

Abstract
The design of synthetic routes via retrosynthetic logic is decisively influenced by the transformations available. In this context, transition metal-catalyzed C–H activation has emerged as a powerful strategy for C–C bond formation, with myriad methods developed for diverse substrates and coupling partners. Despite its interest and proven efficacy however, its uptake in the total synthesis has been tepid, partially owing to their apparent synthetic intractability and inapplicability, as well as a lack of comprehensive guidelines for their implementation. These issues are addressed in this review, where we guide the identification of retrosynthetic opportunities to incorporate C–H activation processes for the generation of C–C bonds. By comparing and analyzing total syntheses accomplished using traditional approaches against recent C–H activation methods, this review demonstrates how the successful implementation of C–H activation-enabled C–C bond construction has led to the realization of more efficient retrosynthetic strategies, as well as the execution of previously unattainable tactical maneuvers. Finally, we identify shortcomings due to limitations in existing C–H activation processes, and project how some of the total syntheses highlighted in this review can be further economized by adopting emerging next-generation C–H activation technologies.
Keywords: C–H activation, transition metal, catalysis, total synthesis, C–H functionalization
Graphical Abstract
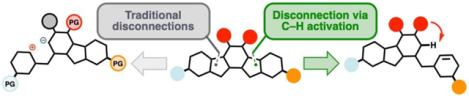
The advent of new synthetic methods has historically motivated the evolution of retrosynthetic logic. A significant recent advance is C–H activation-enabled C–C bond formation, but it has only seen modest application in synthesis. This review compares traditional synthetic routes towards complex targets with reconceptualized ones utilizing C–H activation, highlighting the strategic advantages of this powerful approach.
1. Introduction
1.1. Total synthesis and organic chemistry: a symbiotic relationship
The advancement of organic chemistry is inextricably linked to the pursuit of natural product synthesis. This craft, now known as total synthesis, can be historically traced back to Wöhler’s total synthesis of urea (1) in 1828 (Figure 1A),[1] which fundamentally proved the chemical foundations of nature. In the next 150 years, chemists have achieved syntheses of increasingly more complex natural products, with Robinson’s total synthesis of tropinone (2, 1917) being a particularly elegant example.[2] During this time, society’s demand for bioactive natural products increased in recognition of their beneficial medicinal properties, and it soon became apparent that nature could not offer all of the immediate solutions for their large-scale production. One key player in propelling the utility of organic synthesis lay in Sheehan’s total synthesis of penicillin (3, 1957),[3] which contributed to the development of bespoke artificial alternatives, inaccessible on scale from natural sources, to overcome antibiotic resistance.
Figure 1.
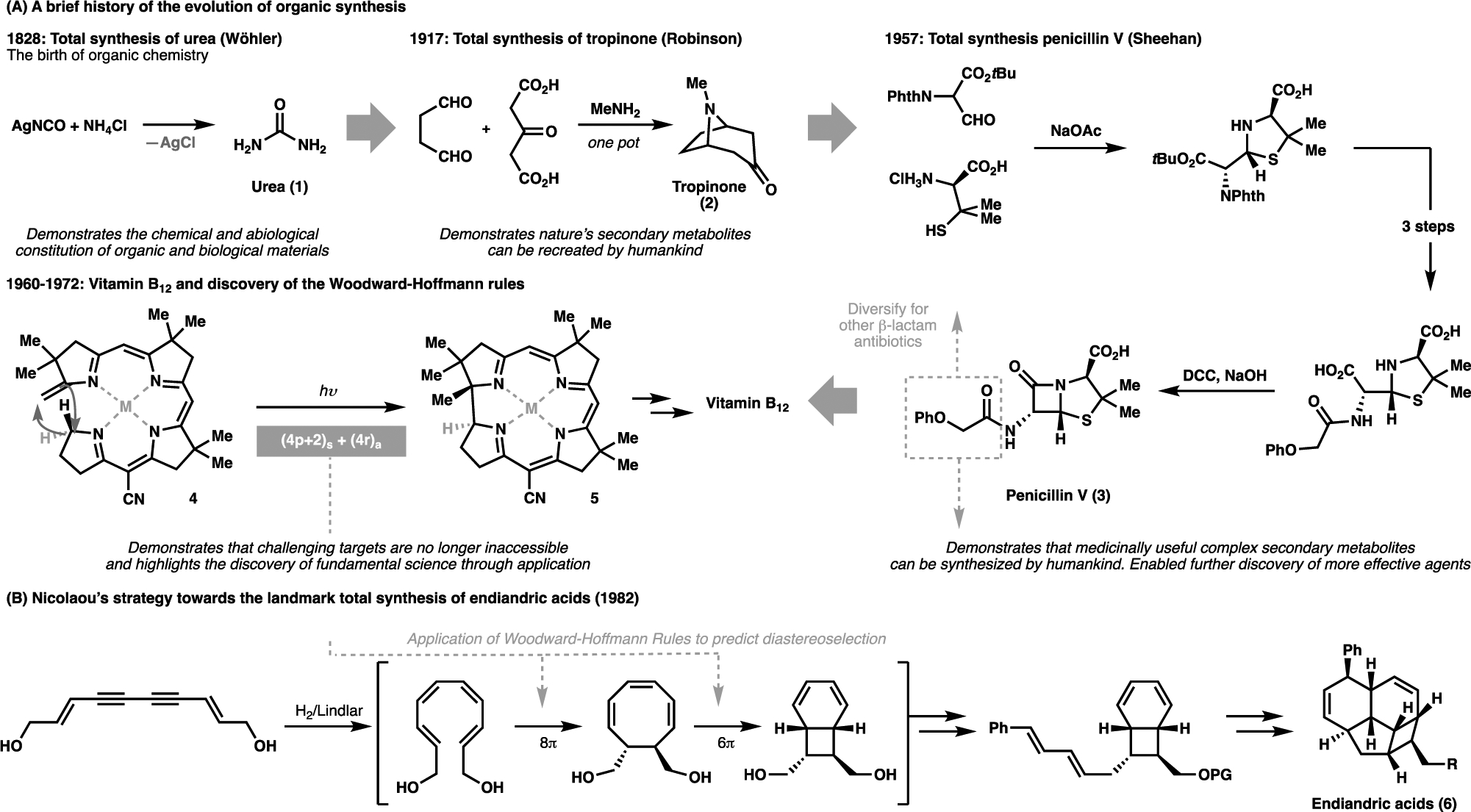
(A) A brief history of the evolution of organic synthesis from Wöhler’s total synthesis of urea (1) to the landmark total synthesis of vitamin B12 by Woodward and Eschenmoser; (B) Application of the newly conceptualized Woodward-Hoffmann rules enabled Nicolaou to access the endiandric acids (6)
Targeting increasingly complex structures necessitated the generation of a vast repertoire of chemical innovations. As the field developed, previously esoteric transformations were repurposed as powerful synthetic strategies, and targeting increasingly complex structures demanded the continual invention of new methods. Moreover, these transformations had to be reliable and robust to withstand the wide array of functionalities present in natural products, making total synthesis the perfect playground to demonstrate the applicability of new chemical innovations. These staggering advancement in our capacity to control reactivity in a space of a few decades enabled chemists to expeditiously target complex structures and illuminate nature’s machinery; the pioneering applications of pericyclic reactions (e.g. from 4 to 5) en route to vitamin B12[4] was a necessary advancements to enable Nicolaou’s elegant biomimetic synthesis of endiandric acids (6) through sequential pericyclic cascades (Figure 1B).[5] At the same time, the conceptualization of the previously-obscure craft of synthesis into defined synthetic logic, as proposed by Corey,[6] enabled the wider community to approach synthetic design in a systematic and structured manner. These discoveries were crucial in setting the foundation of organic chemistry, and are among the numerous achievements enabled through the synthetic interrogation of natural products.
Our increasing proficiency in organic synthesis led to the discovery of the reactivities that extend beyond nature’s chemical repertoire. Advances in transition metal-catalyzed reactions, such as cross coupling and olefin metathesis, have redefined canonically inert functionalities as potentially reactive handles, as well as dramatically simplifying the construction of many C–C bonds.[7,8] The discovery of these transition metal-catalyzed processes represented a paradigm shift in organic synthesis. Over a period of 30 years, what started off as niche findings in organometallic chemistry gradually became more and more integrated into the common parlance of organic chemists as the reactivities of transition metal-catalyzed processes became better understood and their capabilities broadened. This development widened the toolbox available for retrosynthetic analyses, and enabled the facile construction of otherwise challenging motifs, such as polyaryl/alkenyl containing natural products and pharmaceutical agents.[7] These new reactivities redefined the structural cues chemists can mentally anchor their retrosynthetic analyses with, and in doing so, enabled alternative efficacious methods for the construction of secondary metabolites. By way of example, the advent of transition metal-catalyzed coupling processes has led to creative new approaches such as the one employed towards the total synthesis of (+)-lysergol (7) (Figure 2A).[9] Analogously, cross-metathesis cascades can allow for the rapid generation of complexity, as in Smith’s total synthesis of morphine (8) (Figure 2B).[10]
Figure 2.
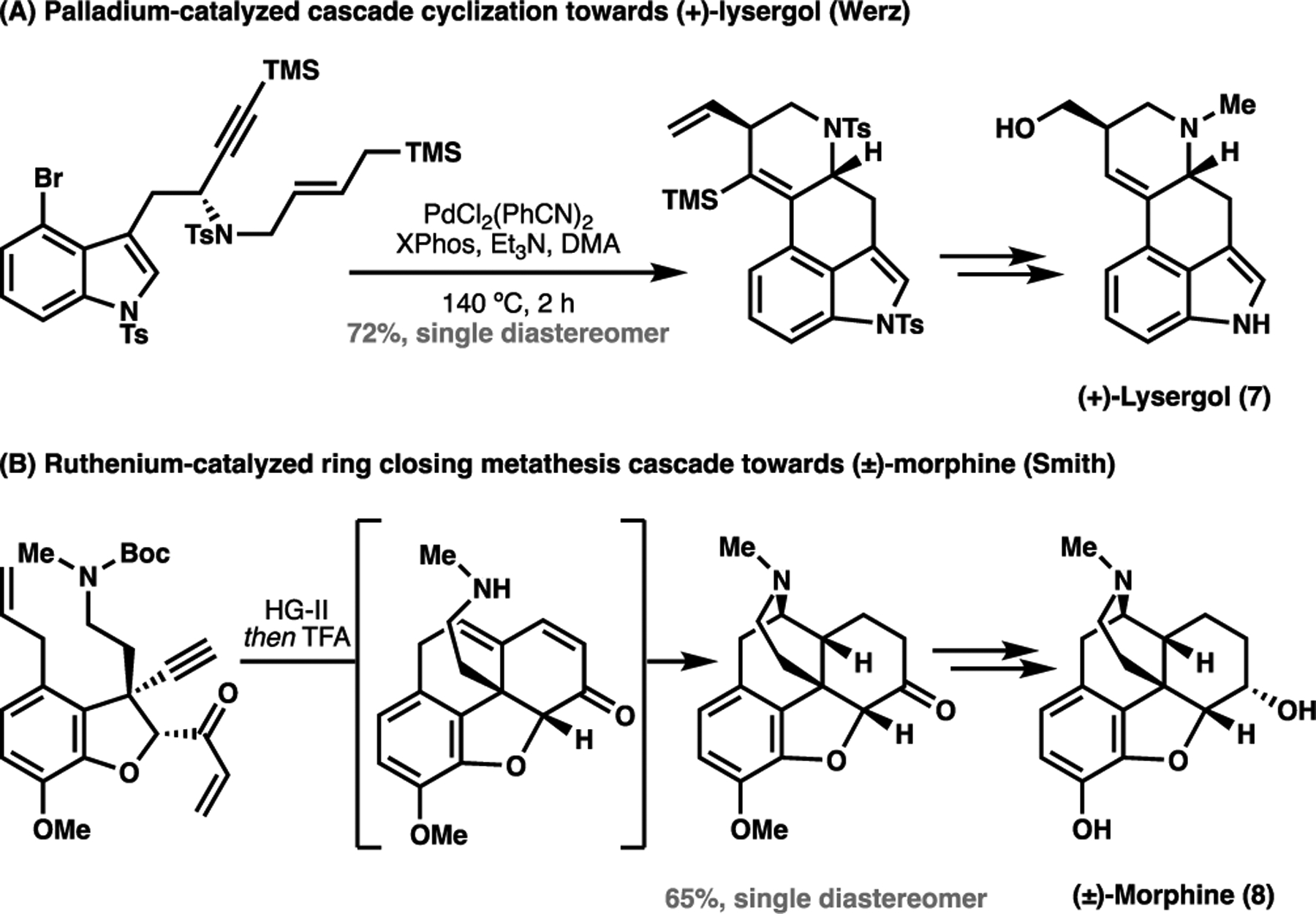
Two examples of total syntheses uniquely enabled by transition-metal catalyzed processes: (A) a palladium-catalyzed cascade cyclization forms the scaffold for (+)-lysergol (7, Werz); (B) a ruthenium-catalyzed cascade ring closing metathesis generates the scaffold for morphine (8, Smith). HG-II: Hoveyda-Grubbs Second Generation Catalyst
These myriad innovations in methodology and strategy have demonstrated that the synthesis of complex molecules is no longer an intractable venture. Therefore, the question that now faces us is how can organic synthesis be better? This multifaceted question underpins the challenging pursuit of synthetic economy[11–13] and synthetic ideality,[14,15] which remain far from being solved despite remarkable recent advances. Furthermore, the pursuit of synthetic economy and ideality often centers on how to implement the existing toolbox of traditional chemical transforms in an optimal manner, which fundamentally constrains the extent that this challenge can be addressed. Another approach to improving the economies of synthesis lies in prospecting and applying new innovations to the craft of complex molecule synthesis. The advent of C–H functionalization — the net conversion of C–H bonds to other functionalities — is one of these innovations. Of the many methods developed to effect this transformation, the most promising strategy, and the method primarily discussed in this review, is transition metal-catalyzed C–H activation/C–C bond formation.
1.2. C–C bond formation via C–H activation as an emerging synthetic paradigm in organic chemistry
The field of transition metal-catalyzed C–H activation arises from the humble observation in organometallic chemistry that transition metals can cleave C–H bonds to generate a metal-carbon bond.[16–19] Such reactivity observed with palladium is particularly lucrative, as it can be intercepted with a range of coupling partners, and has the potential to transform a notionally unreactive C–H bond ubiquitously present in all organic molecules to a variety of functionalized C–C or C–X bonds. In particular, skeletal construction and diversification through C–C bond formation via C–H activation can enable the expeditious generation of molecular complexity by offering conceptually alternative methods towards the synthesis of canonical motifs. Two illustrative examples are highlighted in Figure 3, where C(sp2)–H activation can offer an alternative approach towards transition-metal catalyzed cross-coupling transforms without the need to install a suitable coupling handle (Figure 3A). Similarly, a direct enantioselective β-C(sp3)–H activation process offers a valuable alternative to the widely employed asymmetric conjugate addition transform, which tends to require highly reactive organometallic carbon nucleophiles to concordantly match with a suitable carbon electrophile (Figure 3B).[20] Indeed, the formation of C–C bonds via C–H activation is typified by the use of inert coupling partners, and thus obviates the need to protect reactive functionalities. Furthermore, as the metalated intermediate can be intercepted by a range of nucleophilic and electrophilic coupling partners, a wide range of consonant and dissonant carbon-containing motifs can be generated in a facile manner.[21] In doing so, this method also circumvents the need for polarity matching that typifies organic reactions. These strengths considerably widen the range of potential synthons for a given transformation and allow for greater flexibility to achieve a desired synthetic target.
Figure 3.
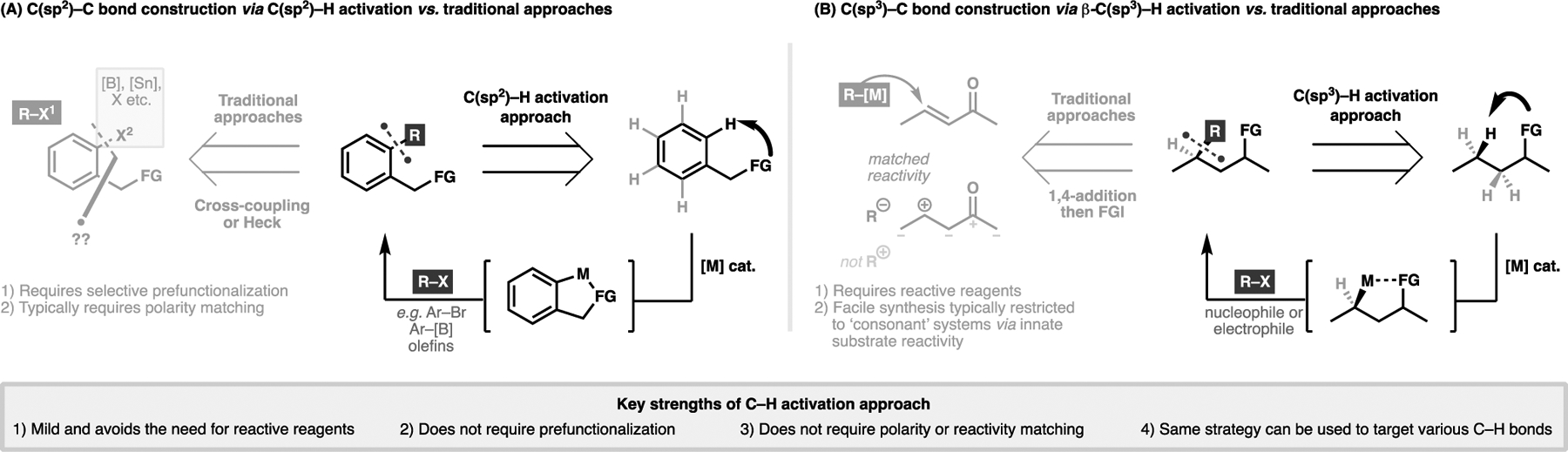
Overview of C–C bond formation via transition-metal catalyzed C–H activation and two examples on how they can offer a conceptually orthogonal approach towards organic synthesis: (A) via C(sp2)–H activation and (B) via β-C(sp3)–H activation
This attractive strategy is not without its risks: the ubiquity of the C–H bond poses challenges in positional (regio-) and spatial (stereo-)selectivity for C–H activation.[22] Over the last twenty years, the community has tackled these aforementioned challenges with a variety of creative innovations (e.g. directing group and ligand design), which has led to an explosion of research interest for this promising methodology. This can be measured by noting that there are more than 7500 reports published over the last ten years (2010–2020) for papers containing the terms C–H activation.[23] Today, C–H activation processes can be tamed to achieve high site-selectivity for sp2 and sp3 systems,[24,25]. Additionally, a greater understanding of catalyst reactivity has meant that site-selectivity for these processes can often be predicted, altered or carefully tuned a priori.
However, compared with cross-coupling and metathesis, a substantial disconnect exists between the plethora of methodological studies on C–H activation and its actual use in total synthesis, such that it is still considered to be a niche method in total synthesis. As an indicator, this is represented by just over 130 reports (containing both the terms ‘C–H activation’ and ‘total synthesis’) out of over 15,000 reports (containing the terms ‘total synthesis’) in the last ten years.[23] In particular, C–C bond construction using C–H activation remains particularly underrepresented. Given that such a technology has the potential impact to redefine how synthesis is conceptualized, the question we then have to ask is why is there such a gap between innovation and application? Is the philosophy of C–H activation methodology fundamentally flawed? Or are there peripheral reasons behind this discordancy? As testified by the diverse examples that have successfully employed this strategy,[26–28] we emphatically argue against the first reason. In fact, these manifold high-profile syntheses, if anything, point towards the potential of this method. While this gap in utility can be partially attributed to limitations of current C–H activation processes (e.g. poor reactivity of native substrates and/or limited scope), we propose that this disconnect primarily arises from our preference to adhere to the familiar, the tried-and-tested and the traditional. We argue that our honed retrosynthetic eye predisposes ourselves to rely on traditional chemical transforms rather than exploring new strategies that offer more efficient routes to targets.
To address this broader issue, this review outlines how to retrosynthetically approach the construction of C–C bonds through transition metal-catalyzed C–H activation as compared with canonical transforms. This is contextualized by discussing how traditional retrosynthetic strategies can be reconceptualized with C–H activation methods by comparing past and present routes taken to complex targets with and without C–H activation. In particular, this review illustrates three unique strengths of C–H activation in a total synthesis context: a) how C–H activation can enable users to adopt a conceptually orthogonal, ‘structurally-obvious’-synthetic strategy; b) how C–C bond construction via C–H activation can challenge traditional C–X disconnections to streamline total syntheses, and c) how native functionalities can be strategically leveraged to direct C–H activation processes. Finally, this review closes by illustrating how newly emerging C–H activation technologies can further improve the step- and overall synthetic economy of total syntheses highlighted in this review, and potentially spur wider adoption of C–H activation disconnections in the synthesis of complex targets.
It is worthy to note that there are several excellent reviews that systematically survey syntheses that have employed C–H functionalization reactions.[26,27,31–33] Our comparative approach instead aims to highlight the key principles and structural cues that can guide the successful implementation of C–H activation processes in the framework of retrosynthetic logic. For this illustrative intent, we have specifically chosen examples with prior historical routes that best represent the strengths we wish to highlight in this review. This will certainly result in omissions, and we apologize in advance for the other elegant works not covered. We hope that the examples highlighted can allow us to critically reexamine any organic structure and understand how C–H activation methods can expedite their syntheses. In doing so, we anticipate that these more efficient and economical strategies will greatly accelerate the synthesis of complex natural products.
2. C(sp2)–H activation and functionalization processes applied in total synthesis
2.1. General and emerging strategies for C–C bond construction via C(sp2)–H activation
The advent of cross-coupling reactions has redefined how chemists conceive the formation of C–C bonds between two C(sp2) motifs. Its prevalence has rendered cross-coupling the prevailing disconnection between these motifs and consequently one of the most performed synthetic transformations in the field. However, these reactions inherently require prefunctionalized substrates in order to engage with the transition metal catalyst and generate the organometallic intermediate. C(sp2)–H activation offers a potential method to streamline this operation in allowing the direct generation of the requisite metal–C(sp2) species without the need for prefunctionalization. Recent developments in this field have allowed chemists to achieve predictable site-selectivity, with many examples utilizing palladium catalysis.[34–36] The divergent reactivity of the organometallic intermediate subsequently allows for direct access to a variety of substitutions in a single step.
Methods developed for site-selective C–H activation fall under two general strategies. The first strategy is directed C–H activation, in which a Lewis basic functionality (e.g. alcohols, amines, acids) directs the cyclometalation event to an energetically favorable site, which typically leads to the preferential functionalization at the ortho position.[34,35,37–39] For more distal C–H bonds, recent developments have also seen the use of covalent, and recently, non-covalent templates[40] to selectively target the farther meta (and in some cases, para) position (Figure 4A).[41,42] The second strategy draws inspiration from the seminal work by Catellani, which leverages the ability for an organopalladium species to be relayed to a site more distal through norbornene relay activation (Figure 4B).[43,44] In the case of native ortho directing motifs, such technology enables a net meta functionalization from a given directing group,[29] and recent reports have demonstrated that meta palladated motifs can be relayed to the para position.[30]
Figure 4.
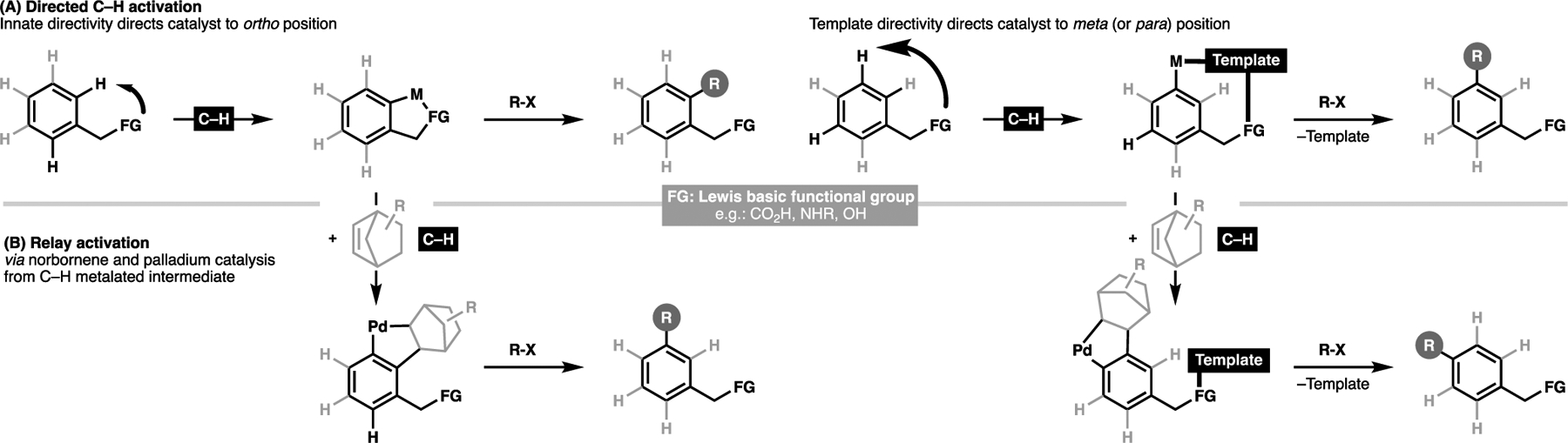
A summary of the two general strategies for the site-selective formation of C–C bonds via transition metal-catalyzed C–H activation process for arenes: (A) directed approaches,[24] or (B) in combination with norbornene-mediated palladation relay[29,30]
Importantly, these approaches are capable of overturning substrate-imposed regioselectivity biases. For example, various examples have been successfully demonstrated for the selective ortho functionalization of electron-poor arene rings (which are typically meta directing),[45,46] as well as the selective meta functionalization of electron-rich systems (which are typically ortho and para directing).[47,48] These are transformations challenging to achieve directly, as the innate reactivity of these systems often impedes site-selective functionalization at these positions. The diverse methods illustrated by these examples demonstrate that such a C–H activation strategy can help overcome substrate-imposed tactical roadblocks encountered in a total synthetic campaign. Finally, emerging studies have shown that non-directed arene C–H activation is a possible venture. In these cases, the site-selectivity of these processes is governed by steric and electronic properties of the substrate, although currently the selectivity tends to be modest at best.[49,50]
Beyond carbocyclic systems, the importance of heteroarenes, in particular N-heterocycles in pharmaceuticals, has motivated development towards the site-selective alkylation, olefination and arylation of these substrates, with key reactions summarized in Figure 5. Compared to their carbocyclic analogs, the intrinsic reactivity of heteroaromatic substrates imparted by the electronegative heteroatom can help guide site-selectivity, with some non-directed C–H activation processes developed for the C3-selective C–H activation of indoles and pyridines.[51,52] For more distal positions, the direct functionalization of the benzocyclic ring in bicyclic N-heteroarenes is a more challenging endeavor owing to the remoteness of the target C–H bond relative to the directive N-functionality. Apart from C7-functionalization of indoles and C8-functionalization of quinolines, the remaining C–H bonds have seen only a handful of methods for their direct C–C bond formation.[53] In recognition of this challenge, we have recently demonstrated the possibility of executing site-selective C5 olefination of quinolines,[40] and in conjunction with norbornene-mediated palladation relay, the C6 arylation of quinolines.[30] While there are still key limitations pertaining to distal activation, these emerging innovations provide a compelling proof-of-concept for the selective targeting of remote C–H bonds in a variety of aryl and heteroaryl substrates.
Figure 5.
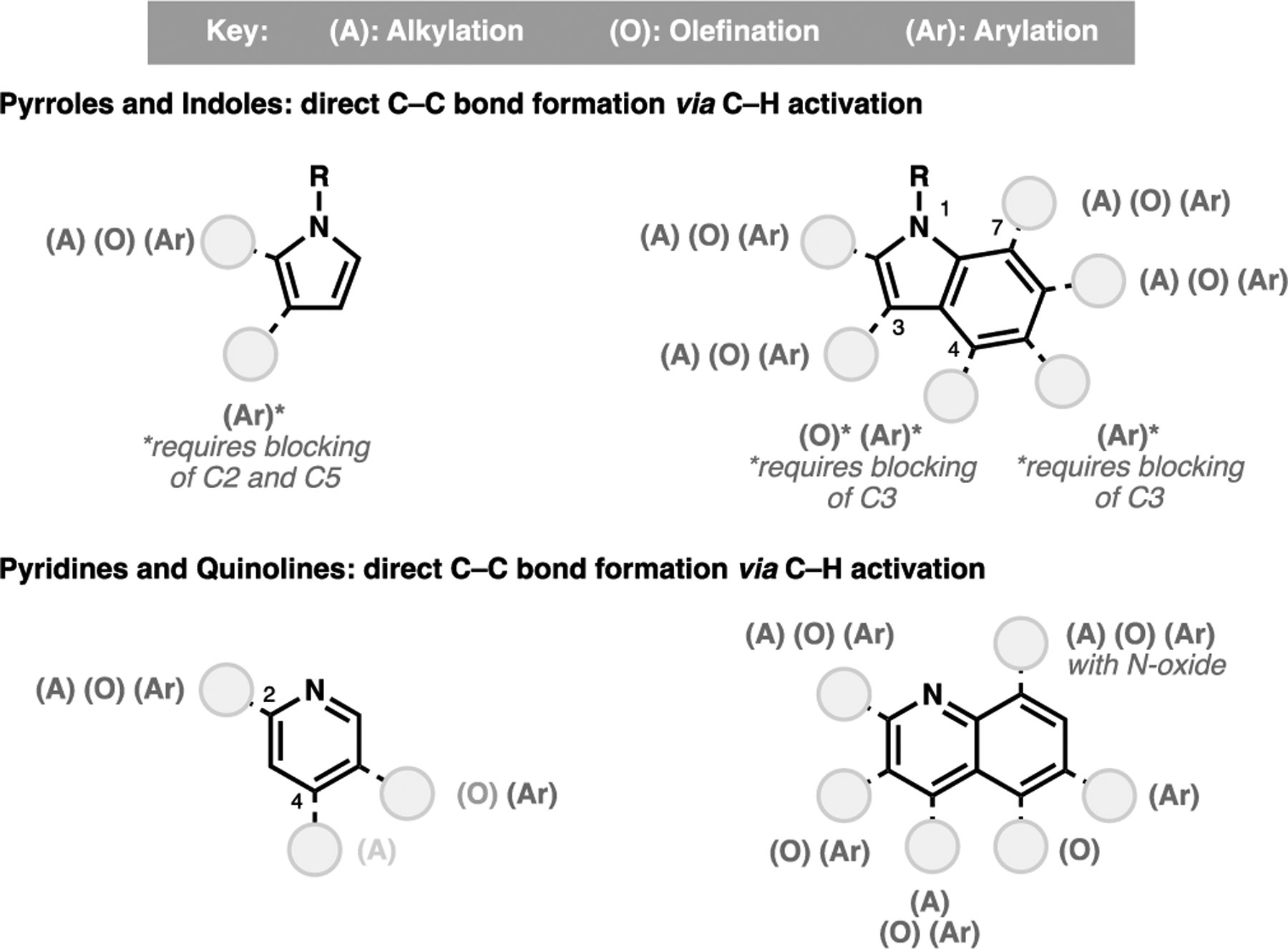
A summary of various direct C–C bond forming reactions via C–H activation processes for a representative class of N-heterocycles: pyrroles,[52] indoles,[53a–b] pyridines[51,53c–h,54] and quinolines[30, 53c,f–g,i]
In the context of complex molecule synthesis, traditional retrosynthetic analyses for C(sp2)–C(sp2) linkages tend to revolve around cross-coupling transforms, which require the installation of coupling handles. Inherent to this approach are three key considerations: 1) which pair of coupling handles, and which permutation, is the most effective for the desired coupling, 2) how can one site-selectively install this handle, and 3) will native functionalities tolerate the conditions for handle installation or do they require protection. If this is implausible, a de novo synthesis of these C(sp2)-containing motifs may be required. C–H activation on the other hand largely bypasses these issues, as a catalyst’s reactivity is principally determined by its proximity to a target C–H bond. This means that C–H activation alters the retrosynthetic analysis of C(sp2)–C(sp2) linkages away from a functional grouporiented approach as described above, to an approach centered on spatial relationships, or its distance and geometry of a C–H bond relative to another functional group (Figure 6).
Figure 6.
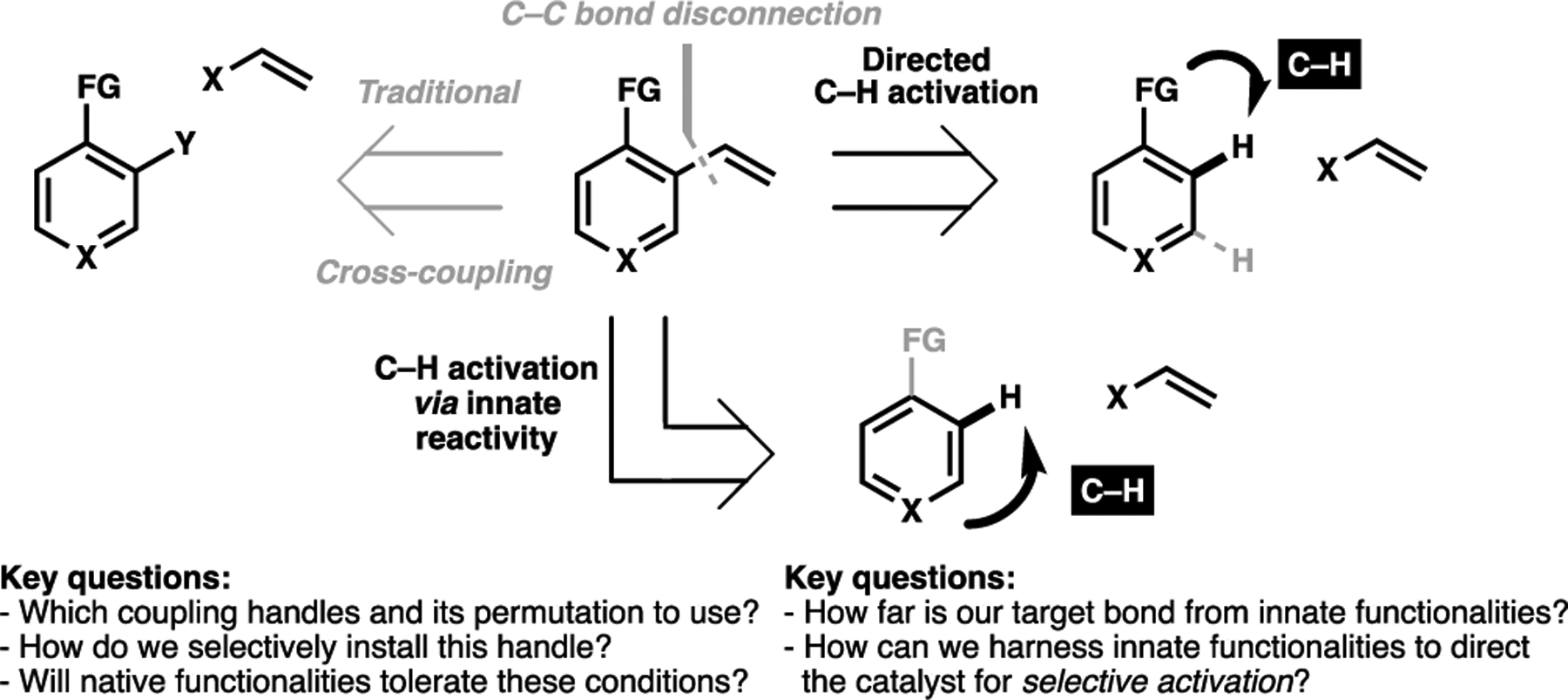
General approach for reconceiving C(sp2)–C bond disconnection via a C(sp2)–H activation, with key considerations outlined
In total synthesis, two approaches have become particularly enabling in the context of C–H activation-mediated C(sp2)–C bond formation. The first harnesses proximate Lewis basic functionalities to direct the active catalyst to a desired C(sp2)–H bond, and remains as the most popular strategy in a total synthetic context. The second approach harnesses the innate reactivity of heteroaromatic substrates. While this tends to be less widely used owing to poorer regioselectivity control, there are several examples that demonstrate that the successful implementation of this approach can be achieved with careful synthetic planning. As demonstrated in the subsequent case studies, both these approaches can enable the expeditious generation of complexity, thus rendering them enabling transforms in total synthesis.
2.2. C–H activation enables a structurally-obvious approach towards total syntheses using innate reactivity: case studies in dictyodendrin B and the lamellarins
Natural products bearing dense carbo-substituted aromatic systems have posed a synthetic challenge as they tend to be difficult to construct through standard aromatic substitution reactions. Typical approaches tend to revolve around a cyclization process (e.g. electrocyclic ring closure) of a suitably functionalized linear precursor, which has been deployed across a range of densely functionalized arene-containing natural products.[55–58] On the other hand, the numerous advances in C–C bond formation technologies via C(sp2)–H activation (vide supra) opens up a conceptually novel approach for the generation of these structures, namely through the direct installation of functionalities onto a carbocyclic scaffold, iteratively generating structural complexity in a modular manner. This alternative retrosynthetic approach can be termed as a ‘structurally-obvious’ approach, as it aims to prune pendant functionalities down to a core scaffold, much like if someone were to build a structure by hand using a molecular Lego® kit. To achieve this using canonical reactivities would be challenging, as this would rely on extensive substrate prefunctionalization. Moreover, the difficulty in executing sequential prefunctionalization escalates in difficulty with increasing substrate complexity owing to chemoselectivity-imposed constraints. On the other hand, a structurally-obvious approach to synthesis can be readily achieved through C–H functionalization processes. In particular, transition metal-catalyzed C–H activation is an especially powerful approach, as the careful tuning can allow for a catalyst to readily discriminate between various C–H bonds, allowing for the modular installation of required functionalities en route to a natural product.
A total synthesis that exemplifies the conceptual power of a structurally-obvious approach lies in the synthesis of the polyheteroaromatic alkaloid dictyodendrin B (9), which demonstrates how a C–H functionalization strategy can aid in reconceiving complex molecule synthesis. In 2005, Fürstner et al. reported a first total synthesis of dictyodendrin B, employing a 6π-electrocyclization as the key step to close the central aromatic ring as part of the carbazole core (Scheme 1A).[59] To achieve this pioneering synthesis, Fürstner commenced the synthesis from a substituted nitrobenzene 10 (Scheme 1B). A two-step sequence involving alcohol protection followed by aldol condensation generated enone 11. This was then cyclized via TosMIC followed by N-alkylation and nitro reduction to afford 12, which was acylated to generate the carbon skeleton for the natural product (13). The use of low-valent titanium species then effected an indole cyclization from the ketoamide substrate in 14, which sets the stage for the pivotal 6π-electrocyclization/oxidation from 14 to furnish the substituted carbazole core in 15. From here, a bromination/lithiation/oxidation sequence furnishes the protected natural product 16, where three further manipulations procedures delivered 9 in 13-steps LLS with an 8% overall yield.
Scheme 1.
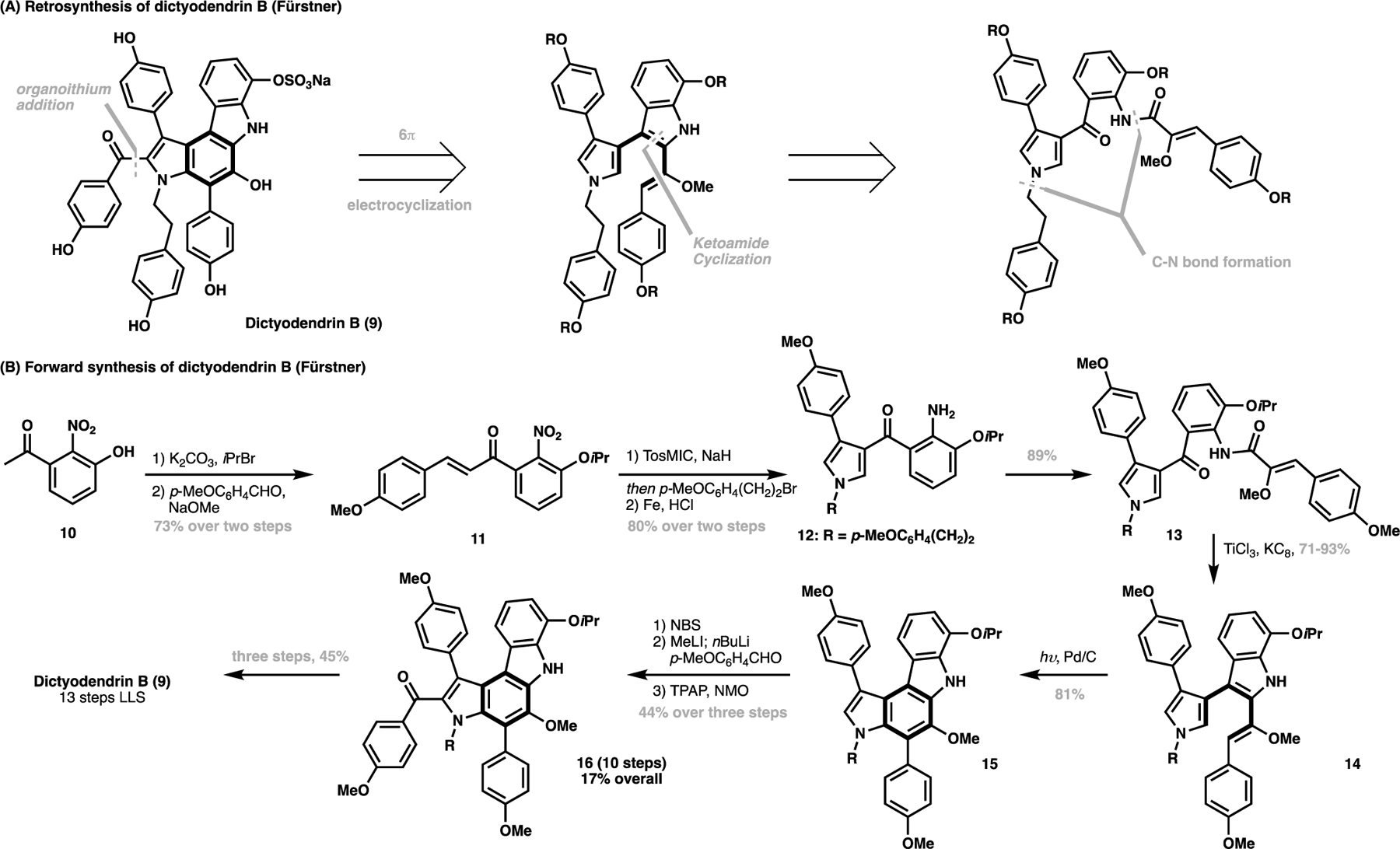
Fürstner’s approach towards the total synthesis of dictyodendrin B (9) (2005): (A) retrosynthesis of dictyodendrin B; (B) forward synthesis
Fürstner’s landmark total synthesis of dictyodendrin B crucially relied on the predecoration of all pendant substitutions for the natural product prior to closing the overall carbon scaffold for the target. Intuitively however, the natural product appears as if several substituted benzyl/phenyl groups are appended onto a central indole/carbazole scaffold, and as such, a ‘structurally-obvious’ approach towards its synthesis would surround the iterative installation of all the required functionalities around this indole scaffold as if one was handmaking the natural product from a central fragment piece-by-piece. This structurally-obvious approach towards dictyodendrin B was first realized by Gaunt et al. in 2015, primarily centered on judicious C(sp2)–H functionalization reactions to install all pendant motifs for the natural product (Scheme 2A).[60] In this revised retrosynthetic analysis, particular attention was drawn to how to best harness the innate nucleophilic reactivity of the pyrrolyl nitrogen motif. This led to the design of a three-step sequence at the exposition of the total synthesis guided on the innate directivity of the indole nitrogen to effect the regioselective C(sp2)–H functionalization sequence.
Scheme 2.
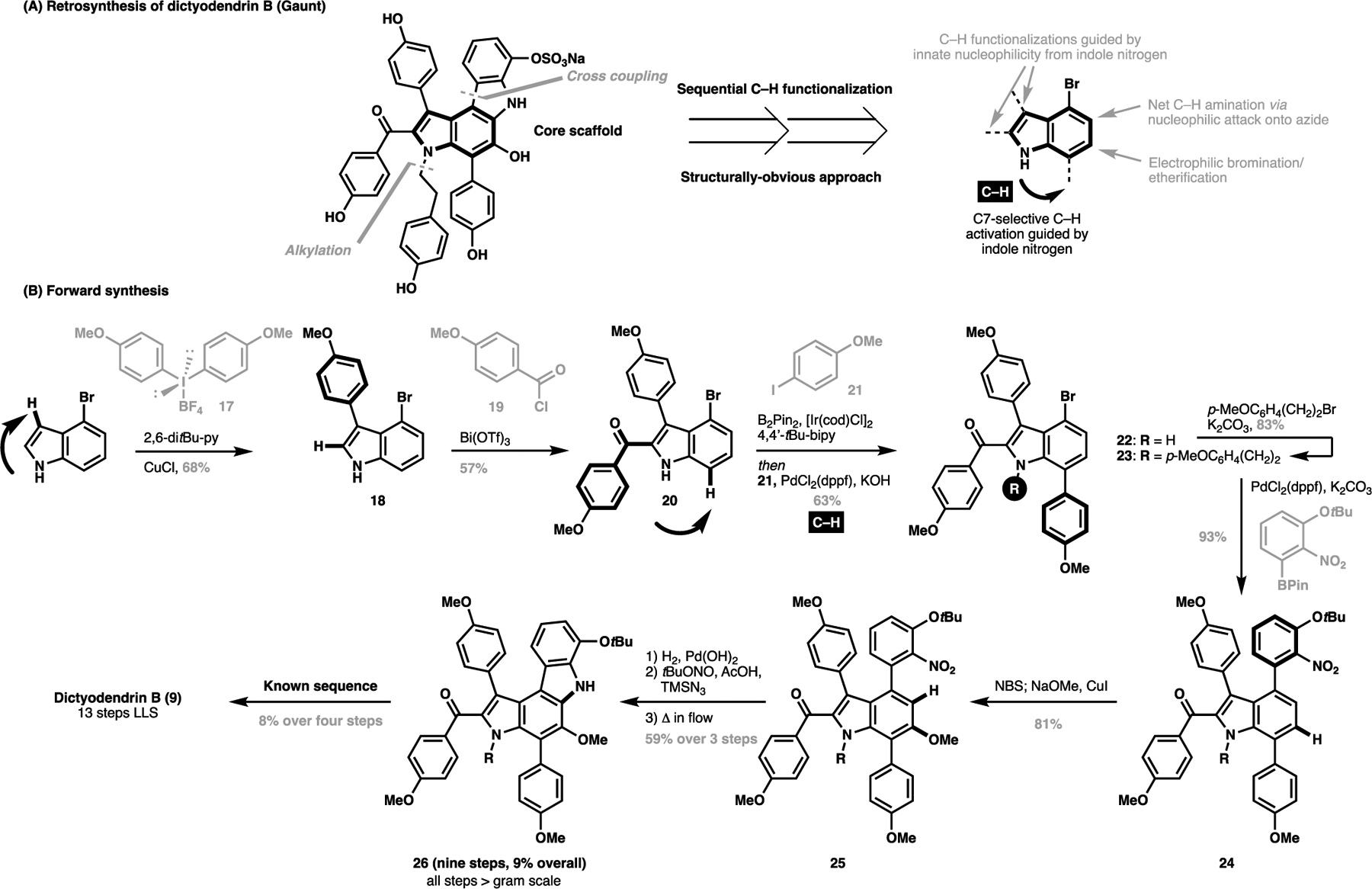
An illustration of a structurally-obvious approach towards dictyodendrin B (9) as reported by Gaunt (2015): (A) retrosynthesis, (B) forward synthesis
Starting from 4-bromoindole, a Cu-catalyzed C3-selective arylation with 17 appended the first substitution in 18 (Scheme 2B). This was followed by a Bi-catalyzed C2-acylation with 19 to give 20, followed by a sequence of N-directed C7-borylation/cross-coupling with 21. At this point, N-alkylation of 22 followed by a cross-coupling of the pre-installed C4 bromide in 23 completes all C–C bond connectivity for the natural product in five operations. From here, a C6-selective bromination/etherification of 24 installed the OMe substituent in 25. Subsequent nitroreduction and diazotization enables a C5-selective amination to close the carbazole ring to furnish the protected natural product 26 in nine steps on a gram scale. At this stage, this intermediate converges to known synthetic routes, which could be converted to the natural product in four more operations. Although not as high yielding as Fürstner’s original synthesis (9% vs. 17%), the synthetic route to protected precursor 26, as enabled by carefully choreographed C(sp2)–H functionalization, is shorter, scalable up to gram quantities and largely consists of catalytic transformations. Importantly, the modularity of this approach with common coupling partners should be amenable for facile late-stage divergent syntheses of analogues for further structure-activity testing valuable in a drug development context.
The lamellarins represents another class of natural product that has benefitted from a structurally-obvious approach. These natural products feature a signature pentacyclic scaffold bearing a pyrrole motif, and have shown to be effective agents to reverse efflux pump-mediated drug resistance in cancer cell lines.[61] Typically, this class of natural product has been synthesized through one of two strategies: a de novo synthesis of the functionalized pyrrole core from linear precursors,[62] or through iterative cross-coupling approaches from a multiply prefunctionalized pyrrole motif (Scheme 3).[63–65] A key characteristic of these syntheses is the requirement of separate functionalization steps (e.g. the site-selective installation of halide coupling handles), leading to concessionary operations required in order to install the required substitutions in a selective manner.
Scheme 3.
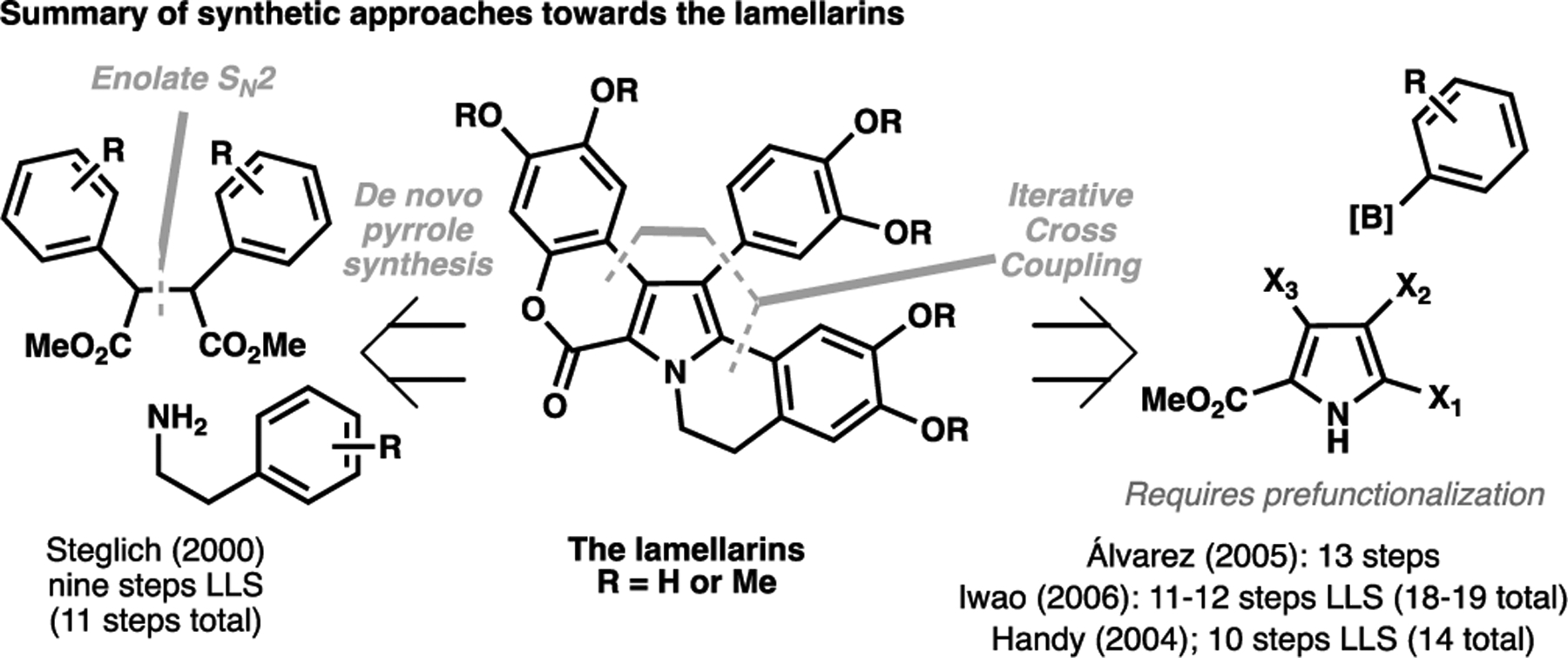
Historical approaches taken towards the lamellarins
In 2014, Yamaguchi et al. reported an expedient total synthesis of lamellarins C and I (27 and 28), effectively harnessing C–H activation processes in a structurally-obvious approach.[61] Starting from substituted pyrrole 29 (derived in three steps), a rhodium-catalyzed C–H arylation selectively appended the aryl motif at C3, guided by the innate directing effect from the pyrrole motif (Scheme 4). A subsequent sequence involving acylation, hydrolysis and esterification with 30 gave 31, which underwent an adventurous double oxidative C–C coupling with the only available positions on the pyrrole ring to complete the pentacyclic scaffold in a single operation. This then underwent a global deprotection to differentially give lamellarin C (27) and I (28) in eight total steps, the shortest synthetic sequence (LLS and in total manipulations) to date for this class of natural product. This expedient route to the lamellarins, as well as other recent syntheses of dictyodendrin congeners,[66,67] demonstrates the efficacy and practicality of a structurally-obvious synthetic approach. Rather than requiring substrate prefunctionalization or reliant on a de novo cyclization to generate the core scaffold, this C(sp2)–H functionalization-based strategy allows for the modular installation of structural motifs in an iterative manner, dramatically simplifying how we conceive of the retrosynthetic analysis of heteroaromatic natural products. Such a structurally-obvious approach cannot be readily achieved through traditional methods and demonstrates the uniquely enabling nature of transition metal-catalyzed C(sp2)–H functionalization processes in natural product synthesis.
Scheme 4.
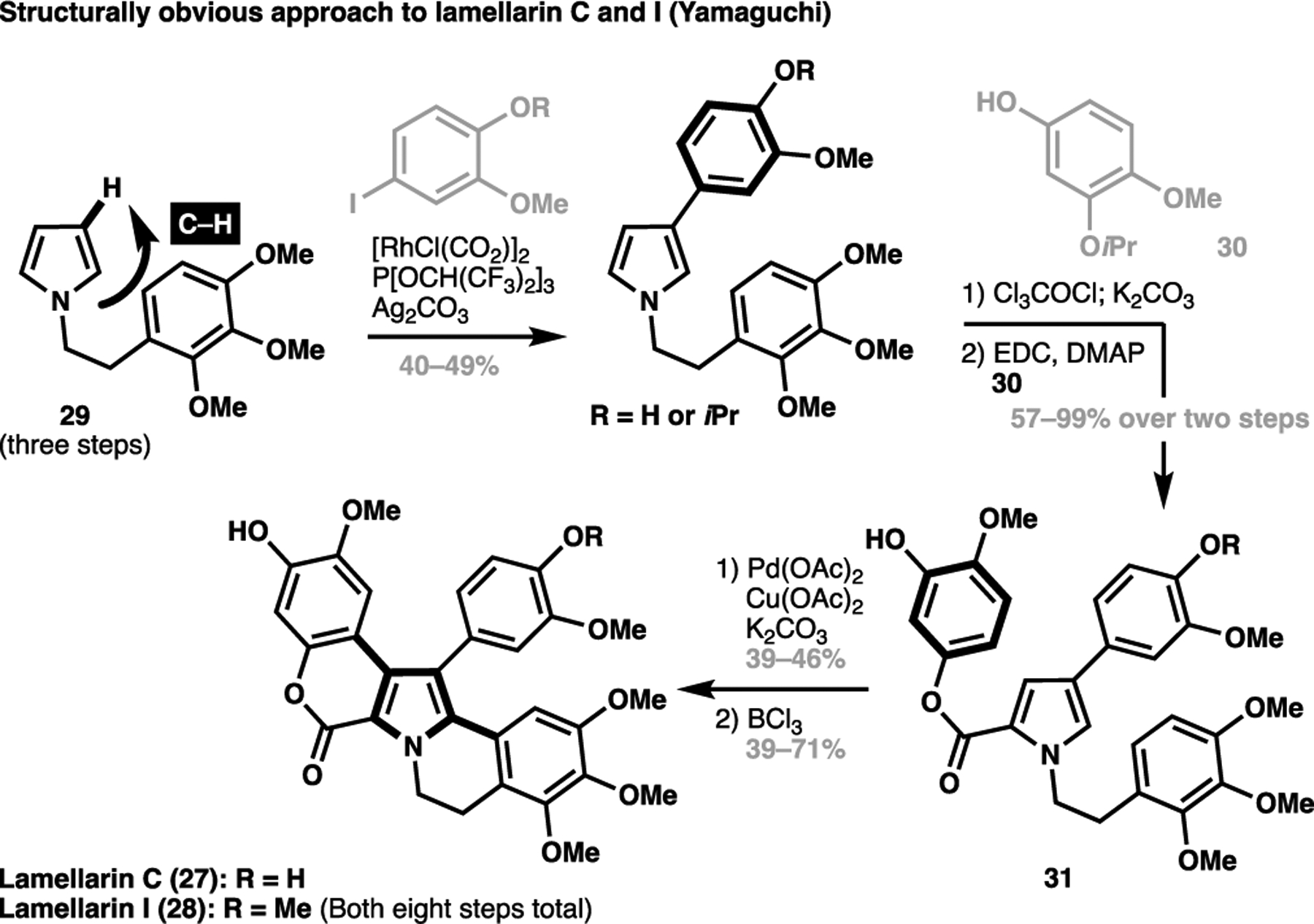
A structurally-obvious and expedient approach towards lamellarin C and I as reported by Yamaguchi (27 and 28, 2014)
2.3. A structurally-obvious approach towards total syntheses using directed C–H activation: case studies in (+)-hongoquercin A and (+)-lithospermic acid
In a similar vein, a directed C–H activation approach can also be highly effective in forging C(sp2)–C bonds in natural products in a structurally-obvious manner. One synthesis that exemplifies the power of this approach is that of the sesquiterpenoid (+)-hongoquercin A (32). The traditional retrosynthetic approach taken for historical syntheses of (+)-hongoquercin A and related congeners tended to revolve around aldehyde addition/cyclisation sequence to forge the pyran ring.[68] Additionally, Hsung et al. recreated a plausible biosynthetic polyene cyclisation cascade from a linear precursor, generating the carbon skeleton for the natural product racemically in modest yields.[69] Notably, all previous syntheses required the de novo synthesis of the phenolic ring in the natural product (Scheme 5).
Scheme 5.
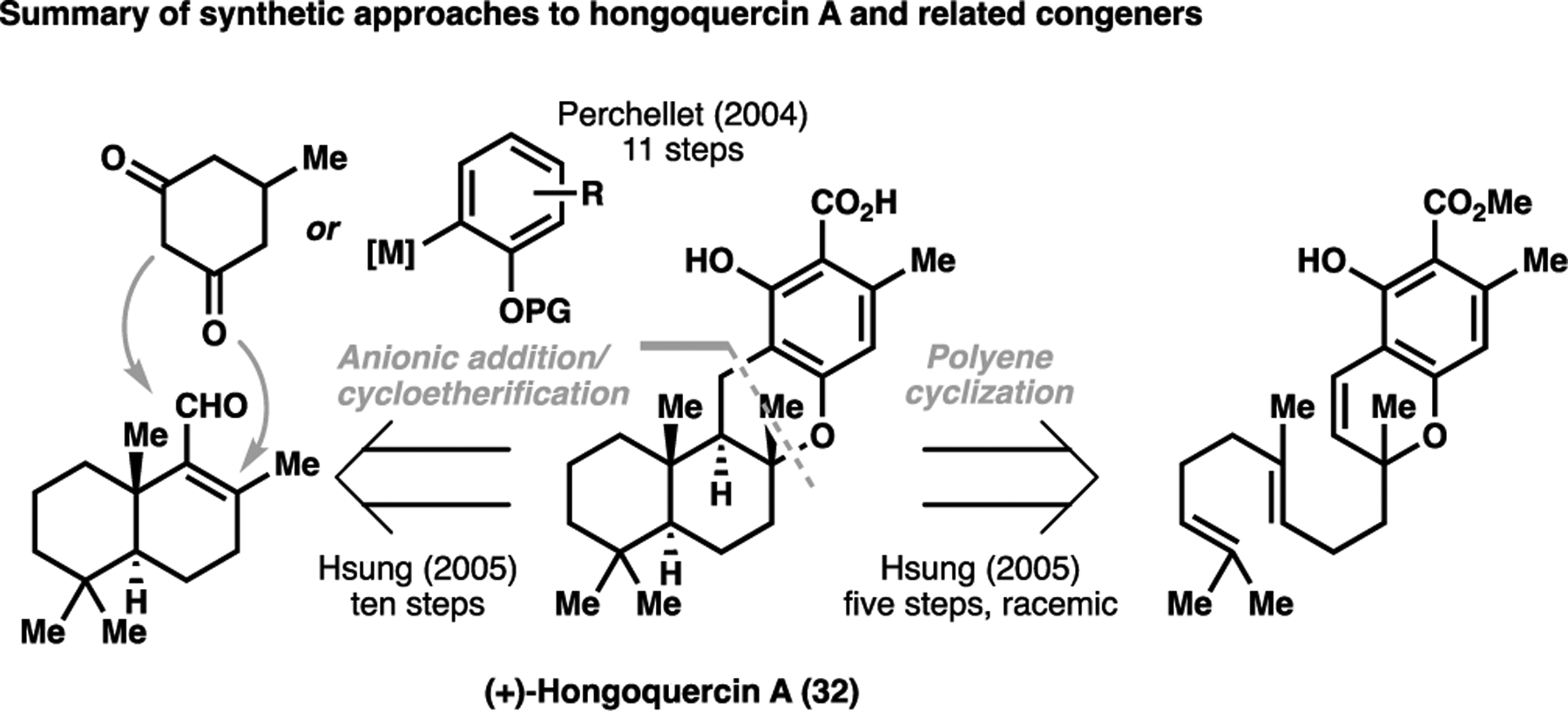
Historical approaches taken towards hongoquercin A (32) and related congeners[68,69]
In 2011, we, in collaboration with the Baran group, achieved the total synthesis of (+)-hongoquercin A (32) from a structurally related natural product (+)-chromazonarol (33).[12] Recognising that the hydroxyl and methyl substitution on the benzoic acid fragment of the natural product lay ortho to the native acid functionality, we envisaged a late-stage installation through palladium-catalyzed C(sp2)–H activation processes to achieve the regioselective installation of both groups, as enabled by the intrinsic ortho directivity of the acid functionality (Scheme 6A). This allowed for the rapid, structurally-obvious total synthesis of the title compound in six steps from a readily prepared natural product (+)-chromazonarol (33), leveraging the common core of the two natural products. The versatility of this method in late-stage diversification was also demonstrated, where seven analogues of hongoquercin A were synthesized by leveraging C–C bond construction via a C(sp2)–H activation strategy (Scheme 6B).
Scheme 6.
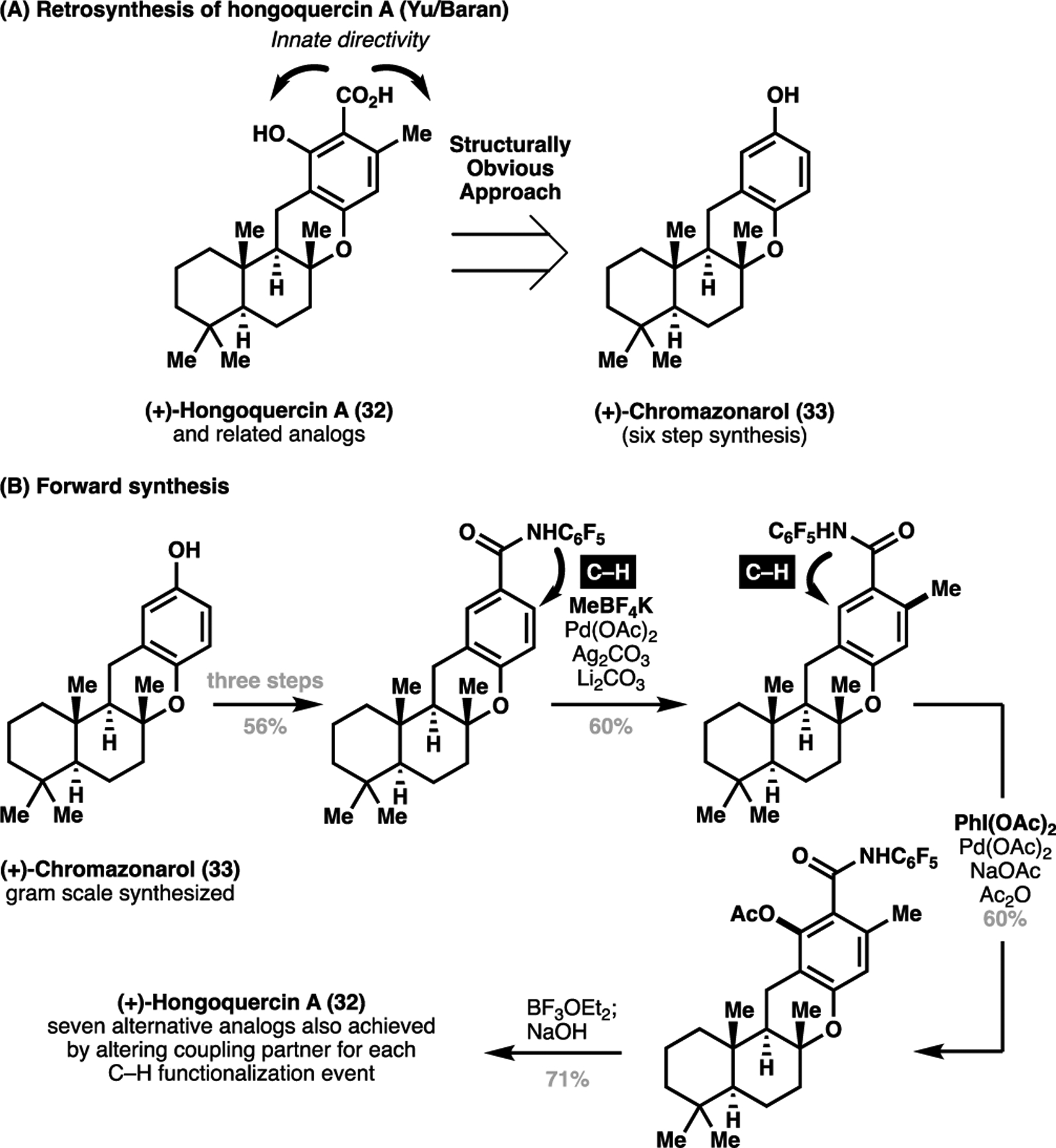
Structurally-obvious approach to the total synthesis of hongoquercin A (32). (A) Retrosynthesis of (+)-hongoquercin A and (B) Forward synthesis of hongoquercin A
This structurally-obvious approach to retrosynthesis can be extended towards C(sp2)–H alkenylations of aromatic motifs. Lithospermic acid for instance, has been another target that has benefitted from the judicious implementation of a C(sp2)–H activation approach for its total synthesis. The first racemic total synthesis of protected lithospermic acid (heptamethyl lithospermate, 34) was conducted by Jacobsen et al., adopting a canonical C–O disconnection strategy for hydrofuran ring construction via an intramolecular oxy-Michael addition of 35, followed by a Knoevanagel condensation to append on the olefin-bearing side chain (Scheme 7).[70] In 2005, Ellman et al. reported the first asymmetric total synthesis of lithospermic acid (36), powerfully demonstrating that the hydrofuran motif can be generated through a C–C disconnection strategy via a rhodium-catalyzed imine-directed C(sp2)–H activation strategy from aldehyde 37 and amine 38.[71] In addition to forming the aryl-alkyl bond, this strategy also succeeded in setting two stereocentres in one operation. Subsequently, aldehyde 39 can then be productively elaborated to the requisite enoic acid 40 through an aldol condensation that can then give the protected natural product 34 after acylation. A final ester hydrolysis and demethylation sequence allowed Ellman et al. to achieve the first enantioselective synthesis of (+)-lithospermic acid in 10 steps LLS (Scheme 8).
Scheme 7.
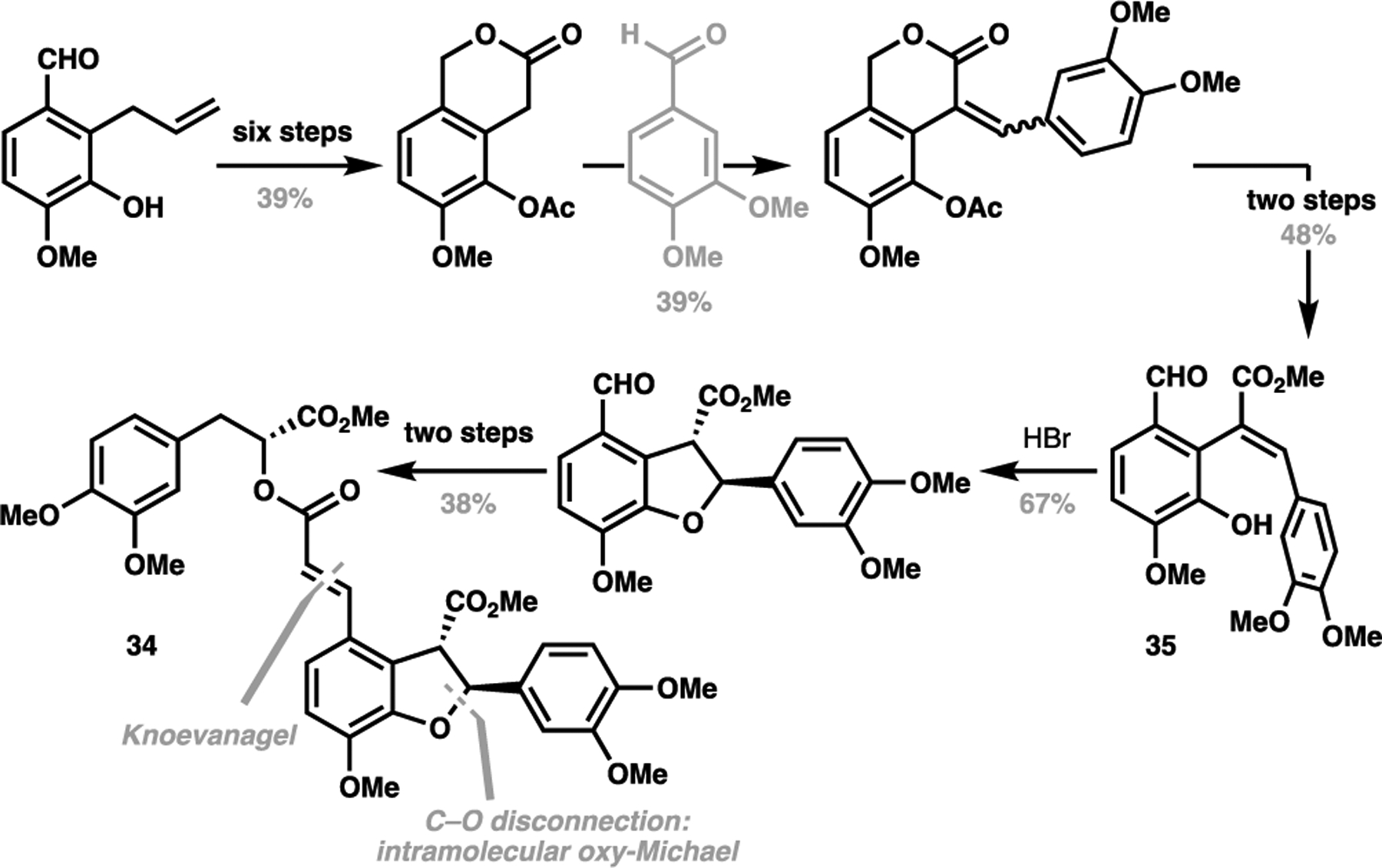
Jacobson’s first synthesis of heptamethyl lithospermate (34, 1979)
Scheme 8.
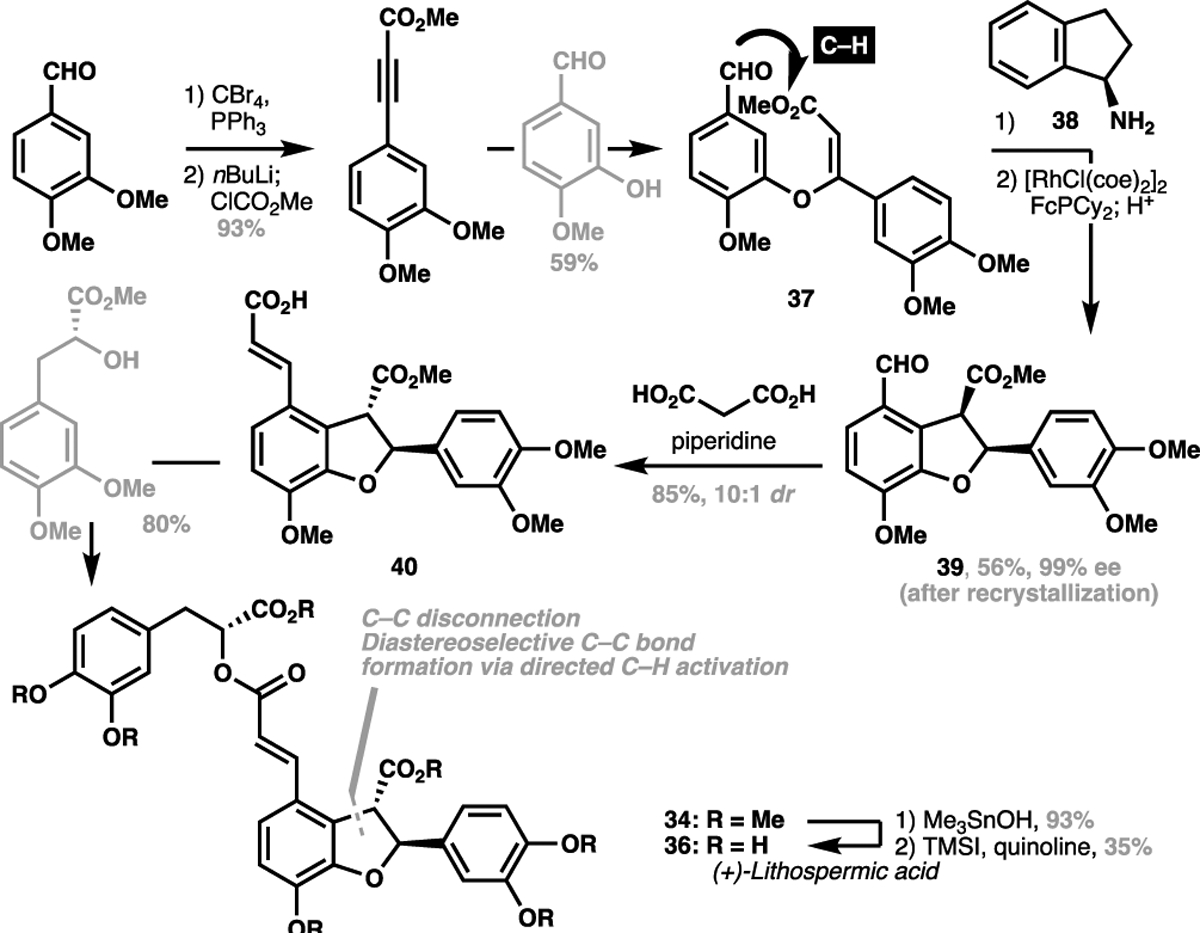
Ellman’s asymmetric total synthesis of (+)-lithospermic acid (36, 2005)
In 2011, we reported a structurally-obvious approach towards the target structure, opting for an ambitious ortho-C(sp2)–H olefination of the benzofuran ring directed by the native carboxyl functionality in the natural product. This would directly append the pendant side chain of lithospermic acid, revealing the core scaffold 41 (Scheme 9) envisaged to be constructed through a C–C disconnective approach for the hydrofuran motif.[72] Our synthesis commenced with a known derivation of o-eugenol to acid 42, which was esterified with chiral hydroxyamide 43 and underwent two-step sequence to give 44. At this point, diazo installation followed by a diastereoselective carbene insertion from 45 configured both stereocentres in the hydrofuran ring. Following ester hydrolysis to give acid 41, the stage was now set for the end game of our synthesis. Gratifyingly, a palladium-catalyzed C(sp2)–H olefination with olefin 46 as directed by the free carboxyl group furnished the protected natural product with excellent yield, and represented the most complex fragment union reactions enabled by C(sp2)–H activation. From here, global demethylation of 34 through a known sequence gave the natural product in 12 steps LLS from o-eugenol. Our expeditious assembly of (+)-lithospermic acid particularly highlights that such C(sp2)–H olefination processes are capable of for the highly efficient union of advanced substrates to aid the realization of convergent total syntheses.
Scheme 9.
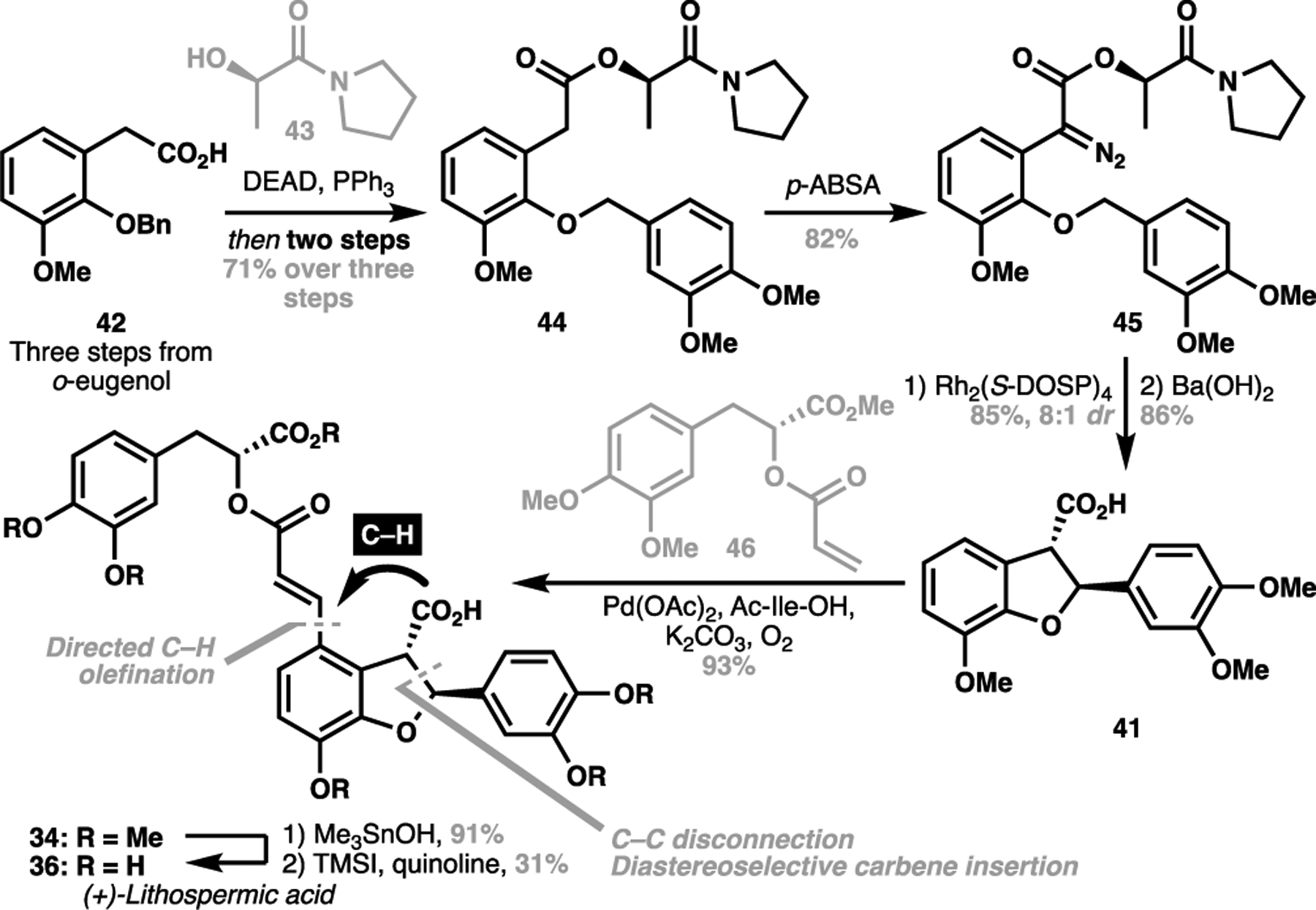
Yu’s total synthesis of (+)-lithospermic acid (36, 2011)
The structurally-obvious approaches towards (+)-lithospermic additionally highlight a second key strength of C–H activation processes — that native functionalities can be strategically leveraged to guide selective functionalization events. In doing so, this adds another dimension to the role a functional group might play in retrosynthetic analyses. This is in contrast with traditional approaches, where a particular functionality is typically created either as part of an intermediary process for further derivatization, or produced as a desired structural motif for the target. Often, the unavoidable chemoselective and reactive constraints as posed by these functional groups render their protection necessary to facilitate canonical C–C bond construction. These reactive features can be increasingly harnessed in C–H activation processes, obviating the need for their protection to facilitate these processes. In the case of Ellman’s synthesis, the aldehyde functionality required for further elaboration was leveraged for a directed C(sp2)–H activation through transient imination. In our approach, the native carboxyl functionality in the natural product was utilized to direct the final olefination in a site-selective manner. These key strengths as illustrated by the case studies in this section serve to further demonstrate the viability and the potential economies generated through the implementation of a structurally-obvious approach towards complex natural product synthesis.
2.4. Coupling C–C bond formation with stereoinduction: case studies in the total syntheses of axially-chiral natural products (+)-steganone, (+)-isoschizandrin and TAN-1085
Transition metal-catalyzed C–H activation processes can often benefit from ligand-mediated rate acceleration arising from careful ligand design. For prochiral substrates, the inclusion of chiral ligand scaffolds can additionally influence the relative energies between diastereomeric transition states and lead to high levels of stereoinduction. This has led C–H activation strategies being employed to desymmetrize substrates that may otherwise require circuitous solutions for their enantioenrichment.
One particular class of natural product that has benefitted from stereoinductive C(sp2)–H activation processes are axially-chiral biaryl containing targets. This non-trivial stereochemical manipulation that has seen decades of research interest towards exerting predictable control over is exemplified by the community’s synthetic interest towards two lignan-derived axially chiral natural products (+)-steganone (47) and (+)-isoschizandrin (48). Over two decades, chemists have devised numerous methods to install the axially chiral biaryl motif that typify these two natural products.[73a–e] The preinstallation of chiral auxiliaries[73f–g] or chiral tethers[74] to bias biaryl conformation represents a common strategy towards accessing these motifs in a stereodefined manner, both of which have been employed for the total synthesis of steganone (Scheme 10A). In addition, a planar-to-axial chirality conversion strategy has been employed by Uemura et al. towards the total synthesis of steganone,[75] while Molander has demonstrated that a chiral nucleophile can add into and kinetically resolve a racemic mixture of biaryl lactone conformers for the stereoselective generation of the chiral axis.[76]
Scheme 10.
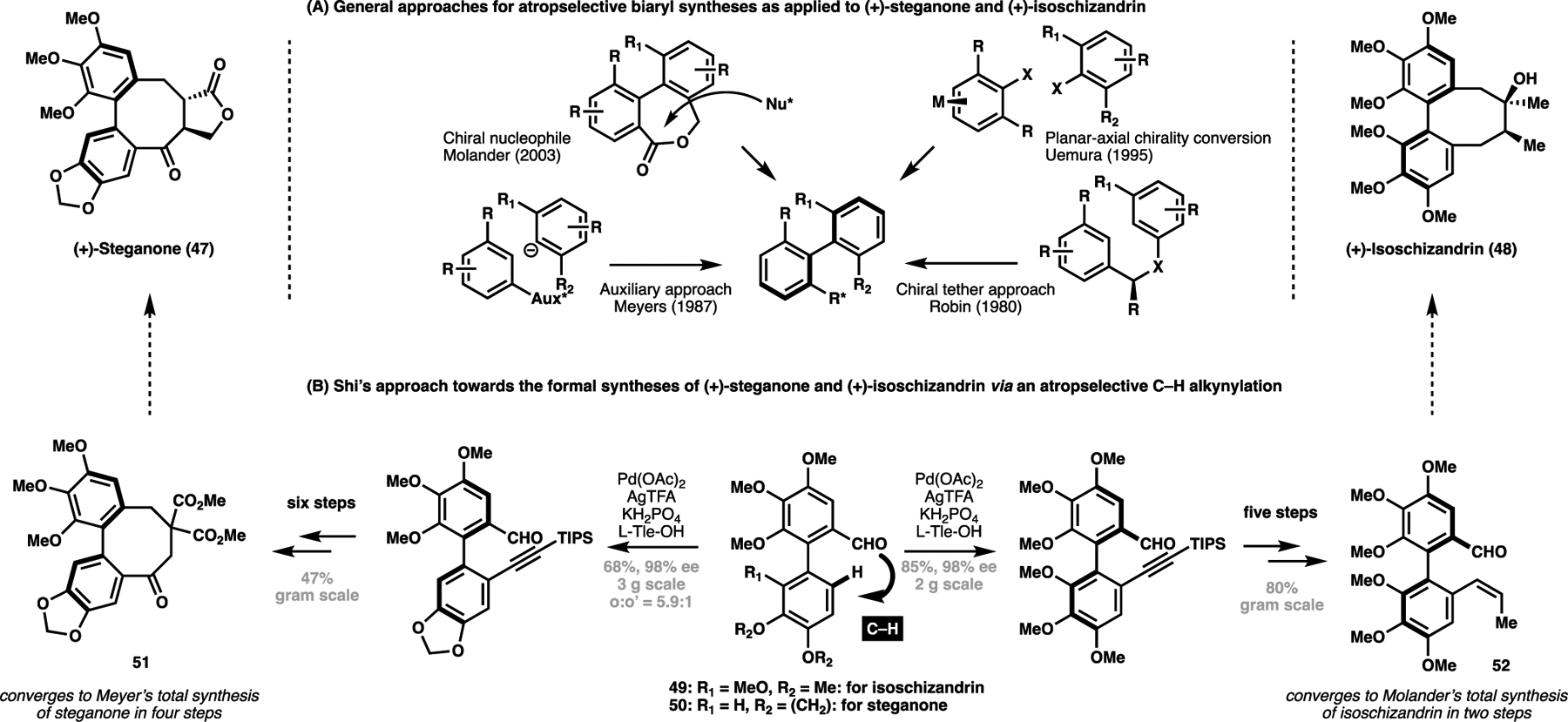
(A) Overview of historical atropselective synthetic approaches towards the total synthesis of (+)-steganone (47) and (+)-isoschizandrin (48); (B) Shi’s formal synthesis of (+)-steganone and (+)-isoschizandrin as enabled by an atropselective C(sp2)–H alkynylation strategy (2018)
In 2018, Shi et al. devised a palladium-catalyzed atropselective C(sp2)–H alkynylation strategy, as directed by a proximate aldehyde-containing functional group. Here, the use of a catalytic chiral amino acid transient directing group, L-tert-leucine, helps strengthen catalyst coordination through the formation of a transient imine functionality.[77] Additionally, the chiral amino acid residue then serves to control the overall conformation of the biaryl ring system. In doing so, the subsequent C(sp2)–H alkynylation event then locks the product enantioselectively from a freely rotatable achiral precursor (Scheme 10B). This novel approach was highly enantioselective and, in the case where two ortho C–H bonds required regioselective discrimination, selective for the least hindered ortho C(sp2)–H bond. From biaryl aldehydes 49 and 50, this process enabled for the highly efficient syntheses to known precursors 51 and 52 towards both (+)-steganone and (+)-isoschizandrin respectively in six to seven steps, with these transformations conducted on a gram scale.
In the previous example, Shi et al. demonstrates that the applicability of C–H activation processes can extend beyond C–C bond construction, and in tandem, can be leveraged to set global conformations to achieve enantiocontrol. This process was also productively demonstrated for the promising tetracyclic antibiotic TAN-1085 (53). Notably, the diol configuration and its hindered rotation around the biaryl axis means that TAN-1085 possesses both point and axially chiral features challenging to its synthesis. In 2004, the first total synthesis of TAN-1085 was reported by Suzuki et al., which relied on an 4π/6π-electrocyclisation/pinacol coupling gambit to install the tetracyclic core in a racemic manner (Scheme 11A).[78] This pericyclic cascade would arise from a functionalized cyclobutane intermediate, arising in turn from a vinyllithium addition to a suitable ketone precursor. The forward synthesis commenced from 54, where an eight-step elaboration via alkyne 55 formed the vinyl iodide precursor 56. A lithiumiodide exchange followed by addition into ketone 57 then gave the cascade precursor 58, however, Suzuki et al. found that oxidation to the aldehyde oxidation state was necessary to ensure the smooth conversion to 59. From here, a diastereoselective pinacol coupling/selective protection gave 60, where six further manipulations allowed the access to separable diastereomers of the natural product upon glycosylation with an enantiomerically pure sugar motif. The first total synthesis of TAN-1085 was achieved in 16 steps LLS, and allowed Suzuki et al. to unambiguously determine the configuration of the natural product.
Scheme 11.
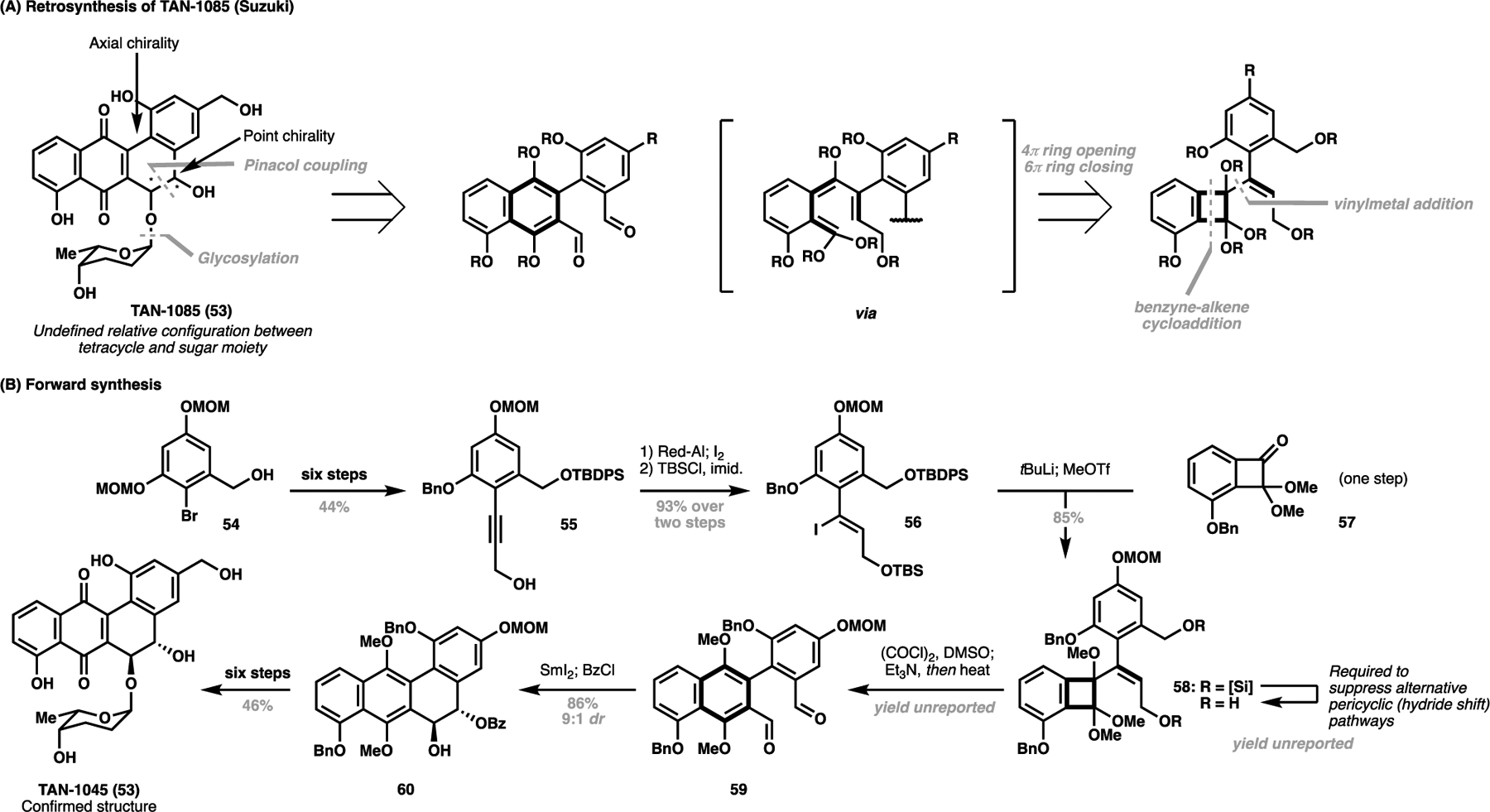
Suzuki’s total synthesis and stereochemical assignment of TAN-1085 (53, 2004); (A) Retrosynthetic analysis of TAN-1085 and (B) forward synthesis
After conclusively determining the overall configuration for TAN-1085, Suzuki et al. embarked an asymmetric total synthesis of the target compound, and posited the possibility of employing an axial-to-point chirality relay from an axially chiral biaryl precursor to configure the diol motif present in the natural product (Scheme 12A).[79] The anti configuration present in the diol motif was proposed to arise from the preferred staggered aldehyde conformation of a chiral biaryl motif. This led to Suzuki et al. to generate the axially chiral aryl precursor 61 via an atropselective cross-coupling mediated by a chiral sulfoxide motif. This sulfoxide auxiliary in 62 was then lithiated and added into ketone 47 to give the axially chiral cyclobutane intermediate 61, which converges onto the previous route to enable a successful asymmetric total synthesis of TAN-1085 (Scheme 12B).
Scheme 12.
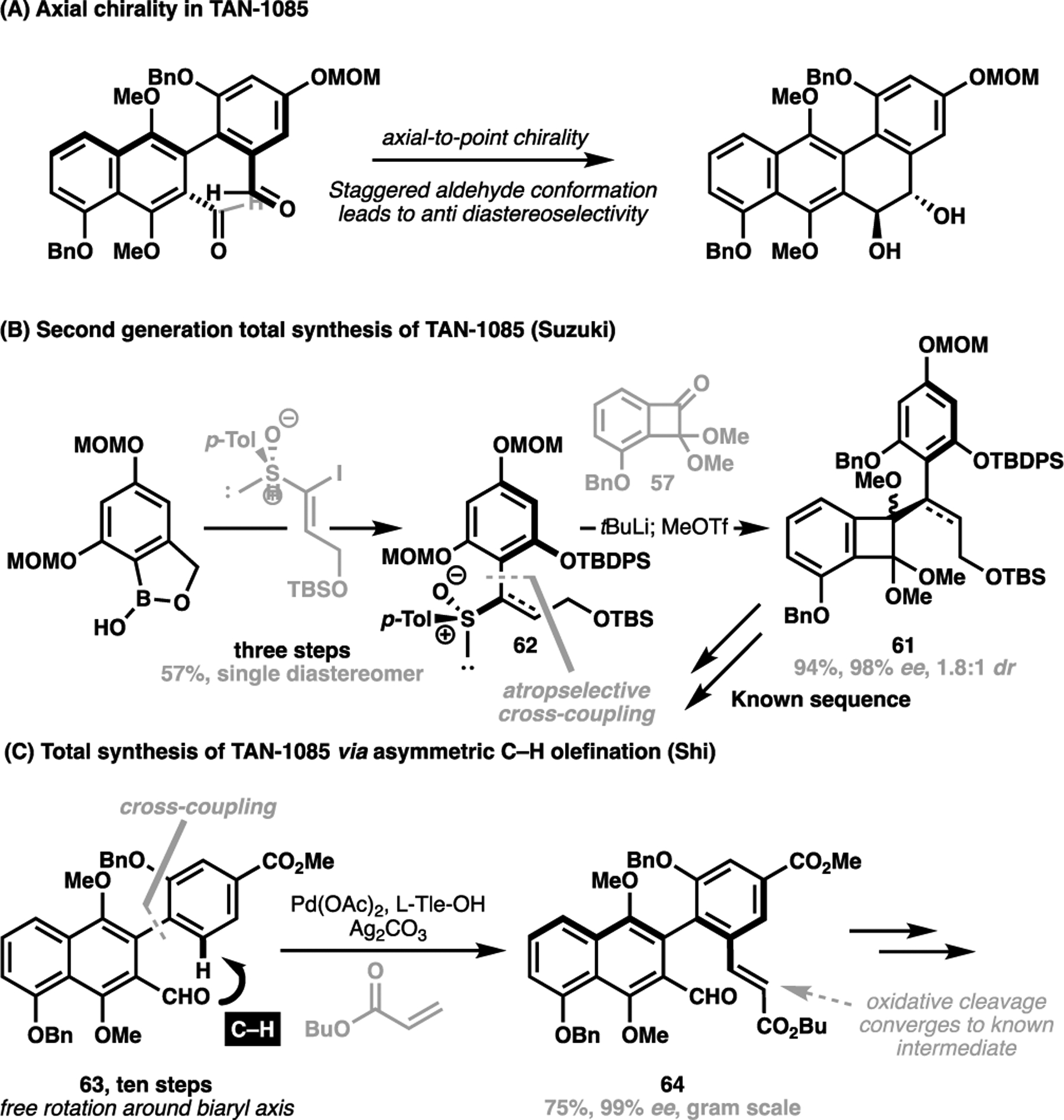
(A) Leveraging axial-to-point chirality in the synthesis of TAN-1085 (2004); (B) Suzuki’s second-generation approach towards TAN-1085 via a point-to-axial chirality relay strategy (2009) (B) Shi’s approach towards TAN-1085 (2019) via an atropselective C–H olefination strategy
In 2019, Shi et al. disclosed an alternative approach towards TAN-1085, leveraging a further development of their C(sp2)–H activation strategy to enantioselectively lock the prochiral biaryl motif via a palladium-catalyzed C(sp2)–H olefination reaction of 63.[80] Again, the reaction was directed through the native carbonyl functionality and mediated through the catalytic use of L-tert-leucine to coordinate to the active catalyst, as well as influence the global conformation of the prochiral biaryl motif (Scheme 12C). This succeeded in generating the C(sp2)–H olefinated product 64 and configured the chiral axis with excellent selectivity on a gram scale. From here, a subsequent oxidative cleavage reveals the dialdehyde substrate, which converges with Suzuki’s route for the total synthesis.
In all three examples, Shi first generated the biaryl scaffold through cross-coupling of two suitably substituted aryl units, before introducing the olefin motif to lock the biaryl geometry prior to its further derivatization to the natural product. Aside from strategically utilizing a native functionality to enable a stereoselective C(sp2)–H functionalization reaction, Shi’s approach towards (+)-steganone (47), (+)-isoschizandrin (48) and TAN-1085 (53) represents a structurally-obvious method for the construction of the natural product. In doing so, this allows for the direct disconnection of the tetracycle to a simple biaryl motif, allowing for a conceptually more straightforward approach than the pericyclic cascade from an axially chiral alkenylarene substrate taken in Suzuki’s total synthesis.
3. C(sp3)–H activation and functionalization processes applied in total synthesis
3.1. General and emerging strategies for C–C bond construction via C(sp3)–H activation
Compared with C(sp2)–H bonds, the corresponding transition-metal catalyzed C(sp3)–H activation process is a significantly more challenging venture owing to the higher bond enthalpies of aliphatic C–H bonds as compared with their C(sp2)–H analogs[81]. In addition, the formation of the requisite cyclometalated complex incurs a greater entropic penalty owing to the inherent flexibility of aliphatic substrates. In addition, competing β-hydride elimination pathways that characterize alkylmetal complexes further complexify the direct C–H activation processes for C–C bond formation. For prochiral methylene substrates, the inherent 3-D nature of tetrahedral carbons poses additional stereoselectivity issues on top of the already challenging task of achieving site-selectivity for a typical C–H activation process (Figure 3, vide supra).[25] Despite these challenges, several strategies have emerged for the site- (and stereo-)selective activation of aliphatic substrates, again largely reliant on palladium-catalysis, to effect the desired transformation.
To overcome the inherent thermodynamic barriers imposed by C(sp3)–H activation processes, the solution, more so than with the corresponding C(sp2)–H bond, critically relies on positioning the metal catalyst proximal to the target C(sp3)–H bond of choice to minimize the entropic penalty required for substrate organization. By far the most reliable method to achieve this relies on the use of directing Lewis basic functionality.[81] While most developments have revolved around specifically designed directing groups to achieve desired regioselectivity, emerging innovations have allowed for direct functionalization as directed by native functionalities, such as carboxylic acids[82–86] and amines.[87–92] The crucial tuning of distance and geometric factors of the directing motif is required to enable the successful cyclometalation of an aliphatic substrate. In most cases, this tends to result in the generation of the kinetically favored five-membered cyclometalated intermediate to give the standard site-selectivity for these processes. Recently, we have demonstrated that this positional selectivity can be overturned through the use of carefully designed bidentate directing groups. The use of transient imine attachments for example, has allowed reliable γ-as well as δ-activation of a variety of amine substrates.[91,93] Additionally, the use of highly rigid bidentate directing groups can disfavor the more strained 5,5-metallabicycle in favor for the less strained 6,5-bicyclic intermediate (Figure 7).[94]
Figure 7.
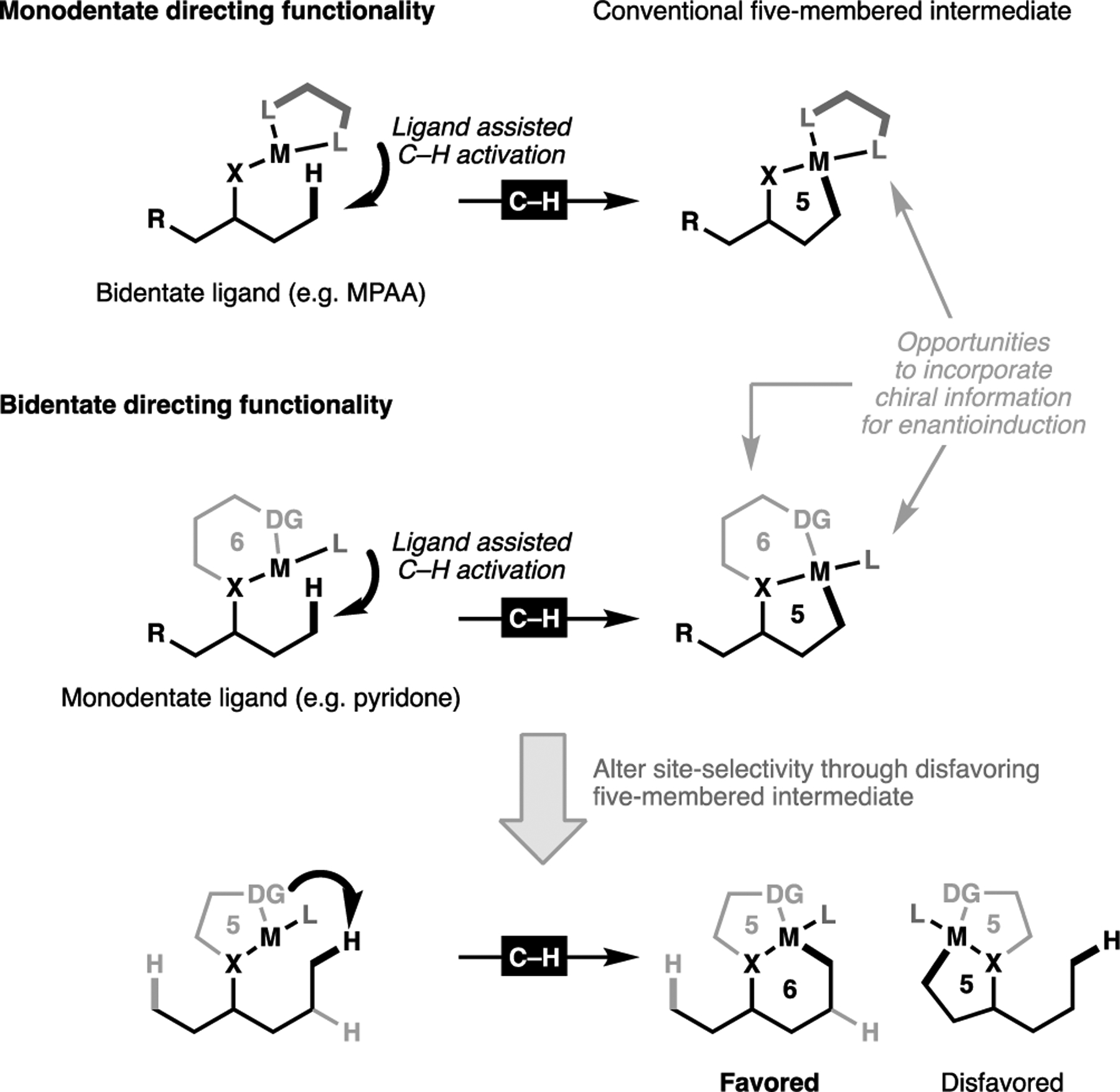
Overview of the strategies to effect site-selective C(sp3)–H activation to form C–C bonds
The strength of the target C(sp3)–H bond has also demanded the development of effective ligands to enable their activation. We have found that the use of monodentate directive functionalities generally benefit from the use of bidentate acetyl-containing ligands, such as mono-protected amino acids.[24] Alternatively, bidentate-directing motifs can benefit from the incorporation pyridone-type ligands.[93] Both ligand classes serve to dramatically accelerate the C–H cleavage step as compared with just the metal catalyst itself, with the resulting ligand-acceleration opening the door to enantioselective transformations that outcompetes against background reactivity.[20,95,96] The recognition of these salient features to enable reactivity has allowed the identification of opportunities to promote enantioinduction. In particular, emerging evidence have shown that enantioselective C(sp3)–H functionalization can be achieved through the incorporation of chiral information on the directing motif or on the ligand scaffold.
The development of effective C(sp3)–H functionalization processes in the context of C–C bond formation is a comparatively nascent field owing to the many inherent challenges that it faces. Despite this, there has been several audacious examples utilized in the context of total synthesis that has proven to be extraordinarily effective. Many of these methods, once again, rely on a proximate directing motif to position the active metal catalyst close to the target C–H bond. The generated alkylmetal species then can undergo cross-coupling-type processes to generate a variety of C–C bond linkages. In essence, one can reconceive C–C bond disconnections away from traditional polar disconnections that relies on a substrate’s intrinsic reactivity (or its reversal, i.e. umpolung processes), and instead viewed in terms of its proximity to another functionality present in the target structure (Figure 8). This proximity-based retrosynthetic generalization underpins almost all the examples discussed in this section, and can serve as a useful guide for identifying C–H activation-mediated C(sp3)–C transforms in the retrosynthesis of complex targets. Finally, it is worth noting that this approach allows for the ready accessibility of a wide variety of C(sp3)–C(sp2) linkages, which typically represent C–C bond disconnections challenging to recreate using canonical polar reactivities.
Figure 8.
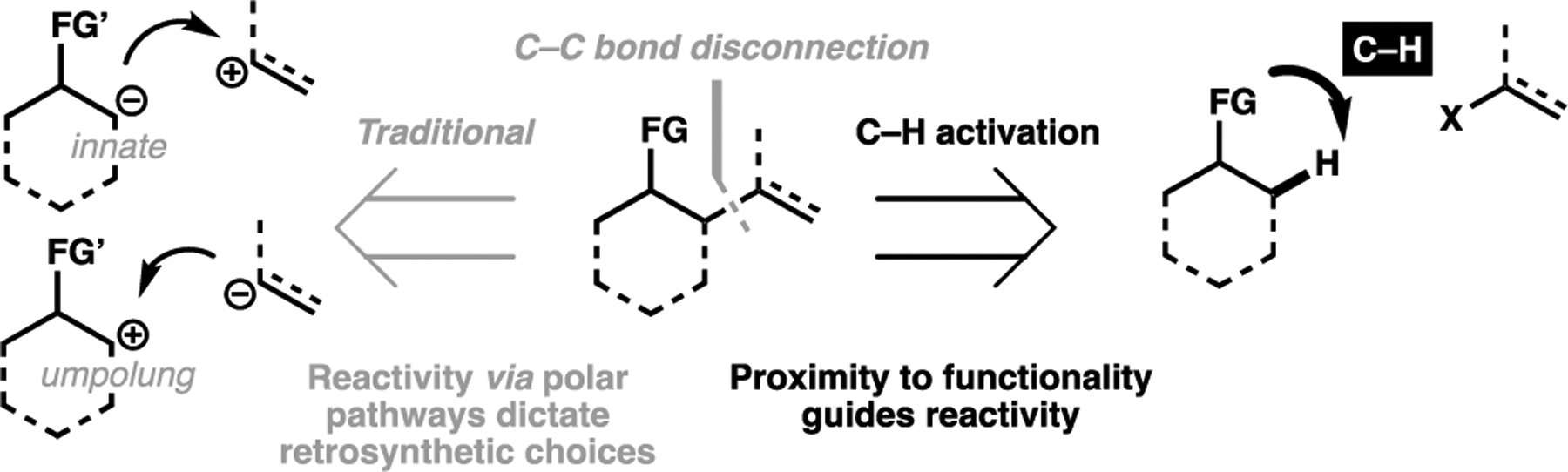
General approach for reconceiving C(sp3)–C bond disconnection via a C(sp3)–H activation approach, distinguishing between the polar reactivities with distance-guided factors that characterize C–H activation processes.
3.2. Structurally-obvious approaches to the total synthesis of a series of cyclobutane-containing natural products
The strength of C–C bond formation via C–H activation truly shines when faced with natural products containing densely functionalized strained ring systems. In nature, cyclobutanebearing natural products are hypothesized to arise from photochemical cycloaddition of two alkene-bearing monomers. This biogenetic hypothesis tends to guide their typical syntheses, with ‘classical’ approaches revolving around photochemical dimerization strategies of functionalized alkenyl precursors.[97] For example, the first total syntheses of the cyclobutane-containing natural products incarvillateine C (65)[98] and dipiperamide A (66)[99] all relied on a photochemical [2+2] cycloaddition to generate the cyclobutane core (Scheme 13). This reaction was particularly capricious to substrate effects to give a wide variety of yields and stereochemical outcomes. This rendered a biomimetic approach unreliable, limited in scope and overall unsuitable for their total syntheses. Furthermore, this strategy tends to be limited to the synthesis of homodimeric cyclobutane natural products, with heterodimeric structures particularly difficult to access in a controlled manner.
Scheme 13.
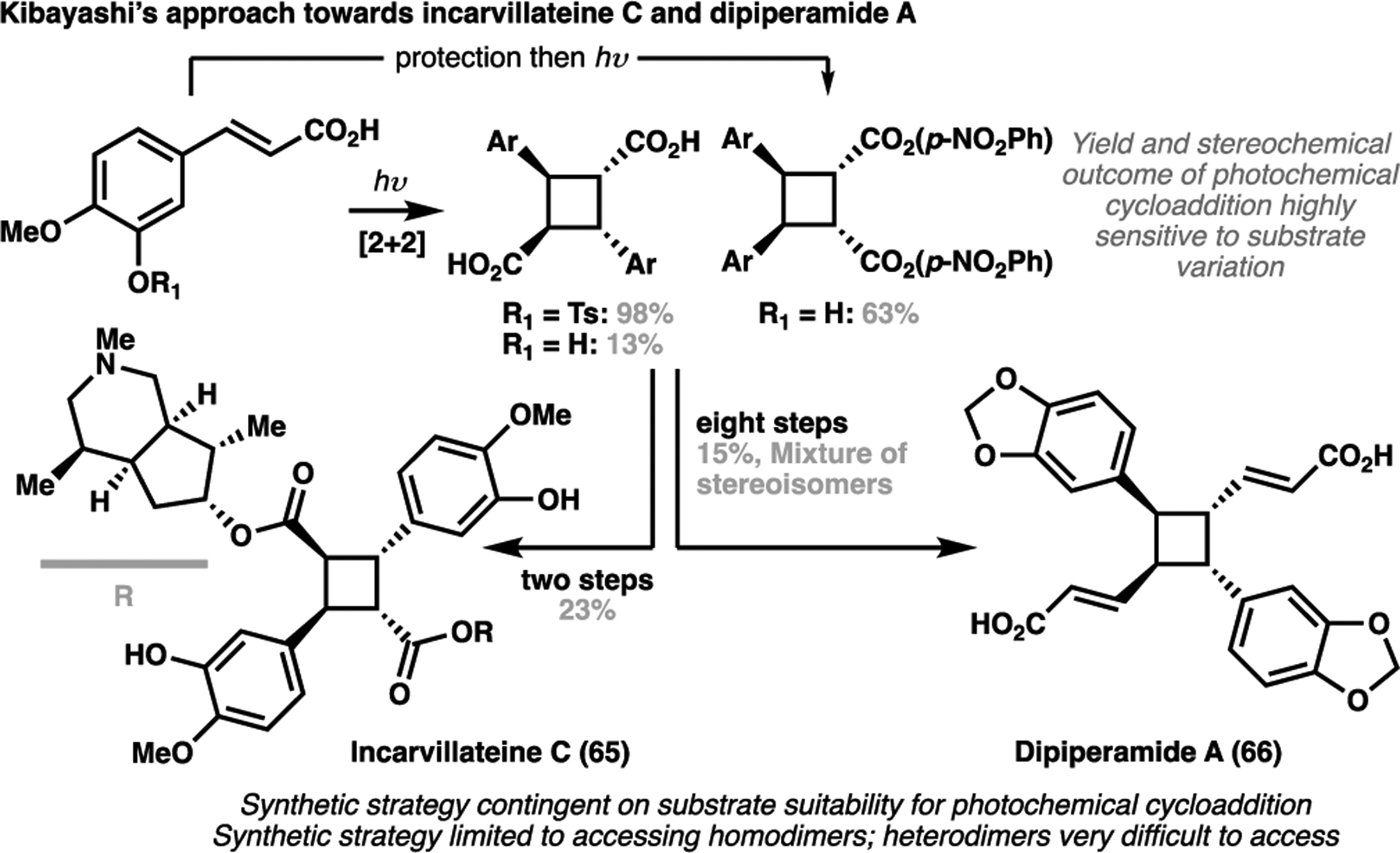
Past routes taken for the total syntheses of cyclobutane-containing natural products incarvillateine C (65, 2004) and dipiperamide A (66, 2005) by Kibayashi
On the other hand, owing to the weaker bond enthalpy of C–H bonds in strained rings, directed C–H functionalization of such bonds then becomes an effective and direct means of installing the otherwise challenging functionalities on these ring systems.[25,100,101] This ‘structurally-obvious’ approach then enables an alternative and concise strategy towards the synthesis of these targets and circumvents the constraints imposed by the canonical photocycloaddition approach. Importantly, the modularity of this approach can then allow the ready access of ‘heterodimeric’ cyclobutane containing natural products. This strategy enabled Baran et al. to achieve the rapid divergent total syntheses of three cyclobutane containing natural products: piperarborenine B (67) and the putative structures for piperarborenine D (68) and pipercyclobutanamide A (69) from a common cyclobutane scaffold.[102] In all instances, synthesis commenced with a common diacylcyclobutane core 70 obtained from 71 (Scheme 14A). For the piperarborenines, an initial directed C–H arylation installed the aryl substituent common to both congeners (72).[100,103] Selective epimerization of the α-stereocenter could then be conducted, with the selectivity dictated by the choice of base employed. From here, a second arylation with 73 from each respective intermediates, followed by ester hydrolysis and acylation with 74 gave piperarborenines in six to seven steps.
Scheme 14.
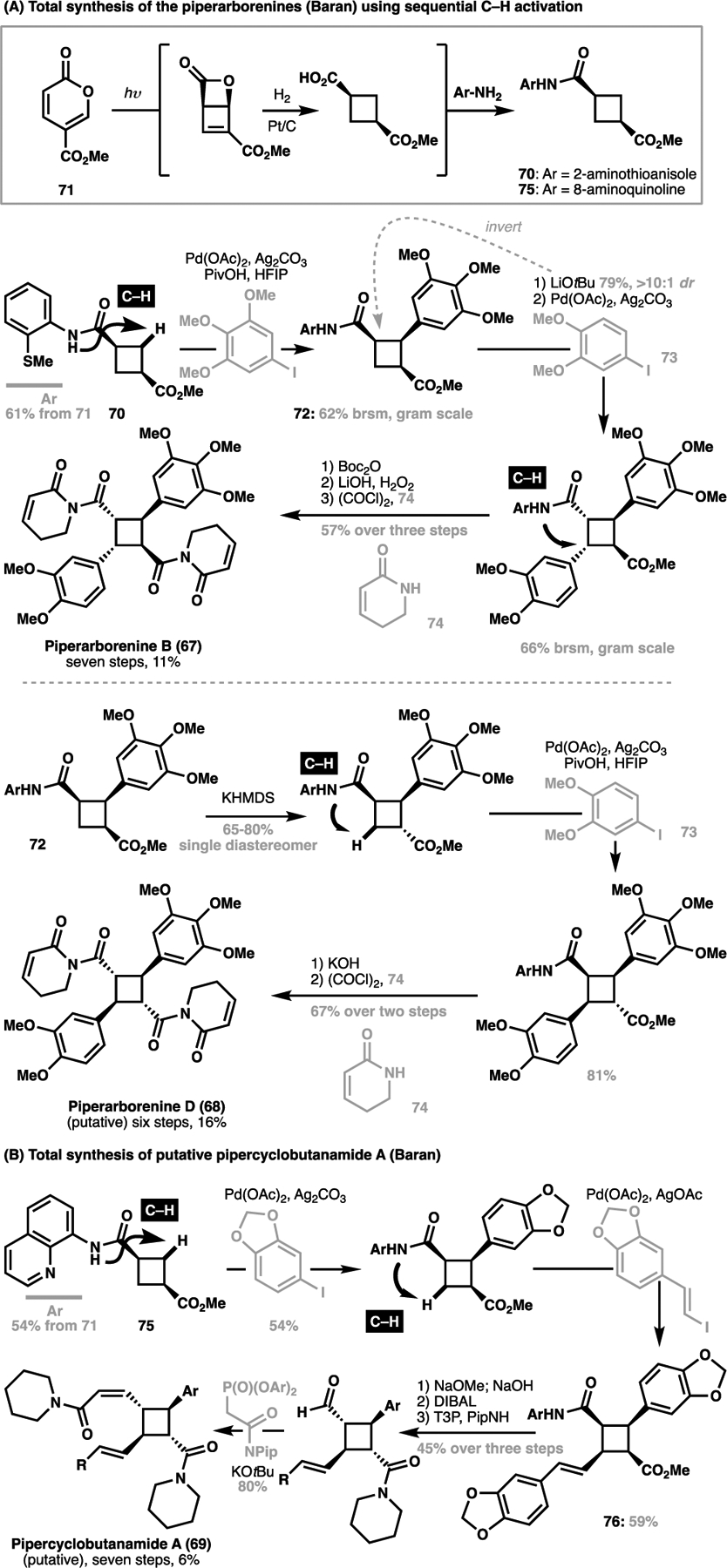
Baran’s total synthesis of (A) the piperarborenines (67 and 68, 2011) and (B) putative pipercyclobutanamide A (69, 2012) as mediated by a sequential C–H functionalization strategy
From an analogous intermediate 75, a diastereoselective arylation followed by olefination generated the required substitution in 76 for pipercyclobutanamide A, which was selectively epimerized to the correct configuration under basic conditions (Scheme 14B).[104] From here, three further steps then gave the putative structure for 69 in seven steps. The brevity of each of these syntheses, enabled by obviating the need for reactive handle installation or protecting group manipulation, demonstrates the power of a structurally-obvious approach towards natural product synthesis uniformly enabled by highly efficient C–C bond formation via C–H activation processes. In the same vein, such an approach has enabled Fox to develop a highly concise enantioselective total synthesis of piperarborenine B (67, Scheme 15A).[105] Similarly, a recent successful execution of this approach was seen in Reisman’s synthesis of (+)-psiguadial B (77), employing a C(sp3)–H alkenylation onto cyclobutane 78 as a key C–C bond forming step towards constructing the overall skeleton for the target molecule (Scheme 15B).[106]
Scheme 15.
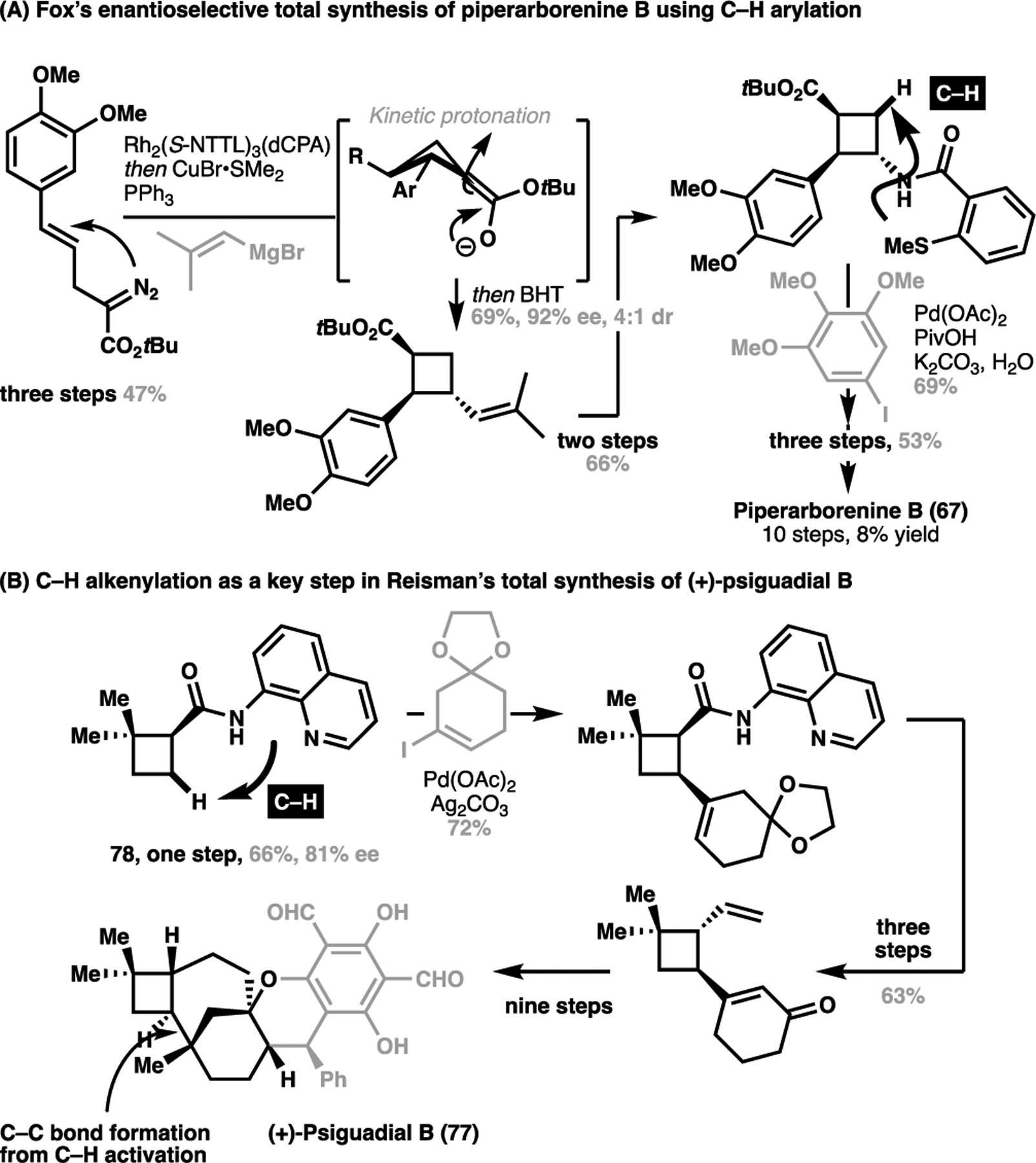
Further applications of C–H activation methods for the synthesis of cyclobutene-containing natural products: (A) Fox’s enantioselective synthesis of piperarborenine B (67); (B) Reisman’s total synthesis of (+)-psiguadial B (77)
3.3. The strategic use of C(sp3)–H alkenylation streamlines the total synthesis of (−)-epiccocin G and the rostratins
Under conventional retrosynthetic logic, the synthesis of saturated heterocycles commonly relies on a C(sp3)–X disconnection. Typically, this gives a nucleophilic heteroatom and an electrophilic carbon center as the key synthons for this strategy. To enable the desired reactivity and to avoid chemoselectivity issues, this approach often requires undesirable redox manipulations and/or protection of other nucleophilic motifs (Figure 9). A much rarer cyclization approach is a C(sp3)–C bond disconnection, typically inaccessible owing to the reactivity of C-centered nucleophiles. By redefining the chemically inert C–H bonds as reactive motifs, transition metal-catalyzed C–H activation processes can circumvent undesired reactivity issues that may plague these C–C bond formation event. The strategic use of which can have the potential to abbreviate syntheses through minimizing concessionary steps required.
Figure 9.
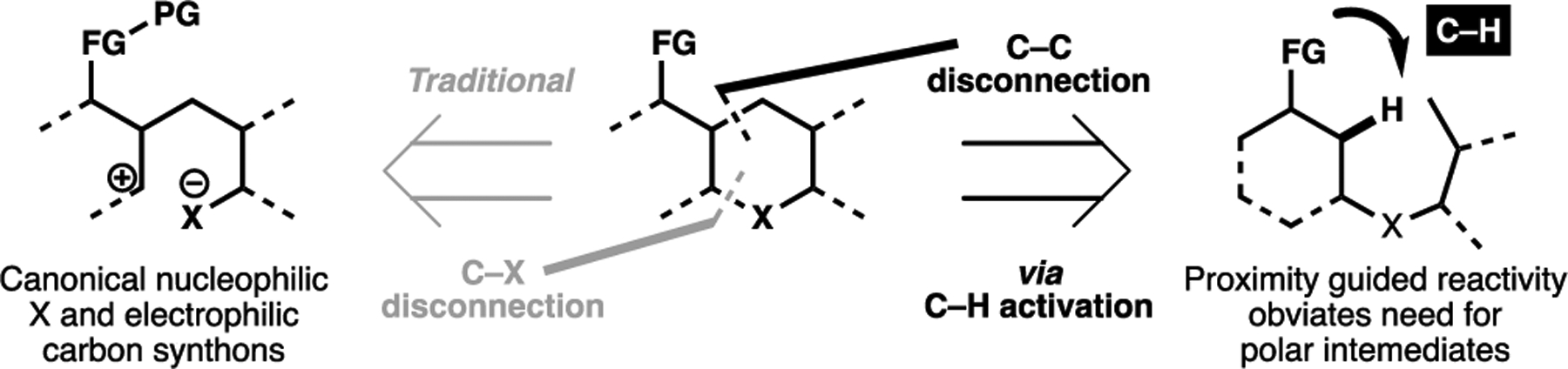
Comparison between a canonical synthetic approach towards saturated heterocycle via a C–X bond disconnection, as compared with an alternative, uncanonical approach via C–C disconnection
Some of the economies achieved through C–C disconnection for saturated heterocycles have been illustrated in a C(sp2)–H context for lithospermic acid (36, vide supra), and in this section, further exemplified in the total syntheses of the dithiodiketopiperazine natural products. In 2011, Nicolaou et al. published the first total synthesis of (−)-epiccocin G (78), and a diastereomer of the related congener 8,8’-epi-ent-rostratin B (79) (Scheme 16).[107,108] Recognizing that the two molecules shared the same core scaffold and possessed C2 symmetry, Nicolaou pursued a dimerization strategy for the synthesis of the core scaffold from two amino acid-derived monomers possessing a 6,5-bicyclic scaffold. Following standard retrosynthetic logic, this bicyclic scaffold could be disconnected at the C–N bond to reveal a protected tyrosine derivative. The synthesis of the bicyclic monomer commenced with an oxidative cyclization of Boc-protected tyrosine (80), followed by a tandem lactone hydrolysis/intramolecular conjugate addition of the amide functionality to give the 6,5-bicycle 81. From here, a five-step sequence encompassing two reductions (deoxygenation and ketone reduction) as well as differential deprotection sequence to reveal two mono-protected monomeric fragments 82 and 83. At this point, amide coupling followed by a Boc deprotection and concomitant cyclization forged the diketopiperazine core in 84. After transforming to the corresponding TFA ester 85, elimination under palladium-catalyzed conditions followed by sulfurization gave 86, which served as the synthetic divergent point for Nicolaou’s total synthesis. Taking 86, a sequence of disulfide reduction/methylation, photooxygenation, Kornblum-DeLaMare rearrangement followed by reduction gave (−)-epiccocin G (78). Alternatively, 86 could be subjected through an analogous sequence of steps to afford 8,8’-epi-ent-rostratin B (79), both in 16 steps LLS from 80.
Scheme 16.
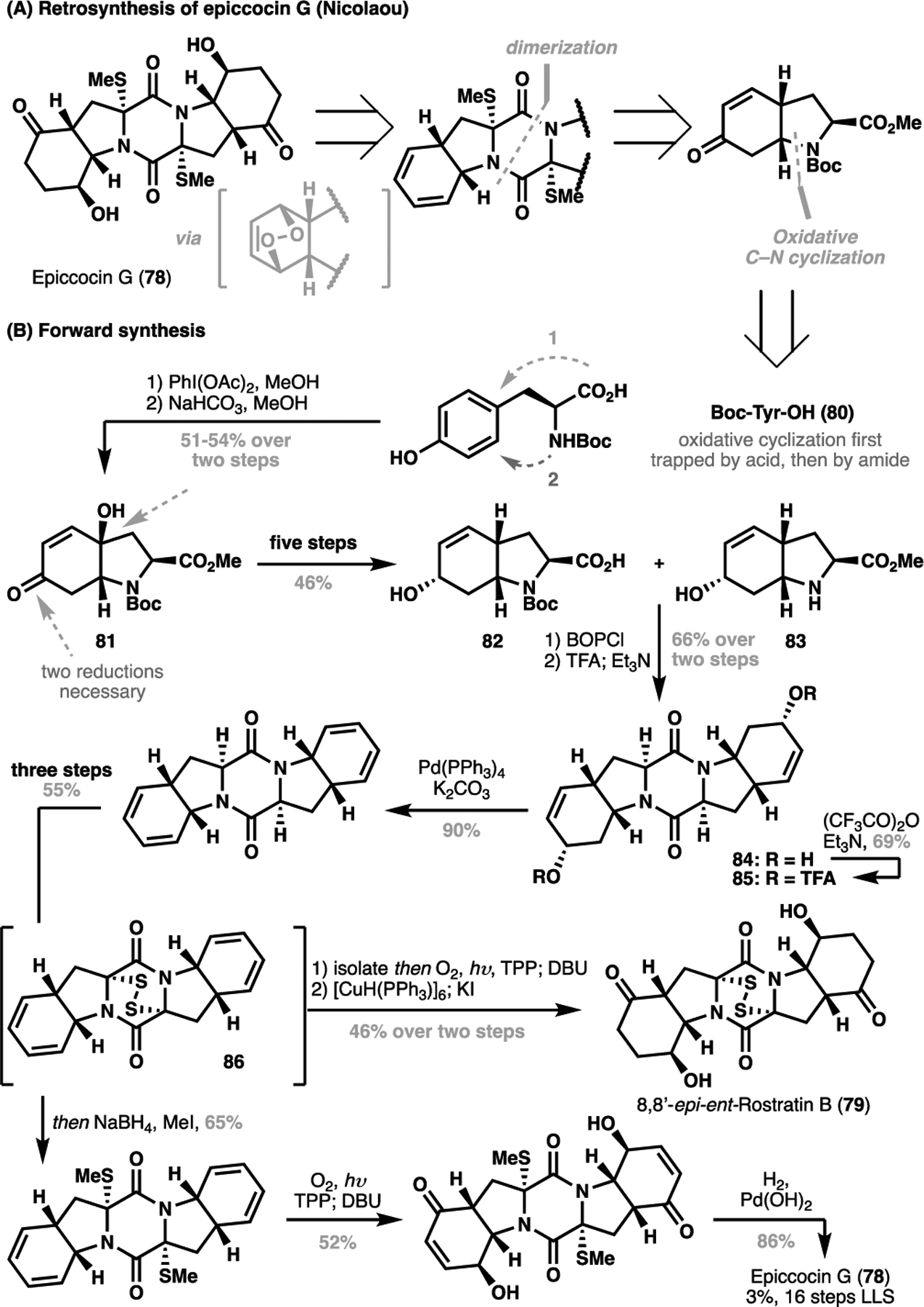
The Nicolaou total synthesis of (−)-epiccocin G and 8,8’-epi-entrostratin B (2011). (A) Retrosynthesis of (−)-epiccocin G; (B) Forward synthesis of (−)-epiccocin G (78) and 8,8’-epi-ent-rostratin B (79)
In 2019, Baudoin et al. reported a total synthesis of (−)-epiccocin G (78), as well as the related congener (−)-rostratin A (87) using a C(sp3)–H alkenylation event as a strategic cyclization steps to efficiently build the core scaffold of these natural products (Scheme 17A).[109] In an analogous symmetry-guided approach, Baudoin et al. elected to disconnect across a C–C bond through a bidirectional C(sp3)–H alkenylation strategy to close the pentacycle.[110] To realize this, Baudoin commenced their synthesis from hexenone (88), which gave 89 stereoselectively after a two-step sequence (Scheme 17B). From here, a regioselective epoxide opening with L-alanine tert-butyl ester, acetylation followed by ester hydrolysis gave the requisite monomeric fragment 90. In the absence of other competing reactivities, Baudoin et al. was able to effect a direct dimerization to give diketopiperazine 91 under amide coupling conditions and set the stage for the pivotal C–H functionalization step. Here, treatment of the alkenyl triflate-bearing substrate 91 with a source of Pd(0) smoothly closed both rings, selectively activating the proximally positioned and most sterically accessible methyl group in a six-membered palladacyclic geometry to give 92 on a multigram scale. After oxidation to 93, Baudoin et al. achieved the synthesis of a common intermediate for both natural products in eight steps. From here, six steps were required to achieve the desired oxidation level for (−)-epiccocin G. Intercepting a common intermediate, the synthesis of (−)-rostratin A could also be achieved in ten manipulations.
Scheme 17.
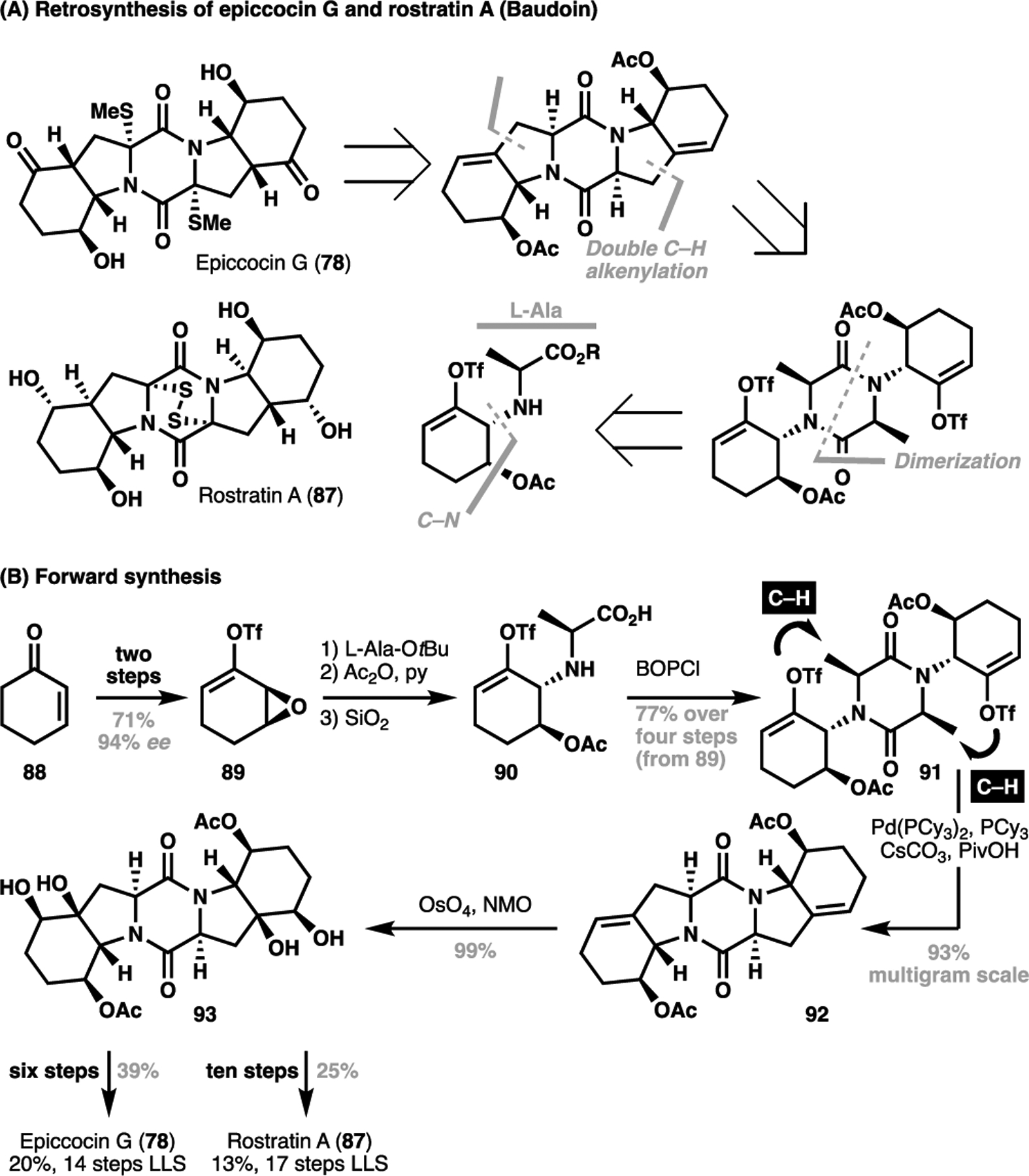
Baudoin’s total synthesis of (−)-epiccocin G (78) and (−)-rostratin A (87), employing a bidirectional C(sp3)–H activation-mediated cyclization as a key step (2019)
Baudoin’s synthesis of the core scaffold is marked by its brevity and efficiency, rapidly generating complexity with minimal redox manipulations. Part of the success of this strategy lay in the counterintuitive retrosynthetic analysis for a cyclization transform, opting for a C–C disconnection rather than across the more conventional disconnection across the C–N bond in the 6,5-bicycle. The execution of this strategy as enabled by C(sp3)–H activation transformed the notionally unreactive alanine residue as a reactive coupling handle while allowing for minimal protection surrounding the amino acid functionalities required for cyclodimerization. As a conceptually different approach, this representative case study illustrates the strengths and viability of C–H activation processes to close heteroatom-bearing ring systems. At this point, it is worth mentioning that there are many more examples where such proximity-driven C–H activation processes have successfully closed challenging ring systems, with two particularly informative accounts published by Chen (2012)[32] and Baudoin (2017).[110]
3.4. New solutions to classical problems: total synthesis of podophyllotoxin and (−)-quinine as enabled by C(sp3)–H activation-mediated C–C bond formation
The direct and diastereoselective functionalization of aliphatic rings can be extended to unstrained systems, though this tends to be a more challenging venture owing to the higher bond enthalpies of the C–H bond. Despite this, a handful of total syntheses have utilized this particularly enabling transformation in elegant syntheses of classical targets. Owing to their potent anticancer properties, one target that has seen enduring synthetic interest for over fifty years are the aryltetralin class of natural products. Podophyllotoxin (94) represents a simpler analogue of this family; it bears the carbon skeleton that typifies the aryltetralins and its total synthesis has been seen to form a synthetic blueprint towards accessing other congeners. A typical retrosynthetic approach for this class of natural products involve disconnecting the aliphatic ring system, although all syntheses require the installation of the methoxy-bearing aryl motif at an early stage using canonical reactivities.[111] In particular, stereoinduction have tended to revolve around the use of auxiliaries to install the required configuration for the pendant aryl motif.[112–114] The singular approach that allowed for a late-stage installation of the aryl motif involved a directed lithiation approach however suffered from unwieldy synthetic sequences, reaching the target in 24 steps (Scheme 18).[112]
Scheme 18.
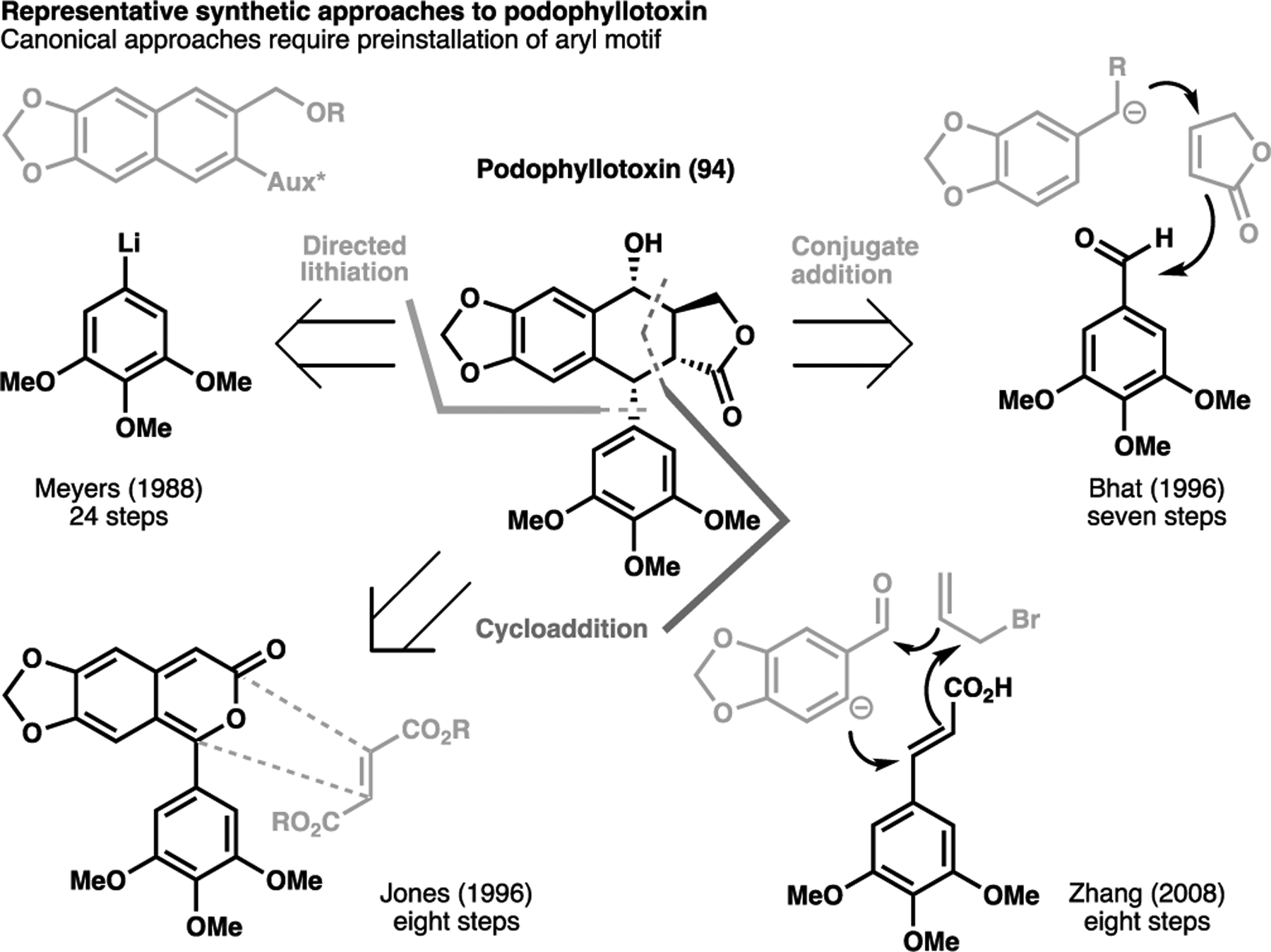
Summary of past synthetic strategies taken towards the total synthesis of podophyllotoxin (94)
Noting that one of the key points of diversity for this class of natural product lie in the pendant aryl motif, a flexible synthetic route that allows for the direct late-stage installation of this group could facilitate divergent syntheses of related congeners. Taking this vision, Maimone published a concise total synthesis of podophyllotoxin in six steps, enabled by a late-stage directed C(sp3)–H arylation transform (Scheme 19).[115] Starting from 95 (accessed in two steps), treatment with base generates a transient ortho-quinone methide intermediate, which underwent a cycloaddition with 96 followed by in situ reduction to afford the crude diol, which could then be protected as the corresponding acetal 97. Here, a C–H arylation installed the requisite aryl motif in 98 as a single diastereomer. This arylated intermediate bearing the acyl directing group which could then be directly cyclized under acidic conditions to give the natural product. Importantly, as this aryl group is a source of structural diversity for this class of natural product, Maimone et al. demonstrated the late-stage installation of this motif via C–H activation can potentially allow for the flexible diversification to generate related congeners, as well as medicinally relevant analogues.[116]
Scheme 19.
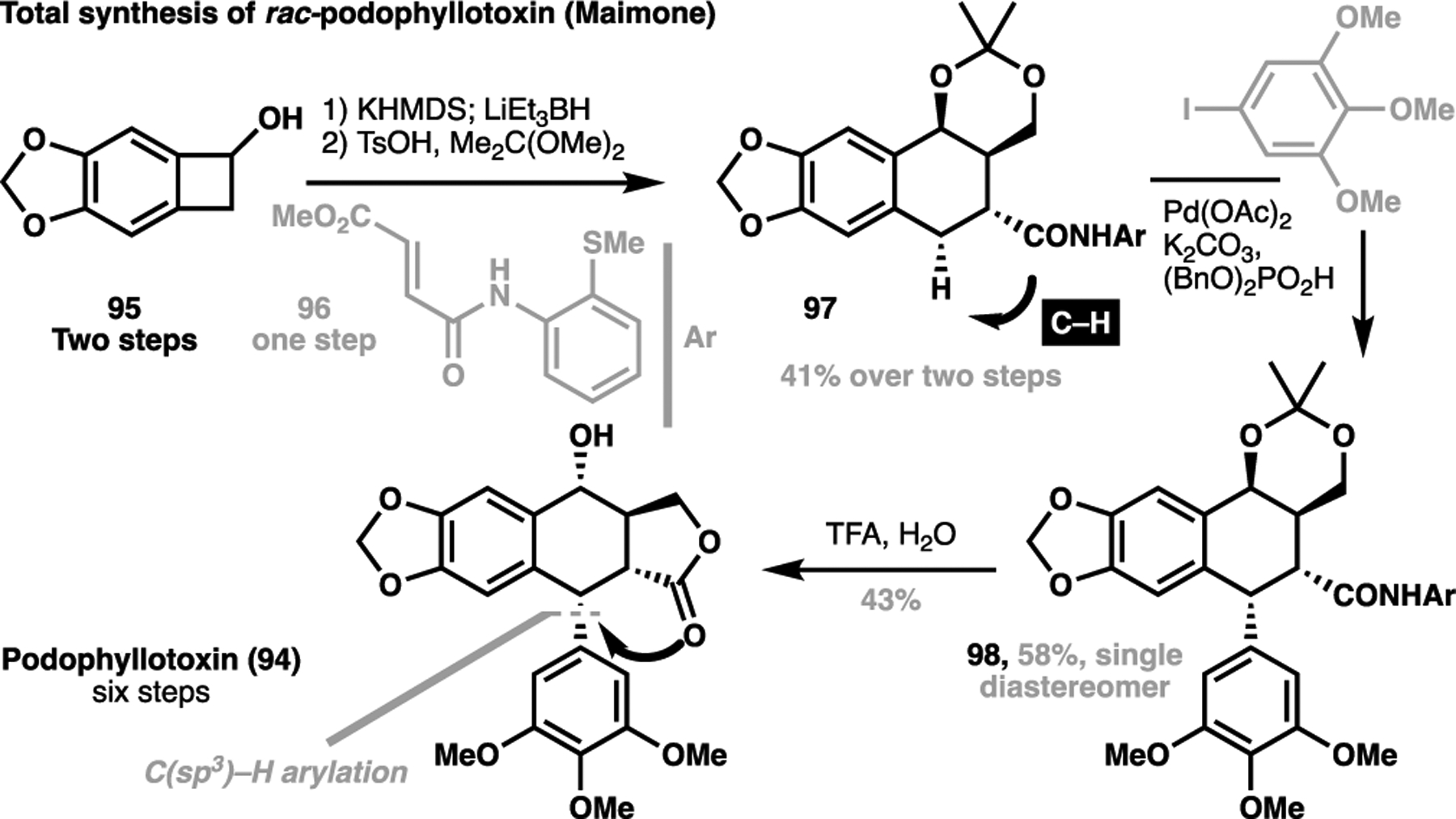
Maimone’s six-step total synthesis of rac-podophyllotoxin via a late-stage C(sp3)–H arylation strategy (2014)
The implementation of a C(sp3)–H activation strategy was crucial to the success of Maulide’s recent approach towards the classical target (−)-quinine (99). Like penicillin, the scarce supply due to the dependence of quinine production on natural sources prompted huge synthetic interest across the community. Since the first formal synthesis proposed by Woodward and Doering in 1944, the deceptively simple structure of quinine has seen no less than eight successful syntheses.[117] Despite this however, quinine has remained an enduring challenge to organic chemists, with the first enantioselective total synthesis only being achieved in 2001 by Stork et al. in 13 steps LLS some sixty years after its first formal synthesis (Scheme 20).[118] All total syntheses to date featured a canonical C–N bond disconnection to forge the quinuclidine ring system, with the vinyl group arising through a ring-cleavage/elimination mechanism,[117] or obtained through degradation of the parent natural product as a ‘relay’ manner.[119]
Scheme 20.
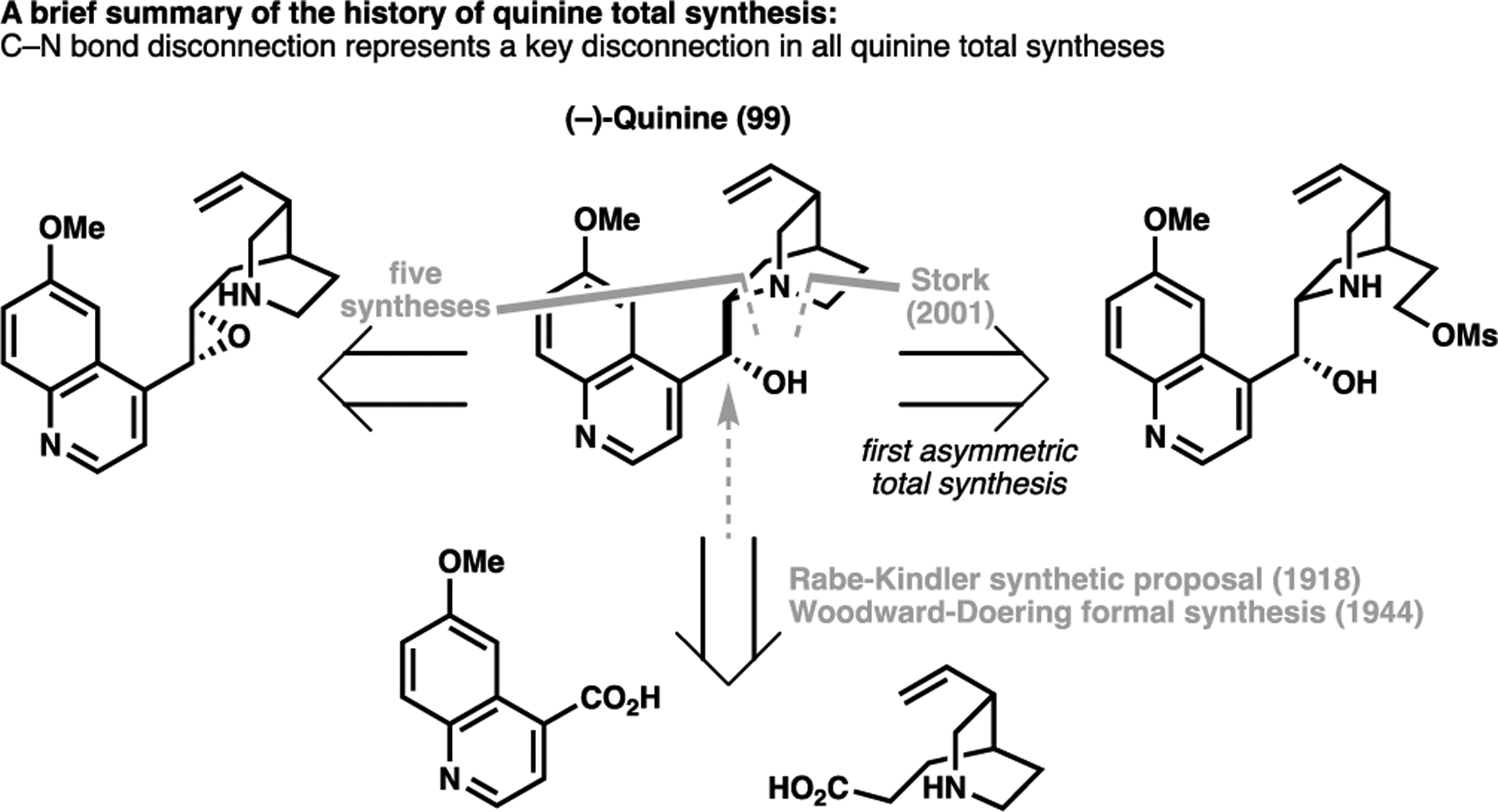
Summary of past synthetic strategies taken towards the total synthesis of (−)-quinine (99)
This changed with a wholly reconceived synthetic approach put forth by Maulide et al. in 2018, where a late-stage diastereoselective aldol addition was conceived to forge the full scaffold, while simultaneously setting the carbinol stereocenter.[120] Importantly, the facial selectivity of the aldol addition would be dictated by the configurationally pure quinuclidine scaffold, with the signature vinyl substituent introduced through a directed C–H activation transform (Scheme 21). Starting with 100, installation of a picolinamide handle to give 101 enabled a diastereoselective C(sp3)–H arylation of the quinuclidine scaffold. The arylated product 102 can be oxidatively manipulated to reveal the free acid and amidated to the corresponding Weinreb amide 103. The authors were unable to execute a direct C(sp3)–H vinylation/olefination of 101 under their optimized methodology, and so required a three-step sequence for the de novo generation of the vinyl group in 104 from amide 103. Amine 104 was then oxidized to the ketone in 105, which underwent a diastereoselective aldol addition via the intermediary mesylhydrazone with known aldehyde 106 to give the complete carbon skeleton for quinine (107). A final Wolff-Kishner reduction of 107 then furnished the natural product in ten steps LLS.
Scheme 21.
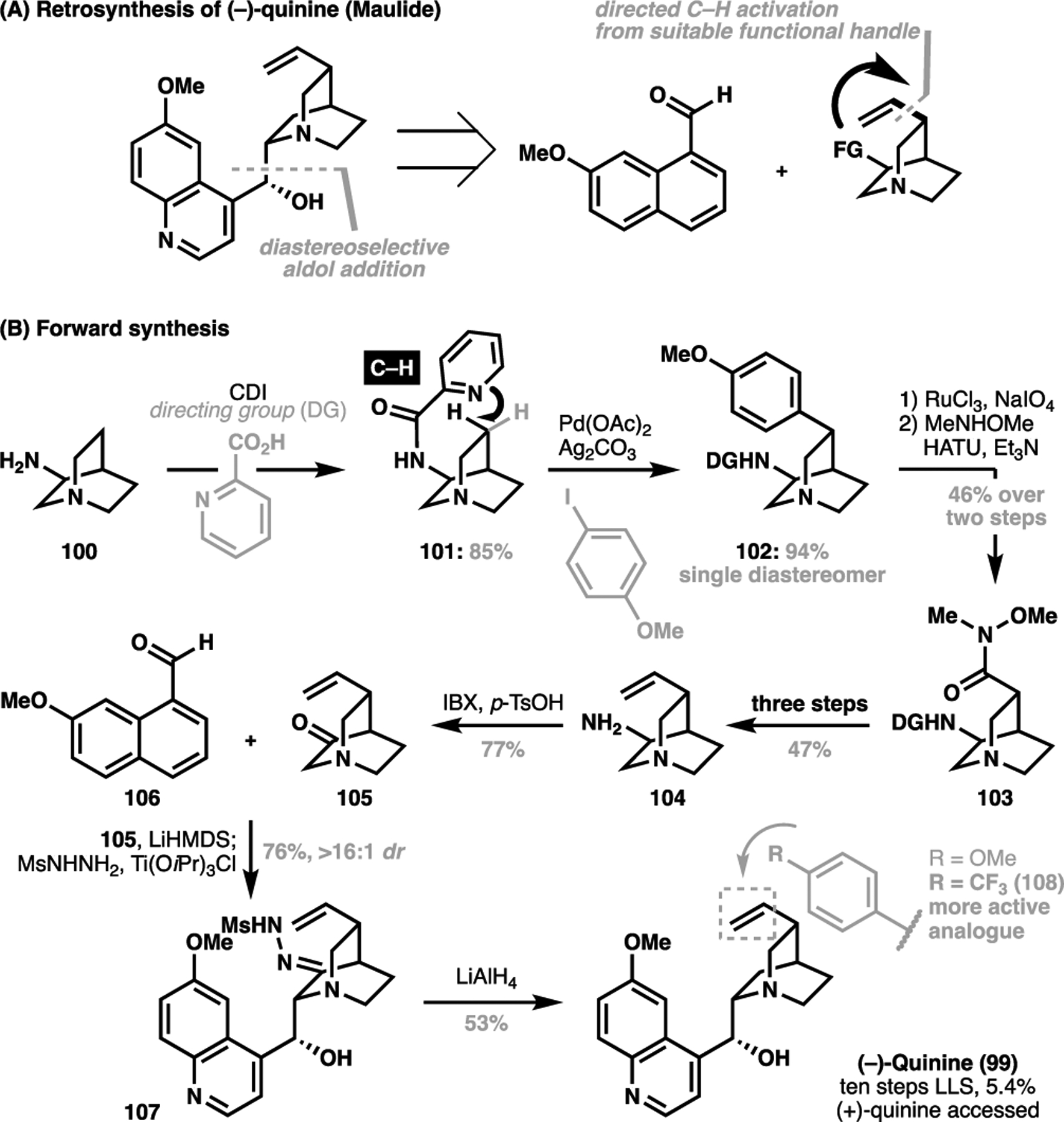
Maulide’s ten-step total synthesis of (−) and (+)-quinine (99), as well as various quinine analogues, enabled by C(sp3)–H arylation reaction (2018)
Alongside with Maimone’s synthesis of podophyllotoxin, the creative implementation of C–H activation strategies in Maulide’s quinine synthesis, while not ideal in that it required further manipulation to reveal the native vinyl group, demonstrated that a structurally-obvious approach towards derivatizing unstrained saturated scaffolds is a possible endeavor. The application in a total synthesis also demonstrates that notionally challenging C(sp3)–C(sp2) bond formation event is possible under a C–H activation strategy. Despite requiring its eventual removal, this synthesis also demonstrated the multipurpose role of the amine motif, where its configuration was relayed in the directed diastereoselective C–H arylation reaction and in the subsequent aldol coupling. This approach to quinine allowed Maulide et al. to easily access a range of designer arylquinine analogues, as well as achieving the first total synthesis of its unnatural antipode (+)-quinine. Interestingly, Maulide et al. found that the quinine analogues bearing an electron-deficient aryl substituent (108) in place of the vinyl motif was a more potent analogue compared to the natural product against the causative parasites for malaria.
4. Looking into the future: strategies for developing effective C–H activation processes for more efficient total syntheses
The examples highlighted in this review begin to highlight some of the economies that can be achieved through C–C bond construction via a C–H activation strategy in natural product total synthesis. At the same time, these examples also demonstrate a lot of the challenges central to the field that merit ongoing attention. These three broad challenges pertain to a) improved scope of reactivity and transformation, b) improved reactivity/reaction design and c) achieving enantioselectivity.[24] On top of these challenges, the development of C–H activation processes that are both high yielding and efficacious across reaction scale remains of paramount importance to enable its continued adoption as a powerful tool in synthesis. Over the last ten years in particular, significant advancements have been made towards addressing some of these methodological gaps, though these are not necessarily translated into a total synthetic context. In this section, we outline some of the key challenges and opportunities of the ongoing development of more efficient C–H activation processes, as applied to how they might further economize the syntheses outlined in this review. Our intent is to highlight methodology gaps that merit attention and how they may be addressed. In doing so, we hope that the ongoing development for these C–H activation transformations can be even more applicable to natural product total syntheses.
4.1. Widening the range of chemical transformations arising from C–H activation processes: Leveraging weak coordination and ligand acceleration
One enduring challenge in the field lies in its often-limited range of chemical transformations that is amenable to C–H activation processes. Intrinsically, the scope of these transformations is deeply rooted in the catalytic cycle in which the transition metal catalyst proceeds, though it is acknowledged that a wide variety of arylation transforms (using either aryl halides or other aryl coupling partners) can be readily achieved. However, the same cannot be said for other C(sp2)-forming transforms, which are often reliant on electronically biased olefins (e.g. with electron withdrawing groups) to induce the required Heck-type reactivity.[121] This renders other transformations such as olefinations, carbonylation or the simple structural act of vinylation highly challenging for C(sp2) and in particular, C(sp3)–H activation. In the last decade, we have found that part of the solution to this problem lies in judicious ligand choices to reduce the barrier for C–H cleavage step,[24,122] as well as particularly harnessing weak coordination of a nearby directing group to render the cyclometalated intermediate more reactive, and therefore more amenable to a diverse set of coupling transforms (Figure 10).[123] This guiding principle has allowed us to continually broaden the scope of substrate and coupling partners competent for C–H activation, but much work remains to be done to achieve synthetic generality.
Figure 10.
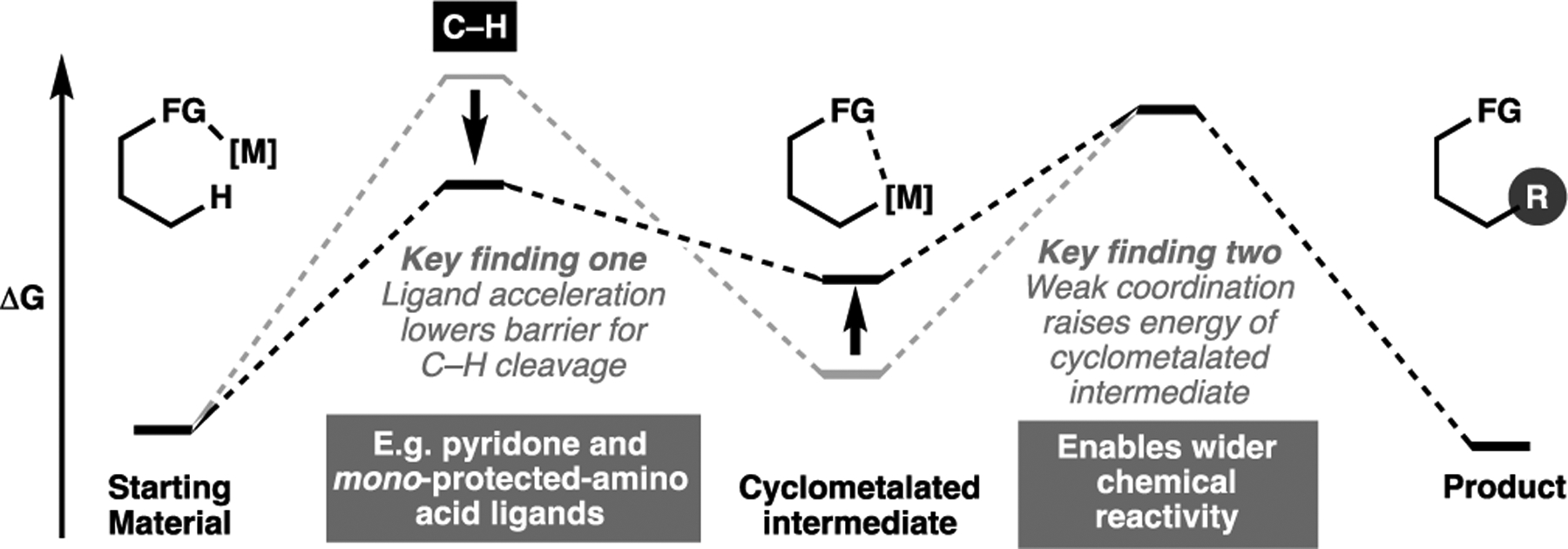
Key findings to help broaden substrate and coupling scope of future C–H activation transformations
Nevertheless, there are several promising studies to demonstrate the viability of these processes. A range of C(sp2)–H olefination have been developed for aromatic systems, and recently, our group has developed a range of C(sp3)–H olefination and cross-coupling reactions enabled by weak coordinative interaction in conjunction with ligand acceleration.[124–126] One example pertains to the triflamide-directed asymmetric γ-C(sp3)–H olefination with vinyl boronates (Scheme 22A),[127] which serves as a promising lead towards bridging the methodology gap faced by Maulide et al. in their conquest of quinine for the direct vinylation of the quinuclidine core. If successful, this approach could enable a six-step asymmetric total synthesis of (−)-quinine (99), constituting a 40% reduction in the total step count from Maulide’s current state-of-the-art approach (Scheme 22B).
Scheme 22.
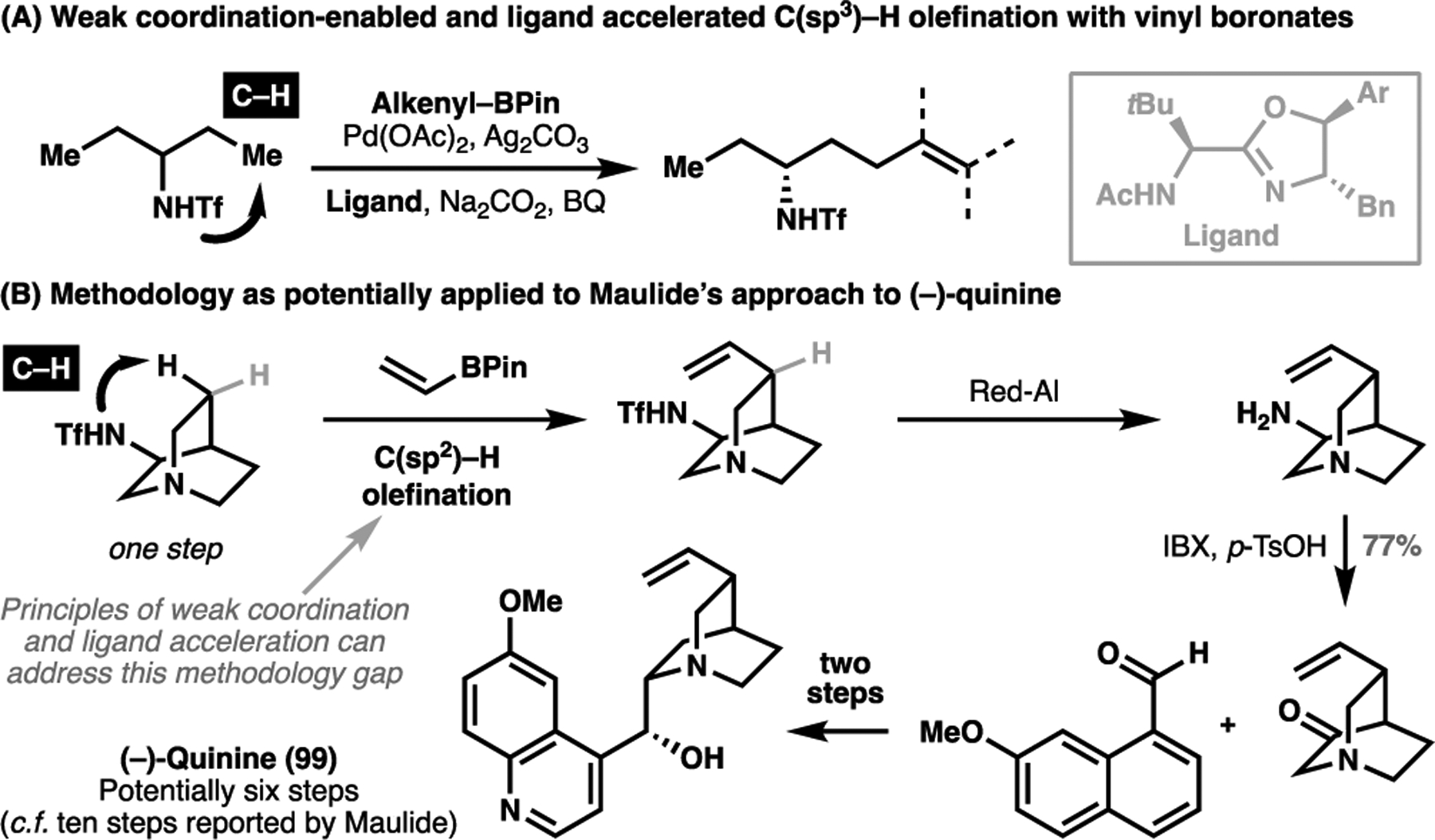
(A) A combination of weak-coordination and ligand acceleration enables a challenging desymmetrizing C(sp3)–H olefination. (B) A potentially more concise total synthesis of (−)-quinine, leveraging a weakly-directing triflimide motif as highlighted in (A)
4.2. Broadening the range of native functionalities for directed C–H activation
To date, the majority of C–H activation reactions employed in total synthesis rely on large and strongly coordinating directing groups to help bind and position the transition metal catalyst towards the target C–H bond in question. Typically, these directing groups are amide-based motifs that require separate installation, and often challenging removal steps. There are instances where clever synthetic design has circumvented this problem, as exemplified by Maimone’s approach to podophyllotoxin, where the directing group was concomitantly removed during the final lactonization process. In many cases however, this is not possible and adds two steps onto the synthetic sequence. Moreover, directing groups contravenes the principles of achieving atom economy, and thus future development of selective C–H activation transforms should harness native functionality to effectively direct the catalyst (Figure 11).
Figure 11.
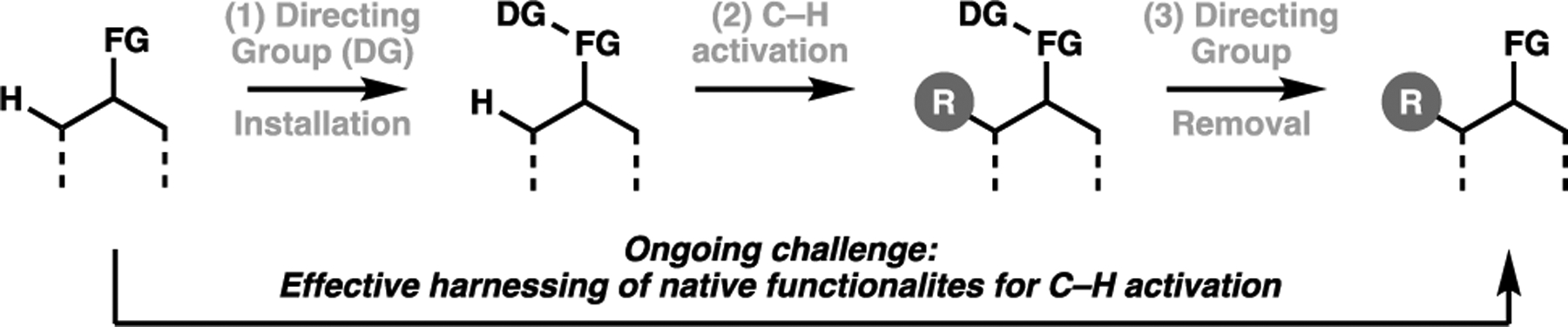
Contrasting canonical approaches for transition metal-catalyzed C–H activation to an ideal process by harnessing native functionalities
Part of the solution to this challenge lie in focusing on the development of native functionality-directed C–H activation processes. To date, a wide variety of carbonyl-bearing functional groups, including acids, amides, and ketones, have proven successful as native directing groups for C–H activations. For amines, aldehydes, and ketones, the concept of reversible imine formation has also permitted the development of transient directing group strategies, which avoid separate installation and removal steps. With ongoing advances, a four-step total synthesis of (+)-hongoquercin A (32, Scheme 23A) may be attainable from (+)-chromazonarol (33) without the need of an extraneous directing group, as well as a more atom-economical method towards generating the functionalized cyclobutane 109 in the total synthesis of psiguadial B (77, Scheme 23B).
Scheme 23.
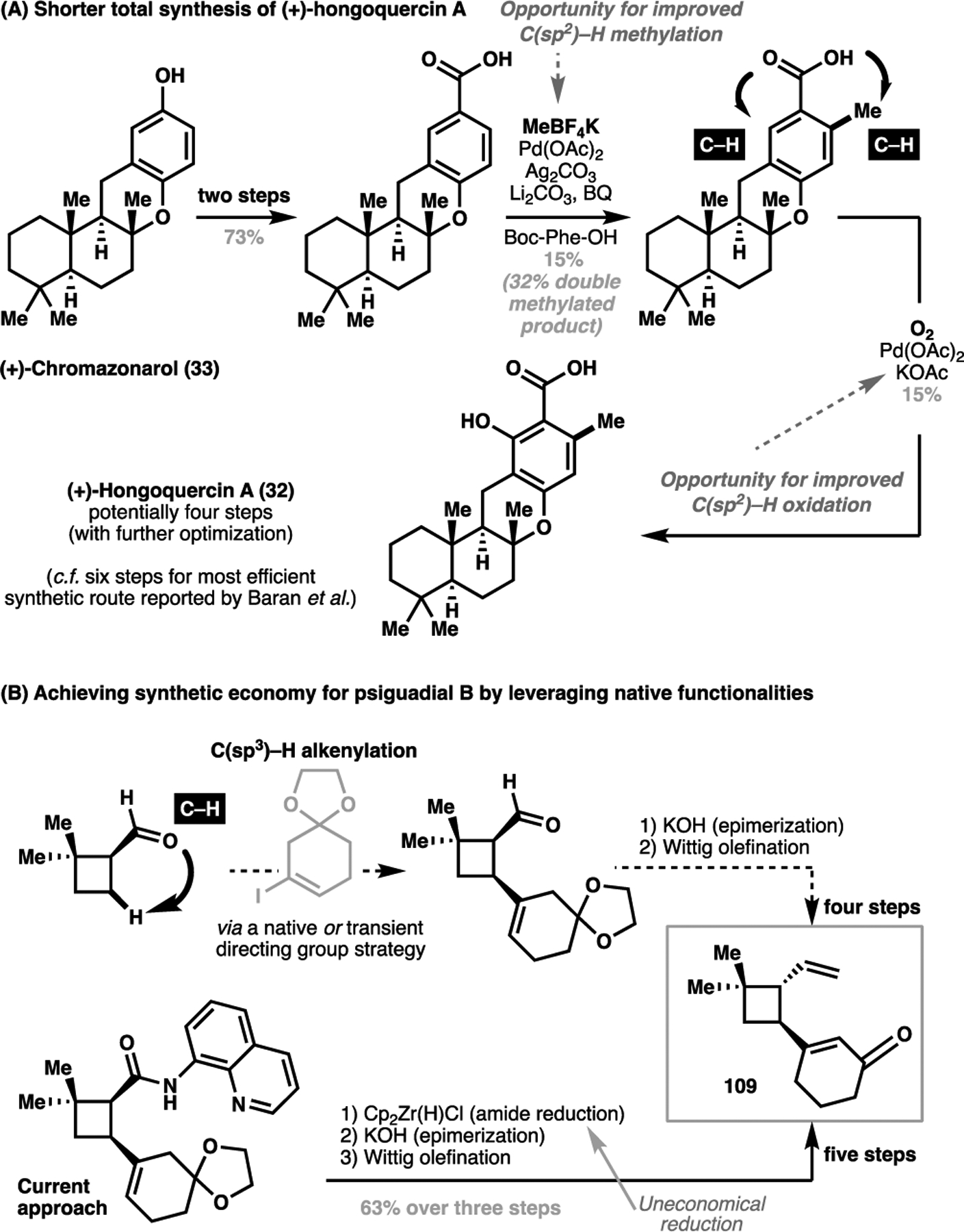
(A) Methodology gaps for a potential four-step total synthesis of hongoquercin A. (B) Opportunities for more atom economical approach towards the synthesis of psiguadial B.
4.3. Achieving enantioselectivity through C–C bond construction via C–H activation
Through achieving ligand acceleration in C–H activation processes, there is a possibility of achieving enantiodiscrimination by virtue of an enantioselective catalytic process outcompeting against a racemic background reaction. In this review, we have illustrated some especially powerful application of C–H activation technologies where the absolute configuration of a structure is also tandemly set. This has been an enduring challenge in the field, and one with tremendous potential to alter the field of how we achieve asymmetric syntheses of complex targets. Owing to the strong reliance of proximity and geometry, we have illustrated several examples in this review of diastereoselective C–H activation processes. We have also illustrated particularly compelling cases of desymmetrizing axially chiral compounds. An emerging body of work have demonstrated that C–H activation processes are capable of desymmetrizing C(sp3) centers, with recent work demonstrated that the use of a chiral ligand can allow for enantioselective discrimination of C(sp3)–H bonds in cyclobutanes.[128] In the case of Baran’s racemic synthesis piperarborenine B (67), the incorporation of such conditions could enable for the enantioselective total synthesis of 67 in six steps; an economy of four synthetic operations as compared with Fox’s enantioselective synthesis of the same target (Scheme 24).
Scheme 24.
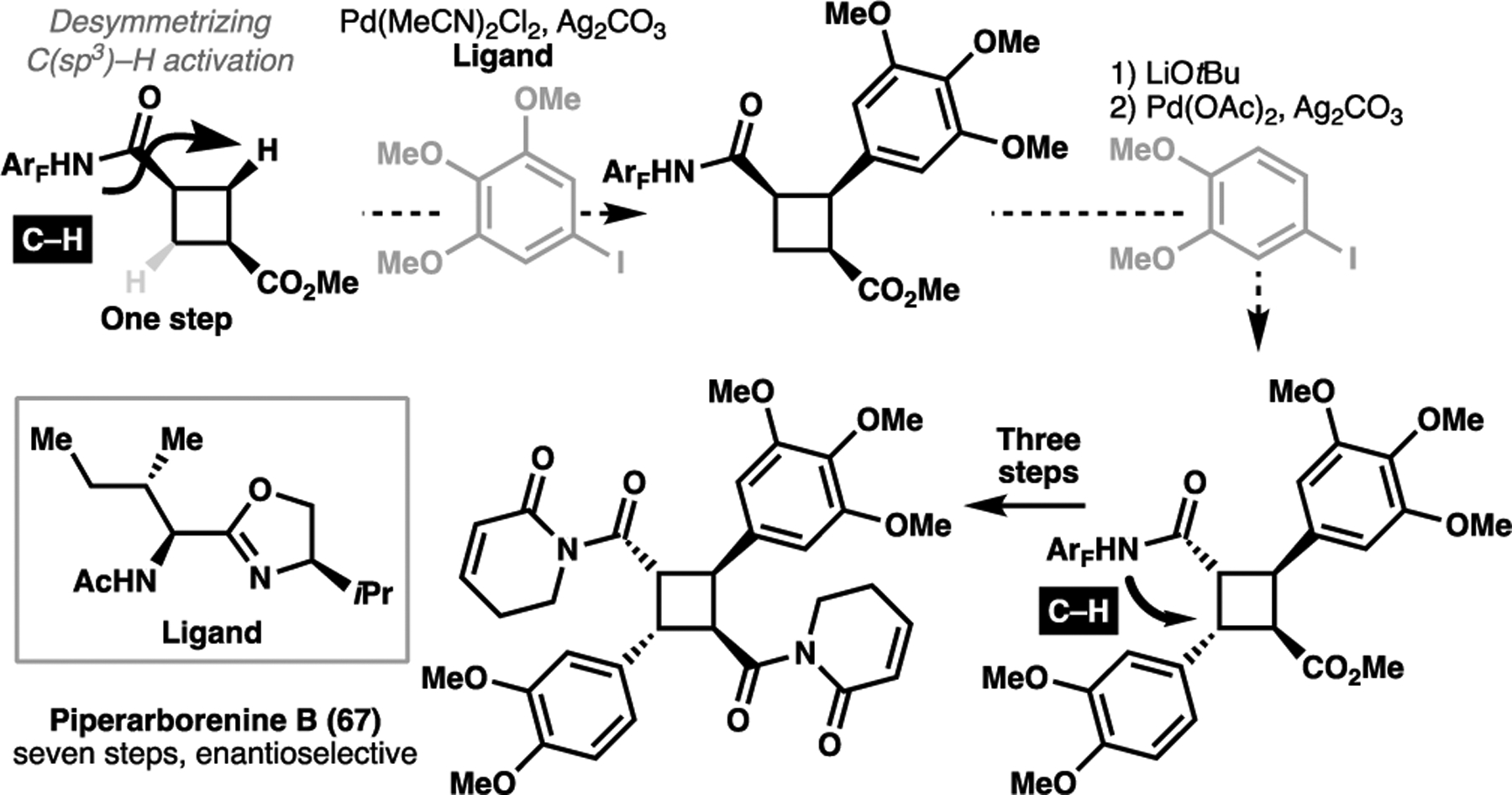
A potentially shorter enantioselective total synthesis of piperarborenine B (67) using a desymmetrizing C(sp2)–H activation transformation
The total synthesis and stereochemical confirmation of delavatine A (110) by Shen, Li, Zhang et al. provides another compelling demonstration of the synthetic potential of these desymmetrative operations.[129] The forward synthesis commenced with a five-step sequence to give indene 111 (Scheme 25). A syn-selective hydrogenation delivered racemic 112, where Shen, Li, Zhang et al. found that optimized C–H olefination conditions was able to kinetically resolve the two enantiomers to obtain enantioenriched 112 as well as the enantioenriched olefinated product 113 in excellent conversion on multigram scale. This triflamide-directed C–H activation step thus elaborates 111, as well as resolving both enantiomers required for the stereochemical verification of the natural product. After an ozonolysis/reduction sequence to 114, a four-step sequence gave the tricyclic coupling partner 115, and a final Stille coupling with pulegone-derived fragment 116 then afforded delavatine A (110). Through this sequence with both enantiomeric series of the tricycle, as well as enantiomers of the pulegone-derived fragment 116, Shen, Li, Zhang et al. were able to expeditiously determine that the stereochemical identity of delavatine A was as shown in 110.
Scheme 25.
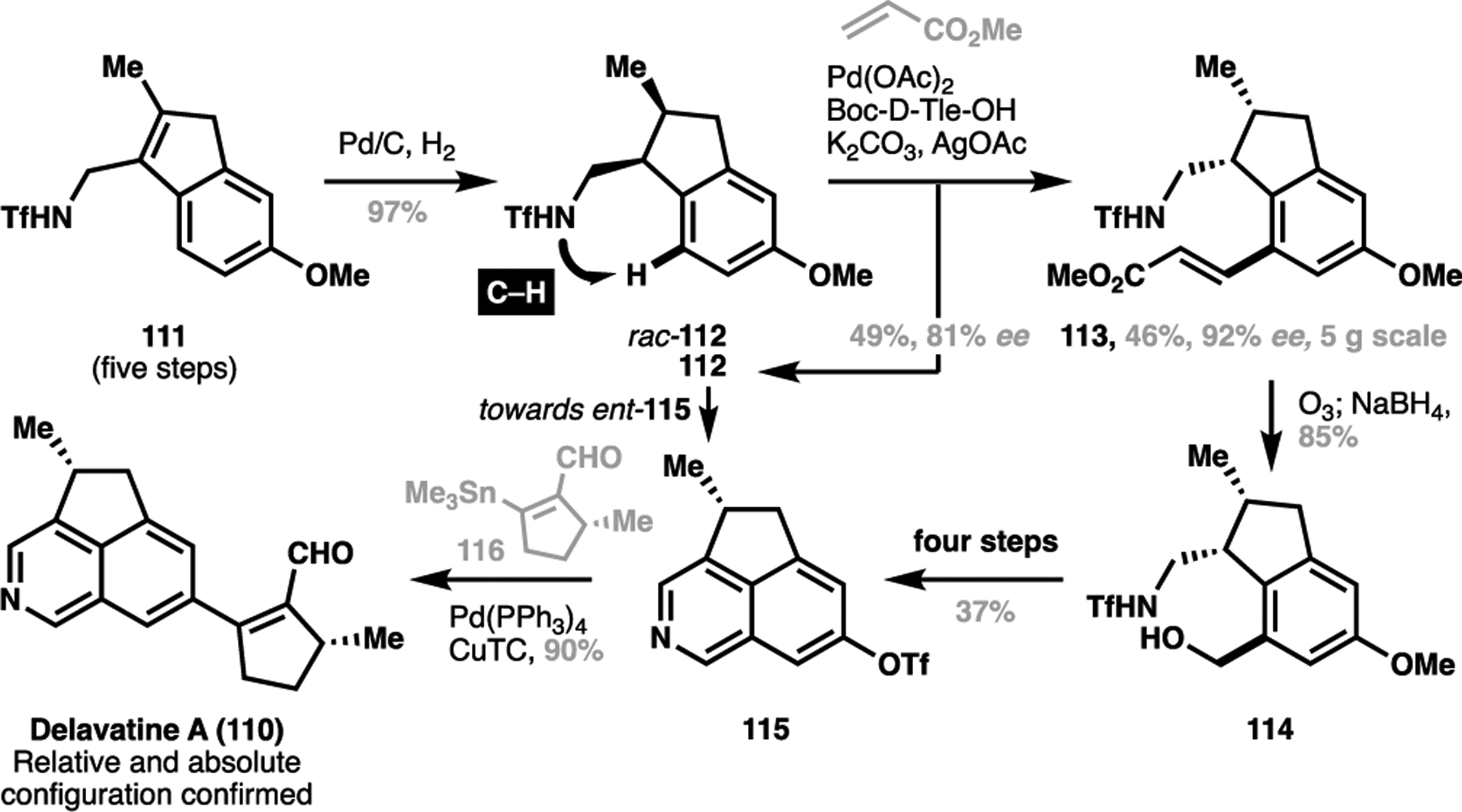
Total synthesis and configurational assignment of delavatine A by Shen, Li, Zhang et al. (110)
4. Summary and outlook
In this review, we illustrate that the use of C–H activation to forge C–C connections in the context of total synthesis need not be a contrived demonstration of methodology, but can instead serve as a powerful synthetic tool in its own right. For each type of C–H bond, this review presents an overview of strategies to effect its selective activation, and compares how traditional C–C bond forming approaches can be reenvisaged using a C–H activation. The comparative case studies analyzed in this review highlight how C–C bond construction via transition metal-catalyzed C(sp2) and C(sp3)–H activation have provided a wholly alternative ‘structurally-obvious’ blueprint for the synthesis of complex natural products, facilitated greater utility of native functionalities, as well as enabled the expeditious realization of alternative and otherwise challenging C–C bond disconnections.
All of these examples demonstrate the synthetic economies achieved by C–H activation, either through the economy of reagent/atom use, economy of synthetic transforms, and/or the potential synthetic divergence. In conjunction with the myriad of successful applications of C–H activation in total synthesis, these examples provide compelling evidence that the adoption of such processes can move us towards the goal of synthetic ideality. While some transformations may not currently represent the most direct method for their functionalization, these limitations only serve to encourage the community to develop new, more effective C–H activation methodologies. Specifically, vast opportunities exist in the design of C(sp2)–H and especially C(sp3)–H-activation processes that can directly use native functionalities without the need of exogenous directing groups, are competent with a broader range of coupling partners, and can achieve high levels of stereoselection. These opportunities challenge the community to innovate and overcome the synthetic hurdles that face us today, and we anticipate our capacity to forge bonds via C–H activation will evolve as new ligands and catalysts are developed.
Finally, it is worth noting that the innovations described in this review represent only one of the many approaches developed by chemists to effect C–H functionalization. Each of these powerful processes delivers not just a variety of novel reactivity, but also opportunities to creatively economize the synthesis of complex targets. It now remains a challenge for practitioners of organic synthesis to broadly embrace these innovations and propel the art and craft of synthesis into the twenty-first century.
Acknowledgements
We gratefully acknowledge The Scripps Research Institute, the NIH (NIGMS, R01GM084019; NIGMS, R01 GM102265), the NSF under the CCI Center for Selective C–H Functionalization (CHE1700982), and the Lindemann Trust (fellowship to NYSL) for their financial support.
Biographies

Nelson Lam received his B.Sc.(Hons) from the University of Auckland, working in the lab of Prof. Christian Hartinger (2015). He recently obtained his Ph.D. at the University of Cambridge (2019) under the supervision of Prof. Ian Paterson as a Woolf Fisher Scholar, where he worked on the total syntheses of polyketide natural products. He is currently a postdoctoral associate and Lindemann fellow with Prof. Jin-Quan Yu at the Scripps Research Institute, developing novel Pd-catalyzed C(sp3)–H functionalization transformations.

Kevin Wu received his B.Sc. in Chemistry from Boston College in 2011, working in Prof. Amir Hoveyda’s lab. He did his Ph.D. studies in Chemistry at Princeton University in Prof. Abigail Doyle’s lab, developing new ligands for Ni catalyzed-cross couplings and studying the mechanism of these cross couplings. He is currently a postdoctoral scientist with Prof. Jin-Quan Yu, developing sustainable Pd-catalyzed C(sp3)–H activation methods.

Jin-Quan Yu received his B.Sc. in Chemistry from East China Normal University and his M.Sc. from the Guangzhou Institute of Chemistry. In 2000, he obtained his Ph.D. at the University of Cambridge with Prof. Jonathan B. Spencer. After some time as a Junior Research Fellow at Cambridge, he joined the laboratory of Prof. E. J. Corey at Harvard University as a postdoctoral fellow. He then began his independent career at Cambridge (2003–2004) before moving to Brandeis University (2004–2007) and finally The Scripps Research Institute, where he is currently the Frank and Bertha Hupp Professor of Chemistry.
References
- [1].Wöhler F, Ann. Phys 1828, 88, 253–256. [Google Scholar]
- [2].Robinson R, J. Chem. Soc. Trans 1917, 111, 762–768. [Google Scholar]
- [3].Sheehan JC, Henbry-Logan KR, J. Am. Chem. Soc 1957, 79, 1262–1263. [Google Scholar]
- [4].Eschenmoser A, Wintner CE, Science 1977, 196, 1410–1420. [DOI] [PubMed] [Google Scholar]
- [5].Nicolaou KC, Petasis NA, Zipkin RE, Uenishi J, J. Am. Chem. Soc 1982, 104, 5555–5557. [Google Scholar]
- [6].Corey EJ, Angew. Chemie Int. Ed. English 1991, 30, 455–465. [Google Scholar]
- [7].Nicolaou KC, Bulger PG, Sarlah D, Angew. Chem. Int. Ed 2005, 44, 4442–4489. [DOI] [PubMed] [Google Scholar]
- [8].Nicolaou KC, Bulger PG, Sarlah D, Angew. Chem. Int. Ed 2005, 44, 4490–4527. [DOI] [PubMed] [Google Scholar]
- [9].Milde B, Pawliczek M, Jones PG, Werz DB, Org. Lett 2017, 19, 1914–1917. [DOI] [PubMed] [Google Scholar]
- [10].Chu S, Münster N, Balan T, Smith MD, Angew. Chem. Int. Ed 2016, 55, 14306–14309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Newhouse T, Baran PS, Hoffmann RW, Chem. Soc. Rev 2009, 38, 3010–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rosen BR, Simke LR, Thuy-Boun PS, Dixon DD, Yu JQ, Baran PS, Angew. Chem. Int. Ed 2013, 52, 7317–7320. [DOI] [PubMed] [Google Scholar]
- [13].Trost BM, Science 1991, 254, 1471–1477. [DOI] [PubMed] [Google Scholar]
- [14].Gaich T, Baran PS, J. Org. Chem 2010, 75, 4657–4673. [DOI] [PubMed] [Google Scholar]
- [15].Hendrickson JB, J. Am. Chem. Soc 1975, 97, 5784–5800. [Google Scholar]
- [16].Gaunt JC, Shaw BL, J. Organomet. Chem 1975, 102, 511–516. [Google Scholar]
- [17].Janowicz AH, Bergman RG, J. Am. Chem. Soc 1983, 105, 3929–3939. [Google Scholar]
- [18].Labinger JA, Bercaw JE, Nature 2002, 417, 507–514. [DOI] [PubMed] [Google Scholar]
- [19].Lavrushko VV, Lermontov SA, Shilov AE, React. Kinet. Catal. Lett 1980, 15, 269–272. [Google Scholar]
- [20].Chen G, Gong W, Zhuang Z, Andrä MS, Chen YQ, Hong X, Yang YF, Liu T, Houk KN, Yu JQ, Science 2016, 353, 1023–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Serratosa F, Chapter 5. Synthesis of Dissonant Systems, Elsevier, 1996. [Google Scholar]
- [22].Giri R, Shi BF, Engle KM, Maugel N, Yu JQ, Chem. Soc. Rev 2009, 38, 3242–3272. [DOI] [PubMed] [Google Scholar]
- [23].Scifinder search.
- [24].Shao Q, Wu K, Zhuang Z, Qian S, Yu JQ, Acc. Chem. Res 2020, 53, 833–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Saint-Denis TG, Zhu RY, Chen G, Wu QF, Yu JQ, Science 2018, 359, DOI 10.1126/science.aao4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Abrams DJ, Provencher PA, Sorensen EJ, Chem. Soc. Rev 2018, 47, 8925–8967. [DOI] [PubMed] [Google Scholar]
- [27].Gutekunst WR, Baran PS, Chem. Soc. Rev 2011, 40, 1976–1991. [DOI] [PubMed] [Google Scholar]
- [28].Yamaguchi J, Yamaguchi AD, Itami K, Angew. Chem. Int. Ed 2012, 51, 8960–9009. [DOI] [PubMed] [Google Scholar]
- [29].Wang XC, Gong W, Fang LZ, Zhu RY, Li S, Engle KM, Yu JQ, Nature 2015, 519, 334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shi H, Lu Y, Weng J, Bay KL, Chen X, Tanaka K, Verma P, Houk KN, Yu JQ, Nat. Chem 2020, 12, 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]; A summary of other methods for the direct site-selective functionalization of quinoline is also detailed in this account.
- [31].Yamaguchi J, Yamaguchi AD, Itami K, Angew. Chem. Int. Ed 2012, 51, 8960–9009. [DOI] [PubMed] [Google Scholar]
- [32].Chen DYK, Youn SW, Chem. Eur. J 2012, 18, 9452–9474. [DOI] [PubMed] [Google Scholar]
- [33].Baudoin O, Angew. Chem. Int. Ed 2020, DOI 10.1002/anie.202001224. [DOI] [Google Scholar]
- [34].Chen X, Engle KM, Wang DH, Yu JQ, Angew. Chem. Int. Ed 2009, 48, 5094–5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lyons TW, Sanford MS, Chem. Rev 2010, 110, 1147–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sigman MS, Werner EW, Acc. Chem. Res 2012, 45, 874–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Daugulis O, Do HQ, Shabashov D, Acc. Chem. Res 2009, 42, 1074–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Daugulis O, Roane J, Tran LD, Acc. Chem. Res 2015, 48, 1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yu JQ, Adv. Synth. Catal 2014, 356, 1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhang Z, Tanaka K, Yu JQ, Nature 2017, 543, 538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bag S, Patra T, Modak A, Deb A, Maity S, Dutta U, Dey A, Kancherla R, Maji A, Hazra A, et al. , J. Am. Chem. Soc 2015, 137, 11888–11891. [DOI] [PubMed] [Google Scholar]
- [42].Li M, Shang M, Xu H, Wang X, Dai HX, Yu JQ, Org. Lett 2019, 21, 540–544. [DOI] [PubMed] [Google Scholar]
- [43].Catellani M, Motti E, Della Ca N’, Acc. Chem. Res 2008, 41, 1512–1522. [DOI] [PubMed] [Google Scholar]
- [44].Della Ca N, Fontana M, Motti E, Catellani M, Acc. Chem. Res 2016, 49, 1389–1400. [DOI] [PubMed] [Google Scholar]
- [45].Wang DH, Mei TS, Yu JQ, J. Am. Chem. Soc 2008, 130, 17676–17677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Fang L, Saint-Denis TG, Taylor BLH, Ahlquist S, Hong K, Liu S, Han L, Houk KN, Yu JQ, J. Am. Chem. Soc 2017, 139, 10702–10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Tang RY, Li G, Yu JQ, Nature 2014, 507, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Liu LY, Qiao JX, Yeung KS, Ewing WR, Yu JQ, J. Am. Chem. Soc 2019, 141, 14870–14877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wang P, Verma P, Xia G, Shi J, Qiao JX, Tao S, Cheng PTW, Poss MA, Farmer ME, Yeung KS, et al. , Nature 2017, 551, 489–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Liu LY, Yeung KS, Yu JQ, Chem. Eur. J 2019, 25, 2199–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ye M, Gao GL, Edmunds AJF, Worthington PA, Morris JA, Yu JQ, J. Am. Chem. Soc 2011, 133, 19090–19093. [DOI] [PubMed] [Google Scholar]
- [52].Beck EM, Gaunt MJ, Pd-Catalyzed C-H Bond Functionalization on the Indole and Pyrrole Nucleus., 2010. and references therein for an overview of site-selective pyrrole functionalization strategies [DOI] [PubMed]
- [53].(a) For a review on transition metal-catalyzed indole C2 and C3 functionalization, see; Sandtorv AH, Adv. Synth. Catal 2015, 357, 2403–2435 and references therein. [Google Scholar]; (b) Leitch JA, Bhonoah Y, Frost CG, ACS Catal. 2017, 7, 5618–562 and references therein for an overview of site-selective indole functionalization strategies beyond C2 and C3. [Google Scholar]; (c) For a review on quinoline C–H functionalization at C3, C4 and C8, see:; Iwai T, Sawamura M, ACS Catal. 2015, 5, 5031–5040 and references therein. For representative examples for azine C2–H functionalization methods, see: [Google Scholar]; (d) Nakao Y, Kanyiva KS, Hiyama T, J. Am. Chem. Soc 2008, 130, 2448–2449. [DOI] [PubMed] [Google Scholar]; (e) Berman AM, Lewis JC, Bergman RG, Ellman JA, J. Am. Chem. Soc 2008, 130, 14926–14927. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Tobisu M, Hyodo I, Chatani N, J. Am. Chem. Soc 2009, 131, 12070–12071. [DOI] [PubMed] [Google Scholar]; (g) Berman AM, Bergman RG, Ellman JA, J. Org. Chem 2010, 75, 7863–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Guan B-T, Hou Z, J. Am. Chem. Soc 2011, 133, 18086–18089. [DOI] [PubMed] [Google Scholar]; (i) Ren X, Wen P, Shi X, Wang Y, Li J, Yang S, Yan H, Huang G, Org. Lett 2013, 15, 5194–5197 [DOI] [PubMed] [Google Scholar]
- [54].Nakao Y, Synthesis 2011, 3209–3219 and references therein for an overview of site-selective pyridine functionalization strategies [Google Scholar]
- [55].Anderson EA, Alexanian EJ, Sorensen EJ, Angew. Chem. Int. Ed 2004, 43, 1998–2001. [DOI] [PubMed] [Google Scholar]
- [56].Lu Z, Li Y, Deng J, Li A, Nat. Chem 2013, 5, 679–684. [DOI] [PubMed] [Google Scholar]
- [57].Yang M, Li J, Li A, Nat. Commun 2015, 6, DOI 10.1038/ncomms7445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yang P, Yao M, Li J, Li Y, Li A, Angew. Chem. Int. Ed 2016, 55, 6964–6968. [DOI] [PubMed] [Google Scholar]
- [59].Fürstner A, Domostoj MM, Scheiper B, J. Am. Chem. Soc 2005, 127, 11620–11621. [DOI] [PubMed] [Google Scholar]
- [60](a).Pitts AK, O’Hara F, Snell RH, Gaunt MJ, Angew. Chem. Int. Ed 2015, 54, 5451–5455. [DOI] [PubMed] [Google Scholar]; For other approaches to dictyodendrin B and related congeners, see:; (b) Fürstner A, Domostoj MM, Scheiper B, J. Am. Chem. Soc 2006, 128, 8087–8094. [DOI] [PubMed] [Google Scholar]; (c) Hirao S, Yoshinaga Y, Iwao M, Ishibashi F, Tetrahedron Lett. 2010, 51, 533–536. [Google Scholar]; (d) Okano K, Fujiwara H, Noji T, Fukuyama T, Tokuyama H, Angew. Chem. Int. Ed 2010, 49, 5925–5929. [DOI] [PubMed] [Google Scholar]; (e) Tokuyama H, Okano K, Fujiwara H, Noji T, Fukuyama T, Chem. Asian J 2011, 6, 560–572. [DOI] [PubMed] [Google Scholar]; (f) Liang J, Hu W, Tao P, Jia Y, J. Org. Chem 2013, 78, 5810–5815. [DOI] [PubMed] [Google Scholar]; For an alternative structurally-obvious approach to the dictyodendrins, see; (g) Yamaguchi AD, Chepiga KM, Yamaguchi J, Itami K, Davies HML, J. Am. Chem. Soc 2015, 137, 644–647. [DOI] [PubMed] [Google Scholar]
- [61].Ueda K, Amaike K, Maceiczyk RM, Itami K, Yamaguchi J, J. Am. Chem. Soc 2014, 136, 13226–13232. [DOI] [PubMed] [Google Scholar]
- [62].Peschko C, Winklhofer C, Steglich W, Chem. Eur. J 2000, 6, 1147–1152. [DOI] [PubMed] [Google Scholar]
- [63].Handy ST, Zhang Y, Bregman H, J. Org. Chem 2004, 69, 2362–2366. [DOI] [PubMed] [Google Scholar]
- [64].Pla D, Marchal A, Olsen CA, Albericio F, Álvarez M, J. Org. Chem 2005, 70, 8231–8234. [DOI] [PubMed] [Google Scholar]
- [65].Fujikawa N, Ohta T, Yamaguchi T, Fukuda T, Ishibashi F, Iwao M, Tetrahedron 2006, 62, 594–604. [Google Scholar]
- [66].Yamaguchi AD, Chepiga KM, Yamaguchi J, Itami K, Davies HML, J. Am. Chem. Soc 2015, 137, 644–647. [DOI] [PubMed] [Google Scholar]
- [67].Fox JC, Gilligan RE, Pitts AK, Bennett HR, Gaunt MJ, Chem. Sci 2016, 7, 2706–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Hua DH, Huang X, Chen Y, Battina SK, Tamura M, Noh SK, Koo SI, Namatame I, Tomoda H, Perchellet EM, et al. , J. Org. Chem 2004, 69, 6065–6078. [DOI] [PubMed] [Google Scholar]
- [69].Kurdyumov AV, Hsung RP, J. Am. Chem. Soc 2006, 128, 6272–6273. [DOI] [PubMed] [Google Scholar]
- [70].Jacobson RM, Raths RA, J. Org. Chem 1979, 44, 4013–4014. [Google Scholar]
- [71].O’Malley SJ, Tan KL, Watzke A, Bergman RG, Ellman JA, J. Am. Chem. Soc 2005, 127, 13496–13497. [DOI] [PubMed] [Google Scholar]
- [72].Wang DH, Yu JQ, J. Am. Chem. Soc 2011, 133, 5767–5769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].These two natural products have seen numerous synthetic studies towards generating the biaryl axis atropselectively. For other total syntheses of (−)-steganone and (+)-isoschizandrin not discussed in this review, see:; (a) Tomioka K, Ishiguro T, Iitaka Y, Koga K, Tetrahedron 1984, 40, 1303; [Google Scholar]; (b) Tanaka M, Itoh H, Mitsuhashi H, Maruno M, Wakamatsu T, Tetrahedron: Asymmetry 1993, 4, 605; [Google Scholar]; (c) Joncour A, Décor A, Thoret S, Chiaroni A, Baudoin O, Angew. Chem. Int. Ed 2006, 45, 4149; [DOI] [PubMed] [Google Scholar]; (d) Yalcouye B, Sabine C, Panossian A, Leroux FR, Colobert F, Eur. J. Org. Chem 2014, 6285; [Google Scholar]; (e) Chen W-W, Zhao Q, Xu M-H, Lin G-Q, Org. Lett 2010, 12, 107; [DOI] [PubMed] [Google Scholar]; (f) Meyers AI, Flisak JR, Aitken RA, J. Am. Chem. Soc 1987, 109, 5446–5452; [Google Scholar]; (g) Warshawsky AM, Meyers AI, J. Am. Chem. Soc 1990, 112, 8090. [Google Scholar]
- [74].Robin JP, Gringore O, Brown E, Tetrahedron Lett. 1980, 21, 2709–2712. [Google Scholar]
- [75].Uemura M, Daimon A, Hayashi Y, J. Chem. Soc. Chem. Commun 1995, 1943–1944. [Google Scholar]
- [76](a).Monovich LG, Le Huérou Y, Rönn M, Molander GA, J. Am. Chem. Soc 2000, 122, 52 [Google Scholar]; (b) Molander GA, George KM, Monovich LG, J. Org. Chem 2003, 68, 9533–9540. [DOI] [PubMed] [Google Scholar]
- [77](a).Liao G, Yao QJ, Zhang ZZ, Wu YJ, Huang DY, Shi BF, Angew. Chem. Int. Ed 2018, 57, 3661–3665. [DOI] [PubMed] [Google Scholar]; (b) An alternative strategy towards (+)-steganone was also disclosed using a chiral sulfoxide-directed atropselective C–H olefination, see:; Dherbassy Q, Wencel-Delord J, Colobert F, Tetrahedron 2016, 72, 5238–5245. [Google Scholar]
- [78].Ohmori K, Mori K, Ishikawa Y, Tsuruta H, Kuwahara S, Harada N, Suzuki K, Angew. Chem. Int. Ed 2004, 43, 3167–3171. [DOI] [PubMed] [Google Scholar]
- [79].Mori K, Ohmori K, Suzuki K, Angew. Chem. Int. Ed 2009, 121, 5743–5747. [Google Scholar]
- [80].Fan J, Yao QJ, Liu YH, Liao G, Zhang S, Shi BF, Org. Lett 2019, 21, 3352–3356. [DOI] [PubMed] [Google Scholar]
- [81].He J, Wasa M, Chan KSL, Shao Q, Yu JQ, Chem. Rev 2017, 117, 8754–8786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Giri R, Maugel N, Li JJ, Wang DH, Breazzano SP, Saunders LB, Yu JQ, J. Am. Chem. Soc 2007, 129, 3510–3511. [DOI] [PubMed] [Google Scholar]
- [83].Chen G, Zhuang Z, Li GC, Saint-Denis TG, Hsiao Y, Joe CL, Yu JQ, Angew. Chem. Int. Ed 2017, 56, 1506–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Ghosh KK, van Gemmeren M, Chem. Eur. J 2017, 23, 17697–17700. [DOI] [PubMed] [Google Scholar]
- [85].Shen PX, Hu L, Shao Q, Hong K, Yu JQ, J. Am. Chem. Soc 2018, 140, 6545–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Zhuang Z, Bin Yu C, Chen G, Wu QF, Hsiao Y, Joe CL, Qiao JX, Poss MA, Yu JQ, J. Am. Chem. Soc 2018, 140, 10363–10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].McNally A, Haffemayer B, Collins BSL, Gaunt MJ, Nature 2014, 510, 129–133. [DOI] [PubMed] [Google Scholar]
- [88].Chen K, Wang D, Li ZW, Liu Z, Pan F, Zhang YF, Shi ZJ, Org. Chem. Front 2017, 4, 2097–2101. [Google Scholar]
- [89].Lin H, Pan X, Barsamian AL, Kamenecka TM, Bannister TD, ACS Catal 2019, 9, 4887–4891. [Google Scholar]
- [90].Rodrigalvarez J, Nappi M, Azuma H, Flodén NJ, Burns ME, Gaunt MJ, Nat. Chem 2020, 12, 76–81. [DOI] [PubMed] [Google Scholar]
- [91].Wu Y, Chen YQ, Liu T, Eastgate MD, Yu JQ, J. Am. Chem. Soc 2016, 138, 14554–14557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Zhuang Z, Yu JQ, J. Am. Chem. Soc 2020, 142, 12015–12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Chen YQ, Wang Z, Wu Y, Wisniewski SR, Qiao JX, Ewing WR, Eastgate MD, Yu JQ, J. Am. Chem. Soc 2018, 140, 17884–17894. [DOI] [PubMed] [Google Scholar]
- [94].Xia G, Weng J, Liu L, Verma P, Li Z, Yu JQ, Nat. Chem 2019, 11, 571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Shi BF, Maugel N, Zhang YH, Yu JQ, Angew. Chem. Int. Ed 2008, 47, 4882–4886. [DOI] [PubMed] [Google Scholar]
- [96].Wu QF, Shen PX, He J, Wang XB, Zhang F, Shao Q, Zhu RY, Mapelli C, Qiao JX, Poss MA, et al. , Science 2017, 355, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Wang M, Lu P, Org. Chem. Front 2018, 5, 254–259. [Google Scholar]
- [98].Ichikawa M, Takahashi M, Aoyagi S, Kibayashi C, J. Am. Chem. Soc 2004, 126, 16553–16558. [DOI] [PubMed] [Google Scholar]
- [99].Takahashi M, Ichikawa M, Aoyagi S, Kibayashi C, Tetrahedron Lett. 2005, 46, 57–59. [Google Scholar]
- [100].Frébault F, Maulide N, Angew. Chem. Int. Ed 2012, 51, 2815–2817. [DOI] [PubMed] [Google Scholar]
- [101].Bach RD, Dmitrenko O, J. Am. Chem. Soc 2004, 126, 4444–4452. [DOI] [PubMed] [Google Scholar]
- [102].Frébault F, Luparia M, Oliveira MT, Goddard R, Maulide N, Angew. Chem. Int. Ed 2010, 49, 5672–5676. [DOI] [PubMed] [Google Scholar]
- [103].Gutekunst WR, Baran PS, J. Am. Chem. Soc 2011, 133, 19076–19079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Gutekunst WR, Gianatassio R, Baran PS, Angew. Chem. Int. Ed 2012, 51, 7507–7510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Panish RA, Chintala SR, Fox JM, Angew. Chem. Int. Ed 2016, 55, 4983–4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Chapman LM, Beck JC, Wu L, Reisman SE, J. Am. Chem. Soc 2016, 138, 9803–9806. [DOI] [PubMed] [Google Scholar]
- [107].Nicolaou KC, Totokotsopoulos S, Giguère D, Sun YP, Sarlah D, J. Am. Chem. Soc 2011, 133, 8150–8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Nicolaou KC, Lu M, Totokotsopoulos S, Heretsch P, Giguère D, Sun YP, Sarlah D, Nguyen TH, Wolf IC, Smee DF, et al. , J. Am. Chem. Soc 2012, 134, 17320–17332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Thesmar P, Baudoin O, J. Am. Chem. Soc 2019, 141, 15779–15783. [DOI] [PubMed] [Google Scholar]
- [110].Baudoin O, Acc. Chem. Res 2017, 50, 1114–1123. [DOI] [PubMed] [Google Scholar]
- [111].Sellars JD, Steel PG, Eur. J. Org. Chem 2007, 3815–3828. [Google Scholar]
- [112].Andrews RC, Teague SJ, Meyers AI, J. Am. Chem. Soc 1988, 110, 7854–7858. [Google Scholar]
- [113].Bush EJ, Jones DW, J. Chem. Soc. - Perkin Trans 1 1996, 151–155. [Google Scholar]
- [114].Wu Y, Zhao J, Chen J, Pan C, Li A, Zhang H, Org. Lett 2009, 11, 597–600. [DOI] [PubMed] [Google Scholar]
- [115].Ting CP, Maimone TJ, Angew. Chem. Int. Ed 2014, 53, 3115–3119. [DOI] [PubMed] [Google Scholar]
- [116].Ting CP, Tschanen E, Jang E, Maimone TJ, Tetrahedron 2019, 75, 3299–3308. [Google Scholar]
- [117].Kaufman TS, Rúveda EA, Angew. Chem. Int. Ed 2005, 44, 854–885. [DOI] [PubMed] [Google Scholar]
- [118].Stork G, Niu D, Fujimoto A, Koft ER, Balkovec JM, Tata JR, Dake GR, J. Am. Chem. Soc 2001, 123, 3239–3242. [DOI] [PubMed] [Google Scholar]
- [119].Illa O, Arshad M, Ros A, McGarrigle EM, Aggarwal VK, J. Am. Chem. Soc 2010, 132, 1828–1830. [DOI] [PubMed] [Google Scholar]
- [120].O’ Donovan DH, Aillard P, Berger M, de la Torre A, Petkova D, Knittl-Frank C, Geerdink D, Kaiser M, Maulide N, Angew. Chem. Int. Ed 2018, 57, 10737–10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Engle KM, Wang DH, Yu JQ, J. Am. Chem. Soc 2010, 132, 14137–14151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Engle KM, Yu JQ, J. Org. Chem 2013, 78, 8927–8955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Engle KM, Mei TS, Wasa M, Yu JQ, Acc. Chem. Res 2012, 45, 788–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Li S, Chen G, Feng CG, Gong W, Yu JQ, J. Am. Chem. Soc 2014, 136, 5267–5270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].He J, Li S, Deng Y, Fu H, Laforteza BN, Spangler JE, Homs A, Yu JQ, Science 2014, 343, 1216–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Park H, Li Y, Yu JQ, Angew. Chem. Int. Ed 2019, 58, 11424–11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Shao Q, Wu QF, He J, Yu JQ, J. Am. Chem. Soc 2018, 140, 5322–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Wu QF, Wang XB, Shen PX, Yu JQ, ACS Catal. 2018, 8, 2577–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Zhang Z, Wang J, Li J, Yang F, Liu G, Tang W, He W, Fu JJ, Shen YH, Li A, et al. , J. Am. Chem. Soc 2017, 139, 5558–5567. [DOI] [PubMed] [Google Scholar]