK with Jun-N-terminal kinase (JNK) result in a positive feedback loop on the oncogenic Ras signaling pathway. Biochemistry 2005; 44: 10784–10795. [DOI] [PubMed] [Google Scholar]
- 55.Feldmann G, Habbe N, Dhara S, Bisht S, Alvarez H, Fendrich V et al. Hedgehog inhibition prolongs survival in a genetically engineered mouse model of pancreatic cancer. Gut 2008; 57: 1420–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Campbell PM, Groehler AL, Lee KM, Ouellette MM, Khazak V, Der CJ. K-Ras promotes growth transformation and invasion of immortalized human pancreatic cells by Raf and phosphatidylinositol 3-kinase signaling. Cancer Res 2007; 67: 2098–2106. [DOI] [PubMed] [Google Scholar]
- 57.Kent OA, Mullendore M, Wentzel EA, Lopez-Romero P, Tan AC, Alvarez H et al. A resource for analysis of microRNA expression and function in pancreatic ductal adenocarcinoma cells. Cancer Biol Ther 2009; 8: 2013–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.