

Abstract
Podoplanin is a key molecule for enhancing tumor‐induced platelet aggregation. Podoplanin interacts with CLEC‐2 on platelets via PLatelet Aggregation–inducing domains (PLAGs). Among our generated antibodies, those targeting the fourth PLAG domain (PLAG4) strongly suppress podoplanin–CLEC‐2 binding and podoplanin‐expressing tumor growth and metastasis. We previously performed a single‐dose toxicity study of PLAG4‐targeting anti‐podoplanin–neutralizing antibodies and found no acute toxicity in cynomolgus monkeys. To confirm the therapeutic efficacy and toxicity of podoplanin‐targeting antibodies, a syngeneic mouse model that enables repeated dose toxicity tests is needed. Replacement of mouse PLAG1‐PLAG4 domains with human homologous domains drastically decreased the platelet‐aggregating activity. Therefore, we searched the critical domain of the platelet‐aggregating activity in mouse podoplanin and found that the mouse PLAG4 domain played a critical role in platelet aggregation, similar to the human PLAG4 domain. Human/mouse chimeric podoplanin, in which a limited region containing mouse PLAG4 was replaced with human homologous region, exhibited a similar platelet‐aggregating activity to wild‐type mouse podoplanin. Thus, we generated knock‐in mice with human/mouse chimeric podoplanin expression (PdpnKI/KI mice). Our previously established PLAG4‐targeting antibodies could suppress human/mouse chimeric podoplanin–mediated platelet aggregation and tumor growth in PdpnKI/KI mice. Repeated treatment of PdpnKI/KI mice with antibody‐dependent cell‐mediated cytotoxicity activity–possessing PG4D2 antibody did not result in toxicity or changes in hematological and biochemical parameters. Our results suggest that anti‐podoplanin–neutralizing antibodies could be used safely as novel anti‐tumor agents. Our generated PdpnKI/KI mice are useful for investigating the efficacy and toxicity of human podoplanin–targeting drugs.
Keywords: CLEC‐2, knock‐in mice, platelet aggregation, podoplanin, toxicity
Podoplanin is a key molecule for enhancing tumor‐induced platelet aggregation. Here, we report a knock‐in mouse with human/mouse chimeric podoplanin expression to examine the therapeutic efficacy and toxicity of podoplanin‐targeting agents. Using this knock‐in mouse model, we confirmed that anti‐podoplanin–neutralizing antibodies could be used safely as novel anti‐tumor agents.

1. INTRODUCTION
The interaction between tumor cells and platelets is the most crucial step to promote distant metastasis of tumor cells. Activated platelets facilitate the survival of circulating tumor cells by secreting various growth factors and cytokines, protecting tumor cells from immunological assault or shear stress in the blood flow and enhancing the formation of tumor cell–platelet aggregates that could enhance embolization in the microvasculature, which results in metastatic formation in distant organs. The importance of platelets in tumor metastasis is also supported by the fact that anti‐platelet agents and thrombocytopenia reduce the incidence of metastasis in some experimental models 1 , 2 and that the administration of anti‐coagulants lowers the mortality rate. 3 , 4 Thus, platelets contribute to tumor malignancy, and platelet‐tumor interaction is a potential target for developing novel anti‐tumor agents.
Podoplanin (PDPN), also known as Aggrus/T1alpha/PA2.26, is a type I transmembrane sialoglycoprotein and was previously identified as a platelet aggregation–inducing factor expressed on highly metastatic tumor cells. 5 The PDPN expression level has been reported to be high in a wide range of tumor cells, including squamous cell carcinoma, 6 mesothelioma, 7 glioblastoma, 8 bladder tumors, 9 and osteosarcoma 10 cells. Soluble PDPN was also detected in the blood samples of patients with adenocarcinoma, squamous cell carcinoma, lung cancer, gastric cancer, and rectal cancer compared with that of healthy individuals. 11 The soluble PDPN levels were reported to be higher in patients with metastatic cancers than in those with non‐metastatic cancer. 11
PDPN induces platelet aggregation by directly interacting with C‐type lectin‐like receptor 2 (CLEC‐2) on platelets, 12 , 13 and its binding to CLEC‐2 transmits platelet‐activation signals through Src family kinases, Syk, and phospholipase Cγ2 in platelets. 14 , 15 Soluble factors such as PDGF and TGF‐β secreted from activated platelets are also known to induce tumor growth and epithelial‐mesenchymal transition. 16 , 17 , 18 Therefore, PDPN expression is well recognized to be correlated with poor prognosis and tumor malignancy in lung carcinomas, oral squamous cell carcinomas, and breast cancers. 19 , 20 , 21 Intriguingly, CLEC‐2–deficient platelets have been reported to respond normally to platelet agonists such as collagen, ADP, U46619, and PAR‐4, which suggests that inhibition of the PDPN–CLEC‐2 interaction may not affect physiological hemostasis. 22
PDPN contains four conserved motif ED(X)XXTs (where X may be any amino acid) in the extracellular domain. These domains were designated as PLatelet AGgregation‐stimulating (PLAG) domains critical for binding to CLEC‐2 and exhibiting PDPN‐mediated platelet‐aggregating ability. 5 , 17 , 23 In particular, the fourth PLAG domain (PLAG4), which shows similarity to other PLAG domains and is highly conserved among mammals, contributed to binding to CLEC‐2 and to its platelet aggregation–inducing ability. Our established mouse anti‐human PLAG4 mAbs, PG4D1 and PG4D2, could almost completely inhibit PDPN–CLEC‐2 binding and suppressed PDPN‐mediated hematogenous metastasis in vivo. 23 Furthermore, PG4D2 mAb could suppress the growth of PDPN‐positive lung squamous cell carcinoma PC‐10 tumors xenografted into NOD/SCID mice. Therefore, PDPN is a promising novel target for suppressing PDPN‐positive tumor growth and metastasis. The antibodies to human PDPN, especially to the PLAG4 domain, might be useful as anti‐metastatic and anti‐tumor agents in clinical situations.
Not only tumors but also PDPN expression was detected in various normal tissues and cells such as lymphatic vessels, kidney podocytes, mesothelium, and alveolar epithelium. 24 , 25 In the absence of PDPN, the perinatal development of lung and lymphatics is lethally hindered. 26 , 27 To evaluate the adverse effects and toxicities caused by blocking PDPN function, we previously identified the monkey PLAG4 domain and established the monkey PLAG4–targeting anti‐PDPN–neutralizing mAb (2F7). 28 2F7 strongly suppressed the platelet aggregation and pulmonary metastasis of monkey PDPN–expressing CHO cells. Single‐dose administration of 2F7 did not exhibit any acute toxicity in cynomolgus monkeys. 28 To assess the toxicity and therapeutic efficacy of PDPN‐targeting antibodies, a syngeneic mouse model that enables repeated dose toxicity tests is needed. 13 In clinical situations, multiple administrations of neutralizing mAb would be recommended to obtain higher therapeutic effects. However, there is no report how toxicity will occur by multiple administration of PDPN‐targeting antibodies.
In this study, we aimed to establish knock‐in mice to examine the therapeutic efficacy and toxicity of anti‐PDPN–neutralizing antibodies and confirmed that our established anti‐PDPN–neutralizing antibodies did not exhibit any severe toxicity in the established knock‐in mice.
2. MATERIALS AND METHODS
2.1. Plasmid construction
Mouse podoplanin cDNA was cloned into the pcDNA3 vector, and the pcDNA3‐mouse podoplanin plasmid (mPDPN) was used to generate mutated mouse podoplanin cDNAs (mPDPN‐ΔPLAG1, mPDPN‐ΔPLAG3, mPDPN‐ΔPLAG4, mPDPN‐ΔPLAG1+4, mPDPN‐ΔPLAG3+4, mPDPN‐ΔPLAG1+3+4, mPDPN‐E81A, mPDPN‐E82A, mPDPN‐L83A, mPDPN‐S84A, and mPDPN‐T85A) or mPDPN with human PLAG1‐PLAG4 domains (mPDPN‐hPLAGs) and human/mouse chimeric PDPN (chiPDPN), which were replaced with human PDPN (Accession No. NM_010329.3), using a QuikChange site‐directed mutagenesis kit (Agilent Technology) according to the manufacturer's instructions. The pcDNA3 vector containing human podoplanin cDNA was generated previously. 5 ChiPDPN was also cloned into the PB‐EF1α‐MCS‐IRES‐Neo cloning and expression vector (PB533A‐2, Systems Biosciences).
2.2. Cell lines and culture conditions
CHO cells were purchased from ATCC and cultured in RPMI 1640 media (Wako) containing 10% FBS (Sigma‐Aldrich). We established CHO cell lines stably transfected with the pcDNA3 vector alone (CHO/mock), pcDNA3 vector containing wild‐type (WT) mouse PDPN (CHO/mPDPN‐WT), WT human PDPN (CHO/hPDPN‐WT), mPDPN mutants with PLAG deletions (CHO/mPDPN‐ΔPLAG1, mPDPN‐ΔPLAG3, mPDPN‐ΔPLAG4, mPDPN‐ΔPLAG1+4, mPDPN‐ΔPLAG3+4, and mPDPN‐ΔPLAG1+3+4), mPDPN point mutants (CHO/mPDPN‐E81A, mPDPN‐E82A, mPDPN‐L83A, mPDPN‐S84A, and mPDPN‐T85A), mPDPN with human PLAG1‐4 domains (CHO/mPDPN‐hPLAGs), or chimeric human/mouse podoplanin (CHO/chiPDPN), according to the procedure described previously. 5 MC38 cells derived from C57BL/6 murine colon adenocarcinoma were purchased from Kerafast and cultured in low‐glucose DMEM (Wako) supplemented with 10% FBS (Sigma). We established MC38 cell lines that were stably transfected with PB‐EF1α‐MCS‐IRES‐Neo vector alone (MC38/mock) or in combination with chiPDPN (MC38/chiPDPN) according to the manufacturer's instructions. Mouse anti‐digoxin mAb (IgG1 subclass)‐producing DIG104.10H.1 hybridoma cells were obtained from the JCRB cell bank (Osaka, Japan) and cultured in DMEM low‐glucose medium (Wako) containing 10% FBS. Mouse anti‐JDP2 mAb (IgG2a subclass)‐producing J#176‐3.2 hybridoma cells 29 were obtained from the Riken BRC cell bank (Ibaraki, Japan) and cultured in RPMI 1640 media (Wako) containing 10% FBS (Sigma).
2.3. Animals
Jcl:ICR (Institute of Cancer Research), C57BL/6N, and BALB/c‐nu/nu were purchased from Charles River. All animal experimental procedures were conducted in accordance with the guidelines of the Japanese Foundation for Cancer Research Animal Care and Use Committee. All mice were housed in specific pathogen‐free conditions.
2.4. Western blot analysis
Each sample was treated with a previously described procedure. 28 Samples were incubated with primary antibodies to PDPN (mouse anti‐human PDPN mAbs, PG4D1 and PG4D2; Syrian hamster anti‐mouse PDPN mAb, ab11936, Abcam) and β‐actin (clone, AC‐15; Sigma‐Aldrich), treated with HRP‐conjugated anti‐mouse IgG (RPN2232, GE Healthcare), anti‐hamster IgG (57003, Cappel), and mouse TrueBlot ULTRA (18‐8817‐33, Rockland), and then reacted with an ECL Prime Western Blotting Detection reagent (GE Healthcare). The proteins were visualized with enhanced chemiluminescence using Amersham Imager 600 (GE Healthcare).
2.5. Flow cytometric analysis
Cells were harvested and treated with 2 μg/mL of anti‐PDPN antibodies (PG4D1, PG4D2, or rat anti‐mouse PDPN mAb, 8F11), followed by incubation with Alexa Flour 488–conjugated anti‐mouse (H+L) or anti‐rat IgG (H+L; Thermo Fisher Scientific). For the CLEC‐2 binding analysis, cells were incubated with 0.4‐10 μg/mL of (His)6‐tagged mouse CLEC‐2 (R&D Systems) or (His)10‐tagged human CLEC‐2 (R&D Systems), followed by incubation with Alexa Flour 488–conjugated anti‐Penta–His antibody (Qiagen). For the antibody inhibition assay, cells were incubated with 100 μg/mL of anti‐PDPN mAbs (PG4D1 and PG4D2) or control IgG for 30 minutes on ice before incubation with 10 μg/mL of mouse CLEC‐2. Fluorescence intensity was measured using a Cytomics FC500 flow cytometry system (Beckman Coulter) and analyzed with the FlowJo software (Treestar).
2.6. Platelet aggregation assay
Washed platelets mixture from Jcl:ICR, C57BL/6N, or PdpnKI/KI mice were prepared according to a previously described procedure. 28 For the antibody inhibition assay, cells were incubated with 30 μg/mL of antibody or control IgG for 30 minutes on ice before incubation with washed platelet mixture. The platelet aggregation rate was estimated using a platelet aggregometer (MCM HEMA TRACER 313M; SSR Engineering).
2.7. Generation of PdpnKI/KI mice with chiPDPN
A targeting vector to generate human/mouse chimeric Pdpn knock‐in mice, in which a part of the mouse PDPN–containing PLAG4 domain was replaced with a human homologous region, was designed so that human/mouse chimeric exon 3 and loxP‐flanked PGK‐neo cassette could be inserted between exons 2 and 3. ES cells (RENKA) from C57BL/6N mice were transfected with the targeting vector and injected into the blastocyst of the host embryo (ICR). F1 heterozygous mice (PdpnKI+neo/+) from homogeneous recombinant ES cells were crossed with CAG‐Cre mice (B6; CBA‐Tg [CAG‐Cre] 47Imeg). F2 mice (PdpnKI/+Tg [CAG‐Cre] 47Imeg) without a Neo cassette transgene were crossed with C57BL/6N to remove the Cre transgene. PdpnKI/KI mice were obtained by crossing between the heterozygous F3 generation (PdpnKI/+). For analysis of the genotypes of the PdpnKI/KI mice, DNA samples from C57BL/6N, PdpnKI/+, and PdpnKI/KI mice were subjected to 30 cycles of amplification, with each cycle consisting of 10 s at 98°C, 30 s at 64°C, and 2 minutes at 68°C, followed by an extension of 2 minutes at 68°C on a thermal cycler by KOD EX (TOYOBO) using the P1 (CAGTCTCCGTGCCTTCTTGTTTTAG) and P2 (CTTTACAGATGGTTATGAGCTTTCACG) primers. The PCR products were electrophoresed in agarose gels. The WT allele provided a 0.3‐kbp band, whereas the Cre‐mediated allele provided a 0.4‐kbp band. All knock‐in mouse generation procedures were performed at Trans Genic Inc.
2.8. Establishment of immortalized cells from PdpnKI/KI mice with chiPDPN
Lungs and lymph nodes were excised from the PdpnKI/KI or C57BL/6 mice, minced, and treated with collagenase/dispase. Adherent cells were infected with the supernatant from the SV40 strains 777 expressing large T antigen and cultured in DMEM media (Wako) containing 10% FBS (Sigma).
2.9. Anti‐tumor effect of neutralizing anti‐PDPN antibodies on chiPDPN‐expressing tumors xenografted into PdpnKI/KI mice
A total of 1 × 106 MC38/chiPDPN cells in 100 µL Hanks' Balnaced salt solutions (HBSS) was subcutaneously inoculated into the right and left flanks of 7‐9‐month‐old female PdpnKI/KI mice. When the average estimated tumor volume was around 50‐100 mm3, mice with comparable tumor volume were randomly grouped. Antibodies were intraperitoneally administered twice a week. Tumor volume was calculated as length × width2 × 0.5 (mm3). Body weight was measured on the first and last days of the experiment.
2.10. Antibody purification
For the toxicity test with PdpnKI/KI mice, large‐scale purifications of control IgG1, control IgG2a, PG4D1, and PG4D2 mAbs were performed. Six‐week‐old female BALB/c‐nu/nu mice were intraperitoneally injected with control IgG‐secreting hybridomas (DIG104.10H.1 for control IgG1 and J#176‐3.2 for control IgG2a), PG4D1‐secreting hybridomas, or PG4D2‐secreting hybridomas. Ascitic fluid was collected, and antibody was purified as previously described. 28
2.11. Repeated administration of anti‐PDPN–neutralizing antibodies in PdpnKI/KI mice
Five‐week‐old male and female PdpnKI/KI mice (each sex, n = 2) were intravenously injected with 10 or 50 mg/kg of PDPN‐neutralizing antibodies (PG4D1 and PG4D2) or control IgG (IgG1 and IgG2a) through the lateral tail vein, five times every other week. The mice were euthanized 1 week after the final injection, tissues and organs were weighed, and hematology and biochemistry analyses were performed. Tissue sections were fixed in 10% neutral buffer formalin and stained with hematoxylin eosin. The tissue specimens were analyzed by two independent pathologists who were blinded to the diagnosis. All animal procedures were conducted in accordance with the guidelines of the Nihon Bioresearch Inc Animal Care and Use Committee.
2.12. Statistical analyses
The Student t test or Mann‐Whitney U test was performed to determine the statistical significance of the comparisons. Statistical significance was assumed for *P <.05 or **P <.01. All statistical tests were two‐sided.
3. RESULTS
3.1. Critical role of the mouse PLAG4 domain in CLEC‐2 binding and platelet activation
PDPN contains four conserved PLAG domains. 5 , 23 Among our identified PLAG domains, the fourth PLAG domain (PLAG4) in human PDPN (hPDPN) was found to play an important role in CLEC‐2 binding and platelet aggregation. 23 , 28 The amino acid sequence alignment in mouse and human revealed that mouse PDPN (mPDPN) had a similar consensus motif (82‐EELST‐86) to human PLAG4 (81‐EDLPT‐85; Figure 1A, see Supplemental materials and methods). To evaluate the contributions of each mouse PLAG domain to CLEC‐2 binding, we established CHO cells expressing PLAG domain–deleted mPDPN mutants (CHO/mPDPN‐ΔPLAG1, ‐ΔPLAG3, ‐ΔPLAG4, ‐ΔPLAG1+4, ‐ΔPLAG3+4, and ‐ΔPLAG1+3+4; Figure 1B) and assessed their binding abilities to recombinant mouse CLEC‐2 (mCLEC‐2). The expression levels of WT and mutant PDPN transfectants were compared using an 8F11 anti‐mPDPN mAb, which can recognize the perimeter structure from Asp39 to Glu47, which are close to the PLAG2 and PLAG3 domains 5 (Figure 1A). We selected clones that expressed almost the same level of cell surface PDPN (Figure 1C, left panels and 1D, upper panels). mCLEC‐2 binding was almost the same between the ΔPLAG1 and ΔPLAG3 mutants and the WT mPDPN (Figure 1C, right panels and 1D, lower panels); almost completely inhibited to the ΔPLAG4 mutant; and completely absent to the ΔPLAG1+4 and ΔPLAG1+3+4 mutants. However, partial binding ability remained to the ΔPLAG 3+4 mutants, such as the ΔPLAG4 mutant (Figure 1C, right panels and 1D, lower panels). Consistent with the CLEC‐2–binding activity, the platelet‐aggregating ability was lowest in the ΔPLAG4 mutant than in the ΔPLAG1 and ΔPLAG3 mutants (Figure 1E).
FIGURE 1.

PLAG4 domain in mouse podoplanin (PDPN) plays an essential role in platelet aggregation. A, Amino acid sequence alignment of human and mouse PDPN. The recognition domains of the rat anti‐mouse PDPN 8F11 mAb and the mouse anti‐human PDPN antibodies PG4D1 and PG4D2 are shown. The GenBank accession numbers were as follows: Homo sapiens, NM_006474.4; Mus musculus, NM_010329.3. B, Schematic representation of wild‐type (WT) mouse PDPN protein and PLAG domain–deleted mouse PDPN protein (ΔPLAG1, ΔPLAG3, ΔPLAG4, ΔPLAG1+4, ΔPLAG3+4, and ΔPLAG1+3+4). C, CHO cells were stably transfected with an empty vector (mock) or expression plasmids containing WT mouse PDPN (mPDPN) or its PLAG domain–deleted mPDPN‐expressing plasmids (ΔPLAG1, ΔPLAG3, ΔPLAG4, ΔPLAG1+4, ΔPLAG3+4, and ΔPLAG1+3+4), followed by incubation with PBS (closed areas) or an anti‐PDPN mAb (8F11; open areas). In some experiments (right panels), these cells were incubated with PBS (closed areas) or (His)6‐tagged mouse C‐type lectin‐like receptor 2 (mCLEC‐2; open areas). After washing, the cells were incubated with Alexa Flour 488–conjugated second antibody, and fluorescence intensity was measured by flow cytometry. D, The quantitative data of C are shown. Each value is presented as the mean ± SD (n = 3) of the peak values (mean fluorescent intensity [MFI]) normalized by that of the WT mPDPN‐expressing CHO cells. *P <.05 using the Mann‐Whitney U test. ns, not significant. E, CHO cells stably transfected with WT or its PLAG domain–deleted mPDPN‐expressing plasmids were incubated with mouse PRP. The platelet aggregation rate was estimated using an aggregometer
To exclude the effects of conformational changes after deletion of the mCLEC‐2–binding region, we established CHO cells that had been transfected with mPDPN harboring each amino acid of the PLAG4 domain to Ala (Figure S1A). Point mutations at Glu81, Glu82, and Thr85 to Ala (E81A, E82A, and T85) drastically decreased their binding capability to mCLEC‐2 (Figures S1B, right panels and S1C, lower panel). This result indicated that the Glu81, Glu82 , and Thr85 residues in the PLAG4 domain were crucial for binding to mCLEC‐2.
3.2. Attenuation of chiPDPN‐mediated platelet aggregation by neutralizing anti‐human PDPN antibodies
To estimate the effects of our previously generated anti‐hPDPN–specific neutralizing mAbs PG4D1 and PG4D2 in a syngeneic mouse model, we generated a chiPDPN that possessed the ability to induce platelet aggregation. Interestingly, replacement of mouse PLAG1‐PLAG4 domains with human PLAG1‐PLAG4 domains (mPDPN‐hPLAGs) drastically decreased the platelet‐aggregating activity (Figures S2A,B). To clarify the cross‐species PDPN binding to CLEC‐2, we first examined the binding affinity of mouse and human CLEC‐2 to hPDPN. The binding affinity to hPDPN was weaker for mCLEC‐2 than for hCLEC‐2 (Figure S2C). Similarly, the binding affinity to mPDPN was weaker for hCLEC‐2 than for mCLEC‐2 (Figure S2D). Therefore, we narrowed the replacement residues and finally obtained the ideal residue containing the mouse PLAG4 domain. We replaced the residue with human counterparts and named the chimeric protein chiPDPN (Figure 2A). CHO cells that were stably transfected with vectors containing chiPDPN exhibited almost the same platelet‐aggregating ability, compared with that of the mPDPN transfectants (Figure 2B). Because the PG4D1 and PG4D2 mAbs recognized the perimeter structure from Arg79 to Leu83 (79‐RIEDL‐83) in hPDPN (Figure 1A), both mAbs could recognize the CHO cells expressing chiPDPN and hPDPN but not mPDPN (Figure 2C) and the synthetic peptides from chiPDPN and hPDPN but not mPDPN (Figures S3A‐E, see Supplemental materials and methods). Preincubation of CHO cells expressing chiPDPN and hPDPN with PG4D1 or PG4D2 mAb attenuated chiPDPN or hPDPN binding to mCLEC‐2 (Figure 2D) and delayed the onset of chiPDPN‐ or hPDPN‐induced platelet aggregation (Figure 2E).
FIGURE 2.

Neutralizing anti‐human podoplanin (PDPN) antibodies could attenuate human/mouse chimeric PDPN (chiPDPN)‐mediated platelet aggregation. A, Schematic representation of amino acid sequences of chiPDPN, in which the murine PDPN (mPDPN) PLAG4 domain–containing region was replaced with a human homologous region. Gray areas indicate mPDPN, and the red areas indicate the region replaced with human PDPN (hPDPN) sequences. B, CHO cells were stably transfected with WT‐mPDPN, WT‐hPDPN, or chiPDPN‐expressing plasmids followed by incubation with mouse platelets. The platelet aggregation rate was estimated using an aggregometer. C, CHO cells that had been stably transfected with empty vector (mock) or WT‐mPDPN, WT‐hPDPN, or chiPDPN‐expressing plasmids were treated with PBS (closed areas) or anti‐PDPN mAbs, PG4D1 (upper panels) or PG4D2 (lower panels). After washing, the cells were incubated with Alexa Flour 488–conjugated anti‐mouse IgG antibody, and fluorescence intensity was measured by flow cytometry. D, Transfected CHO cells as shown in C were incubated with PBS (closed gray areas), control IgG (open areas), or anti‐PDPN mAbs, PG4D1 (upper panels) or PG4D2 (lower panels) (closed green areas), followed by incubation with PBS (closed gray areas) or (His)6‐tagged mouse C‐type lectin‐like receptor 2 (CLEC‐2; open areas and closed green areas). After washing, the cells were incubated with Alexa Flour 488–conjugated anti‐penta‐His antibody, and fluorescence intensity was measured by flow cytometry. E, Transfected CHO cells as shown in B were incubated with control IgG1, control IgG2a, PG4D1, or PG4D2, followed by incubation with mouse platelets. The aggregation rate was estimated using an aggregometer
3.3. Expression pattern and distribution were similar between chiPDPN in PdpnKI/KI mice and WT PDPN in C57BL/6N mice
To estimate the in vivo function of chiPDPN, we generated knock‐in mice with chiPDPN (PdpnKI/KI mice), in which Lys79 to Ser86 of mPDPN was replaced with a human homologous region (Gly78 to Pro85; Figure 3A‐C). A targeting vector for replacing mouse exon 3 with human/mouse chimeric exon 3 and loxP‐flanked PGK‐neo cassette was transfected into ES cells from C57BL/6N mice (Figure 3A). F1 heterozygous mice (PdpnKI+neo/+) from the ES cells were generated and Neo cassette and Cre transgene in F1 mice were removed using the strategy described in Figure 3B. PdpnKI/KI mice were finally selected by genotyping PCR (Figure 3C), and mating with F3 hemizygous mice (PdpnKI/ +) produced 14 offspring, including 3 PdpnKI/KI, 6 PdpnKI/ +, and 5 WT mice with the same sex distribution (Table S1). To confirm chiPDPN expression in the PdpnKI/KI mice, tissue lysates from PdpnKI/KI and C57BL/6N mice were immunoblotted with anti‐hPDPN–specific mAb PG4D2 and anti‐mPDPN–specific mAb ab11936 (Figure 3D). As expected, chiPDPN expression was detected by PG4D2 mAb in male and female PdpnKI/KI mice but not in male and female C57BL/6N mice. The chiPDPN expression and distribution patterns in the organs of PdpnKI/KI mice were almost the same as the mPDPN expression and distribution patterns detected by ab11936 mAb in C57BL/6N mice (Figure 3D).
FIGURE 3.

Generation of knock‐in mouse with human/mouse chimeric podoplanin (chiPDPN). A, Schematic representation of the knock‐in strategy of the human PLAG4 domain–containing region into the mouse podoplanin (mPDPN) homologous region for generating a chiPDPN‐expressing mouse model. The targeting vector contained human/mouse chimeric exon 3 and loxP‐flanked PGK‐neo cassette, which was inserted between exons 2 and 3 of the murine pdpn genomic locus. After successful homologous recombination in ES cells, the recombined allele continued into the germline. B, The Neo cassette was removed by crossing onto CAG‐Cre mice. The Cre transgene was removed by crossing onto C57BL/6N mice after establishment of the PdpnKI/KI mice. C, PCR genotyping of F4 generation as PdpnKI/KI mice mated between F3 generation (PdpnKI/+). PCR genotyping using the P1 and P2 primers revealed the identification of the PdpnKI/KI mouse line. WT mouse (+/+): 0.3 kbp, hemizygous mouse (PdpnKI/+): 0.4 kbp + 0.3 kbp, homozygous mouse (PdpnKI/KI): 0.4 kbp. D, Tissues from homozygous knock‐in mouse (PdpnKI/KI; left panels) or C57BL/6N (right panels), or lysates from CHO cells that had been transfected with empty vector (CHO/mock), WT‐hPDPN–expressing plasmids (CHO/hPDPN) or WT‐mPDPN–expressing plasmids (CHO/mPDPN) were immunoblotted with PG4D2, ab11936, or anti‐β‐actin. Schematic representation of the epitopes of PG4D2 and ab11936 is shown in the top of the panel. PG4D2 mAb could recognize chiPDPN but not WT‐mPDPN and ab11936 mAb could recognize both chiPDPN and WT‐mPDPN
3.4. Usefulness of PdpnKI/KI mice as a syngeneic mouse model to evaluate neutralizing anti‐PDPN mAb
Platelets derived from PdpnKI/KI mice possessed the same aggregating ability (Figure 4A) as that of platelets derived from C57BL/6N mice. Moreover, the anti‐hPDPN–specific PG4D1 and PG4D2 mAbs suppressed platelet aggregation mediated by chiPDPN but not platelet aggregation mediated by mPDPN (Figure 4A). Immortalized lung or lymph node cells from PdpnKI/KI mice expressed chiPDPN on the cell surface (Figure 4B). In contrast, immortalized cells from the lung or lymph node of C57BL/6N mice could be detected only by the anti‐mPDPN–specific 8F11 mAb (Figure 4B). Addition of PG4D1 or PG4D2 mAb delayed the onset of platelet aggregation mediated by the immortalized cells from PdpnKI/KI mice (Figure 4C); this was not observed when the immortalized lung or lymph node cells of C57BL/6N mice were preincubated with PG4D1 or PG4D2 mAb (Figure 4C).
FIGURE 4.

Evaluation of the therapeutic efficacy of neutralizing anti‐human podoplanin antibodies using knock‐in mouse with a chimeric human/murine podoplanin (chiPDPN). A, CHO cells that had been stably transfected with WT mouse podoplanin (mPDPN) or chiPDPN‐expressing plasmids were incubated with control IgG1, control IgG2a, PG4D1, or PG4D2, followed by incubation with PRP from PdpnKI/KI mice. The platelet aggregation rate was estimated using an aggregometer. B, Adherent cells from the lungs and lymph nodes of the PdpnKI/KI or C57BL/6N mice were immortalized with SV40 large T antigen. The immortalized cells from the lungs (left panels) or lymph nodes (right panels) were incubated with PBS (closed areas), anti‐mPDPN–specific mAb 8F11 or anti‐hPDPN–specific mAb PG4D2 (open areas). After washing, the cells were incubated with Alexa Flour 488–conjugated second antibody, and fluorescence intensity was measured by flow cytometry. C, The immortalized cells from the lungs or lymph nodes were incubated with control IgG1, control IgG2a, PG4D1, or PG4D2, followed by incubation with PRP from PdpnKI/KI mice. The platelet aggregation rate was estimated using an aggregometer
The antibody‐dependent cell‐mediated cytotoxicity (ADCC) activity was estimated (see Supplemental materials and methods) because PG4D1 and PG4D2 were previously determined as murine IgG1 and IgG2a subclasses, respectively. As shown in the results of the ADCC reporter bioassay via mouse FcγRIV, which is more closely related to human FcγRIIIa, the PG4D2 mAb of mouse IgG2a subclass and not the PG4D1 mAb of mouse IgG1 subclass induced strong ADCC activity by mediating between mouse FcγRIV and chiPDPN‐expressing immortalized lung cells from PdpnKI/KI mice or hPDPN‐expressing CHO cells (Figure S4A,B). To investigate the therapeutic efficacy of anti‐hPDPN mAbs in vivo, MC38 cells that had been transfected with chiPDPN (MC38/chiPDPN) were generated (Figure 5A) and xenografted into PdpnKI/KI mice. There was no significant difference in the body weight change between the antibody‐treated mice (PG4D2 mAb vs. control IgG2a) (Figure 5B). Administration of PG4D2 mAb significantly suppressed MC38/chiPDPN growth in vivo (Figure 5C). These results suggested that PdpnKI/KI mice were useful for investigating the therapeutic efficacy of anti‐hPDPN mAbs, especially compared with PLAG4‐targeting agents.
FIGURE 5.
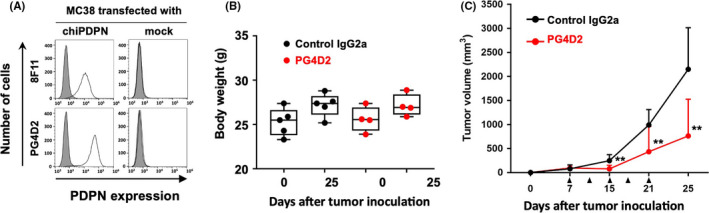
Neutralizing anti‐human podoplanin antibody suppressed tumor growth in vivo. A, MC38 cells were stably transfected with an empty vector (mock) or expression plasmids containing human/mouse chimeric podoplanin (chiPDPN), followed by incubation with PBS (closed areas), anti‐mPDPN–specific mAb 8F11, or anti‐hPDPN–specific mAb PG4D2 (open areas). After washing, the cells were incubated with Alexa Flour 488–conjugated second antibody, and fluorescence intensity was measured by flow cytometry. B and C, PdpnKI/KI mice bearing MC38/chiPDPN tumors were intraperitoneally administered with 500 µg/mouse of control mouse IgG2a (n = 5) or PG4D2 antibody (n = 4) at 7, 11, 15, 18, and 21 d after tumor inoculation (arrow heads). Body weight was measured on days 0 and 25 after tumor inoculation. Tumor volumes were calculated as described in the Materials and Methods. All data are shown as mean ± SD. **P <.01 by the Mann‐Whitney U test
3.5. Assessment of in vivo toxicity of neutralizing anti‐PDPN mAb
We evaluated the toxicity of continuous blockage of PDPN‐mediated platelet aggregation by repeated administration of neutralizing anti‐PDPN mAbs. Male and female PdpnKI/KI mice were intravenously injected with 10 or 50 mg/kg of anti‐hPDPN–specific neutralizing PG4D1 and PG4D2 mAbs through the lateral tail vein five times every other week (Figure 6A) and were evaluated for toxic signs thereafter. Throughout the study, the feeding and body weight changes in all treatment groups were not significantly different compared with those in the control group (Table 1). The relative weights of all tested tissues and organs showed no significant differences between the neutralizing anti‐PDPN mAb‐treated and control groups (Table 1). The creatinine level was significantly lower in the 50 mg/kg of PG4D1 mAb–treated group than in the IgG1‐treated control group (Table 3). Eosinophil differentiation and lactate dehydrogenase (LD) activity were significantly higher in the 50 mg/kg of PG4D2–treated group than in the IgG2a‐treated control group (Tables 2 and 3). Hemoglobin and hematocrit levels were significantly higher in the 50 mg/kg of PG4D2–treated group than in the 10 mg/kg of PG4D2–treated group (Tables 2). Histological evaluation using hematoxylin‐eosin–stained tissue specimens from each group did not show any sign of abnormality (Figure 6B and Figure S5).
FIGURE 6.

Evaluation of the toxicity of the anti‐human podoplanin–neutralizing antibodies using knock‐in mice with a chimeric human/murine podoplanin. A, Schematic representation of the protocol for 5‐wk treatment with mouse anti‐human podoplanin–neutralizing antibodies (PG4D1 and PG4D2) or isotype control mouse antibodies (Control IgG1 and IgG2a). The mice were euthanized 1 wk after the final injection, tissues and organs were weighed, and hematological and biochemistry analyses were performed. B, Representative hematoxylin‐eosin–stained tissue images of the lung, liver, lymph node, kidney, and testis of the 50 mg/kg of antibody–treated male PdpnKI/KI mouse are shown. Bars: 100 µm
TABLE 1.
Relative tissue and organ weights of PdpnKI/KI mice treated with or without anti‐human podoplanin–neutralizing antibodies
| Group | IgG1 | IgG2a | PG4D1 | PG4D2 | ||
|---|---|---|---|---|---|---|
| Dose (mg/kg) | 50 | 50 | 10 | 50 | 10 | 50 |
| Body weights (g) | 23.68 ± 2.47 | 23.23 ± 3.20 | 23.98 ± 2.85 | 23.48 ± 2.59 | 23.00 ± 1.85 | 23.68 ± 3.14 |
| Brain (g%) | 1.91 ± 0.23 | 2.01 ± 0.31 | 1.99 ± 0.38 | 1.96 ± 0.19 | 2.05 ± 0.19 | 1.95 ± 0.25 |
| Pituitary (mg%) | 8.70 ± 0.70 | 10.90 ± 3.53 | 10.20 ± 2.78 | 12.95 ± 4.18 | 9.33 ± 1.93 | 9.73 ± 2.71 |
| Salivary glands (g%) | 0.50 ± 0.08 | 0.58 ± 0.06 | 0.50 ± 0.06 | 0.56 ± 0.09 | 0.60 ± 0.06 | 0.60 ± 0.04 |
| Thymus (g%) | 0.28 ± 0.10 | 0.27 ± 0.07 | 0.24 ± 0.08 | 0.25 ± 0.07 | 0.23 ± 0.07 | 0.25 ± 0.08 |
| Lungs (g%) | 0.67 ± 0.06 | 0.68 ± 0.10 | 0.69 ± 0.11 | 0.71 ± 0.09 | 0.68 ± 0.13 | 0.60 ± 0.06 |
| Heart (g%) | 0.54 ± 0.06 | 0.53 ± 0.06 | 0.51 ± 0.05 | 0.53 ± 0.06 | 0.56 ± 0.06 | 0.49 ± 0.02 |
| Stomach (g%) | 0.73 ± 0.01 | 0.71 ± 0.10 | 0.80 ± 0.16 | 0.79 ± 0.09 | 0.79 ± 0.11 | 0.81 ± 0.15 |
| Liver (g%) | 4.67 ± 1.00 | 5.44 ± 0.20 | 5.08 ± 0.73 | 5.47 ± 0.19 | 5.57 ± 0.46 | 5.29 ± 0.12 |
| Spleen (g%) | 0.39 ± 0.06 | 0.38 ± 0.12 | 0.38 ± 0.06 | 0.41 ± 0.09 | 0.40 ± 0.11 | 0.41 ± 0.10 |
| Kidneys (g%) | 1.42 ± 0.13 | 1.41 ± 0.09 | 1.41 ± 0.17 | 1.40 ± 0.16 | 1.34 ± 0.09 | 1.36 ± 0.07 |
| Pancreas (g%) | 0.95 ± 0.29 | 0.74 ± 0.22 | 0.84 ± 0.11 | 0.79 ± 0.11 | 0.95 ± 0.14 | 0.81 ± 0.14 |
| Adrenals (mg%) | 28.70 ± 8.27 | 31.15 ± 8.53 | 29.93 ± 11.47 | 25.55 ± 6.43 | 28.35 ± 8.08 | 29.60 ± 5.94 |
| Testes (g%) | 0.70 | 0.80 | 0.67 | 0.74 | 0.78 | 0.77 |
| Epididymides (g%) | 0.29 | 0.26 | 0.31 | 0.36 | 0.39 | 0.31 |
| Prostate (mg%) | 48.30 | 44.30 | 38.75 | 42.55 | 57.00 | 48.25 |
| Ovaries (mg%) | 38.75 | 47.60 | 56.80 | 46.50 | 44.80 | 49.35 |
| Uterus (g%) | 0.35 | 0.38 | 0.38 | 0.49 | 0.40 | 0.38 |
Each value shows mean ± SD
TABLE 3.
Comparison of biochemical parameters of PdpnKI/KI mice treated with or without anti‐human podoplanin–neutralizing antibodies
| Group | IgG1 | IgG2a | PG4D1 | PG4D2 | ||
|---|---|---|---|---|---|---|
| Dose (mg/kg) | 50 | 50 | 10 | 50 | 10 | 50 |
| AST (IU/L) | 48.38 ± 6.72 | 41.83 ± 2.96 | 43.90 ± 6.99 | 37.10 ± 5.99 | 40.18 ± 3.05 | 39.58 ± 5.62 |
| ALT (IU/L) | 20.78 ± 2.51 | 20.60 ± 1.99 | 20.15 ± 1.18 | 17.80 ± 4.68 | 20.80 ± 1.56 | 20.93 ± 1.82 |
| ALP (IU/L) | 403.68 ± 84.58 | 377.80 ± 107.44 | 397.45 ± 55.07 | 310.50 ± 147.09 | 392.73 ± 162.38 | 391.55 ± 163.15 |
| LD (IU/L) | 174.18 ± 24.60 | 135.20 ± 7.50* | 147.55 ± 26.63 | 149.28 ± 15.77 | 168.30 ± 37.48 | 172.83 ± 13.48† |
| TP (g/dL) | 4.55 ± 0.24 | 4.72 ± 0.10 | 4.53 ± 0.08 | 4.59 ± 0.07 | 4.63 ± 0.42 | 4.69 ± 0.22 |
| ALB (g/dL) | 2.59 ± 0.19 | 2.67 ± 0.20 | 2.58 ± 0.21 | 2.44 ± 0.51 | 2.77 ± 0.43 | 2.76 ± 0.29 |
| Protein fraction (%) | ||||||
| alb | 57.1 ± 4.62 | 56.53 ± 4.20 | 56.95 ± 5.19 | 53.13 ± 11.05 | 59.53 ± 4.76 | 58.80 ± 4.06 |
| α1‐glb | 9.9 ± 3.08 | 10.03 ± 2.41 | 10.83 ± 3.11 | 8.73 ± 2.78 | 10.03 ± 2.21 | 9.38 ± 2.46 |
| α2‐glb | 9.6 ± 0.58 | 9.63 ± 1.09 | 10.28 ± 0.99 | 11.95 ± 4.94 | 9.33 ± 1.05 | 9.33 ± 0.99 |
| β‐glb | 21.6 ± 1.42 | 20.48 ± 1.44 | 19.93 ± 1.16 | 24.05 ± 7.58 | 19.55 ± 1.64 | 20.15 ± 0.82 |
| γ‐glb | 1.9 ± 0.54 | 3.35 ± 0.29* | 2.03 ± 0.41 | 2.15 ± 0.87 | 1.58 ± 0.48 | 2.35 ± 1.19 |
| A/G | 1.35 ± 0.26 | 1.32 ± 0.22 | 1.35 ± 0.28 | 1.22 ± 0.45 | 1.50 ± 0.28 | 1.44 ± 0.24 |
| T‐Bil (mg/dL) | 0.14 ± 0.04 | 0.11 ± 0.01 | 0.15 ± 0.04 | 0.11 ± 0.02 | 0.11 ± 0.01 | 0.11 ± 0.01 |
| UN (mg/dL) | 25.20 ± 6.62 | 26.80 ± 1.82 | 24.85 ± 3.10 | 25.40 ± 5.76 | 29.70 ± 3.45 | 30.70 ± 4.11 |
| Cre (mg/dL) | 0.12 ± 0.01 | 0.10 ± 0.01* | 0.11 ± 0.01 | 0.11 ± 0.01* | 0.11 ± 0.01 | 0.11 ± 0.01 |
| Glu (mg/dL) | 206.15 ± 41.27 | 209.75 ± 16.12 | 201.30 ± 15.73 | 197.03 ± 16.32 | 215.88 ± 34.19 | 199.85 ± 21.33 |
| T‐Cho (mg/dL) | 67.20 ± 14.63 | 87.00 ± 8.13 | 68.33 ± 12.20 | 79.60 ± 15.77 | 74.13 ± 14.72 | 74.93 ± 6.34 |
| TG (mg/dL) | 40.93 ± 28.53 | 52.93 ± 43.35 | 39.13 ± 19.40 | 40.93 ± 12.98 | 41.60 ± 25.18 | 55.78 ± 22.05 |
| PL (mg/dL) | 155.43 ± 30.17 | 182.95 ± 24.48 | 150.43 ± 15.28 | 161.28 ± 35.99 | 160.85 ± 26.94 | 166.53 ± 13.36 |
| Na (mEq/L) | 146.95 ± 1.66 | 147.30 ± 1.00 | 146.13 ± 1.19 | 147.43 ± 1.72 | 147.80 ± 0.84 | 147.68 ± 1.20 |
| K (mEq/L) | 4.19 ± 0.27 | 4.11 ± 0.28 | 4.41 ± 0.14 | 4.33 ± 0.29 | 4.31 ± 0.15 | 4.31 ± 0.34 |
| Cl (mEq/L) | 116.30 ± 3.25 | 117.45 ± 1.06 | 116.55 ± 2.50 | 117.18 ± 1.45 | 119.20 ± 2.82 | 117.43 ± 1.90 |
| Ca (mg/dL) | 9.20 ± 0.22 | 9.13 ± 0.30 | 9.23 ± 0.13 | 9.30 ± 0.26 | 9.08 ± 0.25 | 9.25 ± 0.06 |
| IP (mg/dL) | 5.30 ± 0.98 | 6.35 ± 0.89 | 4.83 ± 1.24 | 4.98 ± 1.05 | 5.15 ± 1.39 | 5.88 ± 1.30 |
Each value shows mean ± SD.
Significantly different from the control IgG1 group at 50 mg/kg (*: P <.05).
Significantly different from the control IgG2a group at 50 mg/kg (†: P <.05).
α1‐glb, α1‐glboblin; α2‐glb, α2‐glboblin; ALB, albumin; ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; β‐glb, β‐glboblin; γ‐glb, γ‐glboblin; Ca,calcium; Cl, chlorine; Cre, creatinine; Glu, glucose; IP, inorganic phosphate; K, potassium; LD, lactase dehydrogenase; Na, sodium; PL, phospholipid; T‐Bil, total‐bilirubin; T‐Cho, total‐cholesterol; TG, triglyceride; TP, total‐protein; UN, blood urea nitrogen.
TABLE 2.
Comparison of hematological parameters of PdpnKI/KI mice treated with or without anti‐human podoplanin–neutralizing antibodies
| Group | IgG1 | IgG2a | PG4D1 | PG4D2 | ||
|---|---|---|---|---|---|---|
| Dose (mg/kg) | 50 | 50 | 10 | 50 | 10 | 50 |
| RBC (104/μL) | 904.75 ± 23.01 | 894.25 ± 41.60 | 862.25 ± 15.06 | 898.25 ± 26.49 | 869.50 ± 34.90 | 915.00 ± 18.97 |
| HGB (g/dL) | 13.60 ± 0.34 | 13.58 ± 0.43 | 13.18 ± 0.26 | 13.48 ± 0.22 | 13.20 ± 0.50 | 13.75 ± 0.06‡ |
| HCT (%) | 41.03 ± 1.02 | 40.65 ± 1.32 | 39.63 ± 1.05 | 40.55 ± 1.03 | 40.15 ± 1.20 | 41.53 ± 0.34‡ |
| MCV (fL) | 45.35 ± 0.77 | 45.53 ± 1.55 | 45.95 ± 0.51 | 45.18 ± 1.41 | 46.20 ± 1.37 | 45.40 ± 1.07 |
| MCH (pg) | 15.03 ± 0.24 | 15.18 ± 0.31 | 15.30 ± 0.27 | 15.03 ± 0.35 | 15.18 ± 0.43 | 15.03 ± 0.29 |
| MCHC (g/dL) | 33.15 ± 0.24 | 33.43 ± 0.56 | 33.25 ± 0.56 | 33.25 ± 0.39 | 32.90 ± 0.29 | 33.13 ± 0.21 |
| PLT (104/μL) | 119.48 ± 20.79 | 129.88 ± 24.07 | 101.05 ± 16.13 | 126.40 ± 24.44 | 131.40 ± 14.00 | 126.85 ± 20.67 |
| RET (104/μL) | 43.16 ± 2.93 | 46.00 ± 12.42 | 45.82 ± 8.25 | 34.64 ± 16.48 | 53.49 ± 18.27 | 44.01 ± 5.86 |
| RET (%) | 4.77 ± 0.29 | 5.18 ± 1.55 | 5.32 ± 0.95 | 3.88 ± 1.87 | 6.18 ± 2.17 | 4.81 ± 0.63 |
| WBC (102/μL) | 51.90 ± 18.57 | 38.95 ± 4.74 | 47.53 ± 5.30 | 55.58 ± 19.58 | 41.73 ± 5.93 | 44.73 ± 6.65 |
| Differential leukocyte (102/μL) | ||||||
| Lymphocyte (102/μL) | 45.28 ± 15.16 | 34.65 ± 5.47 | 41.33 ± 4.12 | 42.28 ± 12.05 | 37.00 ± 5.53 | 39.20 ± 5.61 |
| Neutrophil (102/μL) | 4.23 ± 2.25 | 3.03 ± 1.39 | 4.08 ± 1.12 | 9.73 ± 10.39 | 3.13 ± 1.50 | 3.23 ± 1.20 |
| Eosinophil (102/μL) | 0.95 ± 0.48 | 0.48 ± 0.13 | 0.73 ± 0.15 | 0.53 ± 0.13 | 0.73 ± 0.25 | 1.25 ± 0.35 † |
| Basophil (102/μL) | 0.18 ± 0.17 | 0.03 ± 0.05 | 0.28 ± 0.31 | 0.08 ± 0.10 | 0.13 ± 0.19 | 0.13 ± 0.25 |
| Monocyte (102/μL) | 1.28 ± 0.84 | 0.78 ± 0.22 | 1.13 ± 0.33 | 2.98 ± 3.36 | 0.75 ± 0.13 | 0.93 ± 0.33 |
| Differential leukocyte (%) | ||||||
| Lymphocyte (%) | 87.80 ± 2.18 | 88.75 ± 4.78 | 87.08 ± 2.85 | 78.43 ± 14.78 | 88.63 ± 2.83 | 87.75 ± 2.27 |
| Neutrophil (%) | 7.80 ± 1.42 | 7.95 ± 4.02 | 8.48 ± 1.65 | 15.53 ± 11.35 | 7.48 ± 3.04 | 7.15 ± 2.61 |
| Eosinophil (%) | 1.83 ± 0.68 | 1.23 ± 0.35 | 1.55 ± 0.26 | 1.15 ± 0.79 | 1.80 ± 0.86 | 2.75 ± 0.42 † |
| Basophil (%) | 0.30 ± 0.22 | 0.05 ± 0.10 | 0.55 ± 0.67 | 0.20 ± 0.34 | 0.30 ± 0.42 | 0.33 ± 0.65 |
| Monocyte (%) | 2.28 ± 0.76 | 2.03 ± 0.67 | 2.35 ± 0.53 | 4.70 ± 3.74 | 1.80 ± 0.38 | 2.03 ± 0.43 |
Each value shows mean ± SD.
Significantly different from the PG4D2 group at 10 mg/kg (‡ P <.05).
Significantly different from the control IgG2a group at 50 mg/kg († P <.05).
HCT, hematocrit; HGB, hemoglobin; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; MCV, mean corpuscular volume; PLT, platelets; RBC, red blood cells; RET, reticulocyte; WBC, white blood cells.
4. DISCUSSION
Several reports, including ours, have demonstrated that mAbs targeting PLAG domains on PDPN could suppress PDPN–CLEC‐2 binding and PDPN‐expressing tumor growth and metastasis. 5 , 17 , 23 In particular, mAbs targeting human PLAG4 were shown to strongly suppress PDPN‐induced platelet aggregation, PDPN‐positive tumor growth, and hematogenous metastasis. 23 Therefore, PLAG4‐recognizing anti‐hPDPN mAb can be used as a novel anti‐tumor drug. However, PDPN is known to be expressed in normal tissues and cells, such as those in the lymphatic vessels, kidney podocytes, mesothelium, alveolar epithelium, and in immune cells, such as follicular dendritic cells, macrophage subsets, and effecter T cells. 24 , 25 , 30 , 31 In addition, the physiologic function of PDPN has been reported in both developmental and postnatal stages. The interaction of endothelial PDPN in the developing lymph sac with circulating platelets from the cardinal vein is critical for separating the lymphatic system from the blood vascular system during embryonic development. 32 , 33 For the immune surveillance of lymphocytes in the lymph node, the interaction between PDPN expressed on high endothelial venules (HEVs) and CLEC‐2 on platelets is essential to maintain HEV integrity in cases of increased lymphocyte trafficking, such as in chronic inflammation. 24 Another report suggested that PDGF‐mediated CLEC‐2 overexpression on dendritic cells promoted regulatory T‐cell differentiation in humans by interacting with PDPN expressed on T cells. 34 Several groups reported that the absence of PDPN in the developmental stage resulted in death at birth secondary to respiratory failure, which was associated with defects such as type II alveolar lung cell differentiation into type I cells, 26 and led to lymphatic defects such as diminished lymphatic transport, congenital lymphedema, and dilation of cutaneous and intestinal lymphatic vessels at birth. 27
Therefore, we have to assess the safety of our generated PDPN‐targeting mAb before clinical application. We previously performed a single‐dose toxicity study in cynomolgus monkeys using anti‐monkey PDPN‐neutralizing mAbs and found no acute toxicity in both blood and histopathological tests. 28 Consistent with our report, a safety study that used a cancer‐specific mouse‐dog chimeric anti‐PDPN mAb, which specifically recognizes canine PDPN‐expressing tumor cells, found no severe adverse effects in normal dogs treated with mAb and in dogs with melanoma. 35 Furthermore, the synthesized small CLEC‐2 inhibitor 2CP did not exhibit any impairment in the physiologic platelet function of hemostasis and cytotoxicity. 36 However, toxicity after repeated administrations of neutralizing anti‐PDPN mAbs remains unclear. The adverse effects of long‐term suppression of PDPN function by our established mAbs must be examined. Therefore, we generated a syngeneic mouse model that enabled the investigation of therapeutic efficacy and toxicity after repeated administrations of neutralizing anti‐human PDPN mAb. Surprisingly, the mouse platelet aggregation–inducing abilities of human PDPN and chiPDPN (mPDPN‐hPLAGs) were weaker, compared with those of mouse WT PDPN (Figures 2B and S2B). With regard to cross‐species reactivity of recombinant CLEC‐2, the binding affinity to hPDPN was weaker for mCLEC‐2 than for hCLEC‐2 (Figure S2C). Similarly, the binding affinity to mPDPN was weaker for hCLEC‐2 than for mCLEC‐2 (Figure S2D). These results suggested that PDPN binding to CLEC‐2 was somehow species‐specific while maintaining cross‐species reactivity.
To overcome the above differences in cross‐species affinity, we tried to generate a novel mouse model that can evaluate the therapeutic efficacy and toxicity of human PDPN–targeting mAbs without changing PDPN‐mediated platelet‐aggregating ability. By minimizing the replacement region, we succeeded to generate PdpnKI/KI mice expressing chiPDPN that contained human PLAG4 domain.
PG4D1 and PG4D2 mAbs attenuated the platelet‐aggregating activity of chiPDPN (Figure 4A and 4C) and significantly suppressed the tumor growth of chiPDPN‐expressing MC38 tumors xenografted into PdpnKI/KI mice (Figure 5C). To our knowledge, there had been no mAbs that can recognize both human and mouse PDPN and can neutralize both human and mouse PDPN–induced platelet aggregation. Our generated PdpnKI/KI mice expressed chiPDPN that could be recognized by anti‐human PDPN antibodies. Therefore, the mouse model was useful for investigating the therapeutic efficacy of human PDPN–neutralizing antibody, especially human PLAG4–targeting antigen.
After repeated treatment with neutralizing anti‐PDPN mAbs, no toxic signs, weight loss, and necropsy findings were observed (Table 1). Although the creatinine level decreased and eosinophil differentiation and LD activity increased in the PG4D1‐ and PG4D2‐treated mice (Tables 2 and 3), these were not outliers that deviated significantly from the normal values. Moreover, although the number of eosinophils in the 50 mg/kg of PG4D2–treated group almost doubled, compared with that in the control IgG2a group, it did not significantly differ from that of the control IgG1 group. Moreover, there were no abnormal signs, such as lymphocyte infiltration or inflammation, in all tested tissues, including the kidney and lymphoid tissues, where PDPN was highly expressed (Figure 6B and Figure S5). Therefore, we supposed that these changes in hematological and biochemical parameters were not critical toxic signs of anti‐PDPN mAb treatment. Surprisingly, no inflammation was observed in mice administered with IgG2a‐subclass PG4D2, which exhibited ADCC activity via chiPDPN‐expressing lung cells from PdpnKI/KI mice (Figure S4A), despite the fact that the murine IgG2a subclass antibody had been well known to induce potent ADCC via murine FcγRIV on immune cells, including neutrophils, monocytes, macrophages, and dendritic cells. 37 The PDPN expression level in various normal tissues might not have been high enough to induce ADCC after in vivo administration of IgG2a‐subclass PG4D2 mAb.
In this study, we generated a novel syngeneic mouse model that expressed chiPDPN (PdpnKI/KI mice) and evaluated the therapeutic efficacy and toxicity of human PLAG4–targeting anti‐PDPN–neutralizing mAbs. We demonstrated that long‐term suppression of PDPN/CLEC‐2 interaction by repeated treatment with neutralizing anti‐PDPN mAb caused no toxicity in the PdpnKI/KI mice. Our results suggested that neutralizing anti‐PDPN mAbs targeting the PLAG4 domain may be used safely and are promising novel anti‐tumor agents in clinical application.
DISCLOSURE
MK is an employee of Api, Co., Ltd. NF is receiving a research grant from Api, Co., Ltd. Other authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Supporting information
Figure S1
{kind=link}
Figure S2
{kind=link}
Figure S3
{kind=link}
Figure S4
{kind=link}
Figure S5
{kind=link}
Table S1
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge Trans Genic Inc (Fukuoka, Japan) for generating PdpnKI/KI mice and Nihon Bioresearch Inc (Gifu, Japan) for performing toxicity study. The authors thank Drs. Kazue Tsuji‐Takayama, Nobuhiko Gyobu, Junya Maeda, Kenji Ichihara, and Shinobu Nakayama of Api, Co., Ltd. for valuable discussions. The authors thank Dr Yoshiyuki Ohsugi, an advisor of Api, Co., Ltd., for valuable suggestions. The authors also thank Ms Fusejima of the Division of Cell Biology, The Cancer Institute, Japanese Foundation for Cancer Research for cryopreserved embryo transplantation and regeneration of PdpnKI/KI mice. This study was financially supported by the Project for Cancer Research and Therapeutic Evolution (P‐CREATE) grant number 20cm0106205h0005 (to NF) and the Acceleration Transformative Research for Medical Innovation (ACT‐M) grant number 20im0210110h0103 (to NF) and grant number 20im0210110h0003 (to Api, Co., Ltd.) from the Japan Agency for Medical Research and Development (AMED). This study was also financially supported by MEXT/JSPS KAKENHI grant number JP17H06327 (to NF) and a grant from the Nippon Foundation grant number 2019516015 (to NF).
Ukaji T, Takemoto A, Shibata H, et al. Novel knock‐in mouse model for the evaluation of the therapeutic efficacy and toxicity of human podoplanin–targeting agents. Cancer Sci. 2021;112:2299–2313. 10.1111/cas.14891
REFERENCES
- 1. Gasic GJ, Gasic TB, Stewart CC. Antimetastatic effects associated with platelet reduction. Proc Natl Acad Sci U S A. 1968;61:46‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kunita A, Kashima TG, Morishita Y, et al. The platelet aggregation‐inducing factor aggrus/podoplanin promotes pulmonary metastasis. Am J Pathol. 2007;170:1337‐1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Klerk CP, Smorenburg SM, Otten HM, et al. The effect of low molecular weight heparin on survival in patients with advanced malignancy. J Clin Oncol. 2005;23:2130‐2135. [DOI] [PubMed] [Google Scholar]
- 4. Lazo‐Langner A, Goss GD, Spaans JN, et al. The effect of low‐molecular‐weight heparin on cancer survival. A systematic review and meta‐analysis of randomized trials. J Thromb Haemost. 2007;5:729‐737. [DOI] [PubMed] [Google Scholar]
- 5. Kato Y, Fujita N, Kunita A, et al. Molecular identification of Aggrus/T1alpha as a platelet aggregation‐inducing factor expressed in colorectal tumors. J Biol Chem. 2003;278:51599‐51605. [DOI] [PubMed] [Google Scholar]
- 6. Martin‐Villar E, Scholl FG, Gamallo C, et al. Characterization of human PA2.26 antigen (T1alpha‐2, podoplanin), a small membrane mucin induced in oral squamous cell carcinomas. Int J Cancer. 2005;113:899‐910. [DOI] [PubMed] [Google Scholar]
- 7. Kimura N, Kimura I. Podoplanin as a marker for mesothelioma. Pathol Int. 2005;55:83‐86. [DOI] [PubMed] [Google Scholar]
- 8. Mishima K, Kato Y, Kaneko MK, et al. Increased expression of podoplanin in malignant astrocytic tumors as a novel molecular marker of malignant progression. Acta Neuropathol. 2006;111:483‐488. [DOI] [PubMed] [Google Scholar]
- 9. Takagi S, Oh‐hara T, Sato S, et al. Expression of Aggrus/podoplanin in bladder cancer and its role in pulmonary metastasis. Int J Cancer. 2014;134:2605‐2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kunita A, Kashima TG, Ohazama A, et al. Podoplanin is regulated by AP‐1 and promotes platelet aggregation and cell migration in osteosarcoma. Am J Pathol. 2011;179:1041‐1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao X, Pan Y, Ren W, et al. Plasma soluble podoplanin is a novel marker for the diagnosis of tumor occurrence and metastasis. Cancer Sci. 2018;109:403‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Suzuki‐Inoue K, Kato Y, Inoue O, et al. Involvement of the snake toxin receptor CLEC‐2, in podoplanin‐mediated platelet activation, by cancer cells. J Biol Chem. 2007;282:25993‐26001. [DOI] [PubMed] [Google Scholar]
- 13. Takagi S, Sato S, Oh‐hara T, et al. Platelets promote tumor growth and metastasis via direct interaction between Aggrus/podoplanin and CLEC‐2. PLoS One. 2013;8:e73609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Suzuki‐Inoue K. Essential in vivo roles of the platelet activation receptor CLEC‐2 in tumour metastasis, lymphangiogenesis and thrombus formation. J Biochem. 2011;150:127‐132. [DOI] [PubMed] [Google Scholar]
- 15. Suzuki‐Inoue K, Fuller GL, Garcia A, et al. A novel Syk‐dependent mechanism of platelet activation by the C‐type lectin receptor CLEC‐2. Blood. 2006;107:542‐549. [DOI] [PubMed] [Google Scholar]
- 16. Miyata K, Takemoto A, Okumura S, et al. Podoplanin enhances lung cancer cell growth in vivo by inducing platelet aggregation. Sci Rep. 2017;7:4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takemoto A, Miyata K, Fujita N. Platelet‐activating factor podoplanin: from discovery to drug development. Cancer Metastasis Rev. 2017;36:225‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Takemoto A, Okitaka M, Takagi S, et al. A critical role of platelet TGF‐beta release in podoplanin‐mediated tumour invasion and metastasis. Sci Rep. 2017;7:42186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pula B, Jethon A, Piotrowska A, et al. Podoplanin expression by cancer‐associated fibroblasts predicts poor outcome in invasive ductal breast carcinoma. Histopathology. 2011;59:1249‐1260. [DOI] [PubMed] [Google Scholar]
- 20. Xu Y, Ogose A, Kawashima H, et al. High‐level expression of podoplanin in benign and malignant soft tissue tumors: immunohistochemical and quantitative real‐time RT‐PCR analysis. Oncol Rep. 2011;25:599‐607. [DOI] [PubMed] [Google Scholar]
- 21. Yuan P, Temam S, El‐Naggar A, et al. Overexpression of podoplanin in oral cancer and its association with poor clinical outcome. Cancer. 2006;107:563‐569. [DOI] [PubMed] [Google Scholar]
- 22. Suzuki‐Inoue K, Inoue O, Ding G, et al. Essential in vivo roles of the C‐type lectin receptor CLEC‐2: embryonic/neonatal lethality of CLEC‐2‐deficient mice by blood/lymphatic misconnections and impaired thrombus formation of CLEC‐2‐deficient platelets. J Biol Chem. 2010;285:24494‐24507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sekiguchi T, Takemoto A, Takagi S, et al. Targeting a novel domain in podoplanin for inhibiting platelet‐mediated tumor metastasis. Oncotarget. 2016;7:3934‐3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Herzog BH, Fu J, Wilson SJ, et al. Podoplanin maintains high endothelial venule integrity by interacting with platelet CLEC‐2. Nature. 2013;502:105‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hoshino A, Ishii G, Ito T, et al. Podoplanin‐positive fibroblasts enhance lung adenocarcinoma tumor formation: podoplanin in fibroblast functions for tumor progression. Cancer Res. 2011;71:4769‐4779. [DOI] [PubMed] [Google Scholar]
- 26. Ramirez MI, Millien G, Hinds A, et al. T1alpha, a lung type I cell differentiation gene, is required for normal lung cell proliferation and alveolus formation at birth. Dev Biol. 2003;256:61‐72. [DOI] [PubMed] [Google Scholar]
- 27. Schacht V, Ramirez MI, Hong YK, et al. T1alpha/podoplanin deficiency disrupts normal lymphatic vasculature formation and causes lymphedema. Embo J. 2003;22:3546‐3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ukaji T, Takemoto A, Katayama R, et al. A safety study of newly generated anti‐podoplanin‐neutralizing antibody in cynomolgus monkey (Macaca fascicularis). Oncotarget. 2018;9:33322‐33336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jin C, Li H, Murata T, et al. JDP2, a repressor of AP‐1, recruits a histone deacetylase 3 complex to inhibit the retinoic acid‐induced differentiation of F9 cells. Mol Cell Biol. 2002;22:4815‐4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kerrigan AM, Navarro‐Nunez L, Pyz E, et al. Podoplanin‐expressing inflammatory macrophages activate murine platelets via CLEC‐2. J Thromb Haemost. 2012;10:484‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Peters A, Pitcher LA, Sullivan JM, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity. 2011;35:986‐996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schacht V, Dadras SS, Johnson LA, et al. Up‐regulation of the lymphatic marker podoplanin, a mucin‐type transmembrane glycoprotein, in human squamous cell carcinomas and germ cell tumors. Am J Pathol. 2005;166:913‐921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Uhrin P, Zaujec J, Breuss JM, et al. Novel function for blood platelets and podoplanin in developmental separation of blood and lymphatic circulation. Blood. 2010;115:3997‐4005. [DOI] [PubMed] [Google Scholar]
- 34. Agrawal S, Ganguly S, Hajian P, et al. PDGF upregulates CLEC‐2 to induce T regulatory cells. Oncotarget. 2015;6:28621‐28632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kamoto S, Shinada M, Kato D, et al. Phase I/II clinical trial of the anti‐podoplanin monoclonal antibody therapy in dogs with malignant melanoma. Cells. 2020;9:2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chang YW, Hsieh PW, Chang YT, et al. Identification of a novel platelet antagonist that binds to CLEC‐2 and suppresses podoplanin‐induced platelet aggregation and cancer metastasis. Oncotarget. 2015;6:42733‐42748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nimmerjahn F, Ravetch JV. Fcgamma receptors: old friends and new family members. Immunity. 2006;24:19‐28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Table S1
Supplementary Material