

Abstract
MicroRNAs (miRNAs) act in concert with Argonaute (AGO) proteins to repress target mRNAs. After AGO loading, miRNAs generally exhibit slow turnover. An important exception occurs when miRNAs encounter highly complementary targets, which can trigger a process termed target-directed microRNA degradation (TDMD). During TDMD, miRNAs undergo tailing and trimming, suggesting that this is an important step in the decay mechanism. Here, we identified a cullin-RING ubiquitin ligase (CRL), containing the substrate adapter ZSWIM8, that mediated TDMD. The ZSWIM8 CRL interacted with AGO proteins, promoted TDMD in a tailing and trimming-independent manner, and regulated miRNA expression in multiple cell types. These findings suggest a model in which the ZSWIM8 ubiquitin ligase mediates TDMD by directing proteasomal decay of miRNA-containing complexes engaged with highly complementary targets.
One Sentence Summary:
The ZSWIM8 ubiquitin ligase mediates target-directed microRNA degradation (TDMD).
MicroRNAs (miRNAs) are ~22 nucleotide noncoding RNAs that post-transcriptionally repress messenger RNAs (mRNAs) (1). After processing, miRNAs are loaded into a family of related Argonaute (AGO) proteins, which they direct to sites of imperfect complementarity in target mRNAs (2). Base-pairing of the miRNA 5’ end, referred to as the seed sequence, is most critical for a productive silencing interaction with a target. Over two decades of study have established the essential biological functions of miRNAs across diverse metazoan species. Consistent with the broad importance of these regulatory RNAs in development and physiology, elaborate mechanisms that regulate rates of miRNA transcription and biogenesis have been elucidated (3). However, much less is known about the mechanisms and effector proteins that carry out regulated miRNA turnover, despite the long-known existence of miRNAs with unusually short half-lives (4–6).
Once loaded into an AGO protein, miRNAs generally exhibit slow turnover kinetics (7). Nevertheless, extended base-pairing with a target can induce accelerated decay of miRNAs, through a process termed target-directed miRNA degradation (TDMD) (8–12). For example, exogenously introduced target mRNAs trigger decay of miRNAs with extensive complementarity in Drosophila (11). Similarly, a Herpesvirus saimiri noncoding RNA that contains a highly complementary binding site for miR-27 directs decay of this miRNA during viral infection (12). Additional viral transcripts that promote decay of host miRNAs, as well as endogenous human and mouse transcripts that induce miRNA degradation, have been identified (8–10, 13–15). In addition to base pairing with the seed sequence at the miRNA 5’ end, TDMD-inducing miRNA:target interactions are characterized by complementarity between the 3’ end of the miRNA and the target, as well as central mismatches. This extensive pairing exposes the 3’ end of the miRNA, which is normally buried in the PAZ domain of AGO (16). The solvent-exposed miRNA 3’ end undergoes a process known as tailing and trimming, in which terminal nucleotidyl transferases add non-templated nucleotides, while exonucleases remove 3’-terminal nucleotides. Although tailing and trimming has been proposed to play an important role in the TDMD pathway (8, 11, 17, 18), recent data suggest that addition of non-templated nucleotides to the miRNA 3’ end is dispensible for decay of certain miRNAs by this mechanism (10, 19). Thus, it remains unresolved whether tailed and trimmed miRNAs are essential intermediates in the pathway or side products formed as a consequence of the distinctive TDMD miRNA:target interaction. Moreover, trans-acting factors that mediate miRNA decay through TDMD have not been identified. Here we employed a genome-wide CRISPR screening strategy to identify the machinery that carries out TDMD and advance our molecular understanding of this important mechanism of miRNA regulation.
A genome-wide CRISPR-Cas9 screen for TDMD factors
We applied a genome-wide CRISPR-Cas9 screening strategy, used previously by our laboratory to identify miRNA regulators (20), to probe the well-established CYRANO-miR-7 TDMD pair (10). CYRANO is a highly conserved long noncoding RNA that harbors a single site with extensive complementarity to miR-7 (Fig. 1A) that has been shown to trigger robust decay of this miRNA. A fluorescent reporter cell line that monitors miR-7 activity was generated by expressing an Enhanced Green Fluorescent Protein (EGFP) transcript with eight perfectly complementary miR-7 binding sites in its 3’ UTR (EGFPmiR−7) in the human cell line K562, which endogenously expressed both CYRANO and miR-7 (10) (Fig. 1B). Validating the ability of this reporter to read-out the efficiency of TDMD, CYRANO loss of function resulted in robust repression of EGFP fluorescence, which mirrored the expected increase in miR-7 abundance.
Fig. 1. A genome-wide CRISPR-Cas9 screen for TDMD factors reveals a cullin-RING E3 ubiquitin ligase.
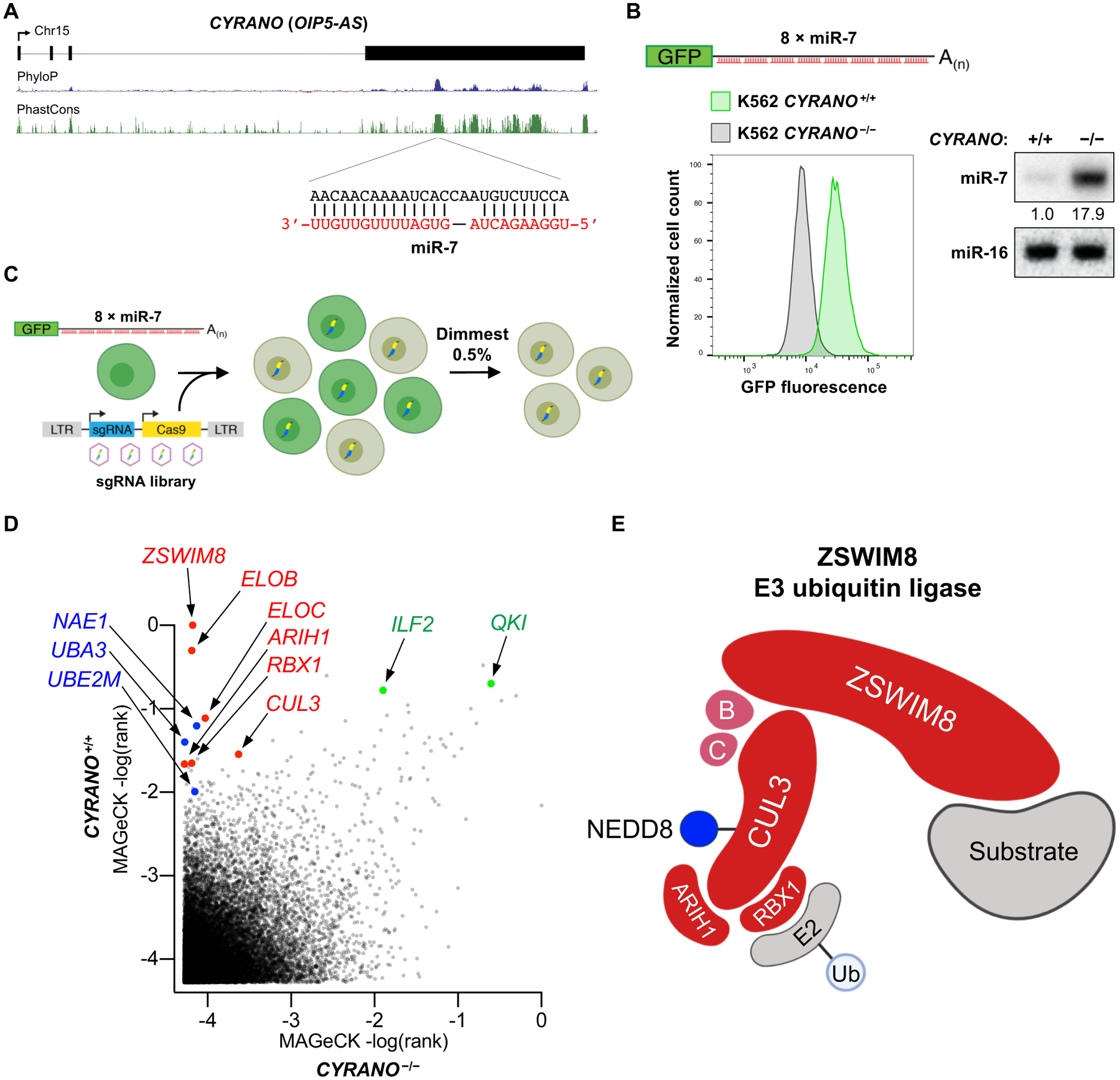
(A) Location of the miR-7 binding site in CYRANO (also known as OIP5-AS). UCSC Genome Browser PhyloP and PhastCons tracks shown (hg38). (B) Loss of CYRANO in the K562 EGFPmiR−7 reporter cell line results in reduced fluorescence, as shown by flow cytometry (left), and increased miR-7 abundance, as shown by northern blotting (right). (C) Overview of the CRISPR-Cas9 screen. (D) Genes plotted by MAGeCK rank in screens performed in CYRANO+/+ and CYRANO−/− EGFPmiR−7 cells. Screens were conducted in two independent clones per genotype. CRL components (red), NEDDylation factors (blue), and known miR-7 regulators (green) highlighted. (E) Proposed components of the ZSWIM8 cullin-RING ubiquitin ligase.
A genome-wide CRISPR-Cas9 screen was performed by infecting EGFPmiR−7 reporter cells with a lentiviral CRISPR library (21) (Fig. 1C). To discern hits in the screen that regulate miR-7 in a TDMD-independent manner, a parallel screen was conducted in CYRANO−/− EGFPmiR−7 reporter cells. After introduction of the library, fluorescence activated cell sorting (FACS) was used to collect cells with reduced fluorescence, thereby enriching for single guide RNAs (sgRNAs) targeting candidate miR-7 regulators. Genes encoding known negative regulators of miR-7, including QKI (22) and ILF2 (23), scored as significant hits in both CYRANO+/+ and CYRANO−/− cells, supporting the sensitivity of this screening approach (Fig. 1D and Table S1).
Among the CYRANO-dependent hits, several components of cullin-RING ubiquitin ligase (CRL) complexes were among the most highly ranked (ZSWIM8, ELOB, ELOC, CUL3, RBX1, and ARIH1) (Fig. 1D). CRLs, the largest superfamily of E3 ubiquitin ligases, are composed of cullin scaffold proteins and associated factors that bring together E2 ubiquitin-conjugating enzymes and substrates for ubiquitylation (24). ZSWIM8 was previously reported to function as a substrate adaptor of a CRL containining ELOB and ELOC (25), and a high-throughput proteomics study identified ZSWIM8 as an interacting partner of CUL3 (26). RBX1 and ARIH1 are important CRL accessory proteins (27, 28). Consistent with the requirement for conjugation of a ubiquitin-like modifier, NEDD8, for CRL activity (29), several genes in the NEDDylation pathway were also highly ranked (Fig. 1D). Thus, we hypothesized that the screen uncovered the major components of an E3 ubiquitin ligase that mediates TDMD (Fig. 1E).
ZSWIM8 E3 ubiquitin ligase components are required for TDMD
Consistent with the results of the screen, knockout of each individual component of the putative ZSWIM8 ubiquitin ligase led to measureable repression of the EGFPmiR−7 reporter in CYRANO+/+, but not CYRANO−/−, cells (Fig. 2A and Fig. S1A–C). ELOB and ELOC are known to associate with CUL2 and CUL5 (30). However, CUL2 and CUL5 were not recovered in the screen, and knockout of CUL2, CUL5, or both did not repress the EGFPmiR−7 reporter (Fig. S1D–E). In contrast, CUL3 knockout strongly repressed the reporter, suggesting that the ZSWIM8 E3 ligase represents a CRL in which ZSWIM8-ELOB-ELOC serve as substrate adapters for CUL3.
Fig. 2. ZSWIM8 ubquitin ligase components are essential for TDMD.
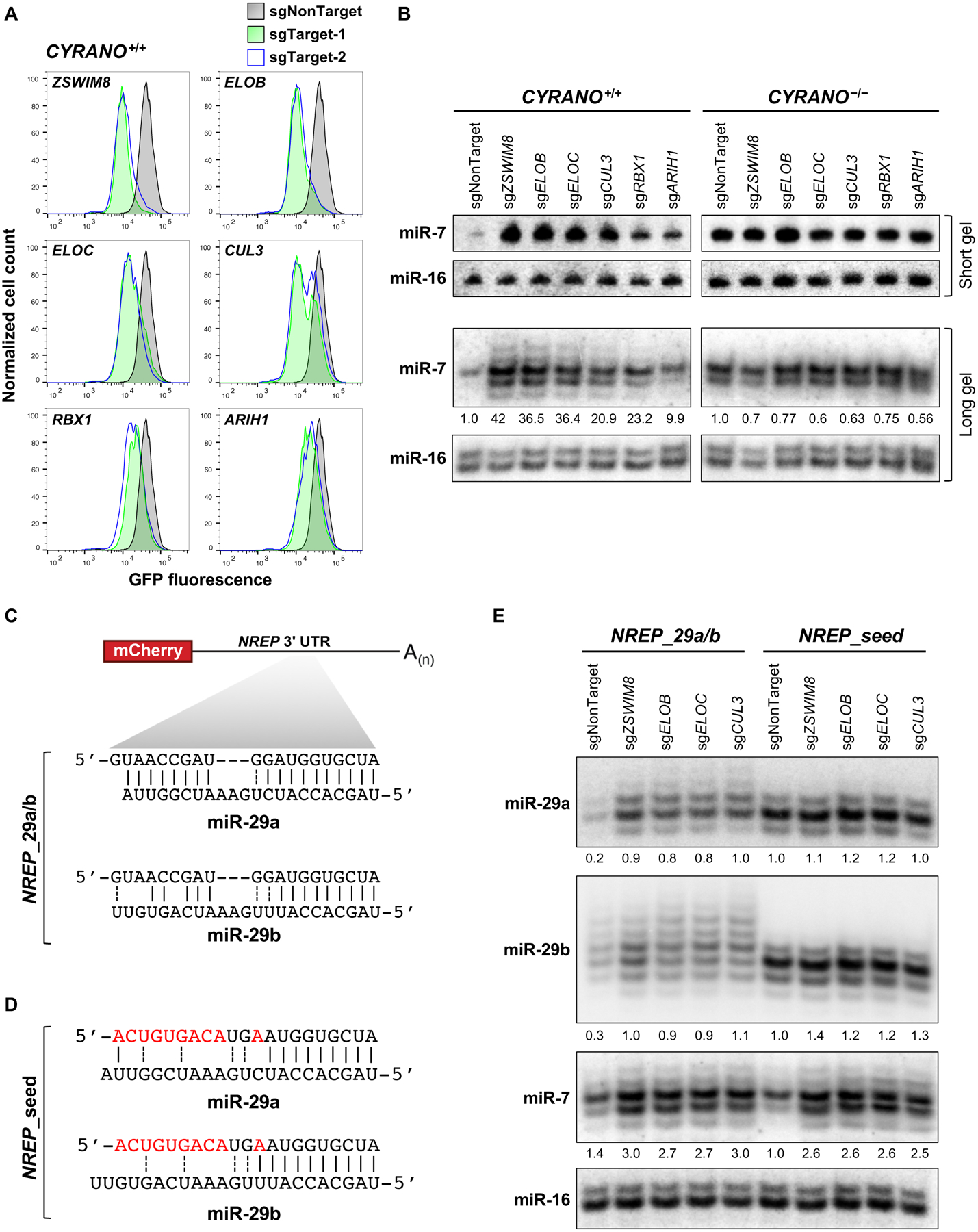
(A) Flow cytometry analysis of EGFP expression in CYRANO+/+ K562 EGFPmiR−7 cells after lentiviral expression of Cas9 and sgRNAs targeting the indicated genes. (B) Northern blot analysis of miRNA expression in CYRANO+/+ and CYRANO−/− K562 cells after expression of sgRNAs targeting ZSWIM8 CRL components. (C) Schematic of reprogrammed mCherry-NREP transcript (NREP_29a/b) and its predicted base-pairing with miR-29a (upper) and miR-29b (lower). (D) Predicted base-pairing of the mutant NREP transcript with seed binding only (NREP_seed) with miR-29a and miR-29b. (E) Northern blot analysis of miRNA expression in HCT116 cells expressing NREP transcripts and the indicated sgRNAs. n=3 biological replicates for northern blot and flow cytometry experiments (representative data shown).
Northern blotting confirmed that depletion of individual ZSWIM8 ubiquitin ligase components increased miR-7 levels as much as 40-fold in CYRANO+/+ K562 cells, but had no effect on miR-7 abundance in CYRANO−/− cells (Fig. 2B). Similar effects were seen in murine embryonic fibroblasts (MEFs), another cell line with active Cyrano-directed decay of miR-7 (7) (Fig. S2A). Expression of V5-ZSWIM8 rescued TDMD of miR-7 in ZSWIM8−/− K562 cells (Fig. S2B). Effects seen upon knockout of the ZSWIM8 complex were not attributable to decreased CYRANO levels (Fig. S3A) or increased biogenesis of miR-7, since pre-miR-7 and miR-7-3p (the miR-7 passenger strand) were not upregulated in knockout cells (Fig. S3B–C).
Northern blot analysis of miR-7 with single-nucleotide resolution revealed a CYRANO-dependent accumulation of tailed and trimmed isoforms in cells lacking the ZSWIM8 CRL (Fig. 2B, long gel). Small RNA sequencing further documented an increase in miR-7 tailing and trimming in ZSWIM8−/− cells and, as reported previously (10), a loss of tailing and trimming of miR-7 in CYRANO−/− cells (Fig. S4). These results agree with prior data indicating that engagement of TDMD-inducing targets stimulates miRNA tailing and trimming (11, 16). However, these data also demonstrate that tailing and trimming is not sufficient for TDMD, and additional steps are required to complete a cycle of miRNA degradation via this mechanism.
The ZSWIM8 ubiquitin ligase mediates TDMD of miR-29
We next tested whether the ZSWIM8 ubiquitin ligase functions specifically in TDMD induced by CYRANO, or was required for TDMD stimulated by other targets. Nrep is a protein-coding gene that harbors a miR-29b binding site in its 3’ UTR that triggers decay of this miRNA in mouse cerebellum (8). To study TDMD mediated by this transcript, we cloned the 3’ UTR of human NREP downstream of mCherry. Additionally, we mutated the miR-29 binding site to increase the 3’ complementarity to miR-29a (NREP_29a/b) (Fig. 2C), which is more highly expressed than miR-29b in HCT116, the cell line used to test this construct due to its low endogenous NREP expression (20) and amenability to genome editing. Expression of this transcript resulted in downregulation of both miR-29a and miR-29b, compared to cells expressing a mutant NREP transcript with complementarity to the miR-29 seed only (NREP_seed) (Fig. 2C–E and Fig. S5A). Depletion of ZSWIM8 CRL components stabilized miR-29a and miR-29b in NREP_29a/b-expressing cells (Fig. 2E and Fig. S5A) without decreasing expression of the mCherry-NREP_29a/b transcript (Fig. S5B) or increasing expression of the corresponding pre-miRNAs or passenger strands (Fig. S5A,D–E). In addition, NREP_29a/b stimulated tailing of miR-29a and miR-29b, and these modified species further accumulated in cells with knockout of ZSWIM8 complex components (Fig. 2E), reinforcing our previous conclusion that 3’ miRNA modification is not sufficient to trigger miRNA decay. Notably, CYRANO and miR-7 are expressed in HCT116 cells, and loss of ZSWIM8 CRL components also stabilized miR-7, but not pre-miR-7 or the miR-7 passenger strand, in this context (Fig. 2E and Fig. S5A,C,F). The requirement for the ZSWIM8 ubquitin ligase in TDMD induced by two unrelated substrates in different cell lines suggests that it functions broadly in this pathway.
The ZSWIM8 CRL interacts with AGO2 and requires proteasome activity for TDMD
Consistent with the genetic requirement for ZSWIM8, ELOB, ELOC, and CUL3 in TDMD, immunoprecipitation of V5-ZSWIM8 demonstrated that these factors form an interacting complex (Fig. 3A). Notably, AGO2 was detectable in this complex, providing a direct link to the miRNA pathway. To further confirm the interaction of the ZSWIM8 complex with AGO2, we employed a proximity labeling assay, in which an enhanced version of biotin ligase, TurboID (TbID), was fused to the C-terminus of ZSWIM8 (31). Biotinylation of AGO2 by V5-ZSWIM8-TbID was clearly detectable in this system (Fig. 3B).
Fig. 3. The ZSWIM8 ubquitin ligase interacts with AGO2 and is required for proteasome-dependent miRNA turnover.
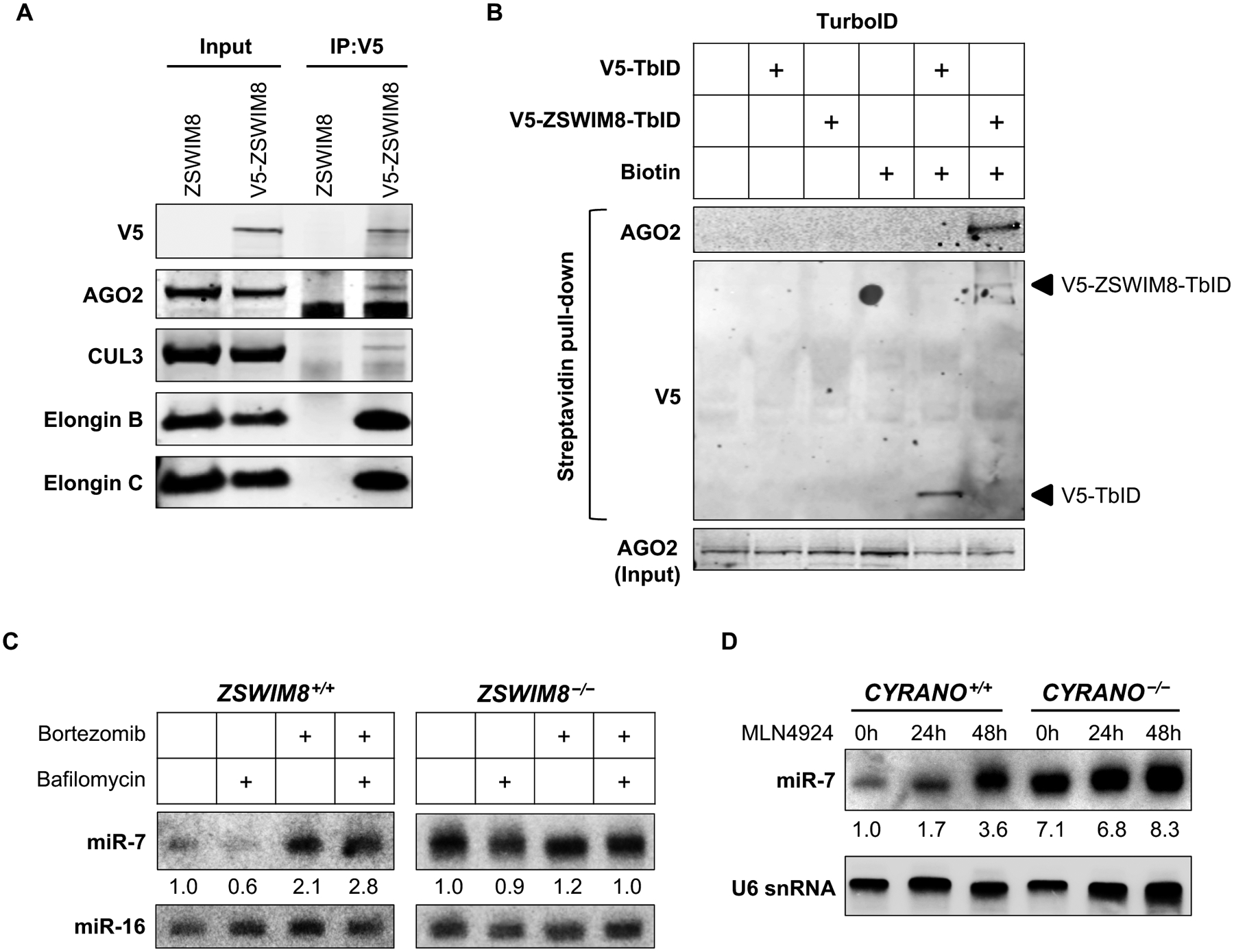
(A) Co-immunoprecipitation of V5-ZSWIM8 with CRL components and AGO2. (B) Western blot analysis of streptavidin pull-downs from cells expressing V5-TbID or V5-ZSWIM8-TbID. (C-D) Northern blot analysis of K562 cells of the indicated genotypes following 24 or 48 hours of treatment with lysosome inhibitor bafilomycin (200 nM) or proteasome inhibitor bortezomib (2 μM) (C) or NEDDylation inhibitor MLN4924 (5 μM) (D). n=3 biological replicates for all experiments (representative results shown).
These data suggest a model in which the ZSWIM8 ubquitin ligase promotes degradation of AGO proteins engaged in a TDMD interaction, leading to release and eventual decay of the targeted miRNA. In support of this model, inhibition of proteasome-, but not lysosome-, mediated decay stabilized miRNAs undergoing TDMD in a ZSWIM8-dependent manner (Fig. 3C). Moreover, since conjugation of the ubiquitin-like modifier NEDD8 to CRLs is necessary for their ubiquitin transfer activity (29), the recovery of several essential NEDDylation factors as significant hits in the CRISPR screen (Fig. 1D) further supported a requirement for ubiquitin transfer in the TDMD mechanism. We confirmed this conclusion by treating with a NEDDylation inhibitor, which abrogated TDMD (Fig. 3D).
Surface-exposed lysines in AGO2 are required for TDMD
Our model that the ZSWIM8 ubiquitin ligase promotes degradation of AGO proteins engaged with TDMD-inducing targets predicts that specific lysines, which serve as acceptors for ubiquitin modification, in AGO should be required for TDMD. To pinpoint such residues, we first identified 25 surface-exposed lysines in human AGO2 that are conserved among all four human AGO proteins as well as in Drosophila Ago1 and C. elegans Alg-1 (Fig. 4A). Reconstitution of AGO2−/− K562 EGFPmiR−7 reporter cells with wild-type AGO2 or an AGO2 mutant with all 25 lysine residues simultaneously mutated to arginine (AGO2KR25) equivalently repressed the EGFP reporter transcript, demonstrating that these residues are not essential for target transcript repression (Fig. S6A). Furthermore, these AGO2 substitutions had no effect on the interaction with ZSWIM8 (Fig. S6B). miR-7 was partially stabilized in AGO2−/− K562 cells reconstituted with AGO2KR25, suggesting that this mutant may be resistant to TDMD (Fig. S6C). We speculated that the presence of endogenous AGO1, AGO3, and AGO4 in these cells may have prevented complete abrogation of TDMD upon expression of mutant AGO2. To circumvent this issue, we introduced AGO2KR25 into previously described HCT116 AGO1/2/3 triple knockout cells (AGO4 is not detectably expressed in this cell line) (32). In these cells, expression of AGO2KR25 stabilized miR-7 to an extent comparable to the level associated with loss of ZSWIM8, and no further stabilization was observed upon ZSWIM8 knockout (Fig. 4B–C and Fig. S6D–G). Thus, conserved, surface exposed lysines in AGO2 are necessary for TDMD.
Fig. 4. Surface-exposed lysines in AGO2 are required for TDMD.
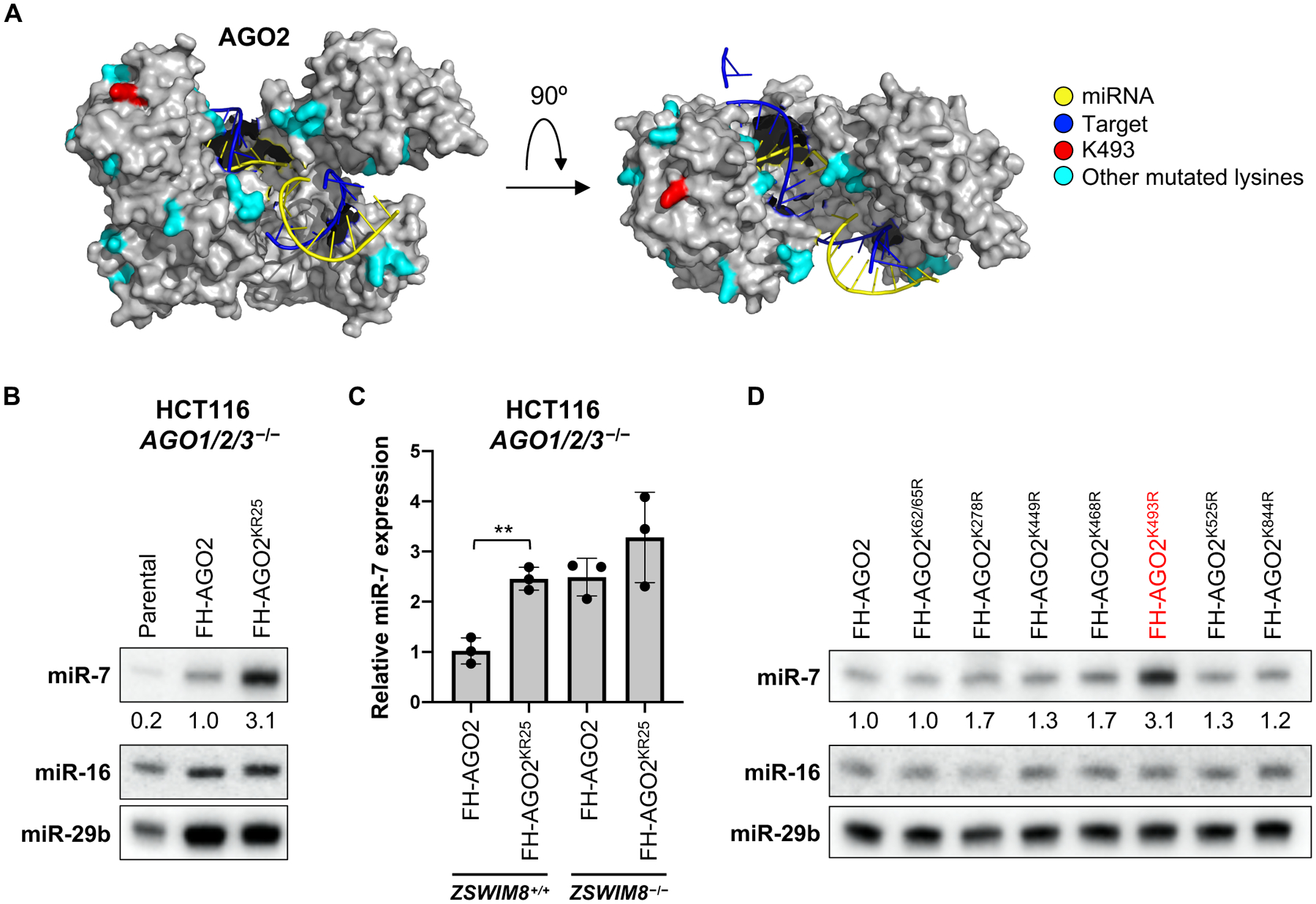
(A) Structure of human AGO2 engaged with a TDMD-inducing target (PDB: 6NIT) (16), with positions of mutated surface exposed lysines highlighted in cyan or red (K493). (B) Northern blot of the indicated miRNAs in HCT116 AGO1/2/3−/− cells reconstituted with wild-type FLAG-HA-tagged AGO2 (FH-AGO2) or FH-AGO2KR25. (C) qRT-PCR analysis of miR-7 levels (normalized to miR-16 levels) in AGO1/2/3−/− cells reconstituted with the indicated FH-AGO2 constructs, with or without knockout of ZSWIM8. Mean +/− SD shown. **p<0.01; Student’s t test. (D) Northern blot of the indicated miRNAs in AGO1/2/3−/− cells reconstituted with wild-type or mutant FH-AGO2. Mutants shown in this panel exhibited a statistically significant increase in miR-7 abundance in qRT-PCR experiments (see Fig. S6H). n=3 biological replicates for all experiments (representative northern blots shown).
To identify the specific lysines that are required for TDMD, we mutated each of the 25 lysines to arginine individually or in pairs and tested their effects on miR-7 levels in AGO1/2/3 triple knockout cells (Fig. S6H). While several individual mutants modestly increased miR-7 abundance, TDMD was strongly impaired upon mutation of K493 (Fig. 4D and Fig. S6H). We confirmed that the AGO2K493R mutant retained the ability to silence the EGFPmiR−7 reporter (Fig. S6A) and interact with ZSWIM8 (Fig. S6B). Thus, K493 of human AGO2 plays a critical role in TDMD and may be an important site of modification by the ZSWIM8 CRL. Further supporting this conclusion, ubiquitylation of AGO2 K493, and the homologous lysine residues in human AGO1 (K491), AGO3 (K494), and AGO4 (K485), has been detected in proteomic surveys of the ubiquitylation landscape in human cells (33–36).
Tailing and trimming is not required for TDMD mediated by the ZSWIM8 ubiquitin ligase
The accumulation of tailed and trimmed intermediates of miRNAs engaged with TDMD substrates in ZSWIM8 knockout cells (Fig. 2B,E and Fig. S4) demonstrated that 3’ miRNA modification is not sufficient to trigger degradation. It remained possible that 3’-modified miRNAs are essential intermediates in the pathway, for example by serving as a signal for recruitment of the ZSWIM8 complex. To determine whether tailing and trimming is necessary for TDMD, we synthesized miR-7 duplexes with or without a 3’-terminal 2’-O-methyl group that blocks 3’ modification (11). Duplexes were introduced into EGFPmiR−7 reporter cells with deletion of the miR-7-1 locus, the major source of endogenous miR-7 in this cell line (Fig. S7A). miR-7 lacking a 2’-O-methyl group initially repressed the EGFPmiR−7 reporter, but fluorescence was recovered over time (Fig. S7B, left). De-repression of the reporter during this time-course was CYRANO and ZSWIM8-dependent, indicating that transfected miR-7 was undergoing TDMD. The 2’-O-methylated miR-7 duplex behaved identically in these assays (Fig. S7B, right), suggesting that 3’ modification is dispensible for TDMD. This conclusion was further substantiated by immunoprecipitating AGO2 and directly examing the fate of transfected AGO2-loaded miR-7. Unmodified miR-7 was stabilized in CYRANO−/− and ZSWIM8−/− cells and tailed species accumulated upon loss of ZSWIM8 (Fig. 5, left). 2’-O-methylated miR-7 also decayed with similar kinetics in a CYRANO and ZSWIM8-dependent manner, despite its inability to undergo tailing and trimming (Fig. 5, right). Thus, 3’ miRNA modification is not an essential step in the degradation of miR-7 by the TDMD pathway.
Fig. 5. miRNA tailing and trimming is not essential for TDMD.
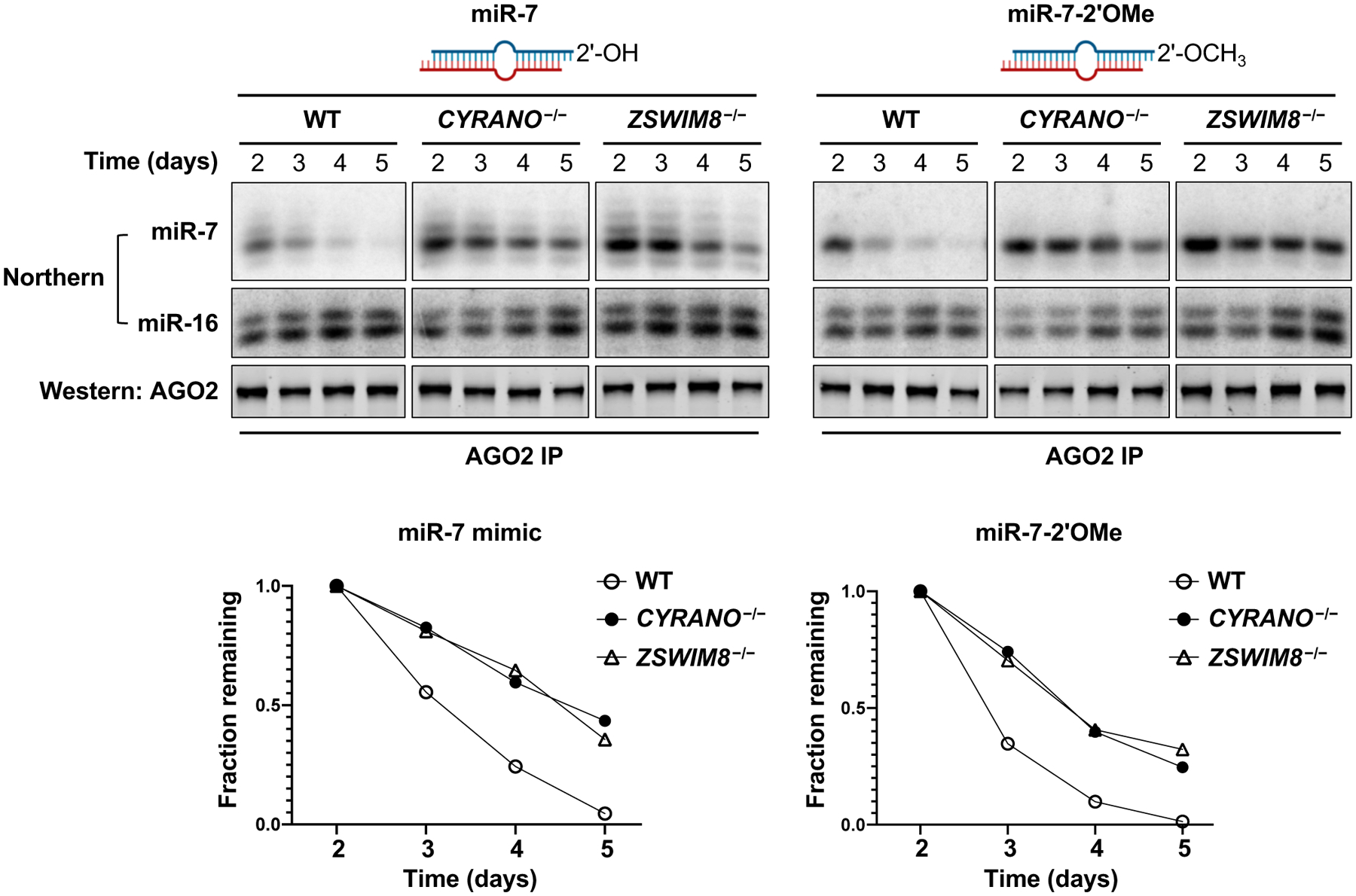
Northern and western blot analyses of AGO2-loaded miRNAs (miR-7, miR-16) and AGO2, respectively, after transfection of the indicated miR-7 duplexes into miR-7–1−/− K562 cells and immunoprecipitation of AGO2 at successive time-points. miR-7 levels were quantified and normalized to miR-16 (bottom panels). Results shown are representative of biological duplicate experiments.
The ZSWIM8 complex regulates miRNA expression in multiple cell types
The discovery of the ZSWIM8 ubquitin ligase complex provided an opportunity to identify additional substrates of the TDMD pathway. As reported previously (10), CYRANO loss of function in K562 cells led to a highly specific increase in miR-7 expression (Fig. 6). Loss of ZSWIM8 in this cell line, however, resulted in the increased expression of several additional miRNAs, without an increase in corresponding passenger strands, which is most consistent with regulation of miRNA stability after AGO loading. Similarly, ZSWIM8 loss of function in HEK293T cells and MEFs resulted in accumulation of miR-7 and several other miRNAs without associated passenger strand upregulation. Together, these data suggest a broad role for the TDMD pathway in shaping the steady-state abundance of the miRNA pool in diverse cell types.
Fig. 6. The ZSWIM8 ubiquitin ligase complex regulates miRNA expression in diverse cell lines.
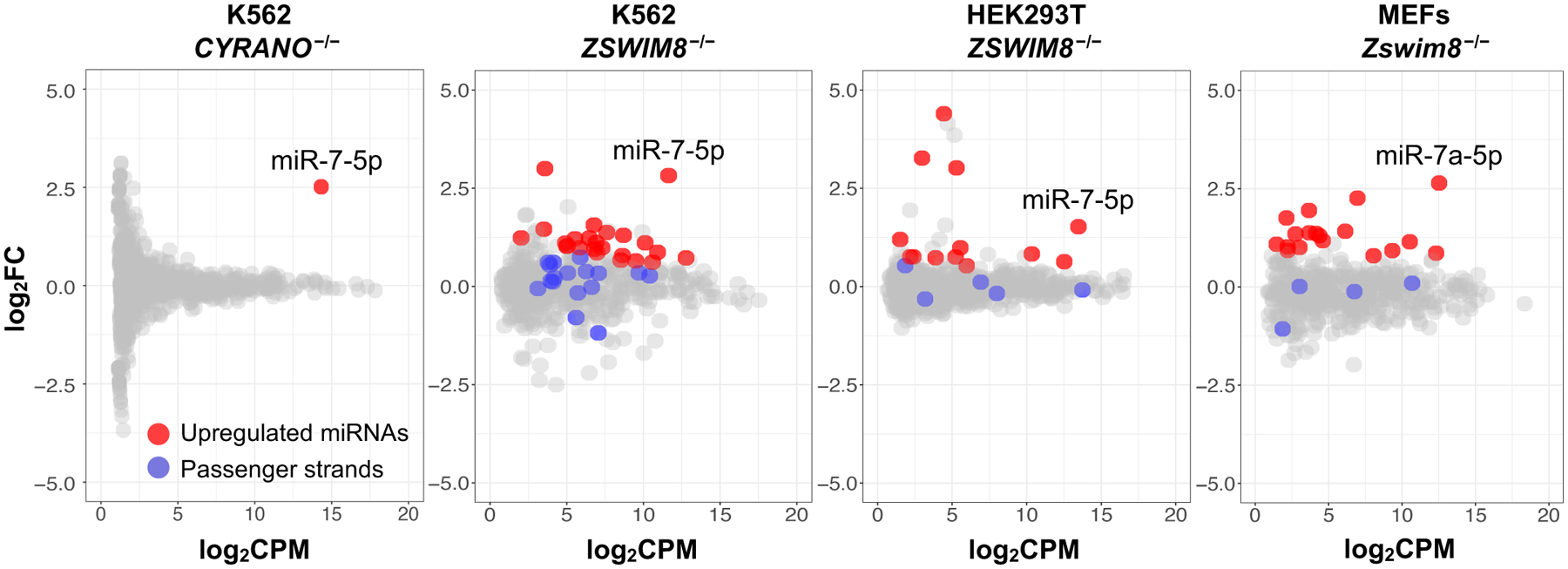
Small RNA sequencing of CYRANO−/− K562, ZSWIM8−/− K562, ZSWIM8−/− HEK293T, or ZSWIM8−/− MEFs, along with associated wild-type cells (n=3 biological replicates per genotype). miR-7–5p is the only significantly upregulated miRNA in CYRANO−/− cells, whereas additional upregulated miRNAs were observed in ZSWIM8−/− cells without an increase in the corresponding passenger strands. FC, fold change; CPM, counts per million.
Discussion
Although TDMD was discovered over ten years ago, the molecular machinery that is responsible for miRNA decay through this pathway has remained elusive. Here we describe the application of an unbiased genetic screening approach to interrogate the mechanism of TDMD, revealing the ZSWIM8 cullin-RING ubiquitin ligase as an essential mediator of this process. This discovery represents an important step towards resolving the molecular basis of miRNA turnover through TDMD and provides new opportunities to study the broad scope and biological role of this mechanism of miRNA regulation.
Base-pairing of a miRNA with a TDMD-inducing transcript promotes broad structural rearrangements of human AGO2, including opening of the central cleft and release of the miRNA 3’ end from the PAZ domain (16). Our data suggest a model in which the ZSWIM8 CRL is able to specifically ubiquitylate AGO in this conformation, possibly along with other associated proteins, leading to the proteasomal degradation of the miRNA-containing complex and release of the miRNA for subsequent decay by cytoplasmic ribonucleases (Fig. 7). Loss of function of components of the ZSWIM8 complex did not result in a consistent increase in expression of TDMD-inducing transcripts. In keeping with these data, loss of miR-7 in mouse brain did not cause upregulation of Cyrano, despite an active TDMD interaction between these RNAs in this setting (10). Likewise, synthetic miRNA targets that induced robust TDMD in rodent primary neuronal cultures were resistant to miRNA-mediated decay (37). Together, these findings suggest that the ZSWIM8 CRL does not induce decay of the target RNA that triggers TDMD. Instead, recognition of the miRNA-containing complex engaged in a TDMD interaction likely results in target release and subsequent recycling for additional rounds of miRNA turnover.
Fig. 7. Proposed model of ZSWIM8 ubquitin ligase activity in TDMD.
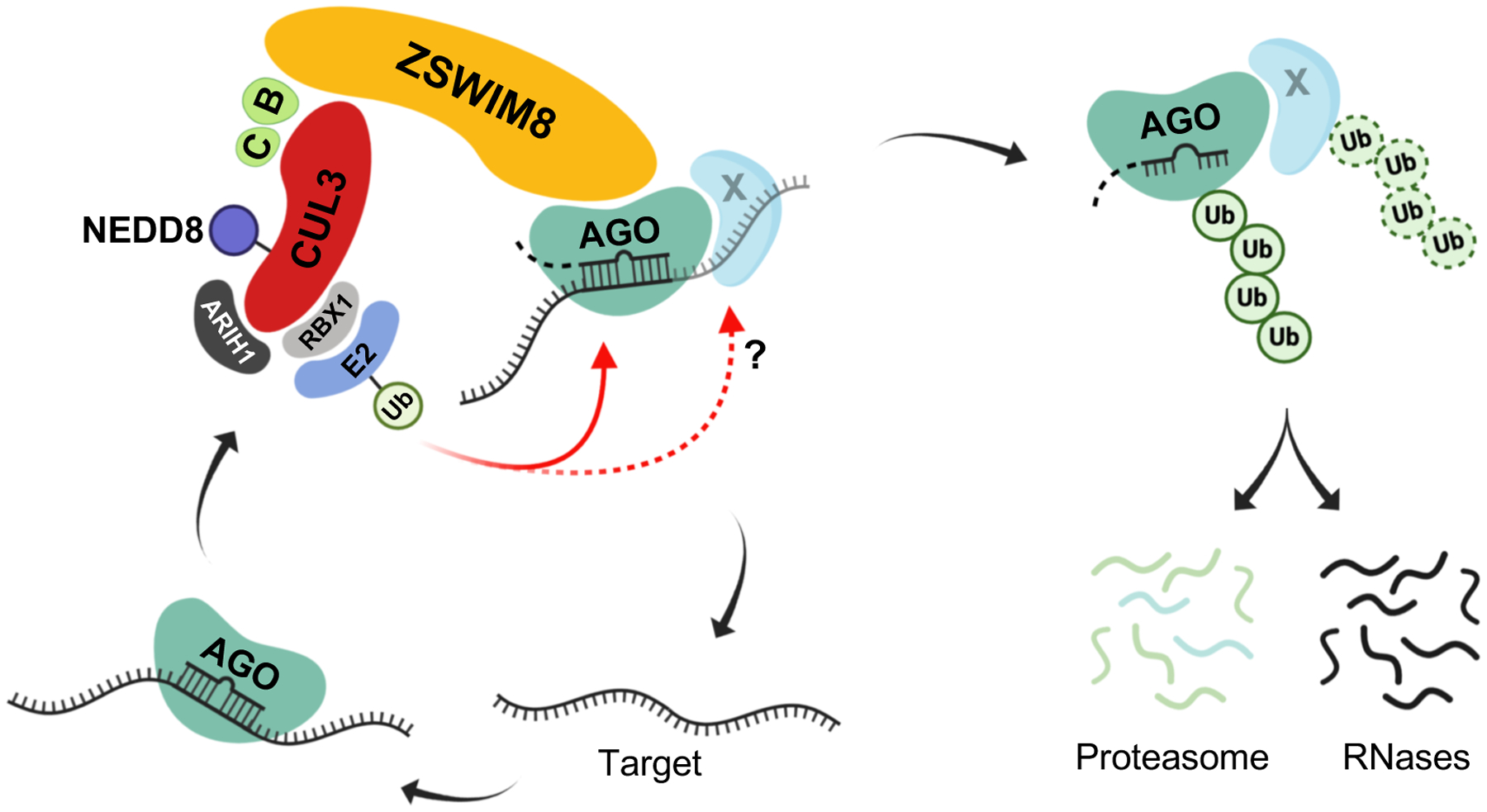
The schematic depicts the ZSWIM8 complex ubiquitylating AGO, and possibly other associated proteins, when they are engaged with a TDMD target, leading to proteasomal degradation of the miRNA-containing complex. Release of the miRNA may lead to its degradation by cytoplasmic RNases while the target transcript is recycled for another round of TDMD. B: Elongin B; C: Elongin C; E2: Ubiquitin-conjugating enzyme; Ub: Ubiquitin.
Although engagement of a miRNA with a TDMD-inducing target is clearly associated with the addition of non-templated nucleotides to the miRNA 3’ end and 3’-to-5’ shortening of the miRNA, a process known as tailing and trimming, there has remained a lack of consensus regarding whether this is an essential step in the TDMD mechanism. The data reported here demonstrated that tailing and trimming of miRNAs is not essential for TDMD mediated by the ZSWIM8 complex. Nevertheless, it remains possible that tailing and trimming may regulate the TDMD pathway or influence its efficiency. For example, 3’ tailing can recruit 3’-to-5’ exonucleases such as DIS3L2 (17, 18), which could assist in disposal of miRNAs after proteasomal targeting by the ZSWIM8 CRL. Tailing and trimming might also destabilize miRNA:target interactions, facilitating release of the target to enable it to participate in additional rounds of TDMD. Notably, miRNAs in plants, as well as short-interfering RNAs (siRNAs) and piwi-interacting RNAs (piRNAs) in flies and other species, are naturally 2’-O-methylated at their 3’ ends (38–40). This modification prevents tailing and trimming when these RNAs engage targets with extensive complementarity, which may be important for maintaining their activity and on-target specificity. Our results demonstrating that the ZSWIM8 complex carries out TDMD in a tailing and trimming independent manner raises the possibility that these classes of silencing RNAs may also be subjected to a TDMD-like mechanism under some conditions. In this scenario, distinct ubiquitin ligase complexes may exist for different classes of small RNAs.
Our small RNA sequencing experiments suggested that the CYRANO-miR-7 pair may represent an exceptionally efficient example of TDMD, while the levels of many miRNAs may be fine-tuned by this decay pathway in diverse cell types. The determinants of TDMD efficiency remain to be established and likely include not only the characteristics of the miRNA:TDMD target base-pairing interaction, but also potentially other sequence elements within a TDMD-inducing transcript that are distinct from the miRNA binding site itself. For example, the miR-7 binding site in Cyrano triggers TDMD much more efficiently in the context of the endogenous Cyrano transcript compared to expression of the site in a heterologous construct (10). Similarly, a sequence motif outside of the miRNA binding site is required for TDMD of miR-17 by a human cytomegalovirus-encoded transcript (13). The results reported here suggest that one mechanism through which a TDMD-promoting sequence element could act is by directly or indirectly recruiting the ZSWIM8 ubiquitin ligase, thereby leading to enhanced recognition and/or ubiquitylation of the associated miRNA-containing complex. Investigation of this possibility, along with the further characterization of TDMD-enhancing sequences, may eventually allow improved prediction of TDMD-inducing targets.
ZSWIM8 homologs are present in distantly related species, including C. elegans and Drosophila (25), suggesting a conserved mechanism of miRNA decay. Although TDMD has been demonstrated to robustly regulate specific miRNAs in mammalian tissues in vivo (8, 10), and disruption of a specific TDMD-inducing target:miRNA pair results in behavioral abnormalities in mice (8), the broad role of the TDMD pathway in metazoan development and physiology remains to be determined. The discovery of the machinery that performs TDMD sets the stage for addressing this question in diverse model systems.
Materials and Methods:
Cell culture
Cell lines were obtained from American Type Culture Collection (ATCC). K562 cells were cultured in RPMI (Invitrogen) supplemented with 10% FBS and Antibiotic-Antimycotic solution. HCT116, HEK293T, and MEFs were cultured in DMEM (Invitrogen) supplemented with 10% FBS and Antibiotic-Antimycotic solution. All cells used in this study were tested for mycoplasma contamination and confirmed to be negative.
Generation of K562 EGFPmiR−7 reporter cell lines
An MSCV-EGFP plasmid with 8 perfectly complementary miR-7-binding sites was constructed by iterative cloning of a synthetic DNA oligo with two miR-7 binding sites (sequence provided in Table S2) into the MSCV-EGFP backbone used previously by our laboratory (20). Retrovirus was generated using the plasmid and recipient K562 cells were transduced by spinoculation with 8 μg/mL polybrene (EMD Millipore) for 1 hour at a multiplicity of infection (MOI) of approximately 0.2. EGFP-positive cells were enriched by FACS followed by derivation of clonal lines. To generate K562 CYRANO−/− EGFPmiR−7, two independent K562 EGFPmiR−7 clones were co-transfected with plasmids (px458; Addgene #48138) expressing Cas9 and two different sgRNAs targeting the 4th exon of CYRANO flanking the miR-7 binding site (sequences provided in Table S2). Clonal cell lines were derived and screened for the desired CYRANO deletion.
Generation of HCT116 NREP_29a/b and NREP_seed cell lines
mCherry was PCR amplified from pCAGImC plasmid (Addgene #92015) (20). The 3’ UTR of NREP was PCR amplified from human genomic DNA. The PCR fragments were cloned into pLJM1 (Addgene #19319) (41), after removal of the puromycin resistance gene, using NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs). The QuikChange Lightning Site-Directed Mutagenesis kit (Agilent) was then used to modify the miR-29 binding site to produce pLJM1-mCherry-NREP_29_a/b and pLJM1-mCherry-NREP_seed. All oligonucleotide sequences are provided in Table S2. HCT116 cells were transduced with lentivirus generated from these plasmids followed by enrichment of mCherry-positive cells by FACS and derivation of clonal lines.
CRISPR-Cas9-mediated gene knockout
Knockout pools were generated by transducing cells with lentiCRISPR_v2 vectors encoding Cas9, sgRNAs, and puromycin- or hygromycin-resistance genes as described previously (Addgene #52961 and #91977) (20). Transduced cells were selected in media containing 0.75–1 μg/mL of puromycin or 800–1000 μg/mL hygromycin for 7–10 days before further analysis. To generate clonal ZSWIM8−/− K562 cell lines, cells were transfected with px458 expressing GFP, Cas9, and sgRNAs targeting ZSWIM8. 24 hours after transfection, cells with the highest 10% of GFP expression were enriched by FACS, followed by generation of clonal cell lines which were screened for indels at the targeted locus. sgRNA sequences are provided in Table S2.
Genome-wide CRISPR-Cas9 screening
Transduction:
The Brunello lentiCRISPR_v2 library (Addgene # 73179) (21) was used for genome-wide CRISPR-Cas9 screens. Two biological replicates were performed for each screen. CYRANO+/+ and CYRANO−/− K562 EGFPmiR−7 cells were spun down, resuspended in fresh media with 8 μg/mL polybrene (EMD Millipore), and mixed with the lentiviral library at multiplicity of infection (MOI) of approximately 0.3. Cells were then plated in 6 well plates and spun at 1000g for 2 hours at room temperature followed by resuspension in fresh media. Beginning 48 hours after transduction, cells were selected in 0.75 μg/mL puromycin and grown for 10 more days before FACS. To achieve ~1000X coverage of the CRISPR library, approximately 2.6 × 108 cells were transduced, and at least 8 × 107 cells were maintained after puromycin selection.
Cell sorting:
Cells were washed with PBS and resuspended in PBS supplemented with 3% FBS and 2 mM EDTA at 1.2 × 107 cells/mL. The 0.5% dimmest cells from each cell line were sorted until 4 × 105 sorted cells were obtained per replicate. Cells were pelleted and frozen at −80 °C for genomic DNA extraction. At least 8 × 107 unsorted cells per replicate were also collected and frozen.
Genomic DNA extraction:
Genomic DNA (gDNA) from unsorted cells was extracted using the MasterPure Complete DNA Purification Kit (Lucigen), and gDNA from sorted cells was extracted using a previously described phenol-chloroform extraction method (20).
Next generation sequencing:
For sequencing library prep, two sequential rounds of PCR were performed using Herculase II Fusion DNA polymerase (Agilent) as described previously (20). For the first PCR, 6.6 μg of gDNA from unsorted cells was used per 100 μL PCR reaction, and a total of 80 reactions were performed per replicate to maintain 1000X coverage. For sorted cells, gDNA was divided into two 100 μL PCR reactions. 18 cycles were performed for PCR 1. After pooling PCR 1 reactions, 5 μL was used for the second PCR (9–11 cycles) with primers containing barcodes and adaptors for Illumina sequencing. Agencourt AMPure XP beads (Beckman Coulter Life Sciences) were used to purify amplicons, which were sequenced on an Illumina NextSeq500 with 75 bp single-end reads. Primer sequences are provided in Table S2.
Sequencing data analysis:
Approximately 4 × 107 sequencing reads were obtained from each replicate. Model-based Analysis of Genome-wide CRISPR/Cas9 Knockout (MAGeCK) was used to identify genes targeted by enriched sgRNAs in sorted populations (42).
Northern blot analysis
Total RNA was isolated using Trizol (Invitrogen). 10–15 μg of total RNA was separated on 15% TBE-Urea polyacrylamide gels. RNAs were transferred to BrightStar-Plus nylon membranes (Invitrogen) followed by UV-crosslinking at 120 mJ/cm2. Membranes were pre-hybridized with ULTRAhyb-Oligo hybridization buffer (Invitrogen) followed by probing with 32P end-labeled oligonucleotide probes. A locked nucleic acid probe was used to detect miR-7 and miR-29b while standard DNA oligonucleotide probes were used for other miRNAs. Probe sequences are provided in Table S2. Densitometry was performed using Quantity One 1-D Analysis software (Bio-Rad), and miR-16 or U6 snRNA was used as a normalization control.
Site-directed mutagenesis and lentiviral expression of AGO2
To generate lysine to arginine substitution mutants of AGO2, site-directed mutagenesis of AGO2 was conducted with the QuikChange Lightning Site-Directed Mutagenesis kit (Agilent), using pLJM1-FH-AGO2 plasmid (20) as a template. Amino acid substitutions and mutagenesis primers are listed in Table S2. For AGO2KR25, the mutant gene fragment was synthesized (GENEWIZ) and cloned into pLJM1. Lentivirus was packaged in HEK293T cells as described previously (20) and K562 AGO2−/− or HCT116 AGO1/2/3−/− cells were transduced with the virus. After infection, cells were selected in media containing puromycin for at least 5 days before analyses.
qRT-PCR assays
cDNA was synthesized using PrimeScript RT (Clontech) and qRT-PCR was performed with SYBR Green Master Mix (Applied Biosystems). TaqMan assays (Applied Biosystems) or miScript PCR system (Qiagen) were used for cDNA synthesis and detection of miRNAs. miR-16 or U6 snRNA was used as a normalizer for all miRNA qRT-PCR measurements while GAPDH was used for all other qRT-PCR normalization. All experiments were performed in biological triplicates. P values were calculated using the Student’s t-test. Primer sequences are provided in Table S2.
Co-immunoprecipitation (Co-IP) assays
ZSWIM8 and V5-ZSWIM8 were PCR amplified from a ZSWIM8 cDNA clone (Transomics) and cloned into a modified AAVS1 hPGK-PuroR-pA donor plasmid (Addgene #22072) for CMV promoter-driven expression (primer sequences in Table S2). For Co-IP, HEK293T cells were transfected with the AAVS1-V5-ZSWIM8 or AAVS1-ZSWIM8 plasmid using FuGENE HD (Promega). 48 hours after transfection, cells were lysed in buffer containing 50 mM Tris-HCl pH 7.5, 100mM NaCl, 1% NP40, and 0.1% sodium deoxycholate. Lysates were cleared by centrifugation at 4 °C for 10 minutes. Protein G Dynabeads were pre-incubated with anti-V5 mouse monoclonal antibody (Invitrogen) for 1 hour and added to lysates for overnight rotation at 4 °C. After washing beads with lysis buffer 5 times, SDS-PAGE sample buffer was added prior to immunoblotting. For testing the interaction of ZSWIM8 with AGO2KR25 or AGO2K493R, AAVS1-V5-ZSWIM8 and FH-AGO2 plasmids were co-transfected into HEK 293T cells, and immunoprecipitation of V5-ZSWIM8 was conducted as described above.
TurboID
V5-TurboID-NES_pCDNA3 was a gift from Alice Ting (Addgene plasmid #107169) (31). V5-TurboID was PCR amplified and cloned into a modified AAVS1 hPGK-PuroR-pA donor plasmid (Addgene #22072) for CMV promoter-driven expression. V5-ZSWIM8 and TurboID were separately PCR amplified and the two amplicons were cloned into AAVS1 hPGK-PuroR-pA donor using the NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs) (primer sequences in Table S2). TurboID plasmids were transiently transfected into biotin-starved HEK293T cells using FuGENE HD (Promega). 48 hours after transfection, Bafilomycin and Bortezomib (Fisher Scientific) were added to final concentrations of 100 nM and 10 nM, respectively. After 2 hours, biotin was added at a final concentration of 500 μM, followed by a 2 hour incubation. The cells were then washed 5 times with ice-cold PBS to stop the reaction and then lysed in RIPA [50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 2mM EDTA, 1% NP-40, 0.5% Sodium deoxycholate, and 0.1% SDS] supplemented with a protease inhibitor tablet (Roche). After clearing by centrifugation, lysates were rotated overnight with Dynabeads MyOne Streptavidin T1 magnetic beads (Invitrogen) at 4°C. Beads were washed as follows (1 mL per wash): 2 times with RIPA, once with 1 M KCl, once with 0.1 M Na2CO3, once with 2 M urea in 10 mM Tris-HCl pH 8, and twice with RIPA. SDS-PAGE sample buffer with 2 mM biotin was used to elute proteins from the beads prior to immunoblotting.
Western blotting and antibodies
NuPAGE 4–12% Bis-Tris protein gels (Thermofisher) were used to separate proteins, which were transferred to nitrocellulose membranes using the iBlot Dry Blotting System (Thermofisher). An infrared fluorescent antibody detection system (LI-COR) was used for protein detection. All antibodies are provided in Table S2.
Treatment of cells with Bortezomib, Bafilomycin, and MLN4924
Bafilomycin, Bortezomib, and MLN4924 were dissolved in DMSO, and cells were treated with DMSO alone, 200 nM Bafilomycin, 2 μM bortezomib, or 5 μM MLN4924 for 24–48 hours before harvesting.
miRNA transfection and immunoprecipitation of AGO2
Approximately 18 × 106 miR-7-1−/−, miR-7-1−/−;CYRANO−/−, or miR-7-1−/−;ZSWIM8−/− EGFPmiR−7 K562 cells were transfected with synthetic unmodified miR-7 duplex or 2’-O-methylated miR-7 duplex (Horizon Discovery) using lipofectamine RNAiMAX reagent (Thermofisher) at a final concentration of 2 nM. 18 × 106 cells were harvested each day for AGO2 immunoprecipitation, and approximately 2 × 105 cells were used for flow cytometry analysis. For AGO2 immunoprecipitation, cells were harvested in 500 μL lysis buffer [25 mM Tris-HCl (pH. 8.0), 150 mM NaCl, 2 mM MgCl2, 0.5% NP-40, and 1 mM DTT] supplemented with a protease inhibitor tablet (cOmplete, EDTA-free, Roche) and 250 U/mL recombinant RNasin Ribonuclease inhibitor (Promega). Cleared lysates were added to 30 μL of Protein G Dynabeads that had been pre-incubated with 4.5 μg anti-AGO2 antibody (Sigma, 11A9). After overnight rotation at 4°C, beads were washed with lysis buffer 5 times before addition of 1 mL Trizol for RNA isolation.
Small RNA-seq
Total RNA was isolated from biological triplicates of each cell line using the miRNeasy Mini RNA isolation kit (Qiagen) including a DNase I digestion step to remove genomic DNA. For all samples except K562 CYRANO+/+ and CYRANO−/− cells, library preparation was performed following a previously published protocol (43) with the following modifications. For each sample, 20 μg of total RNA was combined with 1 μL of 10 nM spike-in control oligos (Bioneer) before size fractionation on a 15% urea-polyacrylamide gel. RNAs were ligated to 3’ randomized adapter using T4 RNA ligase 2 truncated KQ (NEB) supplemented with 20% PEG 8000 (NEB) at 25°C overnight. The PCR-amplified cDNA was gel-purified using an 8% polyacrylamide gel. Library preparation and next generation sequencing for K562 CYRANO+/+ and CYRANO−/− cells was performed by DNA Link using the NEB Next Small RNA Library Prep kit for Illumina (New England Biolabs). Approximately 2 × 107 reads per sample were obtained and the first 18 nt of each read was mapped to mature miRNA sequences downloaded from miRbase (miRbase_v22) as described previously (10, 44). EdgeR was used for differential expression analysis (45), and only miRNAs with a mean RPM > 1 in wild-type cells were used for subsequent analyses.
Supplementary Material
Acknowledgments:
We thank John Doench, Rudolf Jaenisch, David Root, David Sabatini, Alice Ting, and Feng Zhang for plasmids; David Corey for HCT116 AGO1/2/3−/− cells; Ryan Golden for technical advice; Mahmoud Elguindy, Kathryn O’Donnell, and Xiaoquiang Zhu for helpful comments on the manuscript; the McDermott Center Next Generation Sequencing Core for high-throughput sequencing; and the UT Southwestern Flow Cytometry Core facility for FACS.
Funding:
NIH (R35CA197311, P30CA142543, and P50CA196516 to J.T.M.), the Welch Foundation (I-1961-20180324 to J.T.M.), American Heart Association (19POST34380222 to J.H.), and CPRIT (RP150596 to the UTSW Bioinformatics Core). J.T.M. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Competing interests: J.T.M. is a member of the scientific advisory board of Ribometrix
Data and materials availability: All high-throughput sequencing data has been deposited in GEO (accession number GSE151517).
Supplementary Materials:
Figures S1-S7
Tables S1-S2
References and Notes:
- 1.Bartel DP, Metazoan MicroRNAs. Cell 173, 20–51 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gebert LFR, MacRae IJ, Regulation of microRNA function in animals. Nat Rev Mol Cell Biol 20, 21–37 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Treiber T, Treiber N, Meister G, Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat Rev Mol Cell Biol 20, 5–20 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Hwang HW, Wentzel EA, Mendell JT, A hexanucleotide element directs microRNA nuclear import. Science 315, 97–100 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Krol J et al. , Characterizing light-regulated retinal microRNAs reveals rapid turnover as a common property of neuronal microRNAs. Cell 141, 618–631 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Rissland OS, Hong SJ, Bartel DP, MicroRNA destabilization enables dynamic regulation of the miR-16 family in response to cell-cycle changes. Mol Cell 43, 993–1004 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kingston ER, Bartel DP, Global analyses of the dynamics of mammalian microRNA metabolism. Genome Res 29, 1777–1790 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bitetti A et al. , MicroRNA degradation by a conserved target RNA regulates animal behavior. Nat Struct Mol Biol 25, 244–251 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Ghini F et al. , Endogenous transcripts control miRNA levels and activity in mammalian cells by target-directed miRNA degradation. Nat Commun 9, 3119 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kleaveland B, Shi CY, Stefano J, Bartel DP, A Network of Noncoding Regulatory RNAs Acts in the Mammalian Brain. Cell 174, 350–362 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ameres SL et al. , Target RNA-directed trimming and tailing of small silencing RNAs. Science 328, 1534–1539 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cazalla D, Yario T, Steitz JA, Down-regulation of a host microRNA by a Herpesvirus saimiri noncoding RNA. Science 328, 1563–1566 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee S et al. , Selective degradation of host MicroRNAs by an intergenic HCMV noncoding RNA accelerates virus production. Cell Host Microbe 13, 678–690 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Marcinowski L et al. , Degradation of cellular mir-27 by a novel, highly abundant viral transcript is important for efficient virus replication in vivo. PLoS Pathog 8, e1002510 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Libri V et al. , Murine cytomegalovirus encodes a miR-27 inhibitor disguised as a target. Proc Natl Acad Sci U S A 109, 279–284 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheu-Gruttadauria J et al. , Structural Basis for Target-Directed MicroRNA Degradation. Mol Cell 75, 1243–1255 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haas G et al. , Identification of factors involved in target RNA-directed microRNA degradation. Nucleic Acids Res 44, 2873–2887 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang A et al. , AGO-bound mature miRNAs are oligouridylated by TUTs and subsequently degraded by DIS3L2. Nat Commun 11, 2765 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pawlica P, Sheu-Gruttadauria J, MacRae IJ, Steitz JA, How Complementary Targets Expose the microRNA 3’ End for Tailing and Trimming during Target-Directed microRNA Degradation. Cold Spring Harb Symp Quant Biol 84, 179–183 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Golden RJ et al. , An Argonaute phosphorylation cycle promotes microRNA-mediated silencing. Nature 542, 197–202 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doench JG et al. , Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, Vogel G, Yu Z, Richard S, The QKI-5 and QKI-6 RNA binding proteins regulate the expression of microRNA 7 in glial cells. Mol Cell Biol 33, 1233–1243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Higuchi T et al. , Suppression of MicroRNA-7 (miR-7) Biogenesis by Nuclear Factor 90-Nuclear Factor 45 Complex (NF90-NF45) Controls Cell Proliferation in Hepatocellular Carcinoma. J Biol Chem 291, 21074–21084 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen HC, Wang W, Xiong Y, Cullin-RING E3 Ubiquitin Ligases: Bridges to Destruction. Subcell Biochem 83, 323–347 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Z et al. , The EBAX-type Cullin-RING E3 ligase and Hsp90 guard the protein quality of the SAX-3/Robo receptor in developing neurons. Neuron 79, 903–916 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bennett EJ, Rush J, Gygi SP, Harper JW, Dynamics of cullin-RING ubiquitin ligase network revealed by systematic quantitative proteomics. Cell 143, 951–965 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Genschik P, Sumara I, Lechner E, The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): cellular functions and disease implications. EMBO J 32, 2307–2320 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scott DC et al. , Two Distinct Types of E3 Ligases Work in Unison to Regulate Substrate Ubiquitylation. Cell 166, 1198–1214 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merlet J, Burger J, Gomes JE, Pintard L, Regulation of cullin-RING E3 ubiquitin-ligases by neddylation and dimerization. Cell Mol Life Sci 66, 1924–1938 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okumura F, Matsuzaki M, Nakatsukasa K, Kamura T, The Role of Elongin BC-Containing Ubiquitin Ligases. Front Oncol 2, 10 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Branon TC et al. , Efficient proximity labeling in living cells and organisms with TurboID. Nat Biotechnol 36, 880–887 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chu Y et al. , Argonaute binding within 3’-untranslated regions poorly predicts gene repression. Nucleic Acids Res 48, 7439–7453 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oughtred R et al. , The BioGRID interaction database: 2019 update. Nucleic Acids Res 47, D529–D541 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beltrao P et al. , Systematic functional prioritization of protein posttranslational modifications. Cell 150, 413–425 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akimov V et al. , UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat Struct Mol Biol 25, 631–640 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Emanuele MJ et al. , Global identification of modular cullin-RING ligase substrates. Cell 147, 459–474 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de la Mata M et al. , Potent degradation of neuronal miRNAs induced by highly complementary targets. EMBO Rep 16, 500–511 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horwich MD et al. , The Drosophila RNA methyltransferase, DmHen1, modifies germline piRNAs and single-stranded siRNAs in RISC. Curr Biol 17, 1265–1272 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Saito K et al. , Pimet, the Drosophila homolog of HEN1, mediates 2’-O-methylation of Piwi- interacting RNAs at their 3’ ends. Genes Dev 21, 1603–1608 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu B et al. , Methylation as a crucial step in plant microRNA biogenesis. Science 307, 932–935 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sancak Y et al. , Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li W et al. , MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim H et al. , Bias-minimized quantification of microRNA reveals widespread alternative processing and 3’ end modification. Nucleic Acids Res 47, 2630–2640 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kozomara A, Griffiths-Jones S, miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42, D68–73 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robinson MD, McCarthy DJ, Smyth GK, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.