

Abstract
Single‐nucleotide polymorphisms (SNP) and long non‐coding RNAs (lncRNAs) have been involved in the process of lung cancer. Following clues given by lung cancer risk‐associated SNP, we aimed to find novel functional lncRNAs as candidate targets in lung cancer. We identified a lncRNA Oxidative Stress Responsive Serine Rich 1 Antisense RNA 1 (OSER1‐AS1) through a lung cancer risk‐associated SNP rs4142441. OSER1‐AS1 was down‐regulated in tumor tissue and its low expression was significantly associated with poor overall survival among non‐smokers in non‐small cell lung cancer (NSCLC) patients. Gain‐ and loss‐of‐function studies showed that OSER1‐AS1 acted as a tumor suppressor by inhibiting lung cancer cell growth, migration and invasion in vitro. Xenograft tumor assays and a metastasis mouse model confirmed that OSER1‐AS1 suppressed tumor growth and metastasis in vivo. The promoter of OSER1‐AS1 was repressed by MYC, and the 3′‐end of OSER1‐AS1 was competitively targeted by microRNA hsa‐miR‐17‐5p and RNA‐binding protein ELAVL1. Our results indicated that OSER1‐AS1 exerted tumor‐suppressive functions by acting as an ELAVL1 decoy to keep it away from its target mRNAs. Our findings characterized OSER1‐AS1 as a new tumor‐suppressive lncRNA in NSCLC, suggesting that OSER1‐AS1 may be suitable as a potential biomarker for prognosis, and a potential target for treatment.
Keywords: ELAVL1, long non‐coding RNA, miR‐17‐5p, non‐small cell lung cancer, OSER1‐AS1
Our results demonstrated that long non‐coding RNA OSER1‐AS1 performed tumor suppressive function by forming an RNA‐protein complex with ELAVL1 and sequester it from binding and stabilizing its target mRNAs, and it may serve as a potential prognosis markers for patients with NSCLC.
![]()
Abbreviations
- ACTB
beta‐actin
- ceRNA
competitive endogenous RNA
- CI
confidence interval
- CPAT
coding potential assessment tool
- CPC
coding potential calculator
- ELAVL1
ELAV‐Like RNA Binding Protein 1
- EMT
epithelial‐mesenchymal transition
- HR
hazard ratio
- HuR
human antigen R
- lncRNA
long non‐coding RNA
- miRNA
microRNA
- MUT
mutant type
- NC
negative control
- NSCLC
non‐small cell lung cancer
- ORF
open reading frame
- OS
overall survival
- qRT‐PCR
quantitative real‐time PCR
- RBP
RNA binding protein
- RIP
RNA immunoprecipitation
- RISC
RNA‐induced silencing complex
- siRNA
small interfering RNA
- SNP
single‐nucleotide polymorphism
- TSS
transcription start site
- WB
western blot
1. INTRODUCTION
Lung cancer is one of the leading causes of cancer‐associated deaths worldwide. 1 During the past few decades, the 5‐year survival rate has only been 15%. 2 NSCLC accounts for 85% of all lung cancer cases. 3 , 4
Long non‐coding RNAs are a new class of potential biomarkers and therapeutic targets for lung cancer. So far, studies have identified various lncRNAs that contribute to cancerous phenotypes such as proliferation, growth suppression, motility, immortality, and angiogenesis in lung cancer. 5 , 6 , 7 , 8 , 9 , 10 However, in the past decade, numerous SNP have been associated with lung cancer risk. 11 Some of these SNP are located near lncRNAs and they may help to highlight the potential lung cancer‐related lncRNAs in their vicinity.
In this study, we first carried out case‐control studies to identify SNP associated with NSCLC risks. To increase the strength of evidence, we integrated information gathered from various bioinformatics platforms and examined the potential prognostic values of lncRNAs at these NSCLC‐associated loci. Next, we carried out functional studies to investigate the underlying molecular mechanisms of these lncRNAs. LncRNAs have been known to function primarily through their interaction with microRNAs, DNA, RNA or RNA‐binding proteins via the competitive endogenous RNA network (ceRNA network). 12 , 13 , 14 Therefore, we particularly focused on investigating the potential ceRNA interactions in the post‐transcriptional gene regulation process.
We now show that a lncRNA Oxidative Stress Responsive Serine Rich 1 Antisense RNA 1 (OSER1‐AS1), near the NSCLC‐associated SNP rs4142441, was differentially expressed between lung cancer and adjacent normal tissues. We examined the effect of OSER1‐AS1 knockdown and overexpression on cell proliferation, migration and invasion in NSCLC cells. We found that OSER1‐AS1 was transcriptionally repressed by MYC at the promoter, and down‐regulated by microRNA hsa‐miR‐17‐5p at the 3′‐end. Interestingly, OSER1‐AS1 may function as a decoy for ELAVL1, or HuR, which was one of the most widely studied regulators of cytoplasmic mRNA stability.
2. MATERIALS AND METHODS
2.1. Selection of SNP and genotyping
We selected 16 SNP located near potential lung cancer‐related lncRNAs following the procedure described in Figure S1. Additional details on the selecting procedure are available in Document S1. Selected SNP (Table S1 ) were used for case‐control analysis.
2.2. Case‐control analysis
New cases diagnosed with NSCLC were collected from Xinqiao Hospital of the Army Medical University (Third Military Medical University) in Chongqing, China. Healthy controls were collected from the annual physical examination group in the same hospital. Additional details about the inclusion/exclusion criteria for cases and controls are described in Document S1. We matched 645 patients on age and gender with 748 healthy controls. Demographic and other risk information was obtained from subjects via a combination of a structured subject interview and medical records. Blood samples were collected from controls and patients prior to any treatment. All subjects provided written informed consent. Research protocol was approved by the ethics committee of the Army Medical University.
2.3. Human tissue samples
Tissue specimens were collected from lung cancer patients prior to any radiation or chemotherapy during operation. The freshly frozen lung tumors and matched normal lung tissues were sectioned and reviewed by a pathologist to confirm the diagnosis of lung cancer, histological grade, tumor purity, and lack of tumor contamination in the normal lung. Tumor samples with ≥70% tumor‐cell content and matched normal lung tissues were used in the study.
2.4. Cell culture and treatment
The lung cancer cell lines A549, SPCA1, and H1299 were obtained from the Cell Bank of the Chinese Academy of Science (Shanghai, China) and the ATCC (Manassas, VA, USA), cultured in RPMI‐1640 (HyClone, Logan, UT, USA) supplemented with 10% FBS (HyClone).
2.5. qRT‐PCR analysis
Total RNA was extracted using the TRIzol reagent (TaKaRa, Dalian, China). The cDNAs were amplified using PrimeScript RT reagent Kit with gDNA Eraser (TaKaRa). Real‐time RT‐PCR assay was performed using a SYBR PrimeScript RT‐PCR kit according to the standard manufacturer’s instructions (TaKaRa). Results were normalized to the housekeeping gene β‐actin. Nuclear and cytoplasmic RNA were isolated using PARIS Kit (Invitrogen, NY, USA). Primer sequences are provided in Table S2.
2.6. RNA fluorescence in situ hybridization
Cy3‐labeled OSER1‐AS1 probes were synthesized by GenePharma (Shanghai, China). RNA‐FISH was carried out as described by Zhou et al 15 following the manufacturer’s instructions. U6 snRNA and 18S rRNA probes were purchased from GenePharma (Shanghai, China) and were used as nuclear and cytoplasmic localization control, respectively. Briefly, A549 cells were fixed on coverslips with 3.7% paraformaldehyde for 15 minutes. Cells were washed once with wash buffer A, PBS and buffer C, followed by incubation at 37°C for 30 minutes, permeabilization with 70% ethanol, 85% ethanol and 100% ethanol each for 5 minutes, then incubated with CY3‐labeled antisense oligonucleotide probes with a final concentration of 1.7 μM in hybridization buffer at 37°C overnight. Sequences of FISH probes are shown in Table S2.
2.7. Plasmid construction and cell transfection
To construct plasmids expressing OSER1‐AS1, MYC and ELAVL1, the full‐length human OSER1‐AS1 sequence (Transcript ID: ENST00000442383.1), MYC (Transcript ID: ENST00000621592.7) and ELAVL1 (Transcript ID: ENST00000407627.7) were synthesized and subcloned into the pcDNA3.1 vector (Invitrogen, New York, USA). Stable transfected cells were selected under Geneticin (G418 sulfate) (Sangon, Shanghai, China). OSER1‐AS1 siRNA sequences are shown in Table S2. Hsa‐miR‐17‐5p mimics, inhibitors, negative controls and inhibitor‐controls were purchased from GenePharma (Table S2). To avoid the interference of hsa‐miR‐17‐5p (the reverse complementary sequence of hsa‐miR‐17‐5p), 16 we used single‐strand hsa‐miR‐17‐5p mimics for this study.
2.8. Cell Counting Kit‐8 assay, colony formation assay, cell migration and invasion assays in vitro
Cell Counting Kit‐8 (CCK‐8) assay, colony formation assay, cell migration and invasion assays were carried out as described in Yuan et al, 2016. 17
2.9. Animal experiments in vivo
For in vivo tumorigenicity, 10 male BALB/c‐nude mice (4 weeks old) were randomly divided into two groups, with five mice in each group. Stable transfected A549 cells (5.0 × 106) were injected s.c. into the left flanks of the nude mice (100 µL per mouse). Tumor volume was calculated using the equation V = 0.5 × D × d 2 (V, volume; D, longitudinal diameter; d, latitudinal diameter). We observed tumor growth for 5 weeks. For the metastasis model, 10 male BALB/c mice (4 weeks old) were randomly divided into two groups, with five mice in each group. Stable transfected A549 cells (1 × 106) were injected into their tail veins (100 µL per mouse). The mice were killed 5 weeks after injection and the lungs were removed for further analysis. All experimental animal procedures were approved by the Institutional Animal Care and Use Committee of Third Military Medical University.
2.10. Luciferase reporter assay
To evaluate the miRNA/ELAVL1‐lncRNA interaction by luciferase reporter assay, the hsa‐miR‐17‐5p binding sites of the OSER1‐AS1 3′‐end region were inserted into the Pischeck‐2 vector (Promega), and ELAVL1 was inserted into pcDNA3.1 vector (Sangon). Firefly and Renilla luciferase activities were measured 48 hours after transfection using the Dual‐Luciferase Assay System (Promega). Relative luciferase activity was calculated using Renilla/firefly luciferase activity.
To evaluate the binding of MYC to the promoter of OSER1‐AS1, the OSER1‐AS1 promoter sequence (−1000 bp ~ +1000 bp) was synthesized and subcloned into the luciferase reporter vector pGL3‐basic (Promega), for which two versions were constructed: WT with allele A for rs4142441 and MUT with allele G for rs4142441. The plasmids were co‐transfected with pcDNA3.1‐MYC as well as pRL‐SV40 Renilla luciferase plasmid (Promega) for internal control.
2.11. Western blot
Western blot (WB) was carried out as described previously (Yuan et al, 2016). 17 The antibodies used in this study were rabbit monoclonal to MYC (Abcam Inc., Cambridge, MA, USA) (1:2000; Abcam (ab32072)), rabbit monoclonal to ELAVL1 (1:5000; Abcam (ab200342)), rabbit monoclonal to AGO2 (1:2000; Abcam (ab186733)), rabbit monoclonal to BCL2L11 (1:2000; Abcam (ab32158)), rabbit monoclonal to Histone H3 (1:5000; Abcam (ab176842)), rabbit polyclonal to β‐actin (1:5000; Abcam (ab8227)). Immunohistochemistry was carried out as described in Liu et al 18 using the rabbit monoclonal to Ki‐67 (1:500; Abcam).
2.12. Chromatin immunoprecipitation assay
Chromatin immunoprecipitation assay was performed following the protocol of ChIP Assay Kit (Beyotime Institute of Biotechnology, Jiangsu, China). Briefly, crosslinked chromatin was prepared with 1% formaldehyde for 10 minutes at 37°C and the DNA was shredded to an average length of 200‐1000 bp by sonication. Immunoprecipitation were conducted using rabbit polyclonal to MYC (Abcam) or IgG control. Precipitated DNA was amplified by PCR. Primer sequences are provided in Table S2.
2.13. Electrophoretic mobility shift assay
Electrophoretic mobility shift assay was performed following the protocol of Gel Shift Assay Core System E3050 Kit (Promega, Madison, WI, USA). Biotin‐labeled DNA probes were derived from the 30 nt sequence flanking SNP rs4142441; two unlabeled competitor probes were designed as 21 nt sequence flanking SNP rs4142441 (rs4142441‐A & rs4142441‐G) (Beyotime Institute of Biotechnology, Jiangsu, China). The SP1 Consensus Oligo was used as unlabeled non‐competitor probe. DNA probe sequences are shown in Table S2. Briefly, each unlabeled DNA probe (12 pmol) was incubated with H1299 cell nuclear extracts (12 µg) for 10 minutes at room temperature in a 10 µL reaction mixture, then 60 femtomole of biotin‐labeled DNA probes was added to each reaction, and loaded onto a 6% acrylamide gel (0.75‐mm thick) in 0.5 × TBE buffer and run at 100 V for 1 hour at 4°C. For the supershift assay, anti‐MYC (ab32072) or anti‐POLR2A (ab193468) antibodies were add to the reaction mixture (conc. 5 μg/mL) prior to the addition of biotin‐labeled DNA probes and incubated for 15 minutes at room temperature (Abcam Inc.). Then EMSA gel was transferred to nylon membranes in 0.5 × TBE buffer at 380 mA for 30 minutes at 4°C. Oligos in the nylon membrane were fixed by exposing to UV light (254 nm) for 10 minutes at 15‐cm distance. After 15‐min incubation with streptavidin‐HRP conjugate (1:2000) at room temperature, biotin‐labeled DNA bands were detected by the chemiluminescence method using ChemiDoc MP Imaging System (Bio‐Rad, Hercules, CA, USA).
2.14. RNA‐binding protein immunoprecipitation assay
RNA‐binding protein immunoprecipitation assay was carried out following the protocol of Magna RIP RNA‐Binding Protein Immunoprecipitation Kit (Millipore; Sigma, St Louis, MO, USA). Nuclear and cytoplasmic RNA was separated by using the Membrane, Nuclear and Cytoplasmic Protein Extraction kit (Sangon) with added RNase Inhibitor (Beyotime Institute of Biotechnology) to a final concentration of 1 U/μL. After the lysate was prepared, 100 µL whole/nuclear/cytoplasmic lysate was incubated at 4°C overnight with 50 µL of magnetic beads precoated with 5 µg ELAVL1 antibody or 5 µg of AGO2 antibody. Another 100 μL aliquot of cell lysate was incubated with 50 µL of magnetic beads precoated with 5 μg of IgG antibody as a negative control.
2.15. Bioinformatics analysis for The Cancer Genome Atlas‐Lung Adenocarcinoma data
Additional descriptions of the bioinformatic analysis for The Cancer Genome Atlas Lung Adenocarcinoma (TCGA‐LUAD) data are shown in Document S1.
2.16. Statistical analysis
We performed a multivariate logistic regression analysis on the association of SNP with lung cancer by adjusting for age, gender, and smoking. Cell growth and migration results were evaluated using the two‐tailed Student’s t test. Gene expression analyses were evaluated using the two‐tailed Student’s t test or Mann‐Whitney U test. Pearson correlation was performed to analyze the correlation between miRNA and mRNA expressions. A two‐sided P‐value less than 0.05 was taken as statistically significant. Statistical analyses were carried out using the software STATA version 16.0 (STATA Corp., College Station, TX, USA). and software R (version 3.4.2).
3. RESULTS
3.1. Rs4142441 associated with NSCLC risk in non‐smokers is located in the promoter of OSER1‐AS1
We carried out case‐control analysis for the 16 SNP (Table S1 ) selected through the bioinformatic pipeline as described in Figure S1 in 626 cases and 736 controls. Baseline characteristics of cases and controls are shown in Table S3. We found no significant associations between any of the SNP and lung cancer risks (Table 1). However, after stratifying by smoking status, we identified one significant association in the non‐smoker group at rs4142441 in the promoter region of lncRNA OSER1‐AS1 and lung cancer risk (OR = 1.71; 95% CI: 1.14‐2.58; P = .01) (Table 1).
Table 1.
Association between SNP and lung cancer risks stratified by smoking status
| SNP | Effect/Other allele | All (N = 1347) a | Smoker (N = 735) b | Non‐smoker (N = 612) c | |||
|---|---|---|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | ||
| rs10134980 | C/A | 0.91 (0.75,1.11) | .36 | 0.84 (0.63,1.12) | .23 | 1.05 (0.77,1.45) | .75 |
| rs1059292 | T/C | 0.98 (0.79,1.23) | .89 | 1.18 (0.86,1.61) | .30 | 0.74 (0.51,1.08) | .12 |
| rs11655237 | C/T | 1.08 (0.89,1.31) | .42 | 1.15 (0.88,1.51) | .30 | 0.90 (0.66,1.22) | .48 |
| rs1549334 | G/A | 0.98 (0.82,1.18) | .84 | 1.09 (0.84,1.41) | .52 | 0.79 (0.58,1.07) | .13 |
| rs2239895 | G/C | 1.13 (0.90,1.42) | .28 | 1.06 (0.77,1.48) | .71 | 1.38 (0.95,1.99) | .09 |
| rs2275159 | G/A | 1.02 (0.85,1.24) | .82 | 1.11 (0.85,1.45) | .44 | 0.89 (0.66,1.21) | .45 |
| rs2295441 | T/C | 0.99 (0.84,1.16) | .88 | 0.98 (0.78,1.24) | .87 | 1.04 (0.81,1.35) | .74 |
| rs2302177 | T/G | 1.04 (0.89,1.22) | .61 | 1.10 (0.88,1.37) | .41 | 0.92 (0.70,1.19) | .51 |
| rs2720660 | G/A | 0.96 (0.74,1.24) | .75 | 0.97 (0.63,1.42) | .87 | 0.80 (0.54,1.19) | .28 |
| rs28631713 | G/A | 1.17 (0.95,1.45) | .14 | 1.10 (0.81,1.49) | .55 | 1.24 (0.88,1.73) | .22 |
| rs3093873 | T/C | 1.10 (0.89,1.35) | .38 | 1.12 (0.82,1.52) | .48 | 0.95 (0.69,1.30) | .73 |
| rs3200401 | T/C | 1.10 (0.88,1.38) | .39 | 0.90 (0.66,1.24) | .53 | 1.19 (0.84,1.69) | .34 |
| rs3783358 | G/C | 1.16 (0.98,1.37) | .09 | 1.19 (0.94,1.50) | .15 | 1.12 (0.85,1.47) | .43 |
| rs3788632 | T/C | 0.99 (0.79,1.25) | .94 | 1.13 (0.82,1.56) | .45 | 0.90 (0.60,1.34) | .60 |
| rs3806357 | G/A | 0.96 (0.81,1.15) | .69 | 0.87 (0.67,1.12) | .27 | 1.15 (0.86,1.55) | .35 |
| rs4142441 | G/A | 1.18 (0.91,1.53) | .22 | 0.79 (0.54,1.14) | .21 | 1.71 (1.14,2.58) | .01* |
Abbreviations: CI, confidence interval; N, number of patients; OR, odds ratio; SNP, single‐nucleotide polymorphism.
Logistic regression adjusted for age, gender and smoking status (additive model).
Logistic regression adjusted for age and gender (additive model).
Logistic regression adjusted for age and gender (additive model).
Effective allele vs. other allele, 2‐tailed P‐value < .05.
3.2. Rs4142441 is associated with the expression of OSER1‐AS1 in vitro
To investigate whether rs4142441 was associated with the expression level of OSER1‐AS1, we carried out luciferase reporter assays in H1299 cells. We constructed a luciferase reporter vector containing the promoter region of OSER1‐AS1 (± 1000 bp on both sides of rs4142441). Two versions of the promoter sequences were constructed: rs4142441‐WT (with allele A for rs4142441) or rs4142441‐MUT (with allele G for rs4142441) (Figure 1A). We observed that the expression of firefly luciferase fused to the promoter carrying rs4142441‐MUT(G) was significantly lower than that of firefly luciferase fused to the promoter carrying rs4142441‐WT(A) (Mann‐Whitney U test, P = .0005) (Figure 1B). To further validate the results, we constructed a pcDNA3.1 vector containing the WT and MUT rs4142441 alleles of OSER1‐AS1 promoter sequence and the full‐length cDNA sequence of OSER1‐AS1 (Transcripts ID: ENST00000442383.1) (Figure 1C). We transiently transfected the plasmids into H1299 cells and measured OSER1‐AS1 expression levels using qRT‐PCR. OSER1‐AS1 expression levels were significantly lower with vectors carrying the rs4142441‐MUT(G) allele (Mann‐Whitney U test, P = .0041) (Figure 1D). Taken together, these pieces of evidence indicated that the allele G of rs4142441 was associated with downregulation of OSER1‐AS1.
Figure 1.
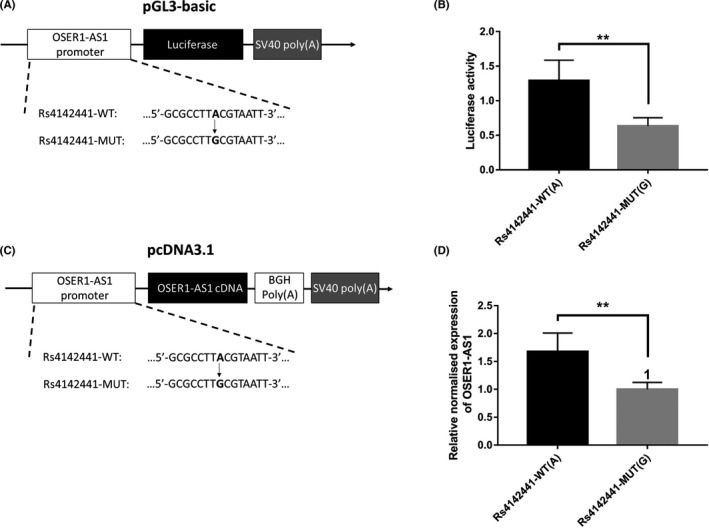
Rs4142441‐MUT is associated with decreased expression of long non‐coding RNA (lncRNA) OSER1‐AS1. A, pGL3 firefly luciferase reporter plasmids with wild‐type or mutant allele for rs4142441. Rs4142441‐WT represents the sequence with the wild‐type allele (A) of rs4142441. Rs4142441‐MUT represents the sequence with the mutant allele (G) of rs4142441. B, The pGL3 firefly luciferase reporter plasmids with wild‐type or mutant allele for rs4142441 were transiently transfected into H1299 cells with a Renilla luciferase reporter for normalization. Luciferase activities were measured after 48 h. Data are presented as the means and standard deviations (SD) of separate transfections (n = 3). Error bars represent the SD. **Mann‐Whitney U test, P < .01. C, The pcDNA3.1 plasmid with its CMV promoter replaced by the promoter region of OSER1‐AS1 (± 1000 bp on both sides of rs4142441). Rs4142441‐WT represents the sequence with the wild‐type allele (A) of rs4142441. Rs4142441‐MUT represents the sequence with the mutant allele (G) of rs4142441. The cDNA sequence of OSER1‐AS1 (transcript ID: ENST00000442383.1) was inserted into the multiple cloning site. D, OSER1‐AS1 expression levels were measured after 48 h of transfection. The relative normalized expression level was calculated as the fold change of the expression level of rs4142441‐WT(A) vector relative to rs4142441‐MUT(G), normalized to the expression level of β‐actin. Data are presented as the means and SD of separate transfections (n = 3). Error bars represent the SD. **Mann‐Whitney U test, P < .01
3.3. OSER1‐AS1 downregulation is associated with poor overall survival in TCGA‐LUAD patients
We measured the expression level of OSER1‐AS1 in 129 paired NSCLC tumor and adjacent normal lung tissues using qRT‐PCR. Clinical characteristics of the 129 NSCLC patients are summarized in Table S4. We first examined the correlation between the expression level of OSER1‐AS1 and clinical characteristics of the NSCLC patients. OSER1‐AS1 was significantly lower among smokers (P = .001, Table S5). Between the paired tumor and adjacent normal tissue, OSER1‐AS1 expression levels were significantly downregulated in NSCLC tumor tissues (P = .0001) (Figure 2A, left panel). After stratifying samples by smoking status, the difference remained significant in the paired tumor and adjacent normal lung tissues from the 90 smokers (P = .0018) (Figure 2A, middle panel) and 39 non‐smokers (P = .0052) (Figure 2A, right panel).
Figure 2.
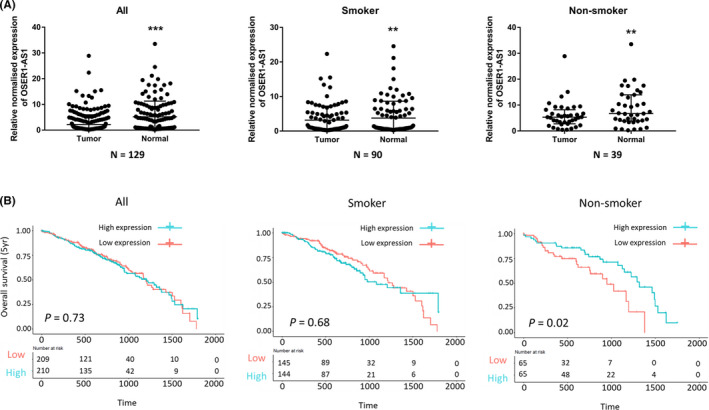
OSER1‐AS1 expression is downregulated in non‐small cell lung cancer (NSCLC) tissues and associated with poor overall survival in The Cancer Genome Atlas Lung Adenocarcinoma (TCGA‐LUAD) patients. A, qRT‐PCR analysis of relative OSER1‐AS1 expression in NSCLC tissues compared with paired adjacent normal lung tissues. Left panel: all groups. Middle panel: smoker group. Right panel: non‐smoker group. The differential expression between tumor and adjacent normal lung tissue was compared using Wilcoxon matched pairs signed rank test: **P < .01; ***P < .001. B, Kaplan‐Meier curves for overall survival (OS) of TCGA‐LUAD patients expressing high and low expression levels of OSER1‐AS1. Left panel: overall group. Middle panel: smoker group. Right panel: non‐smoker group. Difference in survival between high‐ and low‐expression groups (divided by median OSER1‐AS1 expression value) was compared using the log‐rank test
We further assessed the clinical significance of OSER1‐AS1 in NSCLC using the TCGA‐LUAD RNA‐seq dataset. Kaplan‐Meier survival analysis was carried out to investigate the association between OSER1‐AS1 expression and patient outcome. We observed that, in non‐smokers, low expression of OSER1‐AS1 was significantly associated with poor overall survival (n = 130, HR = 0.49 [95% CI 0.26‐0.90]; P = .022) (Figure 2B, right panel). However, we found no significant association of OSER1‐AS1 levels with overall survival in smokers or in the overall group. The association with overall survival in non‐smoker patients remained marginally significant in multivariate Cox proportional‐hazards regression after adjustment for age, gender, and tumor stage (n = 130, HR = 0.560 [95% CI 0.30‐1.04]; P = .066) (Table S6). Collectively, these results showed that the low expression levels of OSER1‐AS1, which were associated with the allele G of rs4142441, were also associated with poor overall survival in TCGA‐LUAD patients among non‐smokers.
3.4. Characterization of OSER1‐AS1
OSER1‐AS1 is located at chromosome 20q13.12 and is coded on the positive strand. It consists of two exons with a full length of 1482 nt (Figure S2A) (Transcript ID: ENST00000442383.1).
We analyzed OSER1‐AS1 expression levels in three NSCLC cell lines (H1299, A549 and SPCA1) by qRT‐PCR, normalized to the expression level of β‐actin. The H1299 cell line showed the highest OSER1‐AS1 expression level, while A549 cell line showed the lowest OSER1‐AS1 expression level (Figure S2B). Therefore, we constructed a knocked down cell model of OSER1‐AS1 expression in the H1299 and SPCA1 cell lines in the following experiments, and constructed a gain‐of‐function cell model by transfecting an OSER1‐AS1‐overexpressing vector into the H1299, SPCA1 and A549 cell lines.
ORFFinder predicted 4 short ORF on the positive strand, which only codes short peptides of 29 to 66 amino acids (Figure S2C) (https://www.ncbi.nlm.nih.gov/orffinder/). However, OSER1‐AS1 lacks a Kozak sequence, which is important for translation initiation. Moreover, OSER1‐AS1 has no coding potential according to Coding Potential Calculator (CPC) (Figure S2D) and Coding Potential Assessment Tool (CPAT) (coding probability [CP] = 0.024) (CP >=0.364 indicates coding sequence, CP <0.364 indicates non‐coding sequence) (Figure S2E).
We further used an RNA‐FISH assay to investigate the subcellular localization of OSER1‐AS1. OSER1‐AS1 was predominantly observed in the cell cytoplasm (Figure S3A). The qRT‐PCR qualification of nuclear and cytoplasmic RNA fractions further validated that OSER1‐AS1 was mainly located in the cytoplasm (cytoplasm : nucleus ratio, 7:1) (Figure S3B).
3.5. OSER1‐AS1 inhibits proliferation, migration, and invasion of NSCLC cells in vitro
To evaluate the potential role of OSER1‐AS1 in NSCLC, we established gain‐of‐function cell models by transfecting pcDNA3.1‐OSER1‐AS1 expressing vectors into the H1299 and SPCA1 cells (Figure S4A). In addition, we transfected H1299 and SPCA1 cells with two siRNAs targeting independent regions of OSER1‐AS1 to investigate the effect of downregulation of OSER1‐AS1 on cell proliferation, migration and invasion (Figure S4B). We examined the effects of OSER1‐AS1 overexpression on cell proliferation, migration and invasion. CCK‐8 assays showed that overexpression of OSER1‐AS1 significantly decreased the proliferation of H1299 and SPCA1 cells (Figure S5A). Moreover, the in vitro Transwell assays showed that overexpression of OSER1‐AS1 significantly decreased the migration and invasion of H1299 and SPCA1 cells compared with vector control (Figure S5C, D). The downregulation of OSER1‐AS1 significantly increased the proliferation, migration and invasion of H1299 and SPCA1 cells compared with vector control (Figure S5B, E, F).
3.6. OSER1‐AS1 represses tumor growth and metastasis in vivo
To investigate whether OSER1‐AS1 suppresses tumorigenesis in vivo, we implanted A549 cells stably transfected with OSER1‐AS1 or control vector s.c. in immunodeficient mice. Tumor mass and volume in the OSER1‐AS1 overexpression group were significantly smaller and lighter than those in the control group (Figure 3A, B and C). To validate the anti‐metastatic effects of OSER1‐AS1 in vivo, A549 cells stably transfected with OSER1‐AS1 or control vector were injected into the tail veins of nude mice. The number of metastatic nodules on the surface of the lung was significantly decreased in mice receiving OSER1‐AS1 stably overexpressing A549 cells compared with the control group (Figure 3D). H&E staining of the mice lung tissue slice confirmed that fewer metastatic nodules were present in the OSER1‐AS1 overexpression group than in the control group. Furthermore, proliferation marker Ki‐67 was evaluated through immunohistochemistry in tumor tissues. Ki‐67 expression levels were also significantly lower in the OSER1‐AS1 overexpression group than those in the control vector group (Figure 3E).
Figure 3.
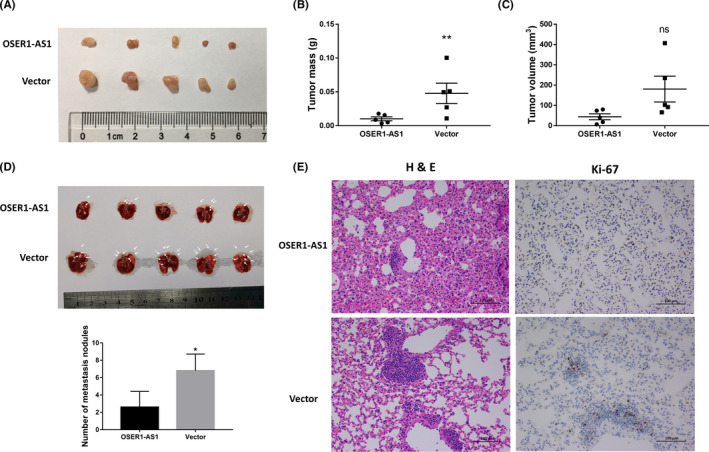
Overexpression of OSER1‐AS1 inhibits lung cancer cell growth and metastasis in vivo. A, Tumors from the OSER1‐AS1 and vector control groups upon resection from BALB/c‐nude mice 5 weeks after s.c. injection with stable transfected A549 cells (5.0 × 106) into the left flanks. B, Tumor weight from the OSER1‐AS1 and vector control groups. Lines represent the mean and SEM of five independent experiments. **, OSER1‐AS1 vs vector control, Mann‐Whitney U test, P < .01. C, Tumor volume was calculated using the equation V = 0.5 × D × d 2 (V, volume; D, longitudinal diameter; d, latitudinal diameter). Lines represent the mean and SEM of five independent experiments. ns (nonsignificant), OSER1‐AS1 vs vector control, Mann‐Whitney U test, P > .05. D, Lungs from nude mice in each group 5 weeks after injection of stable transfected A549 cells (1.0 × 106). Top panel: Arrows point to tumor nodules on the lung surface. Bottom panel: number of lung metastatic nodules on lung surfaces was counted. *OSER1‐AS1 vs vector control, Mann‐Whitney U test, P < .05. E, Left panels: H&E staining of lung tissue slices showed that more metastatic nodules were present in the vector control group than in the OSER1‐AS1 overexpression group. Magnification, ×200. Right panels: immunohistochemical analysis of proliferation marker Ki‐67 shows that Ki‐67 expression levels were significantly lower in the OSER1‐AS1 overexpression group than those in the control vector group
3.7. OSER1‐AS1 promoter is suppressed by MYC
To explore the potential transcription factor that regulates the expression of OSER1‐AS1, we searched the Transcription Factor ChIP‐seq from ENCODE (Txn Factor ChIP) track in the UCSC Genome Browser. We found a binding site for MYC within the OSER1‐AS1 promoter region (−333 bp ~ +116 bp TSS of OSER1‐AS1), covering rs4142441 (Figure 4A). Thus, we then examined the regulation effect of MYC on OSER1‐AS1 expression. By using ChIP experiments, we confirmed direct interaction of MYC to the OSER1‐AS1 promoter using two pairs of primers binding to different positions of the potential binding region of MYC (Figure 4B & C). We next carried out luciferase reporter assays to further investigate the effect of the WT and MUT alleles of rs4142441 on the regulatory efficiency of MYC. The sequences of the OSER1‐AS1 promoter region (−1000 bp ~ +1000 bp) were inserted into the reporter plasmid. Cotransfection of the MYC overexpression vectors with OSER1‐AS1 promoter luciferase construct significantly repressed the luciferase activity. Moreover, the luciferase activity from the promoter carrying rs4142441‐MUT(G) was significantly lower than that of firefly luciferase fused to the promoter carrying rs4142441‐MUT(A), both with or without MYC cotransfection (P < .0001) (Figure 4D ). In addition, we transiently transfected the MYC overexpression plasmids into H1299 cells and measured the OSER1‐AS1 expression levels using qRT‐PCR. The results from qRT‐PCR showed that MYC overexpression vectors significantly decreased the expression of OSER1‐AS1 in comparison to the control vector (P < .05) (Figure 4E ). These lines of evidence demonstrated that MYC suppressed the transcription of OSER1‐AS1, and the allele G of rs4142441 was associated with downregulation of OSER1‐AS1 regardless of the binding status of MYC.
Figure 4.
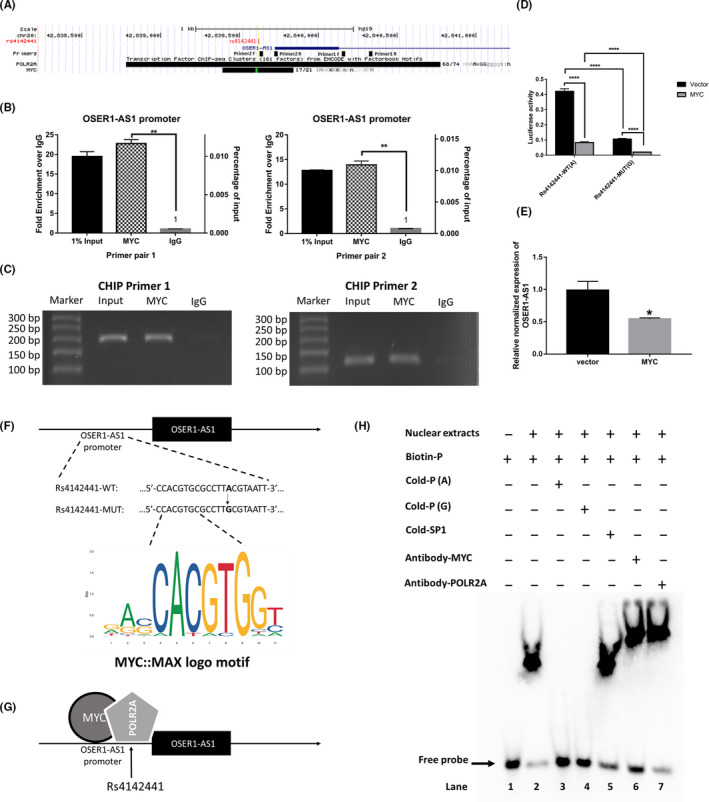
MYC directly binds to the promoter region of long non‐coding RNA (lncRNA) OSER1‐AS1. A, The UCSC Genome Browser representation of the genomic organization of the promoter region of the OSER1‐AS1 gene in the human chromosome (hg19, chr20:42839726‐42854667), where exons are indicated by solid blue boxes and introns by the blue line. Rs4142441 is represented as a vertical red bar. The Primer track shows the binding position of two pairs of primers that were used to detect the promoter sequence of OSER1‐AS1. F primer, forward primer; R primer, reverse primer. The Transcription Factor ChIP‐seq Clusters track shows regions of transcription factor binding derived from a large collection of ChIP‐seq experiments performed by the ENCODE project. The POLR2A box represents the peak cluster of transcription factor occupancy of RNA polymerase II subunit A, whereas the MYC box represents the peak cluster of transcription factor occupancy of MYC, with the darkness of the box being proportional to the maximum signal strength observed in any cell line contributing to the cluster. B, ChIP‐PCR assays were carried out to detect whether MYC directly bound to the promoter of OSER1‐AS1. Left panel: primer pair 1. Right panel: primer pair 2. Error bars represent the SD of three independent experiments. Two‐tailed Student’s t test: **P < .01. C, qRT‐PCR products of the promoter region of OSER1‐AS1. qRT‐PCR was conducted after ChIP, using two pairs of primers amplifying the promoter region of OSER1‐AS1. Input: input control of 1% total DNA without adding any antibody for enrichment. MYC: DNA fragments pulled down by MYC antibody. IgG: DNA fragments pulled down by IgG antibody (used as a negative control). D, Luciferase assays of H1299 cells co‐transfected with pGL3‐basic‐OSER1‐AS1 reporter with rs4142441‐WT(A) or rs4142441‐MUT(G) promoter sequence and MYC overexpression vectors or control vector. E, qRT‐PCR analysis of relative OSER1‐AS1 expression in H1299 cells after transfection with MYC overexpression vector. Data are presented as the means and the standard deviations (SD) of separate transfections (n = 3). Error bars represent the SD. Two‐tailed Student’s t test: *P < .05. F, The MYC binding motif sequence and its position relative to SNP rs4142441 in the promoter region of OSER1‐AS1. G, Schematic drawing summarizing the hypothetical binding mechanism of MYC and POLR2A in the promoter region of OSER1‐AS1. H, EMSA showing the influence of SNP rs4142441 on the binding ability of transcription factors. Biotin‐P: biotin‐labeled DNA probe. Cold‐P (A/G): unlabeled competitor probes (with one nucleotide change at rs4142441 (A/G). Cold‐SP1, unlabeled non‐competitor probes (SP1) used as negative control. Antibody‐MYC, specific antibodies to MYC. Antibody‐POLR2A, specific antibodies to POLR2A
Next, we performed DNA EMSA to further investigate the effect of rs4142441 wild‐type and mutant alleles to the binding affinity of MYC. Biotin‐labeled DNA fragment (30 nt) derived from the flanking sequences of rs4142441 was used as probe. Unlabeled competitor probes with rs4142441(A) or (G) were examined for their ability to compete away the labeled complex (Table S2).
The results showed that the biotin‐labeled probe was able to bind the nuclear extract purified from H1299 cells and cause a mobility shift (lane 2 in Figure 4H), which was abolished by either adding the unlabeled competitors with wild‐type (A) or the mutant (G) alleles of rs4142441 (lanes 3 & 4 in Figure 4H).
Judging by the fact that the position of rs4142441 was 6 bp away from the transcription factor binding motifs (TFBM) of MYC (Figure 4F), it was unlikely for rs4142441 to alter the motif score of MYC. However, we considered that rs4142441 may still interfere with DNA‐protein binding of MYC if MYC binds in complex with other regulatory proteins. We hypothesized that rs4142441 may affect the binding affinity of RNA polymerase2A (POLR2A) (Figure 4G). Therefore, a supershift assay was carried out to identify the specific transcription factors in the DNA‐protein complex by using anti‐MYC and anti‐POLR2A antibodies (lanes 6‐7 in Figure 4H). After the addition of antibodies, the DNA‐protein complex showed extra retardation of mobility, demonstrating that a larger complex formed including the antibodies of MYC and POLR2A. Collectively, the results of the EMSA assay indicated that a DNA‐protein complex of MYC and POLR2A formed at the position of rs4142441. However, there was no detectable change of binding affinity caused by a single‐nucleotide variation.
3.8. OSER1‐AS1 3′‐end is bound by microRNA hsa‐miR‐17‐5p and RNA‐binding protein ELAVL1
Previous studies have reported that MYC could upregulate hsa‐miR‐17‐5p, which in turn mediated MEK inhibitor resistance in AZD6244 treatment in lung cancer cell lines. 19 , 20 , 21 Therefore, we searched the public database (http://starbase.sysu.edu.cn/) to look for the potential binding site of hsa‐miR‐17‐5p in the 3′‐end of OSER1‐AS1. One binding site to hsa‐miR‐17‐5p in the 3′‐end of OSER1‐AS1 was supported by high‐throughput sequencing of RNA isolated by crosslinking immunoprecipitation (HITS‐CLIP) experiment (GEO accession: GSM2065794. Target site (hg19): chr20:42854367‐42854385[+] (Figure S6A).
In addition, up‐ and downstream in close proximity of this hsa‐miR‐17‐5p target site, 4 binding sites of RNA‐binding protein ELAVL1 (ELAV‐like protein 1 or ELAVL1) were reported by independent CLIP‐seq experiments (Figure S6B). ELAVL1 is a widely studied RNA‐binding protein that plays a role in promoting cell proliferation by stabilizing mRNAs of oncogenes involved in cell cycle regulation. 22 Therefore, we next explored the impacts of hsa‐miR‐17‐5p and ELAVL1 binding on the expression levels of OSER1‐AS1. We first investigated the differential luciferase activity of Pischeck‐2 vector containing the OSER1‐AS1 3′‐end region cotransfected with hsa‐miR‐17‐5p mimic, NC mimic, inhibitor and NC‐inhibitor. We found out that hsa‐miR‐17‐5p mimics significantly reduced the luciferase activities of the Pischeck‐2 reporter containing the OSER1‐AS1 3′‐end region, whereas inhibitors of hsa‐miR‐17‐5p caused the opposite effect (Figure S6C). Subsequently, we investigated the expression levels of OSER1‐AS1 after transfection with hsa‐miR‐17‐5p mimic, NC mimic, inhibitor and NC‐inhibitor. The miR‐17‐5p inhibitors significantly increased OSER1‐AS1 expression levels, whereas the miR‐17‐5p mimics significantly decreased OSER1‐AS1 in H1299 cells compared with control (Figure S6D). In contrast, after overexpression or knockdown of OSER1‐AS1 in H1299 cells, miR‐17‐5p was downregulated or upregulated, respectively (Figure S6E). Then we cotransfected ELAVL1 plasmids with hsa‐miR‐17‐p mimics in luciferase assays. ELAVL1 alone significantly increased the luciferase activities of the OSER1‐AS1 3′‐end reporter vector, but when co‐transfecting ELAVL1 plasmids with hsa‐miR‐17‐5p mimics, the luciferase activities were downregulated in comparison to transfecting with ELAVL1 plasmids alone (Figure S6F).
3.9. OSER1‐AS1 sequesters ELAVL1 by direct lncRNA‐protein interaction
We next investigated the biological mechanism by which OSER1‐AS1 suppresses tumorigenesis. The activity of ELAVL1 has been known to be dependent on its subcellular distribution: it predominantly localizes within the nucleus of resting cells. Under certain biological conditions, it transports to the cytoplasm and binds to its target mRNA’s 3′UTR. 23 The ELAVL1‐mRNA complex prevents the mRNA from degradation and thereby increases its translational level. 24 As OSER1‐AS1 was distributed predominantly in the cell cytoplasm (Figure S3), we hypothesized that it might function as a natural sponge to prevent ELAVL1 from binding its target mRNAs in the cytoplasm.
We carried out RIP experiment to pull down the endogenous OSER1‐AS1‐protein complex in the H1299 cell line. We used both ELAVL1 and AGO2 antibodies to pull down OSER1‐AS1, because OSER1‐AS1 was not only bound by ELAVL1 but also by hsa‐miR‐17‐5p, and AGO2 was the catalytic center of the RISC. 25 To determine the relative binding abundances of the OSER1‐AS1‐ELALV1 complex and the OSER1‐AS1‐AGO2 complex in the subcellular compartment, we separated the nuclear and cytoplasmic fractions from RIP lysates. Results showed that the OSER1‐AS1‐ELAVL1 complex was approximately fourfold more enriched than the OSER1‐AS1‐AGO2 complex in the whole cell. While in the nucleus, OSER1‐AS1 was also preferentially bound to ELAVL1 (OSER1‐AS1‐ELAVL1 complex to OSER1‐AS1‐AGO2 complex ratio 5:1). However, the cytoplasmic OSER1‐AS1 was predominantly bound by the AGO2 (OSER1‐AS1‐ELAVL1 complex to the OSER1‐AS1‐AGO2 complex ratio 1:70) (Figure 5A).
Figure 5.
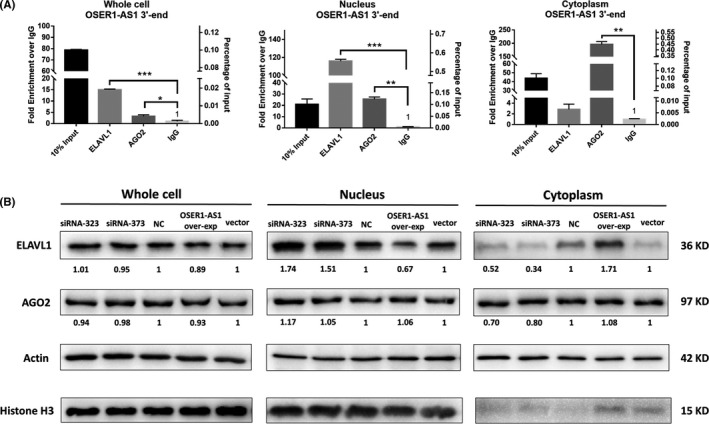
Long non‐coding RNA (lncRNA)‐protein interaction between OSER1‐AS1 and ELAVL1. A, RNA immunoprecipitation (RIP) was performed in H1299 cells using either control IgG, anti‐ELAVL1 or anti‐AGO2 antibodies. Immunoprecipitated OSER1‐AS1 RNA was quantified by qRT‐PCR. Relative abundance of RNA‐protein complex in cell lysate was expressed as either fold enrichment normalized to the control IgG (Left Y‐axis) or percentage of input (Right Y‐axis). Error bars represent the SD of three independent experiments. Two‐tailed Student’s t test: *P < .05; **P < .01; ***P < .001. B, Protein expression levels of ELAVL1 and AGO2 in H1299 cells were detected by western blot after knockdown and overexpression of OSER1‐AS1. Cell lysates were collected for western blotting 48 h post‐transfection. The numbers below the bands indicate the fold change of quantitative analysis results. Beta‐actin was used as positive control for whole cell and cytoplasmic cell lysate. Histone H3 was used as positive controls for nucleus lysate
To further investigate the potential influence of OSER1‐AS1 on the subcellular localization of ELAVL1, we carried out western blot in the whole cell, nuclear and cytoplasmic cell lysate prepared from H1299 cells transfected with scramble siRNA (NC), two separate siRNAs targeting OSER1‐AS1 (siRNA‐323 and siRNA‐373), stable empty vector and OSER1‐AS1 overexpression vector. Consistent with the previous studies, our results showed that ELAVL1 was predominantly distributed in the nucleus, and AGO2 was in both the nucleus and the cytoplasm. 23 Although we found no significant alteration of ELAVL1 and AGO2 expressions in the whole cell, OSER1‐AS1 changed the subcellular localization of ELAVL1. Knockdown of OSER1‐AS1 attenuated the translocation of ELAVL1 from nucleus to cytoplasm in H1299 cells, whereas OSER1‐AS1 overexpression increased the amount of cytoplasmic ELAVL1 proteins (Figure 5B).
Our findings in RIP and western blot experiments together suggested that OSER1‐AS1 directly bound with ELAVL1 and the upregulation of OSER1‐AS1 caused the accumulation of ELAVL1 proteins in the cytoplasm.
3.10. OSER1‐AS1 regulates the target genes of ELAVL1 and miR‐17‐5p
MYC has been reported to stimulate the expression of microRNAs in the miR‐17‐92 cluster, including miR‐17‐5p, 26 , 27 , 28 which, in turn, suppressed the expression of the pro‐apoptotic protein BCL2L11 in lymphoma 29 and in NSCLC. 30 In contrast, the RBP ELAVL1 was known to upregulate the translational level of MYC. 31 , 32 , 33 Considering that the 3′‐end of OSER1‐AS1 was bound by both miR‐17‐5p and ELAVL1, we hypothesized that OSER1‐AS1 could act as a miR‐17‐5p sponge to change the expression of its target gene BCL2L11. By the same theory, OSER1‐AS1 may also sponge ELAVL1 away from its target MYC mRNAs, leading to decreased translational efficiency and decreased MYC protein levels. Hence, we performed western blot to assess MYC and BCL2L11 protein level after knockdown and overexpression of OSER1‐AS1. Results of western blotting indicated that knockdown of OSER1‐AS1 increased MYC expression, whereas overexpression of OSER1‐AS1 decreased MYC expression. However, the change of MYC expression was only detectable in the nucleus (Figure 6A), consistent with the knowledge that MYC was a transcription factor that mainly carried out its functions in the nucleus. 28 The mRNA and protein level of BCL2L11 were both affected by OSER1‐AS1: knockdown of OSER1‐AS1 decreased BCL2L11 expression, whereas overexpression of OSER1‐AS1 increased BCL2L11 expression (Figure 6A & B). The pattern of BCL2L11 expression change was more profound in the cytoplasm. We also observed that, in the cytoplasm, knockdown of OSER1‐AS1 increased hsa‐miR‐17‐5p expression, whereas overexpression of OSER1‐AS1 decreased hsa‐miR‐17‐5p expression (Figure 6C). However, the change of hsa‐miR‐17‐5p expression was not significant in the nucleus.
Figure 6.
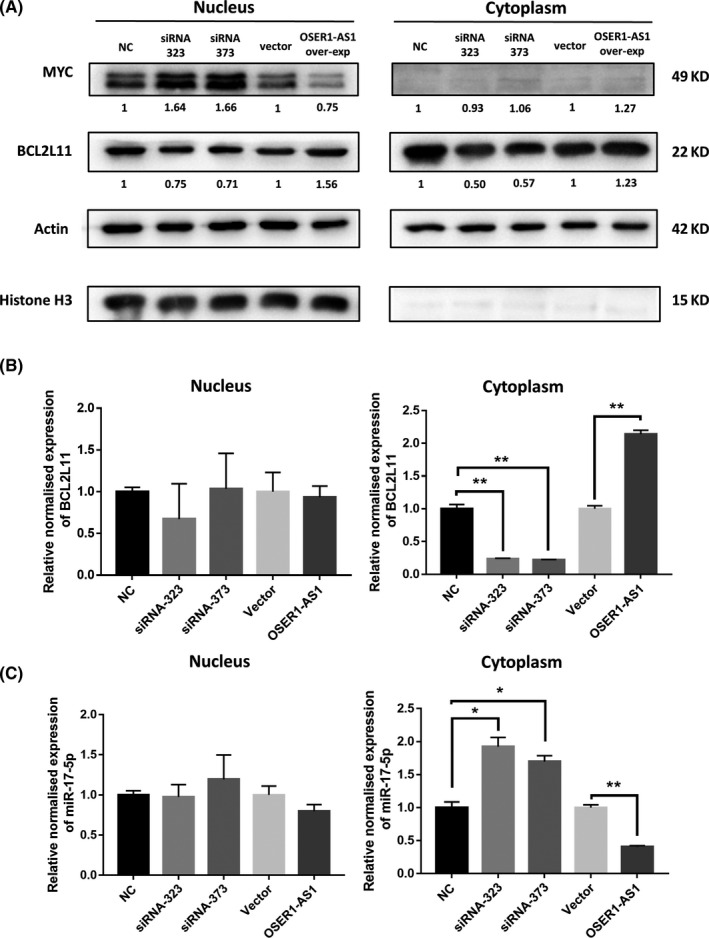
Influence of OSER1‐AS1 on the expression levels of MYC, BCL2L11 and miR‐17‐5p in H1299 cells. A, Protein expression levels of MYC and BCL2L11 in H1299 cells were detected by western blot after knockdown and overexpression of OSER1‐AS1. Cell lysates were collected for western blotting 48 h post‐transfection. Numbers below the bands indicate the fold change of quantitative analysis results. Beta‐actin was used as positive control for cytoplasmic cell lysate. Histone H3 was used as positive control for nuclear cell lysate. B, MiR‐17‐5p expression levels in nucleus and cytoplasmic cell lysate after overexpression or knockdown of OSER1‐AS1. C, BCL2L11 expression levels in nuclear and cytoplasmic cell lysate after overexpression or knockdown of OSER1‐AS1. Error bars represent the SD of three independent experiments. Two‐tailed Student’s t test: *P < .05; **P < .01
4. DISCUSSION
In the present study, we tested a list of SNP located near lncRNAs to detect their associations with lung cancer risk. From one significantly associated SNP rs4142441, we narrowed down our focus to lncRNA OSER1‐AS1. We demonstrated that the mutant allele (G) of rs4142441 was associated with higher lung cancer risk and lower expression level of OSER1‐AS1. Rs4142441 was previously reported in a large‐scale GWAS study as associated with monocyte count in whole blood. The allele G was associated with increased monocyte count (beta coefficient = 0.033; [95% CI 0.023‐0.043]; P = 4 × 10‐11). 34 An elevated peripheral monocyte count has been reported to have a poor prognosis impact on lung adenocarcinoma. 35
The qRT‐PCR analysis confirmed that OSER1‐AS1 was significantly downregulated in lung adenocarcinoma tissues in comparison to adjacent normal tissues. Moreover, differential expression was more profound among non‐smokers. Therefore, we hypothesized that OSER1‐AS1 played a tumor‐suppressive role in NSCLC. We then investigated the prognostic value of OSER1‐AS1 using clinical data from TCGA public database by Kaplan‐Meier survival analysis. We found that high expression of OSER1‐AS1 was associated with better overall survival in TCGA‐LUAD non‐smoker patients. In addition, OSER1‐AS1 suppressed lung cancer cell proliferation, migration, and invasion in vitro and promoted tumorigenesis in vivo.
Furthermore, our results showed that transcription factor MYC suppressed OSER1‐AS1 expression. Transcription factor MYC is a commonly known regulator which activated growth‐related genes and suppressed genes involved in cell cycle arrest, cell adhesion, and cell‐cell communication. 36 In addition, MYC was upregulated in human NSCLC cells 37 , 38 and MYC depletion has been reported to be able to reverse immune evasion and enables effective treatment of lung cancer. 39 MYC was known to bind to the core promoter of the genes it repressed directly, 40 and it has been known to be regulated by a number of lncRNAs. 41 The results from our ChIP experiment confirmed the directing binding of MYC at the promoter region of OSER1‐AS1. The binding site of MYC was near SNP rs4142441, but EMSA assay results indicated that its wild‐type/mutant allelic status did not interfere with MYC‐binding capacity. One possible explanation was that the mutant allele of rs4142441 has only a moderate effect on protein‐binding ability; therefore, EMSA assay may not be sensitive enough to characterize the difference of DNA‐protein binding affinity in vitro. Our results also suggested that rs4142441 might influence the expression level of OSER1‐AS1 by other transcription factors such as RNA polymerase II. The exact biological mechanism of the association between allele G of rs4142441 and the lower expression level of OSER1‐AS1 remains to be explored.
In contrast, the 3′‐end of OSER1‐AS1 was bound competitively by hsa‐miR‐17‐5p and RNA‐binding protein ELAVL1. The RNA sequence of OSER1‐AS1 contained more than 9 ARE motifs required for ELAVL1 binding, and the nearest ARE motif was only 51 bp 5' upstream of the miR‐17‐5p binding site.
Some studies have implicated the roles of ELAVL1 in affecting the binding capacity of AGO2 and miRNAs, 23 and the effects could be either competition 42 or cooperation, 43 , 44 probably depending on the conformational changes of the 3′UTR caused by the initial binding. In the study of Chang et al (2013), ELAVL1 has been reported to antagonize the suppressive effect of miR‐200b on vascular endothelial growth factor A (VEGF‐A) expression by competitive binding. 45 However, unlike VEGF‐A, OSER1‐AS1 as a lncRNA cannot be stabilized by ELAVL1 and translated into proteins. Therefore, we hypothesized that OSER1‐AS1 may execute its biological function as a decoy by forming a stable RNA‐protein complex with ELAVL1 and sequestering it from its target mRNAs.
Several studies have shown that some lncRNAs could serve as a sponge to restrict the availability of RBP to its target mRNAs. 46 , 47 , 48 The post‐transcriptional regulations by ELAVL1 mainly took place in the cytoplasm. 49 Our RNA FISH assays showed that OSER1‐AS1 is located mainly in the cytoplasm (cytoplasm : nucleus ratio 7:1), so the subcellular localization of OSER1‐AS1 was consistent with our “ELAVL1 sponge” hypothesis.
To test this hypothesis, we carried out RIP assays in the whole cell lysate, nuclear lysate, and cytoplasmic lysate separately. The results from RIP experiments confirmed that physical interaction happened between OSER1‐AS1 and ELAVL1. In the whole cell, OSER1‐AS1 preferentially formed an RBP‐RNA complex with ELAVL1. Moreover, OSER1‐AS1 was also bound by AGO2, which was consistent with our finding that OSER1‐AS1 was targeted by hsa‐miR‐17‐5p. Our results of RIP experiments showed that, in the nucleus, OSER1‐AS1 was predominantly bound by ELAVL1, whereas in the cytoplasm OSER1‐AS1 was mainly bound by AGO2. However, this did not suggest that OSER1‐AS1 has higher affinity with AGO2 than with ELAVL1. Because ELAVL1 was known to stay at low levels in the cytoplasm: ELAVL1 mainly localized in the nucleus and only moved from nucleus to cytoplasm when it was performing its mRNA stabilizer function24. Therefore, low cytoplasmic level of ELAVL1‐OSER1‐AS1 complex may not represent low binding affinity between ELAVL1 and OSER1‐AS1.
We then adjusted the expression level of OSER1‐AS1 to further explore whether OSER1‐AS1 had any impact on ELAVL1 protein level. Our results indicated that although OSER1‐AS1 had no significant influence on the overall protein level of ELAVL1, it changed its subcellular localization. Overexpression of OSER1‐AS1 resulted in the accumulation of ELAVL1 proteins in the cytoplasm, whereas the knockdown of OSER1‐AS1 caused the opposite effect. However, this was in conflict with the findings from previous studies which showed a higher cytoplasmic level of ELAVL1 associated with increased tumorigenic activity and poor prognostic outcome in NSCLC. 50 , 51 One possible explanation might be that even though OSER1‐AS1 increased the cytoplasmic level of ELAVL1, these ELAVL1 were not functional because they were not able to bind their target mRNAs, and they could not be transported back to the nucleus.
Subsequently, we investigated whether OSER1‐AS1 changed the level of MYC, which was both a regulator of OSER1‐AS1 and a target gene of ELAVL1. We found that knockdown of OSER1‐AS1 increased MYC expression, whereas the overexpression of OSER1‐AS1 decreased MYC expression. Taken together with the finding that MYC suppressed the transcriptional level of OSER1‐AS1, we concluded that MYC and OSER1‐AS1 controlled each other in a negative feedback loop (Figure 7). In addition, we found that OSER1‐AS1 positively regulates the expression level of BCL2L11, which was targeted by miR‐17‐5p, further validating the hypothesis that OSER1‐AS1 functioned as a miRNA sponge of miR‐17‐5p.
Figure 7.
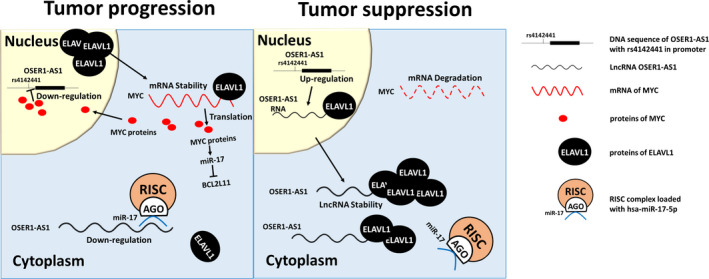
Proposed model depicting the potential mechanisms of OSER1‐AS1 as a tumor suppressor ong non‐coding RNA (lncRNA) in non‐small cell lung cancer (NSCLC). Left panel: in a scenario of tumor progression, OSER1‐AS1 is suppressed by transcription factor MYC in the nucleus and inhibited by miR‐17‐5p in the cytoplasm. ELAVL1 are transported from the nucleus to the cytoplasm to stabilize mRNAs of MYC and increase its translational efficiency. The MYC proteins, in turn, downregulate OSER1‐AS1 and BCL2L11, and upregulate miR‐17‐5p. Right panel: in a scenario of tumor suppression, the cytoplasmic level of OSER1‐AS1 increases to a level to be able to overcome the inhibitory effect of miR‐17‐5p. Free cytoplasmic OSER1‐AS1 forms RBP‐RNA complexes with ELAVL1 to sequester it from its target MYC mRNAs and possibly other oncogenic genes regulated by ELAVL1
Taken together, we demonstrated that OSER1‐AS1 exercised tumor‐suppressive functions in NSCLC by acting as an ELAVL1 decoy and sequestered ELAVL1 from its target mRNAs involved in cell proliferation, migration and tumorigenicity. In company with previous studies, 22 , 23 , 52 , 53 we showed that ELAVL1 can bind to the 3′‐end of lncRNA OSER1‐AS1 in competition with a microRNA hsa‐miR‐17‐5p. These findings highlighted that, other than sponging microRNAs, another possible mechanism of lncRNAs to exercise its tumor‐suppressing function might be sponging oncogenic RBP away from its target mRNAs.
Our study shows that OSER1‐AS1 is downregulated in tumor and acts as a tumor suppressor in NSCLC. OSER1‐AS1 is co‐regulated by SNP rs4142441 and MYC at the promoter, and competitively targeted by both microRNA hsa‐miR‐17‐5p and RBP ELAVL1 at the 3′‐end. It carries out tumor‐suppressive function by forming an RNA‐protein complex with ELAVL1 and sequesters it from binding and stabilizing its target mRNAs, large amounts of which are implicated in carcinogenesis. Our results suggest that OSER1‐AS1 could play important roles in ELAVL1‐regulated mRNA stability. These findings could provide biological insight into the regulation of the ceRNA network in NSCLC, and give new clues for future development of new therapeutic targets and biomarkers.
DISCLOSURE STATEMENT
The authors declare no conflicts of interest.
ETHICAL APPROVAL AND CONSENT TO PARTICIPATE
All experimental animal procedures were approved by the Institutional Animal Care and Use Committee of Third Military Medical University. All subjects provided informed written consent. Research protocol was approved by the ethics committee of the Army Medical University.
Supporting information
Fig S1‐S6
Table S1‐S6
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank all members of the Department of Epidemiology of the Army Medical University for their professional suggestions, and all members of the Department of Respiratory Disease of the Xinqiao Hospital for sample collection. This work was supported by the National Natural Science Foundation of China (No. 81602933 to W. X.; Nos. 81672316, 81472190 and 81171903 to Y. L.; No. 81370139 to L. B.) and foundation for Clinical Research from Xinqiao Hospital, Third Military Medical University (No. 2014YLC13 to L. B., No. 2015YLC21 to Z. Y.)
Xie W, Wang Y, Zhang Y, et al. Single‐nucleotide polymorphism rs4142441 and MYC co‐modulated long non‐coding RNA OSER1‐AS1 suppresses non‐small cell lung cancer by sequestering ELAVL1. Cancer Sci.2021;112:2272–2286. 10.1111/cas.14713
Weijia Xie, Youhao Wang and Yao Zhang contributed equally to this work.
Zubin Yu, Li Bai and Yafei Li jointly directed the project.
DATA AVAILABILITY STATEMENT
The case‐control datasets generated and/or analyzed during the current study are not publicly available due to individual privacy but are available from the corresponding author on reasonable request and with permission of the Department of Respiratory Disease of Xinqiao Hospital. TCGA‐LUAD datasets analyzed in the study are openly available in the GDC data portal (https://portal.gdc.cancer.gov/repository).
REFERENCES
- 1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87‐108. [DOI] [PubMed] [Google Scholar]
- 2. Ettinger DS, Akerley W, Borghaei H, et al. Non‐small cell lung cancer, version 2.2013. J Natl Compr Canc Netw. 2013;11:645–653; quiz 653. [DOI] [PubMed] [Google Scholar]
- 3. Borczuk AC, Toonkel RL, Powell CA. Genomics of lung cancer. Proc Am Thorac Soc. 2009;6:152‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Davidson MR, Gazdar AF, Clarke BE. The pivotal role of pathology in the management of lung cancer. J Thorac Dis. 2013;5(Suppl 5):S463‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xie W, Yuan S, Sun Z, Li Y. Long noncoding and circular RNAs in lung cancer: advances and perspectives. Epigenomics. 2016;8:1275‐1287. [DOI] [PubMed] [Google Scholar]
- 6. Mascaux C, Tsao MS, Hirsch FR. Genomic Testing in Lung Cancer: Past, Present, and Future. J Natl Compr Canc Netw. 2018;16:323‐334. [DOI] [PubMed] [Google Scholar]
- 7. Yuan S, Liu Q, Hu Z, et al. Long non‐coding RNA MUC5B‐AS1 promotes metastasis through mutually regulating MUC5B expression in lung adenocarcinoma. Cell Death Dis. 2018;9:450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yuan S, Xiang Y, Wang G, et al. Hypoxia‐sensitive LINC01436 is regulated by E2F6 and acts as an oncogene by targeting miR‐30a‐3p in non‐small cell lung cancer. Mol Oncol. 2019;13:840‐856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen S, Gu T, Lu Z, et al. Roles of MYC‐targeting long non‐coding RNA MINCR in cell cycle regulation and apoptosis in non‐small cell lung cancer. Respir Res. 2019;20:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hua Q, Jin M, Mi B, et al. LINC01123, a c‐Myc‐activated long non‐coding RNA, promotes proliferation and aerobic glycolysis of non‐small cell lung cancer through miR‐199a‐5p/c‐Myc axis. J Hematol Oncol. 2019;12:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bosse Y, Amos CI. A Decade of GWAS Results in Lung Cancer. Cancer Epidemiol Biomarkers Prev. 2018;27:363‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun C, Li S, Zhang F, et al. Long non‐coding RNA NEAT1 promotes non‐small cell lung cancer progression through regulation of miR‐377‐3p‐E2F3 pathway. Oncotarget. 2016;7:51784‐51814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schmitt AM, Chang HY. Long Noncoding RNAs in Cancer Pathways. Cancer Cell. 2016;29:452‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fang J, Sun CC, Gong C. Long noncoding RNA XIST acts as an oncogene in non‐small cell lung cancer by epigenetically repressing KLF2 expression. Biochem Biophys Res Commun. 2016;478:811‐817. [DOI] [PubMed] [Google Scholar]
- 15. Zhou X, Zhu W, Li H, et al. Diagnostic value of a plasma microRNA signature in gastric cancer: a microRNA expression analysis. Sci Rep. 2015;5:11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu X, Ji Q, Zhang C, et al. miR‐30a acts as a tumor suppressor by double‐targeting COX‐2 and BCL9 in H. pylori gastric cancer models. Sci Rep. 2017;7:7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yuan S, Yu Z, Liu Q, et al. GPC5, a novel epigenetically silenced tumor suppressor, inhibits tumor growth by suppressing Wnt/beta‐catenin signaling in lung adenocarcinoma. Oncogene. 2016;35:6120‐6131. [DOI] [PubMed] [Google Scholar]
- 18. Liu WB, Liu JY, Ao L, et al. Epigenetic silencing of cell cycle regulatory genes during 3‐methylcholanthrene and diethylnitrosamine‐induced multistep rat lung cancer. Mol Carcinog. 2010;49:556‐565. [DOI] [PubMed] [Google Scholar]
- 19. Li Y, Choi PS, Casey SC, Dill DL, Felsher DW. MYC through miR‐17‐92 suppresses specific target genes to maintain survival, autonomous proliferation, and a neoplastic state. Cancer Cell. 2014;26:262‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aguda BD, Kim Y, Piper‐Hunter MG, Friedman A, Marsh CB. MicroRNA regulation of a cancer network: consequences of the feedback loops involving miR‐17‐92, E2F, and Myc. Proc Natl Acad Sci USA. 2008;105:19678‐19683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen L, Li C, Zhang R, et al. miR‐17‐92 cluster microRNAs confers tumorigenicity in multiple myeloma. Cancer Lett. 2011;309:62‐70. [DOI] [PubMed] [Google Scholar]
- 22. Wang W, Caldwell MC, Lin S, Furneaux H, Gorospe M. HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. EMBO J. 2000;19:2340‐2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Srikantan S, Tominaga K, Gorospe M. Functional interplay between RNA‐binding protein HuR and microRNAs. Curr Protein Pept Sci. 2012;13:372‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang J, Guo Y, Chu H, Guan Y, Bi J, Wang B. Multiple functions of the RNA‐binding protein HuR in cancer progression, treatment responses and prognosis. Int J Mol Sci. 2013;14:10015‐10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Martinez NJ, Gregory RI. Argonaute2 expression is post‐transcriptionally coupled to microRNA abundance. RNA. 2013;19:605‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kumar P, Luo Y, Tudela C, Alexander JM, Mendelson CR. The c‐Myc‐regulated microRNA‐17~92 (miR‐17~92) and miR‐106a~363 clusters target hCYP19A1 and hGCM1 to inhibit human trophoblast differentiation. Mol Cell Biol. 2013;33:1782‐1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ventura A, Young AG, Winslow MM, et al. Targeted deletion reveals essential and overlapping functions of the miR‐17 through 92 family of miRNA clusters. Cell. 2008;132:875‐886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Psathas JN, Thomas‐Tikhonenko A. MYC and the art of microRNA maintenance. Cold Spring Harb Perspect Med. 2014;4:a014175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xiao C, Srinivasan L, Calado DP, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR‐17‐92 expression in lymphocytes. Nat Immunol. 2008;9:405‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dai B, Meng J, Peyton M, et al. STAT3 mediates resistance to MEK inhibitor through microRNA miR‐17. Cancer Res. 2011;71:3658‐3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hatanaka T, Higashino F, Tei K, Yasuda M. The neural ELAVL protein HuB enhances endogenous proto‐oncogene activation. Biochem Biophys Res Commun. 2019;517:330‐337. [DOI] [PubMed] [Google Scholar]
- 32. Perrone EE, Liu L, Turner DJ, Strauch ED. Bile salts increase epithelial cell proliferation through HuR‐induced c‐Myc expression. J Surg Res. 2012;178:155‐164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kakuguchi W, Kitamura T, Kuroshima T, et al. HuR knockdown changes the oncogenic potential of oral cancer cells. Mol Cancer Res. 2010;8:520‐528. [DOI] [PubMed] [Google Scholar]
- 34. Astle WJ, Elding H, Jiang T, et al. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell. 2016;167:1415‐1429 e1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kumagai S, Marumo S, Shoji T, et al. Prognostic impact of preoperative monocyte counts in patients with resected lung adenocarcinoma. Lung Cancer. 2014;85(3):457‐464. [DOI] [PubMed] [Google Scholar]
- 36. Eilers M, Eisenman RN. Myc's broad reach. Genes Dev. 2008;22:2755‐2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li H, Liu J, Cao W, et al. C‐myc/miR‐150/EPG5 axis mediated dysfunction of autophagy promotes development of non‐small cell lung cancer. Theranostics. 2019;9:5134‐5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mustachio LM, Roszik J, Farria AT, Guerra K, Dent SY. Repression of GCN5 expression or activity attenuates c‐MYC expression in non‐small cell lung cancer. Am J Cancer Res. 2019;9:1830‐1845. [PMC free article] [PubMed] [Google Scholar]
- 39. Topper MJ, Vaz M, Chiappinelli KB, et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell. 2017;171(1284–1300):e1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Herkert B, Eilers M. Transcriptional repression: the dark side of myc. Genes Cancer. 2010;1:580‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Swier L, Dzikiewicz‐Krawczyk A, Winkle M, van den Berg A, Kluiver J. Intricate crosstalk between MYC and non‐coding RNAs regulates hallmarks of cancer. Mol Oncol. 2019;13:26‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen Y, Yang F, Fang E, et al. Circular RNA circAGO2 drives cancer progression through facilitating HuR‐repressed functions of AGO2‐miRNA complexes. Cell Death Differ. 2019;26:1346‐1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Balkhi MY, Iwenofu OH, Bakkar N, et al. miR‐29 acts as a decoy in sarcomas to protect the tumor suppressor A20 mRNA from degradation by HuR. Sci Signal. 2013;6:ra63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sun Q, Tripathi V, Yoon JH, et al. MIR100 host gene‐encoded lncRNAs regulate cell cycle by modulating the interaction between HuR and its target mRNAs. Nucleic Acids Res. 2018;46:10405‐10416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chang SH, Lu YC, Li X, et al. Antagonistic function of the RNA‐binding protein HuR and miR‐200b in post‐transcriptional regulation of vascular endothelial growth factor‐A expression and angiogenesis. J Biol Chem. 2013;288:4908‐4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim J, Abdelmohsen K, Yang X, et al. LncRNA OIP5‐AS1/cyrano sponges RNA‐binding protein HuR. Nucleic Acids Res. 2016;44:2378‐2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang F, Ni H, Li X, Liu H, Xi T, Zheng L. LncRNA FENDRR attenuates adriamycin resistance via suppressing MDR1 expression through sponging HuR and miR‐184 in chronic myelogenous leukaemia cells. FEBS Lett. 2019;593:1993‐2007. [DOI] [PubMed] [Google Scholar]
- 48. Shen L, Han J, Wang H, et al. Cachexia‐related long noncoding RNA, CAAlnc1, suppresses adipogenesis by blocking the binding of HuR to adipogenic transcription factor mRNAs. Int J Cancer. 2019;145:1809‐1821. [DOI] [PubMed] [Google Scholar]
- 49. Tirosh I. Transcriptional priming of cytoplasmic post‐transcriptional regulation. Transcription. 2011;2:258‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang J, Wang B, Bi J, Zhang C. Cytoplasmic HuR expression correlates with angiogenesis, lymphangiogenesis, and poor outcome in lung cancer. Med Oncol. 2011;28(Suppl 1):S577‐585. [DOI] [PubMed] [Google Scholar]
- 51. Wang J, Zhao W, Guo Y, et al. The expression of RNA‐binding protein HuR in non‐small cell lung cancer correlates with vascular endothelial growth factor‐C expression and lymph node metastasis. Oncology. 2009;76:420‐429. [DOI] [PubMed] [Google Scholar]
- 52. Tominaga K, Srikantan S, Lee EK, et al. Competitive regulation of nucleolin expression by HuR and miR‐494. Mol Cell Biol. 2011;31:4219‐4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Epis MR, Barker A, Giles KM, Beveridge DJ, Leedman PJ. The RNA‐binding protein HuR opposes the repression of ERBB‐2 gene expression by microRNA miR‐331‐3p in prostate cancer cells. J Biol Chem. 2011;286:41442‐41454. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S6
Table S1‐S6
Supplementary Material
Data Availability Statement
The case‐control datasets generated and/or analyzed during the current study are not publicly available due to individual privacy but are available from the corresponding author on reasonable request and with permission of the Department of Respiratory Disease of Xinqiao Hospital. TCGA‐LUAD datasets analyzed in the study are openly available in the GDC data portal (https://portal.gdc.cancer.gov/repository).