

Abstract
Chemotherapy plays an important role in the treatment of patients with gynecological cancers. Delivering anticancer drugs effectively to tumor cells with just few side effects is key in cancer treatment. Lipid bubbles (LB) are compounds that increase the vascular permeability of the tumor under diagnostic ultrasound (US) exposure and enable the effective transport of drugs to tumor cells. The aim of our study was to establish a novel drug delivery technique for chemotherapy and to identify the most effective anticancer drugs for the bubble US‐mediated drug delivery system (BUS‐DDS) in gynecological cancer treatments. We constructed xenograft models using cervical cancer (HeLa) and uterine endometrial cancer (HEC1B) cell lines. Lipid bubbles were injected i.v., combined with either cisplatin (CDDP), pegylated liposomal doxorubicin (PLD), or bevacizumab, and US was applied to the tumor. We compared the enhanced chemotherapeutic effects of these drugs and determined the optimal drugs for BUS‐DDS. Tumor volume reduction of HeLa and HEC1B xenografts following cisplatin treatment was significantly enhanced by BUS‐DDS. Both CDDP and PLD significantly enhanced the antitumor effects of BUS‐DDS in HeLa tumors; however, volume reduction by BUS‐DDS was insignificant when combined with bevacizumab, a humanized anti‐vascular endothelial growth factor mAb. The BUS‐DDS did not cause any severe adverse events and significantly enhanced the antitumor effects of cytotoxic drugs. The effects of bevacizumab, which were not as dose‐dependent as those of the two drugs used prior, were minimal. Our data suggest that BUS‐DDS technology might help achieve “reinforced targeting” in the treatment of gynecological cancers.
Keywords: chemotherapy, drug delivery system, microbubble, ultrasonography, uterine cervical neoplasm
In vivo application of the bubble ultrasound (US)‐mediated drug delivery system in mouse xenograft tumors. Anticancer drugs were injected with lipid bubbles into the tail vein. The US probe was placed on the tumor and US exposure was simultaneously initiated.

Abbreviations
- BBB
blood‐brain barrier
- BUS‐DDS
bubble US‐mediated drug delivery system
- CCRT
concurrent chemoradiotherapy
- CDDP
cisplatin
- DDS
drug delivery system
- DSPC
1,2‐distearoyl‐sn‐glycero‐3‐phosphocholine
- DSPE‐PEG2000
N‐(carbonyl‐methoxypolyethyleneglycol2000)‐1,2‐distearoyl‐sn‐glycero‐3‐phosphoethanolamine
- DSPG
1,2‐distearoylsn‐glycero‐3‐phosphoglycerol
- EMEM
Eagle’s minimal essential medium
- LB
lipid bubbles
- MB
microbubbles
- MI
mechanical index
- PECAM‐1
platelet‐endothelial cell adhesion molecule‐1
- PLD
pegylated liposomal doxorubicin
- US
ultrasound
- VEGF
vascular endothelial growth factor
1. INTRODUCTION
Cervical and endometrial cancers are major causes of mortality among women, and common treatments for both include surgery, radiation therapy, and chemotherapy, 1 , 2 , 3 and among these, chemotherapy plays an important role. However, anticancer drugs have various side effects, such as bone marrow suppression and body weight loss. Moreover, anticancer drugs often exert less pronounced effects on cancer cells due to the development of interstitial tissues in solid tumors and increase in tumor interstitial fluid pressure. 4 , 5
The combination of US and US contrast agents is considered a novel cancer therapy. Moreover, the US contrast agents generally used for the diagnosis of liver tumors are also known to serve as therapeutic agents. 6 , 7 These agents have played important roles in diagnosis, with several studies reporting findings on contrast agents, such as nanobubbles or MB. Nanobubbles also function as therapeutic agents. 8 , 9 , 10 Although MB are a type of contrast agent, they also function as therapeutic agents when used in the form of drug‐loaded MB. 11 , 12 , 13 , 14
In clinical settings, various types of US contrast agents are used; however, they are usually removed from the bloodstream due to their instability and blood clearance after administration. 15 To overcome this, a novel contrast agent (lipid‐based MB) composed of DSPC, DSPG, and DSPE‐PEG2000 has been developed. 16 The gas loaded in the LB is called perfluoropropane (C3F8), and MB loaded with perfluoropropane are more stable than other contrast agents under US application. Moreover, US contrast agents have been used as diagnostic agents in clinical settings. Under US application, the contrast agents oscillate and/or cavitate.
The combination of MB and US is believed to enhance the permeability of the BBB, 17 , 18 which prevents drugs in the bloodstream from reaching the brain parenchyma. Several types of MB under the application of US were evaluated in terms of their efficiency and safety for crossing the BBB. 19 , 20 , 21 Microbubbles enhanced the transportation of molecules between the brain and can be applied for thrombolysis. 22 , 23 Similar to thrombolysis or transportation across the BBB, this mechanism can potentially be applied to cancer tissues, and was recently applied as a novel drug delivery system for cancer therapy 24 , 25 that could allow anticancer drugs to reach the target tissue effectively and reduce the systemic dose. 26
The application of US for local cancer lesions and the continuous administration of MB and anticancer drugs enables the drugs to reach the target lesion. Some reports suggest that the combination of MB and US therapy has a tumor‐suppressive effect in clinical settings. 27 , 28
Ultrasound is commonly divided into two types: diagnostic US, which is generally used in clinical settings and also enables accurate diagnosis in gynecology, and therapeutic US, which has low frequency and high intensity. High‐intensity focused US was developed for the treatment of prostate cancer, uterine fibroids, and several cancer types. 29 , 30
Although the diagnostic US has a lower intensity compared to therapeutic US, using the diagnostic US, clinicians can visually identify the tumor with reduced effects on normal tissue around the tumor. As a US parameter, the MI is known as an indicator of the cavitation bio‐effect. The MI of US is defined as the peak negative pressure divided by the square root of the center frequency. 31 The MI is generally limited to below 1.9 to avoid excessive effects on the tissue during therapy. 32 A high MI leads to higher cavitation activity. When MI is set as high as possible, it maximizes the effect of US exposure on the MB. Generally, at higher acoustic power (MI > 0.5), a series of rapid and strong oscillations of MB occurs, followed by disruption. 33
Even under low MI, MB oscillate and produce microstreaming, which induces physical effects and opens intercellular junctions. Moreover, under the higher pressure of US, MB produce a push‐pull effect and form transient pores. 34 In a clinical study, it was reported that the administration of MB might enhance the chemotherapeutic effect under the exposure of diagnostic US. 27
In this study, we assessed whether the anticancer effects of chemotherapy on gynecological cancers could be enhanced by the combination of diagnostic US and MB (BUS‐DDS). Chemotherapy often induces various side effects, such as neutropenia, febrile neutropenia, cardiotoxicity, and renal toxicity. These myelosuppressive chemotherapy side effects often lead to reduced dosage or treatment delays in subsequent cycles, adversely affecting treatment outcomes. 35 We hypothesized that BUS‐DDS would maintain anticancer effects even when the dose is reduced.
We assessed the effect of BUS‐DDS by administering various types of anticancer drugs used in gynecological cancer treatment. We employed three types of anticancer drugs commonly used in gynecological cancer treatment: CDDP, PLD, and bevacizumab, for our experiment. Cisplatin is a key drug for adjuvant chemotherapy in both endometrial and cervical cancers. 2 , 3 , 36 , 37 , 38 Pegylated liposomal doxorubicin is a macromolecular drug (80‐90 nm) and has unique characteristics of extended circulation and stable retention. 39 Although in the gynecological clinical setting, PLD is generally known as a recommended therapy for recurrent tumors, it is considered to have a high efficiency in cervical cancer treatment. 40 Bevacizumab is an anti‐VEGF mAb that efficiently treats cervical cancer. 41 , 42
The antitumor activity of BUS‐DDS in gynecological cancers and under exposure to diagnostic US is not known. Moreover, determination of the efficiency of BUS‐DDS depending on the use of various anticancer drugs will improve the understanding of their pharmacokinetics and serve as a novel strategy for gynecological cancer treatment.
2. MATERIALS AND METHODS
2.1. Materials
We purchased DSPC, DSPG, and DSPE‐PEG2000 from NOF Corporation. Perfluoropropane (C3F8) was obtained from Takachiho Chemical Industrial Co. Ltd.
The cervical cancer HeLa cell line and endometrial cancer HEC1B cell line were purchased from ATCC. Cisplatin was purchased from Nichi‐Iko Pharmaceutical Company. Pegylated liposomal doxorubicin was purchased from Mochida Pharmaceutical Co., Ltd. Bevacizumab was obtained from Chugai Pharmaceutical Co., Ltd. Both DMEM and EMEM were purchased from Fujifilm Wako Pure Chemical Corporation. Fetal bovine serum was purchased from Thermo Fisher Scientific. Hematoxylin (Mayer’s hematoxylin; Wako) and 0.5% eosin Y ethanol solution were purchased from FUJIFILM Wako Pure Chemical Corporation. WST‐8 was purchased from Dojindo. Anti‐mouse CD31 Ab and anti‐mouse CD31 (PECAM‐1) were purchased from Dianova. Takara DAB substrate was purchased from Takara Bio Inc. BALB/c nude female mice and C57BL/6 mice were purchased from CLEA Japan, Inc. SP‐10 tubes were purchased from Natsume Seisakusho Co., LTD. Acuson Sequoia 512 (diagnostic US) was purchased from Siemens. An in situ apoptosis detection kit was purchased from Takara Bio Inc. All animal experiments were carried out in accordance with the Guidelines for the Care and Use of Laboratory Animals, as stated by The University of Tokyo.
2.2. Preparation of LB
Lipid bubbles were prepared as described previously. 43 , 44 Briefly, liposomes composed of DSPC, DSPG, and DSPE–PEG(2k)–OMe (30/60/10 [mol/mol/mol]) were prepared using the lipid film hydration method. The liposomes (lipid concentration, 4 mmol/L) were vigorously mixed with perfluoropropane using a homogenizer (Labolution Mark II 2.5; Primix Corporation). The LB suspension was mixed with a sucrose solution (1/1 [v/v]), and the mixture was placed in a 5‐mL vial. The mixtures were freeze‐dried, and the perfluoropropane was injected into the freeze‐drying chamber. After recovering pressure, the vials were sealed, and the freeze‐dried LB were reconstituted with ultrapure water (MilliQ) before each experiment.
2.3. Cell culture
HeLa cell lines were cultured in DMEM supplemented with 10% FBS in humidified air with 5% CO2 at 37°C. HEC1B cells were cultured in EMEM supplemented with 10% FBS in humidified air with 5% CO2 at 37°C.
2.4. In vitro cytotoxicity assay
HeLa or HEC1B cells were seeded into 96‐well plates (5 × 104 cells/mL) and incubated for 24 hours in 0.5 mL DMEM or EMEM supplemented with 10% FBS/penicillin/streptomycin at 37°C with 5% CO2. Cisplatin (0.1, 1.2, 2.5, or 5.0 µmol/L), PLD (0.01, 0.1, 0.5 or 1.0 µmol/L), or bevacizumab (0.001, 0.01, 0.1 or 1.0 µmol/L), and LB (0.5%) were added to the cell suspension, followed by exposure to diagnostic US. For US, the MI was 1.6 and the frequency level was 6 MHz in B‐mode. Thereafter, the cell viability in each cell line was quantitatively evaluated using the MTT assay 72 hours post administration and exposure to US.
The MTT assay was carried out using CCK‐8 with tetrazolium salt WST‐8 (Dojindo, Tokyo, Japan). Proliferation was quantified by monitoring the changes in absorbance using a microplate reader (BioTek Instruments Inc.). 45 , 46
2.5. In vivo antitumor activity assay
Subcutaneous xenograft models were established by transplanting HeLa and HEC1B cells into BALB/c nude mice to evaluate the in vivo antitumor activity of BUS‐DDS. Donor tumors were prepared by s.c. injecting cells (HeLa, 2.0 × 106 cells; HEC1B, 2.0 × 106 cells) into 6‐week‐old female mice and allowing maturation for 2‐3 weeks. When the donor tumors grew up to 7 mm in diameter, they were excised and cut into 2 × 2‐mm pieces. Tumor fragments were s.c. inoculated into the bilateral flank of 6‐8‐week‐old female mice. At 7‐21 days after implantation, the mice were treated with anticancer drugs and LB with or without exposure to US.
Mice were anesthetized with isoflurane, and an i.v. catheter was placed in the tail vein using a 30‐gauge needle and Sp‐10 tube. The US probe was positioned on the tumor for 30 minutes. The transducer was 7v3c (Figure 1A). Pulse intensity of US in B‐mode was estimated at 249 W/cm2. Cisplatin (0.05 or 0.5 mg/kg) was i.v. injected into the mice bearing HEC1B or HeLa tumors, and 40 µL LB was repeatedly injected i.v. every 3 minutes, 10 times. The total dose of LB was 400 µL (Figure 1B).
FIGURE 1.
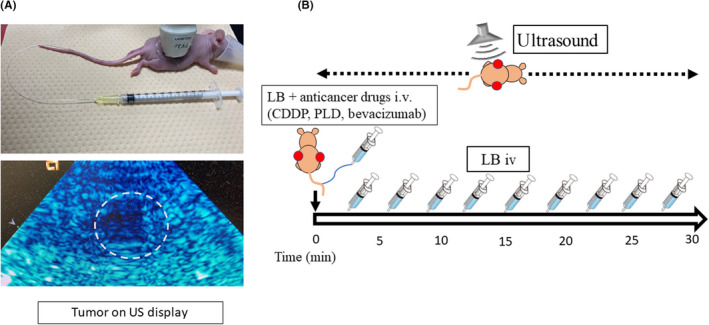
In vivo application of the bubble ultrasound (US)‐mediated drug delivery system in mouse xenograft tumors. (A) Drug injection under diagnostic US application. The mice were anesthetized, and the catheter was placed in the tail vein (top photograph). The US probe was placed on the tumor, which was observed on the US display (dotted circle, bottom photograph). (B) Treatment protocol indicating that anticancer drugs were injected with 40 µL lipid bubbles (LB) from the tail vein. The US probe was placed on the tumor and US exposure was simultaneously initiated. LB (40 µL) were repeatedly injected every 3 min (total dose: 400 µL). The duration of US exposure was 30 min. CDDP, cisplatin; PLD, pegylated liposomal doxorubicin
Pegylated liposomal doxorubicin (0.2 or 2.0 mg/kg) and bevacizumab (2.0 or 20 mg/kg) were given to mice bearing HeLa tumors. All treatment procedures were undertaken on a thermal table at 37°C. Intravenous injection and US application were carried out once on day 0, and tumor sizes were measured two to three times per week. Tumor volume was calculated as 1/2a × b2, where a represents the major axis, and b represents the minor axis.
2.6. In vivo histological damage assay
Application of the BUS‐DDS technology to uterine cancers might affect normal ovary and uterus and neighboring organs, such as the bladder and colon. Therefore, a histological assay was undertaken to evaluate the presence of organ damage. Six‐week‐old C57BL/6 female mice were treated using the method described for the in vivo antitumor activity assay, with 0.5 mg/kg CDDP, 2.0 mg/kg PLD, and 20 mg/kg bevacizumab. The methods of anesthetization and administration of anticancer drugs and LB, as well as the settings for US exposure, were the same as those used in the in vivo antitumor activity assay. The US probe was placed in the lower abdomen of the mice to include the uterus, ovaries, bladder and sigmoid colon. The uterus, ovaries, bladder, and sigmoid colon were excised on day 7 and embedded in paraffin, to evaluate the damage to normal tissue after treatment. Thereafter, sections were deparaffinized, treated with hematoxylin, washed with PBS, and treated with 0.5% eosin Y ethanol solution for 5 minutes at room temperature. 47
2.7. In vivo bone marrow suppression assay
Additionally, we analyzed the blood data of mice treated with BUS‐DDS to observe bone marrow suppression. Ten‐week‐old C57BL/6 female mice were treated using the same method as that described for the in vivo antitumor activity assay with 0.5 mg/kg CDDP, 2.0 mg/kg PLD, and 20 mg/kg bevacizumab. The methods used, including those for anesthetization, administration of anticancer drugs and LB, and the US settings, were identical. Blood sampling was carried out on day 7 from the heart after anesthetization.
2.8. Apoptosis assay
Tumor tissue apoptosis was evaluated using TUNEL staining to assess the antitumor effect of BUS‐DDS. We examined whether the enhanced antitumor effect was associated with the rate of apoptotic tumor cells. Six‐week‐old BALB/c nude female mice were inoculated s.c. with HeLa tumors on the bilateral flank. After 3 weeks, LB and 0.5 mg/kg CDDP or 2.0 mg/kg PLD were given, and mice were exposed to US. The anesthesia method, administration of CDDP, PLD, and LB, and the US settings were the same as those used in the in vivo antitumor activity assay. On day 5, the mice were killed, the tumors were excised, and paraffin embedding was carried out on the excised tumor. Tumor apoptosis was evaluated using TUNEL staining and the In Situ Apoptosis Detection Kit (Takara Bio Inc., Shiga, Japan) was used for coloring apoptotic cells. Paraffin‐embedded tumor sections were deparaffinized, treated with proteinase K for 15 minutes, and washed with PBS. After an endogenous peroxidase block in H2O2 methanol, the sections were treated with TdT enzyme and labeling safe buffer for 60 minutes at 37°C, followed by the administration of anti‐FITC HRP conjugate for 30 minutes at 37°C. Finally, tumors were stained with 3,3‐diaminobenzidine and hematoxylin. 45 The number of stained cells was counted at 400× in three random fields for each tumor.
2.9. Drug biodistribution assay
Platinum concentration was measured to assess CDDP distribution in the tumor. Cisplatin accumulates and can be detected almost 8 hours after administration. 48 , 49 Six‐week‐old BALB/c nude female mice were inoculated s.c. on the bilateral flank with HeLa tumors and treated for 3 weeks. The method of anesthetization, administration of CDDP and LB, and US settings were the same as those used in the in vivo antitumor activity assay, with 0.5 mg/kg CDDP. Mice were killed 4 hours after systemic administration and exposure to US. Thereafter, the tumors before and after treatment were excised. The platinum concentration in the tumor was measured by inductively coupled plasma mass spectrometry (Japan Testing Laboratories Inc).
2.10. Immunohistochemistry of CD31
CD31 PECAM‐1 is an adhesion molecule that is highly expressed in endothelial cells. Anti‐VEGF drugs, such as bevacizumab, inhibit angiogenesis and suppress the expression of CD31. 50 We undertook a quantitative analysis of CD31 to assess the antitumor effect and angiogenesis of bevacizumab under BUS‐DDS. BALB/c nude mice were s.c. injected on the bilateral flank with HeLa tumor cells. The method of anesthetization, systemic administration, and US settings were the same as those used in the in vivo antitumor activity assay. Bevacizumab was given at a dose of 20 mg/kg. Seven days after systemic administration and exposure to US, the mice were killed and tumors were excised. Immunohistochemical localization of CD31 was assessed. Excised tumors were paraffin‐embedded. Thereafter, tumor sections were deparaffinized and antigen retrieval was achieved using citric acid buffer (pH 6) at pressure within a microwave oven. Hydrated paraffin sections were incubated in blocking solution for 30 minutes at room temperature and then at 4°C overnight with CD31 (1:20) and observed using 3,3‐diaminobenzidine as the chromogen. The tumors were counterstained with hematoxylin. Three fields per tumor were observed at 200× magnification for each tumor. Fields were selected from among highly vascularized tumor areas. 51
2.11. Statistical analysis
Data are presented as mean ± SD. Significance was determined by Student’s t test using a two‐tailed distribution and two‐sample unequal variance using the t test function on Microsoft Excel for the positive proportion of TUNEL staining, cisplatin concentration, and immunohistochemistry. Statistically significant changes in tumor volume were determined by repeated measure ANOVA using GraphPad Prism version 5 (GraphPad Software). P < .05 was considered statistically significant.
3. RESULTS
3.1. In vitro anticancer effect of chemotherapy by BUS‐DDS
In the in vitro study, HEC1B and HeLa cell lines were treated with diagnostic US. The cell viability of the “cell only” group was used as the 100% viability standard and described as control. The combination of LB, anticancer drugs (CDDP, PLD, or bevacizumab), and diagnostic US did not affect the viability of HEC1B or HeLa cells at any concentration of the anticancer drugs used (Figure 2). These data indicated that our BUS‐DDS using diagnostic US did not affect toxicity against nearby cells. In order to use diagnostic US for BUS‐DDS, generally in the gynecological clinical setting, it is important for this technology to exert minimal damage to the surrounding cells in the bloodstream.
FIGURE 2.
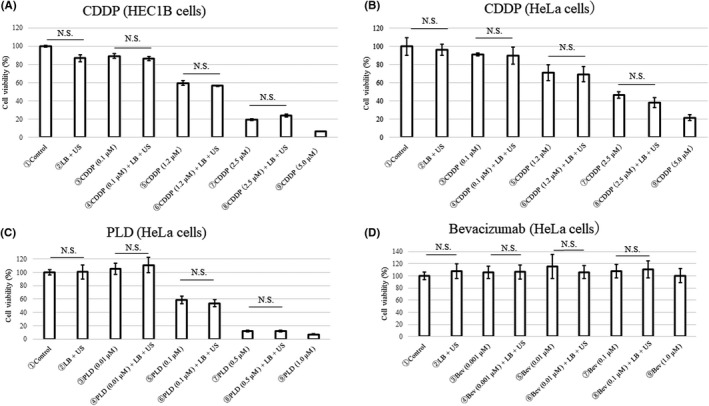
In vitro effect of bubble ultrasound (US)‐mediated drug delivery system. Cytotoxicity on (A) HEC1B cells after cisplatin (CDDP) treatment and on (B) HeLa cells after treatment with CDDP, (C) pegylated liposomal doxorubicin (PLD), or (D) bevacizumab. The viability of each type of cell was quantitatively evaluated by MTT assay 72 h after exposure to the treatments and application of US. The anticancer drugs, lipid bubbles (LB), and US did not enhance the anticancer effects in vitro
3.2. In vivo tumor‐suppressive effect of the combination of LB, US, and anticancer drugs (BUS‐DDS)
Considering the characteristics and strategy, BUS‐DDS technology could potentially be effective for cancer cells when they are in tumor form with microvessels. In order to prove that BUS‐DDS is effective in treating gynecological cancers, especially cervical and endometrial cancers, which are both easily detected by diagnostic US, we applied and validated the enhanced anticancer activity of BUS‐DDS in these cancers.
Mice inoculated with HEC1B tumors were treated with LB and 0.05 mg/kg CDDP with (BUS‐DDS) or without US. The tumor volumes were significantly smaller in the BUS‐DDS group (P = .013). The enhanced antitumor effect of BUS‐DDS was also shown under treatment with 0.5 mg/kg CDDP with (BUS‐DDS) or without US (P = .0151). The efficacy of BUS‐DDS was comparable to treatment with a 10‐fold higher CDDP dosage (Figure 3A). In the same way, in vivo analysis of HeLa tumors treated with 0.05 mg/kg or 0.5 mg/kg CDDP showed that BUS‐DDS significantly enhanced antitumor effects (P = .0013 and P = .0139, respectively; Figure 3B). Therefore, it was shown that BUS‐DDS significantly suppressed the tumor growth of two different gynecological cancer cell lines, regardless of CDDP concentration.
FIGURE 3.
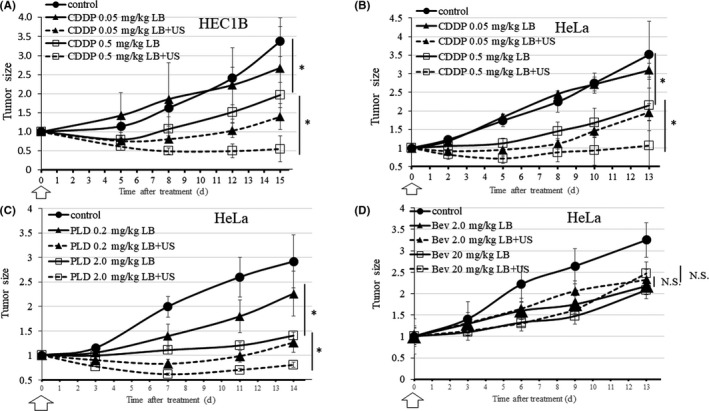
Effects of bubble ultrasound (US)‐mediated drug delivery system (BUS‐DDS) on in vivo antitumor activity of drugs applied to HeLa and HEC1B cells on day zero (white arrow). *P < .05. (A) BUS‐DDS (CDDP), HEC1B. Growth of HEC1B cell xenografts. upon treatment with 0.05 mg/kg and 0.5 mg/kg CDDP under BUS‐DDS (B) BUS‐DDS (CDDP), HeLa. Growth of HeLa cells xenografts upon treatment with 0.05 mg/kg and 0.5 mg/kg CDDP under BUS‐DDS (C) BUS‐DDS (PLD). Growth of HeLa cell xenografts upon treatment with 0.2 mg/kg and 2.0 mg/kg of PLD under BUS‐DDS. (D) BUS‐DDS (bevacizumab). Growth of HeLa cell xenografts upon treatment with 2 mg/kg and 20 mg/kg of bevacizumab under BUS‐DDS
Next, we analyzed whether BUS‐DDS was similarly effective with different types of antitumor drugs using HeLa tumors. Lipid bubbles and PLD were i.v. injected into HeLa tumor‐bearing mice simultaneously under the same procedure with 0.2 mg/kg or 2.0 mg/kg PLD. HeLa tumors treated with LB, PLD, and US (BUS‐DDS) were significantly suppressed compared with those treated without US (P = .0031 and P = .0365, respectively; Figure 3C). From this result, BUS‐DDS was effective not only with micromolecular drugs, such as CDDP, but also with macromolecular drugs designed using a DDS, such as PLD. 39
Furthermore, we examined BUS‐DDS combined with 2.0 mg/kg and 20 mg/kg bevacizumab, a molecular targeting cytostatic drug that affects the microenvironment by trapping VEGF‐A, 52 , 53 using the same procedure. Unlike the results of CDDP and PLD, BUS‐DDS did not show significant tumor suppression (Figure 3D).
3.3. In vivo assessment of local accumulation of anticancer drugs (CDDP concentration, TUNEL staining, immunohistochemistry)
Apoptosis in the HeLa tumor tissue was observed by TUNEL staining in mice 5 days after treatment with a combination of LB, 0.5 mg/kg CDDP or 2.0 mg/kg PLD, and US. In this experiment, the positive rate of apoptotic tumor cells in mice treated with a combination of LB, 0.5 mg/kg CDDP, and US (BUS‐DDS) was significantly higher than that in mice treated with LB and 0.5 mg/kg CDDP alone (non‐US) (P = .0026; Figure 4A). Platinum distribution originating from CDDP was analyzed using the biodistribution assay. The average concentration level of platinum in the BUS‐DDS group was higher than that in the non‐US group (P = .34; Figure 4B). Regarding PLD administration, we found that the positive rate of apoptotic tumor cells in mice treated with a combination of LB, 2.0 mg/kg PLD, and US (BUS‐DDS) was significantly higher than that in mice treated with LB and 2.0 mg/kg PLD alone (non‐US) (P = .0019; Figure 4C). In bevacizumab administration, we compared angiogenesis between BUS‐DDS and non‐US by immunohistostaining of CD31. The quantitative expression of CD31 was comparable between the two groups (Figure 4D). It has been shown that a combination of LB, US, and bevacizumab did not suppress tumor growth compared with administration of LB and bevacizumab alone.
FIGURE 4.
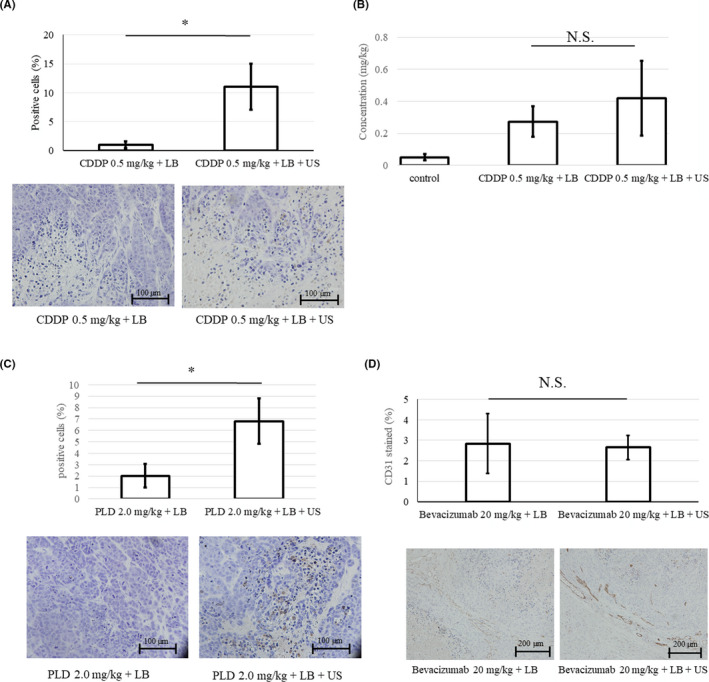
(A) Positive rate of TUNEL staining of HeLa tumor tissue. The rate of TUNEL‐positive cells (%) treated with 0.5 mg/kg cisplatin (CDDP) was significantly increased under the bubble ultrasound (US)‐mediated drug delivery system. (B) CDDP distribution in the tumor. CDDP accumulation was assessed by measuring the concentration of platinum in the tumor tissue (mg/kg). The tumors were excised 4 h after treatment with CDDP 0.5 mg/kg + lipid bubbles (LB) (±US). (C) Rate of TUNEL‐positive staining. Rate of TUNEL‐positive cells (%) treated with 2.0 mg/kg pegylated liposomal doxorubicin (PLD). (D) CD31 staining in the tumor. Immunohistochemical staining of CD31 to assess the density of vascular endothelial cells after treatment with 20 mg/kg bevacizumab + LB (±US). The distribution area of CD31 (%) was comparable between the two groups. *p = 0.86. N.S., not significant
3.4. Assessment of side effects on normal tissue (In vitro cytotoxicity assay of BUS‐DDS with anticancer drugs)
It is already known that there were no fetal cases and no histological or biochemical abnormalities were observed when LB were used. 44 Furthermore, we assessed whether toxicity of anticancer drugs was enhanced by the use of LB and exposure to US by histologically assessing the damage to the organ around the tumor and measuring the complete blood count of the mice 7 days post‐therapy. There were no histological abnormalities, such as hemorrhage or necrosis (Figure 5A). Bone marrow suppression observed between the combination of LB, anticancer drugs (including CDDP, PLD, and bevacizumab), and US (BUS‐DDS) and treatment with LB and anticancer drugs (non‐US) was similar (Figure 5B).
FIGURE 5.
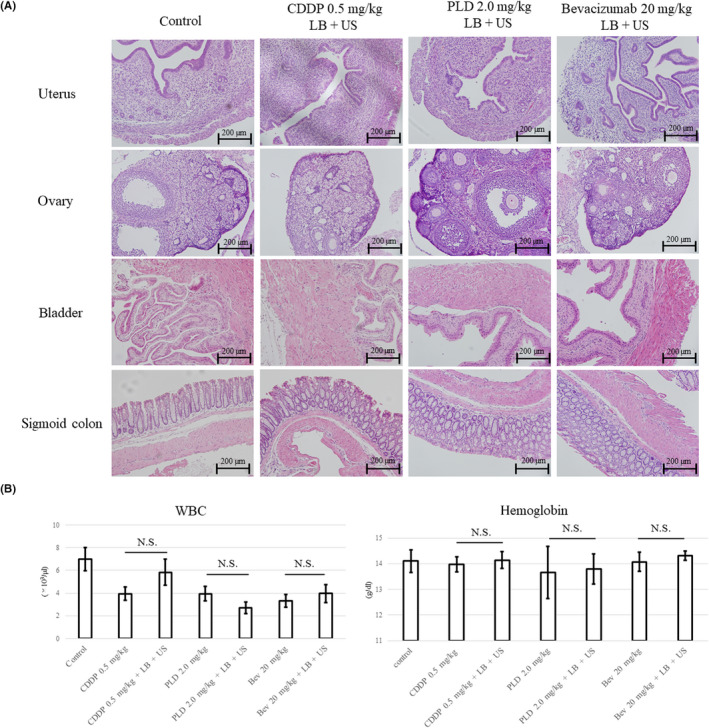
(A) Effects of bubble ultrasound (US)‐mediated drug delivery system (BUS‐DDS) on normal tissue. Anticancer drugs (0.5 mg/kg cisplatin [CDDP], 2.0 mg/kg pegylated liposomal doxorubicin [PLD], or 20 mg/kg bevacizumab) and lipid bubbles (LB) were injected under US exposure to the lower abdomen of the mice. (B) Bone marrow suppression by BUS‐DDS. Assessment of side effects caused by a combination of LB, US, and anticancer drugs. Bone marrow suppression was not observed in mice. N.S., not significant
4. DISCUSSION
In this study, LB were prepared from H8 gas, PEG, and phospholipids, and were observed to enhance the anticancer effects of CDDP and PLD under US exposure.
The treatment with US, which has a low frequency level with high intensity and produces higher energy, is known to enhance the damage to cancer cells in vitro when applied with anticancer drugs. Strong acoustic power from the US treatment induces LB collapse, known as cavitation, and also enables the gene or drugs to be transferred into the cells. 25 This phenomenon indicated that treatment with US could damage blood cells in the bloodstream with LB or surrounding normal tissues and organs. Under the low intensity of diagnostic US, LB were not expected to cavitate. Therefore, LB did not induce direct damage to cancer cells. However, the antitumor effect was enhanced in vivo. Even with the low intensity of diagnostic US, vibrations known as oscillation are induced in the LB. The physical force of the oscillation or stable cavitation would temporarily enhance the permeability of the blood vessel wall of the tumor and enable the anticancer drugs to reach the tumor cells. 34 Both CDDP and PLD are dose‐dependent and cytotoxic drugs. Moreover, PLD is a macromolecular drug that displays characteristic pharmacokinetics of increased circulation. Therefore, it is expected to accumulate in cancers. 39 In this study, although tumor accumulation enhancement of PLD was not clear, an increase of apoptotic cells was observed. It is unknown whether the additional permeability potential of cytotoxic drugs is always proportional to the enhancement of antitumor effects. Therefore, in order to find the ideal combination of drugs for BUS‐DDS, further studies will be needed. In contrast, bevacizumab is a cytostatic drug that affects the microenvironment. We assumed that, although larger amounts of bevacizumab were accumulated in tumor tissue, it did not directly correspond to an anticancer effect.
In conclusion, although we applied diagnostic US to the tumor, treatment with BUS‐DDS resulted in significant growth inhibition of endometrial and cervical cancer tumors. Our data suggest that the combination of LB and US resulted in oscillation and enhanced permeability of the blood vessel wall of the tumor tissue, which led to increased uptake of anticancer drugs. The LB were optimized for treatment by improving their retention in blood flow; therefore, these might serve as a suitable treatment tools. Cisplatin or PLD inhibited tumor growth by BUS‐DDS; in contrast, treatment with bevacizumab did not. The obtained results suggest that BUS‐DDS therapy could be a promising strategy for endometrial or cervical cancer, especially for local lesions. The BUS‐DDS therapy itself does not exert any adverse effects; therefore, it might serve as an appropriate therapy for patients with renal dysfunction or poor general condition. In clinical settings, as CCRT is known as one of the standard treatments for locally advanced cervical cancer, CDDP is given as a radiation sensitizer. But when BUS‐DDS is applied with CCRT, cisplatin could also have antitumor effects or BUS‐DDS could reduce the CDDP dosage (that results in reduction of adverse events). 2 The combination of chemoradiotherapy and BUS‐DDS might enhance the therapeutic effects and improve outcomes in patients with cervical cancer.
DISCLOSURE
The authors declare no conflicts of interest.
ACKNOWLEDGMENTS
This work was supported by MEXT KAKENHI Grant Number 17K11269 (Yoko Matsumoto), a research Grant from Nozawa Memorial Foundation (Yoko Matsumoto), JSPS KAKENHI Grant Numbers JP17K19501 (Ryo Suzuki), JP17H00864 (Ryo Suzuki), JP18K19939 (Kazuo Maruyama), JP16H0319 (Kazuo Maruyama), and MEXT Support Program for the Strategic Research Foundation at Private Universities Grant Number S1311015 (Kazuo Maruyama).
Yamaguchi K, Matsumoto Y, Suzuki R, et al. Enhanced antitumor activity of combined lipid bubble ultrasound and anticancer drugs in gynecological cervical cancers. Cancer Sci. 2021;112:2493–2503. 10.1111/cas.14907
Funding information
MEXT KAKENHI, Grant/Award Number: 17K11269; Nozawa Memorial Foundation; JSPS KAKENHI, Grant/Award Numbers: JP17K19501, JP17H00864, JP18K19939, JP16H0319; MEXT Support Program for the Strategic Research Foundation at Private Universities, Grant/Award Number: S1311015.
REFERENCES
- 1. Cibula D, Pötter R, Planchamp F, et al. The European Society of Gynaecological Oncology/European Society for Radiotherapy and Oncology/European Society of Pathology guidelines for the management of patients with cervical cancer. Int J Gynecol Cancer. 2018;28:641‐655. [DOI] [PubMed] [Google Scholar]
- 2. Koh W‐J, Abu‐Rustum NR, Bean S, et al. Cervical Cancer, version 3.2019, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2019;17:64‐84. [DOI] [PubMed] [Google Scholar]
- 3. Koh W‐J, Abu‐Rustum NR, Bean S, et al. Uterine neoplasms, version 1.2018, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2018;16:170‐199. [DOI] [PubMed] [Google Scholar]
- 4. Jain RK. Delivery of molecular medicine to solid tumors: lessons from in vivo imaging of gene expression and function. J Control Release. 2001;74:7‐25. [DOI] [PubMed] [Google Scholar]
- 5. Hofmann M, McCormack E, Mujic M, et al. Increased plasma colloid osmotic pressure facilitates the uptake of therapeutic macromolecules in a xenograft tumor model 1. Neoplasia. 2009;11:812‐822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brown E, Lindner JR. Ultrasound molecular imaging: principles and applications in cardiovascular medicine. Curr Cardiol Rep. 2019;21:30. [DOI] [PubMed] [Google Scholar]
- 7. Song K‐H, Harvey BK, Borden MA. State‐of‐the‐art of microbubble‐assisted blood‐brain barrier disruption. Theranostics. 2018;8:4393‐4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chandan R, Banerjee R. Pro‐apoptotic liposomes‐nanobubble conjugate synergistic with paclitaxel: a platform for ultrasound responsive image‐guided drug delivery. Sci Rep. 2018;8:2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu H, Rognin NG, Krupka TM, et al. Acoustic characterization and pharmacokinetic analyses of new nanobubble ultrasound contrast agents. Ultrasound Med Biol. 2013;39:2137‐2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suzuki R, Takizawa T, Negishi Y, et al. Gene delivery by combination of novel liposomal bubbles with perfluoropropane and ultrasound. J Control Release. 2007;117:130‐136. [DOI] [PubMed] [Google Scholar]
- 11. Endo‐Takahashi Y, Negishi Y, Nakamura A, et al. Systemic delivery of miR‐126 by miRNA‐loaded bubble liposomes for the treatment of hindlimb ischemia. Sci Rep. 2014;4:3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lentacker I, Geers B, Demeester J, De Smedt SC, Sanders NN. Design and evaluation of doxorubicin‐containing microbubbles for ultrasound‐triggered doxorubicin delivery: cytotoxicity and mechanisms involved. Mol Ther. 2010;18:101‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao Y‐Z, Dai D‐D, Lu C‐T, et al. Using acoustic cavitation to enhance chemotherapy of DOX liposomes: experiment in vitro and in vivo. Drug Dev Ind Pharm. 2012;38:1090‐1098. [DOI] [PubMed] [Google Scholar]
- 14. Tinkov S, Winter G, Coester C, Bekeredjian R. New doxorubicin‐loaded phospholipid microbubbles for targeted tumor therapy: part I—formulation development and in‐vitro characterization. J Control Release. 2010;143:143‐150. [DOI] [PubMed] [Google Scholar]
- 15. Yanagisawa K, Moriyasu F, Miyahara T, Yuki M, Iijima H. Phagocytosis of ultrasound contrast agent microbubbles by Kupffer cells. Ultrasound Med Biol. 2007;33:318‐325. [DOI] [PubMed] [Google Scholar]
- 16. Omata D, Maruyama T, Unga J, et al. Effects of encapsulated gas on stability of lipid‐based microbubbles and ultrasound‐triggered drug delivery. J Control Release. 2019;311–312:65‐73. [DOI] [PubMed] [Google Scholar]
- 17. Aryal M, Arvanitis CD, Alexander PM, McDannold N. Ultrasound‐mediated blood‐brain barrier disruption for targeted drug delivery in the central nervous system. Adv Drug Deliv Rev. 2014;72:94‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hynynen K, McDannold N, Vykhodtseva JFA. Noninvasive MR imaging‐guided focal opening of the blood‐brain barrier in rabbits. Radiology. 2001;220:640‐646. [DOI] [PubMed] [Google Scholar]
- 19. Wang S, Samiotaki G, Olumolade O, Feshitan JA, Konofagou EE. Microbubble type and distribution dependence of focused ultrasound‐induced blood‐brain barrier opening. Ultrasound Med Biol. 2014;40:130‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tung Y‐S, Vlachos F, Feshitan JA, Borden MA, Konofagou EE. The mechanism of interaction between focused ultrasound and microbubbles in blood‐brain barrier opening in mice. J Acoust Soc Am. 2011;130:3059‐3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu S‐Y, Chen CC, Tung Y‐S, Olumolade OO, Konofagou EE. Effects of the microbubble shell physicochemical properties on ultrasound‐mediated drug delivery to the brain. J Control Release. 2015;212:30‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang X, Gkanatsas Y, Palasubramaniam J, et al. Thrombus‐targeted theranostic microbubbles: a new technology towards concurrent rapid ultrasound diagnosis and bleeding‐free fibrinolytic treatment of thrombosis. Theranostics. 2016;6:726‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li H, Lu Y, Sun Y, et al. Diagnostic ultrasound and microbubbles treatment improves outcomes of coronary no‐reflow in canine models by Sonothrombolysis. Crit Care Med. 2018;46:e912‐e920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Feng G, Hao L, Xu C, et al. High‐intensity focused ultrasound‐triggered nanoscale bubble‐generating liposomes for efficient and safe tumor ablation under photoacoustic imaging monitoring. Int J Nanomed. 2017;12:4647‐4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Suzuki R, Oda Y, Omata D, et al. Tumor growth suppression by the combination of nanobubbles and ultrasound. Cancer Sci. 2016;107:217‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tzu‐Yin W, Wilson KE, Machtaler S, Willmann JK. Ultrasound and microbubble guided drug delivery: mechanistic understanding and clinical implications. Curr Pharm Biotechnol. 2013;14:743‐752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dimcevski G, Kotopoulis S, Bjånes T, et al. A human clinical trial using ultrasound and microbubbles to enhance gemcitabine treatment of inoperable pancreatic cancer. J Control Release. 2016;243:172‐181. [DOI] [PubMed] [Google Scholar]
- 28. Kotopoulis S, Dimcevski G, Gilja OH, Hoem D, Postema M. Treatment of human pancreatic cancer using combined ultrasound, microbubbles, and gemcitabine: a clinical case study. Med Phys. 2013;40:072902. [DOI] [PubMed] [Google Scholar]
- 29. Poissonnier L, Chapelon J‐Y, Rouvière O, et al. Control of prostate cancer by transrectal HIFU in 227 patients. Eur Urol. 2007;51:381‐387. [DOI] [PubMed] [Google Scholar]
- 30. Hou R, Wang L, Li S, et al. Pilot study: safety and effectiveness of simple ultrasound‐guided high‐intensity focused ultrasound ablating uterine leiomyoma with a diameter greater than 10 cm. Br J Radiol. 2018;91:20160950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Apfel RE, Holland CK. Gauging the likelihood of cavitation from short‐pulse, low‐duty cycle diagnostic ultrasound. Ultrasound Med Biol. 1991;17:179‐185. [DOI] [PubMed] [Google Scholar]
- 32. Meltzer RS. Food and Drug Administration ultrasound device regulation: the output display standard, the ‘mechanical index’, and ultrasound safety. J Am Soc Echocardiogr. 1996;9:216‐220. [DOI] [PubMed] [Google Scholar]
- 33. Correas JM, Bridal L, Lesavre A, Claudon M, Hélénon O. Ultrasound contrast agents: properties, principles of action, tolerance, and artifacts. Eur Radiol. 2001;11:1316‐1328. [DOI] [PubMed] [Google Scholar]
- 34. Snipstad S, Sulheim E, de Lange DC, et al. Sonopermeation to improve drug delivery to tumors: from fundamental understanding to clinical translation. Expert Opin Drug Deliv. 2018;15:1249‐1261. [DOI] [PubMed] [Google Scholar]
- 35. Crawford J, Dale DC, Kuderer NM, et al. Risk and timing of neutropenic events in adult cancer patients receiving chemotherapy: the results of a prospective nationwide study of oncology practice. J Natl Compr Canc Netw. 2008;6:109‐118. [DOI] [PubMed] [Google Scholar]
- 36. Bestvina CM, Fleming GF. Chemotherapy for endometrial cancer in adjuvant and advanced disease settings. Oncologist. 2016;21:1250‐1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kumar L, Harish P, Malik PS, Khurana S. Chemotherapy and targeted therapy in the management of cervical cancer. Curr Probl Cancer. 2018;42:120‐128. [DOI] [PubMed] [Google Scholar]
- 38. Rose PG, Bundy BN, Watkins EB, et al. Concurrent cisplatin‐based radiotherapy and chemotherapy for locally advanced cervical cancer. N Engl J Med. 1999;340:1144‐1153. [DOI] [PubMed] [Google Scholar]
- 39. Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal doxorubicin: review of animal and human studies. Clin Pharmacokinet. 2003;42:419‐436. [DOI] [PubMed] [Google Scholar]
- 40. Lê LH, Swenerton KD, Elit L, et al. Phase II multicenter open‐label study of carboplatin and pegylated liposomal doxorubicin in uterine and cervical malignancies. Int J Gynecol Cancer. 2005;15:799‐806. [DOI] [PubMed] [Google Scholar]
- 41. Suzuki K, Nagao S, Shibutani T, et al. Phase II trial of paclitaxel, carboplatin, and bevacizumab for advanced or recurrent cervical cancer. Gynecol Oncol. 2019;154:554‐557. [DOI] [PubMed] [Google Scholar]
- 42. Tewari KS, Sill MW, Long HJ, et al. Improved survival with bevacizumab in advanced cervical cancer. N Engl J Med. 2014;370:734‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Suzuki R, Takizawa T, Negishi Y, Utoguchi N, Maruyama K. Effective gene delivery with liposomal bubbles and ultrasound as novel non‐viral system. J Drug Target. 2007;15:531‐537. [DOI] [PubMed] [Google Scholar]
- 44. Unga J, Kageyama S, Suzuki R, Omata D, Maruyama K. Scale‐up production, characterization and toxicity of a freeze‐dried lipid‐stabilized microbubble formulation for ultrasound imaging and therapy. J Liposome Res. 2020;30:297‐304. [DOI] [PubMed] [Google Scholar]
- 45. Makii C, Ikeda Y, Oda K, et al. Anti‐tumor activity of dual inhibition of phosphatidylinositol 3‐kinase and MDM2 against clear cell ovarian carcinoma. Gynecol Oncol. 2019;155:331‐339. [DOI] [PubMed] [Google Scholar]
- 46. Ishiyama M, Miyazono Y, Sasamoto K, Ohkura Y, Ueno K. A highly water‐soluble disulfonated tetrazolium salt as a chromogenic indicator for NADH as well as cell viability. Talanta. 1997;44:1299‐1305. [DOI] [PubMed] [Google Scholar]
- 47. Fischer AH, Jacobson KA, Rose J, Zeller R. Hematoxylin and eosin staining of tissue and cell sections. CSH Protoc. 2008;2008:pdb.prot4986. 10.1101/pdb.prot4986 [DOI] [PubMed] [Google Scholar]
- 48. Nishiyama N, Kato Y, Kataoka K. Cisplatin‐loaded polymer‐metal complex micelle with time‐modulated decaying property as a novel drug delivery system. Pharm Res. 2001;18:1035‐1041. [DOI] [PubMed] [Google Scholar]
- 49. Nishiyama N, Okazaki S, Cabral H, et al. Novel cisplatin‐incorporated polymeric micelles can eradicate solid tumors in mice. Cancer Res. 2003;63:8977‐8983. [PubMed] [Google Scholar]
- 50. McCrudden KW, Hopkins B, Frischer J, et al. Anti‐VEGF antibody in experimental hepatoblastoma: suppression of tumor growth and altered angiogenesis. J Pediatr Surg. 2003;38:308–314; discussion 308‐314. [DOI] [PubMed] [Google Scholar]
- 51. Decio A, Taraboletti G, Patton V, et al. Vascular endothelial growth factor C promotes ovarian carcinoma progression through paracrine and autocrine mechanisms. Am J Pathol. 2014;184:1050‐1061. [DOI] [PubMed] [Google Scholar]
- 52. Ferrara N, Hillan KJ, Gerber H‐P, Novotny W. Discovery and development of bevacizumab, an anti‐VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391‐400. [DOI] [PubMed] [Google Scholar]
- 53. Ferrara N, Hillan KJ, Novotny W. Bevacizumab (Avastin), a humanized anti‐VEGF monoclonal antibody for cancer therapy. Biochem Biophys Res Commun. 2005;333:328‐335. [DOI] [PubMed] [Google Scholar]