

Abstract
Within pituitary gonadotropes, the gonadotropin-releasing hormone receptor (GnRHR) receives hypothalamic input from GnRH neurons that is critical for reproduction. Previous studies have suggested that androgens may regulate GnRHR, although the mechanisms remain unknown. In this study, we demonstrated that androgens positively regulate Gnrhr mRNA in mice. We then investigated the effects of androgens and androgen receptor (AR) on Gnrhr promoter activity in immortalized mouse LβT2 cells, which represent mature gonadotropes. We found that AR positively regulates the Gnrhr proximal promoter, and that this effect requires a hormone response element (HRE) half site at −159/−153 relative to the transcription start site. We also identified nonconsensus, full-length HREs at −499/−484 and −159/−144, which are both positively regulated by androgens on a heterologous promoter. Furthermore, AR associates with the Gnrhr promoter in ChIP. Altogether, we report that GnRHR is positively regulated by androgens through recruitment of AR to the Gnrhr proximal promoter.
Keywords: Androgen receptor, GnRH receptor, Pituitary, Gonadotrope
1. Introduction
Gonadotropin-releasing hormone (GnRH) acts through the GnRH receptor (GnRHR, encoded by Gnrhr) on pituitary gonadotropes to stimulate synthesis and secretion of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) (Seeburg et al., 1987; Vale et al., 1977). LH and FSH then act on the gonads to regulate a variety of processes, including folliculogenesis, ovulation, spermatogenesis, and steroidogenesis (Thackray et al., 2010). Gonadal steroids in turn feedback on the hypothalamic-pituitary-gonadal (HPG) axis at both the hypothalamus and the pituitary. Because GnRH neurons make up the final common pathway for central control of reproduction, GnRHR is critical to receive hypothalamic input that regulates downstream reproductive function. Due to the indispensable role of this receptor within the HPG axis, a thorough understanding of the mechanisms underlying its expression is key to our understanding of normal reproductive physiology.
Many cis-regulatory elements have previously been identified that are essential for basal and tissue-specific expression of Gnrhr, including an activator protein-1 (AP-1) binding site at −276/−269 relative to the transcriptional start site (TSS) and a steroidogenic factor-1 (SF-1) binding site at −181/−173 (Schang et al., 2012). An additional cis-regulatory element at −329/−318, termed GnRH receptor activating sequence (GRAS) (Duval et al., 1997b), binds to SMAD4, FOXL2, and AP-1 proteins (Ellsworth et al., 2003; Norwitz et al., 2002b). GRAS not only contributes to basal expression of Gnrhr, but is also involved in hormonal regulation of the proximal promoter by both activin and GnRH (Duval et al., 1999). GnRH also activates transcription of Gnrhr at cis elements termed sequence underlying responsiveness to GnRH-1 (SURG), located at −292/−285 (SURG-1), and the AP-1 site located at −276/−269 (SURG-2) (Norwitz et al., 1999). A downstream activin regulatory element (DARE) at −298/−295 binds LIM-homeodomain (LHX) proteins and is required for positive regulation of Gnrhr by activin in αT3-1 cells (Fortin et al., 2015; McGillivray et al., 2005), which represent immature gonadotropes (Alarid et al., 1998; Xie et al., 2015).
In addition to activin and GnRH, sex steroids may contribute to hormonal regulation of Gnrhr transcription. Estradiol has been shown to increase Gnrhr, both in ewes (Hamernik et al., 1995; Turzillo et al., 1995) and in female mice (Naik et al., 1985). Although the manner in which estradiol regulates Gnrhr has not been elucidated, it appears to occur through nonclassical mechanisms in ovine pituitary cells (Davis et al., 2011). Androgens have several known roles in pituitary gonadotropes, where they differentially regulate expression of the gonadotropin subunit genes, increasing expression of Fshb, while repressing Lhb and the common gonadotropin subunit Cga (Gharib et al., 1990; Paul et al., 1990; Thackray et al., 2006). Thus, regulation of GnRHR may be an additional, indirect mechanism through which androgens affect gonadotropin subunit gene expression, by modulating the sensitivity of gonadotropes to GnRH. Furthermore, in mice, it has been reported that removal of testicular androgen production by castration decreases GnRHR number in whole-cell radioligand binding assays, and that this effect is reversed with androgen treatment (Naik et al., 1984a, 1984b). However, it is unclear whether this result represents internalized GnRHR due to rapid GnRH pulse frequency in the castrated state, or transcriptional downregulation of Gnrhr. Supporting the latter possibility, we previously demonstrated that the proximal Gnrhr promoter is positively regulated by the nonaromatizable androgen dihydrotestosterone (DHT) in the LβT2 immortalized cell line, which represents mature pituitary gonadotropes (Spady et al., 2004). However, the molecular mechanisms through which androgens may act to positively regulate Gnrhr have not yet been elucidated.
Androgens classically act by binding AR, a member of the nuclear receptor superfamily, to regulate the expression of target genes. AR shares homology with the progesterone, glucocorticoid, and mineralocorticoid receptors, all of which recognize and bind to hormone response elements (HREs) containing inverted, palindromic repeats of AGAACA separated by a 3-basepair spacer (Roche et al., 1992). Within HREs, the correct positioning of G and C residues is critical for receptor binding (Wilson et al., 2016). Like other steroid receptors, AR contains discrete functional domains including an amino-terminal transactivation domain, a DNA-binding domain (DBD), and a ligand-binding domain (LBD) (Chang et al., 1988; Gaspar et al., 1990). The gene encoding AR (Ar) is located on the X chromosome, is indispensable for male reproduction (Batch et al., 1992; Gehring and Tomkins, 1974a, 1974b; Hiort, 2013; Ohno et al., 1971a, 1971b), and is broadly expressed in many tissues including throughout the HPG axis.
In the present study, we first sought to determine whether androgen status regulates Gnrhr mRNA in vivo, and found that androgens positively regulate Gnrhr mRNA in both male and female mice. We next aimed to characterize the molecular mechanisms through which AR induces GnRHR in vitro using LβT2 cells as a model system. Because LβT2 cells endogenously express Ar, Gnrhr, and the gonadotropin subunits Lhb, Fshb, and Cga, they are an appropriate model system to study the effects of AR on GnRHR expression (Lawson et al., 2001; McGillivray et al., 2007; Spady et al., 2004; Thackray et al., 2006, 2009). We found that positive regulation of the Gnrhr promoter by AR requires an HRE half site in the Gnrhr proximal promoter at −159/−153 relative to the TSS. We further identified two putative, full-length HREs at −499/−484 and −159/−144 that are sufficient for robust AR induction of luciferase on a heterologous promoter. We also found that AR interacts with the −159 locus within the endogenous Gnrhr promoter in chromatin immunoprecipitation (ChIP) assays. Altogether, we showed that Gnrhr is positively regulated by AR in mice, and that this regulation occurs through recruitment of AR to the Gnrhr proximal promoter.
2. Material and methods
2.1. Gonadectomy and DHT pellet implantation
All animal procedures were performed in accordance with the UCSD Institutional Animal Care and Use Committee regulations. Wildtype C57BL/6 mice were obtained from Harlan Laboratories. Mice were group-housed by treatment on a 12-h light, 12-h dark cycle with ad libitum standard chow and water. Gonadectomy and DHT pellet implantation surgeries were performed on 10–12 week old male mice and 6-month old female mice. Pellets were made in-house using Silastic tubing (interior diameter 1.47 mm, exterior diameter 1.96 mm) containing 8 mm DHT (Sigma) and were sealed with silicone sealant. These pellets have previously been shown to produce physiological DHT levels in male mice (2.8 ± 0.7 ng/mL) (Poling et al., 2012). Empty pellets filled with silicone sealant were used as control treatment.
2.2. Tissue collection and quantitative real-time PCR
One week after surgery, mice were euthanized by CO2 inhalation and decapitation. Pituitaries were collected, rapidly frozen on dry ice, and stored at −80 °C. Total RNA from pituitary was isolated using a RNeasy Mini kit (Qiagen). Genomic DNA was removed using an Ambion TURBO DNA-free kit (Thermo Fisher Scientific). Complementary DNA was made by reverse transcription of total RNA using an iScript cDNA synthesis kit (Bio-Rad Laboratories). cDNA products were detected on a Bio-Rad CFX Connect quantitative real-time PCR (qRT-PCR) system (Bio-Rad Laboratories) using SYBR Green Supermix (Bio-Rad Laboratories). A dissociation curve was performed following PCR to ensure the presence of a single product. All data were analyzed by the 2−ΔΔCt method (Livak and Schmittgen, 2001) by normalizing the gene of interest to Ppia or Gapdh as indicated in the figure legends. Data are represented as mean fold change compared to control ± SEM. Primer sequences used for qRT-PCR have previously been reported (Hoffmann et al., 2016; Kauffman et al., 2015).
2.3. Cell lines and reagents
LβT2 cells (Alarid et al., 1996) were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Mediatech Inc., Herndon, VA) supplemented with 10% fetal bovine serum (Gemini Bio, West Sacramento, CA) and 1% penicillin/streptomycin (Life Technologies, Inc./Invitrogen, Grand Island, NY) at 37 °C in humidified 5% CO2 in air as previously described (Cherrington et al., 2008). 1x Trypsin-EDTA (Sigma) was used to dissociate cells. Testosterone (T; 17β-Hydrox-y-3-oxo-4-androstene) and dihydrotestosterone (DHT; 5α-androstan-17β-ol-3-one) were obtained from Sigma-Aldrich (St. Louis, MO). Methyltrienolone (R1881) was purchased from NEN Life Sciences (Boston, MA). T, DHT, and R1881 were dissolved in absolute ethanol. Activin A was obtained from Calbiochem and diluted in 0.1% bovine serum albumin.
2.4. Plasmids and cloning
The following expression plasmids were used: rat AR-pSG5 (rAR) (Ikonen et al., 1998), human AR-pSG5 (hAR) (Ceraline et al., 2004), hAR with a truncated ligand-binding domain (mut-LBD AR) (Ceraline et al., 2004), and rAR with a point mutation in the DNA-binding domain (mut-DBD AR) (Ikonen et al., 1994). A luciferase reporter containing the mouse −600 bp Gnrhr promoter was used (GnRHR-luc), which was created from a 1.1 kb mouse Gnrhr promoter-luciferase plasmid (Albarracin et al., 1994; Spady et al., 2004). Within GnRHR-luc, mutations in two HRE half sites at −499 bp and −159 bp were created using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla). Multimer constructs were created by annealing and inserting oligonucleotides into a thymidine kinase (TK)/pGL3-luciferase backbone. Multimers of the −499/−493 and −159/−153 HRE half sites each contain four repeats of the respective HRE, each separated by a 3-bp spacer. A double multimer was also created containing four repeats each of the −499/−493 and −159/−153 HRE half sites, with each repeat separated by a 3-bp spacer. Multimer constructs were also created containing the −499/−493 or −159/−153 HRE half site with 9 additional nucleotides from the endogenous Gnrhr promoter on the 3′ or 5′ end, repeated four times with a 3-basepair spacer between each repeat. All oligonucleotides were synthesized by Integrative DNA Technologies. Forward (5′ to 3’) oligonucleotide sequences used for cloning are listed in Table 1.
Table 1.
Oligonucleotide sequences used for cloning.
| Construct | Sequence (5′-3′) |
|---|---|
| −499 multimer | AGAACATTTAGAACATTTAGAACATTTAGAACATTTAGAACATTT |
| −159 multimer | TGTTCTAAATGTTCTAAATGTTCTAAATGTTCTAAATGTTCTAAA |
| −159/−499 multimer | TGTTCTAAATGTTCTAAAAGAACATTTAGAACATTTTGTTCTAAATGTTCTAAAAGAACATTTAGAACATTT |
| −508/−493 multimer | ATTGGTATTAGAACAAAAATTGGTATTAGAACAAAAATTGGTATTAGAACAAAAATTGGTATTAGAACA |
| −499/−484 multimer | AGAACAGGCTGCTTAAAAAGAACAGGCTGCTTAAAAAGAACAGGCTGCTTAAAAAGAACAGGCTGCTTA |
| −168/−153 multimer | GGCTTGGCATGTTCTAAAGGCTTGGCATGTTCTAAAGGCTTGGCATGTTCTAAAGGCTTGGCATGTTCT |
| −159/−144 multimer | TGTTCTGTTAGCACTAAATGTTCTGTTAGCACTAAATGTTCTGTTAGCACTAAATGTTCTGTTAGCACT |
2.5. Transient transfections and luciferase assays
Cells were plated in 12-well plates at a density of 4.5 × 105 cells per well and then transfected using PolyJet transfection reagent (SignaGen Laboratories) according to the manufacturer’s instructions. To ensure consistent and adequate expression of AR, AR or the empty vector, pSG5, were transfected in all experiments as indicated. 6 h after transfection, cells were changed to serum-free media containing 0.1% bovine serum albumin (Sigma) and 1% penicillin/streptomycin (Life Technologies, Inc./Invitrogen, Grand Island, NY). Cells were treated 24 h after transfection as indicated below (Spady et al., 2004). For dose response experiments, cells were treated with T, DHT, or R1881 at concentrations ranging from 10−5 M to 10−9 M as indicated. In all subsequent transfection experiments, cells were treated with 10−7 M R1881. For activin treatments, cells were treated with 10 ng/mL activin. Ethanol was used as vehicle control for steroid treatments, and 0.1% BSA was used as vehicle control for activin treatments. Cells were lysed 24 h after treatment and assayed for luciferase and β-galactosidase activity as previously described (Brayman et al., 2012b). In all experiments, luciferase values were normalized to the values from a co-transfected TK-β-galactosidase reporter to control for transfection efficiency, and data represent the mean ± SEM of at least three independent experiments performed in triplicate.
2.6. Electrophoretic mobility shift assays (EMSA)
Full-length rat AR (Ikonen et al., 1998) was made from a pSG5 expression plasmid by in vitro transcription and translation using a transcription/translation (TNT)-coupled reticulocyte lysate system (Promega). Empty pSG5 was transcribed/translated as a control. TNT reaction products were incubated with 2 fmol of 32P-labeled double stranded oligonucleotide at room temperature for 20 min in a DNA-binding buffer [10 mM HEPES (pH 7.8), 50 mM KCl, 5 mM MgCl2, 0.1% Nonidet P-40, 1 mM dithiothreitol, 2 μg polydeoxyinosinic deoxycytidylic acid, and 10% glycerol]. The oligonucleotides were end-labeled with T4 DNA polymerase and [γ-32p]-ATP. After 30 min, the DNA binding reactions were run on a 5% polyacrylamide gel (30:1 acrylamide-bisacrylamide) containing 2.5% glycerol in a 0.5 × Tris-acetate-EDTA buffer. A rabbit polyclonal AR antibody (Santa Cruz C-19X) was used to supershift AR. Mouse IgG was used as a control for nonspecific binding (Santa Cruz) and a 100-fold excess of the relevant oligonucleotide was used for competition. The following oligonucleotide sequences were used for EMSA: HRE consensus, 5′ ACGGGTGGAACGCGGTGTTCTTTTGGCACG 3’; −499/−484 HRE, 5′ ATCTAGAATAATTGGTATTAGAACAGGCTGCTTAAAACAGTTAA 3’; −159/−144 HRE, 5′ GGGCTTGGCATGTTCTGTTAGCACTCTTTTAGATT 3’.
2.7. Analysis of protein binding microarray data for AR
Protein Binding Microarray (PBM) data for AR were obtained from the Universal PBM Resource for Oligonucleotide Binding Evaluation (UniPROBE) database (Hume et al., 2015; Mariani et al., 2017). The C4-type zinc finger DBD of AR was evaluated for its ability to bind all possible 8-mer DNA sequences (Mariani et al., 2017). From this dataset, we extracted the raw fluorescence intensities generated by the AR DBD binding to 1) all 8-mer sequences containing the HRE consensus motif 5′ AGAACA 3’ (n = 48), 2) all 8-mer sequences containing the −490/−484 nonconsensus half site 5′ TGCTTA 3’ (n = 48), 3) all 8-mer sequences containing the −150/−144 nonconsensus half site 5′ AGCACT 3’ (n = 48), and 4) 48 randomly-selected 8-mer sequences. The raw fluorescence intensity for each sequence was normalized to the average intensity of all 48 8-mer sequences containing the HRE consensus motif. Normalized fluorescence values are represented as mean ± SEM for each set of 48 8-mer sequences listed above.
2.8. Chromatin immunoprecipitation and quantitative PCR
LβT2 cells were plated at a density of 7.3 × 105 cells in 10 cm tissue culture dishes in DMEM supplemented with 10% charcoal-stripped FBS (GenminiBio, Cat # 100–119) and 1% penicillin/streptomycin. 24 h after plating, cells were transfected with 2ug rAR/10 cm culture dish using Lipofectamine™ 3000 (ThermoFisher Scientific, cat #L3000001). DNA was mixed with opti-MEM (Gibco, Cat # 31985062), 4 μL P3000 reagent, and 3 μL Lipofectamine 3000 reagent, vortexed briefly, incubated 15 min at room temperature, and dispensed dropwise onto the cells. Approximately 16 h after transfection, cells were treated with 10−7 M R1881 for 24 h. ChIP was performed using the ChIP-IT High Sensitivity kit (Active Motif) according to the manufacturer’s instructions with some minor modifications: Prior to fixation, adherent cells were washed twice with PBS. 10 mL of PBS supplemented with 1 mM MgCl2 was added to the plate before adding 40 μL of ChIP Cross-link Gold (Diagenode) for a 45-min incubation at room temperature. Cells were then washed twice with PBS and fixed for an additional 10 min in PBS with 1% formaldehyde. Cells were lysed by passing through a 25-gauge needle four times. Nuclei were diluted with “ChIP Buffer” to a density of approximately 3 × 106/100 μL and sonicated in 300 μL aliquots in 1.5 mL Bioruptor Pico Microtubes (Diagenode) using the Bioruptor Pico (Diagenode), 30 s on/30 s off for 15 min total. Chromatin was sheared to an average fragment size of 150–1000 bp.
50 μg of chromatin and 10 μg each of either Santa Cruz AR antibody (Santa Cruz C-19X), Active Motif AR antibody (39781), or normal rabbit IgG (Cell Signaling, cat # 2729) were used per immunoprecipitation reaction. Input was collected from the flow-through of the IgG immunoprecipitation. Input and samples were treated with Proteinase K, purified using the QIAquick PCR Purification kit (Qiagen) and eluted in a total volume of 100 μL. Three independent ChIP experiments were performed.
For qPCR analysis, DNA from input and immunoprecipitated samples were measured using iQ SYBR Green Supermix (Bio-Rad Laboratories) in a CFX Connect Detection System (Bio-Rad Laboratories). A standard curve of serial input dilutions was constructed for each plate and used to compute the concentration of each sample as % input. A dissociation curve was performed following PCR to ensure the presence of a single product. Three technical replicates were included per experiment. Primer sequences are as follows: 499 GnRHR HRE Fwd, 5′ TGAATAGTCACACGACGGAC 3’; −499 GnRHR HRE Rev, 5′ TCAGCATGGATAGTCTGATC 3’; −159 GnRHR HRE Fwd, 5′ AGAGTGCTAACAGAACATGCC 3’; −159 GnRHR HRE Rev, 5′ CAAGTGTCCTTCCTCACCTAC 3’; Ch14 desert Fwd, 5′ GTCACAGAAACGCAAAGGTTTA 3’; Ch14 desert Rev, 5′ CCCAAAGTCATGTTGTACTTGATAG 3’.
2.9. Statistical analysis
All data are presented as mean ± SEM. Statistical analyses were performed by unpaired two-tailed Student t-test, one-way ANOVA, two-way ANOVA, or multivariate ANOVA followed by Tukey post hoc analysis, as indicated in the figure legends, with p < 0.05 considered statistically significant.
3. Results
3.1. Androgens positively regulate Gnrhr mRNA in mouse pituitary
We first sought to determine whether androgen status regulates Gnrhr mRNA in mice. It has been reported that castration decreases GnRH binding to whole pituitary cells from male mice (Naik et al., 1984a), but it is unknown whether this effect represents changes in Gnrhr transcription. We found that castration (Cx) significantly decreased expression of pituitary Gnrhr mRNA compared to males who received sham surgery (Fig. 1A, p < 0.01). Next, to determine whether this effect of castration could be attributed to removal of testicular androgens, we compared castrated males (Cx) to males that were castrated and received physiological DHT replacement (Cx + DHT) (Poling et al., 2012). Pituitary Gnrhr was significantly elevated in Cx + DHT males compared to males that were castrated without androgen replacement (Fig. 1B, p < 0.05), confirming a role of androgens in positive regulation of Gnrhr mRNA in vivo. Interestingly, we observed a similar effect in females, with DHT positively regulating pituitary expression of Gnrhr in ovariectomized (Ovx) mice (Fig. 1C, p < 0.05).
Fig. 1.
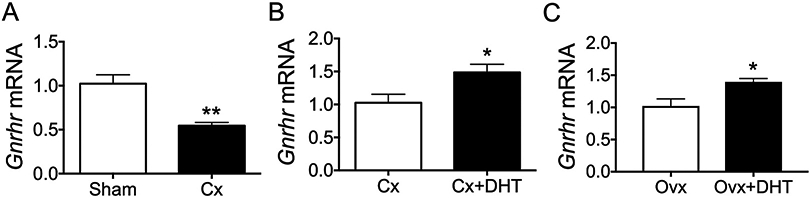
Androgen status regulates Gnrhr mRNA in vivo. (A) Wildtype male mice received sham or castration (Cx) surgery, and pituitaries were collected 1 week later for analysis of Gnrhr mRNA by qRT-PCR. Data are normalized to Gapdh and represented as mean fold change relative to sham ± SEM. n = 6 animals per group. **, p < 0.01 by Student t-test. (B) Wildtype male mice were Cx with or without physiological DHT replacement, and pituitaries were collected 1 week later. Data are normalized to Ppia and represented as fold change relative to Cx ± SEM. n = 5–6 animals per group. *, p < 0.05 by Student t-test. (C) 6-month old female mice were ovariectomized (Ovx) with or without DHT and pituitaries were collected 1 week later for analysis of Gnrhr mRNA by qRT-PCR. Data are normalized to Gapdh and represented as mean fold change relative to Ovx ± SEM. n = 5–6 animals per group. *, p < 0.05 by Student t-test.
3.2. Androgens regulate Gnrhr promoter activity in LβT2 cells
Having found that androgen status positively regulates pituitary expression of Gnrhr in vivo, we next used immortalized LβT2 cells, which represent male mouse pituitary gonadotropes, to investigate the mechanism through which androgens regulate Gnrhr (Alarid et al., 1996, 1998). We utilized a −600 bp Gnrhr promoter-luciferase reporter (GnRHR-luc), which contains all of the cis-regulatory elements known to be required for basal and hormonal regulation of Gnrhr (Duval et al., 1997a, 1999; Ellsworth et al., 2003; McGillivray et al., 2005; Norwitz et al., 1999, 2002a, 2002b). LβT2 cells were transiently transfected with GnRHR-luc and rat AR (rAR), and then treated for 24 h with the synthetic androgen R1881 using doses ranging from 10−5 M to 10−9 M. R1881 significantly induced Gnrhr promoter activity compared to untreated control at concentrations of 10−9 M, 10−8 M, 10−7 M, and 10−5 M (Fig. 2A, p < 0.05), but not at 10−6 M (Fig. 2A, p = 0.1149). Because R1881 is a synthetic androgen, we next sought to determine whether the physiological androgens testosterone (T) and dihydrotestosterone (DHT) could similarly induce GnRHR-luc in vitro. DHT treatment resulted in significant induction of GnRHR-luc at 10−8 M and 10−7 M compared to untreated control (Fig. 2B, p < 0.01), but not at concentrations of 10−9 M, 10−6 M, or 10−5 M. T treatment induced GnRHR-luc activity only at higher concentrations (Figs. 2C and 10−7 M, 10−6 M, and 10−5 M, p < 0.05), likely due to its weaker affinity for AR relative to DHT and R1881 (Saartok et al., 1984; Turcotte et al., 1988). We also treated cells containing no transfected AR (empty vector control) with the highest concentration used of each hormone (10−5 M). We did not observe any effects of androgen treatment on GnRHR-luc without transfected AR, suggesting that AR endogenously expressed in LβT2 cells (Lawson et al., 2001) (Thackray et al., 2006) is not sufficient for regulation of Gnrhr. While T, DHT, and R1881 all significantly induced GnRHR-luciferase, we chose to use 10−7 M R1881 for all subsequent experiments due to its increased metabolic stability (Brayman et al., 2012a; Ripple et al., 1997; Thackray et al., 2006) and consistent ability to positively regulate GnRHR-luc over a wide range of concentrations.
Fig. 2.
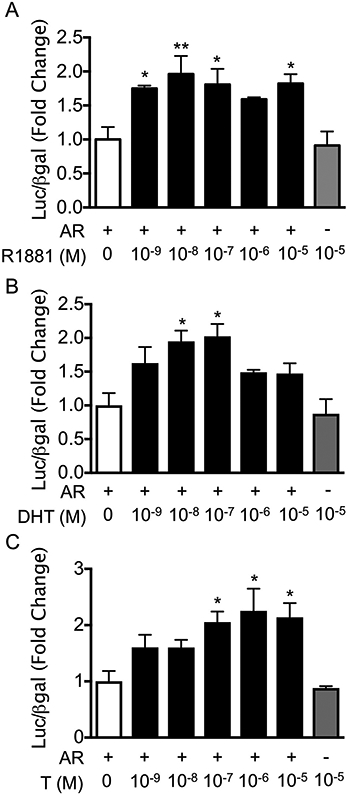
GnRHR-luciferase is positively regulated by androgens. LβT2 cells were transiently transfected with rAR (AR+) or empty vector (AR−) and the mouse −600 bp GnRHR-luciferase reporter. Cells were treated with vehicle control or (A) R1881, (B) DHT, or (C) T at concentrations ranging from 10−9 M to 10−5 M for 24 h before harvest for luciferase and β-galactosidase assays. For each hormone treatment, data are represented as mean fold change relative to AR+/ vehicle ± SEM for three independent experiments performed in triplicate. *, p < 0.05; **, p < 0.01 by one-way ANOVA.
3.3. Both the ligand- and DNA-binding domains of AR are required for regulation of Gnrhr
We next asked whether AR regulation of Gnrhr requires the ligand- and DNA-binding domains of AR. First, we transiently transfected LβT2 cells with GnRHR-luc along with an empty vector, wildtype AR (WT AR), or AR with a deleted ligand-binding domain (mut-LBD AR) (Ceraline et al., 2004) and then treated with R1881 for 24 h. We found that R1881 significantly induced luciferase expression compared to vehicle only in the presence of WT AR (Fig. 3A, p < 0.05), but not in the absence of AR or with mut-LBD AR, indicating that an intact LBD is necessary for AR induction of the Gnrhr promoter. We then transfected LβT2 cells with GnRHR-luc and an empty vector, WT AR, or AR with a point mutation in the DNA-binding domain (mut-DBD AR) (Ikonen et al., 1994), and found that R1881 treatment significantly induced promoter activity in the presence of wildtype AR (Fig. 3B, p < 0.001), but not with empty vector or mut-DBD AR. This demonstrates that androgen induction of GnRHR-luc also requires the DNA-binding domain of AR, suggesting that AR is classically acting as a ligand-inducible transcription factor to regulate expression of Gnrhr.
Fig. 3.
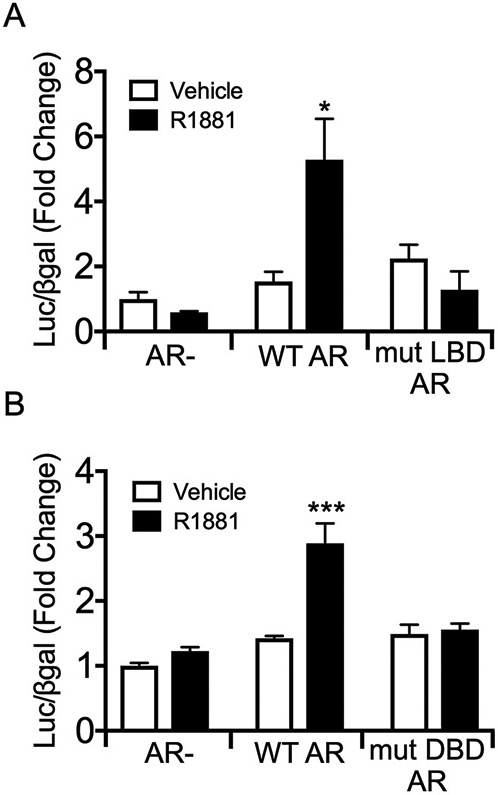
Both the ligand- and DNA-binding domains of AR are required for AR induction of GnRHR-luciferase. LβT2 cells were transiently transfected with the GnRHR-luciferase reporter and (A) empty vector, hAR, or hAR with a mutated ligand-binding domain (mut LBD AR), or (B) empty vector, rAR, or rAR with a point mutation in the DNA-binding domain (mut DBD AR). Cells were treated with vehicle control or 10−7 M R1881 for 24 h before harvest for luciferase and β-galactosidase assays. For each experiment, all data are represented as mean fold change relative to empty vector/vehicle ± SEM for three independent experiments performed in triplicate. *, p < 0.05 by two-way ANOVA.
3.4. HRE half site at −159/−153 is required for AR regulation of Gnrhr
Having found that AR induction of Gnrhr requires the DNA-binding domain of AR, we searched within the −600 bp region of the Gnrhr proximal promoter for putative hormone response elements (HREs). Two consensus HRE half sites located at −499/−493 (5′-AGAACA-3′) and at −159/−153 (5′-TGTTCT-3′) relative to the transcription start site (TSS) were identified (Fig. 4A). We mutated these half sites both individually and together within GnRHR-luc to evaluate whether they are necessary for AR activation of Gnrhr. The G and C residues within these half sites, which are critical for AR binding to HREs, were changed to T residues for the −499/−493 site and to A residues for the −159/−153 site. As expected, wildtype GnRHR-luc was significantly induced by R1881 treatment (Fig. 4B, p < 0.01). With mutation of the −499/−493 consensus HRE half site there was a nearly significant increase in luciferase expression with androgen treatment and AR (Fig. 4B, p = 0.0561), suggesting that this site is likely not required for AR regulation of Gnrhr. Induction of Gnrhr by AR was lost when the −159/−153 consensus HRE half site was mutated, suggesting that this site is necessary for positive regulation of the Gnrhr promoter by androgens (Fig. 4B). Similarly, when both consensus HRE half sites were mutated, luciferase expression was no longer inducible by androgens or AR (Fig. 4B), likely due to mutation of the −159/−153 HRE half site. Together, these results demonstrate that the −159/−153 consensus HRE half site, but likely not the −499/−493 consensus HRE half site, is necessary for AR regulation of Gnrhr.
Fig. 4.
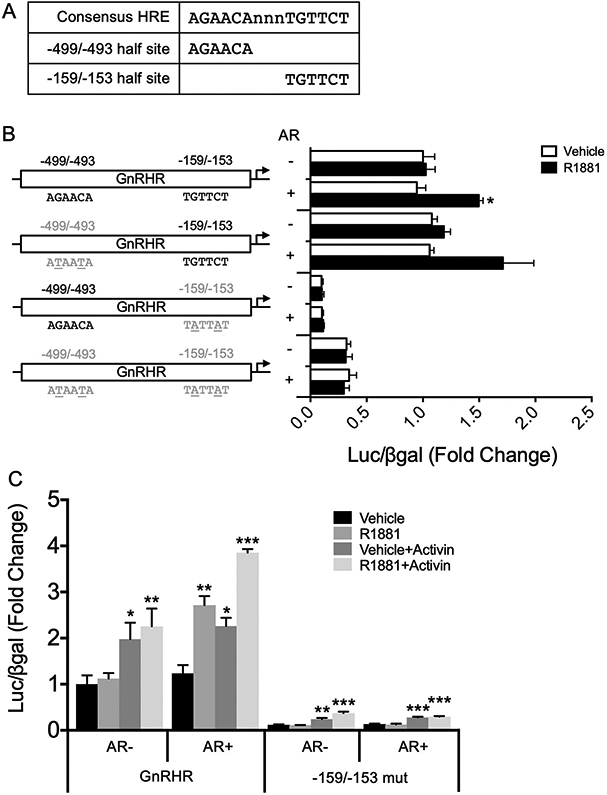
An HRE half site at −159/−153, but not −499/−493, is required for AR induction of GnRHR-luciferase. (A) Consensus HRE half sites identified at −499/−493 and −159/−153 within the mouse Gnrhr promoter, compared to a full-length consensus HRE. (B) Within the mouse −600 bp Gnrhr promoter driving luciferase, HRE half sites at −159/−153 and −499/−493 were mutated such that critical G and C residues were mutated to A or T residues. Mutated half sites are indicated by gray text, and the specific nucleotides mutated are underlined. LβT2 cells were transiently transfected with wildtype or mutant GnRHR-luciferase reporter constructs with empty vector or rAR. Cells were treated with vehicle control or 10−7 M R1881 for 24 h before harvest for luciferase and β-galactosidase assays. Data are represented as mean fold change relative to empty vector/vehicle/GnRHR-luc ± SEM for three independent experiments performed in triplicate. For each reporter, data were analyzed by two-way ANOVA with Tukey post hoc analysis. Stars indicate significant results compared to empty vector/vehicle for each reporter. *, p < 0.05. (C) LβT2 cells were transiently transfected with wildtype or −159 mut GnRHR-luciferase with empty vector or rAR. Cells were treated with vehicle or 10−7 M R1881, with or without 10 ng/mL activin, for 24 h before harvest for luciferase and β-galactosidase assays. Data are represented as mean fold change relative to empty vector/vehicle/GnRHR-luc ± SEM for three independent experiments performed in triplicate. For each reporter, data were analyzed by multivariate ANOVA with Tukey post hoc analysis. Stars indicate significant results compared to empty vector/vehicle for each reporter. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Interestingly, luciferase expression was markedly reduced with mutation of the −159/−153 consensus HRE half site (Fig. 4B, p < 0.001 by multivariate ANOVA), even under basal conditions (empty vector/vehicle treatment), suggesting that this site may be important for basal regulation of Gnrhr. To determine whether mutation of this consensus HRE half site affects expression of GnRHR-luc in a different context, we next asked whether the −159/−153 mutant construct was inducible by activin, a known positive regulator of Gnrhr (Duval et al., 1999; McGillivray et al., 2005). LβT2 cells were transiently transfected with the WT or −159/−153 mutant GnRHR-luc along with empty vector or AR, and then treated with vehicle or R1881, with or without activin. We found that both the WT and −159/−153 mutant GnRHR-luc constructs were induced approximately two-fold with activin treatment (Fig. 4C, p < 0.05), regardless of the presence of AR or androgen treatment, although basal expression of luciferase remained decreased with mutation of the −159/−153 consensus HRE half site.
3.5. HRE half sites at −159/−153 and −499/−493 are induced by AR
Having shown that the −159/−153 HRE half site is required for AR regulation of GnRHR-luciferase, we next sought to determine whether the −159/−153 or −499/−493 consensus HRE half sites, alone or in combination, are sufficient for AR induction of luciferase. LβT2 cells were transfected with a thymidine kinase (TK)-luciferase reporter containing multimer repeats of the −499/−493 consensus HRE half site (−499 multimer), the −159/−153 consensus HRE half site (−159 multimer), or both together (−159/−499 multimer), along with wildtype AR or empty expression vector. Despite significantly repressing basal TK-luciferase activity (Fig. 5, p < 0.001 by multivariate ANOVA), both the −499 multimer and the −159 multimer were induced by androgen treatment in the presence of AR (Fig. 5B, p < 0.01 and Fig. 5C, p < 0.001, respectively). The −159/−499 multimer exhibited basal expression similar to TK-luciferase (Fig. 5D, p > 0.54 by multivariate ANOVA). However, unlike TK-luciferase, the −159/−499 multimer was strongly activated by androgen treatment and AR (Fig. 5D, p < 0.01), suggesting that AR may act at these consensus HRE half sites to regulate Gnrhr.
Fig. 5.
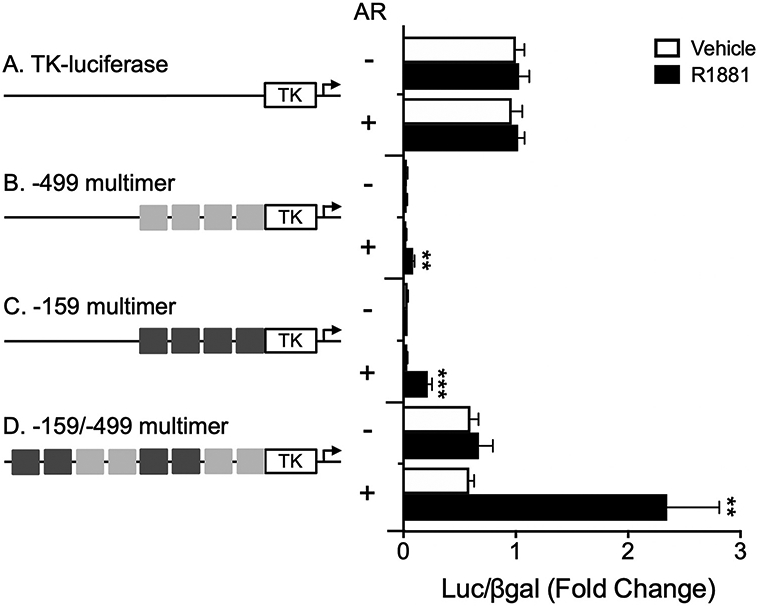
HRE half sites at −499/−493 and −159/−153 are sufficient for androgen induction of luciferase. LβT2 cells were transiently transfected with empty TK-luciferase reporter or TK-luciferase containing 6 base-pair long multimer repeats of the −499/−493 consensus HRE half site (−499 multimer), the −159/−153 consensus HRE half site (−159 multimer), or both consensus HRE half sites (−499/−159 multimer), with empty vector or rAR. Cells were treated with vehicle or 10−7 M R1881 for 24 h before harvest for luciferase and β-galactosidase assays. For each experiment, all data are represented as mean fold change relative to empty vector/vehicle/TK-luciferase ± SEM for three independent experiments performed in triplicate. For each reporter, data were analyzed by two-way ANOVA with Tukey post hoc analysis. Stars indicate significant results compared to empty vector/vehicle for each reporter construct. **, p < 0.01; ***, p < 0.001.
3.6. Nonconsensus full-length HREs at −159/−144 bp and −499/−484 bp are sufficient for robust AR induction of luciferase
Because AR typically acts as a homodimer, we next interrogated whether additional sequence flanking the consensus HRE half sites at −499/−493 and −159/−153 might constitute nonconsensus full-length HREs. To find putative full-length HREs, we allowed for a 3 bp spacer on either side of the −499/−493 and −159/−153 HRE half sites and then looked for a 6-mer sequence containing G and C residues positioned properly for steroid receptor binding (Roche et al., 1992; Wilson et al., 2016). We identified putative full-length HREs consisting of the −499/−493 HRE half site with additional sequence 5’ (−508/−493) or 3’ (−499/−484), each with one G residue in the correct position compared to the HRE consensus sequence (Fig. 6A). We also identified putative full-length HREs consisting of the −159/−153 HRE half site with additional sequence 5’ (−168/−153) having a G in the correct position and 3’ (−159/−144) having both a G and a C residue in the consensus positions (Fig. 6A). LβT2 cells were transfected with TK-luciferase reporters containing multimer repeats of these four putative nonconsensus HREs, individually, along with an empty expression vector or wildtype AR. We found that the putative HREs at −508/−493 bp and −168/−153 bp did not respond to androgen treatment (Fig. 6C, E). Conversely, the nonconsensus HREs at −499/−484 and −159/−144 were sufficient for robust luciferase activity in the presence of androgen treatment and AR (Fig. 6D, F, p < 0.01 for each reporter construct), suggesting that AR may bind to these HREs as a homodimer to positively regulate the GnRHR promoter.
Fig. 6.
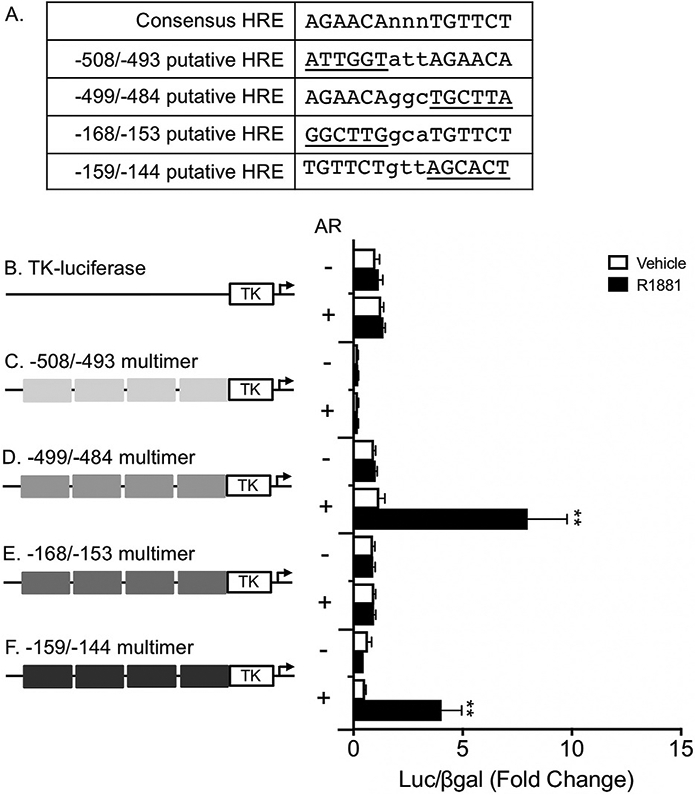
Full-length, nonconsensus HREs at −499/−484 and −159/−144 are sufficient for robust androgen induction of luciferase. (A) Putative full-length HREs compared to a consensus HRE sequence. Nonconsensus 6mer sequences contained within each putative full-length HRE are underlined and 3 bp spacers between half sites are shown in lowercase. (B–F) LβT2 cells were transiently transfected with a TK-luciferase reporter containing 15 base-pair long multimer repeats of putative HREs located at −508/−493 bp, −499/−484 bp, −168/−153 bp, or −159/−144 bp relative to the Gnrhr TSS. Cells were cotransfected with empty vector or rAR and treated with vehicle or 10−7 M R1881 for 24 h before harvest for luciferase and β-galactosidase assays. For each experiment, all data are represented as mean fold change relative to empty vector/vehicle/TK-luciferase ± SEM for three independent experiments performed in triplicate. For each reporter, data were analyzed by two-way ANOVA with Tukey post hoc analysis. Stars indicate significant results compared to empty vector/vehicle for each reporter construct. **, p < 0.01.
3.7. AR binding to nonconsensus HRE motifs is not detected in electrophoretic mobility shift or protein binding microarray assays
To test the ability of AR to bind nonconsensus HREs within the Gnrhr promoter, we first performed electrophoretic mobility shift assays (EMSA). In contrast to AR binding to the consensus HRE sequence, we did not observe AR binding to either the −499/−484 HRE or the −159/−144 HRE (Fig. 7). Furthermore, these nonconsensus HREs were unable to outcompete AR bound to the consensus HRE sequence (Fig. 7). Because the −499/−484 HRE and −159/−144 HRE each contain a nonconsensus half site (located at −490/−484 and −150/−144, respectively), and EMSA is most likely to capture high affinity protein-DNA binding, we next asked whether these sites may have decreased affinity for AR compared to the consensus HRE.
Fig. 7.
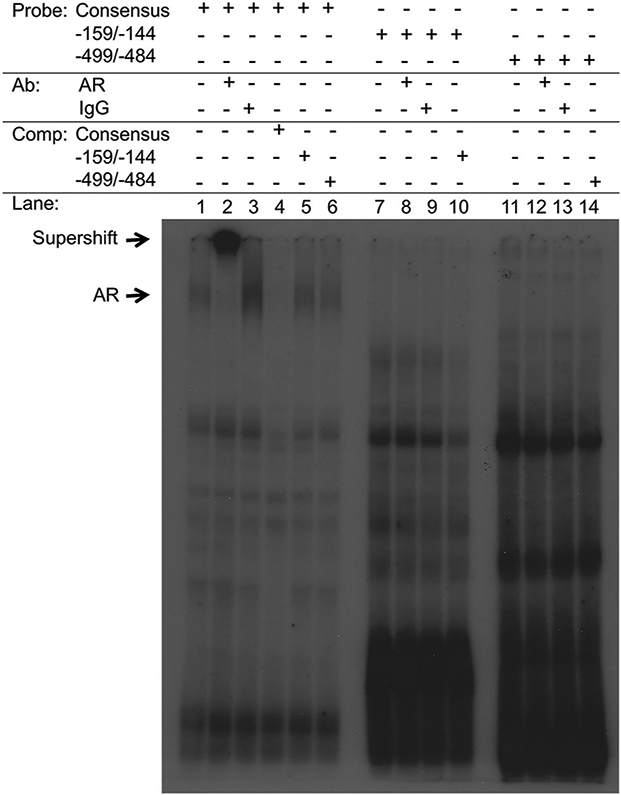
AR does not bind directly to HREs at −159/−144 or −499/−484 in electrophoretic mobility shift assays. Rat AR was in vitro transcribed and translated from a pSG5 expression vector, and incubated with the consensus HRE sequence, the −159/−144 HRE, or the −499/−484 HRE and tested for protein-DNA complex formation by EMSA. rAR bound the consensus HRE sequence (lane 1) and was supershifted with an AR antibody (lane 2) but not with control IgG (lane 3). rAR binding was not observed for either the −159/−144 HRE or the −499/−484 HRE. “Ab” indicates antibody and “comp” indicates cold competitor at 100-fold excess.
We obtained protein binding microarray (PBM) data for AR from the Universal PBM Resource for Oligonucleotide Binding Evaluation (UniPROBE) database (Hume et al., 2015; Mariani et al., 2017). The DBD of AR was tested for its ability to bind all possible 8mer DNA sequences. Compared to 8mer sequences containing the consensus HRE half site (5′ AGAACA 3′), binding of AR DBD to 8mer sequences containing the −490/−484 nonconsensus half site (5′ TGCTTA 3′) or the −150/−144 nonconsensus half site (5′ AGCACT 3’) was significantly reduced (Fig. 8, p < 0.0001 by one-way ANOVA). Furthermore, binding of AR DBD to 8mer sequences containing the −490/−484 or −150/−144 nonconsensus half site was similar to its binding of random 8mers (Fig. 8). Indeed, enrichment scores for AR DBD binding 8mer sequences containing the −490/−484 or −150/−144 nonconsensus half site largely fell within the range of background noise reported for PBM assays (Berger and Bulyk, 2009; Mariani et al., 2017). Thus, while AR has significantly decreased affinity for these nonconsensus sequences compared to the consensus HRE, PBM lacks the sensitivity to distinguish specific, low-affinity binding from nonspecific signal. Altogether, while AR induction of GnRHR-luciferase requires an intact DBD (Fig. 3B), EMSA and PBM were unable to detect direct or specific binding to nonconsensus HRE motifs within the GnRHR promoter, which may be due to decreased affinity of AR for these motifs compared to the HRE consensus sequence.
Fig. 8.
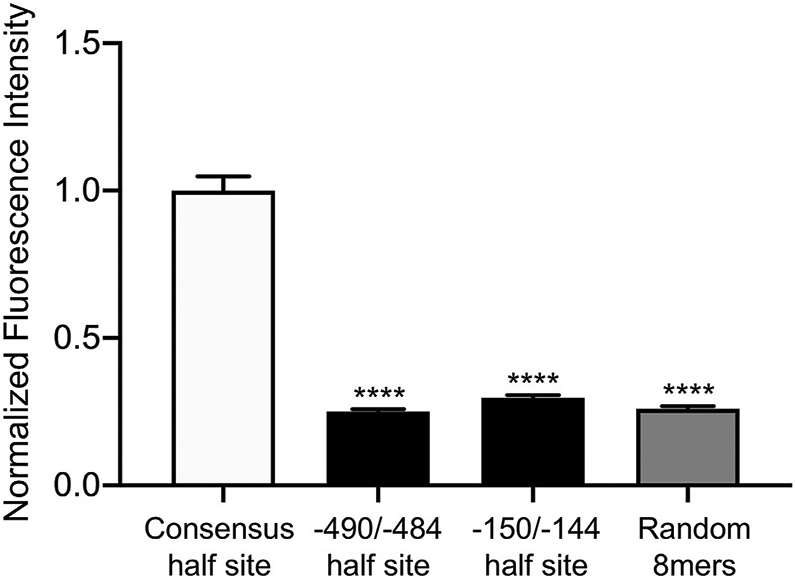
Nonconsensus half sites within the −499/−484 and −159/−144 HREs have decreased affinity for AR compared to the consensus half site. We obtained in vitro binding data for AR from the UniPROBE database (Hume et al., 2015; Mariani et al., 2017). The DBD of AR was tested for its ability to bind every possible 8mer DNA sequence. From this dataset, we extracted the raw fluorescence intensities generated by the AR DBD binding to all 8-mer sequences containing, from left to right, the HRE consensus motif 5′ AGAACA 3′, the −490/−484 nonconsensus half site 5′ TGCTTA 3′, the −150/−144 nonconsensus half site 5′ AGCACT 3′, and randomly-selected 8-mer sequences (n = 48 sequences per category). The raw fluorescence intensity for each sequence was normalized to the average intensity of all 48 sequences containing the HRE consensus motif. Normalized fluorescence values are represented as mean ± SEM for each category of 8-mer sequences listed above. Data were analyzed by one-way ANOVA with Tukey post hoc analysis. Stars indicate significant results compared to consensus half site. ****, p < 0.0001.
3.8. AR is recruited to the −159 HRE within the endogenous Gnrhr promoter in LβT2 cells
While AR binding to nonconsensus HRE motifs was not detectable in EMSA and PBM assays, we reasoned that the endogenous Gnrhr promoter context in gonadotropes may facilitate AR binding. For example, if AR has low affinity for nonconsensus HREs within the Gnrhr promoter, as suggested by PBM data, transcriptional cofactors expressed in gonadotropes may stabilize its interaction with these sites. We utilized ChIP to ask whether AR interacts with the endogenous Gnrhr promoter in LβT2 cells. Cells were transfected with AR and treated with R1881 prior to chromatin extraction. Chromatin was immunoprecipitated using a Santa Cruz AR antibody (SC AR), an Active Motif AR antibody (AM AR), or IgG control. Enrichment of AR was measured by qPCR as percent input using primers specifically targeting the −499 Gnrhr HRE locus, the −159 Gnrhr HRE locus, or a gene desert located on mouse chromosome 14 (negative control). As expected, AR enrichment was not detected at the chromosome 14 gene desert with either AR antibody (Fig. 9, p > 0.99), and background IgG signal was similar at each locus tested (p > 0.89). At the −499 Gnrhr locus, we observed a slight but statistically insignificant enrichment with SC AR (Fig. 9, p = 0.306) and no enrichment with AM AR (p > 0.99). In contrast, we found significant enrichment of AR at the −159 Gnrhr locus with both SC AR (Fig. 9, p < 0.0001) and AM AR (p = 0.037), indicating that AR is recruited to this region of the endogenous Gnrhr promoter in LβT2 cells.
Fig. 9.
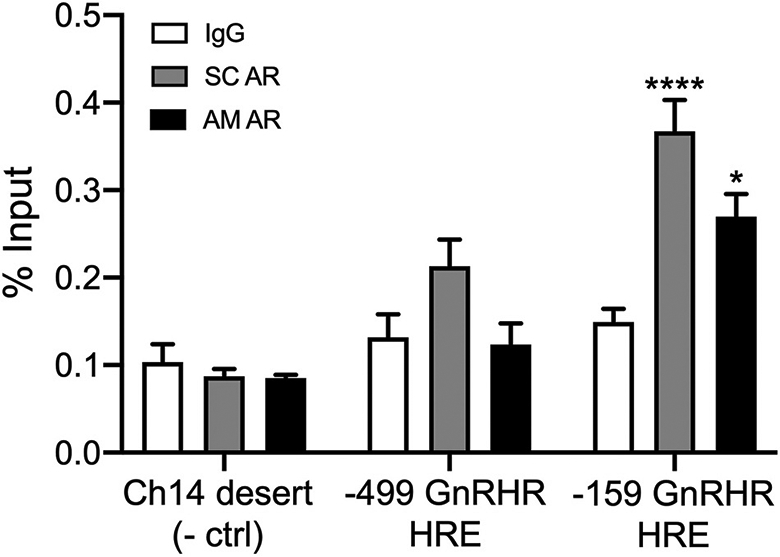
AR is recruited to the −159 HRE locus within the endogenous Gnrhr promoter in LβT2 cells. LβT2 cells were transiently transfected with rAR and then treated with 10−7 M R1881 for 24 h. Chromatin was collected and immunoprecipitated using Santa Cruz AR antibody (SC AR), Active Motif AR antibody (AM AR), or normal rabbit IgG (negative control). qPCR was performed using primers specifically targeting the −499 HRE or −159 HRE within the Gnrhr proximal promoter or a gene desert within mouse chromosome 14 (negative control). Data are represented as mean percent input ± SEM for three independent experiments performed in triplicate. Data were analyzed by two-way ANOVA with Tukey post hoc analysis. Stars indicate significant enrichment compared to IgG for each locus tested. *, p < 0.05; ****, p < 0.0001.
4. Discussion
In the present study, we sought to determine whether androgen and AR regulate pituitary expression of Gnrhr, both in vivo and in vitro. Using the LβT2 line of immortalized gonadotropes, we identified two nonconsensus, androgen-responsive HREs at −499/−484 and −159/−144 relative to the Gnrhr transcription start site. While both HREs are sufficient for robust androgen induction of luciferase on a heterologous promoter, only the −159 locus appears to be required for AR regulation of Gnrhr or recruit AR (discussed more below). These nonconsensus HREs consist of consensus half sites at −499/−493 and −159/−153, with additional 3′ sequence. While it has been suggested that AR may bind to consensus HRE half sites to direct the expression of target genes (Massie et al., 2007), our study and others demonstrate that AR can utilize full-length, imperfect HREs (Gnanapragasam et al., 2000; Riegman et al., 1991; Takayama et al., 2007). The −159/−144 HRE contains a nonconsensus half site (5′ AGCACT 3′) that has previously been identified within the prostate-specific antigen (PSA) androgen response element (ARE) (Gnanapragasam et al., 2000; Riegman et al., 1991), although it is typically 5′ rather than 3′ relative to a consensus half site, and thus has a novel orientation within the Gnrhr proximal promoter. While the PSA ARE has been described as a high affinity but nonspecific site that can bind other steroid receptors (Gnanapragasam et al., 2000), PBM data suggest that AR binds the 5′ AGCACT 3′ motif with low affinity compared to the consensus HRE half site. Nevertheless, although AR was recruited to the −159 locus within the endogenous Gnrhr promoter, and the −159/−144 HRE was strongly inducible only in the presence of androgen treatment and AR, we have not ruled out whether this nonconsensus HRE may also mediate the effects of other steroid receptors. To our knowledge, the nonconsensus HRE half site (5′-TGCTTA-3′) within the −499/−484 HRE has not previously been described. Interestingly, the putative −508/−493 HRE construct we created was not inducible by AR, even though it differs by just one base-pair from an ARE that has previously been described to regulate the probasin gene (Rennie et al., 1993).
While both the −499/−493 and −159/−153 HRE half sites, and the full-length HREs containing them, were sufficient for AR induction of luciferase in our in vitro assays, only the −159/−153 half site appeared to be required for AR induction of GnRHR-luc. Furthermore, we observed significantly reduced basal expression of GnRHR-luc when the critical G and C residues in this half site were mutated to A, suggesting that this site may contribute to basal regulation of the Gnrhr promoter. While the −159/−153 HRE half site does not overlap with sequences that are known to be important for basal regulation of Gnrhr, there is a nearby SF-1 site at −181/−173 (Schang et al., 2012). Interactions between AR and SF-1 have previously been reported to occur at the Lhb and Amh proximal promoters; however, adjacent SF-1 and AR binding sites were not identified in these studies, perhaps due to a focus on high-affinity binding sites (Edelsztein et al., 2018; Jorgensen and Nilson, 2001). Given the proximity of an SF-1 site to the −159/−144 HRE, possible interactions between SF-1 and AR at the GnRHR promoter would be an interesting area of future investigation. Besides SF-1, other transcription factors known to regulate the Gnrhr promoter bind >100 basepairs away from either the −159/−144 or −499/−484 HRE and are thus unlikely to interact directly with AR. Indeed, many of these sites are responsive to activin and we did not observe any interaction between activin treatment and AR on GnRHR-luc. Another explanation for decreased basal activity of the mutant −159/−153 Gnrhr construct is that we may have introduced a cryptic repressive element into the promoter by mutating this site. This could be further investigated by randomizing the sequence at −159/−153 within the Gnrhr proximal promoter and then screening variants for basal and androgen-induced luciferase activity.
We observed that androgen regulation of GnRHR-luc requires both the LBD and the DBD of AR to be intact, suggesting that AR acts in a classical, genomic manner to regulate Gnrhr. Interestingly, while deletion of the LBD confers constitutive activity to AR on the mouse mammary tumor virus (MMTV) promoter (data not shown), we did not find this mutant AR to be constitutively active on the Gnrhr promoter, likely due to differences in promoter context. Despite the requirement of an intact DBD for AR regulation of Gnrhr, EMSA did not show evidence that AR directly binds to either the −499/−484 or the −159/−144 HRE in vitro. Because these sites represent nonconsensus HREs, and EMSA is most likely to capture high affinity protein-DNA binding, we predicted that the AR DBD may weakly interact with these sites. This idea was supported by PBM data for AR (Mariani et al., 2017), which showed that binding of AR DBD to the −490/−484 and −159/−144 nonconsensus half sites is greatly reduced compared to the consensus HRE half site. Because the PBM utilized 8mer sequence motifs, we were not able to score the relative affinities of full-length −499/−484, −159/−144, and consensus HRE sequences. Furthermore, at affinities as low as those measured for the −490/−484 and −150/−144 nonconsensus half sites, PBM loses sensitivity to distinguish specific, low-affinity binding from nonspecific signal (Berger and Bulyk, 2009).
To address the question of whether AR may interact with the −499/−484 or the −159/−144 GnRHR HRE in the endogenous cellular context, we performed ChIP assays and found significant enrichment of AR at the −159 locus, indicating that AR acts at this region to regulate Gnrhr. Although ChIP lacks the resolution to determine whether AR binds precisely to the −159/−144 HRE, the requirement of the −159/−153 HRE half site and the AR DBD for induction of GnRHR-luc, together with AR recruitment to the −159 locus, suggest that AR may act directly at this site to regulate Gnrhr. However, we note that ChIP cannot distinguish between indirect versus direct protein-DNA interactions. Furthermore, the DBD of AR is required in some contexts where AR regulation of target genes is thought to be indirect (Heckert et al., 1997; Jorgensen and Nilson, 2001), although low-affinity binding sites, which can confer increased specificity for transcription factor binding (Kribelbauer et al., 2019), were ignored in these studies. Indeed, the present study highlights known challenges in identifying and characterizing functional low-affinity and nonconsensus binding sites (Jindal and Farley, 2021; Kribelbauer et al., 2019). Our ability to detect AR interacting with the endogenous Gnrhr promoter, but not oligonucleotide sequences, suggests that transcriptional cofactors expressed in LβT2 cells may be required for AR interaction with the −159 locus.
In contrast to the −159 locus, we did not observe significant recruitment of AR to the −499 region of the endogenous Gnrhr promoter. Given the resolution of ChIP, the slight but insignificant enrichment of AR we observed with the SC AR antibody at −499 is likely due to signal from the −159 site. A lack of AR recruitment to the −499 region of the Gnrhr promoter is consistent our finding that the −499/−493 HRE half site is likely not required for AR induction of GnRHR-luciferase. Although the full-length nonconsensus −499/−484 HRE responded robustly to AR and androgen treatment on a heterologous promoter, this may be irrelevant within the context of the Gnrhr promoter. It is also possible that the −499/−484 GnRHR HRE may respond to indirect or nongenomic actions of AR, or bind other sex steroid receptors. The physiological significance of both HREs, and especially the −159/−144 HRE, could be addressed in future studies by creating mice in which critical nucleotides within these sites are mutated. The physiological roles of nonconsensus HREs are of particular interest given many reports that low-affinity transcription factor binding sites can be required for cis-regulatory element specificity and function (Crocker et al., 2015; Farley et al., 2015, 2016; Hosokawa et al., 2018; Ramos and Barolo, 2013; Rowan et al., 2010; Zandvakili et al., 2018).
Several lines of evidence have pointed toward in vivo roles of AR in regulating GnRHR. Data from radioligand binding assays demonstrated that removal of androgens by castration decreases GnRH binding to pituitary cell membranes, and that this effect could be reversed with androgen replacement. However, it remained unclear whether decreased GnRHR in these assays represented internalized GnRHR due to rapid GnRH stimulation following castration, or transcriptional downregulation of Gnrhr mRNA. Confirming the latter scenario, we found that castration decreases pituitary expression of Gnrhr mRNA, while DHT treatment increases Gnrhr expression in both male and female gonadectomized mice.
Interestingly, the effects of castration on regulation of GnRHR appear to significantly differ among species. Castration increases GnRHR in rabbits (Limonta et al., 1986), perhaps to accommodate for increased GnRH stimulation with loss of androgen negative feedback. Similarly, castration increases GnRHR in rats (Conne et al., 1982; Duncan et al., 1983; Frager et al., 1981; Naess et al., 1981) both at the levels of mRNA and membrane-bound receptor. However, one study reported that in long-term castrated rats, androgen treatment can increase GnRHR content and enhance the effect of GnRH on serum LH levels, suggesting that the timing of androgen treatment in castrated rats may modulate GnRHR expression (Wilson et al., 1986). In golden hamsters, GnRHR is increased by castration, but pituitary volume increases such that there is no effect of gonadectomy on receptor concentration (Pieper, 1984). At present, it is unclear why these species differences might exist. Since GnRH is increased in all of these species following gonadectomy, the effects of castration on GnRHR may be independent of GnRH. Conversely, because GnRH potently upregulates its own receptor, it is possible that any effects of androgens on GnRHR in rabbits, rats, or hamsters are masked by increased GnRH following castration, which does not represent normal physiology.
It was previously reported that deletion of AR from whole pituitary results in decreased expression of both Gnrhr and Fshb in male mice (O’Hara et al., 2015). Although the Cre allele used in that study is known to be ectopically expressed in many tissues (Hebert and McConnell, 2000; O’Hara et al., 2011), our in vitro data support AR acting directly in pituitary gonadotropes to regulate Gnrhr. While loss of AR from the male pituitary did not significantly impact circulating gonadotropins, pituitary AR may be important for the preovulatory LH surge in female mice. Indeed, it has previously been reported that specific deletion of AR from pituitary gonadotropes results in decreased amplitude of the LH surge on the evening of proestrus (Wu et al., 2014). This impaired LH surge may be due to decreased pituitary sensitivity to GnRH. In the present study, we found that DHT significantly increased Gnrhr mRNA in ovariectomized females, indicating that androgens upregulate Gnrhr in both male and female mice. While females lacking pituitary AR had normal basal expression of Gnrhr compared to controls, Gnrhr was decreased with ovariectomy both in WT females and in females with gonadotrope-specific deletion of AR (Wu et al., 2014). This effect was not rescued by treatment with estradiol (Wu et al., 2014), raising the possibility that another gonadal factor, perhaps androgen, plays a role in regulation of Gnrhr. Although the cycle stage of these animals was not reported, androgen levels are highest at proestrus (Belanger et al., 1981), and thus if androgens have a normal physiological role in regulation of Gnrhr in intact female mice, we would expect to see positive regulation of this transcript at proestrus. In line with this idea, LH surge amplitude is lower in ovariectomized females with estradiol replacement (Ovx + E2) compared to intact proestrus females (Wagenmaker and Moenter, 2017), despite similar increases in firing of kisspeptin neurons in the anteroventral periventricular nucleus (Wang et al., 2016), suggesting that pituitary sensitivity to GnRH may be impaired in the Ovx + E2 model. The mechanism through which pituitary AR contributes to the LH surge warrants further investigation, and may involve positive regulation of Gnrhr to increase pituitary sensitivity to GnRH.
Altogether, we have found that AR positively regulates pituitary expression of Gnrhr mRNA in both male and female mice. This regulation appears to occur through recruitment of AR to the Gnrhr proximal promoter, where AR may act at a nonconsensus HRE we identified at −159/−144 bp relative to the transcription start site. Thus, regulation of Gnrhr by AR may provide an additional mechanism of control over the expression and secretion of gonadotropins, which would then contribute to the overall function of the HPG axis.
Acknowledgements
We thank Erica L. Schoeller, Jason D. Meadows, Shaddy Malik, and Jesse Solvason for technical assistance and Hanne M. Hoffmann for helpful discussions.
This work was supported by National Institutes of Health (NIH) Grants R01 HD072754, R01 HD082567, and R01 HD100580 (to P.L.M.). It was also supported by NICHD/NIH P50 HD012303 as part of the National Centers for Translational Research in Reproduction and Infertility (P.L.M. and V.G.T.). P.L.M. was also partially supported by P30 DK063491, P30 CA023100, and P42 ES010337. G.E.R. and E.A.W. were partially supported by P42 ES010337 and T32 GM008666. SCB. was partially supported by NIH F31 HD096838 and NIH T32 NS061847. JC. was partially supported by NIH T32 HD007203 and K12 GM026584. E. V.H. was partially supported by T32 GM008666 and T32 GM007198. E. A.W. was also partially supported by T32 DA007315.
The sponsors were not involved in the study design, the collection, analysis and interpretation of data, the writing of the report and the decision to submit the article for publication.
Abbreviations
- AP-1
activator protein-1 androgen receptor
- ARE
androgen response element
- Cx
castration
- ChIP
chromatin immunoprecipitation
- DARE
downstream activin regulatory element
- DBD
DNA-binding domain
- DHT
dihydrotestosterone
- LBD
ligand-binding domain
- FSH
follicle-stimulating hormone
- GnRHR
gonadotropin-releasing hormone receptor
- GRAS
GnRH receptor activating sequence
- HPG
hypothalamic-pituitary-gonadal
- HRE
hormone response element
- LHX
LIM-homeodomain proteins
- LH
luteinizing hormone
- Luc
luciferase reporter
- Ovx
ovariectomized
- MMTV
mouse mammary tumor virus
- PSA
prostate-specific antigen
- PBM
Protein Binding Microarray
- SF-1
steroidogenic factor-1
- SURG
sequence underlying responsiveness to GnRH
- T
Testosterone
- TK
thymidine kinase
- TSS
transcriptional start site (TSS)
- TNT
transcription/translation
Footnotes
Declaration of competing interest
The authors declare no conflict of interest.
References
- Alarid ET, Holley S, Hayakawa M, Mellon PL, 1998. Discrete stages of anterior pituitary differentiation recapitulated in immortalized cell lines. Mol. Cell. Endocrinol 140, 25–30. [DOI] [PubMed] [Google Scholar]
- Alarid ET, Windle JJ, Whyte DB, Mellon PL, 1996. Immortalization of pituitary cells at discrete stages of development by directed oncogenesis in transgenic mice. Development 122, 3319–3329. [DOI] [PubMed] [Google Scholar]
- Albarracin CT, Kaiser UB, Chin WW, 1994. Isolation and characterization of the 5’-flanking region of the mouse gonadotropin-releasing hormone receptor gene. Endocrinology 135, 2300–2306. [DOI] [PubMed] [Google Scholar]
- Batch JA, Williams DM, Davies HR, Brown BD, Evans BA, Hughes IA, Patterson MN, 1992. Role of the androgen receptor in male sexual differentiation. Horm. Res 38, 226–229. [DOI] [PubMed] [Google Scholar]
- Belanger A, Cusan L, Caron S, Barden N, Dupont A, 1981. Ovarian progestins, androgens and estrogen throughout the 4-day estrous cycle in the rat. Biol. Reprod 24, 591–596. [DOI] [PubMed] [Google Scholar]
- Berger MF, Bulyk ML, 2009. Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nat. Protoc 4, 393–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayman MJ, Pepa PA, Berdy SE, Mellon PL, 2012a. Androgen receptor repression of GnRH gene transcription. Mol. Endocrinol 26, 2–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayman MJ, Pepa PA, Mellon PL, 2012b. Androgen receptor repression of gonadotropin-releasing hormone gene transcription via enhancer 1. Mol. Cell. Endocrinol 363, 92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceraline J, Cruchant MD, Erdmann E, Erbs P, Kurtz JE, Duclos B, Jacqmin D, Chopin D, Bergerat JP, 2004. Constitutive activation of the androgen receptor by a point mutation in the hinge region: a new mechanism for androgen-independent growth in prostate cancer. Int. J. Cane 108, 152–157. [DOI] [PubMed] [Google Scholar]
- Chang CS, Kokontis J, Liao ST, 1988. Structural analysis of complementary DNA and amino acid sequences of human and rat androgen receptors. Proc. Natl. Acad. Sci. U. S. A. 85, 7211–7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherrington BD, Bailey JS, Diaz AL, Mellon PL, 2008. NeuroD1 and Mash1 temporally regulate GnRH receptor gene expression in immortalized mouse gonadotrope cells. Mol. Cell. Endocrinol 295, 106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conne BS, Scaglioni S, Lang U, Sizonenko PC, Aubert ML, 1982. Pituitary receptor sites for gonadotropin-releasing hormone: effect of castration and substitutive therapy with sex steroids in the male rat. Endocrinology 110, 70–79. [DOI] [PubMed] [Google Scholar]
- Crocker J, Abe N, Rinaldi L, McGregor AP, Frankel N, Wang S, Alsawadi A, Valenti P, Plaza S, Payre F, Mann RS, Stern DL, 2015. Low affinity binding site clusters confer hox specificity and regulatory robustness. Cell 160, 191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis TL, Whitesell JD, Cantlon JD, Clay CM, Nett TM, 2011. Does a nonclassical signaling mechanism underlie an increase of estradiol-mediated gonadotropin-releasing hormone receptor binding in ovine pituitary cells? Biol. Reprod 85, 770–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JA, Dalkin AC, Barkan A, Regiani S, Marshall JC, 1983. Gonadal regulation of pituitary gonadotropin-releasing hormone receptors during sexual maturation in the rat. Endocrinology 113, 2238–2246. [DOI] [PubMed] [Google Scholar]
- Duval DL, Ellsworth BS, Clay CM, 1999. Is gonadotrope expression of the gonadotropin releasing hormone receptor gene mediated by autocrine/prarcrine stimulation of an activin response element? Endocrinology 140, 1949–1952. [DOI] [PubMed] [Google Scholar]
- Duval DL, Nelson SE, Clay CM, 1997a. A binding site for steroidogenic factor-1 is part of a complex enhancer that mediates expression of the murine gonadotropin-releasing hormone receptor gene. Biol. Reprod 56, 160–168. [DOI] [PubMed] [Google Scholar]
- Duval DL, Nelson SE, Clay CM, 1997b. The tripartite basal enhancer of the gonadotropin-releasing hormone (GnRH) receptor gene promoter regulates cell-specific expression through a novel GnRH receptor activating sequence. Mol. Endocrinol 11, 1814–1821. [DOI] [PubMed] [Google Scholar]
- Edelsztein NY, Racine C, di Clemente N, Schteingart HF, Rey RA, 2018. Androgens downregulate anti-Mullerian hormone promoter activity in the Sertoli cell through the androgen receptor and intact steroidogenic factor 1 sites. Biol. Reprod 99, 1303–1312. [DOI] [PubMed] [Google Scholar]
- Ellsworth BS, Burns AT, Escudero KW, Duval DL, Nelson SE, Clay CM, 2003. The gonadotropin releasing hormone (GnRH) receptor activating sequence (GRAS) is a composite regulatory element that interacts with multiple classes of transcription factors including Smads, AP-1 and a forkhead DNA binding protein. Mol. Cell. Endocrinol 206, 93–111. [DOI] [PubMed] [Google Scholar]
- Farley EK, Olson KM, Zhang W, Brandt AJ, Rokhsar DS, Levine MS, 2015. Suboptimization of developmental enhancers. Science 350, 325–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley EK, Olson KM, Zhang W, Rokhsar DS, Levine MS, 2016. Syntax compensates for poor binding sites to encode tissue specificity of developmental enhancers. Proc. Natl. Acad. Sci. U. S. A. 113, 6508–6513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin J, Ongaro L, Li Y, Tran S, Lamba P, Wang Y, Zhou X, Bernard DJ, 2015. Minireview: activin signaling in gonadotropes: what does the FOX say… To the SMAD? Mol. Endocrinol 29, 963–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frager MS, Pieper DR, Tonetta SA, Duncan JA, Marshall JC, 1981. Pituitary gonadotropin-releasing hormone receptors. Effects of castration, steroid replacement, and the role of gonadotropin-releasing hormone in modulating receptors in the rat. J. Clin. Invest 67, 615–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar ML, Meo T, Tosi M, 1990. Structure and size distribution of the androgen receptor mRNA in wild-type and Tfm/Y mutant mice. Mol. Endocrinol 4, 1600–1610. [DOI] [PubMed] [Google Scholar]
- Gehring U, Tomkins GM, 1974a. Characterization of a hormone receptor defect in the androgen-insensitivity mutant. Cell 3, 59–64. [DOI] [PubMed] [Google Scholar]
- Gehring U, Tomkins GM, 1974b. A new mechanism for steroid unresponsiveness: loss of nuclear binding activity of a steroid hormone receptor. Cell 3, 301–306. [DOI] [PubMed] [Google Scholar]
- Gharib SD, Leung PC, Carroll RS, Chin WW, 1990. Androgens positively regulate follicle-stimulating hormone beta-subunit mRNA levels in rat pituitary cells. Mol. Endocrinol 4, 1620–1626. [DOI] [PubMed] [Google Scholar]
- Gnanapragasam VJ, Robson CN, Leung HY, Neal DE, 2000. Androgen receptor signalling in the prostate. BJU Int. 86, 1001–1013. [DOI] [PubMed] [Google Scholar]
- Hamernik DL, Clay CM, Turzillo A, Van Kirk EA, Moss GE, 1995. Estradiol increases amounts of messenger ribonucleic acid for gonadotropin-releasing hormone receptors in sheep. Biol. Reprod 53, 179–185. [DOI] [PubMed] [Google Scholar]
- Hebert JM, McConnell SK, 2000. Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev. Biol 222, 296–306. [DOI] [PubMed] [Google Scholar]
- Heckert LL, Wilson EM, Nilson JH, 1997. Transcriptional repression of the alpha-subunit gene by androgen receptor occurs independently of DNA binding but requires the DNA-binding and ligand-binding domains of the receptor. Mol. Endocrinol 11, 1497–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiort O, 2013. The differential role of androgens in early human sex development. BMC Med. 11, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann HM, Trang C, Gong P, Kimura I, Pandolfi EC, Mellon PL, 2016. Deletion of Vax1 from GnRH neurons abolishes GnRH expression and leads to hypogonadism and infertility. J. Neurosci 36, 3506–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa H, Ungerback J, Wang X, Matsumoto M, Nakayama KI, Cohen SM, Tanaka T, Rothenberg EV, 2018. Transcription factor PU.1 represses and activates gene expression in early T cells by redirecting partner transcription factor binding. Immunity 49, 782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume MA, Barrera LA, Gisselbrecht SS, Bulyk ML, 2015. UniPROBE, update 2015: new tools and content for the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res. 43, D117–D122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonen T, Palvimo JJ, Janne OA, 1998. Heterodimerization is mainly responsible for the dominant negative activity of amino-terminally truncated rat androgen receptor forms. FEBS Lett. 430, 393–396. [DOI] [PubMed] [Google Scholar]
- Ikonen T, Palvimo JJ, Kallio PJ, Reinikainen P, Janne OA, 1994. Stimulation of androgen-regulated transactivation by modulators of protein phosphorylation. Endocrinology 135, 1359–1366. [DOI] [PubMed] [Google Scholar]
- Jindal GA, Farley EK, 2021. Enhancer grammar in development, evolution, and disease: dependencies and interplay. Dev. Cell 56, 575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen JS, Nilson JH, 2001. AR suppresses transcription of the LHbeta subunit by interacting with steroidogenic factor-1. Mol. Endocrinol 15, 1505–1516. [DOI] [PubMed] [Google Scholar]
- Kauffman AS, Thackray VG, Ryan GE, Tolson KP, Glidewell-Kenney CA, Semaan SJ, Poling MC, Iwata N, Breen KM, Duleba AJ, Stener-Victorin E, Shimasaki S, Webster NJ, Mellon PL, 2015. A novel letrozole model recapitulates both the reproductive and metabolic phenotypes of Polycystic Ovary Syndrome in female mice. Biol. Reprod 93, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kribelbauer JF, Rastogi C, Bussemaker HJ, Mann RS, 2019. Low-affinity binding sites and the transcription factor specificity paradox in eukaryotes. Annu. Rev. Cell Dev. Biol 35, 357–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson MA, Li D, Glidewell-Kenney CA, Lopez FJ, 2001. Androgen responsiveness of the pituitary gonadotrope cell line LbetaT2. J. Endocrinol 170, 601–607. [DOI] [PubMed] [Google Scholar]
- Limonta P, Ladizhenskaya A, Gunsalus GL, Bardin CW, Thau RB, 1986. Regulation of pituitary gonadotropin-releasing hormone receptors by androgens in the male rabbit. Endocrinology 118, 340–347. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD, 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Mariani L, Weinand K, Vedenko A, Barrera LA, Bulyk ML, 2017. Identification of human lineage-specific transcriptional coregulators enabled by a glossary of binding modules and tunable genomic backgrounds. Cell Syst 5, 654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massie CE, Adryan B, Barbosa-Morais NL, Lynch AG, Tran MG, Neal DE, Mills IG, 2007. New androgen receptor genomic targets show an interaction with the ETS1 transcription factor. EMBO Rep. 8, 871–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGillivray SM, Bailey JS, Ramezani R, Kirkwood BJ, Mellon PL, 2005. Mouse GnRH receptor gene expression is mediated by the LHX3 homeodomain protein. Endocrinology 146, 2180–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGillivray SM, Thackray VG, Coss D, Mellon PL, 2007. Activin and glucocorticoids synergistically activate follicle-stimulating hormone β-subunit gene expression in the immortalized LβT2 gonadotrope cell line. Endocrinology 148, 762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naess O, Cusan L, Brekke I, Purvis K, Torjesen P, Hansson V, 1981. Effects of castration, sex steroids, LHRH and glucocorticoids on LHRH binding in the anterior pituitary of male rats. Int. J. Androl 4, 685–690. [DOI] [PubMed] [Google Scholar]
- Naik SI, Young LS, Charlton HM, Clayton RN, 1984a. Pituitary gonadotropin-releasing hormone receptor regulation in mice. I: Males. Endocrinology 115, 106–113. [DOI] [PubMed] [Google Scholar]
- Naik SI, Young LS, Charlton HM, Clayton RN, 1984b. Pituitary gonadotropin-releasing hormone receptor regulation in mice. II: Females. Endocrinology 115, 114–120. [DOI] [PubMed] [Google Scholar]
- Naik SI, Young LS, Charlton HM, Clayton RN, 1985. Evidence for a pituitary site of gonadal steroid stimulation of GnRH receptors in female mice. J. Reprod. Fertil 74, 615–624. [DOI] [PubMed] [Google Scholar]
- Norwitz ER, Cardona GR, Jeong KH, Chin WW, 1999. Identification and characterization of the gonadotropin-releasing hormone response elements in the mouse gonadotropin-releasing hormone receptor gene. J. Biol. Chem 274, 867–880. [DOI] [PubMed] [Google Scholar]
- Norwitz ER, Xu S, Jeong KH, Bedecarrats GY, Winebrenner LD, Chin WW, Kaiser UB, 2002a. Activin A augments GnRH-mediated transcriptional activation of the mouse GnRH receptor gene. Endocrinology 143, 985–997. [DOI] [PubMed] [Google Scholar]
- Norwitz ER, Xu S, Xu J, Spiryda LB, Park JS, Jeong KH, McGee EA, Kaiser UB, 2002b. Direct binding of AP-1 (Fos/Jun) proteins to a SMAD binding element facilitates both GnRH-and activin-mediated transcriptional activation of the mouse GnRH receptor gene. J. Biol. Chem 277, 37469–37478. [DOI] [PubMed] [Google Scholar]
- O’Hara L, Curley M, Tedim Ferreira M, Cruickshanks L, Milne L, Smith LB, 2015. Pituitary androgen receptor signalling regulates prolactin but not gonadotrophins in the male mouse. PloS One 10, e0121657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hara L, Welsh M, Saunders PT, Smith LB, 2011. Androgen receptor expression in the caput epididymal epithelium is essential for development of the initial segment and epididymal spermatozoa transit. Endocrinology 152, 718–729. [DOI] [PubMed] [Google Scholar]
- Ohno S, Dofuku R, Tettenborn U, 1971a. More about X-linked testicular feminization of the mouse as a noninducible(is)mutation of a regulatory locus: 5-alpha-androstan-3-alpha-17-beta-diol as the true inducer of kidney alcohol dehydrogenase and beta-glucuronidase. Clin. Genet 2, 128–140. [DOI] [PubMed] [Google Scholar]
- Ohno S, Tettenborn U, Dofuku R, 1971b. Molecular biology of sex differentiation. Hereditas 69, 107–124. [DOI] [PubMed] [Google Scholar]
- Paul SJ, Ortolano GA, Haisenleder DJ, Stewart JM, Shupnik MA, Marshall JC, 1990. Gonadotropin subunit messenger RNA concentrations after blockade of gonadotropin-releasing hormone action: testosterone selectively increases follicle-stimulating hormone beta-subunit messenger RNA by posttranscriptional mechanisms. Mol. Endocrinol 4, 1943–1955. [DOI] [PubMed] [Google Scholar]
- Pieper DR, 1984. Effects of photoperiod, castration, and gonadotropin-releasing hormone (GnRH) on the number of GnRH receptors in male golden hamsters. Endocrinology 115, 1857–1862. [DOI] [PubMed] [Google Scholar]
- Poling MC, Kim J, Dhamija S, Kauffman AS, 2012. Development, sex steroid regulation, and phenotypic characterization of RFamide-related peptide (Rfrp) gene expression and RFamide receptors in the mouse hypothalamus. Endocrinology 153, 1827–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos AI, Barolo S, 2013. Low-affinity transcription factor binding sites shape morphogen responses and enhancer evolution. Philos. Trans. R. Soc. Lond. B Biol. Sci 368, 20130018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennie PS, Bruchovsky N, Leco KJ, Sheppard PC, McQueen SA, Cheng H, Snoek R, Hamel A, Bock ME, MacDonald BS, et al. , 1993. Characterization of two cis-acting DNA elements involved in the androgen regulation of the probasin gene. Mol. Endocrinol 7, 23–36. [DOI] [PubMed] [Google Scholar]
- Riegman PH, Vlietstra RJ, van der Korput JA, Brinkmann AO, Trapman J, 1991. The promoter of the prostate-specific antigen gene contains a functional androgen responsive element. Mol. Endocrinol 5, 1921–1930. [DOI] [PubMed] [Google Scholar]
- Ripple MO, Henry WF, Rago RP, Wilding G, 1997. Prooxidant-antioxidant shift induced by androgen treatment of human prostate carcinoma cells. J. Natl. Cancer Inst 89, 40–48. [DOI] [PubMed] [Google Scholar]
- Roche PJ, Hoare SA, Parker MG, 1992. A consensus DNA-binding site for the androgen receptor. Mol. Endocrinol 6, 2229–2235. [DOI] [PubMed] [Google Scholar]
- Rowan S, Siggers T, Lachke SA, Yue Y, Bulyk ML, Maas RL, 2010. Precise temporal control of the eye regulatory gene Pax6 via enhancer-binding site affinity. Genes Dev. 24, 980–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saartok T, Dahlberg E, Gustafsson JA, 1984. Relative binding affinity of anabolic-androgenic steroids: comparison of the binding to the androgen receptors in skeletal muscle and in prostate, as well as to sex hormone-binding globulin. Endocrinology 114, 2100–2106. [DOI] [PubMed] [Google Scholar]
- Schang AL, Querat B, Simon V, Garrel G, Bleux C, Counis R, Cohen-Tannoudji J, Laverriere JN, 2012. Mechanisms underlying the tissue-specific and regulated activity of the Gnrhr promoter in mammals. Front. Endocrinol 3, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg PH, Mason AJ, Stewart TA, Nikolics K, 1987. The mammalian GnRH gene and its pivotal role in reproduction. Recent Prog. Horm. Res 43, 69–98. [DOI] [PubMed] [Google Scholar]
- Spady TJ, Shayya R, Thackray VG, Ehrensberger L, Bailey JS, Mellon PL, 2004. Androgen regulates FSHb gene expression in an activin-dependent manner in immortalized gonadotropes. Mol. Endocrinol 18, 925–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama K, Kaneshiro K, Tsutsumi S, Horie-Inoue K, Ikeda K, Urano T, Ijichi N, Ouchi Y, Shirahige K, Aburatani H, Inoue S, 2007. Identification of novel androgen response genes in prostate cancer cells by coupling chromatin immunoprecipitation and genomic microarray analysis. Oncogene 26, 4453–4463. [DOI] [PubMed] [Google Scholar]
- Thackray VG, Hunnicutt JL, Memon AK, Ghochani Y, Mellon PL, 2009. Progesterone inhibits basal and GnRH induction of luteinizing hormone β-subunit gene expression. Endocrinology 150, 2395–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thackray VG, McGillivray SM, Mellon PL, 2006. Androgens, progestins and glucocorticoids induce follicle-stimulating hormone β-subunit gene expression at the level of the gonadotrope. Mol. Endocrinol 20, 2062–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thackray VG, Mellon PL, Coss D, 2010. Hormones in synergy: regulation of the pituitary gonadotropin genes. Mol. Cell. Endocrinol 314, 192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcotte G, Chapdelaine A, Roberts KD, Chevalier S, 1988. Androgen binding as evidenced by a whole cell assay system using cultured canine prostatic epithelial cells. J. Steroid Biochem 29, 69–76. [DOI] [PubMed] [Google Scholar]
- Turzillo AM, DiGregorio GB, Nett TM, 1995. Messenger ribonucleic acid for gonadotropin-releasing hormone receptor and numbers of gonadotropin-releasing hormone receptors in ovariectomized ewes after hypothalamic-pituitary disconnection and treatment with estradiol. J. Anim. Sci 73, 1784–1788. [DOI] [PubMed] [Google Scholar]
- Vale W, Rivier C, Brown M, 1977. Regulatory peptides of the hypothalamus. Annu. Rev. Physiol 39, 473–527. [DOI] [PubMed] [Google Scholar]
- Wagenmaker ER, Moenter SM, 2017. Exposure to acute psychosocial stress disrupts the luteinizing hormone surge independent of estrous cycle alterations in female mice. Endocrinology 158, 2593–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, DeFazio RA, Moenter SM, 2016. Excitability and burst generation of AVPV kisspeptin neurons are regulated by the estrous cycle via multiple conductances modulated by estradiol action. eNeuro 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CA, Herdon HJ, Bailey LC, Clayton RN, 1986. Effect of testosterone on gonadotrophin-releasing hormone receptors in the castrated rat: preliminary evidence for a stimulatory effect of testosterone on gonadotrophin function in the male rat. J. Endocrinol 108, 441–449. [DOI] [PubMed] [Google Scholar]
- Wilson S, Qi J, Filipp FV, 2016. Refinement of the androgen response element based on ChIP-Seq in androgen-insensitive and androgen-responsive prostate cancer cell lines. Sci. Rep 6, 32611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Chen Y, Fajobi T, DiVall SA, Chang C, Yeh S, Wolfe A, 2014. Conditional knockout of the androgen receptor in gonadotropes reveals crucial roles for androgen in gonadotropin synthesis and surge in female mice. Mol. Endocrinol 28, 1670–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Hoffmann HM, Meadows JD, Mayo SL, Trang C, Leming SS, Maruggi C, Davis SW, Larder R, Mellon PL, 2015. Homeodomain proteins SIX3 and SIX6 regulate gonadotrope-specific genes during pituitary development. Mol. Endocrinol 29, 842–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandvakili A, Campbell I, Gutzwiller LM, Weirauch MT, Gebelein B, 2018. Degenerate Pax2 and Senseless binding motifs improve detection of low-affinity sites required for enhancer specificity. PLoS Genet. 14, e1007289. [DOI] [PMC free article] [PubMed] [Google Scholar]