

Abstract
Milademetan (DS‐3032, RAIN‐32) is an orally available mouse double minute 2 (MDM2) antagonist with potential antineoplastic activity owing to increase in p53 activity through interruption of the MDM2‐p53 interaction. This phase I, dose‐escalating study assessed the safety, tolerability, efficacy, and pharmacokinetics of milademetan in 18 Japanese patients with solid tumors who relapsed after or were refractory to standard therapy. Patients aged ≥ 20 years received oral milademetan once daily (60 mg, n = 3; 90 mg, n = 11; or 120 mg, n = 4) on days 1 to 21 in a 28‐day cycle. Dose‐limiting toxicities, safety, tolerability, maximum tolerated dose, pharmacokinetics, and recommended dose for phase II were determined. The most frequent treatment‐emergent adverse events included nausea (72.2%), decreased appetite (61.1%), platelet count decreased (61.1%), white blood cell count decreased (50.0%), fatigue (50.0%), and anemia (50.0%). Dose‐limiting toxicities (three events of platelet count decreased and one nausea) were observed in the 120‐mg cohort. The plasma concentrations of milademetan increased in a dose‐dependent manner. Stable disease was observed in seven out of 16 patients (43.8%). Milademetan was well tolerated and showed modest antitumor activity in Japanese patients with solid tumors. The recommended dose for phase II was considered to be 90 mg in the once‐daily 21/28‐day schedule. Future studies would be needed to further evaluate the potential safety, tolerability, and clinical activity of milademetan in patients with solid tumors and lymphomas. The trial was registered with Clinicaltrials.jp: JapicCTI‐142693.
Keywords: DS‐3032, MDM2 protein, milademetan, phase I clinical trial, RAIN‐32
We are presenting the first report of a phase I study evaluating the safety, tolerability, efficacy, and pharmacokinetics of milademetan in Japanese patients with advanced solid tumors. The plasma concentrations of milademetan increased in a dose‐dependent manner. Milademetan was well tolerated and showed modest antitumor activity in Japanese patients with solid tumors.
Abbreviations
- AE
adverse event
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- AUC
area under the concentration‐time curve
- CI
confidence interval
- Cmax
maximum plasma concentration
- CTCAE
Common Terminology Criteria for Adverse Events
- DCR
disease control rate
- DLT
dose‐limiting toxicity
- FFPE
formalin‐fixed paraffin‐embedded
- MDM2
mouse double minute 2
- MedDRA
Medical Dictionary for Regulatory Activities
- MIC‐1
macrophage inhibitory cytokine‐1
- MTD
maximum tolerated dose
- ORR
overall response rate
- PFS
progression‐free survival
- PK
pharmacokinetics
- QD
quaque die
- SD
stable disease
- TEAE
treatment‐emergent adverse event
- Tmax
time to maximum concentration
1. INTRODUCTION
The transcription factor p53 which is induced by stress signals including DNA damage, oncogene activation, and nutrient deprivation, plays a prominent role in cell cycle arrest and apoptosis. 1 P53 also has a potential role in DNA repair, regulation of antioxidant response and metabolism, 2 and is frequently inactivated by mutations or deletions in the natural history of cancer. Mutant p53 is a potential target for cancer therapy 3 and a poor prognostic factor in patients with cancer.
The oncoprotein mouse double minute 2 (MDM2) has a tumorigenic role, as it inhibits p53 activity and promotes its degradation. In many cancers, overexpression of MDM2 promotes cell survival and proliferation. 4 Stabilization of p53 by pharmacological inhibition of the MDM2‐p53 interaction is a target in cancer therapy 5 and could result in a sustained increase in p53 activity and subsequent antitumor effects. 5 , 6 , 7
Several small‐molecule inhibitors of the MDM2‐p53 interaction are currently being investigated for cancer therapy 8 and seven MDM2 inhibitors are being evaluated in human clinical trials. 9 Milademetan (DS‐3032, RAIN‐32) is a novel, orally available MDM2 inhibitor with potential antineoplastic activity owing to interruption of the MDM2‐p53 interaction. It restores p53 signaling by inhibiting MDM2‐p53 interaction leading to proteasome‐mediated enzymatic degradation of p53 and reinstating the transcriptional activity of p53. 10 , 11 Milademetan inhibits the growth of cancer cells harboring wild‐type p53 across six human cancer cell lines and also suppresses tumor growth of human osteosarcoma xenografts in nude mice. 10
In the United States, a phase I dose‐escalation study of milademetan in advanced solid tumors or lymphomas is ongoing (NCT01877382, DS3032‐A‐U101). 12 , 13 Here, we present the results of a phase I study (JapicCTI‐142693, DS3032‐A‐J103) of milademetan in Japanese patients with solid tumors who relapsed after or were refractory to standard therapy, or for whom no standard therapy was available.
2. MATERIALS AND METHODS
2.1. Study design and treatment
This was a phase I, single‐arm, open‐label, dose‐escalation study conducted at the National Cancer Center Hospital and the Cancer Institute Hospital of Japanese Foundation for Cancer Research in Japan (Figure 1). Eligible patients received oral milademetan treatment on days 1 to 21 in a 28‐day cycle. Treatment was continued until disease progression, as defined by the Response Evaluation Criteria in Solid Tumors (RECIST version 1.1, Japanese version), or unacceptable toxicity was observed. Milademetan was administered orally as milademetan tosylate at three dose levels: cohort 1 (60 mg as milademetan QD), cohort 2 (90 mg QD), and cohort 3 (120 mg QD). Based on the safety information of the dose‐escalation phase I study (DS3032‐A‐U101) in the United States, 120 mg in the once‐daily 21/28‐day schedule was considered the maximum tolerated dose (MTD), and considering the dose that can be expected to have an antitumor effect, 60 mg/d was selected as the initial dose for this phase I study in Japan. 12
FIGURE 1.
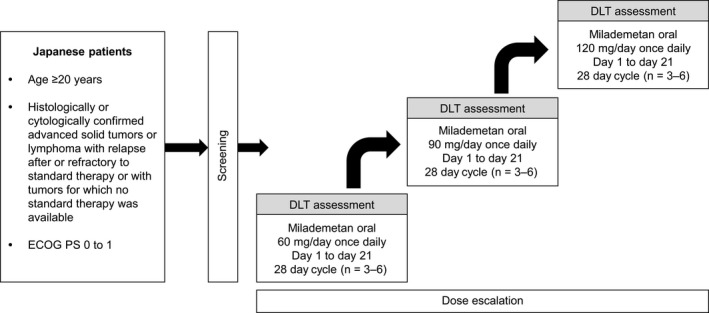
Study design. DLT, dose‐limiting toxicity; ECOG PS, Eastern Cooperative Oncology Group Performance Status
The first 28 days after the first dose of milademetan (cycle 1) were specified as the dose‐limiting toxicity (DLT) evaluation period. Each cohort consisted of three to six patients, and all were assessed for DLT before escalation to the next dose level. Dose‐escalation and de‐escalation were guided by the modified continual reassessment method using a Bayesian logistic regression model following the principle of escalation with overdose control. 14
The study was conducted in compliance with all international and local laws, as well as the principles of the “Declaration of Helsinki” and Good Clinical Practice Guidelines. All patients provided written informed consent prior to the conduct of the study. The study protocol and all subsequent amendments were approved by the relevant institutional review boards or independent ethics committees.
2.2. Study population
This study included adult (age ≥20 years) Japanese patients with histologically or cytologically confirmed advanced solid tumors or lymphoma with relapse after or refractory to standard therapy or with tumors for which no standard therapy was available. Patients with adequate function of major organs and an Eastern Cooperative Oncology Group performance status of 0 or 1 were eligible. Patients were required to have attained treatment‐free periods of at least 3 weeks for previous chemotherapy (6 weeks for nitrosoureas or mitomycin C), immunotherapy, hormone therapy, or radiation therapy; and 3 weeks following major surgery. Patients who had tumors containing nonsynonymous mutation, insertion, or deletion in the TP53 gene as determined at the time of informed consent or earlier; another malignant tumor or lymphoma requiring treatment; symptomatic brain metastasis; or any serious concomitant disease condition were excluded. Tumor samples were tested for TP53 mutational status post enrollment, and patients with TP53 mutations were informed of the result. These patients were allowed to continue or to exit the study, as patients with wild‐type p53 were expected to benefit from milademetan therapy.
2.3. Objectives
The primary objectives of the study were to evaluate the safety and tolerability of milademetan in Japanese patients with advanced solid tumors or lymphomas, to assess the pharmacokinetics (PK), and to determine the MTD or recommended phase II dose of milademetan. The secondary objective was to determine the pharmacodynamic effects of milademetan on macrophage inhibitory cytokine‐1 (MIC‐1) levels in serum. In addition, we performed an exploratory evaluation of tumor response to milademetan based on TP53 genomic status.
2.4. Safety evaluation
All patients who received at least one dose of milademetan constituted the safety analysis set. A DLT was defined as any ≥grade 3 treatment‐emergent adverse event (TEAE) not attributable to disease or disease‐related processes that occurred during the DLT evaluation period (cycle 1 [days 1‐28]). For hematological events, DLTs included grade 4 neutropenia lasting more than 7 days, ≥grade 3 febrile neutropenia, ≥grade 3 anemia that required blood transfusion, and grade 4 thrombocytopenia or ≥grade 3 thrombocytopenia lasting >7 days or associated with bleeding or requiring blood transfusion. For elevations in hepatic function enzymes, DLTs included grade 4 aspartate aminotransferase/alanine aminotransferase (AST/ALT) level, AST/ALT > five times upper limit of normal (ULN) lasting more than 3 days, or if accompanied by ≥grade 2 elevation in bilirubin. Patients who could not complete at least 75% of the prescribed doses (i.e., 16 days) in cycle 1 as a result of non–disease‐related ≥grade 2 adverse events (AEs) were considered to have a DLT. A dose delay of more than 7 days secondary to myelosuppression or a delay of ≥1 week in initiating cycle 2 secondary to a non–disease‐related ≥grade 2 AE were also considered DLTs. Grade 3 fatigue lasting less than 3 days; grade 3 nausea, vomiting, or diarrhea that resolved to ≤grade 2 within 48 hours after standard therapy; and isolated laboratory findings not associated with signs or symptoms and for which continuous treatment was not required were not considered DLTs.
Additional assessments included body weight, vital signs, Eastern Cooperative Oncology Group performance status, laboratory tests, and 12‐lead electrocardiogram. All AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA, Version 18.0) and graded by the Common Terminology Criteria for Adverse Events (CTCAE), version 4.0, Japanese version. Patients with AEs were followed up until resolution.
2.5. Pharmacokinetics assessments
Blood samples were collected prior to dose and at 1, 2, 3, 6, 8, 10 to 12 hours post dose on cycle 1 day 1, and prior to dose and at 1, 2, 3, 6 to 8, 10 to 12, 22 to 24, 30 to 32, 46 to 48 hours post dose on cycle 1 day 21. Additional blood samples were collected prior to dose on cycle 1 days 2, 8, and 15, as well as cycle 2 day 1. Plasma concentrations of milademetan were determined at a central laboratory (Q2 Solutions) using a prevalidated high‐performance liquid chromatography–tandem mass spectrometry method. Standard PK parameters including maximum concentration (Cmax), time to reach Cmax (Tmax), and area under the plasma concentration‐time curve (AUC) up to infinity (AUCinf) for cycle 1 day 1 dosing, for a 24‐hour dosing interval (AUCtau), and up to the last quantifiable concentration (AUClast) for cycle 1 day 21 dosing were calculated using a standard noncompartmental analysis approach (Phoenix® WinNonlin® 8.1; Certara).
2.6. Efficacy assessments
Efficacy assessments included best overall response, overall response rate (ORR) (proportion of patients demonstrating complete and partial response), disease control rate (DCR) (proportion of patients demonstrating complete response, partial response, or stable disease [SD; minimum criterion for duration of SD was defined as 8 weeks after the first dosing date]), progression‐free survival (PFS), and maximum percentage tumor reduction. Efficacy was evaluated using RECIST version 1.1. 15 Efficacy assessments for solid tumors were performed every 8 weeks until the first eight cycles, and every 12 weeks thereafter using computed tomography and magnetic resonance imaging scanning.
Tumor TP53 genotyping was performed using tumor tissue sample collected before the study drug administration. TP53 genomic status was determined using targeted next‐generation sequencing based on the reference human genome sequence (hg19). For tumor content analysis, formalin‐fixed paraffin‐embedded (FFPE) tissue specimens were processed for macrodissection. DNA was extracted using QIAamp® DSP FFPE Kit (Qiagen). Quantified DNA samples were processed by PCR amplification using custom primer sets of all 11 exons of TP53. Libraries from purified PCR pools using Illumina TruSeq kits™ (Illumina) were prepared and analyzed by MiSeq platform™ (Illumina). Raw reads were aligned against hg19, and data sets were analyzed with an in‐house–developed program to detect genomic aberrations (mutations, deletions, and insertions).
Serum MIC‐1 samples were collected prior to dose of cycle 1 day 1 (including 6 hours after administration), day 2, day 8, day 15, and day 21. Serum MIC‐1 also known as growth and differentiation factor‐15 (GDF‐15) was determined using the Quantikine® ELISA Human GDF‐15 immunoassay (R&D Systems, Inc), which employs the quantitative sandwich enzyme immunoassay technique.
2.7. Statistical methods
TEAEs were tabulated by event, relationship, and CTCAE grade. The safety analysis set including patients who received at least one dose of milademetan was used to summarize all safety endpoints. For DLT assessment, a summary was provided for all patients who received at least one dose of milademetan and were evaluable for DLTs.
The PK and efficacy endpoints were summarized using the PK and efficacy analysis set, which included all patients who had one post‐treatment plasma concentration and efficacy data, respectively. No hypothesis testing was performed and data were summarized using descriptive statistics. Continuous variables were summarized using mean, standard deviation, minimum, median, and maximum values, and categorical variables were summarized as a frequency table. The geometric mean and geometric coefficient of variation were used to describe the PK parameters in addition to the descriptive statistics. All analyses were performed using SAS® (version 9.3; SAS Institute Inc).
3. RESULTS
The patients were enrolled between December 03, 2014 and February 02, 2016, and the last patient's last visit was on January 23, 2019. All patients (n = 18) had solid tumors and none of the patients had lymphoma. The median age (range) of the patients was 49.0 (31‐74) years, and two patients were aged ≥ 65 years. All but one patient had received prior chemotherapy for cancer. Table 1 shows the demographic and baseline characteristics of the patients who received milademetan in daily doses of 60, 90, and 120 mg.
TABLE 1.
Patient demographics and baseline characteristics
| Characteristic |
Cohort 1 (60 mg/d) (n = 3) |
Cohort 2 (90 mg/d) (n = 11) |
Cohort 3 (120 mg/d) (n = 4) |
Total (N = 18) |
|---|---|---|---|---|
| Age (y) | 57.0 (38‐67) | 50.0 (31‐74) | 45.5 (42‐59) | 49.0 (31‐74) |
| Male, n (%) | 3 (100.0) | 4 (36.4) | 3 (75.0) | 10 (55.6) |
| Body weight (kg) | 67.50 (60.4‐80.0) | 58.50 (44.1‐75.9) | 53.15 (45.2‐70.2) | 59.45 (44.1‐80.0) |
| Previous treatment, n (%) | ||||
| Cancer therapy (excluding radiation and surgery) | 3 (100.0) | 10 (90.9) | 4 (100.0) | 17 (94.4) |
| Cancer surgery | 3 (100.0) | 7 (63.6) | 2 (50.0) | 12 (66.7) |
| Radiation therapy | 0 (0.0) | 4 (36.4) | 2 (50.0) | 6 (33.3) |
| Number of prior cancer drugs, n (%) | ||||
| 0 | 0 (0.0) | 1 (9.1) | 0 (0.0) | 1 (5.6) |
| 1 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 2 | 0 (0.0) | 1 (9.1) | 0 (0.0) | 1 (5.6) |
| 3 or more | 3 (100.0) | 9 (81.8) | 4 (100.0) | 16 (88.9) |
| Cancer type, n (%) | ||||
| Sarcoma | 0 (0.0) | 5 (45.5) | 0 (0.0) | 5 (27.8) |
| Dedifferentiated liposarcoma | 0 (0.0) | 2 (18.2) | 0 (0.0) | 2 (11.1) |
| Leiomyosarcoma | 0 (0.0) | 2 (18.2) | 0 (0.0) | 2 (11.1) |
| Unknown | 0 (0.0) | 1 (9.1) | 0 (0.0) | 1 (5.6) |
| Breast | 0 (0.0) | 0 (0.0) | 1 (25.0) | 1 (5.6) |
| Stomach | 0 (0.0) | 1 (9.1) | 0 (0.0) | 1 (5.6) |
| Other | 3 (100.0) | 4 (36.4) | 2 (50.0) | 9 (50.0) |
| Epipharynx | 0 (0.0) | 0 (0.0) | 1 (25.0) | 1 (5.6) |
| Gallbladder | 0 (0.0) | 1 (9.1) | 0 (0.0) | 1 (5.6) |
| Parotid gland | 0 (0.0) | 1 (9.1) | 0 (0.0) | 1 (5.6) |
| Submandibular gland | 1 (33.3) | 1 (9.1) | 0 (0.0) | 2 (11.1) |
| Thymic carcinoma | 1 (33.3) | 1 (9.1) | 0 (0.0) | 2 (11.1) |
| Urachal | 1 (33.3) | 0 (0.0) | 0 (0.0) | 1 (5.6) |
| Vater's Papilla | 0 (0.0) | 0 (0.0) | 1 (25.0) | 1 (5.6) |
| Unknown primary | 0 (0.0) | 1 (9.1) | 1 (25.0) | 2 (11.1) |
| Cancer stage, n (%) | ||||
| II | 0 (0.0) | 0 (0.0) | 1 (25.0) | 1 (5.6) |
| IV | 3 (100.0) | 11 (100.0) | 2 (50.0) | 16 (88.9) |
| Other | 0 (0.0) | 0 (0.0) | 1 (25.0) | 1 (5.6) |
| ECOG PS, n (%) | ||||
| 0 | 3 (100.0) | 9 (81.8) | 2 (50.0) | 14 (77.8) |
| 1 | 0 (0.0) | 2 (18.2) | 2 (50.0) | 4 (22.2) |
| TP53 genotype, n (%) | ||||
| Wild‐type | 1 (33.3) | 7 (63.6) | 3 (75.0) | 11 (61.1) |
| Mutant | 2 (66.7) | 3 (27.3) | 1 (25.0) | 6 (33.3) |
| Indeterminate a | 0 (0.0) | 1 (9.1) | 0 (0.0) | 1 (5.6) |
| Number of prior cancer drugs, n (%) | ||||
| 0 | 0 (0.0) | 1 (9.1) | 0 (0.0) | 1 (5.6) |
| 1 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 2 | 0 (0.0) | 1 (9.1) | 0 (0.0) | 1 (5.6) |
| ≥3 | 3 (100.0) | 9 (81.8) | 4 (100.0) | 16 (88.9) |
Values of continuous variables are presented as median (range).
Abbreviation: ECOG PS, Eastern Cooperative Oncology Group Performance Status.
Indeterminate with some reasons such as the sample cannot be amplified sufficiently and the allele frequency is below the detection limit.
3.1. Safety
Patients received a median of two cycles of milademetan (range, 1‐50), and the median duration of treatment was 6.5 (1.3‐201.1) weeks. All 18 patients had at least one drug‐related TEAE. Table 2 lists the TEAEs occurring in at least 20% of patients. The most frequent TEAEs were nausea (72.2%), decreased appetite (61.1%), platelet count decreased (61.1%), fatigue (50.0%), anemia (50.0%), and white blood cell count decreased (50.0%). Of these, all except anemia (38.9%) were drug‐related TEAEs.
TABLE 2.
Most frequent treatment‐emergent adverse events (TEAEs) [≥20% (all grades) or any (grade ≥ 3)] (safety analysis set)
| TEAE, n (%) |
Cohort 1 60 mg/d (n = 3) |
Cohort 2 90 mg/d (n = 11) |
Cohort 3 120 mg/d (n = 4) |
Total (n = 18) | ||||
|---|---|---|---|---|---|---|---|---|
| All grades | Grade ≥ 3 | All grades | Grade ≥ 3 | All grades | Grade ≥ 3 | All grades | Grade ≥ 3 | |
| Nausea | 3 (100.0) | 0 (0.0) | 7 (63.6) | 0 (0.0) | 3 (75.0) | 0 (0.0) | 13 (72.2) | 0 (0.0) |
| Platelet count decreased | 0 (0.0) | 0 (0.0) | 8 (72.7) | 1 (9.1) | 3 (75.0) | 3 (75.0) | 11 (61.1) | 4 (22.2) |
| Decreased appetite | 2 (66.7) | 0 (0.0) | 6 (54.5) | 0 (0.0) | 3 (75.0) | 0 (0.0) | 11 (61.1) | 0 (0.0) |
| Fatigue | 2 (66.7) | 0 (0.0) | 6 (54.5) | 0 (0.0) | 1 (25.0) | 0 (0.0) | 9 (50.0) | 0 (0.0) |
| Anemia | 0 (0.0) | 0 (0.0) | 7 (63.6) | 3 (27.3) | 2 (50.0) | 0 (0.0) | 9 (50.0) | 3 (16.7) |
| White blood cell count decreased | 1 (33.3) | 0 (0.0) | 6 (54.5) | 0 (0.0) | 2 (50.0) | 1 (25.0) | 9 (50.0) | 1 (5.6) |
| Hypoalbuminemia | 1 (33.3) | 0 (0.0) | 6 (54.5) | 0 (0.0) | 1 (25.0) | 0 (0.0) | 8 (44.4) | 0 (0.0) |
| Hyponatremia | 0 (0.0) | 0 (0.0) | 3 (27.3) | 1 (9.1) | 2 (50.0) | 1 (25.0) | 5 (27.8) | 2 (11.1) |
| Neutrophil count decreased | 0 (0.0) | 0 (0.0) | 3 (27.3) | 0 (0.0) | 2 (50.0) | 1 (25.0) | 5 (27.8) | 1 (5.6) |
| Constipation | 2 (66.7) | 0 (0.0) | 2 (18.2) | 0 (0.0) | 1 (25.0) | 0 (0.0) | 5 (27.8) | 0 (0.0) |
| Diarrhea | 0 (0.0) | 0 (0.0) | 3 (27.3) | 0 (0.0) | 2 (50.0) | 0 (0.0) | 5 (27.8) | 0 (0.0) |
| Vomiting | 0 (0.0) | 0 (0.0) | 3 (27.3) | 0 (0.0) | 2 (50.0) | 0 (0.0) | 5 (27.8) | 0 (0.0) |
| Aspartate aminotransferase increased | 0 (0.0) | 0 (0.0) | 2 (18.2) | 0 (0.0) | 2 (50.0) | 1 (25.0) | 4 (22.2) | 1 (5.6) |
| Alanine aminotransferase increased | 1 (33.3) | 0 (0.0) | 1 (9.1) | 1 (9.1) | 2 (50.0) | 1 (25.0) | 4 (22.2) | 2 (11.1) |
| Lymphocyte count decreased | 1 (33.3) | 0 (0.0) | 1 (9.1) | 0 (0.0) | 2 (50.0) | 1 (25.0) | 4 (22.2) | 1 (5.6) |
| Pyrexia | 0 (0.0) | 0 (0.0) | 3 (27.3) | 0 (0.0) | 1 (25.0) | 0 (0.0) | 4 (22.2) | 0 (0.0) |
| Sepsis | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (25.0) | 1 (25.0) | 1 (5.6) | 1 (5.6) |
| Urinary tract infection | 0 (0.0) | 0 (0.0) | 1 (9.1) | 1 (9.1) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 1 (5.6) |
| Lymphangiosis carcinomatosa | 1 (33.3) | 1 (33.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 1 (5.6) |
| Hypophosphataemic rickets | 0 (0.0) | 0 (0.0) | 1 (9.1) | 1 (9.1) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 1 (5.6) |
| Jaundice cholestatic | 0 (0.0) | 0 (0.0) | 1 (9.1) | 1 (9.1) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 1 (5.6) |
| Disease progression | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (25.0) | 1 (25.0) | 1 (5.6) | 1 (5.6) |
Coded with Medical Dictionary for Regulatory Activities Version 18.0.
Four DLTs were observed in four patients who received 120 mg/day milademetan. These included three events of platelet count decreased (grade 2, 3, or 4, with grade 4 being serious and grade 2 leading to dose interruption) and one event of grade 2 nausea. The drug was withdrawn in the patient who experienced grade 2 nausea, although the nausea was resolved with antiemetics. The patients who experienced grade 2 platelet count decreased and grade 2 nausea were unable to complete at least 75% of the prescribed doses of milademetan. No DLTs occurred in the 60‐mg and 90‐mg cohorts. The MTD was determined to be 90 mg/day for days 1 to 21 of a 28‐day cycle.
Six patients (four receiving 90‐mg dose, one receiving 60‐mg dose, and one receiving 120‐mg dose) developed eight serious TEAEs, including lymphangiosis carcinomatosa, anemia, tumor pain, urinary tract infection, jaundice cholestatic, disease progression, sepsis, and platelet count decreased. Of these, one patient, who received a dose of 120 mg/day, died as a result of disease progression, although this was not related to the study drug. This patient also experienced sepsis and platelet count decreased, with the platelet count decreased (grade 4, day 28) being the only serious drug‐related TEAE which lasted until death (day 35).
Grade ≥ 3 TEAEs were experienced by eight of the 18 patients (44.4%). Grade ≥ 3 TEAEs occurring in at least two patients included platelet count decreased (n = 4), anemia (n = 3), and ALT increased and hyponatremia (n = 2, each) (Table 2).
Three patients withdrew from the study because of TEAEs. One patient in the 90‐mg cohort discontinued study treatment because of grade 3 jaundice cholestatic, which was not considered to be related to the study drug. Two patients in the 120‐mg cohort discontinued study treatment because of drug‐related grade 3 platelet count decreased or grade 2 nausea. All three events were confirmed to have resolved after discontinuation of the study drug.
No patients required a dose reduction due to TEAEs. Dose interruptions were reported in ten patients who received 60 mg/day (n = 2), 90 mg/day (n = 5), and 120 mg/day (n = 3) of milademetan. Reasons for dose interruptions included nausea (n = 6), platelet count decreased (n = 4), and decreased appetite (n = 2).
3.2. Pharmacokinetics
A dose‐dependent increase in plasma concentration of milademetan was observed (Figure 2). The mean values of Cmax and AUC (AUCinf, AUCtau, and AUClast) increased with increasing dose of milademetan on both day 1 and day 21 of cycle 1. The median Tmax was approximately 3 hours in all cohorts. The mean half‐life (t1/2) ranged from 15.1 hours in the 60‐mg cohort to 17.9 hours in the 120‐mg cohort on day 1 and from 18.1 hours in the 60‐mg cohort to 26.1 hours in the 120‐mg cohort on day 21. The mean accumulation ratios ranged from 1.18 (120‐mg cohort) to 3.11 (60‐mg cohort). Table 3 shows the overall PK profile of milademetan (Supplementary Figures S1 and S2).
FIGURE 2.

Mean plasma concentration‐time curves of milademetan. Top panel: day 1 of cycle 1; bottom panel: day 21 of cycle 1. Cohort 1: 60 mg 21/28 once daily (n = 3); cohort 2: 90 mg 21/28 once daily (n = 11 for day 1, n = 9 for day 21); cohort 3: 120 mg 21/28 once daily (n = 4 for day 1, n = 2 for day 21). Lower limit of quantification (0.500 ng/mL). Error bars represent standard deviation. QC, quaque die
TABLE 3.
Pharmacokinetics of milademetan
| Pharmacokinetic parameter | Day 1 of cycle 1 | Day 21 of cycle 1 | ||||
|---|---|---|---|---|---|---|
|
Cohort 1 (60 mg/d) (n = 3) |
Cohort 2 (90 mg/d) (n = 11) |
Cohort 3 (120 mg/d) (n = 4) |
Cohort 1 (60 mg/d) (n = 2) |
Cohort 2 (90 mg/d) (n = 9) |
Cohort 3 (120 mg/d) (n = 1) |
|
| Cmax (ng/mL) | 429 (253) | 692 (231) | 1140 (549) | 625 (161) | 995 (415) | 1230 (NC) |
| Ctrough (ng/mL) | ‐ | ‐ | ‐ | 192 (29.7) | 389 (234) | 552 (NC) |
| Tmax (h) | 3.00 (2.02, 6.07) | 3.00 (2.92, 6.05) | 3.01 (2.87, 6.03) | 2.48 (1.95, 3.00) | 3.02 (2.00, 6.07) | 2.95 (2.95, 2.95) |
| AUCtau (ng·h/mL) | 4250 (2340) | 8380 (2660) | 14 900 (6330) | 7390 (2450) | 14 100 (6760) | 17 300 (NC) |
| AUClast (ng·h/mL) | ‐ | ‐ | ‐ | 10 300 (3830) | 20 300 (10 300) | 25 600 (NC) |
| AUCinf (ng·h/mL) | 6320 (3720) | 13 400 (5160) | 25 500 (10 700) | ‐ | ‐ | ‐ |
| t1/2 (h) | 15.1 (2.74) | 15.4 (3.06) | 17.9 (4.09) | 18.1 (6.90) | 20.9 (3.25) | 26.1 (NC) |
| Accumulation rate | ‐ | ‐ | ‐ | 3.11 (2.57) | 1.65 (0.440) | 1.18 (NC) |
Abbreviations: AUC, area under the concentration‐time curve; AUCinf, AUC up to infinity; AUClast, AUC up to the last quantifiable time; AUCtau, AUC during dosing interval; Cmax, maximum plasma concentration; Ctrough, trough plasma concentration; NC, not calculated; ‐, not applicable. Values are means (standard deviations) for all parameters, except Tmax which is median (minimum, maximum).
3.3. Efficacy endpoints
Seventeen patients were included in the efficacy analysis set (one patient in the 120‐mg cohort had no efficacy assessment after the first dose of the study drug). Objective response results were available for 16 patients, while one patient in the 90‐mg cohort was excluded from the objective response analysis because there was no target lesion at baseline. The mean (standard deviation) best (minimum) percentage change from baseline in target lesions was +6.8% (25.3%) in the 16 patients (Supplementary Figure S3). The individual percentage changes ranged widely from –15.1% to 95.0% without a relationship to the dose level of milademetan. No patients achieved complete or partial response. Stable disease was attained in seven out of 16 patients (43.8% [19.8%‐70.1%]), and progressive disease was observed as the best response in five out of 16 patients (31.3% [11.0%‐58.7%]). The remaining four patients were nonevaluable. The DCR was 43.8% (19.8%‐70.1%) (Table S1). The median (range) duration of stable disease was 55.4 (7.3‐202.1) weeks (including censored patients).
3.3.1. Progression‐free survival
Eight of the 17 patients experienced a PFS event. The PFS rate (95% confidence interval [CI]) was 56.8% (24.6%‐79.6%) at 3 months and 35.5% (9.6%‐63.2%) at 6, 9, and 12 months. The median (minimum, maximum) PFS was 15.29 (0.1, 202.1) weeks (including censored patients). The PFS reached 202.1 weeks with one patient in the 60‐mg cohort, who showed SD with best response.
3.4. Biomarker analysis
3.4.1. TP53 genomic status
Patients with TP53 mutations at the time of informed consent were not included in the study. However, six patients were found to have the TP53 mutations post enrollment.
3.4.2. MIC‐1 levels
MIC‐1 is a secreted p53 downstream gene product and is used as a pharmacodynamic biomarker for p53 activation. 16 Serum MIC‐1 samples were collected prior to dose of cycle 1 day 1 (including 6 hours after administration), day 2, day 8, day 15, and day 21. After administration of milademetan, the mean MIC‐1 levels increased from baseline in all cohorts (Supplementary Figure S4). The mean MIC‐1 levels were higher in the 90‐mg and 120‐mg cohorts compared with the 60‐mg cohort. The increase of MIC‐1 levels from baseline to day 21 [mean [standard deviation]) was 2175.7 (1677.40) pg/mL, 9514.5 (6812.78) pg/mL, and 7930.5 (7636.72) pg/mL in the 60‐mg, 90‐mg, and 120‐mg cohort, respectively.
4. DISCUSSION
This study is the first to examine the safety, tolerability, dose setting, and efficacy of milademetan in Japanese patients with advanced solid tumors who relapsed after or were refractory to standard therapy, or for whom no standard therapy was available. Along with other clinical studies, this study is part of a comprehensive worldwide clinical development program for milademetan.
In this phase I study, the MTD of milademetan was determined to be 90 mg in the once‐daily 21/28‐day schedule, although other dose schedules for single‐agent milademetan are also being explored (NCT01877382). In the current study, the most frequent TEAEs included nausea, decreased appetite, and platelet count decreased. Four DLTs were observed in four patients who received 120 mg/d milademetan. No DLTs occurred in the 60‐mg and 90‐mg cohorts. Serious TEAEs occurred in six out of 18 patients (33.3%), including one event of progressive disease that resulted in mortality.
In a US phase I study, four treatment schedules were tested, and the MTD was determined to be 120 mg in the once‐daily 21/28‐day schedule, 90 mg in the once‐daily 28/28‐day schedule, and 260 mg in the once‐daily 3/14‐day two‐time schedule, but the MTD was not determined in the once‐daily 7/28‐day schedule. Although we observed a difference in MTD in the once‐daily 21/28‐day schedule between the US phase I and Japanese phase I trial, the trends in AEs were similar across the studies. Based on the preliminary PK assessment of the US phase I study, milademetan exposure was comparable when 90 mg once daily was administered to Japanese patients and 120 mg once daily was administered to US patients, suggesting that the difference in milademetan exposure could partially explain the difference in MTD in the two populations (unpublished data). In general, patient characteristics such as body weight and BMI were different in the US phase I and Japanese phase I studies; lower body weight and BMI might result in lower MTD in Japanese patients. Further investigation in future clinical trials is needed to explore the factors that are associated with different PK and MTDs between Japanese and US patients. 12
Similar safety profiles for MDM2 inhibitors have been reported in other clinical studies. In a systematic review of 18 prospective clinical studies (January 1947 to November 2018) that investigated 10 MDM2 inhibitors in pediatric or adult cancers (n = 1005), the most common toxicities included gastrointestinal (GI) toxicity (reported in 14 studies), cytopenias (11 studies), fatigue (10 studies), infectious complications (including pneumonia and febrile neutropenia), stomatitis (six studies), and metabolic disturbances (five studies). 17 Cytopenias can be explained by the apoptotic depletion of megakaryocyte precursors by MDM2 inhibitors in in‐vitro models. 18 Hematological toxicities with MDM2 inhibitors support the role of MDM2 in normal hematopoiesis. GI toxicities can be explained by the loss of enterocytes owing to MDM2 inhibitors. 19
In an open‐label, randomized crossover study in patients (n = 76) with solid tumors, RG7112, an oral selective antagonist of MDM2, was generally well tolerated, with GI toxicities being the most commonly reported AEs. The most frequently reported grade ≥ 3 hematological toxicities were lymphocyte count decreased (11 patients) and platelet count decreased (two patients). 20 In a phase I study of 26 patients with wild‐type p53 and RAS or RAF mutations and locally advanced or metastatic solid tumors who were treated with SAR405838, an orally selective antagonist of MDM2, the most common reported AEs were diarrhea, blood creatine phosphokinase increased, nausea, and vomiting. The most common grade ≥ 3 hematological abnormalities were lymphocyte count decreased and platelet count decreased. 21 In an open‐label phase I study (n = 36) evaluating the safety, PK, and MTD of AMG 232, an oral selective antagonist of MDM2, in patients with acute myeloid leukemia, common treatment‐related AEs included nausea, diarrhea, and vomiting. Unacceptable GI AEs at higher doses resulted in the discontinuation of dose‐escalation, and the MTD of AMG 232 was not reached. Treatment‐related hematological toxicities included anemia, white blood cell count decreased, and platelet count decreased. 22
Eight out of 18 patients in the current study experienced grade ≥ 3 TEAEs, including grade 5 disease progression (one patient in the 120‐mg cohort) and grade 4 events of anemia and platelet count decreased (one patient each in the 90‐mg cohort), as well as sepsis, platelet count decreased, and lymphocyte count decreased (one patient each in the 120‐mg cohort). Three patients reported TEAEs leading to drug withdrawal: one in cohort 2 (cholestatic jaundice) and two patients in cohort 3 (platelet count decreased and nausea). Ten patients had TEAEs leading to dose interruption, and none of the patients experienced TEAEs that resulted in dose reduction. Other than platelet count decreased, we observed no clinically relevant changes in laboratory parameters including hematology, blood chemistry, coagulation, urinalysis, vital signs, or electrocardiogram parameters.
Although the sample size was small (or limited), milademetan showed a linear PK in the dose range of 60 to 120 mg/day in 21‐day cycles. The increase in mean Cmax and AUC (AUCtau and AUClast) was approximately dose proportional on both day 1 and day 21 of cycle 1. In our analysis, the mean t1/2 was 15.1 to 17.9 hours on day 1 and 18.1 to 26.1 hours on day 21. The mean (standard deviation) accumulation rate at the MTD (90 mg once daily; n = 9) was 1.65 (0.440).
Although no patients achieved complete or partial response, SD and DCR were attained in 43.8% patients. The median (range) duration of SD and PFS was 55.4 (7.3+, 202.1+) and 15.3 (0.1+, 202.1+) weeks, respectively. When compared with baseline, the mean (standard deviation) best percent change in target lesions for the 16 patients was 6.8% (25.3%). The change in target lesions ranged widely from –15.1% to 95.0% without a relationship to the milademetan dose level.
All seven patients who attained SD, including one who achieved 202.1 weeks in PFS had tumors with wild‐type TP53. Patients with wild‐type TP53 generally had better efficacy as measured by percent tumor shrinkage, DCR, and PFS, compared with those having mutant TP53. According to a small sample size limited the interpretation of these data, further investigation whether future clinical studies should be enriched with wild‐type TP53 patients might be needed. MIC‐1 is a transcriptional target of p53 and thus serves as a pharmacodynamic biomarker for p53 reactivation. The mean MIC‐1 levels increased exposure‐dependently with milademetan. The mean MIC‐1 levels in cohorts 2 and 3 were higher than those in cohort 1 at all time points. Together, these data suggest that the MDM2 inhibitor milademetan demonstrated on‐target inhibition of MDM2 to elicit reactivation of wild‐type p53. This study provides initial evidence of antitumor activity of milademetan in patients with wild‐type TP53 solid tumors who are intolerant to or failed to respond to standard therapy. However, it remains to be seen whether efficacy will be higher in select patients with high MDM2 protein expression or gene amplification or when milademetan is administered in combination with other anticancer agents.
MDM2 status was not defined as eligibility criteria in this study, as there was no robust evidence that MDM2 status was a suitable predictive biomarker when this study started. Milademetan was determined to be generally safe and well tolerated in a dose level of up to 90 mg in the once‐daily 21/28‐day schedule. The increase in Cmax and AUC was generally dose‐proportional in a dose range of 60 to 120 mg once daily. The MTD in the once‐daily 21/28‐day schedule was determined to be 90 mg in Japanese patients with solid tumors. Milademetan was well tolerated and showed modest antitumor activity in Japanese patients with solid tumors. Future studies would be needed to further evaluate the potential safety, tolerability, and clinical activity of milademetan in patients with solid tumors and lymphomas.
DISCLOSURE
Shunji Takahashi reports grants and personal fees from Daiichi Sankyo during the conduct of the study, and grants and personal fees from Ono, Bristol‐Myers‐Squib, Chugai, MSD, Novartis, Taiho, Bayer, AstraZeneca, and Eisai outside the submitted work. Yutaka Fujiwara reports honoraria from AstraZeneca and MSD, and research funds from AstraZeneca, AbbVie, Bristol‐Myers Squibb, Chugai, Eisai, Eli Lilly, Incyte, Merck Serono, MSD, and Novartis outside the submitted work. Toshio Shimizu reports grants and personal fees from Novartis, Pfizer, Eli Lilly, Loxo Oncology, Daiichi Sankyo, AstraZeneca, Eisai, AbbVie, Incyte, Bristol‐Myers Squibb, Takeda, Symbio, 3D‐Medicine, Chordia Therapeutics, Five Prime, PharmaMar, and Astellas outside the submitted work. Junichi Tomomatsu reports his family's annual remuneration for occupational physician from Eisai outside the submitted work. Takafumi Koyama reports honoraria from Cysmex and Chugai outside the submitted work. Noboru Yamamoto reports research funds from Daiichi Sankyo during the conduct of the study, research funds from Astellas, Chugai, Eisai, Taiho, Bristol‐Myers‐Squib, Pfizer, Novartis, Eli Lilly, AbbVie, Bayer, Boehringer Ingelheim, Kyowa‐Hakko Kirin, Takeda, Ono, Janssen, MSD, Merck, GSK, Sumitomo Dainippon, and Chiome Bioscience, and honoraria from Bristol‐Myers‐Squib, Pfizer, AstraZeneca, Eli Lilly, Ono, Chugai, and Sysmex outside the submitted work. Masaya Tachibana, Yasuyuki Kakurai, and Tomonari Yamashita are employees of Daiichi Sankyo Co., Ltd., Tokyo, Japan. Sakura Sakajiri was an employee of Daiichi Sankyo Co., Ltd, Tokyo, Japan during the conduct of the study. Kenji Nakano and Mariko Ogura have no conflicts of interest to declare.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Table S1
ACKNOWLEDGMENTS
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work, and have given approval for the final version to be published. Funding for this research was provided by Daiichi Sankyo Co., Ltd., Tokyo, Japan. The authors thank Taiga Takagi, Shohei Osawa, Hiroyuki Sumi, Sachiko Sakakibara, Hiroyuki Inoue, Nobuaki Adachi, and Daisuke Kuroki from Daiichi Sankyo Co., Ltd. for supporting the medical review and editing, and Kenji Nakamaru for the biomarker strategy. Medical writing assistance was provided by Dr Tarveen Jandoo and Amit Garg from Enago Life Sciences, India, which was funded by Daiichi Sankyo Co., Ltd., Tokyo, Japan in accordance with the Good Publication Practice (GPP3) guidelines.
Takahashi S, Fujiwara Y, Nakano K, et al. Safety and pharmacokinetics of milademetan, a MDM2 inhibitor, in Japanese patients with solid tumors: A phase I study. Cancer Sci. 2021;112:2361–2370. 10.1111/cas.14875
REFERENCES
- 1. Blagih J, Buck MD, Vousden KH. p53, cancer and the immune response. J Cell Sci. 2020;133:jcs237453. [DOI] [PubMed] [Google Scholar]
- 2. Chen J. The cell‐cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb Perspect Med. 2016;6:a026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rivlin N, Brosh R, Oren M, Rotter V. Mutations in the p53 tumor suppressor gene: important milestones at the various steps of tumorigenesis. Genes Cancer. 2011;2:466‐474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li W, Peng X, Lang J, Xu C. Targeting mouse double minute 2: current concepts in DNA damage repair and therapeutic approaches in cancer. Front Pharmacol. 2020;11:631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vassilev LT. In vivo activation of the p53 pathway by small‐molecule antagonists of MDM2. Science. 2004;303:844‐848. [DOI] [PubMed] [Google Scholar]
- 6. Shangary S, Qin D, McEachern D, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci USA. 2008;105:3933‐3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol Med. 2007;13:23‐31. [DOI] [PubMed] [Google Scholar]
- 8. Liao G, Yang D, Ma L, et al. The development of piperidinones as potent MDM2‐P53 protein‐protein interaction inhibitors for cancer therapy. Eur J Med Chem. 2018;159:1‐9. [DOI] [PubMed] [Google Scholar]
- 9. Wang S, Zhao Y, Aguilar A, Bernard D, Yang C‐Y. Targeting the MDM2‐p53 protein‐protein interaction for new cancer therapy: progress and challenges. Cold Spring Harb Perspect Med. 2017;7:a026245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arnhold V, Schmelz K, Proba J, et al. Reactivating TP53 signaling by the novel MDM2 inhibitor DS‐3032b as a therapeutic option for high‐risk neuroblastoma. Oncotarget. 2017;9:2304‐2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. NCBI. National Center for Biotechnology Information . PubChem Compound Summary for CID 73297272, Milademetan. Milademetan. https://pubchem.ncbi.nlm.nih.gov/compound/Milademetan. Accessed Jul. 19, 2020. 2020.
- 12. Bauer TM, Gounder MM, Weise AM, et al. A phase 1 study of MDM2 inhibitor DS‐3032b in patients with well/de‐differentiated liposarcoma (WD/DD LPS), solid tumors (ST) and lymphomas (L). J Clin Oncol. 2018;36:11514. [Google Scholar]
- 13. Gounder MM, Bauer TM, Schwartz GK, et al. Milademetan, an oral MDM2 inhibitor, in well‐differentiated/ dedifferentiated liposarcoma: results from a phase 1 study in patients with solid tumors or lymphomas. Eur J Cancer. 2020;138:S3‐S4. [Google Scholar]
- 14. Neuenschwander B, Branson M, Gsponer T. Critical aspects of the Bayesian approach to phase I cancer trials. Stat Med. 2008;27:2420‐2439. [DOI] [PubMed] [Google Scholar]
- 15. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228‐247. [DOI] [PubMed] [Google Scholar]
- 16. Jeay S, Ferretti S, Holzer P, et al. Dose and schedule determine distinct molecular mechanisms underlying the efficacy of the p53‐MDM2 inhibitor HDM201. Cancer Res. 2018;78:6257‐6267. [DOI] [PubMed] [Google Scholar]
- 17. Pi L, Rooprai J, Allan DS, et al. Evaluating dose‐limiting toxicities of MDM2 inhibitors in patients with solid organ and hematologic malignancies: a systematic review of the literature. Leuk Res. 2019;86:106222. [DOI] [PubMed] [Google Scholar]
- 18. Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discovery. 2014;13:217‐236. [DOI] [PubMed] [Google Scholar]
- 19. Tisato V, Voltan R, Gonelli A, Secchiero P, Zauli G. MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. J Hematol Oncol. 2017;10:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Patnaik A, Tolcher A, Beeram M, et al. Clinical pharmacology characterization of RG7112, an MDM2 antagonist, in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2015;76:587‐595. [DOI] [PubMed] [Google Scholar]
- 21. de Weger VA, de Jonge M, Langenberg MHG, et al. A phase I study of the HDM2 antagonist SAR405838 combined with the MEK inhibitor pimasertib in patients with advanced solid tumours. Br J Cancer. 2019;120:286‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Erba HP, Becker PS, Shami PJ, et al. Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv. 2019;3:1939‐1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Table S1