

Abstract
Human OAT1 and OAT3 play major roles in renal drug elimination and drug-drug interactions. However, there is little information on the interactions of drug metabolites with transporters. The goal of this study was to characterize the interactions of drug metabolites with OAT1 and OAT3 and compare their potencies of inhibition with those of their corresponding parent drugs. Using HEK293 cells stably transfected with OAT1 and OAT3, 25 drug metabolites and their corresponding parent drugs were screened for inhibitory effects on OAT1-and OAT3-mediated 6-carboxyfluorescein uptake at a screening concentration of 200 μM for all but 3 compounds. 20 and 24 drug metabolites were identified as inhibitors (inhibition > 50%) of OAT1 and OAT3, respectively. Seven drug metabolites were potent inhibitors of either or both OAT1 and OAT3 with Ki values less than 1 μM. 22 metabolites were more potent inhibitors of OAT3 than OAT1. Importantly, one drug and four metabolites were predicted to inhibit OAT3 at unbound plasma concentrations achieved clinically (Cmax,u/Ki values ≥ 0.1). In conclusion, our study highlights the potential interactions of drug metabolites with OAT1 and OAT3 at clinically relevant concentrations, suggesting that drug metabolites may modulate therapeutic and adverse drug response by inhibiting renal drug transporters.
Keywords: Organic anion transporters (OAT), Drug-drug interaction(s), Metabolism
Introduction
Drug-drug interactions (DDIs), through modulating drug levels, play important roles in drug safety and efficacy. In fact, the use of concomitant medications resulting in DDIs has been associated with serious adverse drug reactions, which have led to the removal of a number of drugs from the market (e.g., terfenadine1 and cerivastatin2). Many DDIs are mediated by drug metabolizing enzymes,3 but recently transporters in the intestine, liver and kidney have been recognized as important targets for DDIs.4,5 Concomitant medications that inhibit drug transporters in these organs may interfere with drug absorption, distribution or elimination, leading to high drug levels and accordingly, adverse drug reactions.
In the kidney, transporters located in the tubular epithelium are involved in the disposition and excretion of prescription drugs and their metabolites. In particular, human organic anion transporter 1 and 3, OAT1 (SLC22A6) and OAT3 (SLC22A8), expressed on the basolateral membrane of the renal proximal tubule, are secretory transporters. These transporters contribute to the tubular secretion of a variety of drugs belonging to various therapeutic classes, including beta-lactam antibiotics, loop diuretics, non-steroidal anti-inflammatory drugs, and antiviral nucleoside analogs.6–9 Because of their roles in drug accumulation in the kidney as well as overall drug elimination, OAT1 and OAT3 are major determinants of the efficacy, adverse drug reactions, and renal toxicities of a number of drugs.4,10,11 For example, probenecid, a known inhibitor of OAT1, is used clinically in conjunction with cidofovir in order to reduce its accumulation in renal proximal tubular cells and prevent its toxicity.12 Historically, probenecid was coadministered with penicillin in World War II to decrease its overall renal secretion, mediated by organic anion transporters, and therefore increase its systemic levels.13 Based on clear implications of the involvement of OAT1 and OAT3 in drug efficacy and safety, evaluation of a new molecular entity’s interaction potential with OAT1 and OAT3 has become an integral part of drug development and regulatory review before its market approval.14–16
To date, DDI studies have focused primarily on parent compounds, although more than 75% of the top 200 marketed drugs are eliminated by metabolism.17 Because drug metabolites are generally inactive or substantially less active than their parent compounds, they are largely ignored during drug development. In fact, few studies have examined the role of drug metabolites as inhibitors of clinically important drug transporters and mediators of DDIs. It is reasonable to speculate that drug metabolites derived from parent compounds which are known ligands of particular transporters may contribute to DDIs mediated by these transporters. For example, glucuronide conjugates of nonsteroidal anti-inflammatory drugs (NSAIDs), which are known ligands of OATs, inhibit OAT-mediated uptake of methotrexate in cellular studies18 consistent with the notion that metabolites of ligands for OAT1 and OAT3 may potentially cause DDIs.
The goal of this study was to determine whether drug metabolites inhibit human OAT1 and OAT3 and characterize their interactions with the two transporters. We selected metabolites of drugs that are highly metabolized. In particular, we examined the inhibitory effect of 25 drug metabolites that are commercially available and measurable in the systemic circulation on the transport activities of human OAT1 and OAT3. We further characterized the kinetics of inhibition to understand the comparative potency of the metabolites versus their respective parent compounds towards the two renal transporters. Inhibition potencies were compared to clinical unbound concentrations of drugs and metabolites (where available) to predict their potential to cause clinically relevant DDIs. Our studies revealed that many drug metabolites are potent inhibitors of OAT1 and OAT3 with the majority of metabolites being more potent inhibitors of OAT3 over OAT1. Notably, some metabolites, such as sulindac sulfide, sulindac sulfone and abiraterone sulfate, are predicted to inhibit OAT3 at clinically relevant unbound plasma concentrations, underscoring the potential impact of drug metabolites on OAT1-and OAT3-mediated clinical DDIs.
Materials and Methods
Chemicals and Reagents
Sources of drugs and drug metabolites are listed in Supplementary Table S1. 6-Carboxyfluorescein (6CF) was purchased from Sigma-Aldrich (St. Louis, MO).
Cell Line and Cell Culture
HEK293-Flp-In cells stably transfected with the empty vector, SLC22A6, or SLC22A8 were grown as described previously.19 In brief, cells were cultured in DMEM supplemented with 10% fetal bovine serum, penicillin (100 U/mL), streptomycin (100 μg/mL), sodium pyruvate (110 μg/mL) and hygromycin B (100 μg/mL) at 37 °C in a humidified incubator with 5% CO2.
Transporter Uptake Studies
The uptake assays have been described previously.19 In brief, empty vector-transfected cells, OAT1-and OAT3-overexpressing cells (8 × 104 cells/well) were seeded in black wall poly-D-lysine-coated 96 well plates for 24 h to reach 95% confluence. Before the uptake experiment, cell culture medium was removed, and the cells were washed with Hank’s balanced salt solution (HBSS). The uptake was initiated by incubating cells with HBSS containing desired concentrations of 6CF. The uptake was performed at 37 °C and was terminated by washing the cells two times with ice-cold HBSS. Fluorescence in cells was measured using a fluorescence microplate reader with excitation and emission wavelengths at 485 nm and 535 nm, respectively. Transporter-mediated 6CF uptake was determined by subtracting background uptake from the empty vector-transfected cells. The known inhibitor of OAT1 and OAT3, probenecid, was used at 200 μM as a positive control.
Inhibition of human OAT1-and OAT3-mediated 6CF uptake by parent drugs and drug metabolites. The methods used have been described previously19 and were modified slightly. Stably transfected cells were seeded in black-wall poly-D-lysine-coated 96-well plates for 24 h to reach 95% confluence. Before the uptake experiment, cell culture medium was removed, and the cells were washed with Hank’s balanced salt solution (HBSS). For screening, inhibition studies of the uptake of 2 μM 6CF by OAT1 and 10 mM 6CF by OAT3 were performed in triplicate at 37 °C in the presence of parent drugs and drug metabolites at 200 μM (or specified concentrations for those with limited solubility) (Supplementary Table S3). The uptake was terminated at 3 min. Cells were washed twice with icecold HBSS buffer. The IC50 values were calculated as previously described by GraphPad Prism software.20 The Ki value was calculated based on the following equation, where S and Km values for OAT1-mediated 6CF uptake are 2 μM and 15.1 μM, respectively, and S and Km values for OAT3-mediated 6CF uptake are 10 μM and 10.7 μM, respectively.
Statistical Analysis
In general, in the screening, three replicates of the effects of each drug or metabolite on 6CF uptake were performed. For the inhibition potency studies, IC50 values were obtained from one experiment with three replicate determinations for each concentration. Statistical analyses, as specified in the legends of the figures, were performed to determine significant differences between controls and study groups. The data were analyzed using GraphPad Prism 8 (La Jolla, CA). A p-value <0.05 was considered statistically significant. Linear regression was also done by GraphPad Prism 8 showing R2 for OAT1 and OAT3.
Results
Drug and drug metabolites screened were from many pharmacological classes. In total, 25 drug metabolites were tested for their inhibitory effects on human OAT1-and OAT3-mediated 6carboxyfluorescein (6-CF) uptake19 (Supplementary Fig. S1). The inhibitory effect of 19 parent drugs, from which the drug metabolites were derived, was also determined for comparison. These drugs were from 9 pharmacologic classes, including nonsteroidal anti-inflammatory drugs (NSAIDs, N = 9), antidiabetic agents (N = 3), antineoplastic agents (N = 2), and others (Fig. 1, Supplementary Table S2). The metabolites studied were the products of Phase I (e.g., hydroxylation) and Phase II (e.g., glucuronidation and sulfation) metabolism of the 19 parent compounds.
Fig. 1.
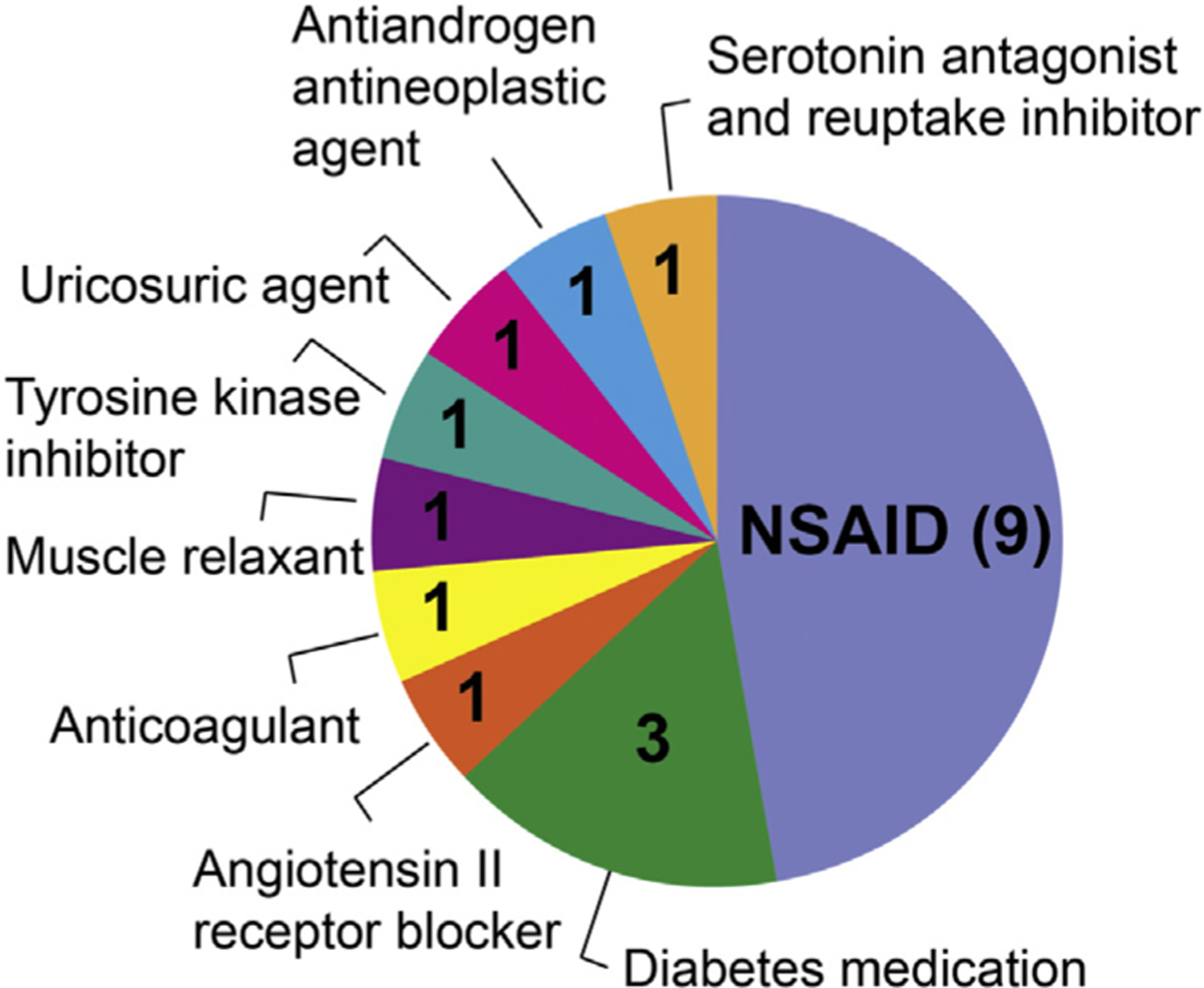
Therapeutic categories of parent drugs used for testing as OAT1 and OAT3 inhibitors. In total 19 drugs were characterized for their inhibitory effects on OAT1-and OAT3-mediated 6CF uptake in cells.
Drug metabolites were generally more potent inhibitors of OAT3-than OAT1-mediated 6CF uptake. At the screening concentration of 200 μM except for those with limited solubility (Supplementary Table S3), 20 drug metabolites were identified as OAT1 inhibitors and 24 were identified as OAT3 inhibitors (Table 1). Potency studies were conducted for each inhibitor by determining their effects at various concentrations on OAT1-or OAT3-mediated 6CF uptake. Two metabolites were potent inhibitors of OAT1 whereas seven metabolites were potent OAT3 inhibitors (Ki values less than 1 μM) (Table 1). In addition, 22 of the 25 drug metabolites were more potent inhibitors of OAT3 than of OAT1 (Fig. 2A, Table 1, Supplementary Fig. S2). Abiraterone N-oxide sulfate, (S)-naproxen acyl-β-D-glucuronide, 4-hydroxy-trazodone, and imatinib (piperidine)-4-oxide were inhibitors of OAT3 with Ki values ranging between 1.36 μM and 64.2 μM; however, none of these four metabolites inhibited OAT1 at concentrations of 100 μM or 200 μM. In addition, the Ki values of nine metabolites for OAT3, such as sulindac sulfone and celecoxib carboxylic acid, were at least 12 times lower in comparison with the Ki values for OAT1, indicating that these drug metabolites are more potent inhibitors of OAT3 than of OAT1 (Table 1). From a chemical perspective, the average molecular weight of drug metabolites having >10-fold higher potency towards OAT3 (over OAT1) was significantly higher than the average molecular weight of the drug metabolites having ≤ 10-fold potency towards OAT3 (p < 0.05) (Fig. 2B).
Table 1.
Ki Values for 25 Drug Metabolites.
| Drug Metabolite | Molecular Weight (g/mol)a | Ki (μM) |
Potency (hOAT3/hOAT1) | |
|---|---|---|---|---|
| OAT1 | OAT3 | |||
| Abiraterone N-oxide sulfate (DBMET00951) | 445.572 | >100 | 1.36 | NA |
| (S)-naproxen acyl-β-D-glucuronide (DBMET00160) | 400.4648 | >200 | 5.43 | NA |
| 4-hydroxy-trazodone (DBMET00405) | 387.863 | >200 | 16.9 | NA |
| Imatinib (piperidine)-4-oxide (DBMET00027) | 510.622 | >200 | 64.2 | NA |
| Sulindac sulfone (DBMET01003) | 372.41 | 23.0 | 0.60 | 38.4 |
| Celecoxib carboxylic acid (DBMET00810) | 411.355 | 262 | 8.43 | 31.1 |
| N-desmethyl rosiglitazone (DBMET00295) | 343.4 | 33.3 | 1.15 | 28.9 |
| 5-Carboxy-meloxicam (DBMET00754) | 381.384 | 19.6 | 0.70 | 27.9 |
| 5-OH-rosiglitazone sulfate (DBMET01112) | 453.489 | 33.7 | 1.31 | 25.8 |
| Abiraterone sulfate (DBMET00788) | 429.572 | 38.2 | 1.83 | 20.9 |
| Hydroxy pioglitazone (DBMET01306) | 372.438 | 27.6 | 1.37 | 20.2 |
| Keto pioglitazone (DBMET01307) | 370.422 | 12.5 | 0.73 | 17.2 |
| 3-cis-hydroxycyclohexyl glyburide (DBMET01806) | 510.003 | 10.1 | 0.81 | 12.5 |
| (S)-7-hydroxy warfarin (DBMET01042) | 324.3273 | 14.5 | 1.93 | 7.48 |
| Losartan carboxylic acid (DBMET00303) | 436.9 | 0.69 | 0.10 | 6.66 |
| Probenecid acyl β-D-glucuronide | 461.5 | 130 | 19.9 | 6.56 |
| O-desmethyl indomethacin (DBMET00156) | 343.8 | 1.62 | 0.27 | 6.01 |
| Sulindac sulfide (DBMET01002) | 340.411 | 3.68 | 0.66 | 5.56 |
| (S)-ketoprofen β-D-glucuronide (DBMET00589) | 430.4047 | 40.2 | 8.07 | 4.98 |
| Tolmetin β-D-glucuronide (DBMET00652) | 433.4086 | 7.17 | 2.92 | 2.46 |
| (S)-O-desmethyl naproxen (DBMET00159) | 216.2326 | 4.70 | 3.44 | 1.37 |
| 4-Hydroxy flurbiprofen (DBMET00151) | 260.2603 | 1.60 | 1.39 | 1.15 |
| 6-Hydroxy chlorzoxazone (DBMET00327) | 185.565 | 3.60 | 3.21 | 1.12 |
| 4-Hydroxy diclofenac (DBMET00144) | 312.148 | 0.48 | 2.03 | 0.24 |
| 5-Hydroxy Rosiglitazone β-D-Glucuronide (DBMET00981) | 549.55 | >200 | >200 | NA |
| Estrone sulfate | 350.4 | 170 | 1.00 | 170 |
NA: Not available.
Data from Drugbank or PubChem.
Fig. 2.

Comparison of inhibition potency of drug metabolites for OAT1 and OAT3. (a) The Ki values of 20 drug metabolites plus estrone sulfate (positive control) for OAT1 and OAT3 were plotted. Each dot represents Ki values of each drug metabolite. The line was forced through x = 0 and y = 0 with an angle of 45° (indicating a slope of 1). The further a point is away from the line, the more different between the Ki values for OAT1 and OAT3. Five drug metabolites were not shown in the plot because they are not inhibitors of OAT1 and/or OAT3, including abiraterone N-oxide sulfate, (S)-naproxen acyl-β-D-glucuronide, 4-hydroxy-trazodone, imatinib (piperidine)-4-oxide and 5-hydroxy rosiglitazone-β-D-glucuronide. (b) Comparison of the averaged molecular weight of drug metabolites showing >10-fold inhibition potency ratio (OAT3/OAT1) with molecular weight of drug metabolites showing 10-fold inhibition potency ratio. *p < 0.05 (student’s unpaired t-test). The inhibition potency ratio is calculated by dividing the Ki value of a drug metabolite for OAT1 by the Ki value of the same drug metabolite for OAT3. The higher the ratio, the higher the inhibition potency for OAT3.
Drug metabolite Ki values were more correlated with parent drug Ki values for OAT3 than for OAT1. IC50 studies were also performed for the 19 parent drugs corresponding to the tested drug metabolites. Our study confirmed that 11 drugs previously identified as OAT1 and OAT3 inhibitors were indeed inhibitors of the two transporters, e.g., losartan, probenecid, and diclofenac (Table 2). In addition, we determined that meloxicam, rosiglitazone, imatinib, warfarin, chlorzoxazone, and trazodone were inhibitors of the transporters (Table 2). These drugs are not listed as OAT1 or OAT3 inhibitors in Drugbank (https://www.drugbank.ca/). Of the 17 parent compounds that inhibited OAT1 or OAT3, 16 were more potent inhibitors of OAT3 than OAT1, with the ratio of Ki values for OAT1 over OAT3 greater than 1 (Fig. 3A, Table 2). The average molecular weight of the parent drugs having >10-fold higher potency towards OAT3 (over OAT1) was significantly higher than the average molecular weight of the drugs having ≤ 10-fold potency towards OAT3 (p < 0.05) (Fig. 3B). These data agree with the inhibitory characteristics of drug metabolites, which also show greater potencies for inhibiting OAT3 compared with OAT1 (Fig. 2A, Table 1) and higher molecular weights (Fig. 2B). In fact, the Ki values of drug metabolites were better correlated with the Ki values of parent drugs for OAT3 (R2 = 0.556) (Fig. 4A) than for OAT1 (R2 = 0.018) (Fig. 4B).
Table 2.
Ki Values for 19 Parent Drugs.
| Drug | Molecular Weight (g/mol)a | Ki (μM) |
Potency (hOAT3/hOAT1) | |
|---|---|---|---|---|
| OAT1 | OAT3 | |||
| Indomethacin | 357.788 | 4.35 | 0.11 | 39.5 |
| Sulindac | 356.411 | 56.8 | 1.65 | 34.4 |
| Losartan | 422.911 | 11.4 | 0.36 | 31.7 |
| Glyburide | 494.004 | 5.7 | 0.21 | 27.1 |
| Meloxicam | 351.401 | 6.88 | 0.54 | 12.7 |
| Rosiglitazone | 357.427 | 4.2 | 0.59 | 7.12 |
| Trazodone | 371.864 | 19.9 | 2.94 | 6.77 |
| Probenecid | 285.359 | 7.74 | 1.25 | 6.19 |
| Pioglitazone | 356.439 | 6.3 | 1.02 | 6.18 |
| Chlorzoxazone | 169.565 | 52.3 | 10.2 | 5.13 |
| Warfarin | 308.3279 | 15.8 | 3.11 | 5.08 |
| Tolmetin | 257.2845 | 3.01 | 0.81 | 3.72 |
| Diclofenac | 296.149 | 1.67 | 0.62 | 2.69 |
| Imatinib | 493.6027 | 29.2 | 14.5 | 2.01 |
| Ketoprofen | 254.2806 | 1.73 | 1.61 | 1.07 |
| Flurbiprofen | 244.2609 | 1.91 | 1.8 | 1.06 |
| Naproxen | 230.2592 | 3.23 | 4.75 | 0.68 |
| Celecoxib | 381.372 | >200 | >200 | NA |
| Abiraterone | 349.509 | >20 | >20 | NA |
NA: Not available.
Data from Drugbank or PubChem.
Fig. 3.

Comparison of inhibition potency of parent drugs for OAT1 and OAT3. (a) The Ki values of 17 drugs for both OAT1 and OAT3 were plotted. Each dot represents Ki values of each unique drug. The line was forced through x = 0 and y = 0 with the angle of 45° (indicating a slope of 1). The further a point is away from the line, the more different between the Ki values for OAT1 and OAT3. Two drugs were not shown in the plot because they are not inhibitors of OAT1 and OAT3, including celecoxib and abiraterone. (b) Comparison of molecular weight of parent drugs showing >10-fold inhibition potency ratio (OAT3/OAT1) with molecular weight of parent drugs showing ≤ 10-fold inhibition potency ratio. *p < 0.05 (student’s unpaired t-test).
Fig. 4.

Correlation of Ki values between parent drugs and drug metabolites for OAT3 (A) and OAT1 (B). For both (a) and (b) each point represents a pair of drug and drug metabolite with the parent drug Ki value showing on the x-axis and the drug metabolite Ki value showing on the y-axis. The line was forced through x = 0 and y = 0 with the angle of 45° consistent with a slope of 1. The further a dot is away from the line, the more different between the Ki values of a parent drug and the corresponding drug metabolite.
Some drug metabolites were more potent inhibitors of OAT1 or OAT3 compared to their corresponding parent compounds. Although drug metabolites generally had higher Ki values than their parent drugs (Fig. 4A and B), we found that eight drug metabolites were equal to or more potent in inhibiting OAT1-mediated 6CF uptake than the corresponding parent drugs. (Supplementary Table S4). Similarly, seven drug metabolites were equal to or more potent in inhibiting OAT3 than their parent molecules (Supplementary Table S5). For example, losartan carboxylic acid was 16.5 times more potent in inhibiting OAT1 (Supplementary Table S4) and 3.6 times more potent in inhibiting OAT3 (Supplementary Table S5) than losartan. In addition, abiraterone and celecoxib were not inhibitors of OAT1 and OAT3 (Supplementary Table S5) while abiraterone N-oxide sulfate, abiraterone sulfate and celecoxib carboxylic acid were potent inhibitors of OAT3 with Ki values of 1.36 μM, 1.83 μM and 8.43 μM, respectively (Supplementary Table S5). Similarly, abiraterone sulfate and celecoxib carboxylic acid were inhibitors of OAT1 with Ki values of 38.2 μM and 262 μM, respectively (Supplementary Table S4).
Some drug metabolites are predicted to inhibit OAT3 at clinically relevant unbound plasma concentrations. According to the FDA guidance,21 any drug with Cmax,u/IC50 ≥ 0.1 for OAT1 and OAT3 should be considered for a clinical DDI study. Cmax,u is the maximal unbound plasma concentration of a drug. Consideration of conducting a clinical DDI study depends on many other factors such as therapeutic window of the “victim” drug, and likelihood of use of the inhibitor with concomitant medications need to be considered.22 Nevertheless, using these criteria, the potential clinical significance of drug and drug metabolites was examined by determining Cmax,u/Ki values for OAT1 and OAT3. Notably, sulindac, its active metabolite, sulindac sulfide, and its inactive metabolite, sulindac sulfone, showed Cmax,u/Ki ≥ 0.1 for OAT3 (Table 3). In addition, losartan had a Cmax,u/Ki value < 0.1 for OAT3, but its active metabolite, losartan carboxylic acid, had a Cmax,u/Ki value ≥ 0.1 (Table 3), suggesting that the metabolite is more likely to inhibit the transporter clinically than the parent drug. Similarly, abiraterone did not inhibit OAT3 but its inactive metabolite, abiraterone sulfate, had a Cmax,u/Ki value of 0.137 (Table 3).
Table 3.
List of Drugs and Drug Metabolites with Cmax,u/Ki Values ≥ 0.1.
| Compound | Cmax (μM) | Unbound Fraction 100, % | Cmax,u (μM) | Ki(OAT1) (μM) | Cmax,u/Ki(OAT1) | Ki(OAT3) (μM) | Cmax,u/Ki(OAT3) |
|---|---|---|---|---|---|---|---|
| Sulindac | 32.0a | 6.78%a | 2.24 | 56.8 | 0.039 | 1.65 | 1.36 |
| Sulindac sulfide | 22.6a | 6.01%a, 4.6%b | 0.14e1.5b | 3.68 | 0.038e0.408 | 0.66 | 0.212e2.27 |
| Sulindac sulfone | 4.48a | 2.1%b | 0.1e0.4b | 23.0 | 0.004e0.017 | 0.60 | 0.167e0.667 |
| Losartan | 0.814e1.44c | 1.4%d | 0.011e0.02 | 11.4 | 0.00096e0.0018 | 0.36 | 0.03e0.056 |
| Losartan carboxylic acid | 0.554e1.13c | 1%d,e | 0.005e0.011 | 0.69 | 0.007e0.016 | 0.10 | 0.05e0.11 |
| Abiraterone | 0.37f | <1%e | 0.0037 | >20 | NA | >20 | NA |
| Abiraterone sulfate | 25f | <0.15%e | 0.25 | 38.2 | 0.0065 | 1.83 | 0.137 |
| Abiraterone N-oxide sulfate | 9.2f | 0.22%e | 0.092 | >100 | NA | 1.36 | 0.068 |
Values ≥ 0.1 are highlighted in bold.
NA: Not available.
Drugbank (https://www.drugbank.ca/drugs/DB00678).
According to FDA guidance (https://www.fda.gov/media/108130/download), the unbound fraction should be set to 1% if experimentally determined to be less than 1%.
Discussion
Many drugs undergo biotransformation by Phase I and Phase II metabolizing enzymes to form more polar and less membranepermeable drug metabolites.23 Thus, to enter cells or subcellular organelles, many drug metabolites require membrane transporters.24 However, our understanding of the interaction of drug metabolites with transporters is extremely limited. In particular, many drug metabolites are poorly characterized during drug development because they are inactive with respect to their pharmacological targets. However, it is reasonable to postulate that drug metabolites may be ligands (substrates or inhibitors) of membrane transporters, particularly metabolites of parent drugs which are ligands of the transporters. That is, drug metabolites contain the chemical scaffold of the parent molecules and therefore may harbor the necessary structural elements that confer transporter specificity. In this study, we evaluated the interaction of a number of drug metabolites and their corresponding parent compounds with human OAT1 and OAT3, key determinants of drug excretion and major targets for drug-drug interactions.
Our study led to three major findings. First, metabolites of the drugs studied were inhibitors of OAT1 and OAT3, and consistent with the inhibitory characteristics of their respective parent compounds, their potency for inhibition of OAT3 was generally greater than for OAT1. Second, though parent compounds were generally more potent inhibitors of OAT1 and OAT3 than their metabolites, this was not always the case. That is, some drug metabolites were considerably more potent inhibitors than their parent compounds. Third, some metabolites are predicted to cause clinically relevant DDIs based on current criteria used by the U.S. Food and Drug Administration.23 Below we discuss each of our findings.
First, we observed that drug metabolites were inhibitors of OAT1 and OAT3, and consistent with the inhibitory characteristics of their respective parent compounds, their potency for inhibition of OAT3 was generally greater than for OAT1. Though there are no high-resolution crystal structures of the transporters or indeed any transporter in the human SLC22 family,25 several studies using homology modeling have demonstrated structural differences between OAT1 and OAT3, including differences in the substrate binding sites of the two transporters.26 Further, the molecular features distinguishing inhibitors from non-inhibitors, such as molecular weight and volume are distinct for the two transporters.26–28 Consistent with the notion that higher molecular weight is an important feature of OAT3, but not OAT1 inhibitors,28 we observed that the average molecular weight was significantly larger for both parent drugs and metabolites having >10-fold inhibition potency ratio (OAT3/OAT1) compared to those having ≤10-fold inhibition potency ratio (Figs. 2B and 3B). In addition, whereas naproxen, trazodone and imatinib inhibited OAT1 with Ki values of 3.23 μM, 19.9 μM and 29.2 μM, respectively (Table 2), their bulkier (higher molecular weight) metabolites did not inhibit OAT1 at 200 μM (Table 1). In contrast, these metabolites remained inhibitors of OAT3 with Ki values within 0.7–5.7-fold of the respective Ki values for their parent drugs. These results further support the idea that compounds with bulkier groups or larger volumes generally prefer to interact with OAT3 rather than OAT1.26,28
Second, though parent compounds were generally more potent inhibitors of OAT1 and OAT3 than their metabolites, there were some notable exceptions. The most striking examples were celecoxib and abiraterone, both of which were non-inhibitors of OAT1 and OAT3 at screening concentrations of 200 μM and 20 μM, respectively (Table 2). Celecoxib is a neutral compound, which may be the reason why it did not inhibit OAT1 and OAT3, which prefer anionic compounds. In contrast, the metabolite, celecoxib carboxylic acid, has an acidic pKa of 3.9829 and will thus form an anion at pH 7.4 that interacts with OAT1 and OAT3 (Table 1). For abiraterone, the basic pKa is 5.19,30 which indicates that most of the drug is in the unionized form at pH 7.4. Due to the low water solubility of abiraterone in DMSO, concentrations of 20 μM (rather than 200 μM) were used to screen the transporters, and at 20 μM, abiraterone did not inhibit OAT1 or OAT3 (Table 2). However, its metabolites, abiraterone N-oxide sulfate and abiraterone sulfate, are anions and therefore likely form more favorable interactions with the OAT1 and OAT3 binding pockets (Table 1). These results suggest that even if a drug is uncharged at physiological pH and therefore, not an inhibitor of OAT1 and OAT3, biotransformation may produce negatively charged metabolites that inhibit OAT1 and OAT3.
Our third finding has implications to clinical DDIs. That is, some metabolites are predicted to cause clinically relevant DDIs based on current criteria used by the U.S. FDA.21 Our studies identified several drug metabolites, including sulindac sulfide (active metabolite), losartan carboxylic acid (active metabolite), sulindac sulfone (inactive metabolite) and abiraterone sulfate (inactive metabolite), that have Cmax,u/Ki values ≥ 0.1 for OAT3 (Table 3). It is worth noting that many drug metabolites may be generally more easily and rapidly eliminated from the body compared to parent drugs. In addition, for losartan carboxylic acid, abiraterone and its metabolites, their plasma unbound fraction was lower than 1% and according to FDA guidance, their Cmax,u values were calculated assuming that the unbound fraction is 1%. The use of 1% as the fraction unbound will result in a conservative estimate of the actual Cmax,u, which may be considerably lower. Thus, though application of the criteria (with a 1% fraction unbound) suggests that a clinical study is warranted, the likelihood of a clinical DDI will depend on the actual unbound drug concentrations and the time course of the metabolite in the body. However, a conservative approach is warranted, given that the parent compounds of these three metabolites are commonly prescribed drugs used in the treatment of chronic disorders in individuals who are likely to be on concomitant medications. For example, losartan is an anti-hypertensive drug, listed in the top 200 most highly prescribed drugs.31 Sulindac is a commonly prescribed NSAID used in the treatment of inflammatory conditions such as osteoarthritis,32 and abiraterone is used chronically to prevent recurrence of prostate cancer.33 Thus, there is a risk for the metabolites of these drugs to cause or contribute to DDIs in patients on concomitant medications. Although speculative, abiraterone sulfate may have contributed to rosuvastatin-induced rhabdomyolysis in a 76-year-old Caucasian man with metastatic prostate cancer treated with abiraterone.34 Renal clearance of rosuvastatin accounts for 28% of its total plasma clearance, with 90% of the renal clearance thought to result from OAT3-mediated tubular secretion.35,36 Thus, higher plasma levels of rosuvastatin may have resulted from inhibition of renal tubular secretion by abiraterone sulfate in this individual, increasing his risk for rosuvastatin-induced rhabdomyolysis.
In conclusion, our study identified drug metabolites as potent inhibitors of OAT1 and OAT3, indicating that these transporters, and especially OAT3, may be targets for drug-drug metabolite interactions. Clinical pharmacologic studies are clearly needed to demonstrate proof of concept that drug metabolites may reduce renal tubule secretion of concomitantly administered drugs in vivo, and to determine if routine prospective characterization of drug metabolites as inhibitors of renal transporters is needed during drug development to ensure drug safety and efficacy.
Supplementary Material
Acknowledgement
We acknowledge that this publication was supported in part by Grant U01FD004979/U01FD005978 from the US Food and Drug Administration (FDA), which supports the University of California, San Francisco–Stanford Center of Excellence in Regulatory Sciences and Innovation (UCSF-Stanford CERSI). We are pleased to publish this manuscript in an issue of Journal of Pharmaceutical Sciences dedicated to Per Artursson, who is a leader in the pharmaceutical sciences and a friend.
Footnotes
Supporting Information
The supporting information is available online. Two figures and five tables including chemical structures of drugs and drug metabolites; IC50 curves for drugs and drug metabolites; Chemical purchase information; Therapeutic categories of drugs for screening; Screening concentrations for drugs and drug metabolites. Drug metabolites that are ≥2 times more potent inhibitors of OAT3 compared to parent drugs.
Appendix A. Supplementary Data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.xphs.2020.09.004.
References
- 1.Ma G, Ks E. Pharmacokinetic overview of oral second-generation H1 antihistamines. Int J Clin Pharmacol Ther. 1998;36(5):292–300. [PubMed] [Google Scholar]
- 2.Staffa JA, Chang J, Green L. Cerivastatin and reports of fatal rhabdomyolysis. N Engl J Med. 2002;346(7):539–540. [DOI] [PubMed] [Google Scholar]
- 3.Krogstad V, Peric A, Robertsen I, et al. A comparative analysis of cytochrome P450 activities in paired liver and small intestinal samples from patients with obesity. Drug Metab Dispos. 2020;48(1):8–17. [DOI] [PubMed] [Google Scholar]
- 4.International Transporter Consortium, Giacomini KM, Huang S-M Tweedie DJ, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9(3):215–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsson P, Fenu LA, Lundquist P, Wisniewski JR, Kansy M, Artursson P. Quantifying the impact of transporters on cellular drug permeability. Trends Pharmacol Sci. 2015;36(5):255–262. [DOI] [PubMed] [Google Scholar]
- 6.Fujita T, Brown C, Carlson EJ, et al. Functional analysis of polymorphisms in the organic anion transporter, SLC22A6 (OAT1). Pharmacogenet Genomics. 2005;15(4):201–209. [DOI] [PubMed] [Google Scholar]
- 7.Yee SW, Nguyen AN, Brown C, et al. Reduced renal clearance of cefotaxime in asians with a low-frequency polymorphism of OAT3 (SLC22A8). J Pharm Sci. 2013;102(9):3451–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watanabe T, Kusuhara H, Watanabe T, et al. Prediction of the overall renal tubular secretion and hepatic clearance of anionic drugs and a renal drug-drug interaction involving organic anion transporter 3 in humans by in vitro uptake experiments. Drug Metab Dispos. 2011;39(6):1031–1038. [DOI] [PubMed] [Google Scholar]
- 9.Antonescu IE, Karlgren M, Pedersen ML, et al. Acamprosate is a substrate of the human organic anion transporter (OAT) 1 without OAT3 inhibitory properties: implications for renal acamprosate secretion and drugedrug interactions. Pharmaceutics. 2020;12(4):390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morrissey KM, Stocker SL, Wittwer MB, Xu L, Giacomini KM. Renal transporters in drug development. Annu Rev Pharmacol Toxicol. 2013;53(1):503–529. [DOI] [PubMed] [Google Scholar]
- 11.Nozaki Y, Kusuhara H, Kondo T, et al. Species difference in the inhibitory effect of nonsteroidal anti-inflammatory drugs on the uptake of methotrexate by human kidney slices. J Pharmacol Exp Ther. 2007;322(3):1162–1170. [DOI] [PubMed] [Google Scholar]
- 12.Li M, Anderson GD, Wang J. Drugdrug interactions involving membrane transporters in the human kidney. Expert Opin Drug Metab Toxicol. 2006;2(4): 505–532. [DOI] [PubMed] [Google Scholar]
- 13.Yin J, Wang J. Renal drug transporters and their significance in drugedrug interactions. Acta Pharm Sin B. 2016;6(5):363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zamek-Gliszczynski MJ, Taub ME, Chothe PP, et al. Transporters in drug development: 2018 ITC recommendations for transporters of emerging clinical importance. Clin Pharmacol Ther. 2018;104(5):890–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giacomini KM, Galetin A, Huang SM. The international transporter consortium: summarizing advances in the role of transporters in drug development. Clin Pharmacol Ther. 2018;104(5):766–771. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L, Huang S-M, Reynolds K, Madabushi R, Zineh I. Transporters in drug development: scientific and regulatory considerations. Clin Pharmacol Ther. 2018;104(5):793–796. [DOI] [PubMed] [Google Scholar]
- 17.Appendix: drug metabolizing enzymes and biotransformation reactions. In: ADME-enabling Technologies in Drug Design and Development. John Wiley & Sons, Ltd; 2012:545–565. [Google Scholar]
- 18.Iwaki M, Shimada H, Irino Y, Take M, Egashira S. Inhibition of methotrexate uptake via organic anion transporters OAT1 and OAT3 by glucuronides of nonsteroidal anti-inflammatory drugs. Biol Pharm Bull. 2017;40(6):926–931. [DOI] [PubMed] [Google Scholar]
- 19.Zou L, Stecula A, Gupta A, et al. Molecular Mechanisms for Species Differences in Organic Anion Transporter 1, OAT1: Implications for Renal Drug Toxicity. Mol Pharmacol. 2018;94(1):689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zou L, Pottel J, Khuri N, et al. Interactions of oral molecular excipients with breast cancer resistance protein, BCRP. Mol Pharm. 2020;17(3):748–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.In vitro drug interaction studies - cytochrome P450 enzyme- and transporter-mediated drug interactions guidance for industry. 2020. Available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/vitro-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions. Accessed June 7, 2020.
- 22.Clinical drug interaction studies - cytochrome P450 enzyme- and transporter-mediated drug interactions guidance for industry. Avaiable at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions; 2020. Accessed June 7, 2020. [Google Scholar]
- 23.Zamek-Gliszczynski MJ, Chu X, Polli JW, Paine MF, Galetin A. Understanding the transport properties of metabolites: case studies and considerations for drug development. Drug Metab Dispos. 2014;42(4):650–664. [DOI] [PubMed] [Google Scholar]
- 24.Matsson P, Lundquist P, Artursson P. The need for speed–kinetic limits of drug transporters. Trends Pharmacol Sci. 2016;37(4):243–245. [DOI] [PubMed] [Google Scholar]
- 25.César-Razquin A, Snijder B, Frappier-Brinton T, et al. A call for systematic research on solute carriers. Cell. 2015;162(3):478–487. [DOI] [PubMed] [Google Scholar]
- 26.Astorga B, Wunz TM, Morales M, Wright SH, Pelis RM. Differences in the substrate binding regions of renal organic anion transporters 1 (OAT1) and 3 (OAT3). Am J Physiol Renal Physiol. 2011;301(2):F378–F386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nigam AK, Li JG, Lall K, et al. Unique metabolite preferences of the drug transporters OAT1 and OAT3 analyzed by machine learning. J Biol Chem. 2020;295(7):1829–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duan P, Li S, Ai N, Hu L, Welsh WJ, You G. Potent inhibitors of human organic anion transporters 1 and 3 from clinical drug libraries: discovery and molecular characterization. Mol Pharm. 2012;9(11):3340–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Compound report card. Avaiable at: https://www.ebi.ac.uk/chembl/compound_report_card/CHEMBL743/. Accessed May 20, 2020.
- 30.Abiraterone. Avaiable at: https://www.ebi.ac.uk/chembl/compound_report_card/CHEMBL254328/. Accessed May 20, 2020.
- 31.Top 200 drugs by search results and sales. Drugs.com. Avaiable at: https://www.drugs.com/top200. Accessed May 18, 2020.
- 32.Sulindac: MedlinePlus Drug Information. Avaiable at: https://medlineplus.gov/druginfo/meds/a681037.html. Accessed May 18, 2020.
- 33.Abiraterone: MedlinePlus Drug Information. Avaiable at: https://medlineplus.gov/druginfo/meds/a611046.html. Accessed May 18, 2020.
- 34.Neyra JA, Rocha NA, Bhargava R, Vaidya OU, Hendricks AR, Rodan AR. Rhabdomyolysis-induced acute kidney injury in a cancer patient exposed to denosumab and abiraterone: a case report. BMC Nephrol. 2015;16(1):118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin PD, Warwick MJ, Dane AL, Brindley C, Short T. Absolute oral bioavailability of rosuvastatin in healthy white adult male volunteers. Clin Ther. 2003;25(10):2553–2563. [DOI] [PubMed] [Google Scholar]
- 36.Windass AS, Lowes S, Wang Y, Brown CDA. The contribution of organic anion transporters OAT1 and OAT3 to the renal uptake of rosuvastatin. J Pharmacol Exp Ther. 2007;322(3):1221–1227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.