

Abstract
Harnessing the immune system to treat cancer through inhibitors of Cytotoxic T-Lymphocyte Antigen-4 (CTLA-4) and Programmed Death Ligand-1 (PD-L1) has revolutionalized the landscape of cancer. Rational combination strategies aim to enhance the antitumor effects of immunotherapies, but require a deep understanding of the mechanistic underpinnings of the immune system and robust preclinical and clinical drug development strategies. We review the current approved immunotherapy combinations, before discussing promising combinatorial approaches in clinical trials and detail innovative preclinical model systems being used to develop rational combinations. We also discuss the promise of high-order immunotherapy combinations, and novel biomarker and combinatorial trial strategies.
Introduction
For most of the last century, research focused on exploiting the antitumor effects of different classes of therapeutics exclusively on cancer cells (1). Over the past decade, however, we have gained a deeper undersrtanding of the role of the immune system in mediating such responses. This has led to a revolution in preclinical, translational and clinical efforts dedicated to harnessing the immune system for the development of novel immunotherapeutics in cancer medicine (2). Recent studies have led to a vast range of immunotherapies being assessed in clinical trials and subsequently achieving U.S. Food and Drug Administration (FDA) approval.
Immunotherapy has allowed the field of oncology to turn a critical corner where long term survival and even durable cures are achievable for patients with metastatic solid tumors. The current reality, however, is that the majority of patients enter care with immune “cold” tumors which respond poorly, if at all, to existing checkpoint therapies (3) (Figure 1). Immune suppression in these cancers resists reversal with checkpoint blockade due to its multi-modal nature encompassing suppressive cytokines, lack of antigen presentation, apoptotic triggering of T-cells, and hostile metabolic states and nutrient deprivation. These additional layers of tumor immune privilege must be peeled back therapeutically in order to reveal the benefits of T-cell checkpoint blockade and drive tumor regression. Thus, combinations of multiple immune interventions are necessary to reverse the “cold” tumor state, yet most of the existing “backbone” immunotherapies already approach the ceiling of tolerability even when used at doses that are clearly below their maximum efficacy (4–6).
Figure 1: Immune checkpoint blockade and ‘hot’ vs ‘cold’ tumor microenvironments.
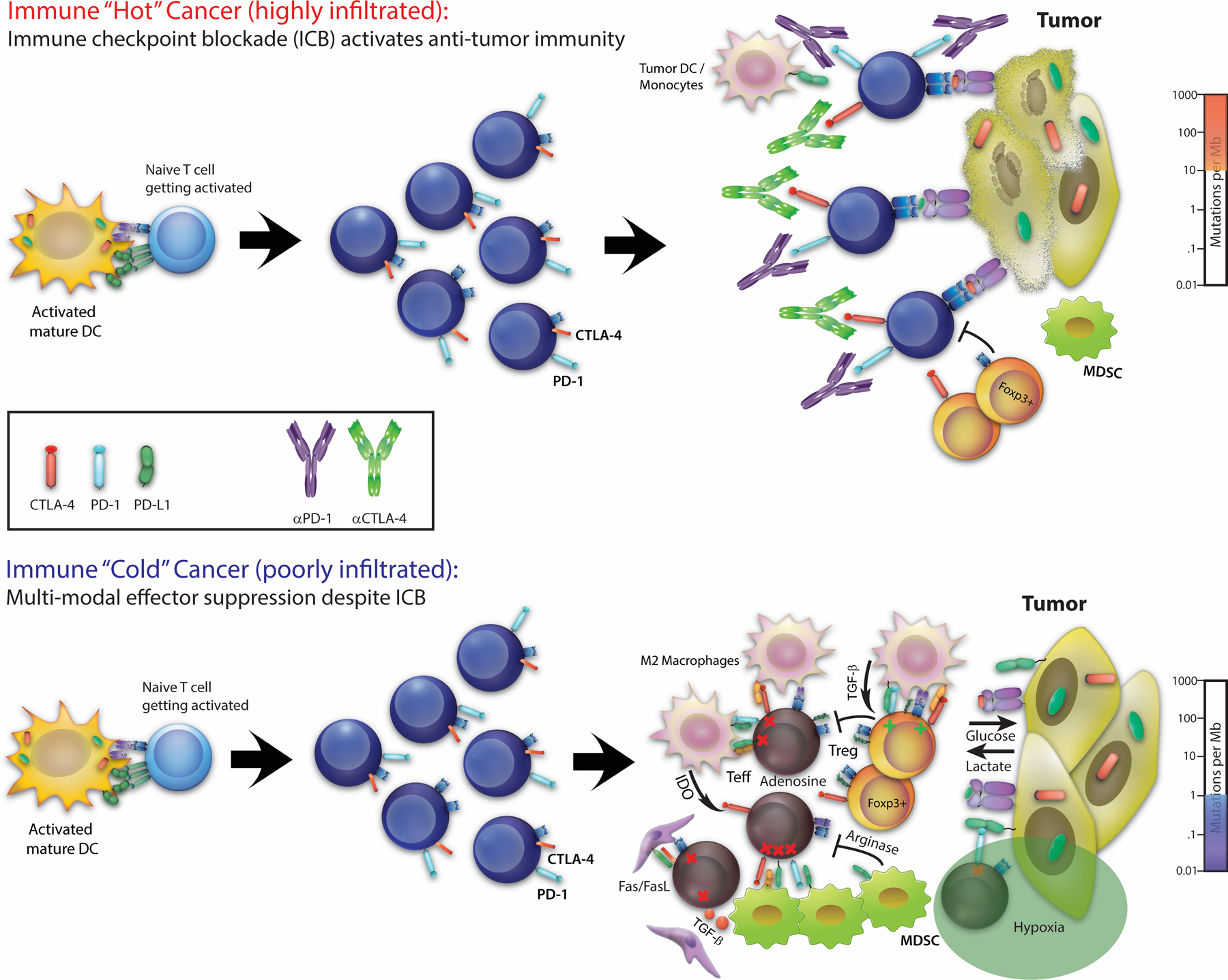
Immune checkpoint blockade frees T cells in ‘hot’ tumor microenvironments (top panel), but fails in ‘cold’ tumors due to dominant, multi-model suppressive mechanisms (bottom panel).
The limitations in efficacy observed with the first wave of approved immunotherapies, primarily involving immune checkpoint inhibitors, have illustrated a need to improve our understanding of the mechanistic underpinnings of the immune system and thereby develop more robust preclinical and clinical drug development strategies (2). A better understanding of primary and secondary resistance is also required to improve patient outcomes to single agent immunotherapy strategies (7). Improving our insights into mechanisms of response and resistance are the crucial next steps for the future development of immunotherapeutics.
In this article, we begin by detailing the successes observed to date with FDA-approved immunotherapy combinations in different tumor indications, before reviewing promising strategies currently in clinical trial testing. We then describe the use of preclinical models to optimize the development of rational combinations. Finally, we discuss the development of high-order immunotherapy combination strategies and novel biomarker and clinical trial strategies to support the development of combination approaches.
Current progress in the clinic with FDA approved IO combinations
A number of immune checkpoint-based combination treatments are now FDA approved (Table 1; Figure 2), with nivolumab (anti-PD-1) and ipilimumab (anti-CTLA4) the earliest immuno-oncology (IO) combination to receive FDA approval in September 2015 for the first-line treatment of metastatic melanoma (8,9). Although the treatment of metastatic melanoma had already been transformed by single-agent immune checkpoint blockade, the nivolumab-ipilimumab combination improved objective response rates to 58% and median progression-free survival (mPFS) to 11.5 months compared to nivolumab monotherapy (ORR 45%, mPFS 6.9 months) or ipilimumab alone (ORR 19%, mPFS 2.9 months). Despite a high rate of discontinuation due to toxicities, a survival benefit was apparent even for patients who discontinued treatment with median overall survival (OS) not reached at 60 months (10,11).
Table 1:
FDA approved immune checkpoint combinations
| Combination | Control arm | Tumor type(s) | No of patients | Gd 3–5 toxicity | Median PFS (95% CI) (months) |
Median OS (95% CI) (months) |
|---|---|---|---|---|---|---|
| Metastatic melanoma | ||||||
| Nivolumab 1mg/kg and Ipilimumab 3mg/kg (N+I) CHECKMATE067 |
Nivolumab 1mg/kg (N) Or Ipilimumab (3mg/kg) (I) |
Metastatic melanoma: BRAF wildtype or mutant | 945 (n = 314 in combination arm) |
N+I: 60% N: 24% I: 28% † |
N+I: 11.5 (8.7–19.3) N: 6.9 (5.1–10.2) I: 2.9 (2.8–3.2) |
N+I: NR (38.2-NR) N: 36.9 (28.2–58.7) I: 19.9 (16.8–24.6) (8–11) |
| Atezolizumab, Cobimetinib, Vemurafenib (ACV) IMspire150 |
Cobimetinib, Vemurafenib, and Placebo (CV) |
BRAF V600E-mutated advanced melanoma | 514 (n=256 in ACV) |
ACV 80% CV 73% † |
ACV: 15.1 (11.4–18.4) CV: 10.6 (9.3–12.7) |
NR 24-months: ACV: 60% CV: 53% (162) |
| Colorectal cancer | ||||||
| Nivolumab 3mg/kg and Ipilimumab 1mg/kg (N+I) CHECKMATE142 |
N/A | dMMR colorectal cancer, disease progression after 5FU, irinotecan or oxaliplatin based treatment | 119 | 32% | NR 12 month PFS = 71% (61.4–78.7) |
NR 12 month OS = 85% (77.0–90.2) (13) |
| Head and Neck Squamous Cell Cancer | ||||||
| Pembrolizumab and Chemotherapy (5-FU and platinum) (P + chemo) KEYNOTE-048 |
Cetuximab +chemotherapy (5-FU and platinum) (C + Chemo) | Untreated Recurrent or metastatic head and squamous cell cancer | ITT: 882 (n=281 for P + chemo; n=301 C + Chemo) |
P + Chemo: 85% C + Chemo: 83% |
P + Chemo: 4·9 (4·7−6·0) C + Chemo: 5·1 (4·9−6·0) |
C + Chemo: 10.7 (9.3–11.7) P + Chemo: 13.0 (10.9–14.7) |
| CPS ≥1: P + Chemo:242 C + Chemo: 235 |
P + Chemo: 5·0 (4·7−6·2) C + Chemo: 5·0 (4·8−5·8) |
C + Chemo: 10.4 (9.1–11.7) P + chemo: 13.6 (10.7–15.5) (42) |
||||
| Non-Small Cell Lung Cancer | ||||||
| Nivolumab 3mg/kg and Ipilimumab 1mg/kg (N+I) CHECKMATE-227 |
Nivolumab (N), Nivolumab + Platinum Doublet (N + Chemo) or Chemotherapy (chemo) |
Stage IV or Recurrent NSCLC with WT EGFR and ALK | PD-L1% ≥ 1%: 1189 (n = 396 in N; n = 396 in N+I) |
N+I: 36.8% N:19.9% Chemo:37.7 † |
N+I: 5.1 (4.1–6.3) N: 4.2 (3.0–5.3) Chemo: 5.6 (4.6–5.8) |
N+I: 17.1 (15.0–20.1) N: 15.7 (13.3–18.1) Chemo:14.9 (12.7–16.7) |
| PD-L1% ≤ 1%: 550 (n=187 for N+I, n=177 for N+Chemo) |
N+I:28.6% N+Chemo:58.1% Chemo:35.5% † |
N+I: 5.1 (3.2–6.4) N+Chemo: 5.6 (4.6–6.9) Chemo: 4.7 (4.2–5.6) |
N+I: 17.2 (12.8–22.0) N+Chemo: 15.2 (12.3–19.8) Chemo: 12.2 (9.2–14.3) (232) |
|||
| Nivolumab 3mg/kg q 3w, Ipilimumab 1mg/kg q 6w + chemo (2 cycles) (IO) CHECKMATE-9LA |
Chemotherapy – 4 cyles (Chemo) | Stage IV or recurrent NSCLC (EGFR/ALK non-mutated) – treatment naive | IO: 361 Chemo: 358 |
IO: 49% Chemo: 40% † |
NE | IO: 15.6 (13.9–20.0) Chemo: 10.9 (9.5–12.5) (233) |
| Pembrolizumab, Pemetrexed and Platinum (PPP) KEYNOTE-189 |
Pemetrexed and platinum (Chemo) | Metastatic nonsquamous NSCLC (EGFR/ALK non-mutated) | 616 (n = 410 in IO combination arm) |
PPP: 71.9% Chemo: 66.8% |
PPP: 9.0(8.1–9.9) Chemo: 4.9 (4.7–5.5) |
PPP: 22.0 (19.5–25.2) Chemo: 10.7 (8.7–13.6) 24month OS = PPP: 45.5% Chemo: 29.9% (22,24) |
| Pembrolizumab, Chemotherapy (Carboplatin, Taxane) (PC) KEYNOTE-407 |
Carboplatin, Taxane (Chemo) | Metastatic squamous NSCLC | 559 (n = 278 in IO combination arm) |
PC: 74.1% Chemo: 69.6% |
PC: 8.0 (6.3–8.4) Chemo: 5.1 (4.3–6.0) |
PC: 17.1 (14.4–19.9) Chemo: 11.6 (10.1–13.7) (25,26) |
| Chemo-radiotherapy – durvalumab consolidation (IO) PACIFIC |
Chemo-radiotherapy – placebo (Pl) | Stage III NSCLC | 713 (n = 476 in IO combination arm) |
IO: 34.9% Pl: 32.1% † |
IO: 17.2 (13.1–23.9) Pl: 5.6 (4.6–7.7) |
IO: NR (38.4-NR) Pl: 29.1 (22.1–35.1) 36-month OS: IO: 57.0 (52.3–61.4%) Pl:43.5% (37.0–49.9) (27–29) |
| Atezolizumab, Bevacizumab, Carboplatin, Paclitaxel (ABCP)* or Atezolizumab, Carboplatin, Paclitaxel (ACP) IMpower150 |
Bevacizumab, Carboplatin, Paclitaxel (BCP) |
Metastatic non-squamous NSCLC (inc. EGFR/ALK mutated)** | Non-EGFR/ALK mutated 1045 (n = 359 in ABPC, n = 349 in ACP) |
ACP: 43.8% ABCP: 58.5% BCP: 50.0% † |
ABCP: 8.3 (7.7–9.8) BCP: 6.8 (6.0–7.1) |
ABCP: 19.2 (17.0–23.8) BCP: 14.7 (13.3–16.9) (234,235) |
| EGFR mutated 124 (n = 34 in ABCP and n = 45 in ACP)** |
ABCP: 10.2 (7.9–15.2) BCP: 6.9 (5.7–8.5) ACP: 6.9 (5.7–8.2) |
ABCP:NE (17.0-NE) BCP:18.7 (11.7-NE) ACP: 21.4 (13.8-NE) |
||||
| Atezolizumab, Carboplatin, nab-paclitaxel (IO) IMpower130 |
Platinum-based chemotherapy (Chemo) | Metastatic non-squamous NSCLC (inc. EGFR/ALK mutated)$ | Non-EGFR/ALK mutated 723 (n = 451 in IO, n = 228 in Chemo) |
IO: 74.8% Chemo: 60.8%# † |
IO: 7.0 (6.2–7.3) Chemo: 5.5 (4.4–5.9) |
IO: 18.6 (16.0–21.2) Chemo: 13.9 (12.0–18.7) (236) |
| Extensive Stage Small Cell Lung Cancer (SCLC) | ||||||
| Atezolizumab, Carboplatin, Etoposide (APE) IMpower133 |
Carboplatin, Etoposide (Chemo) | Extensive Stage-SCLC | 403 (n = 201 in IO combination arm) |
APE: 69.2% Chemo: 64.8% † |
APE: 5.2 (4.4–5.6) Chemo: 4.3 (4.2–4.5) |
APE: 12.3 (10.8–15.8) Chemo: 10.3 (9.3–11.3) (32,33) |
| Durvalumab, Etoposide, Platinum (DPE) CASPIAN |
Platinum Etoposide (chemo) | Extensive Stage -SCLC | 805 (n = 268 for DPE) |
DPE:66.4 % Chemo:68.0% † |
DPE:5.1 (4.7–6.2) Chemo:5.4 (4.8–6.2) |
DPE:13.0(11.5–14.8) Chemo: 10.3 (9.3–11.2) (34) |
| Triple negative breast cancer | ||||||
| Atezolizumab and nab-paclitaxel (AP) Impassion130 |
Nab-paclitaxel (Chemo) | Advanced triple negative breast cancer | ITT Population: 902 (n = 451 in IO combination arm) |
AP: 50.1% Chemo: 43.0% † |
AP: 7.2 (5.6–7.4) Chemo: 5.5 (5.3–5.6) |
AP: 21.0 (19.0–22.6) Chemo: 17.5 (16.9–20.3) |
| PD-L1 ≥ 1% 369 (n = 185 in IO combination arm) |
AP: 7.5 (6.7–9.2) Chemo: 5.3 (3.8–5.6) |
AP: 25.0 (19.6–30.7) Chemo: 18.0 (13.6–20.1) (39,127) |
||||
| Renal cell cancer | ||||||
| Nivolumab 3mg/kg and Ipilimumab 1mg/kg (N+I) CHECKMATE 214 |
Sunitinib (S) | Metastatic clear cell renal cancer – int. - poor risk (IMDC score) | 847 (n= 425 in combination arm) |
N+I: 48% S: 65% |
N+I: 8.2 (8.7–15.5) S: 8.3 (7.0–8.8) |
N+I: NR (35.6-NR) S: 26.6 (22.1–33.4) 30 month OS = N+I: 60% (55–64%) S: 47% (43–52%) |
| Metastatic clear cell renal cancer - favorable risk (IMDC score) | 249 (n = 125 in combination arm) |
N+I: 13.9 (9.9–17.9) S: 19.9 (15.1–23.5) |
N+I: NR S: 32.9 (NE) 30 month OS = N+I: 80% (72–86-%) S: 85% (77–90%) (16,17) |
|||
| Pembrolizumab, Axitinib (PA) KEYNOTE-426 |
Sunitinib (S) | Untreated Advanced Clear-Cell Renal Cell Carcinoma | 861 (n = 432 in IO combination arm) |
PA:75.8% S: 70.6% |
PA: 15.4 (12.7–18.9) S: 11.1 (9.1–12.5) |
mOS: PA: NR (NR-NR) S: 35.7 (33.3-NR) 24-month OS: PA: 74% S:66% (51,237) |
| Avelumab, Axitinib (A+A) JAVELIN RENAL 101 |
Sunitinib (S) | Untreated Renal Cell Carcinoma | Overall: 886 (n = 442 for IO combination) |
A+A: 71.2% S: 71.5% |
Overall Population: A+A: 13.3 (11.1–15.3) S: 8.0 (6.7–9.8) |
NE |
| PD-L1 ≥ 1% 560 |
PD-L1 Positive: A+A: 13.8 (10.1–20.7) S: 7.0 (5.7–9.6) |
NE (52,238) |
||||
| Hepatocellular cancer | ||||||
| Nivolumab 1mg/kg + Ipilimumab 3mg q 4w (A) Nivolumab 1mg/kg + Iipilimumab 3mg/kg q 3w (B) Nivolumab 3mg/kg q 2w + Ipilimumab 1mg/kg q 6w (C) CHECKMATE 040 |
none | Sorafenib treated advanced hepatocellular cancer | 148 (n = 50 for A, n = 49 for B, n = 49 for C) |
A: 53% B: 29% C: 31 % |
A: 23 (9-NR) B: 12 (8–15) C:13 (7–33) (21) |
|
| Atezolizumab, Bevacizumab (AB) | Sorafenib (S) | Unresectable Hepatocellular Cancer | 501 (n=336 in IO combination) | AB:61.1% S:60.9% † |
AB: 6.8 (5.7–8.3) S:4.3 (4.0–5.6) |
12-month OS: AB: 67.2%(61.3–73.1) S: 54.6% (45.2–64.0) (239) |
| Endometrial Cancer | ||||||
| Pembrolizumab, Lenvatinib | N/A | Advanced endometrial cancer (inc.MSI and MSS)& | 108 | 68.5% † |
Total: 7.4 (5.3–8.7) MSI: 18.9 (4.0-NE) MSS: 7.4 (5.0–7.6) |
Total: 16.7 (15.0-NE) MSI: NE (7.4- NE) MSS: 16.4 (13.5–25.9) (44,45) |
Notes:
NR = not reached
NE = not estimatable
IMDC score = International Metastatic Renal Cell Cancer Database Consortium score
MSI = Microsatellite instable
MSS = Microsatellite stable
Analysis of ACP vs. BCP has not yet been reported in NSCLC.
EGFR and ALK mutated non-squamous NSCLC are excluded from the FDA approval of ABCP.
EGFR and ALK mutated non-squamous NSCLC are excluded from the FDA approved of Atezolizumab, nab-paclitaxel and carboplatin.
Safety data includes EGFR and ALK mutated cancers.
FDA approval encompasses microsatellite stable cancers only.
- Grade 3–4 and Grade 5 reported sepearately in original paper, percentages calculated by combination of grade 3–4 and grade 5.
Figure 2: Different Classes of Immunotherapy Combination Strategies.
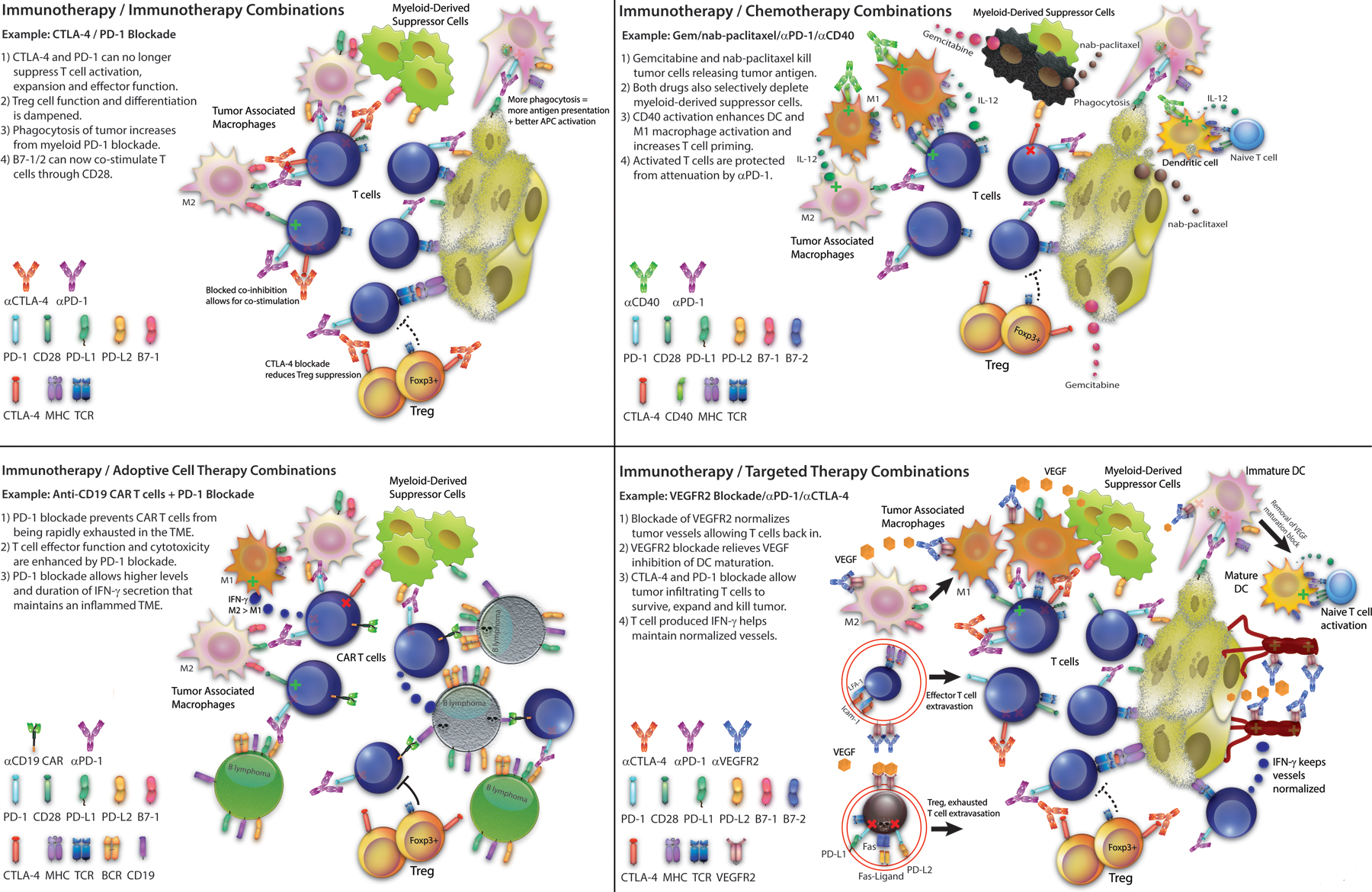
(1) Immunotherapy/Immunotherapy Combinations: Example: CTLA-4/PD-1 Blockade: 1) CTLA-4 and PD-1 can no longer suppress T cell activation, expansion and effector function; 2) Treg cell function and differentiation is dampened; 3) Phagocytosis of tumor increases from myeloid PD-1 blocakde; 4) B7–1/2 can now co-stimulate T cells through CD28. (2) Immunotherapy/Chemotherapy Combinations: Example: Gemcitabine/nab-paclitaxel/PD-1 blockade/CD40 agonist: 1) Gemcitabine and nab-paclitaxel kill tumor cells releasing tumor antigen; 2) Both drugs also selectively deplete myeloid-derived suppressor cells; 3) CD40 activation enhances DC and M1 macrophage activation and increases T cell priming; 4) Activated T cells are protected from attenuation by PD-1 blockade. (3) Immunotherapy/Adoptive Cell Therapy Combinations: Example: Anti-CD19 CAR T cells/PD-1 Blockade: 1)PD-1 blockade prevents CAR T cells from being rapidly exhausted in the tumor microenvironment; 2) T cell effector function and cytotoxicity are enhanced by PD-1 blockade; 3) PD-1 blockade allows higher levels and duration of IFN-γ secretion that maintains an inflamed tumor microenvironment. (4) Immunotherapy/Targeted Therapy Combinations: Example: VEGFR2/PD-1/CTLA-4 blockade: 1) Blockade of VEGFR2 normalizes tumor vessels allowing T cell back in; 2) VEGFR2 blockade relieves VEGF inhibition of DC maturation; 3) PD-1 and CTLA-4 blockade allow tumor infiltrating T cells to survive, expand and kill tumor; 4) T cell produced IFN-γ helps maintain normalized vessels.
Following approval for metastatic melanoma, the combination of nivolumab and ipilimumab has been similarly successful for other tumor sites. While single-agent immune checkpoint blockade has been strikingly effective in mismatch repair deficient (dMMR) cancers (12), this is improved by a combination approach. Nivolumab with lower dose ipilimumab (1mg/kg) for the second-line treatment of metastatic dMMR colorectal cancer showed a 60% objective response rate (ORR) (13,14) compared to 31% ORR with nivolumab alone (15). There was a relatively lower toxicity rate than that seen with higher dose ipilimumab (3mg/kg) in melanoma, with 32% patients experiencing grade 3 – 5 toxicities.
Similarly, in the first-line treatment of intermediate or poor-risk advanced clear cell renal cell cancer, nivolumab-ipilimumab studied in CheckMate 213 resulted in improved patient outcomes, with an ORR of 42%, in contrast to 29% with sunitinib (16,17). As 1mg/kg ipilimumab appeared to reduce toxicity without losing efficacy, clinical trials are investigating this dose of ipilimumab with nivolumab (3mg/kg)in metastatic melanoma, with early data indicating comparable efficacy and lower grade 3 to 5 toxicity rates (18).
Multikinase inhibitors have been the only effective and approved treatments in advanced unresectable HCC until the recent approval of immune checkpoint inhibitors (19). The CheckMate 040 study included comparison of nivolumab/ipilimumab combinations as second line treatment: comprising Arm A (nivolumab 1mg/kg plus ipilimumab 3mg/kg every 3 weeks for 4 doses), Arm B (nivolumab 3mg/kg plus ipilimumab 1mg/kg every 3 weeks for 4 doses followed by maintenance nivolumab every 2 weeks), or Arm C (nivolumab 3mg/kg every 2 weeks plus ipilimumab 1mg/kg every 6 weeks). ORR was similar between arms A, B, and C at 32%, 31%, and 31%, respectively. Arm A experienced the longest survival at 23 months (95% CI, 9-NA), versus 12 (95% CI, 9–15), and 13 months (95% CI, 7–33), with Arms B and C, respectively. Arms B and C, however, at 1mg/kg ipilimumab carried fewest Grade 3–4 toxicities (29% Arm B/31% Arm C vs. 53% Arm A) (20). FDA approval has been granted to combination IO using the Arm A dosing.
In the same trial in HCC, the triplet combination of cabozantinib, nivolumab and ipilimumab was compared to cabozantinib and nivolumab, including in the first line setting. While median OS has not been reported, the triplet arm had a mPFS of 6.8 months versus 5.5 months in the doublet arm, albeit with higher Grade 3–4 toxicities of 71% versus 42%, respectively (21). While results from this study are awaited, a number of IO plus other anti-cancer treatments have already been granted FDA approval, as discussed below.
IO combinations with chemotherapy
Combinations with chemotherapy have, to date, been the most numerous approved regimens in various indications. A number of studies in non-small cell lung cancer (NSCLC) have demonstrated a survival advantage when immune checkpoint blockade is administered in combination with chemotherapy. Pembrolizumab given in combination with pemetrexed and platinum chemotherapy as first line treatment of non-squamous NSCLC without EGFR or ALK mutations resulted in a median OS of 22.0 months (95% CI 19.5–25.2) versus 10.7 months (95% CI 8.7–13.6) (22–24). Similarly in squamous NSCLC, pembrolizumab in combination with carboplatin and taxane (paclitaxel or nab-paclitaxel) chemotherapy demonstrated an improved mOS at 17.1 months (95% CI 14.4–19.9) compared to chemotherapy alone (11.6 months, 95% CI 10.1–13.7) (25,26). Moreover, following consolidation chemo-radiotherapy for stage III NSCLC, durvalumab was shown to lead to a significantly improved mPFS (17.2 vs. 5.6 months) and OS (NR vs. 29.1 months), respectively (27–29).
While combination IO treatments were early successes in the treatment of NSCLC, small cell lung cancer (SCLC) initially proved challenging – studies of maintenance anti-PD-1 and anti-CTLA4-chemotherapy combination treatment did not result in improved PFS (30,31). However, a study of the PD-L1 inhibitor atezolizumab in combination with carboplatin and etoposide for extensive stage SCLC resulted in improved median OS of 12.3 months (10.8–15.8) versus 10.3 months (9.3–11.3) in the placebo arm (32,33), leading to FDA approval in the first line setting. Similarly, the CASPIAN Phase 3 trial assessed durvalumab in combination with platinum and etoposide chemotherapy as first-line treatment of extensive stage small-cell lung cancer, demonstrating an improvement in overall survival when compared to platinum etoposide alone (13.0 months v 10.3 months, HR: 0.73; p=0.005) (34).
Successful approaches have similarly been reported in breast cancer. Triple negative breast cancer (TNBC) is the most immunogenic of breast cancers, with high TILs particularly in tumors with features of “BRCAness” (35–37). Initially, however, single agent anti-PD-1 treatment was disappointing with a response rate of only 5.3% (38). More recently, atezolizumab in combination with nab-paclitaxel resulted in improved OS (25.0 v 18.0 months) in PD-L1 positive tumors (with PD-L1≥ 1% on tumor infiltrating immune cells) although not in unselected TNBC (39). Interestingly, a similar approach with paclitaxel (as opposed to nab-paclitaxel, a regimen which does not require steroid immunosuppression for potential allergic reactions) did not improve patient outcomes (https://www.fda.gov/drugs/resources-information-approved-drugs/fda-issues-alert-about-efficacy-and-potential-safety-concerns-atezolizumab-combination-paclitaxel). These conflicting results highlight the need for robust preclinical studies and the development of rational combinations.
Head and neck squamous cell carcinoma (HNSCC) was an early success in single agent checkpoint studies with combination approaches now approved in the frontline HNSCC setting (40). Recurrent or metastatic HNSCC has a dismal prognosis of 6–9 months and standard chemotherapy combination regimens lead to significant comorbidity and involve taxing treatment schedules (41). Pembrolizumab plus chemotherapy was compared to standard of care 5-fluorouracil plus platinum chemotherapy with cetuximab given as first line therapy for incurable or metastatic HNSCC. The immunotherapy combination resulted in improved median OS of 13.0 versus 10.7 months in the standard of care group (HR, 0.77, p=0.0067) (42). This OS benefit was observed despite no significant change in PFS, highlighting the importance of this clinical endpoint in trials of immunotherapy.
IO combinations with antiangiogenics
Anti-angiogenic agents, targeting vascular endothelial growth factor (VEGF) or its receptor (VEGFR), are promising agents in combination with immune checkpoint blockade. The increased density of high endothelial venules (typically surrounded by tertiary lymphoid structures) in the tumor microenvironment promotes T-cell trafficking to the tumor and subsequent response to anti-PD-1 therapy (43). This has been borne out in the clinical setting, with many combinations of antiangiogenics and immune checkpoint blockade now approved. Accelerated FDA approval was granted to the combination of pembrolizumab plus lenvatinib for the treatment of patients with advanced endometrial carcinoma that is not microsatellite instability high (MSI-H) or mismatch repair deficient (dMMR). Efficacy was demonstrated in 108 patients with metastatic endometrial carcinoma that had progressed following at least one prior line of therapy on the KEYNOTE-146 (NCT02501096) trial (44,45). The overall response rate at 24 weeks was 38.0% (63.6% in patients with MSI-H tumors (n=11) and 36.2% in patients with MSS tumors (n=94)). For previously treated patients, the overall median DOR was 21.2 months, median PFS was 7.4 months and median OS was 16.7 months. Of note, grade 3 or 4 treatment-related adverse events occurred in 83/124 (66.9%) patients.
A quadruple combination regimen (ABCP) with the anti-angiogenic bevacizumab with atezolizumab (anti-PD-L1) and carboplatin-paclitaxel chemotherapy in nonsquamous NSCLC demonstrated improved mPFS compared to bevacizumab-carboplatin-paclitaxel (8.3 vs 6.8 months). This study also reported a PFS benefit in patients with EGFR or ALK mutant NSCLC, which is recognized to have a lower benefit rate from single agent PD-1/PD-L1 inhibitors (46–48). The FDA approval however did not include the EGFR and ALK mutant patient subgroup.
Successful strategies to treating RCC have focused on the sensitivity of RCC to immune checkpoint inhibitors and anti-angiogenic agents, especially given its insensitivity to cytotoxic chemotherapy (49). The combination of bevacizumab with atezolizumab in renal cell cancer did not lead to an improvement in overall survival at interim analysis (33.6 months v 34.9 months) despite demonstrating a superior median PFS versus sunitinib alone (11.2 months v 8.4 months, p<0.02) (50). However, single agent checkpoint inhibition has also been added to the oral small molecule axitinib in two phase III trials. Compared to sunitinib, the combination of pembrolizumab and axitinib led to a benefit in 12-month OS of 89.9% versus 78.3% (HR, 0.53; 95%CI 0.38–0.74; p<0.01) (51). Axitinib has also been assessed in combination with avelumab in untreated RCC and compared to sunitinib. While OS for this combination is yet to be reported, there was PFS benefit of 13.8 months (95% CI 11.1-NR) versus 8.4 months (6.9–11.1) with sunitinib (52). Both combinations have subsequently been FDA approved.
In the IMBrave150 open-label phase 3 trial randomized trial in patients with resectable HCC naive to systemic therapy, the combination of the angiogenesis inhibitor bevacizumab added to atezolizumab was compared to sorafenib in a 2:1 fashion. While the median OS has not been published, overall survival at 12 months was 67.2% for the IO combination vs. 54.6% for sorafenib. Median PFS was improved in the IO group (6.8 months (95%CI, 5.7–8.3) vs. 4.3 months (95%CI, 4.0–5.6)), respectively. Grade 3 or 4 advers events were comparable between both arms, at 56.5% with the IO combination and 55.1% with sorafenib (53).
Also in HCC, the Phase 1b/2 trial combining lenvatinib and pembrolizumab regardless of PD-L1 status resulted in an ORR of 36.4% (8/22) but with 91% experiencing grade 3 or 4 toxicities (54). This combination is now undergoing evaluation in a randomized phase 3 study (NCT04199104).
IO combinations with targeted agents
The combination of BRAF and MEK inhibitors for the treatment of BRAF mutant metastatic melanoma transformed outcomes in this aggressive subgroup, achieving rapid disease control, and now also approved in the adjuvant setting(55–57). In IMspire150, vemurafenib, cobimetinib given in combination with atezolizumab improved progression free survival for patients with unresectable or metastatic melanoma (15.1 months, 95% CI 11.4 – 18.4) versus vemurafenib, cometinib with placebo (10.6 months, 95% CI 9.3 – 12.7). DOR was also improved with the addition of checkpoint inhibition (21.0 (95% CI 15.1 – NE) vs 12.6 months (95% CI 10.5 – 16.6)) with, importantly, little change in Grade 3 – 4 toxicity rate (79% vs 73%). FDA approval was granted in July 2020, with a first cycle of vemurafenib and cobimetinib, and atezolizumab from cycle 2 onwards. Interestingly, a subsequent Phase 3 trial of the anti-PD1 antibody spartalizumab in combination with BRAFi dabrafenib and MEKi trametinib in patients with BRAF-mutated metastatic melanoma was reported at the 2020 ESMO Annual Meeting failed to reach its primary endpoint and raised questions regarding the utility of triplet combinations in melanoma (58).
These encouraging results with regulatory approval in multiple indications with combination immunotherapy strategies have provided the continued impetus for a large number of combination studies currently in pre-clinical trial testing, and selected studies are detailed in Table 2.
Table 2:
Selected preclinical studies of IO combinations
| ICB | Combination | Model | Treatment schedule | Key Results | Ref |
|---|---|---|---|---|---|
| PD-1 | Targeting ANGPT2 and VEGF | Breast cancer (MMTV-PyMT and 4T1), colon cancer (MC38-OVA), melanoma (B16-OVA) | Anti-PD-1 (RMP1–14; 10mg/kg), anti-ANGPT2 (LC06, 10mg/kg), and anti-VEGF (B20–4.1, 10mg/kg),Once per week; Concurrent treatment | Antiangiogenic treatment induced PD-L1 expression in tumor endothelial cells. The combination enhanced activation and cytokine production of CD8+ T cells. |
(240) |
| PD-1 | Targeting VEGF | Colon cancer (CT26) | Anti-PD-1 (RMP1–14; 0.25mg/kg twice a week), and anti-VEGF (Genentech); Concurrent treatment | VEGF enhanced the expression of inhibitory immune checkpoints (PD-1, CTLA4, Tim3 and LAG3) on CD8 T cells. VEGF mediated resistance to anti-PD-1 treatment. Anti-VEGF synergized with anti-PD-1 treatment in controlling tumor growth. |
(241) |
| PD-1 | Targeting ANG2 and VEGF | Breast cancer (EMT-6/CDDP) | Anti-PD-L1 (10F.9G2; 5mg/kg; twice a week), and anti-VEGF /Ang (CVX-241; 30mg/Kg/Week); Concurrent treatment | The combination slightly extend mice survival. | (242) |
| PD-1/ PD-L1 | Cytotoxic chemotherapy | Bladder cancer (MBT-2 and MB49), breast cancer (4T1), colon cancer (MC38) | Anti-PD-1/ anti-PD-L1 (RMP1–14/10F.9G2; 12.5mg/kg; Once per week); Cytotoxic reagents including methotrexate + vinblastine +doxorubicin+ cisplatin(MB49 and MBT-2), cyclophosphamide+ doxorubicin (4T1), capecitabine+ oxaliplatin (MC38); Concurrent treatment | The combination of cytotoxic chemotherapy with ICIs may result in enhanced (MC38 and MB49), similar (4T1) or reduced antitumor activity (MBT-2), in a model- and regimen-dependent fashion. | (243) |
| PD-1/ CTLA-4 | Cytotoxic chemotherapy | Mesothelioma (RN5) | Anti-PD-1/ anti-CTLA-4 (RMP1–14/UC10–4F10–11; 250ug; Once per week), Gemcitabine; Concurrent treatment | The combination significantly extended the survival of tumor-bearing mice and increased tumor infiltration of immune cells. | (244) |
| ICOS | Cytotoxic chemotherapy | Breast cancer (MDA-MB-231) in humanized NSG mice | Anti-ICOS (314.8; 250ug; Once per week), Cyclophosphamide; Concurrent treatment | This combination reduced tumor growth and increased the ratio CD8 /Treg. Depletion of human CD8+ T cells or of murine myeloid cells blunted antitumor effect of this combination treatment. | (245) |
| CTLA-4 | Targeting DNA methyl transferase | Ovarian cancer (BR5FVB1-Akt) | Anti-CTLA-4 (9H10), Decitabine; Concurrent treatment | This combination increased the percentage of effector memory T cells at tumor sites, enhanced the cytokine production by T cells and NK cells, and prolonged the duration of high cytokine production by immune cells. | (246) |
| PD-1 | Targeting DNA methyl transferase and histone deacetylase | Ovarian cancer (ID8) | Anti-PD-1 (200ug twice per week for two weeks), Vidaza (AZA), entinostat (MS17) and/or givinostat (ITF17); Pretreatment of epigenetic reagents for two week and followed by the combination of anti-PD-1 | MS17/ITF17+ α-PD-1 is ineffective, while AZA+α-PD-1 is effective. The longest overall survival was found in the group treated with AZA, ITF and anti-PD-1. Through upregulating the type I IFN signaling, AZA+ITF treatment increased T and NK cell activation, and reduced the number of macrophages. |
(247) |
| PD-L1 | Targeting EZH2 and DNMT1 | Ovarian cancer (ID8) | Anti-PD-L1 (10F.9G2, 10mg/kg twice per week for two weeks); Concurrent treatment | The triple combination displayed synergistic antitumor effect and increased the number of tumor-infiltrating CD8+ T cells and Th1-type chemokine expression. | (248) |
| CTLA-4 | Oncolytic vaccinia virus | Colon cancer (MC38), kidney cancer (Renca) | Αnti-CTLA-4 (9D9, 100ug, twice per week for two weeks), double-deleted western reserve viral strains; Concurrent and sequential | Delayed anti-CTLA-4 treatment enhanced the antitumor effects of the oncolytic vaccinia virus, which are dependent on CD8+ and NK cells, but not CD4+ cells. Concurrent treatment did not improve the efficacy of oncolytic vaccinia. | (249) |
| PD-1/ CTLA-4 | Oncolytic herpes simplex virus (oHSV) | Glioma Cell (005 GSCs and CT-2A) | Anti-PD-1 and anti-CTLA-4 (RMP1–14/9D9, 10mg/Kg and 5mg/Kg respectively, twice per week for two weeks), oHSV-mIL12; Concurrent | The triple combination significantly extended survival, facilitated M1-like polarization of macrophage, and increased the ratio of Teff /Treg. CD4+ and CD8+ T cells as well as macrophages are required for synergistic activity. |
(250) |
| PD-1/ CTLA-4 | Maraba Oncolytic virus(oHSV) | Breast cancer (4T1, EM6 and E0771) | Anti-PD-1 and anti-CTLA-4 (RMP1–14/9D9, 100ug, every two days for a total of five injections), Maraba treatment first, then antibody treatment | Maraba treatment sensitized breast tumor cells to immune checkpoint blockade. | (251) |
| PD-1 | Aglatimagene besadenovec (Adv-tk) | Glioblastoma (GL26 and CT-2) | α -PD-1 (29F.1A12; 200ug; every two days for a total of four injections), intratumoral injection of Adv-tk; Concurrent treatment | Anti-PD-1 enhanced the effectiveness of this oncolytic viral treatment by overcoming interferon-induced PD-L1-mediated inhibitory signals. | (252) |
| PD-1/ CTLA-4 | Oncolytic vaccinia | Kidney cancer (Renca) Breast cancer (MMTV-PyMT transgenic mice) | Anti-PD-1 and anti-CTLA-4 (J43/9D9, 10mg/Kg and 4mg/Kg respectively, every three days), mJX-594 (JX), a targeted and GM-CSF-armed oncolytic vaccinia virus;Concurrent versus Sequential treatment | This combination delayed tumor growth and increased the number and effector function of tumor infiltrating T-cells regardless of the treatment schedule. | (253) |
| PD-1/ CD137/ CD40 | Stereotactic body radiation (SBRT) | Breast cancer (4T1.2 and AT-3) | Anti-CD137 (100 μg) and/or anti-CD40 (100 μg), anti-PD-1 (100 μg) on days 0, 4, 8, and 12 relative to radiotherapy, 12 Gy radiotherapy | Tumor eradication can be achieved by the triple combination of IR, anti-PD-1 and anti-CD137, but not the combination of IR, anti-CD40 and anti-CD137. The profound therapeutic efficacy of the triple combination of IR+ anti-PD-1 + anti-CD137 is associated with the capacity of radiotherapy to enrich the tumor microenvironment of critical tumor-reactive CD8+ T cells that coexpress CD137 and PD-1. |
(254) |
| PD-L1/ CTLA-4 | SBRT | Melanoma (B16), Pancreatic cancer (PDA.4662) and Breast cancer (TSA) | Anti-PD-L1 and anti-4–1BB (10F.9G2/9H10, 200ug, two day before tumor inoculation and every 4 days); Concurrent or sequential treatment | Sequential treatment of anti-CTLA-4 and RT has similar antitumor effect as concurrent treatment. Adding anti-PD-L1 reinvigorated tumor infiltrating T-cells and synergize with the combination of anti-CTLA-4 and RT. |
(255) |
| PD-1/4–1BB | SBRT | Braf mutant and PTEN-deficient melanoma GEMM | Anti-PD-1 and anti-4–1BB (RMP1–14/3H3, 100ug, twice per week for two weeks); SBRT first and followed by antibody treatment | The triple combination significantly prolonged overall survival by increasing immunogenicity of tumors. | (256) |
| PD-1/CTLA-4 | Irreversible electroporation (IRE) | Pancreatic cancer (KRAS*) | Anti-PD-1 and anti-CTLA-4 (J43/9D9, 100ug, every two days for a total of six injections); IRE first and followed by antibody treatment | The combination of IRE and anti-PD-1 promoted tumor infiltration by CD8+ cells and enhanced long-term memory immune response. Adding anti-CTLA-4 to this double combination cannot further improve antitumor activity. |
(257) |
| PD-1 | Adoptive T-cell therapy | Melanoma (B16) and Colon cancer (MC38) | Anti-PD-1 (RMP1–14; 100ug; twice a week), IL-2 and gp100-pulsed DCs; Concurrent treatment | Anti-PD-1 treatment enhances tumor trafficking of transferred T cells via elevating IFN-γ inducible chemokines. | (258) |
| PD-1 | Adoptive T cell therapy | Human Lung cancer (A549) | Anti-PD-1 (10mg/Kg; every five days), Human T cells engineered to target NY-ESO-1; Concurrent treatment | Anti-PD-1 treatment augmented antitumor activity of transferred tumor-reactive T-cells by enhancing effector function of T-cells. | (259) |
| PD-1/CTLA-4 | Targeting Hypoxia | Prostate cancer (TRAMP-c2, and a PTEN and SMAD4-deficient GEMM) | Anti-PD-1 (RMP1–14, 250ug) and anti-CTLA-4 (9H10, 100ug, every three days for a total of six injections), TH-302 (evofosfamide); Concurrent treatment | The triple combination can achieve profound antitumor activity by enhancing the ratio of T-cells/ myeloid-derived suppressor cells. | (260) |
| PD-L1 | Targeting CDK4/6 | Breast cancer (a MMTV-rtTA/tetO-HER2 GEMM) | α -PD-L1 (10F.9G2, 200ug, every 3 days), Abemaciclib/ palbociclib; Concurrent treatment | CDK4/6 inhibition synergize with anti-PD-1 by stimulating the IFN signaling pathway. | (261) |
| PD-1/CTLA-4 | Targeting KIT | Colon cancer (Colon 26) | Anti-PD-1 (5 mg/kg on days 3, 6, 10 and 13), anti-CTLA-4 (5 mg/kg on day 8, 2.5 mg/kg on days 11 and 14), anti-KIT (1, 3, 10, or 15 mg/kg on days 3, 6, 10, and 13) |
The combination of anti-KIT and anti-CTLA-4 reduced tumor growth by reducing the number of M-MDSCs. The combination of anti-KIT with anti–PD-1 delayed tumor development, but without statistical significance. |
(262) |
| PD-1/PD-L1 | Targeting KIT | Gastrointestinal stromal cancer (a Kit V558 Δ/+ GEMM) | Anti-PD-1 (RMP1–14, 250ug ) or anti-PD-L1 (10F.9G2, 200ug) every 4 days for a total of four treatments, Imatinib; Concurrent treatment | Combining imatinib with either anti–PD-1 or anti–PD-L1 can achieve better antitumor activity by suppressing multiple immunosuppressive mechanisms, including IDO pathway. | (263) |
| PD-1/ CTLA-4 | Targeting PI3Kβ | A Braf-mutant and PTEN-deficient melanoma GEMM | Anti-PD-1 (29F.1A12, 100ug)/anti–CTLA-4 (9H10, 100ug) every 2 days for a total of three treatments, GSK2636771; Concurrent treatment | GSK2636771 synergizes with ICIs by enhancing tumor infiltration of T-cells and reducing VEGF expression. | (189) |
| PD-1/CTLA4 | Targeting PI3Kγ | Melanoma and Breast cancer (B16-GMCSF and 4T1) | Anti-PD-1 (RMP1–14, 250ug)/ anti–CTLA-4 (9H10, 100ug) every 3 days for a total of six treatments,IPI-549; Concurrent treatment | The triple combination significantly improved overall survival of tumor-bearing mice. | (264) |
| PD-1 | Targeting PI3Kγ | Head and neck squamous cell cancer (MEER and SCCVII) | α -PD-1 (RMP1–14, 250ug) every 4 days for a total of four treatments, IPI-549; Concurrent treatment | This combination inhibited the expression of immunosuppressive genes, promoted CD8+ T cell recruitment, and induced sustained tumor regression. | (265) |
| PD-1/ CD137/ CD134 | Targeting BRAF and MEK | Melanoma (SM1) | α -PD-1/ α -PD-L1/α –CD137/ α –CD134 (200ug) every 4 days for a total of four treatments, dabrafenib and trametinib; Concurrent | Adding immune-stimulating reagents such as α –CD137 and α –CD134 can further improve the efficacy of the combination of dabrafenib, trametinib and PD-1 inhibition. | (266) |
| PD-1 | Targeting PI3K | Breast cancer (MMTV-PyMT and 4T1) | α-PD-1 (100ug) Twice per week; BKM120 and genetic approach, Concurrent treatment | Inhibiting the PI3K pathway improved tumor sensitivity of α-PD-1 treatment, and increased tumor infiltration of T cells. | (267) |
| PD-1 | Targeting COX2 pathway | Melanoma (a cell line derived from a Braf+/LSL-V600E;Tyr::CreERT2+/o;p16INK4a/ mouse) | α-PD-1(RMP1–14, 200ug)/ Twice per week for two weeks, a genetic approach to silencing PGES1; Concurrent treatment | Inhibiting the PGES1, an effector of the COX2 pathway enhanced the sensitivity of tumor α -PD-1 treatment. | (268) |
| PD-1/CTLA-4 | Targeting MEK | Kras mutant Non-small-cell lung cancer (LLC) | α -PD-1(RMP1–14, 250ug)/ α -CTLA -4(9H10 or 9D9, 100ug) Twice per week for three weeks, Selumetinib (continuous treatment versus pulsatile treatment); concurrent treatment | Only the combination of CTLA-4 blockade with pulsatile MEKi can significantly prolong overall survival of tumor-bearing mice. This synergistic effect is dependent on immune cells. | (269) |
| PD-1 | Targeting PARP | BRAC1-deficient Ovarian cancer (a Trp53−/−,Brca1−/−,Myc GEMM) | α -PD-1(332.8H3, 250ug) every three days, olaparib; concurrent treatment | PARP inhibition augmented antitumor activity of α -PD-1 through a STING-dependent manner. | (144) |
| PD-1/CD134 | STING-based Neoantigen vaccine | Pancreatic cancer (Pan02) | Anti-PD-1 (RMP1–14, 100ug), α-OX40 (OX86, 50ug) ADU-V16 (sting adjuvant), peptide vaccine; concurrent treatment | The triple combination can induce a durable antitumor immune response and eliminate majority of tumor development. | (270) |
| CTLA-4 | Neoantigen vaccine | Pancreatic cancer (Pan02) | Anti–CTLA-4 (9H10, 100ug, every two days for a total of three treatments), pIC and anti– CD40 (adjuvant), peptide vaccine;concurrent treatment | Anti-CTLA-4 significantly increased the number of vaccine-induced effector T cells within tumor, and this effect is dependent on T cell-derived IL-3. | (271) |
Promising immunotherapy combination strategies in clinical trials
The generally safe and durable responses observed with single agent PD-1/L1 inhibitors has established this class of agents as the therapeutic backbone of the majority of immune checkpoint inhibitor combinations (59). The tantalising promise of durable antitumor responses has understandably fed enthusiasm for further combination studies, with 2,251 active PD-1/L1 inhibitor-based combination trials of 295 targets in 2019 (source www.cancerresearch.org) (Figure 2).
In metastatic melanoma, high dose anti-CTLA-4 agent ipilimumab at 10mg/kg versus 3mg/kg was found to have increased median OS (15.7 months versus 11.5 months; p=0.04) at a significant cost of increased treatment-related adverse events (37% versus 18%) (5). Conversely, the anti-PD-1 pembrolizumab has been found to have similar efficacy with similar rates of toxicity at two different weight-based dosing schemes in NSCLC (60) and melanoma (61). Exposure-response modeling in melanoma for nivolumab at doses of 0.1mg/kg-10mg/kg every two weeks has shown linear pharmacokinetics with time-varying clearance and without exposure being a significant predictor of response or survival. The FDA has since recommended a flat-dosing scheme for both nivolumab and pembrolizumab.
PD-L1 antibodies have been found to more effectively block PD-1 signaling in vivo (62) and have less PD-L2 interaction (63). This difference has led some to propose differential efficacy and AEs, although prospective head to head comparisons have not been made. A meta-analysis of 11,379 clinical trial patients found improved survival and progression free survival with anti-PD-1 inhibitors compared to anti-PD-L1 while adverse events were similar (64). PD-1 inhibitors have associated with increased risk of grade 3–4 colitis (0.85% vs. 0.34%, relative risk; 2.52) compared to PD-L1 inhibitors (65).
In preclinical studies, the combination of anti-CTLA4 and anti-PD-1 results in an approximate doubling of the tumor rejection rate compared to anti-PD-1 alone. In the tumor microenvironment of immune checkpoint inhibitor combination treated tumors, increased CD8+ cytotoxic T-cell and CD4+ T effector (Teff) cell tumor infiltration can be observed, along with reduced immunosuppressive T-regulatory (Treg) and myeloid derived suppressor cells (MDSCs) (66,67). As discussed, this combination regimen has led to improved response rates and overall survival in metastatic melanoma, dMMR colorectal cancer and HCC.
Expanding dual checkpoint blockade approaches
Building on the principle of dual checkpoint inhibition with anti-CTLA-4 and anti-PD-1/L1 to enhance responses, targeting other checkpoints in combination with PD-1/L1 inhibition has been an area of intense investigation with the aim of improving responses and optimizing toxicity rates. The immune checkpoint indoleamine 2, 3-dioxygenase 1 (IDO1) results in suppression of Teff and natural killer (NK) cells in the tumor microenvironment, and increases Treg and MDSC activity, leading to immunosuppression (68,69). The combination of anti-CTLA-4 and IDO1 inhibition in mice bearing B16 melanoma resulted in improved survival compared to anti-CTLA-4 alone; however, single agent IDO1 inhibition did not demonstrate antitumor effects in vivo (70). Similarly, in clinical trials assessing IDO1 inhibitors, single agent therapy led to limited or no responses (71,72). A number of phase II studies of IDO1 inhibitors in combination with anti-PD-1 therapy across solid tumors demonstrated promising response rates and reduced toxicities (13–24% grade 3–4 toxicity) compared to CTLA-4 plus PD-1/L1 inhibitor combinations (73). However, research enthusiasm was significantly reduced when a phase III trial in checkpoint-naïve metastatic melanoma demonstrated no PFS benefit of the IDO1 inhibitor (epacadostat) in combination with pembrolizumab versus the PD-1 alone (mPFS: 4.7 vs 4.9 months) (74). This study also highlighted the need for a better understanding of the underlying biology, and the need for improved biomarkers for therapeutic efficacy and patient selection.
T-cells in a chronically exhausted state can co-express multiple checkpoints, suggesting that the targeting of a number of these, concurrently or sequentially, may be required to invoke an antitumorigenic response (75). Studies have reported that the upregulation of the TIM-3 immune checkpoint correlates with resistance to anti-PD-1 agents in both in vivo and clinical samples (76–78). Early results from the combination of anti-TIM3 anti-PD1/L1 therapies have been promising. The AMBER trial of a TIM-3 inhibitor in combination with an anti-PD-1 inhibitor reported partial responses in 4 of 31 patients with advanced NSCLC who had previously progressed on PD-1/L1 inhibitors (79). A trial of the anti-TIM-3 agent LY3321367 given as a single agent reported one partial response out of 23 in a patient who had previously progressed on CTLA-4 plus PD-1 inhibitor combination therapy, with results from the combination of LY3321367 and PD-L1 inhibitior LY3300054 awaited (80).
The immune checkpoint LAG-3 mediates the suppressive activity of Tregs and regulates T-cell expansion and homeostasis, with blockade of LAG-3 resulting in an antitumorigenic TH1 phenotype (81–83). In preclinical models, the combination of PD-1 and LAG-3 blockade resulted in improved tumor rejection compared to PD-1 inhibition alone (84–86). A phase II combination trial of LAG525, targeting LAG-3, and the PD-1 inhibitor spartalizumab in 121 heavily pretreated patients with a range of solid tumors demonstrated 1 complete response and 11 partial responses (9.9%) with combination therapy, although no responses were observed with single agent LAG-3 blockade (87). Another LAG-3 inhibitor relatlimab in combination with nivolumab demonstrated a response rate of 11.5% in 61 patients with melanoma whose disease had progressed on prior anti–PD-1/L1 therapy. This response rate appeared to be 3.5-fold higher in patients with LAG-3 expression of at least 1% versus those with less than 1% LAG-3 expression (88). This relatlimab combination is now being assessed in a Phase 2/3 trial in patients with advanced melanoma in the first line setting. In a smaller study of 15 patients with the anti-LAG-3 agent MK-4280 with pembrolizumab, 4 of 15 patients achived a partial response (89). The lack of single agent LAG-3 inhibitor activity has led to caution; however, there is a clear and urgent clinical need to understand mechanisms of resistance to current immunotherapy regimens so as to develop new options for patients who develop progressive disease on immunotherapy. The PLATforM study aims to address this critical question with the combination of LAG525 and spartalizumab, as well as spartalizumab in combination with c-MET or CDK4/6 inhibition or anti-IL-1β in patients with advanced metastatic melanoma (90). This adaptive trial design represents a rational approach and may be a model for future clinical trials in the post-immunotherapy space.
Bispecific Antibodies Development and Potential
The concept of dual checkpoint inhibition may lead to higher order combinations and will be expanded upon later in this review. An emerging approach that promises further specificity in targeting multiple checkpoints is the development of bispecific antibodies (91). This evolving technology allows a monovalent antibody that can target 2 or more receptors opens up multiple potential approaches to drug development in this space. Specifically, some of the first emerging bispecific antibodies are targeting 2 checkpoints such as bispecifics to PD-1 and CTLA-4, allowing a more specific targeting of the CTLA-4 receptor on PD-1 positive cells and promises to uncouple efficacy from toxicity of this combination. The interesting characteristic of this approach is that depending on which receptor the bispecific has higher affinity to (e.g., PD-1) will define which cells will be targeted for inhibition of the second receptor (e.g., CTLA-4), and sometimes at hundreds of folds higher affinity (92) (NCT03517488).
From an immunotherapy resistance perspective, using bispecifics can allow targeting specific mechanisms of resistance such as TGFß or MET inhibition (93). For instance, a particulary interesting molecule that has a PD-L1 antibody and TGF beta-TRAP as the second part of the bispecific allows TGFß levels, a well described mediator of resistance in the tumor micronenvironment, to be modulated favorably, and this approach is currently in clinical testing alone or in combination (94) (NCT04349280). Virtually every mechanism of resistance described in this review could potentially be amenable to targeting by bispecific antibodies. Bispecifics allow targeting specific cells as well, with CD3 bearing bispecifics called T cell engagers (TCE) or BiTE that could attach a T cell directly to a tumor cell and initiate an immune response that is MHC-independent (94). Trispecifics are also being develops with a focus on NK cells in so called TriNKETs (trispecific NK cell engager therapeutics). Those novel agents are entering the therapeutic armamentarium and will likely be used alone or in combination with other therapeutics including chemotherapy and TKIs allowing multi-pronged targeting of immunotherapy resistance.
Neoadjuvant combination immune blockade
Delivering combination immune checkpoint inhibition in the neoadjuvant setting takes advantage of the in situ tumor to stimulate immune responses, resulting in increased clonal expansion of T cells compared to adjuvant IO (95). Systemic immunosuppression is observed in the metastatic setting (96). and patients with metastatic disease have often experienced a prior treatment course including immunosuppressive treatments. It could be hypothesized, therefore, that treating at the earliest stage possible not only results in increased exposure to tumor antigens but also utilizes an active, responsive immune system which may improve long term systemic immunity.
In keeping with this, combination nivolumab-ipilimumab results in improved pathlogical complete response (pCR) rates in resectable melanoma compared to nivolumab alone (45% vs 25% in a 23 patient study), although with an increased toxicity cost (73% Grade 3 – 4 toxicities vs 8%) (97). A randomized trial examined 3 different doses and schedules of the combination of ipilimumab and nivolumab in the neoadjuvant therapy of melanoma and conclude that 2 doses of ipilimumab at 1mg/kg and nivolumab at 3 mg/kg IV 3 weeks apart gave the optimal outcome of over 70% pCR rate and 30% toxicity and is the currently accepted neo-adjuvant regimen (98). Similarly in resectable colorectal cancer, neoadjuvant nivolumab-ipilimumab for 4 weeks resulted in pathological responses in 100% of MSI cancers, and 27% of MSS disease (99). Grade 3 – 4 toxicities were experienced by 13% with one patient (out of 40) requiring infliximab for checkpoint-induced colitis. In head and neck cancer, where palliative immunotherapy has been effective, neoadjuvant nivolumab-ipilimumab resulted in responses in 69% of patients, with limited toxicity from this short course of treatment (100). Neoadjuvant combinations with chemotherapy or other standard of care agents are also promising, with combinations in breast cancer discussed below.
Exploiting co-stimulatory molecules to enhance antitumorigenic activity
Activating the immune system using co-stimulatory molecules in combination with anti-PD-1/L1 has been shown to lead to improved outcomes by enhancing antitumorigenic activity. For example, activation of IL-2 signalling via CD122 results in activation of naïve CD8+ T-cells and also increases NK cell activity (101,102). The combination of the CD122 agonist NKTR-214 and anti-PD-1 demonstrated promising results in preclinical models of NSCLC and colorectal cancer (103). In treatment-naïve metastatic melanoma, the combination of NKTR-214 and nivolumab resulted in an objective response rate of 53%, with 10 (24%) of 41 patients experiencing a complete response (104). This combination has now been granted breakthrough therapy designation from the FDA, expected to expedite this combination through ongoing phase III studies. Other co-stimulatory molecules, such as 4–1BB, OX-40 and GITR, similarly result in increased CD8+ and NK cell activity and reduced Treg mediated immunosuppression, with multiple phase I/II trials ongoing in combination with immune checkpoint blockade (105–107).
Innate immune stimulating agents, such as TLR9 or STING (STimulator of INterferon Genes) agonists, delivered intratumorally can prime the tumor microenvironment prior to immune checkpoint therapy. A number of TLR9 agonists (CpG-oligodeoxynucleotides) are in ongoing phase I – III studies in combination with anti-PD-1 and anti-CTLA4 immune checkpoint inhibition. A Phase Ib study of the TLR9 agonist SD-101 in combination with pembrolizumab resulted in an ORR of 78% in IO naïve patients with melanoma and 15% of checkpoint (anti-PD-1) refractory disease (108), with CD8+ and natural killer (NK) cell trafficking to the tumor site. A similar approach with lefitolimod and ipilimumab is ongoing in an all-comer study (109). Following promising results in Phase II, with 38% ORR in anti-PD-1-refractory melanoma (110), the combination of tilsotolimod and ipilimumab is now under investigation in the Phase III study ILLUMINATE301 (111). Initial studies of intratumoral STING agonists have also demonstrated the ability of these agents to activate local immune responses and synergize with anti-PD-1 therapy (112,113), although mixed results from trials to date have demonstrated the need for patient selection for these approaches, and further pre-clinical studies to understand potential resistance mechanisms.
Immune checkpoint inhibition and CAR-T cell therapy
Chimeric antigen receptor (CAR)-T cells directly target tumor-specific antigens, independent of major histocompatibility complex expression, with successful outcomes in a number of haematological malignancies(114). However, replicating this success in solid tumors has proved challenging. One potential resistance mechanism is the upregulation of immune checkpoint expression common to solid tumors, causing CAR-T cells to become exhausted and ineffective. In mesothelioma, which has already demonstrated positive efficacy data with nivolumab-ipilimumab combination therapy (115), CAR-T cells targeting mesothelin followed by anti-PD-1 resulted in an ORR of 50% (7/14 patients) (116). CAR-T cells can also be manipulated using CRISPR-cas9 to disrupt PD-1 expression. The advantages of this approach include potential reduction in systemic effects of PD-1 inhibition with a highly-specific PD-1 knockdown in tumor-targeted T cells only, as well as avoiding the need for repeated administrations of systemic anti-PD-1 (117). Both PD-1 disrupted CAR-T cell therapy and combinations with systemic anti-PD-1 therapy are the subject of ongoing study in the early phase setting (118).
Improving patient outcomes with chemotherapy and radiotherapy combinations
The role of the immune system in response to chemotherapy has been noted since the 1970s, when studies assessing the mechanisms of action of anthracyclines demonstrated an improved response in immuno-competent murine models (119). Immunogenic cell death (ICD) has been proposed as a key mechanism resulting in an immune-infiltrated tumor microenvironment favorable for immunotherapy, suggesting that ICD-inducing regimens may synergize with immune checkpoint inhibitors (120). Ongoing chemotherapy-immunotherapy combination trials are discussed in greater detail in other review articles (121). The combination of platinum chemotherapy and immune checkpoint blockade has been successful in non-small cell lung cancer (22), with the immunogenic response generated by chemotherapy overcoming the lower response rates observed with anti-PD-1 treatment alone in low PD-L1 expressing tumors (31). DNA damaging chemotherapy additionally activates the innate immune cGAS-STING pathway via cytosolic DNA, resulting in an immune rich microenvironment that may predict responses to checkpoint blockade (122–124).
Although taxanes do not directly cause immunogenic cell death, they decrease intratumoral immunosuppressive MDSCs and Tregs, augmenting cytotoxic T-lymphocyte activity (125,126). The combination of nab-paclitaxel and atezolizumab is approved for PD-L1 positive metastatic triple negative breast cancer, but was not effective in the PD-L1 negative population (127). Alternative chemotherapy regimens may further improve patient outcomes, as illustrated by the ISPY-2 studies in neoadjuvant breast cancer with pembrolizumab and standard anthracycline-based neoadjuvant chemotherapy (128). In these studies, neoadjuvant pembrolizumab and paclitaxel followed by anthracycline-cyclophosphamide resulted in pathological complete response (pCR) rates of 60% in triple negative breast cancer compared to 22% without pembrolizumab, and 30% vs 13% in hormone receptor positive/HER2 negative disease (129). In a similar setting, 4 cycles of pembrolizumab in combination with paclitaxel-carboplatin chemotherapy, followed by anthracycline-cyclophosphamide in triple negative breast cancer resulted in a pCR of 64.8% vs 51.2% without pembrolizumab. In this study, patients received adjuvant pembrolizumab or placebo for a further 9 cycles following resection, with follow-up ongoing (130).
DNA damage by radiotherapy is associated with a range of immunogenic effects, including activation of the cGAS-STING pathway, increasing neoantigen expression and upregulation of PD-L1 expression (131–133). Sequential chemoradiotherapy-IO has been shown to lead to significantly improved patient outcomes in stage III NSCLC (27,28), while a phase II study of concurrent anti-CTLA-4 plus radiotherapy in NSCLC suggested improved responses in patients previously resistant to anti-CTLA-4 given alone or in combination with chemotherapy (134). While over 100 trials of anti-PD-1/L1 therapy in combination with chemo/radiotherapy are ongoing (135), questions remain over the optimal dose and schedule of radiotherapy-immunotherapy combinations. For example, serial low doses of radiation have been shown to activate a STING-mediated immune response, while higher doses (i.e. ≥ 12–18 Gy) instead activate TREX1, removing cytosolic DNA and preventing STING activation (136). Concurrent radiation may result in enhanced responses, but this may be dependent on the target (CTLA-4 vs. PD-1/L1), dose and delivery of the radiation using proton-beam or other approaches (137). While the abscopal effect of radiotherapy may be enhanced by immune targeting therapies, clinical evidence for this approach has thus far been largely confined to case reports, rather than large scale trials (138,139).
Activating innate immunity with DNA damage response inhibitors
DNA damage response (DDR) deficiency results in activation of the innate immune system via the critical cGAS-STING pathway, which is required for interferon expression in response to cytoplasmic DNA (140–142). A number of studies have now demonstrated cytoplasmic DNA and tumor-cell intrinsic activation of the STING pathway following treatment with agents targeting the DDR, in particular poly(ADP-ribose) polymerase (PARP) inhibitors (143–146). PARP inhibition leads to activation of intratumoral dendritic cells and increased CD8+ infiltration via activation of the STING pathway. Interestingly, PARP inhibition also reduces the PARylation of STAT3, while increasing STAT3 transcriptional activity and expression of PD-L1 (147). These studies suggest that PARP inhibition acts synergistically with immune checkpoint blockade in both DNA repair deficient (eg. BRCA1/2 mutant) and proficient cancers (148).
Early phase trials of PARP inhibitor plus immune checkpoint inhibitor combinations have been promising, with activity demonstrated in patients with advanced castration resistant prostate (149,150), triple negative breast (151) and ovarian cancers (152). This combination has demonstrated antitumor activity in BRCA1/2-wildtype TNBC, and platinum-resistant ovarian cancer, populations that are typically resistant to single agent PARP inhibitors. Importantly, bone marrow suppression typical of PARP inhibitor toxicity was shown to occur at similar rates to that observed with single agent therapy, and there were also no significant increase in immune-related toxicities.
Targeting the ATR/CHK1 pathway also results in activation of a STING-dependent immune response (145) and ATR inhibitors have demonstrated promising clinical activity in combination with durvalumab (153). There is also growing evidence for a number of DDR inhibitors in combination with immune checkpoint inhibitors, including those targeting WEE1 (154), ATM (155,156) and DNA-PK (157). With improved understanding of the impact of these novel DDR targeting agents on the immune response in specific patient subgroups, it is tempting to propose that future DDR inhibitor combination therapies will focus on targeting specific patient populations, based on the DNA repair capacity identified in individual tumors, in combination with immune targeting agents designed to appropriately modulate the tumor microenvironment.
Increasing tumor immunogenicity with molecularly targeted agents
A number of targeted therapies are recognized to increase the immunogenicity of tumors, and therefore are predicted to work synergistically with immune checkpoint blockade. For example, inhibitors of the mitogen activated protein kinase (MAPK) pathway result in increased expression of major histocompatibility complex 1 (MHC-I) on the tumor cell surface, with subsequent increase in antigen-specific T-effector cells and T-cell mediated cytotoxicity (122,158). This combination has been hampered in early clinical trials by associated increases in toxicity rates, with a trial of anti-CTLA-4 and the BRAF inhibitor vemurafenib terminated due to a high rate of hepatotoxicity (159). The addition of the MEK inhibitor trametinib, while reducing hepatotoxicity, resulted in colitis and bowel perforation in 2 of 7 patients receiving this triplet combination (160). More promisingly, a phase II study of BRAF and MEK inhibition with anti-PD-1/L1 therapy in treatment-naïve BRAF mutant melanoma reported tolerability (58% grade 3–5 toxicities vs. 27% in patients receiving BRAF and MEK inhibition alone) and improved PFS (12 month PFS 59% vs. 45%)(161). A phase III randomized trial in patients with unresectable stage IIIC-IV BRAF v600 mutant melanoma assessed vemurafenib plus cobimetinib with atezolizumab versus placebo with atezolizumab. In the triplet therapy group, progression-free survival was superior versus the reference arm (15.1 vs. 10.6 months; HR 0.78; 95% CI 0.63–0.97; p=0.025), with minimal increases in grade 3–4 toxicities (79% v 73%), this study resulted in FDA approval of the triplet combination in the first line therapy of BRAF mutant metastatic melanoma (162). However, a phase III study of MEK inhibition with atezolizumab in heavily pretreated MSS colorectal cancer did not result in improved survival compared to standard-of-care regorafenib therapy alone (163).
Epigenetic modification by histone deacetylase (HDAC) inhibitors reduces the number of myeloid derived suppressor cells (MDSCs) in the tumor microenvironment in preclinical models, resulting in improved response to anti-PD-1 blockade (164,165). In addition, treatment with HDAC inhibition leads to upregulation of chemokines CXCL10 and CCL5 in the tumor microenvironment that, in turn, drives recruitment of antitumorigenic CD8+ T cells (166). A phase Ib/II trial of the HDAC inhibitor etinostat with pembrolizumab in patients with metastatic melanoma who had previously progressed on immune checkpoint therapy demonstrated responses in 10 (18.9%) of of 53 patients, including one complete response (167). Treatment with demethylating agents such as azacitdine upregulates MHC-I on tumor cells and also unmasks endogenous retroviruses, resulting in cytoplasmic dsRNA that activates innate immune responses, increasing tumor immunogenicity (37,168,169). A combination study of azacitidine and nivolumab demonstrated responses in relapsed or refractory acute myeloid leukaemia (170). However, a phase II study of azacitidine and pembrolizumab in microsatellite stable colorectal cancer demonstrated only 1 partial response out of 31 patients to the combination therapy (171).
Exploiting murine tumor models for optimal combinatorial development
Given our current superficial knowledge of the therapeutic effects of different combinations on the immune system, multiple preclinical tumor models have been developed to provide a deeper understanding of the mechanisms of action and to help predict the efficacy and safety of different immunotherapeutic interventions in cancer patients. As summarized in Table 2, many proof-of-principle preclinical studies involving a wide range of immunotherapy combinations have been conducted with different murine models. The results from these studies provide a compendium of immunotherapy combinations for future clinical assessment. While none of these murine models completely mirror the heterogeneity and adaptability of the antitumor immune network in cancer patients, each one has its unique value in the cancer immunotherapy research field. Therefore, understanding the pros and cons of each model can guide the optimal selection of appropriate murine tumor models for preclinical assessment of different immunotherapy combinations. Current murine tumor models may be summarized into three categories based on the types of immune systems involved: syngeneic, chimeric and humanized tumor models.
Syngeneic tumor models
Mouse models in this category are fully immunocompetent. Tumor development in these mice is induced either by inoculation of murine cell lines or by tissue-specific activation of oncogenic pathways. Due to the general ease of use, inoculating wild-type mice with established syngeneic tumor cell lines is by far the most commonly used approach to generate syngeneic tumor models. Several inbred strains such as C57BL/6, BABL/C, 129/sv and DBA/2 have been used to generate a variety of murine cell lines to represent different cancer types, including but not limited to B16 (melanoma) (172), MCA205 (sarcoma) (173), A20 (lymphoma) (174), P815 (mastocytoma) (175), 4T1 (breast cancer) (176), 344SQ (lung cancer) (177), ID8 (ovarian cancer) (178), MB49 (bladder cancer) (179), and MC38 (180) and CT26 (181) (colon cancer). The majority of these tumor cell lines display a certain degree of genome instability, which results in the expression of tumor-specific antigens that may be recognized by the adaptive immune system, particularly T-cells (182,183). Since mice challenged with these tumor cells have intact murine immune systems, a “physiologically-relevant” tumor immune microenvironment can be achieved in these tumor-bearing mice.
However, host mice used in syngeneic cell line models are commonly young and healthy inbred mice, which are housed in specific pathogen-free conditions. In addition, there is mounting evidence supporting a high divergence in immune responses among inbred mouse strains and cell lines. For example, the C57BL/6 strain is TH1-biased, while BALB/c and DBA/2 strains are TH2-biased (184). Discrepancies in the antitumor effects of IO combinations may thus be observed depending on the murine tumor cell line model chosen. Therefore, to increase the likelihood of successful clinical translation, the use of multiple murine tumor cell line models on a range of genetic backgrounds is encouraged. Moreover, introducing fundamental cancer-driven mutations into tumor cell lines or perturbing environmental factors in housing conditions, such as diet and microbiome, should be used to more broadly recapitulate interpatient heterogeneity.
Recent advances in technologies of in vivo genetic manipulation allow us to precisely control the timing, duration and tissue type of gene expression. Using these approaches, genetically engineered mouse models (GEMMs) have been applied in immunotherapy studies to better mimic the heterogeneity of the natural steps of tumor development in cancer patients. The Mouse Tumor Biology Database (http://tumor.informatics.jax.org) provides a comprehensive list of GEMM models for a variety of human cancer types (185). As tumor development in GEMMs is mainly driven by one or two genetic alterations in dominant oncogenes, these models generally have a low tumor mutation burden. Furthermore, de novo tumor growth in GEMMs provides sufficient time to trigger a broad range of immunosuppressive mechanisms within the tumor microenvironment. This lack of neoantigens and the profound immunosuppression make GEMMs typically highly resistant to cancer immunotherapy, sometimes even more so than clinically equivalent malignancies. For example, melanomas derived from the Tyr gene promoted BRAF-mutant PTEN loss model (186) have been demonstrated to be resistant to immune checkpoint inhibitors (187). Depleting Tregs also failed to control the development of tumors with transgenic expression of the RET receptor tyrosine kinase gene, suggesting that other immunosuppressive cells play an important role and replace immunosuppressive, tumor-promoting functions of Tregs (188). Therefore, effective immunotherapy strategies should consider including the inhibition of Treg migration into the tumor combined with neutralization of other immunosuppressive cells and factors in the tumor microenvironment (188). These models have now been used to address the challenges identified in cancer patients with low immunogenic tumors or immune-resistant tumors. Peng and colleagues utilized the BRAF mutant-PTEN loss GEMM to demonstrate that inhibition of the oncogenic activation of the PI3K pathway by PTEN loss can sensitize tumors to immune checkpoint blockade therapy (189). Despite the expensive and time-consuming procedures required, GEMMs are currently the optimal models to use to evaluate the potential of IO combinations to overcome immune resistance, particularly resistance associated with genetic alterations.
Chimeric tumor models
Although syngeneic murine models provide an opportunity to characterize the immunological changes associated with IO combinations in the presence of an intact immune system, they cannot be used to test the human version of IO reagents due to cross-species differences. To circumvent this limitation, chimeric murine tumor models have been developed by knocking in human immune-related genes. In these immunocompetent tumor models, the majority of immune compartments are murine-based, while the knocked-in human genes allow a portion of their immune system to be controlled by certain human immune factors. The first human knock in (KI) model was the HLA-A2 murine strain. Mice carrying a human MHC class I gene (HLA-A2.1) were created to evaluate the antitumor activity of HLA-restricted vaccination strategies (190). Since then, further models have been generated to study clinical grade immunomodulatory agents including anti-human CTLA-4, OX40 or Tim3 (191–193). These studies successfully confirmed the in vivo efficacy of immunomodulatory agents alone or in combination with other treatment modalities. Chimeric models, therefore, are valuable in validating combination effects of clinical grade reagents targeting novel immune regulators in vivo, with commercial services now available to generate customized models.
Humanized tumor models
To better represent tumor heterogeneity within cancer patients, efforts have been made to generate humanized tumor models. In these models, immunocompromised mice are partially reconstituted with the human immune system by transplantation of CD34+ hematopoietic progenitor cells (HPCs) from human umbilical cord blood, bone marrow or peripheral blood. The implantation of tumor tissues or cell lines derived from cancer patients in humanized host mice is used to induce tumor development. The NSG strain, which carries the NOD/SCID IL2Rγ chain knockout and lacks functional T, B and NK cells, is most frequently employed as the host strain for humanized models. Additional genetic modifications have been recently performed in NSG and other immunodeficient mice to express human cytokines, including stem cell factor, M-CSF, GM-CSF and/or IL-3, which are essential for the growth and differentiation of HPCs and to support human myeloid reconsitution (194,195). The expression of human cytokines in these new strains, namely NSG-SGM3, NOG-ExL, MSTRG and MISTRG, significantly increases the engraftment rate of the human immune system (196). The efficacy of pembrolizumab in a CD8+ T cell-dependent manner was successfully reproduced in these humanized models (197,198).
However, antitumor immune responses in these models come largely from allogenic immune cells. Compared with natural tumor rejection through tumor-associated antigens, the antitumor immune responses mediated by allogenic rejection may be more intense. Moreover, the cost of humanized tumor models is dramatically higher than other two types of models described above. Therefore, humanized tumor models are more suited for validating the efficacy and safety of the human versions of immunotherapy combinations than discovering novel combinations.
High order immunotherapy combinations
Highly active anti-retroviral therapy (HAART) consisting of four-drug combinations that completely suppress Human Immunodeficiency Virus (HIV) replication has evolved to become so efficacious yet tolerable that it can even be used as a routine prophylactic in “at risk” populations. HIV therapy began, however, with single, toxic drugs with limited efficacy which gained efficacy but even greater toxicity in two-drug combinations. It was only with the inception of truly virus-specific therapeutics that high-order, high-efficacy combination therapies became practical. In cancer, high-order (3 or more) immunotherapies have now begun to enter the clinic with promising initial signs against “cold” tumors. Much like HAART, however, the development of widely applicable and broadly efficacious multiple IO combinations will necessitate the development of more tumor-specific modulators of host immunity.
Due to the toxicities inherent in high order combinations of existing immunotherapies, these trials have thus far been limited to patients with metastatic “cold” cancers (Figure 1). In preclinical models of pancreatic ductal adenocarcinoma (PDAC), for example, only high order combinations of chemotherapy, T cell checkpoint blockade, and antigen-presentng cell activation via CD40 agonism could promote durable benefit and tumor regression (199,200). This combination of checkpoint blockade, CD40 agonist antibody and chemotherapy has recently reported promising Phase I clinical trial data in PDAC and final data are eagerly awaited (201). In this case, existing agents were combined successfully through thoughtful sequencing in order to avoid intolerable levels of additive toxicity.
In many cases, the ideal setting for high order IO combinations would neither necessistate dose-reduction to sub-optimal levels nor sequencing of component therapies. To this end, multiple immunomodulatory antibodies have entered the clinic having been engineered to act selectively within the tumor microenvironment, thus sparing the host from adverse events. These novel approaches can be broadly classified into conditionally active versus bi-specific antibodies. The most advanced example of conditionally active antibodies are “probodies”, in which the antigen-binding region of the antibody is blocked by a peptide which is tethered to the antibody with a linker containing multiple cleavage sites for tumor-selective proteases (202). In a Phase I trial of the PD-L1 probody CX-072, only 6% of the 72 patients treated experienced a Grade 3 or greater adverse event and no AE-related discontinuations occurred (203). The alternative approach to tumor selectivity are bispecific antibodies in which one arm of the antibody, usually the higher affinity, binds to a tumor or tumor microenviroment selective antigen thus localizing the effect of the lower affinity active arm. The CD137 (4–1BB) and HER-2 bispecific antibody, PRS-343, in which 4–1BB activation is sequestered in the tumor microenvironment by HER-2 binding is one of the most advanced examples of this approach (204). While CD137 activation can cause liver toxicity in certain patients (4), this construct safely sequesters its activity to the tumor. PRS-343 was safe and well tolerated in the first-in-human phase I trial involving 53 HER2+ patients with advanced solid tumors. Two patients achieved radiological responses, while other patients had stable disease. Importantly, PRS-343 demonstrated a potent increase in CD8+ T-cell numbers and proliferative index in the tumor microenvironment of responders, suggestive of 4–1BB agonism on T-cells. By localizing the effect of checkpoint blocking and co-stimulatory agonist antibodies to the tumor, these approaches set the stage for tolerable combinations of multiple synergistic immunotherapies, e.g. CTLA-4, PD-1 and 4–1BB inhibitor combination therapy.
An alternative emergent approach to multi-modal immunotherapy circumvents systemic toxicity concerns by administering some or all of therapeutics directly into the tumor itself. Innate immune agonists such as activators of Toll-like Receptors (TLR) or the STING pathway are injected intra-tumorally in a single lesion to generate T-cell responses which can then traffic to and eradicate distal sites of cancer (abscopal effect) (205). While clinical combinations with these therapies pair them with a single systemic checkpoint antibody, preclinical studies have demonstrated that as many as three immunomodulatory antibodies can be co-injected with a STING agonist resulting in superior capacity to eliminate non-injected sites of disease (206,207). With this type of non-toxic scaffold to build upon, current efforts can focus on identifying systemic therapies to weaken the immune resistance of uninjected sites of cancer making it easier for the injection-mobilized T-cells to eradicate them.
While novel, less toxic immunotherapies will form the backbone of most emergent high order IO combinations, the effect of these therapies may be augmented and extended through the addition of FDA-approved, even over-the-counter, drugs which can contribute to antitumor immunity without significant toxicities. Extracellular adenosine, for example, plays a clearly established role in immune suppression in the tumor microenvironment (208). Although still pending approval in United States, the adenosine receptor blocking compound istradefylline is approved in Japan for the treatment of Parkinson’s disease where it has shown high tolerability (209). Such adenosine receptor antagonists may potentially be a promising addition to any immunotherapy combination (210). Multiple studies have demonstrated antitumor efficacy of inhibition of the renin-angiotensin system through decreased suppressive polarization and activation of tumor stroma (211). These drugs are commonly used to treat cardiovascular disease and would be a rational addition to immunotherapy combination regimens. Platelets have also been implicated in tumor immune suppression, which can be blocked by clopidogrel and over-the-counter aspirin (212). PDE5 inhibitors may also have value through their capacity to inhibit myeloid derived suppressor cells (213). Further study is warranted to assess the capacity of these non-toxic, widely available drugs to augment tumor immunity; however, one or more could likely become key components of multi-modal immunotherapy combination therapy.
Finally, the greatest challenge to development of these high order combinations may not be toxicity or engineering, but rather finding a system in which the most effective combinations can be screened, so that only the most promising ones enter clinical trials. As discussed above, there are over 2250 trials evaluating various PD-1/ L1 inhibitor combinations, a number which is increasingly straining resources and the availability of appropriate patients to evaluate potential immunotherapies. The number of possible permutations involved in developing five-drug IO combination therapies makes primary screening in patients impractical, if not impossible. In this setting, drugs must first be prioritized based on single agent efficacy, underlying biologic rationale for combination, and lack of additive toxicity. Potential combinations involving such agents can then be screened through appropriate murine tumor models of cancer to identify the most promising candidates for study in patient trials. Using such a defined development and screening approach and leveraging both existing therapies, novel engineering, and selective FDA-approved drugs may provide a path to multi-modal IO combinations, which approach the efficacy and tolerability of HAART regimens for HIV.
Novel biomarker and clinical trial strategies for combination approaches
Drug development to reverse resistance to immunotherapy needs to take into account multiple streams of knowledge being generated in the preclinical, translational, and clinical spaces. For instance, PD-L1 testing seems to predict a better response to immunotherapy across the board. However, due its poor ability to discriminate antitumor efficacy in cancers such as melanoma, it has not entered clinical practice as a useful test. However, given our increasing abilities to pathologically, genomically, and transcriptomally characterize cancers, we have a mandate to now hone drug development into better defined patient populations where therapeutic benefit can be augmented.
A recent effort initiated by SITC provided updated and improved clinical definitions of drug resistance to harmonize and standardize drug development efforts in the PD-1 inhibitor resistance space (214). Specifically, the SITC Resistance Taskforce defined three main clinical patterns of resistance to single agent anti-PD-(L)1 antibodies (1). Primary resistance that occurs while on treatment with single agent therapy; this pattern requires patients to have adequate exposure to the agent (at least 6 weeks), to have shown no evidence of therapeutic benefit (no CR or PR or SD>6 months), and to have a confirmatory scan to confirm progression (and predominantly to rule out pseudo-progression) (2). Secondary resistance is defined as tumor progression after an initial clinical benefit (achievement of radiological complete response (CR), parial response (PR), or stable disease (SD)>6 months), and also requires a confirmatory scan (3). The third pattern defined in this effort, is recurrence or progression of the tumor after halting therapy. This definition assigns the pattern to be similar to either primary or secondary resistance depending on the setting of therapy (adjuvant, neo-adjuvant, or advanced disease) and the time from when therapy was discontinued (<12 weeks or >6 months).
Another clinical pattern of resistance is site-specific resistance, where patients may experience control of the disease in most of their body but have progressive lesions in one or two organs. The central nervous system (CNS) is a particularly relevant location and has emerged as a major therapeutic challenge. For instance, in melanoma, it has been reported that the CNS is the most frequent site of first progression on single agent checkpoint blockade (215). Moving forward, it will be important to include this population of patients with progressing brain metastases in trials of novel agents, so as to enhance our understanding of the intracranial activity of immunotherapeutic agents, as well as to develop more brain-penetrant agents, and approaches that tackle this most devastating complication of cancer progression. This has been recognized by the FDA, which has issued guidance to increase the representation of patients with CNS metastases on clinical trials (216).
Improved patient selection on immunotherapy combination clinical trials may seem like an aspirational goal at this time, but all the elements now appear to be falling in place, and a concerted effort between academia and industry partners could indeed accelerate this approach to achieve improved patient outcomes. Some of the examples listed above provide context to this evolving immunotherapeutic landscape. For instance, the failure of the IDO inhibitor epacadostat in a large Phase III melanoma study despite demonstrating somewhat promising data in early phase trials, has been attributed largely to the fact that this was an unselected population, and that this agent may indeed still have useful activity in a subset of patients (74,217,218). It is therefore encouraging to see how the development of other immunotherapy combination trials seem to be informed by that experience. An example is the anti-LAG3 antibody relatlimab and nivolumab combination already described above. The Phase I data of this combination in the second line setting indicated an ORR of 11% in unselected melanoma patients, but was 18% in LAG-3 positive patients (88). Subsequently, a Phase 2/3 trial of relatlimab was initiated in the first line setting in melanoma, where, while all front line patients are still eligible, does require screening of patient tumor samples obtained within a 3-month period before enrollment and for evaluable results from PD-L1 and LAG-3 IHC staining to be available prior to patients being randomized (NCT03743766). Additionally, the study is stratified for both of these biomarkers, as well as BRAF mutations, which will be incredibly valuable at the time of study analysis since one can start gleaning a specific effect of the combination in a particular subset of patients. In general, such biomarker-driven approaches will of course have to be carefully designed, taking into account all the statistical caveats of subgroup analyses.
This trend is now becoming increasingly visible across the immunotherapeutic landscape, and still needs to be furthered, perhaps even more so in the immunotherapy-refractory setting in cancers where immune checkpoint blockade has become a front line approach. Translational and preclinical studies are continually identifying novel mechanisms of resistance to different immunotherapy agents on an ongoing basis. Investigators and pharma should be emboldened to conduct studies in moleculary-characterized populations of patients where the agent in use specifically targets the biomarker used for patient selection. An example of such a strategy is the use of selective PI3Kβ inhibitors in patients harboring tumors with PTEN loss. Such a thoughtful biomarker-driven approach takes into consideration that certain key signaling networks that can be potently targeted in tumors, such as the PI3K pathway, are also critical for T cell activation and function. Being able to identify specific molecularly targeted agents that may have a differential impact on tumor versus T cells is the new quest for a “therapeutic window”. This is especially true of novel metabolism-directed agents, such as glutaminase inhibitors (219).
There are also opportunities for the development of agents with pleotrophic effects where patient selection may be challenging, but have the potential to impact multiple nodes of immune activation and tolerance. An example of those approaches are agents targeting epigenetic modulation, such as hypomethylating agents (220), HDAC inhibitors (221), bromodomain and extra-terminal domain (BET) inhibitors (222), which have genome-wide effects and affect both tumor cells and T cells.
In 2017, the FDA approved pembrolizumab for treatment of microsatellite instability-high (MSI-H) or deficient DNA mismatch repair (dMMR) regardless of primary tumor site (223), which has subsequently been supported by a randomized trial of pembrolizumab utilized for 27 different tumor types (224). However MSI-H may have been a surrogate for TMB-H (225), which has subsequently used as a biomarker for selection both in tissue and blood based whole exome sequencing (226). A meta-regression analysis of 117 clinical trials for response in TMB-High defined as ≥10 mutations per Megabase, found single agent PD-L1, CTLA-4, and combination to significantly affect ORR (227). Prospectively, the multicohort phase 2 KEYNOTE-158 found an improved overall response rate of pembrolizumab in the TMB-High group compared to the TMB-Low group (29% versus 6%) leading to approval of single-agent IO for TMB-High (228).
In addition, there is an increasing appreciation of the role of host factors on the immune response unrelated to the intrinsic biology of tumor development and resistance. For instance, as gender and diet differences have long been observed to impact patient outcomes to cancer therapy, the microbiome has emerged as a critical element in modulating the immune response. There have been many high impact publications indicating that the diversity and composition of the gut microbiome can indeed predict for higher response rates and better outcomes to therapy. Functional studies, as well as in vivo data, offer solid evidence that this is a causal effect resulting from the intricate interaction of gut bacteria with the immune system (229,230). This has emerged as a novel therapeutic target and multiple studies currently underway, where modulation of the microbiome is being utilized with fecal microbiota transplatantion (FMT), are showing early evidence of activity, including patients with PD-1 resistant disease (231). Other interesting approaches utilizing diet to modulate the microbiome favorably are also underway.
Biomarker development continues to evolve at a rapid pace and emphasis should continue to be placed on the incorporation of tumor biopsy and blood collection from patients on clinical trials, especially at the time of disease progression for the assessment of predictive biomarkers of resistance. This could yield a deeper understanding on how the tumor has evolved clonally and perhaps activated other pathways that could then be subsequently targeted. While there are of course logistical, and occasionally even ethical challenges, the data obtained from such clinical trial biopsies may also be incredibly valuable in identifying areas where the studied agent has failed in exerting its effects, offering potential pharmacokinetic and pharmacodynamic strategies to improve pharmacological outcomes. Comprehensive and longitudinal profiling along the patient journey, within and outside a clinical trial, should become the norm rather than the exception.
Significance.
While immune checkpoint inhibitors are approved as dual checkpoint strategies, and in combination with cytotoxic chemotherapy and angiogenesis inhibitors for multiple cancers, patient benefit remains limited. Innovative approaches are required to guide the development of novel immunotherapy combinations, ranging from improvements in preclinical tumor model systems to biomarker-driven trial strategies.
Author Disclosures
TAY received research support to the institution from Artios, AstraZeneca, Bayer, Clovis, Constellation, Cyteir , Eli Lilly, EMD Serono, Forbius, F-Star, GlaxoSmithKline, Genentech, ImmuneSensor, Ipsen, Jounce, Karyopharm, Kyowa, Merck, Novartis, Pfizer, Ribon Therapeutics, Regeneron, Repare, Sanofi, Scholar Rock, Seattle Genetics, Tesaro, and Vertex Pharmaceuticalsre, and has served as a consultant for Almac, Aduro, AstraZeneca, Atrin, Axiom, Bayer, Bristol Myers Squibb, Calithera, Clovis, Cybrexa, EMD Serono, F-Star, Guidepoint, Ignyta, I-Mab, Jansen, Merck, Pfizer, Repare, Roche, Rubius, Schrodinger, Seattle Genetics, Varian and Zai Labs. EEP has served as a consultant for Boehringer Ingleheim. JTM received travel fund reimbursement from Astellas Pharmaceuticals. MAC received grants from ImmunoMet, Inc. and personal fees from ImmunoGenesis, Inc., Alligator Bioscience, Inc., ImmunoOS, Inc., ImmunoMet, Inc., Oncoresponse, Inc., Pieris, Inc., Nurix, Inc., Apetevo, Inc., Servier, Inc., Kineta, Inc., Salarius, Inc., Xencor, Inc., Agenus, Inc., and Mereo. In addition, MAC also has a patent Methods and Composition for Localized Secretion of Anti-CTLA-4 Antibodies that has been issued and licensed to multiple licensees, and a patent Dual specifically antibodies which bind both PD-L1 and PD-L2 and prevent their binding to PD-1 that is pending and licensed with ImmunoGenesis, Inc. HAT serves as a consultant for BMS, Novartis, Merck, Genentech, Pfizer, Eisai, and Iovance, and receives research funding to the institution from BMS, Novartis, Merck, Genetech, GSK, and Celgene.
Footnotes
WP has no conflict of interest outside of the submitted work.
REFERENCES
- 1.Chen Daniel S, Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39(1):1–10 doi 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 2.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 2015;161(2):205–14 doi 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018;362(6411) doi 10.1126/science.aar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Segal NH, Logan TF, Hodi FS, McDermott D, Melero I, Hamid O, et al. Results from an Integrated Safety Analysis of Urelumab, an Agonist Anti-CD137 Monoclonal Antibody. Clin Cancer Res 2017;23(8):1929–36 10.1158/1078-0432.CCR-16-1272.. [DOI] [PubMed] [Google Scholar]
- 5.Ascierto PA, Del Vecchio M, Robert C, Mackiewicz A, Chiarion-Sileni V, Arance A, et al. Ipilimumab 10 mg/kg versus ipilimumab 3 mg/kg in patients with unresectable or metastatic melanoma: a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol 2017;18(5):611–22 doi 10.1016/s1470-2045(17)30231-0. [DOI] [PubMed] [Google Scholar]
- 6.Lebbe C, Meyer N, Mortier L, Marquez-Rodas I, Robert C, Rutkowski P, et al. Evaluation of Two Dosing Regimens for Nivolumab in Combination With Ipilimumab in Patients With Advanced Melanoma: Results From the Phase IIIb/IV CheckMate 511 Trial. J Clin Oncol 2019;37(11):867–75 doi 10.1200/JCO.18.01998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017;168(4):707–23 doi 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 2015;373(1):23–34 doi 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015;372(21):2006–17 doi 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med 2017;377(14):1345–56 doi 10.1056/NEJMoa1709684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med 2019;381(16):1535–46 doi 10.1056/NEJMoa1910836. [DOI] [PubMed] [Google Scholar]
- 12.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357(6349):409–13 doi 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J Clin Oncol 2018;36(8):773–9 doi 10.1200/JCO.2017.76.9901. [DOI] [PubMed] [Google Scholar]
- 14.Lenz H-J, Lonardi S, Zagonel V, Cutse EV, Limon ML, Wong KYM, et al. Nivolumab plus low-dose ipilimumab as first-line therapy in microsatellite instability-high/DNA mismatch repair deficient metastatic colorectal cancer: Clinical update. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2020;38(suppl 4; abstr 11). [Google Scholar]
- 15.Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 2017;18(9):1182–91 doi 10.1016/S1470-2045(17)30422-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med 2018;378(14):1277–90 doi 10.1056/NEJMoa1712126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Motzer RJ, Rini BI, McDermott DF, Arén Frontera O, Hammers HJ, Carducci MA, et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. The Lancet Oncology 2019;20(10):1370–85 doi 10.1016/S1470-2045(19)30413-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lebbé C, Meyer N, Mortier L, Marquez-Rodas I, Robert C, Rutkowski P, et al. LBA47 - Initial results from a phase IIIb/IV study evaluating two dosing regimens of nivolumab (NIVO) in combination with ipilimumab (IPI) in patients with advanced melanoma (CheckMate 511). Annals of Oncology 2018;29:viii737 doi 10.1093/annonc/mdy424.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Villanueva A Hepatocellular Carcinoma. New England Journal of Medicine 2019;380(15):1450–62 doi 10.1056/NEJMra1713263. [DOI] [PubMed] [Google Scholar]
- 20.He AR, Yau T, Hsu C, Kang Y-K, Kim T-Y, Santoro A, et al. Nivolumab (NIVO) + ipilimumab (IPI) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Subgroup analyses from CheckMate 040. Journal of Clinical Oncology 2020;38(4_suppl):512- doi 10.1200/JCO.2020.38.4_suppl.512. [DOI] [Google Scholar]
- 21.Yau T, Kang Y-K, Kim T-Y, El-Khoueiry AB, Santoro A, Sangro B, et al. Nivolumab (NIVO) + ipilimumab (IPI) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Results from CheckMate 040. Journal of Clinical Oncology 2019;37(15_suppl):4012- doi 10.1200/JCO.2019.37.15_suppl.4012. [DOI] [Google Scholar]
- 22.Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med 2018;378(22):2078–92 doi 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 23.Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol 2016;17(11):1497–508 doi 10.1016/S1470-2045(16)30498-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gadgeel S, Rodríguez-Abreu D, Speranza G, Esteban E, Felip E, Dómine M, et al. Updated Analysis From KEYNOTE-189: Pembrolizumab or Placebo Plus Pemetrexed and Platinum for Previously Untreated Metastatic Nonsquamous Non-Small-Cell Lung Cancer. J Clin Oncol 2020;38(14):1505–17 doi 10.1200/jco.19.03136. [DOI] [PubMed] [Google Scholar]
- 25.Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gumus M, Mazieres J, et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N Engl J Med 2018;379(21):2040–51 doi 10.1056/NEJMoa1810865. [DOI] [PubMed] [Google Scholar]
- 26.Paz-Ares L, Vicente D, Tafreshi A, Robinson A, Parra HS, Mazières J, et al. A Randomized, Placebo-Controlled Trial of Pembrolizumab Plus Chemotherapy in Patients With Metastatic Squamous Non-Small-Cell Lung Cancer: Protocol-Specified Final Analysis of KEYNOTE-407. J Thorac Oncol 2020. doi 10.1016/j.jtho.2020.06.015. [DOI] [PubMed] [Google Scholar]
- 27.Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. N Engl J Med 2018;379(24):2342–50 doi 10.1056/NEJMoa1809697. [DOI] [PubMed] [Google Scholar]
- 28.Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N Engl J Med 2017;377(20):1919–29 doi 10.1056/NEJMoa1709937. [DOI] [PubMed] [Google Scholar]
- 29.Gray JE, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Three-Year Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC—Update from PACIFIC. Journal of Thoracic Oncology 2020;15(2):288–93 doi 10.1016/j.jtho.2019.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gadgeel SM, Pennell NA, Fidler MJ, Halmos B, Bonomi P, Stevenson J, et al. Phase II Study of Maintenance Pembrolizumab in Patients with Extensive-Stage Small Cell Lung Cancer (SCLC). J Thorac Oncol 2018;13(9):1393–9 doi 10.1016/j.jtho.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reck M, Luft A, Szczesna A, Havel L, Kim SW, Akerley W, et al. Phase III Randomized Trial of Ipilimumab Plus Etoposide and Platinum Versus Placebo Plus Etoposide and Platinum in Extensive-Stage Small-Cell Lung Cancer. J Clin Oncol 2016;34(31):3740–8 doi 10.1200/JCO.2016.67.6601. [DOI] [PubMed] [Google Scholar]
- 32.Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med 2018;379(23):2220–9 doi 10.1056/NEJMoa1809064. [DOI] [PubMed] [Google Scholar]
- 33.Reck M, Liu SV, Mansfield AS, Mok TSK, Scherpereel A, Reinmuth N, et al. IMpower133: Updated overall survival (OS) analysis of first-line (1L) atezolizumab (atezo) + carboplatin + etoposide in extensive-stage SCLC (ES-SCLC). Annals of Oncology 2019;30(suppl_5):v710–v1 doi 10.1093/annonc/mdz264. [DOI] [Google Scholar]
- 34.Paz-Ares L, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet 2019;394(10212):1929–39 doi 10.1016/s0140-6736(19)32222-6. [DOI] [PubMed] [Google Scholar]
- 35.Denkert C, von Minckwitz G, Brase JC, Sinn BV, Gade S, Kronenwett R, et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J Clin Oncol 2015;33(9):983–91 doi 10.1200/JCO.2014.58.1967. [DOI] [PubMed] [Google Scholar]
- 36.Loi S, Michiels S, Salgado R, Sirtaine N, Jose V, Fumagalli D, et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: results from the FinHER trial. Ann Oncol 2014;25(8):1544–50 doi 10.1093/annonc/mdu112. [DOI] [PubMed] [Google Scholar]
- 37.Luo N, Nixon MJ, Gonzalez-Ericsson PI, Sanchez V, Opalenik SR, Li H, et al. DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nat Commun 2018;9(1):248 doi 10.1038/s41467-017-02630-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adams S, Schmid P, Rugo HS, Winer EP, Loirat D, Awada A, et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: cohort A of the phase II KEYNOTE-086 study. Ann Oncol 2019;30(3):397–404 doi 10.1093/annonc/mdy517. [DOI] [PubMed] [Google Scholar]
- 39.Schmid P, Rugo HS, Adams S, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2020;21(1):44–59 doi 10.1016/s1470-2045(19)30689-8. [DOI] [PubMed] [Google Scholar]
- 40.Le X, Ferrarotto R, Wise-Draper T, Gillison M. Evolving Role of Immunotherapy in Recurrent Metastatic Head and Neck Cancer. Journal of the National Comprehensive Cancer Network : JNCCN 2020;18(7):899–906 doi 10.6004/jnccn.2020.7590. [DOI] [PubMed] [Google Scholar]
- 41.Chow LQM. Head and Neck Cancer. N Engl J Med 2020;382(1):60–72 doi 10.1056/NEJMra1715715. [DOI] [PubMed] [Google Scholar]
- 42.Burtness B, Harrington KJ, Greil R, Soulières D, Tahara M, de Castro G Jr., et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. The Lancet 2019;394(10212):1915–28 doi 10.1016/S0140-6736(19)32591-7. [DOI] [PubMed] [Google Scholar]
- 43.Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V, et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med 2017;9(385) doi 10.1126/scitranslmed.aak9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Makker V, Taylor MH, Aghajanian C, Oaknin A, Mier J, Cohn AL, et al. Lenvatinib Plus Pembrolizumab in Patients With Advanced Endometrial Cancer. J Clin Oncol 2020:JCO1902627 doi 10.1200/JCO.19.02627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Makker V, Rasco D, Vogelzang NJ, Brose MS, Cohn AL, Mier J, et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: an interim analysis of a multicentre, open-label, single-arm, phase 2 trial. The Lancet Oncology 2019;20(5):711–8 doi 10.1016/S1470-2045(19)30020-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, Piotrowska Z, et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non-Small Cell Lung Cancer: A Retrospective Analysis. Clin Cancer Res 2016;22(18):4585–93 10.1158/1078-0432.CCR-15-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 2016;387(10027):1540–50 doi 10.1016/S0140-6736(15)01281-7. [DOI] [PubMed] [Google Scholar]
- 48.Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet 2017;389(10066):255–65 doi 10.1016/S0140-6736(16)32517-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choueiri TK, Motzer RJ. Systemic Therapy for Metastatic Renal-Cell Carcinoma. The New England journal of medicine 2017;376(4):354–66 doi 10.1056/NEJMra1601333. [DOI] [PubMed] [Google Scholar]
- 50.Rini BI, Powles T, Atkins MB, Escudier B, McDermott DF, Suarez C, et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): a multicentre, open-label, phase 3, randomised controlled trial. Lancet 2019;393(10189):2404–15 doi 10.1016/s0140-6736(19)30723-8. [DOI] [PubMed] [Google Scholar]
- 51.Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med 2019;380(12):1116–27 doi 10.1056/NEJMoa1816714. [DOI] [PubMed] [Google Scholar]
- 52.Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med 2019;380(12):1103–15 doi 10.1056/NEJMoa1816047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim T-Y, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. New England Journal of Medicine 2020;382(20):1894–905 doi 10.1056/NEJMoa1915745. [DOI] [PubMed] [Google Scholar]
- 54.Taylor M, Rasco D, Brose M. A phase 1b/2 trial of lenvatinib plus pembrolizumab in patients with squamous cell carcinoma of the head and neck [abstract]. J Clin Oncol 2018;36 doi 10.1200/JCO.2018.36.15_suppl.6016. [DOI] [Google Scholar]
- 55.Robert C, Grob JJ, Stroyakovskiy D, Karaszewska B, Hauschild A, Levchenko E, et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N Engl J Med 2019;381(7):626–36 doi 10.1056/NEJMoa1904059. [DOI] [PubMed] [Google Scholar]
- 56.Dummer R, Hauschild A, Santinami M, Atkinson V, Mandalà M, Kirkwood JM, et al. Five-Year Analysis of Adjuvant Dabrafenib plus Trametinib in Stage III Melanoma. N Engl J Med 2020;383(12):1139–48 doi 10.1056/NEJMoa2005493. [DOI] [PubMed] [Google Scholar]
- 57.Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2018;19(5):603–15 doi 10.1016/s1470-2045(18)30142-6. [DOI] [PubMed] [Google Scholar]
- 58.Nathan P, Dummer R, Long GV, Ascierto PA, Tawbi HA, Robert C, et al. LBA43 Spartalizumab plus dabrafenib and trametinib (Sparta-DabTram) in patients (pts) with previously untreated BRAF V600-mutant unresectable or metastatic melanoma: Results from the randomized part 3 of the phase III COMBI-i trial. Annals of Oncology 2020;31:S1172 doi 10.1016/j.annonc.2020.08.2273. [DOI] [Google Scholar]
- 59.Melero I, Berman DM, Aznar MA, Korman AJ, Pérez Gracia JL, Haanen J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat Rev Cancer 2015;15(8):457–72 doi 10.1038/nrc3973. [DOI] [PubMed] [Google Scholar]
- 60.Herbst RS, Baas P, Kim DW, Felip E, Pérez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 2016;387(10027):1540–50 doi 10.1016/s0140-6736(15)01281-7. [DOI] [PubMed] [Google Scholar]
- 61.Ribas A, Puzanov I, Dummer R, Schadendorf D, Hamid O, Robert C, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol 2015;16(8):908–18 doi 10.1016/s1470-2045(15)00083-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.De Sousa Linhares A, Battin C, Jutz S, Leitner J, Hafner C, Tobias J, et al. Therapeutic PD-L1 antibodies are more effective than PD-1 antibodies in blocking PD-1/PD-L1 signaling. Scientific Reports 2019;9(1):11472 doi 10.1038/s41598-019-47910-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deng R, Bumbaca D, Pastuskovas CV, Boswell CA, West D, Cowan KJ, et al. Preclinical pharmacokinetics, pharmacodynamics, tissue distribution, and tumor penetration of anti-PD-L1 monoclonal antibody, an immune checkpoint inhibitor. MAbs 2016;8(3):593–603 doi 10.1080/19420862.2015.1136043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duan J, Cui L, Zhao X, Bai H, Cai S, Wang G, et al. Use of Immunotherapy With Programmed Cell Death 1 vs Programmed Cell Death Ligand 1 Inhibitors in Patients With Cancer: A Systematic Review and Meta-analysis. JAMA Oncology 2020;6(3):375–84 doi 10.1001/jamaoncol.2019.5367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miyashita H, Mikami T, Satoi S, Cruz C, Galsky MD. Incidence and Risk of Colitis With Programmed Death 1 Versus Programmed Death Ligand 1 Inhibitors for the Treatment of Cancer. J Immunother 2020;43(9):291–8 doi 10.1097/CJI.0000000000000339. [DOI] [PubMed] [Google Scholar]
- 66.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 2010;107(9):4275–80 doi 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res 2013;73(12):3591–603 doi 10.1158/0008-5472.CAN-12-4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chung DJ, Rossi M, Romano E, Ghith J, Yuan J, Munn DH, et al. Indoleamine 2,3-dioxygenase-expressing mature human monocyte-derived dendritic cells expand potent autologous regulatory T cells. Blood 2009;114(3):555–63 doi 10.1182/blood-2008-11-191197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med 1999;189(9):1363–72 doi 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med 2013;210(7):1389–402 doi 10.1084/jem.20130066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu M, Wang X, Wang L, Ma X, Gong Z, Zhang S, et al. Targeting the IDO1 pathway in cancer: from bench to bedside. J Hematol Oncol 2018;11(1):100 doi 10.1186/s13045-018-0644-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Prendergast GC, Malachowski WP, DuHadaway JB, Muller AJ. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res 2017;77(24):6795–811 doi 10.1158/0008-5472.CAN-17-2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Labadie BW, Bao R, Luke JJ. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy via Downstream Focus on the Tryptophan-Kynurenine-Aryl Hydrocarbon Axis. Clin Cancer Res 2019;25(5):1462–71 10.1158/1078-0432.CCR-18-2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol 2019;20(8):1083–97 doi 10.1016/S1470-2045(19)30274-8. [DOI] [PubMed] [Google Scholar]
- 75.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol 2009;10(1):29–37 doi 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun 2016;7:10501 doi 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Oweida A, Hararah MK, Phan A, Binder D, Bhatia S, Lennon S, et al. Resistance to Radiotherapy and PD-L1 Blockade Is Mediated by TIM-3 Upregulation and Regulatory T-Cell Infiltration. Clin Cancer Res 2018;24(21):5368–80 10.1158/1078-0432.CCR-18-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, Ferris RL. Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology 2017;6(1):e1261779 doi 10.1080/2162402X.2016.1261779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Davar D BP, Eroglu Z, Falchook G, Gainor J, Hamilton E, Hecht JP, LukeJ J, Pishvaian M, Ribas A, Wang JS, McEachern K, Waszak A, Lu S, Li Y, Wang Y, LoRusso P,. A phase 1 study of TSR-022, an anti-TIM-3 monoclonal antibody, in combination with TSR-042 (anti-PD-1) in patients with colorectal cancer and post-PD-1 NSCLC and melanoma. J Immunother Cancer 2018;6:O21. [Google Scholar]
- 80.Harding JJ, Patnaik A, Moreno V, Stein M, Jankowska AM, de Mendizabal NV, et al. A phase Ia/Ib study of an anti-TIM-3 antibody (LY3321367) monotherapy or in combination with an anti-PD-L1 antibody (LY3300054): Interim safety, efficacy, and pharmacokinetic findings in advanced cancers. DOI: 101200/JCO2019378_suppl12 Journal of Clinical Oncology 37, no 8_suppl (March 10, 2019) 12–12. [Google Scholar]
- 81.Durham NM, Nirschl CJ, Jackson CM, Elias J, Kochel CM, Anders RA, et al. Lymphocyte Activation Gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS One 2014;9(11):e109080 doi 10.1371/journal.pone.0109080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity 2004;21(4):503–13 doi 10.1016/j.immuni.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 83.Workman CJ, Vignali DA. Negative regulation of T cell homeostasis by lymphocyte activation gene-3 (CD223). J Immunol 2005;174(2):688–95 doi 10.4049/jimmunol.174.2.688. [DOI] [PubMed] [Google Scholar]
- 84.Goding SR, Wilson KA, Xie Y, Harris KM, Baxi A, Akpinarli A, et al. Restoring immune function of tumor-specific CD4+ T cells during recurrence of melanoma. J Immunol 2013;190(9):4899–909 doi 10.4049/jimmunol.1300271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wierz M, Pierson S, Guyonnet L, Viry E, Lequeux A, Oudin A, et al. Dual PD1/LAG3 immune checkpoint blockade limits tumor development in a murine model of chronic lymphocytic leukemia. Blood 2018;131(14):1617–21 doi 10.1182/blood-2017-06-792267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res 2012;72(4):917–27 doi 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hong DS, Schoffski P, Calvo A, Sarantopoulos J, Ochoa De Olza M, Carvajal RD, et al. Phase I/II study of LAG525 ± spartalizumab (PDR001) in patients (pts) with advanced malignancies. Journal of Clinical Oncology 2018;36(15_suppl):3012- doi 10.1200/JCO.2018.36.15_suppl.3012. [DOI] [Google Scholar]
- 88.Ascierto PA, Bono P, Bhatia S, Melero I, Nyakas MS, Svane IM, et al. Efficacy of BMS-986016, a monoclonal antibody that targets lymphocyte activation gene-3 (LAG-3), in combination with nivolumab in pts with melanoma who progressed during prior anti–PD-1/PD-L1 therapy (mel prior IO) in all-comer and biomarker-enriched populations. Annals of Oncology 2017;28:v611–v2 doi 10.1093/annonc/mdx440.011. [DOI] [Google Scholar]
- 89.Lakhani N BT, Abraham AK, Luddy J, Palcza J, Chartash E, Healy J, Patnaik A, The anti–LAG-3 antibody MK-4280 as monotherapy and in combination with pembrolizumab for advanced solid tumors: first-in-human phase 1 dose-finding study. J Immunother cancer 2018(6):O114. [Google Scholar]
- 90.Weber J, Long GV, Haanen JB, Arance A, Dummer R, Nathan P, et al. 1304TiP - A randomized, open-label, phase II open platform study evaluating the efficacy and safety of novel spartalizumab (PDR001) combinations in previously treated unresectable or metastatic melanoma (PLATForM). Annals of Oncology 2018;29:viii465–viii6 doi 10.1093/annonc/mdy289.060. [DOI] [Google Scholar]
- 91.Yu S, Li A, Liu Q, Yuan X, Xu H, Jiao D, et al. Recent advances of bispecific antibodies in solid tumors. J Hematol Oncol 2017;10(1):155 doi 10.1186/s13045-017-0522-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hickingbottom B, Clynes R, Desjarlais J, Li C, Ding Y. Preliminary safety and pharmacodynamic (PD) activity of XmAb20717, a PD-1 x CTLA-4 bispecific antibody, in a phase I dose escalation study of patients with selected advanced solid tumors. Journal of Clinical Oncology 2020;38(15_suppl):e15001–e doi 10.1200/JCO.2020.38.15_suppl.e15001. [DOI] [Google Scholar]
- 93.Hou W, Yuan Q, Yuan X, Wang Y, Mo W, Wang H, et al. A novel tetravalent bispecific antibody targeting programmed death 1 and tyrosine-protein kinase Met for treatment of gastric cancer. Investigational new drugs 2019;37(5):876–89 doi 10.1007/s10637-018-0689-3. [DOI] [PubMed] [Google Scholar]
- 94.Li S, Liu M, Do MH, Chou C, Stamatiades EG, Nixon BG, et al. Cancer immunotherapy via targeted TGF-β signalling blockade in T(H) cells. Nature 2020;587(7832):121–5 doi 10.1038/s41586-020-2850-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Blank CU, Rozeman EA, Fanchi LF, Sikorska K, van de Wiel B, Kvistborg P, et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat Med 2018;24(11):1655–61 doi 10.1038/s41591-018-0198-0. [DOI] [PubMed] [Google Scholar]
- 96.Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018;560(7718):382–6 doi 10.1038/s41586-018-0392-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Amaria RN, Reddy SM, Tawbi HA, Davies MA, Ross MI, Glitza IC, et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat Med 2018;24(11):1649–54 doi 10.1038/s41591-018-0197-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rozeman EA, Menzies AM, van Akkooi ACJ, Adhikari C, Bierman C, van de Wiel BA, et al. Identification of the optimal combination dosing schedule of neoadjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma (OpACIN-neo): a multicentre, phase 2, randomised, controlled trial. Lancet Oncol 2019;20(7):948–60 doi 10.1016/s1470-2045(19)30151-2. [DOI] [PubMed] [Google Scholar]
- 99.Chalabi M, Fanchi LF, Dijkstra KK, Van den Berg JG, Aalbers AG, Sikorska K, et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat Med 2020;26(4):566–76 doi 10.1038/s41591-020-0805-8. [DOI] [PubMed] [Google Scholar]
- 100.Schoenfeld JD, Hanna GJ, Jo VY, Rawal B, Chen YH, Catalano PS, et al. Neoadjuvant Nivolumab or Nivolumab Plus Ipilimumab in Untreated Oral Cavity Squamous Cell Carcinoma: A Phase 2 Open-Label Randomized Clinical Trial. JAMA Oncol 2020;6(10):1563–70 doi 10.1001/jamaoncol.2020.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Caudana P, Núñez NG, De La Rochere P, Pinto A, Denizeau J, Alonso R, et al. IL2/Anti-IL2 Complex Combined with CTLA-4, But Not PD-1, Blockade Rescues Antitumor NK Cell Function by Regulatory T-cell Modulation. Cancer Immunology Research 2019;7(3):443–57 doi 10.1158/2326-6066.Cir-18-0697. [DOI] [PubMed] [Google Scholar]
- 102.Cho JH, Boyman O, Kim HO, Hahm B, Rubinstein MP, Ramsey C, et al. An intense form of homeostatic proliferation of naive CD8+ cells driven by IL-2. J Exp Med 2007;204(8):1787–801 doi 10.1084/jem.20070740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Langowski JL, Addepalli M, VanderVeen L, Pena R, Nutakki R, Kirksey Y, et al. Abstract B057: The CD122-biased immunostimulatory cytokine NKTR-214 combined with checkpoint blockade leads to mobilization of antitumor immunity and synergistic activity. Cancer Immunology Research 2016;4(11 Supplement):B057–B doi 10.1158/2326-6066.Imm2016-b057. [DOI] [Google Scholar]
- 104.Diab A TS, Curti B, Cho D, Wong M, Puzanov I, Lewis KD, Maio M, Daniels GA, Spira AI, Tagliaferri MA,Hannah AI, Clemens W, Imperiale M,Bernatchez C,Haymaker C, Bentebibel SE, Zalevsky J, Hoch U, Fanton C, Rizwan AN, Aung S, Cattaruzza F, Iaccucci E, Sawka D, Bilen MA, Lorigan P,Grignani G,Larkin J, Jang S, Kalinka-Warzocha E, Sznol M, Hurwitz ME, O4 Immune monitoring after NKTR-214 plus nivolumab (PIVOT-02) in previously untreated patients with metastatic Stage IV melanoma. J Immunother cancer 2018;6(114). [Google Scholar]
- 105.Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res 2013;73(24):7189–98 doi 10.1158/0008-5472.CAN-12-4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Goodwin RG, Din WS, Davis-Smith T, Anderson DM, Gimpel SD, Sato TA, et al. Molecular cloning of a ligand for the inducible T cell gene 4-1BB: a member of an emerging family of cytokines with homology to tumor necrosis factor. Eur J Immunol 1993;23(10):2631–41 doi 10.1002/eji.1830231037. [DOI] [PubMed] [Google Scholar]
- 107.Zappasodi R, Sirard C, Li Y, Budhu S, Abu-Akeel M, Liu C, et al. Abstract PR01: Mechanistic rationale to combine GITR agonism with PD-1 blockade in cancer patients. Cancer Immunology Research 2019;7(2 Supplement):PR01–PR doi 10.1158/2326-6074.Cricimteatiaacr18-pr01. [DOI] [Google Scholar]
- 108.Ribas A, Medina T, Kummar S, Amin A, Kalbasi A, Drabick JJ, et al. SD-101 in Combination with Pembrolizumab in Advanced Melanoma: Results of a Phase Ib, Multicenter Study. Cancer Discov 2018;8(10):1250–7 doi 10.1158/2159-8290.Cd-18-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Reilley M, Tsimberidou AM, Piha-Paul SA, Yap TA, Fu S, Naing A, et al. Phase 1 trial of TLR9 agonist lefitolimod in combination with CTLA-4 checkpoint inhibitor ipilimumab in advanced tumors. Journal of Clinical Oncology 2019;37(15_suppl):TPS2669–TPS doi 10.1200/JCO.2019.37.15_suppl.TPS2669. [DOI] [Google Scholar]
- 110.Diab A, Haymaker C, Bernatchez C, Andtbacka RHI, Shaheen M, Johnson D, et al. 1245PD - Intratumoral (IT) Injection of the TLR9 agonist tilsotolimod (IMO-2125) in combination with ipilimumab (ipi) triggers durable responses in PD-1 inhibitor refractory metastatic melanoma (rMM): Results from a multicenter, phase I/II study. Annals of Oncology 2018;29:viii442 doi 10.1093/annonc/mdy289.001. [DOI] [Google Scholar]
- 111.Butler MO, Robert C, Negrier S, In GK, Walker JW, Krajsova I, et al. ILLUMINATE 301: A randomized phase 3 study of tilsotolimod in combination with ipilimumab compared with ipilimumab alone in patients with advanced melanoma following progression on or after anti-PD-1 therapy. Journal of Clinical Oncology 2019;37(15_suppl):TPS9599–TPS doi 10.1200/JCO.2019.37.15_suppl.TPS9599. [DOI] [Google Scholar]
- 112.Meric-Bernstam F, Sandhu SK, Hamid O, Spreafico A, Kasper S, Dummer R, et al. Phase Ib study of MIW815 (ADU-S100) in combination with spartalizumab (PDR001) in patients (pts) with advanced/metastatic solid tumors or lymphomas. Journal of Clinical Oncology 2019;37(15_suppl):2507– doi 10.1200/JCO.2019.37.15_suppl.2507. [DOI] [Google Scholar]
- 113.Harrington KJ, Brody J, Ingham M, Strauss J, Cemerski S, Wang M, et al. LBA15 - Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Annals of Oncology 2018;29:viii712 doi 10.1093/annonc/mdy424.015. [DOI] [Google Scholar]
- 114.Curran KJ, Pegram HJ, Brentjens RJ. Chimeric antigen receptors for T cell immunotherapy: current understanding and future directions. The journal of gene medicine 2012;14(6):405–15 doi 10.1002/jgm.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Scherpereel A, Mazieres J, Greillier L, Lantuejoul S, Dô P, Bylicki O, et al. Nivolumab or nivolumab plus ipilimumab in patients with relapsed malignant pleural mesothelioma (IFCT-1501 MAPS2): a multicentre, open-label, randomised, non-comparative, phase 2 trial. Lancet Oncol 2019;20(2):239–53 doi 10.1016/s1470-2045(18)30765-4. [DOI] [PubMed] [Google Scholar]
- 116.Adusumilli PS, Zauderer MG, Rusch VW, O’Cearbhaill R, Zhu A, Ngai D, et al. Regional delivery of mesothelin-targeted CAR T cells for pleural cancers: Safety and preliminary efficacy in combination with anti-PD-1 agent. Journal of Clinical Oncology 2019;37(15_suppl):2511– doi 10.1200/JCO.2019.37.15_suppl.2511.31154918 [DOI] [Google Scholar]
- 117.Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep 2017;7(1):737 doi 10.1038/s41598-017-00462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Grosser R, Cherkassky L, Chintala N, Adusumilli PS. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 2019;36(5):471–82 10.1016/j.ccell.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schwartz HS, Grindey GB. Adriamycin and daunorubicin: a comparison of antitumor activities and tissue uptake in mice following immunosuppression. Cancer Res 1973;33(8):1837–44. [PubMed] [Google Scholar]
- 120.Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 2016;44(2):343–54 doi 10.1016/j.immuni.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Heinhuis KM, Ros W, Kok M, Steeghs N, Beijnen JH, Schellens JHM. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann Oncol 2019;30(2):219–35 doi 10.1093/annonc/mdy551. [DOI] [PubMed] [Google Scholar]
- 122.Erdal E, Haider S, Rehwinkel J, Harris AL, McHugh PJ. A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev 2017;31(4):353–69 doi 10.1101/gad.289769.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med 2015;7(283):283ra52 doi 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. JNCI: Journal of the National Cancer Institute 2016;109(1) doi 10.1093/jnci/djw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kodumudi KN, Weber A, Sarnaik AA, Pilon-Thomas S. Blockade of myeloid-derived suppressor cells after induction of lymphopenia improves adoptive T cell therapy in a murine model of melanoma. J Immunol 2012;189(11):5147–54 doi 10.4049/jimmunol.1200274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 2013;38(4):729–41 doi 10.1016/j.immuni.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 127.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med 2018;379(22):2108–21 doi 10.1056/NEJMoa1809615. [DOI] [PubMed] [Google Scholar]
- 128.Nanda R, Liu MC, Yau C, Asare S, Hylton N, Veer LVt, et al. Pembrolizumab plus standard neoadjuvant therapy for high-risk breast cancer (BC): Results from I-SPY 2. Journal of Clinical Oncology 2017;35(15_suppl):506– doi 10.1200/JCO.2017.35.15_suppl.506.28029304 [DOI] [Google Scholar]
- 129.Nanda R, Liu MC, Yau C, Shatsky R, Pusztai L, Wallace A, et al. Effect of Pembrolizumab Plus Neoadjuvant Chemotherapy on Pathologic Complete Response in Women With Early-Stage Breast Cancer: An Analysis of the Ongoing Phase 2 Adaptively Randomized I-SPY2 Trial. JAMA Oncol 2020;6(5):676–84 doi 10.1001/jamaoncol.2019.6650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Schmid P, Cortes J, Pusztai L, McArthur H, Kümmel S, Bergh J, et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N Engl J Med 2020;382(9):810–21 doi 10.1056/NEJMoa1910549. [DOI] [PubMed] [Google Scholar]
- 131.Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014;41(5):843–52 doi 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun 2017;8(1):1751 doi 10.1038/s41467-017-01883-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015;348(6230):69–74 doi 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 134.Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat Med 2018;24(12):1845–51 doi 10.1038/s41591-018-0232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Shevtsov M, Sato H, Multhoff G, Shibata A. Novel Approaches to Improve the Efficacy of Immuno-Radiotherapy. Front Oncol 2019;9:156 doi 10.3389/fonc.2019.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun 2017;8:15618 doi 10.1038/ncomms15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ko EC, Raben D, Formenti SC. The Integration of Radiotherapy with Immunotherapy for the Treatment of Non-Small Cell Lung Cancer. Clin Cancer Res 2018;24(23):5792–806 10.1158/1078-0432.CCR-17-3620. [DOI] [PubMed] [Google Scholar]
- 138.Komatsu T, Nakamura K, Kawase A. Abscopal Effect of Nivolumab in a Patient with Primary Lung Cancer. J Thorac Oncol 2017;12(9):e143–e4 doi 10.1016/j.jtho.2017.05.004. [DOI] [PubMed] [Google Scholar]
- 139.Yuan Z, Fromm A, Ahmed KA, Grass GD, Yang GQ, Oliver DE, et al. Radiotherapy Rescue of a Nivolumab-Refractory Immune Response in a Patient with PD-L1-Negative Metastatic Squamous Cell Carcinoma of the Lung. J Thorac Oncol 2017;12(9):e135–e6 doi 10.1016/j.jtho.2017.04.029. [DOI] [PubMed] [Google Scholar]
- 140.Gratia M, Rodero MP, Conrad C, Bou Samra E, Maurin M, Rice GI, et al. Bloom syndrome protein restrains innate immune sensing of micronuclei by cGAS. J Exp Med 2019;216(5):1199–213 doi 10.1084/jem.20181329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hartlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015;42(2):332–43 doi 10.1016/j.immuni.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 142.Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J Natl Cancer Inst 2017;109(1) doi 10.1093/jnci/djw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chabanon RM, Muirhead G, Krastev DB, Adam J, Morel D, Garrido M, et al. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J Clin Invest 2019;129(3):1211–28 doi 10.1172/JCI123319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ding L, Kim HJ, Wang Q, Kearns M, Jiang T, Ohlson CE, et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep 2018;25(11):2972–80 e5 doi 10.1016/j.celrep.2018.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov 2019;9(5):646–61 doi 10.1158/2159-8290.CD-18-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, et al. PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Research 2018:canres.1003.2018 doi 10.1158/0008-5472.CAN-18-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ding L, Chen X, Xu X, Qian Y, Liang G, Yao F, et al. PARP1 Suppresses the Transcription of PD-L1 by Poly(ADP-Ribosyl)ating STAT3. Cancer Immunol Res 2019;7(1):136–49 doi 10.1158/2326-6066.CIR-18-0071. [DOI] [PubMed] [Google Scholar]
- 148.Stewart RA, Pilie PG, Yap TA. Development of PARP and Immune-Checkpoint Inhibitor Combinations. Cancer Res 2018;78(24):6717–25 doi 10.1158/0008-5472.CAN-18-2652. [DOI] [PubMed] [Google Scholar]
- 149.Karzai F, VanderWeele D, Madan RA, Owens H, Cordes LM, Hankin A, et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J Immunother Cancer 2018;6(1):141 doi 10.1186/s40425-018-0463-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yu EY, Massard C, Retz M, Tafreshi A, Carles Galceran J, Hammerer P, et al. Keynote-365 cohort a: Pembrolizumab (pembro) plus olaparib in docetaxel-pretreated patients (pts) with metastatic castrate-resistant prostate cancer (mCRPC). Journal of Clinical Oncology 2019;37(7_suppl):145– doi 10.1200/JCO.2019.37.7_suppl.145. [DOI] [Google Scholar]
- 151.Vinayak S, Tolaney SM, Schwartzberg LS, Mita MM, McCann GA-L, Tan AR, et al. TOPACIO/Keynote-162: Niraparib + pembrolizumab in patients (pts) with metastatic triple-negative breast cancer (TNBC), a phase 2 trial. Journal of Clinical Oncology 2018;36(15_suppl):1011– doi 10.1200/JCO.2018.36.15_suppl.1011. [DOI] [Google Scholar]
- 152.Konstantinopoulos PA, Waggoner SE, Vidal GA, Mita MM, Fleming GF, Holloway RW, et al. TOPACIO/Keynote-162 (NCT02657889): A phase 1/2 study of niraparib + pembrolizumab in patients (pts) with advanced triple-negative breast cancer or recurrent ovarian cancer (ROC)—Results from ROC cohort. Journal of Clinical Oncology 2018;36(15_suppl):106– doi 10.1200/JCO.2018.36.15_suppl.106. [DOI] [Google Scholar]
- 153.Yap TAK MG; Postel-Vinay S; Bang YJ; El-Khoueiry A; Abida W; Harrington K; Sundar R; Carter L; Castanon-Alvarez E; Im SA; Berges A; Khan M; Stephens C; Ross G; Soria JC Phase I modular study of AZD6738, a novel oral, potent and selective ataxia telangiectasia Rad3-related (ATR) inhibitor in combination (combo) with carboplatin, olaparib or durvalumab in patients (pts) with advanced cancers. European Journal of Cancer 2016;69(Suppl. 1) doi 10.1016/S0959-8049(16)32607-7. [DOI] [Google Scholar]
- 154.Friedman J, Morisada M, Sun L, Moore EC, Padget M, Hodge JW, et al. Inhibition of WEE1 kinase and cell cycle checkpoint activation sensitizes head and neck cancers to natural killer cell therapies. J Immunother Cancer 2018;6(1):59 doi 10.1186/s40425-018-0374-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jin MH, Nam AR, Park JE, Bang JH, Bang YJ, Oh DY. Therapeutic Co-targeting of WEE1 and ATM Downregulates PD-L1 Expression in Pancreatic Cancer. Cancer Res Treat 2020;52(1):149–66 doi 10.4143/crt.2019.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhang L, Xu LJ, Zhu J, Li J, Xue BX, Gao J, et al. ATMJAKPDL1 signaling pathway inhibition decreases EMT and metastasis of androgenindependent prostate cancer. Mol Med Rep 2018;17(5):7045–54 doi 10.3892/mmr.2018.8781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tsai AK, Khan AY, Worgo CE, Wang LL, Liang Y, Davila E. A Multikinase and DNA-PK Inhibitor Combination Immunomodulates Melanomas, Suppresses Tumor Progression, and Enhances Immunotherapies. Cancer Immunol Res 2017;5(9):790–803 doi 10.1158/2326-6066.CIR-17-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Brea EJ, Oh CY, Manchado E, Budhu S, Gejman RS, Mo G, et al. Kinase Regulation of Human MHC Class I Molecule Expression on Cancer Cells. Cancer Immunol Res 2016;4(11):936–47 doi 10.1158/2326-6066.CIR-16-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med 2013;368(14):1365–6 doi 10.1056/NEJMc1302338. [DOI] [PubMed] [Google Scholar]
- 160.Minor DR, Puzanov I, Callahan MK, Hug BA, Hoos A. Severe gastrointestinal toxicity with administration of trametinib in combination with dabrafenib and ipilimumab. Pigment Cell Melanoma Res 2015;28(5):611–2 doi 10.1111/pcmr.12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ascierto PA, Ferrucci PF, Stephens R, Del Vecchio M, Atkinson V, Schmidt H, et al. 1244O - KEYNOTE-022 Part 3: Phase II randomized study of 1L dabrafenib (D) and trametinib (T) plus pembrolizumab (Pembro) or placebo (PBO) for BRAF-mutant advanced melanoma. Annals of Oncology 2018;29:viii442 doi 10.1093/annonc/mdy289. [DOI] [Google Scholar]
- 162.Gutzmer R, Stroyakovskiy D, Gogas H, Robert C, Lewis K, Protsenko S, et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF(V600) mutation-positive melanoma (IMspire150): primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020;395(10240):1835–44 doi 10.1016/s0140-6736(20)30934-x. [DOI] [PubMed] [Google Scholar]
- 163.Bendell J, Ciardiello F, Tabernero J, Tebbutt N, Eng C, Di Bartolomeo M, et al. LBA-004 - Efficacy and safety results from IMblaze370, a randomised Phase III study comparing atezolizumab+cobimetinib and atezolizumab monotherapy vs regorafenib in chemotherapy-refractory metastatic colorectal cancer. Annals of Oncology 2018;29:v123 doi 10.1093/annonc/mdy208.003. [DOI] [Google Scholar]
- 164.Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A 2014;111(32):11774–9 doi 10.1073/pnas.1410626111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye-Ogala R, Arisa S, et al. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin Cancer Res 2017;23(17):5187–201 10.1158/1078-0432.CCR-17-0741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zheng H, Zhao W, Yan C, Watson CC, Massengill M, Xie M, et al. HDAC Inhibitors Enhance T-Cell Chemokine Expression and Augment Response to PD-1 Immunotherapy in Lung Adenocarcinoma. Clinical Cancer Research 2016;22(16):4119–32 10.1158/1078-0432.Ccr-15-2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sullivan RJ, Moschos SJ, Johnson ML, Opyrchal M, Ordentlich P, Brouwer S, et al. Abstract CT072: Efficacy and safety of entinostat (ENT) and pembrolizumab (PEMBRO) in patients with melanoma previously treated with anti-PD1 therapy. Cancer Research 2019;79(13 Supplement):CT072–CT doi 10.1158/1538-7445.Am2019-ct072. [DOI] [Google Scholar]
- 168.Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015;162(5):974–86 doi 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ørskov AD, Saini SK, Bjerregaard A-M, Otani H, Holmberg S, Hansen JW, et al. Activation of T Cells Specific to Endogenous Retroviral Peptides: Possible Association with Clinical Response to Azacitidine in Myeloid Malignancies. Blood 2017;130(Supplement 1):4243– doi 10.1182/blood.V130.Suppl_1.4243.4243. [DOI] [Google Scholar]
- 170.Daver N, Garcia-Manero G, Basu S, Boddu PC, Alfayez M, Cortes JE, et al. Efficacy, Safety, and Biomarkers of Response to Azacitidine and Nivolumab in Relapsed/Refractory Acute Myeloid Leukemia: A Nonrandomized, Open-Label, Phase II Study. Cancer Discov 2019;9(3):370–83 doi 10.1158/2159-8290.CD-18-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lee JJ, Sun W, Bahary N, Ohr J, Rhee JC, Stoller RG, et al. Phase 2 study of pembrolizumab in combination with azacitidine in subjects with metastatic colorectal cancer. Journal of Clinical Oncology 2017;35(15_suppl):3054– doi 10.1200/JCO.2017.35.15_suppl.3054. [DOI] [Google Scholar]
- 172.Alvarez E B16 Murine Melanoma: Historical Perspective on the Development of a Solid Tumor Model. In: Teicher BA, editor. Tumor Models in Cancer Research. Totowa, NJ: Humana Press; 2011. p 79–95. [Google Scholar]
- 173.Barth RJ Jr., Bock SN, Mule JJ, Rosenberg SA. Unique murine tumor-associated antigens identified by tumor infiltrating lymphocytes. J Immunol 1990;144(4):1531–7. [PubMed] [Google Scholar]
- 174.Kim KJ, Kanellopoulos-Langevin C, Merwin RM, Sachs DH, Asofsky R. Establishment and characterization of BALB/c lymphoma lines with B cell properties. J Immunol 1979;122(2):549–54. [PubMed] [Google Scholar]
- 175.Gajewski TF, Markiewicz MA, Uyttenhove C. The p815 mastocytoma tumor model. Curr Protoc Immunol 2001;Chapter 20:Unit 20 4 doi 10.1002/0471142735.im2004s43. [DOI] [PubMed] [Google Scholar]
- 176.Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res 1992;52(6):1399–405. [PubMed] [Google Scholar]
- 177.Sutherland KD, Proost N, Brouns I, Adriaensen D, Song JY, Berns A. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 2011;19(6):754–64 10.1016/j.ccr.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 178.Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, et al. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis 2000;21(4):585–91 doi 10.1093/carcin/21.4.585. [DOI] [PubMed] [Google Scholar]
- 179.Summerhayes IC, Franks LM. Effects of donor age on neoplastic transformation of adult mouse bladder epithelium in vitro. J Natl Cancer Inst 1979;62(4):1017–23. [PubMed] [Google Scholar]
- 180.Corbett TH, Griswold DP Jr., Roberts BJ, Peckham JC, Schabel FM Jr. Tumor induction relationships in development of transplantable cancers of the colon in mice for chemotherapy assays, with a note on carcinogen structure. Cancer Res 1975;35(9):2434–9. [PubMed] [Google Scholar]
- 181.Wang M, Bronte V, Chen PW, Gritz L, Panicali D, Rosenberg SA, et al. Active immunotherapy of cancer with a nonreplicating recombinant fowlpox virus encoding a model tumor-associated antigen. J Immunol 1995;154(9):4685–92. [PMC free article] [PubMed] [Google Scholar]
- 182.Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res 2012;72(5):1081–91 doi 10.1158/0008-5472.CAN-11-3722. [DOI] [PubMed] [Google Scholar]
- 183.Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015;520(7549):692–6 doi 10.1038/nature14426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sellers RS, Clifford CB, Treuting PM, Brayton C. Immunological Variation Between Inbred Laboratory Mouse Strains:Points to Consider in Phenotyping Genetically Immunomodified Mice. Veterinary Pathology 2012;49(1):32–43 doi 10.1177/0300985811429314. [DOI] [PubMed] [Google Scholar]
- 185.Krupke DM, Begley DA, Sundberg JP, Richardson JE, Neuhauser SB, Bult CJ. The Mouse Tumor Biology Database: A Comprehensive Resource for Mouse Models of Human Cancer. Cancer Research 2017;77(21):e67–e70 doi 10.1158/0008-5472.Can-17-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr., et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet 2009;41(5):544–52 doi 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hooijkaas A, Gadiot J, Morrow M, Stewart R, Schumacher T, Blank CU. Selective BRAF inhibition decreases tumor-resident lymphocyte frequencies in a mouse model of human melanoma. Oncoimmunology 2012;1(5):609–17 doi 10.4161/onci.20226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kimpfler S, Sevko A, Ring S, Falk C, Osen W, Frank K, et al. Skin Melanoma Development in ret Transgenic Mice Despite the Depletion of CD25+Foxp3+ Regulatory T Cells in Lymphoid Organs. The Journal of Immunology 2009;183(10):6330–7 doi 10.4049/jimmunol.0900609. [DOI] [PubMed] [Google Scholar]
- 189.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov 2016;6(2):202–16 doi 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Le AX, Bernhard EJ, Holterman MJ, Strub S, Parham P, Lacy E, et al. Cytotoxic T cell responses in HLA-A2.1 transgenic mice. Recognition of HLA alloantigens and utilization of HLA-A2.1 as a restriction element. J Immunol 1989;142(4):1366–71. [PubMed] [Google Scholar]
- 191.Lute KD, May KF Jr., Lu P, Zhang H, Kocak E, Mosinger B, et al. Human CTLA4 knock-in mice unravel the quantitative link between tumor immunity and autoimmunity induced by anti-CTLA-4 antibodies. Blood 2005;106(9):3127–33 doi 10.1182/blood-2005-06-2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Huang X, Zheng L, Ouyang W, Zhang M, An AX, Zhao J, et al. Abstract A207: Utilizing human OX40 knock-in mice (HuGEMMTM) to assess antitumor efficacy of OX40-agonistic antibodies. Molecular Cancer Therapeutics 2018;17(1 Supplement):A207–A doi 10.1158/1535-7163.Targ-17-a207. [DOI] [Google Scholar]
- 193.Zheng L, Huang X, Ouyang W, Chen G, An AX, Dong X, et al. Abstract A203: Generation of human TIM3 knock-in mice for preclinical efficacy assessment of therapeutic antibodies. Molecular Cancer Therapeutics 2018;17(1 Supplement):A203–A doi 10.1158/1535-7163.Targ-17-a203. [DOI] [Google Scholar]
- 194.Jangalwe S, Shultz LD, Mathew A, Brehm MA. Improved B cell development in humanized NOD-scid IL2Rgamma(null) mice transgenically expressing human stem cell factor, granulocyte-macrophage colony-stimulating factor and interleukin-3. Immun Inflamm Dis 2016;4(4):427–40 doi 10.1002/iid3.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Saito Y, Ellegast JM, Rafiei A, Song Y, Kull D, Heikenwalder M, et al. Peripheral blood CD34(+) cells efficiently engraft human cytokine knock-in mice. Blood 2016;128(14):1829–33 doi 10.1182/blood-2015-10-676452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, et al. Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol 2014;32(4):364–72 doi 10.1038/nbt.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Wang M, Yao LC, Cheng M, Cai D, Martinek J, Pan CX, et al. Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. FASEB J 2018;32(3):1537–49 doi 10.1096/fj.201700740R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Capasso A, Lang J, Pitts TM, Jordan KR, Lieu CH, Davis SL, et al. Characterization of immune responses to anti-PD-1 mono and combination immunotherapy in hematopoietic humanized mice implanted with tumor xenografts. J Immunother Cancer 2019;7(1):37 doi 10.1186/s40425-019-0518-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer AR, Bajor DL, et al. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol Res 2015;3(4):399–411 doi 10.1158/2326-6066.CIR-14-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011;331(6024):1612–6 doi 331/6024/1612 [pii] 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.O’Hara MH, O’Reilly EM, Rosemarie M, Varadhachary G, Wainberg ZA, Ko A, et al. Abstract CT004: A Phase Ib study of CD40 agonistic monoclonal antibody APX005M together with gemcitabine (Gem) and nab-paclitaxel (NP) with or without nivolumab (Nivo) in untreated metastatic ductal pancreatic adenocarcinoma (PDAC) patients. Cancer Research 2019;79(13 Supplement):CT004–CT doi 10.1158/1538-7445.Am2019-ct004. [DOI] [Google Scholar]
- 202.Desnoyers LR, Vasiljeva O, Richardson JH, Yang A, Menendez EE, Liang TW, et al. Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci Transl Med 2013;5(207):207ra144 doi 10.1126/scitranslmed.3006682. [DOI] [PubMed] [Google Scholar]
- 203.Naing A, Thistlethwaite FC, Spira AI, Garcia-Corbacho J, Randhawa M, Eskens F, et al. CX-072, a PD-L1 Probody therapeutic, as monotherapy in patients with advanced solid tumors: Preliminary results of PROCLAIM-CX-072. Journal of Clinical Oncology 2019;37(15_suppl):2513– doi 10.1200/JCO.2019.37.15_suppl.2513. [DOI] [Google Scholar]
- 204.Hinner MJ, Aiba RSB, Jaquin TJ, Berger S, Dürr MC, Schlosser C, et al. Tumor-Localized Costimulatory T-Cell Engagement by the 4-1BB/HER2 Bispecific Antibody-Anticalin Fusion PRS-343. Clinical Cancer Research 2019;25(19):5878–89 10.1158/1078-0432.Ccr-18-3654. [DOI] [PubMed] [Google Scholar]
- 205.Aznar MA, Tinari N, Rullan AJ, Sanchez-Paulete AR, Rodriguez-Ruiz ME, Melero I. Intratumoral Delivery of Immunotherapy-Act Locally, Think Globally. J Immunol 2017;198(1):31–9 doi 10.4049/jimmunol.1601145. [DOI] [PubMed] [Google Scholar]
- 206.Sagiv-Barfi I, Czerwinski DK, Levy S, Alam IS, Mayer AT, Gambhir SS, et al. Eradication of spontaneous malignancy by local immunotherapy. Sci Transl Med 2018;10(426) doi 10.1126/scitranslmed.aan4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Ager CR, Reilley MJ, Nicholas C, Bartkowiak T, Jaiswal AR, Curran MA. Intratumoral STING Activation with T-cell Checkpoint Modulation Generates Systemic Antitumor Immunity. Cancer Immunol Res 2017;5(8):676–84 doi 10.1158/2326-6066.CIR-17-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Leone RD, Emens LA. Targeting adenosine for cancer immunotherapy. J Immunother Cancer 2018;6(1):57 doi 10.1186/s40425-018-0360-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Park A, Stacy M. Istradefylline for the treatment of Parkinson’s disease. Expert Opin Pharmacother 2012;13(1):111–4 doi 10.1517/14656566.2012.643869. [DOI] [PubMed] [Google Scholar]
- 210.Congreve M, Brown GA, Borodovsky A, Lamb ML. Targeting adenosine A2A receptor antagonism for treatment of cancer. Expert Opin Drug Discov 2018;13(11):997–1003 doi 10.1080/17460441.2018.1534825. [DOI] [PubMed] [Google Scholar]
- 211.Pinter M, Jain RK. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Sci Transl Med 2017;9(410) doi 10.1126/scitranslmed.aan5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Rachidi S, Metelli A, Riesenberg B, Wu BX, Nelson MH, Wallace C, et al. Platelets subvert T cell immunity against cancer via GARP-TGFbeta axis. Sci Immunol 2017;2(11) doi 10.1126/sciimmunol.aai7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med 2006;203(12):2691–702 doi 10.1084/jem.20061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kluger HM, Tawbi HA, Ascierto ML, Bowden M, Callahan MK, Cha E, et al. Defining tumor resistance to PD-1 pathway blockade: recommendations from the first meeting of the SITC Immunotherapy Resistance Taskforce. J Immunother Cancer 2020;8(1) doi 10.1136/jitc-2019-000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Schvartsman G, Ma J, Bassett RL Jr., Haydu LE, Amaria RN, Hwu P, et al. Incidence, patterns of progression, and outcomes of preexisting and newly discovered brain metastases during treatment with anti-PD-1 in patients with metastatic melanoma. Cancer 2019;125(23):4193–202 doi 10.1002/cncr.32454. [DOI] [PubMed] [Google Scholar]
- 216.Food and Drug Administration H. Cancer Clinical Trial Eligibility Criteria: Brain Metastases Guidance for Industry. Federal Register 2020;85(134):41992–3. [Google Scholar]
- 217.Mitchell TC, Hamid O, Smith DC, Bauer TM, Wasser JS, Olszanski AJ, et al. Epacadostat Plus Pembrolizumab in Patients With Advanced Solid Tumors: Phase I Results From a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J Clin Oncol 2018;36(32):3223–30 doi 10.1200/jco.2018.78.9602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Gibney GT, Hamid O, Lutzky J, Olszanski AJ, Mitchell TC, Gajewski TF, et al. Phase 1/2 study of epacadostat in combination with ipilimumab in patients with unresectable or metastatic melanoma. J Immunother Cancer 2019;7(1):80 doi 10.1186/s40425-019-0562-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019;366(6468):1013–21 doi 10.1126/science.aav2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Daver N, Boddu P, Garcia-Manero G, Yadav SS, Sharma P, Allison J, et al. Hypomethylating agents in combination with immune checkpoint inhibitors in acute myeloid leukemia and myelodysplastic syndromes. Leukemia 2018;32(5):1094–105 doi 10.1038/s41375-018-0070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wang X, Waschke BC, Woolaver RA, Chen Z, Zhang G, Piscopio AD, et al. Histone Deacetylase Inhibition Sensitizes PD1 Blockade-Resistant B-cell Lymphomas. Cancer Immunol Res 2019;7(8):1318–31 doi 10.1158/2326-6066.Cir-18-0875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Mao W, Ghasemzadeh A, Freeman ZT, Obradovic A, Chaimowitz MG, Nirschl TR, et al. Immunogenicity of prostate cancer is augmented by BET bromodomain inhibition. J Immunother Cancer 2019;7(1):277 doi 10.1186/s40425-019-0758-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Lemery S, Keegan P, Pazdur R. First FDA Approval Agnostic of Cancer Site — When a Biomarker Defines the Indication. New England Journal of Medicine 2017;377(15):1409–12 doi 10.1056/NEJMp1709968. [DOI] [PubMed] [Google Scholar]
- 224.Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord JP, et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol 2020;38(1):1–10 doi 10.1200/jco.19.02105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Hierro C, Matos I, Martin-Liberal J, Ochoa de Olza M, Garralda E. Agnostic-Histology Approval of New Drugs in Oncology: Are We Already There? Clin Cancer Res 2019;25(11):3210–9 10.1158/1078-0432.Ccr-18-3694. [DOI] [PubMed] [Google Scholar]
- 226.Gandara DR, Paul SM, Kowanetz M, Schleifman E, Zou W, Li Y, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med 2018;24(9):1441–8 doi 10.1038/s41591-018-0134-3. [DOI] [PubMed] [Google Scholar]
- 227.Osipov A, Lim SJ, Popovic A, Azad NS, Laheru DA, Zheng L, et al. Tumor Mutational Burden, Toxicity, and Response of Immune Checkpoint Inhibitors Targeting PD(L)1, CTLA-4, and Combination: A Meta-regression Analysis. Clinical Cancer Research 2020;26(18):4842–51 10.1158/1078-0432.Ccr-20-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol 2020;21(10):1353–65 doi 10.1016/S1470-2045(20)30445-9. [DOI] [PubMed] [Google Scholar]
- 229.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018;359(6371):91–7 doi 10.1126/science.aan3706. [DOI] [PubMed] [Google Scholar]
- 230.Pitt JM, Vétizou M, Waldschmitt N, Kroemer G, Chamaillard M, Boneca IG, et al. Fine-Tuning Cancer Immunotherapy: Optimizing the Gut Microbiome. Cancer Research 2016;76(16):4602–7 doi 10.1158/0008-5472.Can-16-0448. [DOI] [PubMed] [Google Scholar]
- 231.Davar D, Vetizou MA, Dzutsev A, Badger J, McCullogh J, Menna C, et al. Abstract IA38: Manipulating the gut microbiome to improve immunotherapy of melanoma. Cancer Immunology Research 2019;7(2 Supplement):IA38–IA doi 10.1158/2326-6074.Cricimteatiaacr18-ia38. [DOI] [Google Scholar]
- 232.Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, et al. Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. N Engl J Med 2019;381(21):2020–31 doi 10.1056/NEJMoa1910231. [DOI] [PubMed] [Google Scholar]
- 233.Reck M, Ciuleanu T-E, Dols MC, Schenker M, Zurawski B, Menezes J, et al. Nivolumab (NIVO) + ipilimumab (IPI) + 2 cycles of platinum-doublet chemotherapy (chemo) vs 4 cycles chemo as first-line (1L) treatment (tx) for stage IV/recurrent non-small cell lung cancer (NSCLC): CheckMate 9LA. Journal of Clinical Oncology 2020;38(15_suppl):9501– doi 10.1200/JCO.2020.38.15_suppl.9501. [DOI] [Google Scholar]
- 234.Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N Engl J Med 2018;378(24):2288–301 doi 10.1056/NEJMoa1716948. [DOI] [PubMed] [Google Scholar]
- 235.Reck M, Mok TSK, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. The Lancet Respiratory medicine 2019;7(5):387–401 doi 10.1016/s2213-2600(19)30084-0. [DOI] [PubMed] [Google Scholar]
- 236.West H, McCleod M, Hussein M, Morabito A, Rittmeyer A, Conter HJ, et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol 2019;20(7):924–37 doi 10.1016/s1470-2045(19)30167-6. [DOI] [PubMed] [Google Scholar]
- 237.Plimack ER, Rini BI, Stus V, Gafanov R, Waddell T, Nosov D, et al. Pembrolizumab plus axitinib versus sunitinib as first-line therapy for advanced renal cell carcinoma (RCC): Updated analysis of KEYNOTE-426. Journal of Clinical Oncology 2020;38(15_suppl):5001– doi 10.1200/JCO.2020.38.15_suppl.5001. [DOI] [Google Scholar]
- 238.Choueiri TK, Motzer RJ, Rini BI, Haanen J, Campbell MT, Venugopal B, et al. Updated efficacy results from the JAVELIN Renal 101 trial: first-line avelumab plus axitinib versus sunitinib in patients with advanced renal cell carcinoma. Ann Oncol 2020;31(8):1030–9 doi 10.1016/j.annonc.2020.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. The New England journal of medicine 2020;382(20):1894–905 doi 10.1056/NEJMoa1915745. [DOI] [PubMed] [Google Scholar]
- 240.Schmittnaegel M, Rigamonti N, Kadioglu E, Cassara A, Wyser Rmili C, Kiialainen A, et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med 2017;9(385) doi 10.1126/scitranslmed.aak9670. [DOI] [PubMed] [Google Scholar]
- 241.Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med 2015;212(2):139–48 doi 10.1084/jem.20140559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Wu FT, Man S, Xu P, Chow A, Paez-Ribes M, Lee CR, et al. Efficacy of Cotargeting Angiopoietin-2 and the VEGF Pathway in the Adjuvant Postsurgical Setting for Early Breast, Colorectal, and Renal Cancers. Cancer Res 2016;76(23):6988–7000 doi 10.1158/0008-5472.CAN-16-0888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Grasselly C, Denis M, Bourguignon A, Talhi N, Mathe D, Tourette A, et al. The Antitumor Activity of Combinations of Cytotoxic Chemotherapy and Immune Checkpoint Inhibitors Is Model-Dependent. Front Immunol 2018;9:2100 doi 10.3389/fimmu.2018.02100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Tallon de Lara P, Cecconi V, Hiltbrunner S, Yagita H, Friess M, Bode B, et al. Gemcitabine Synergizes with Immune Checkpoint Inhibitors and Overcomes Resistance in a Preclinical Model and Mesothelioma Patients. Clin Cancer Res 2018;24(24):6345–54 10.1158/1078-0432.CCR-18-1231. [DOI] [PubMed] [Google Scholar]
- 245.Burlion A, Ramos RN, Kc P, Sendeyo K, Corneau A, Menetrier-Caux C, et al. A novel combination of chemotherapy and immunotherapy controls tumor growth in mice with a human immune system. Oncoimmunology 2019;8(7):1596005 doi 10.1080/2162402X.2019.1596005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Wang L, Amoozgar Z, Huang J, Saleh MH, Xing D, Orsulic S, et al. Decitabine Enhances Lymphocyte Migration and Function and Synergizes with CTLA-4 Blockade in a Murine Ovarian Cancer Model. Cancer Immunol Res 2015;3(9):1030–41 doi 10.1158/2326-6066.CIR-15-0073. [DOI] [PubMed] [Google Scholar]
- 247.Stone ML, Chiappinelli KB, Li H, Murphy LM, Travers ME, Topper MJ, et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proceedings of the National Academy of Sciences 2017;114(51):E10981–E90 doi 10.1073/pnas.1712514114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015;527(7577):249–53 doi 10.1038/nature15520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Rojas JJ, Sampath P, Hou W, Thorne SH. Defining Effective Combinations of Immune Checkpoint Blockade and Oncolytic Virotherapy. Clin Cancer Res 2015;21(24):5543–51 10.1158/1078-0432.CCR-14-2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Saha D, Martuza RL, Rabkin SD. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 2017;32(2):253–67 e5 10.1016/j.ccell.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Bourgeois-Daigneault M-C, Roy DG, Aitken AS, El Sayes N, Martin NT, Varette O, et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Science Translational Medicine 2018;10(422):eaao1641 doi 10.1126/scitranslmed.aao1641. [DOI] [PubMed] [Google Scholar]
- 252.Speranza MC, Passaro C, Ricklefs F, Kasai K, Klein SR, Nakashima H, et al. Preclinical investigation of combined gene-mediated cytotoxic immunotherapy and immune checkpoint blockade in glioblastoma. Neuro Oncol 2018;20(2):225–35 doi 10.1093/neuonc/nox139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Chon HJ, Lee WS, Yang H, Kong SJ, Lee NK, Moon ES, et al. Tumor Microenvironment Remodeling by Intratumoral Oncolytic Vaccinia Virus Enhances the Efficacy of Immune-Checkpoint Blockade. Clin Cancer Res 2019;25(5):1612–23 10.1158/1078-0432.CCR-18-1932. [DOI] [PubMed] [Google Scholar]
- 254.Verbrugge I, Hagekyriakou J, Sharp LL, Galli M, West A, McLaughlin NM, et al. Radiotherapy increases the permissiveness of established mammary tumors to rejection by immunomodulatory antibodies. Cancer Res 2012;72(13):3163–74 doi 10.1158/0008-5472.CAN-12-0210. [DOI] [PubMed] [Google Scholar]
- 255.Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015;520:373 doi 10.1038/nature1429210.1038/nature14292https://www.nature.com/articles/nature14292#supplementary-informationhttps://www.nature.com/articles/nature14292#supplementary-information . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Kroon P, Gadiot J, Peeters M, Gasparini A, Deken MA, Yagita H, et al. Concomitant targeting of programmed death-1 (PD-1) and CD137 improves the efficacy of radiotherapy in a mouse model of human BRAFV600-mutant melanoma. Cancer Immunol Immunother 2016;65(6):753–63 doi 10.1007/s00262-016-1843-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Zhao J, Wen X, Tian L, Li T, Xu C, Wen X, et al. Irreversible electroporation reverses resistance to immune checkpoint blockade in pancreatic cancer. Nat Commun 2019;10(1):899 doi 10.1038/s41467-019-08782-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Peng W, Liu C, Xu C, Lou Y, Chen J, Yang Y, et al. PD-1 blockade enhances T-cell migration to tumors by elevating IFN-gamma inducible chemokines. Cancer Res 2012;72(20):5209–18 doi 10.1158/0008-5472.CAN-12-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Moon EK, Ranganathan R, Eruslanov E, Kim S, Newick K, O’Brien S, et al. Blockade of Programmed Death 1 Augments the Ability of Human T Cells Engineered to Target NY-ESO-1 to Control Tumor Growth after Adoptive Transfer. Clin Cancer Res 2016;22(2):436–47 10.1158/1078-0432.CCR-15-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Jayaprakash P, Ai M, Liu A, Budhani P, Bartkowiak T, Sheng J, et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J Clin Invest 2018;128(11):5137–49 doi 10.1172/JCI96268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017;548(7668):471–5 doi 10.1038/nature23465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Garton AJ, Seibel S, Lopresti-Morrow L, Crew L, Janson N, Mandiyan S, et al. Anti-KIT Monoclonal Antibody Treatment Enhances the Antitumor Activity of Immune Checkpoint Inhibitors by Reversing Tumor-Induced Immunosuppression. Molecular Cancer Therapeutics 2017;16(4):671–80 doi 10.1158/1535-7163.Mct-16-0676. [DOI] [PubMed] [Google Scholar]
- 263.Seifert AM, Zeng S, Zhang JQ, Kim TS, Cohen NA, Beckman MJ, et al. PD-1/PD-L1 Blockade Enhances T-cell Activity and Antitumor Efficacy of Imatinib in Gastrointestinal Stromal Tumors. Clinical Cancer Research 2017;23(2):454–65 10.1158/1078-0432.Ccr-16-1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature 2016;539:443 doi 10.1038/nature20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016;539:437 doi 10.1038/nature1983410.1038/nature19834https://www.nature.com/articles/nature19834#supplementary-informationhttps://www.nature.com/articles/nature19834#supplementary-information . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Homet Moreno B, Mok S, Comin-Anduix B, Hu-Lieskovan S, Ribas A. Combined treatment with dabrafenib and trametinib with immune-stimulating antibodies for BRAF mutant melanoma. OncoImmunology 2016;5(7):e1052212 doi 10.1080/2162402X.2015.1052212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Sai J, Owens P, Novitskiy SV, Hawkins OE, Vilgelm AE, Yang J, et al. PI3K Inhibition Reduces Mammary Tumor Growth and Facilitates Antitumor Immunity and Anti-PD1 Responses. Clinical Cancer Research 2017;23(13):3371–84 10.1158/1078-0432.Ccr-16-2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Kim SH, Roszik J, Cho SN, Ogata D, Milton DR, Peng W, et al. The COX2 Effector Microsomal PGE2 Synthase 1 is a Regulator of Immunosuppression in Cutaneous Melanoma. Clin Cancer Res 2019;25(5):1650–63 10.1158/1078-0432.CCR-18-1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Choi H, Deng J, Li S, Silk T, Dong L, Brea EJ, et al. Pulsatile MEK Inhibition Improves Anti-tumor Immunity and T Cell Function in Murine Kras Mutant Lung Cancer. Cell Rep 2019;27(3):806–19 e5 doi 10.1016/j.celrep.2019.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Kinkead HL, Hopkins A, Lutz E, Wu AA, Yarchoan M, Cruz K, et al. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight 2018;3(20) doi 10.1172/jci.insight.122857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Zaidi N, Quezada SA, Kuroiwa JMY, Zhang L, Jaffee EM, Steinman RM, et al. Anti-CTLA-4 synergizes with dendritic cell-targeted vaccine to promote IL-3-dependent CD4(+) effector T cell infiltration into murine pancreatic tumors. Ann N Y Acad Sci 2019;1445(1):62–73 doi 10.1111/nyas.14049. [DOI] [PMC free article] [PubMed] [Google Scholar]