

Abstract
Germline variation and smoking are independently associated with pancreatic ductal adenocarcinoma (PDAC). We conducted genome-wide smoking interaction analysis of PDAC using genotype data from four previous genome-wide association studies in individuals of European ancestry (7,937 cases and 11,774 controls). Examination of expression quantitative trait loci data from the Genotype-Tissue Expression Project followed by colocalization analysis was conducted to determine if there was support for common SNP(s) underlying the observed associations. Statistical tests were two sided and P-values < 5 x 10-8 were considered statistically significant. Genome-wide significant evidence of qualitative interaction was identified on chr2q21.3 in intron 5 of the transmembrane protein 163 (TMEM163) and upstream of the cyclin T2 (CCNT2). The most significant SNP using the Empirical Bayes method, in this region which included 45 significantly associated SNPs, was rs1818613 (per allele OR in never smokers 0.87, 95% CI 0.82-0.93; former smokers 1.00, 95 CI 0.91-1.07; current smokers 1.25, 95%CI 1.12-1.40, interaction P-value=3.08x10-9). Examination of the Genotype-Tissue Expression Project data demonstrated an expression quantitative trait locus in this region for TMEM163 and CCNT2 in several tissue types. Colocalization analysis supported a shared SNP, rs842357, in high LD with rs1818613 (r2=0. 94) driving both the observed interaction and the expression quantitative trait loci signals. Future studies are needed to confirm and understand the differential biologic mechanisms by smoking status that contribute to our PDAC findings.
Keywords: pancreatic cancer, smoking, genetics, interaction, epidemiology
Introduction
Pancreatic cancer is the seventh leading cause of cancer death worldwide (1). In 2018, 458,918 new cases of pancreatic cancer were diagnosed, and 432,242 individuals died from this disease in the world (1). The incidence of pancreatic cancer has significantly increased since the mid-1990s in the United States and worldwide (2,3). The risk of pancreatic cancer increases with age, with the majority of cases diagnosed after age 55 years (1). Pancreatic ductal adenocarcinoma (PDAC) is the most common subtype and represents > 85% of all pancreatic cancers (1).
Inherited susceptibility plays an important role, as demonstrated by the high-risk of PDAC in individuals with a family history of pancreatic cancer, particularly those with multiple affected relatives (4). Pathogenic variants in BRCA1, BRCA2, PALB2, ATM, CDKN2A, STK11, and DNA mismatch repair genes are associated with increased PDAC (5), with recent studies demonstrating up to 10% of people with PDAC carry variants in these genes (6). Common variants also play a role in PDAC, with array-based heritability estimates of up to 21.2% (7). Our genome-wide association studies (GWAS) have identified 18 regions associated with PDAC. Associated gene regions include 1p36.33 (NOC2L), 2 independent loci at 1q32.1 (NR5A2), 2p13.3 (ETAA1), 3q29 (TP63), 3 loci at 5p15.33 (CLPTM1L-TERT), 7p12 (TNS3), 7p13 (SUGCT), 7q32.3 (LINC-PINT), 8q21.11 (HNF4G), 8q24.21(MYC), 9q34.2 (ABO), 13q12.2 (PDX1), 13q22.1 (KLF5), 16q23.1 (BCAR1), 17q12 (HNF1B), 17q25.1 (LINC00673), 18q21.32 (GRP) and 22q12.1 (ZNRF3) (8–13).
Other risk factors for PDAC include cigarette smoking, diabetes, chronic pancreatitis, heavy alcohol use, and excess body weight (1). In particular, the association between smoking and PDAC is well established, with an estimated population attributable fraction in the United States of 12.1% (14). Both case-control and cohort studies have demonstrated an approximately 2-fold elevated risk for current smokers compared with never smokers (15,16). A pooled analysis of data from 12 case-control studies within the Pancreatic Cancer Case-Control Consortium (PanC4) showed that compared with never smokers, the odds ratio (OR) of PDAC was 1.17 (95% confidence interval [CI] 1.02-1.34) for former smokers and 2.20 (95% CI 1.71-2.83) for current smokers (15,16). A pooled nested case-control study within 12 cohorts in the Pancreatic Cancer Cohort Consortium (PanScan) showed an increased PDAC risk for current smokers compared with never smokers (OR = 1.77, 95% CI 1.38- 2.2) (15). Although no overall association was observed for former smokers, former smokers who had quit less than 10 years had an elevated risk (OR=2.19, 95% CI 1.25-3.83) with the risk attenuating with time since cessation and approaching that of never smokers after 15 years (15).
Cigarette exposure has also been shown to cluster within families, and nicotine addiction has a strong heritable component (17,18). Several genome-wide significant loci have been associated with distinct smoking-related traits (19–21). Established associations include a cluster of nicotinic acetylcholine receptor (nAChR) genes, CHRNA5-CHRNA3-CHRNB4 located on chromosome 15q24 (20,21). Despite the large number of associated loci, altogether the common genetic variants account only for 0.1% of the phenotypic variation in smoking cessation and 2.9% of the phenotypic variation in age at smoking initiation, indicating the highly polygenic nature of these traits (21).
Given the known the importance of genetic variation in modulating both pancreatic cancer and cigarette smoking, understanding whether the association of specific genetic variants differs by smoking status could increase our understanding of both of these traits. Candidate gene studies, mostly related to carcinogen metabolism, DNA repair, oxidative stress, and inflammation, have examined risk smoking interactions for PDAC with inconsistent results (22). A previous genome-wide gene-smoking interaction analysis for PDAC that included 2,028 cases and 2,109 controls from PanC4 did not find significant interactions (23); however, the small sample size did not have sufficient power to detect modest effect sizes. Therefore, in the present study, we conducted genome-wide smoking interaction analysis of PDAC risk using genotype data from four previous GWAS studies conducted by the PanScan and PanC4 Consortia (8–12) and three alternative statistical methods that have robust power to detect gene-environment interactions (24). We also examined if smoking modified the association of established PDAC susceptibility variants (8–13).
Material and Methods
Study sample
Study participants were selected from four previously conducted GWAS from the Pancreatic Cancer Cohort Consortium (PanScan) and the Pancreatic Cancer Case-Control Consortium (PanC4). Details of these studies have been previously published (8–12). Each participating study obtained written informed consent from participants and approval from their local Institutional Review Board. The studies were conducted in accordance with recognized ethical guidelines (e.g., Declaration of Helsinki, CIOMS, Belmont Report, U.S. Common Rule). Both the Johns Hopkins School of Medicine Institutional Review Board and the National Cancer Institute (NCI) Special Studies Institutional Review Board approved the consortia study. Our study was based on 9,038 primary PDAC cases (ICD-O-3 code C250-C259) and 12,389 controls free of known PDAC. Participants with non-exocrine pancreatic tumors were excluded (histology types 8150, 8151, 8153, 8155 and 8240). We included only participants of European ancestry, to reduce confounding due to population stratification, and those with complete smoking data (Supplemental figure 1). Our final analytic dataset included 7,937 PDAC cases and 11,774 controls (Supplemental table 1).
Genome-wide association study genotype data
Genotyping was conducted in four phases, PanScan I, PanScan II, PanScan III and PanC4. The PanScan studies were genotyped at the NCI Cancer Genomics Research Laboratory at the National Institutes of Health and genotyped on the Illumina HumanHap series arrays (Illumina HumanHap550 Infinium II (8), Human 610-Quad (11) for PanScan I and II, respectively, and the Illumina Omni series arrays (OmniExpress, Omni1M, Omni2.5 and Omni5M) for PanScan III (12). PanC4 was genotyped on the Illumina HumanOmniExpressExome-8v1 array at the Johns Hopkins Center for Inherited Disease Research (9). The data from the PanScan and PanC4 GWAS are available through dbGAP (accession numbers phs000206.v5.p3 and phs000648.v1.p1, respectively). Details on imputation and quality controls have been described (10). In brief, SNPs were excluded if they had call rates ≤ 98%, MAF ≤ 0.05 and Hardy-Weinberg equilibrium P-values, < 1.0 x 10−6 in the controls. SNPs were pre-phased using SHAPEIT2 software (25). Genotype imputation was conducted using IMPUTE2 (26) with 1000 genomes Phase 3 (27). Imputation was conducted separately for the PanScan I/II, PanScan III and PanC4 GWAS. After imputation, we retained only SNPs with imputation quality scores >0.5 and MAFs >0.05. Our final analysis included 6,769,447 variants.
Smoking and demographic assessment
Smoking status was assessed through self- or proxy-report via self-administered questionnaires (15) or in-person interviews (15,16). Baseline smoking status was used for the cohort studies. For the case-control studies, smoking status at diagnosis (for cases) or when the questionnaire was administered (for controls) was used (16). For these analyses smoking was categorized as never, former, and current cigarette smoker. Never smokers were individuals who smoked fewer than 100 cigarettes in their lifetime or for less than 6 months. Former smokers were individuals who reported quitting cigarette smoking > 1 year prior to the administration of the questionnaire. Current smokers were individuals who reported current smoking at the time of the questionnaire or who reported quitting cigarette smoking within the past year. Data on age, sex, and other possible confounders were collected from questionnaires at baseline from each cohort study and when smoking was assessed from the case-control studies (15,16).
Statistical analyses
We used the unconstrained maximum-likelihood (UML), constrained maximum likelihood (CML), and Empirical Bayes (EB) methods to evaluate the interaction between smoking status and each individual genetic variant (24). The UML corresponds to standard logistic regression analysis of binary outcome data (case-control) which allows the joint distribution of underlying covariates of the model to remain completely unspecified. The CML exploits an assumption of independence between SNP and smoking status in the underlying population (28) The method, similar to the case-only method (29), can gain in inferential efficiency of the interaction parameter and yet it can be used to test or estimate all of the parameters of an underlying logistic model, including the main effect of a SNP and the exposure of interest. The EB is an intermediate between the two methods and allows for a data-adaptive relaxation of the gene-environment independence assumption. Because the EB provides a good compromise between bias and variance (30,31), we used this as the primary method for evaluating interaction and the other two methods for sensitivity analysis.
The association analysis was conducted using CGEN software (Version 3.5.0) (https://dceg.cancer.gov/tools/analysis/cgen), an R package for logistic regression analyses of SNP-environment interactions (32), using the ‘snp.score’ option to incorporate the genotype probabilities from the imputed data in the analysis. This option calculates a score test (JScore), which tests for the joint effect of SNP and SNP-by-environment interaction under a logistic regression model.
The analyses were first conducted separately for the PanScan and PanC4 data sets and results were combined using meta-analysis. Smoking was included as categorical dummy variable with never smokers as reference. Association magnitude of each SNP was modeled according to additive SNP-dosage. Imputed SNPs were incorporated through expected dosage using the snp.score function of the CGEN package (32). Interaction between SNP and smoking was modeled using two parameters (current, never) and (former, never). Each SNP genotype was coded using an underlying dosage model, coded in terms of observed/impute allele counts. The analysis was adjusted for age in decade, sex, and top eigenvectors (5 for PanScan and 9 for PanC4) from principal components analysis to control for ancestry (10). PanScan analyses were also adjusted for study and geographic region of the parent studies (12). For each SNP we obtained the one-step maximum-likelihood estimate of SNP and SNP-smoking interaction effects along with the associated variance-covariance matrix from the SNP-score function (24). We implemented a fixed-effects meta-analysis using these summary statistics. Meta-analysis was performed for the joint association magnitudes of the SNP and the SNP by smoking, and interaction effect only. We then performed a 2 degree-of-freedom tests for SNP by smoking interaction terms of the model as a way of identifying SNPs/regions the effect of which may be modified by smoking. In addition, we performed 3 degree-of-freedom joint tests (33) that simultaneously test for both the main effect of a SNP and two SNP by smoking interaction terms. We also conducted Linkage disequilibrium (LD) clumping at an r2 threshold of 0.05 to identify independent regions of association (34). Statistical tests were two sided and P-values ≤ 5 x 10−8 statistically significant.
Expression quantitative trait locus (eQTL) analysis
Using the NIH Genotype-Tissue Expression (GTEx) v8 (dbGaP Accession phs000424.v8.p2, https://gtexportal.org/home/) (35), we examined eQTL to assess the cis effects of the SNPs in the 2q21.3 region on gene expression across multiple tissues. In addition, we created regional plots of the 2q21.3 region using the SNP2GENE function of FUMA (36).
The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from: (https://gtexportal.org/home/) the Genotype-Tissue Expression (GTEx) Project Portal on 06/30/19 and 7/30/20 and/or dbGaP accession number phs000424.vN.pN on 06/30/2019.
Colocalization analysis
For each SNP, we first meta-analyzed estimate of interaction parameters across current and former smokers to obtain a single estimate of SNP by smoking interaction under a dose-response model for smoking with never former and current coded as 0, 1 and 2, respectively. We used single statistics to summarize the evidence of interaction of individual SNPs with respect to smoking status in the colocalization analysis for the ease of interpretation of final results. Estimates of interaction separately by smoking categories indicate the dose-response model is adequate. To perform colocalization analysis, we matched the reference and alternate allele across our genome-wide interaction study and the eQTL results using GTEx v7. We performed colocalization analysis using two methods, co-loc and eCAVIAR (37,38). For eCAVIAR, we investigated the locus by considering 500Kb upstream and downstream of the most significant SNP, rs1818613, from the genome-wide interaction scan. In addition, we chose genes with at least one significant variant and set the maximum number causal variants to 3.
Results
We found genome-wide significance (P-value < 5X10−8) evidence for interaction between smoking and multiple SNPs located in a region on chromosome 2q21.2 on pancreatic cancer risk. Figure 1 shows the Q-Q and Manhattan plot associated with genome-wide test for interaction using the EB method. Compared with the theoretical distributions, the lambda value of 0.93 showed reasonable control of type-1 errors.
Figure 1:
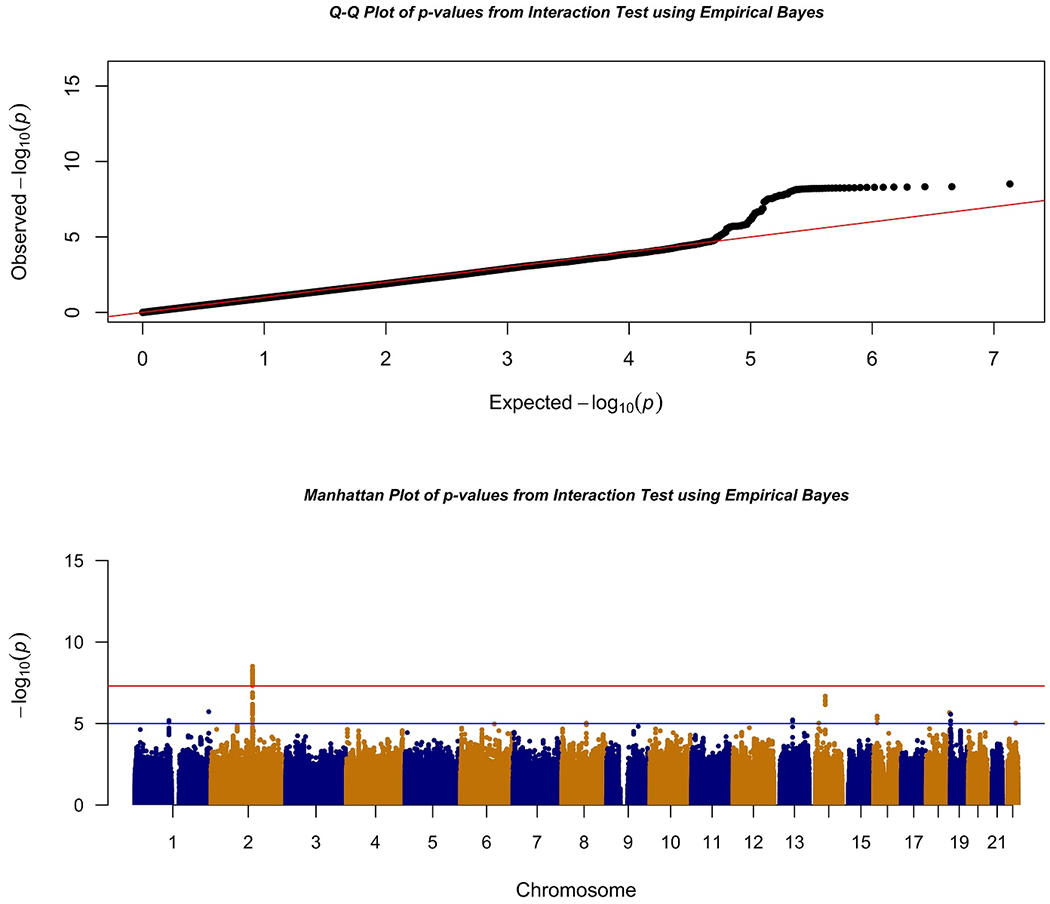
Q-Q and Manhattan Plots of Interaction Analysis using Empirical Bayes Approach
Supplemental figure 2 shows the Q-Q and Manhattan plots for the CML and UML methods. The most significant SNP in this region was rs1818613 (P-value = 3.08x10−9). The SNP also achieved genome-wide significance (p-value=2.7x10−9) for interaction in CML and was strong but not at the genome-wide significance threshold using UML (Table 1).
Table 1:
Region with genome wide significant evidence for SNP by smoking interaction on risk of PDAC
| Chromosome Physical Position SNP Ref/Effect Alleles Ref Allele Frequency Imputation Quality Gene |
Analytical Method |
Odds Ratio for rs1818613 (95% Confidence Interval) P-value |
Interaction P-value# |
||
|---|---|---|---|---|---|
| Never Smokers | Former Smokers | Current Smokers | |||
| 2q21.3 135356285 rs1818613 G/T 0.39 0.99 TMEM163 (intronic) |
CML | 0.87 (0.82, 0.92) 1.19x10−6 |
0.97 (0.91,1.02) 0.24 |
1.16 (1.07,1.25) 3.3x10−4 |
2.7x10−9 |
| EB | 0.87 (0.82,0.93) 0.001 |
1.00 (0.93,1.07) 0.94 |
1.25 (1.12,1.40) 1x10−4 |
3.08x10−9 | |
| UML | 0.89 (0.84,0.96) 2.04x10−5 |
0.99 (0.92,1.06) 0.74 |
1.17 (1.08,1.28) 2.6x10−4 |
1.02x10−6 | |
Physical position in Build 37: CML, Constrained maximum-likelihood; EB, Empirical Bayes; UML, Unconstrained maximum-likelihood
Based on 2 degrees of freedom chi-square test. Analysis was adjusted for age, sex, ancestry (via principle components) and for PanScan study phase and site
More than 45 additional SNPs within the ~100Kb region of high LD (r2>0.9) also showed evidence of interaction (2 degree-of-freedom interaction EB P-value < 5x10−8) (Figure 2, (36)). This region is located in intron 5 of the transmembrane protein 163 gene (TMEM163) and 100kb upstream of transcription factor cyclin T2 (CCNT2). The pattern of the association was similar across the GWAS phases and in both cohorts and case-control study designs Supplemental table 2 and 3).
Figure 2:
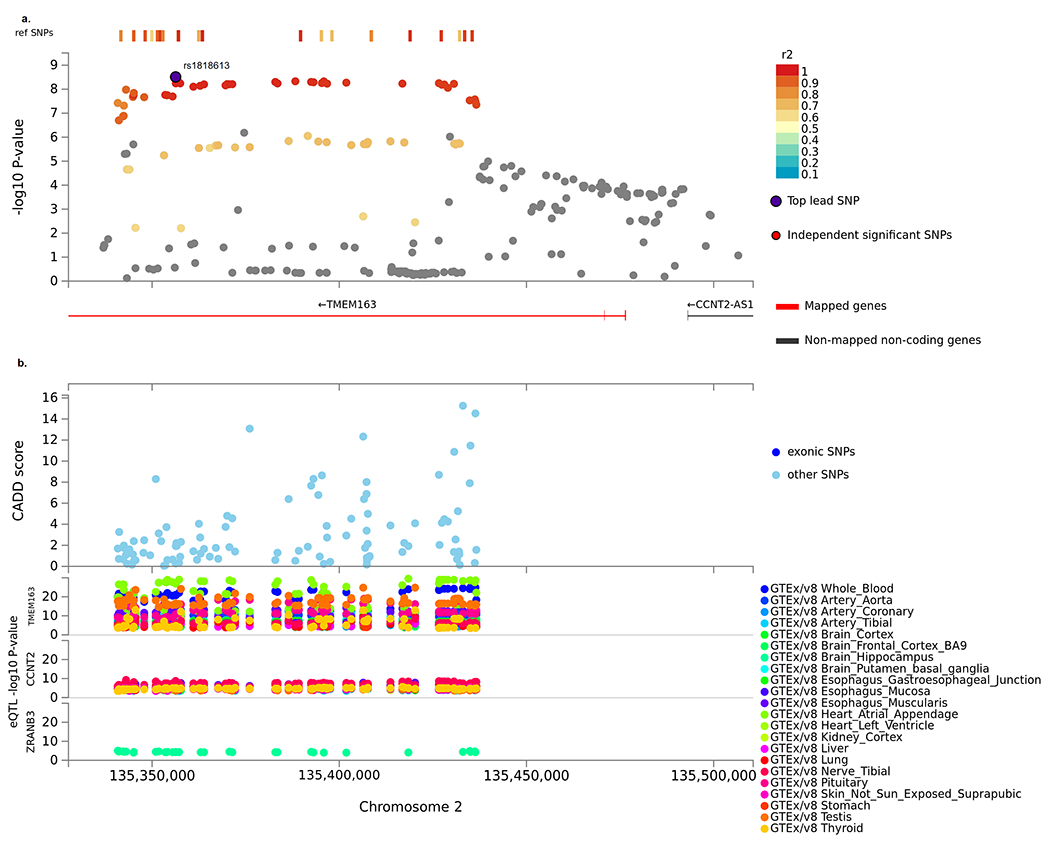
Locus plot of 2q21.3 region for the interaction GWAS of pancreatic cancer by smoking using FUMA. a. Extended region of the TMEM163 locus that prioritizes the TMEM163 gene. b. Zoomed in regional plot of TMEM163 locus with GWAS interaction P-values (SNPs are colored based on r2), Combined Annotation Dependent Depletion (CADD score), and Expression quantitative trait loci (eQTL) P-value for the most significant associations. eQTLs are plotted per gene and colored based on tissue types.
Three genomic regions, 14q22.1 (lead SNP, rs8003600) , 1q12 (rs3765814 OPN3 intronic), and 16q12.1 (rs78013006 LOC105371082 intronic) showed suggestive evidence for interactions using the empirical Bayes approach (2 degree-of-freedom interaction EB P-value ≤ 5x10−6, Supplemental table 4).
Established GWAS regions
Overall, no significant interactions by smoking status were observed for any of the 20 previously identified GWAS SNPs for PDAC in European populations (8–12). One SNP (rs16986825 ZNRF3 on chr.22q12.1) had a P-value = 0.005 under the CML method (Supplemental table 5).
Expression quantitative trait locus (eQTL) and co-localization analysis
Examination of the GTEx8 data identified an eQLT (False Discovery Rate ≤0.05) in the same chromosome 2q21 region as our SNP-by-smoking interaction (Figure 2, Supplemental table 6). For rs1818613, under the additive model each copy of the T compared to the G allele was associated with increased expression of TMEM163 in heart atrial appendage (β=0.55, P-value =1.2x10−27), whole blood (β=0.40, P-value =1.1x10−21), esophagus muscularis (β=0.41, P-value =7.0x10−15) and pituitary (β=0.28, P-value=1.8 X 10−11) and decreased expression in testis (β=−0.45, P-value =3.1 X10−17) tissue. In addition, there was lower CCNT2 expression in tibial nerve tissue (β=−0.13, P-value=1.1X 10−9) and lung tissue (β=−0.14, P-value=1.5X 10−7) for the T allele compared with the G allele. At a lower significance, the T allele compared to the G allele of rs1818613 was also associated with expression in the brain, including higher expression of TMEM163 in the frontal cortex (β=0.21, P-value=3.8 X 10−6), CCNT2-AS1 in the cerebellar hemisphere (β=0.41, P-value=2.3 X 10−5) and cerebellum (β=0.35, P-value=4.9 X 10−5), and VDAC2P4 in the cerebellar hemisphere (β=0.38 P-value=6.6 X 10−5), and lower expression for ZRANB3 in the hippocampus (β=−0.30, P-value=4.1 X 10−5) and CCNT2 in the cortex (β=−0.15, P-value=6.1 X 10−5).
Both co-loc and eCAVIAR colocalization analyses using GTEx 7 showed the posterior probability of a shared functional locus was extremely high. The most significant evidence was for rs842357, which is in strong LD with rs1818613 (r2=0.94), and also had significant evidence of interaction with smoking (EB p=1.75 x 10−08) (Figure 3).
Figure 3:
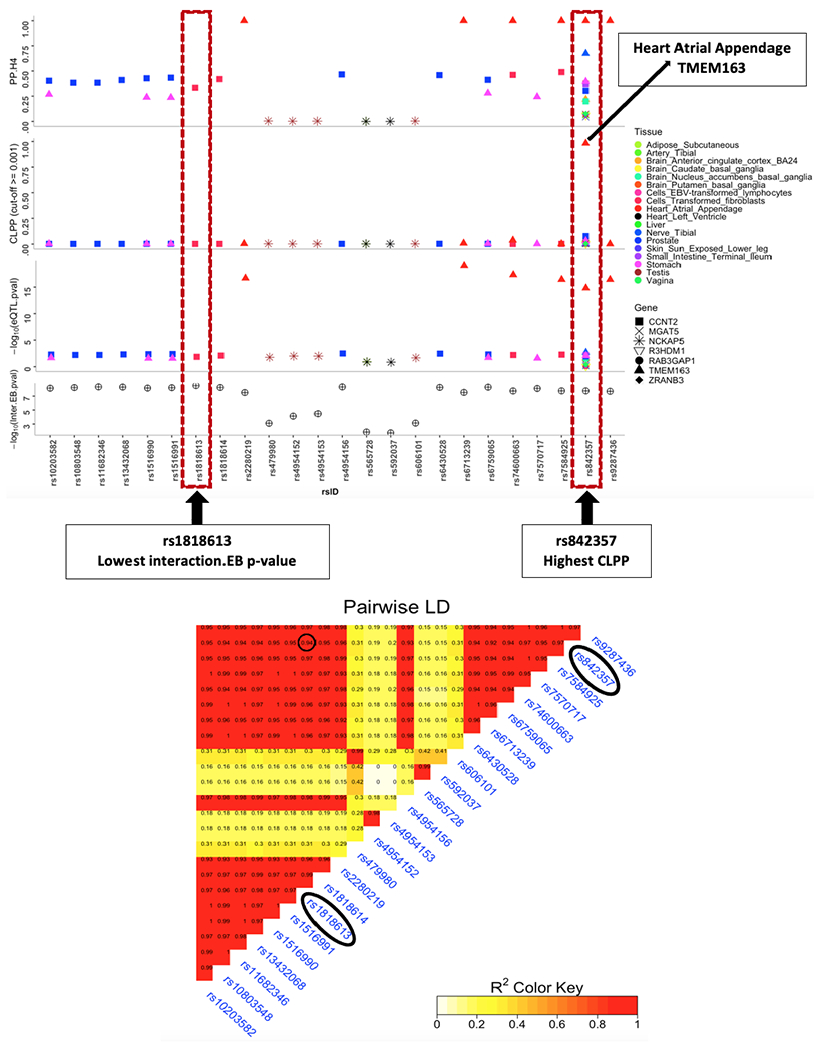
Results of colocalization analysis using eCAVIAR and co-loc. SNPs with colocalization probability (CLPP) >= 0.001 are shown in this plot. PP.H4 denotes posterior probability of having a common causal snp across eQTL and SNP X Smoking loci.
The A compared to the G allele of rs842357 was associated with lower expression of TMEM163 in heart atrial appendage, tibial nerve, and stomach. The eCAVIAR posterior probability of a shared locus underlying both the SNP-by-smoking association and eQTL results for heart atrial appendage was 0.98. Additional evidence of colocalization (CLPP >0.01) was observed for rs842357 and TMEM163 in brain anterior cingulate cortex BA24 and for CCNT2 in prostate, cells transformed fibroblasts, and small intestine terminal ileum (Supplemental table 7). Interestingly, the A allele of rs843257 associated with decreased TMEM163 expression and increased CCNT2 expression compared with the T allele.
We used HaploReg (39) to inspect the region in LD (r2>0.8) with rs842357 as the anchoring SNP, in order to identify variants altering transcription factor bindings (Supplemental table 8). HaploReg includes a wide variety of functional annotations collected from Roadmap Epigenomics Project (40) as chromatin, DNase and promoter and enhancer histones marks. We observed H3K4me1 marks in this region, indicating this is an active enhancer region for tissues including pancreas as well as H3K4me3 marks in blood, heart and thymus.
Discussion
We observed a qualitative interaction by cigarette smoking status for genetic variation and PDAC in a large LD block on chromosome 2 (2q21.3) in intron 5 of TMEM163 and upstream of CCNT2 such that alleles were associated with increased risk among current smokers and demonstrated a decreased risk among never smokers. The pattern of the interaction was consistent across three analytical methods and across study designs and individual GWAS studies. Our colocalization results support a single locus at the 2q21.3 region that underlies the interaction and an eQLT for TMEM163 and CCNT2 in this region. Given the qualitative nature of this interaction, it is not surprising we did not observe an association in this region when examining the main effect of SNPS in this region GWAS without modeling the interacting effect of smoking as the differing associations for smokers and non-smokers would result in no overall association. To the best of our knowledge, this is the largest gene-by-smoking interaction study for PDAC conducted to date.
The TMEM163 gene is conserved across many vertebrate species; it is highly expressed in specific brain regions and neuronal populations (glutamatergic and γ-aminobutyric acid (GABA)-ergic) (41), and is modestly expressed in other tissues including the pancreas, pituitary, and testis (35). TMEM163 is a zinc binding and transporter protein involved in cellular zinc homeostasis and whose putative interaction with other zinc transporters and role in health and disease is not well understood (42). Zinc mediates a wide range of cellular processes and alternations in its homeostasis can disrupt cellular function (43). Dysregulation of other zinc transporters has been observed in PDAC such that zinc transporter upregulation has been associated with enhanced cancer cell migration and worse patient prognosis (43). Interestingly, a genome-wide association study reported an association in the same 2q21.3 region in an intron of TMEM163 gene for nicotine withdrawal in heavy smokers in a population-based Finnish Twin Cohort (19). Germline variation in 2q21 region has also been associated with Parkinson’s disease (44) and hematocrit concentrations (45) in populations of European ancestry and Type 2 diabetes in Asian Indians (46) and a Mongolian population in China (47).
Any hypothesis regarding the underly mechanism of the observed interaction between genetic variation at the 2q21.3 region and cigarette smoking is speculative. Its qualitative nature suggests either a single mechanism that has protective effects in never smokers compared to adverse in current smokers or given the role of this region in regulating multiple proteins, the protective effect observed in never-smokers is overwhelmed by a second risk-increasing mechanism in the context of cigarette smoking. Co-localization of our association results with eQLT signals in TMEM163 and CCNT2 in several tissue types suggests gene-regulation beyond the pancreatic gland may play a role. There was colocalization for the eQLT in TMEM163 in heart, nerve and stomach. We observed high levels of H3K4me1 in this region for many tissues including pancreas, indicating this is an active enhancer region. Further work is needed to understand these findings. We also observed multiple eQTL signals in the brain and colocalization at a lower posterior probability for the brain anterior cingulate cortex which functions in impulse control (48) and may play a role in nicotine dependence (49). This is in accord with the Finnish GWAS study which linked variation inTMEM163 to nicotine dependence (19) and might be contributing to our qualitative interaction and increased risk in smokers. Experimental studies have also shown CCNT2 to be differentially altered in the brain, especially the hypothalamus, in response to nicotine, nicotine-withdrawal, and coinciding change in body weight (50). Future studies are needed to explore the 2q21.3 region in individuals with detailed smoking and other phenotype data as this may have utility for smoking cessation and cancer prevention. Metals found in cigarette smoke, such a cadmium, have been shown to compete with other zinc transporters (e.g. metallothionein and ZIP8) and increase chronic toxicity (51). It is possible a similar process could be contributing to the increased risk in smokers and qualitative interaction that we observe. Long term cigarette smoke spreads smoke related chemicals systemically in the bloodstream to target organs (52) and tobacco smoke inhalation causes pancreatic inflammation and damage to β-cells (53,54). It is plausible that the interaction that we observe may be related to pancreatogenic disease processes in smokers (53,55) that are not present in never smokers. Experimental studies are needed to explore these hypothesized mechanisms.
In conclusion, we identified a qualitative interaction for PDAC by cigarette smoking status at 2q21.3 in intron 5 of the TMEM163 region. The co-localization results and eQTLs for TMEM163 and CCNT2 provides evidence of the importance of this gene region. Further studies are needed to replicate and understand the differential functional mechanisms by smoking status that contribute to our findings as these may have implications for cancer prevention.
Supplementary Material
Statement of Significance:
This large genome-wide interaction study identifies a susceptibility locus on 2q21.3 that significantly modified PDAC risk by smoking status, providing insight into smoking-associated PDAC with implications for prevention.
Acknowledgments
Funding section:
This work was supported by RO1CA154823 and P50CA062924 (PI: A.P. Klein), R01 HG010480-01 (PI: N. Chatterjee), Patient-Centered Outcomes Research Institute Award (ME-1602-34530) and the Intramural Research Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute (PI: R.Z. Stolzenberg-Solomon). Funding for the parent studies are listed below.
The IARC/Central Europe study was supported by a grant from the US National Cancer Institute at the National Institutes of Health (R03 CA123546-02) and grants from the Ministry of Health of the Czech Republic (NR 9029-4/2006, NR9422-3, NR9998-3, MH CZ-DRO-MMCI 00209805).
The work at Johns Hopkins University was supported by the NCI Grants P50CA062924 and R01CA97075. Additional support was provided by the Lustgarten Foundation, Susan Wojcicki and Dennis Troper and the Sol Goldman Pancreas Cancer Research Center. This work was supported by RO1 CA154823 and federal funds from the National Cancer Institute (NCI), US National Institutes of Health (NIH) under contract number HHSN261200800001E.
The Mayo Clinic Biospecimen Resource for Pancreas Research study is supported by the Mayo Clinic SPORE in Pancreatic Cancer (P50 CA102701).
The Memorial Sloan Kettering Cancer Center Pancreatic Tumor Registry is supported by P30CA008748, the Geoffrey Beene Foundation, the Arnold and Arlene Goldstein Family Foundation, and the Society of MSKCC. We acknowledge the contribution of Dr. Irene Orlow, MS, PhD to this analysis.
The PACIFIC Study was supported by RO1CA102765, Kaiser Permanente and Group Health Cooperative.
The Queensland Pancreatic Cancer Study was supported by a grant from the National Health and Medical Research Council of Australia (NHMRC) (Grant number 442302). RE Neale is supported by a NHMRC Senior Research Fellowship (#1060183).
The UCSF pancreas study was supported by NIH-NCI grants (R01CA1009767, R01CA109767-S1 and R0CA059706) and the Joan Rombauer Pancreatic Cancer Fund. Collection of cancer incidence data was supported by the California Department of Public Health as part of the statewide cancer reporting program; the NCI’s SEER Program under contract HSN261201000140C awarded to CPIC; and the CDC’s National Program of Cancer Registries, under agreement #U58DP003862-01 awarded to the California Department of Public Health.
The Yale (CT) pancreas cancer study is supported by National Cancer Institute at the U.S. National Institutes of Health, grant 5R01CA098870. The cooperation of 30 Connecticut hospitals, including Stamford Hospital, in allowing patient access, is gratefully acknowledged. The Connecticut Pancreas Cancer Study was approved by the State of Connecticut Department of Public Health Human Investigation Committee. Certain data used in that study were obtained from the Connecticut Tumor Registry in the Connecticut Department of Public Health. The authors assume full responsibility for analyses and interpretation of these data.
Studies included in PANDoRA were partly funded by: the Czech Science Foundation (No. P301/12/1734), the Internal Grant Agency of the Czech Ministry of Health (IGA NT 13 263); the Baden-Württemberg State Ministry of Research, Science and Arts (Prof. H. Brenner), the Heidelberger EPZ-Pancobank (Prof. M.W. Büchler and team:Prof. T. Hackert, Dr. N. A. Giese, Dr. Ch. Tjaden, E. Soyka, M. Meinhardt; Heidelberger Stiftung Chirurgie and BMBF grant 01GS08114), the BMBH (Prof. P. Schirmacher; BMBF grant 01EY1101), the “5x1000” voluntary contribution of the Italian Government, the Italian Ministry of Health (RC1203GA57, RC1303GA53, RC1303GA54, RC1303GA50), the Italian Association for Research on Cancer (Prof. A. Scarpa; AIRC n. 12182), the Italian Ministry of Research (Prof. A. Scarpa; FIRB - RBAP10AHJB), the Italian FIMP-Ministry of Health (Prof. A. Scarpa; 12 CUP_J33G13000210001), and by the National Institute for Health Research Liverpool Pancreas Biomedical Research Unit, UK. We would like to acknowledge the contribution of Dr Frederike Dijk and Prof. Oliver Busch (Academic Medical Center, Amsterdam, the Netherlands).
Assistance with genotype data quality control was provided by Cecelia Laurie and Cathy Laurie at University of Washington Genetic Analysis Center
The American Cancer Society (ACS) funds the creation, maintenance, and updating of the Cancer Prevention Study II cohort.
Cancer incidence data for CLUE were provided by the Maryland Cancer Registry, Center for Cancer Surveillance and Control, Department of Health and Mental Hygiene, 201 W. Preston Street, Room 400, Baltimore, MD 21201, http://phpa.dhmh.maryland.gov/cancer,410-767-4055. We acknowledge the State of Maryland, the Maryland Cigarette Restitution Fund, and the National Program of Cancer Registries of the Centers for Disease Control and Prevention for the funds that support the collection and availability of the cancer registry data.” We thank all the CLUE participants.
Melbourne Collaborative Cohort Study (MCCS) cohort recruitment was funded by VicHealth and Cancer Council Victoria. The MCCS was further augmented by Australian National Health and Medical Research Council grants 209057, 396414 and 1074383 and by infrastructure provided by Cancer Council Victoria. Cases and their vital status were ascertained through the Victorian Cancer Registry and the Australian Institute of Health and Welfare, including the National Death Index and the Australian Cancer Database.
The NYU study (AZJ and AAA) was funded by NIH R01 CA098661, UM1 CA182934 and center grants P30 CA016087 and P30 ES000260.
The PANKRAS II Study in Spain was supported by research grants from Instituto de Salud Carlos III-FEDER, Spain: Fondo de Investigaciones Sanitarias (FIS) (#PI13/00082 and #PI15/01573) and Red Temática de Investigación Cooperativa en Cáncer, Spain (#RD12/0036/0050); and European Cooperation in Science and Technology (COST Action #BM1204: EU_Pancreas), Ministerio de Ciencia y Tecnología (CICYT SAF 2000-0097), Fondo de Investigación Sanitaria (95/0017), Madrid, Spain; Generalitat de Catalunya (CIRIT - SGR); ‘Red temática de investigación cooperativa de centros en Cáncer’ (C03/10), ‘Red temática de investigación cooperativa de centros en Epidemiología y salud pública‘ (C03/09), and CIBER de Epidemiología (CIBERESP), Madrid.
The Physicians’ Health Study was supported by research grants CA-097193, CA-34944, CA-40360, HL-26490, and HL-34595 from the National Institutes of Health, Bethesda, MD USA.
The Women’s Health Study was supported by research grants CA182913, CA-047988, HL-043851, HL-080467, and HL-099355 from the National Institutes of Health, Bethesda, MD USA.
Health Professionals Follow-up Study is supported by NIH grant UM1 CA167552. from the National Cancer Institute, Bethesda, MD USA
Nurses’ Health Study is supported by NIH grants UM1 CA186107, P01 CA87969, and R01 CA49449 from the National Cancer Institute, Bethesda, MD USA
Additional support from the Hale Center for Pancreatic Cancer Research, U01 CA21017 from the National Cancer Institute, Bethesda, MD USA , and the United States Department of Defense CA130288, Lustgarten Foundation, Pancreatic Cancer Action Network, Noble Effort Fund, Peter R. Leavitt Family Fund, Wexler Family Fund, and Promises for Purple to B.M. Wolpin.
The Shanghai Men’s Health Study is supported by NIH grant UM1CA173640. The Shanghai Women’s Health Study is supported by NIH grant UM1CA182910.
The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts HHSN268201600018C, HHSN268201600001C, HHSN268201600002C, HHSN268201600003C, and HHSN268201600004C. The authors thank the WHI investigators and staff for their dedication, and the study participants for making the program possible. A full listing of WHI investigators can be found at: http://www.whi.org/researchers/Documents%20%20Write%20a%20Paper/WHI%20Investigator%20Long%20List.pdf.
SELECT study is supported by National Institutes of Health grant award number U10 CA37429 (CD Blanke), and UM1 CA182883 (CM Tangen/IM Thompson). The authors thank the site investigators and staff and, most importantly, the participants from PCPT and SELECT who donated their time to this trial.
This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the NIH, Bethesda, MD, USA (http://biowulf.nih.gov).
The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the tissue data from the GTEx Portal on June 2019 and July 2020.
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Rawla P, Sunkara T, Gaduputi V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J Oncol 2019;10(1):10–27 doi 10.14740/wjon1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gordon-Dseagu VL, Devesa SS, Goggins M, Stolzenberg-Solomon R. Pancreatic cancer incidence trends: evidence from the Surveillance, Epidemiology and End Results (SEER) population-based data. Int J Epidemiol 2018;47(2):427–39 doi 10.1093/ije/dyx232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo G, Zhang Y, Guo P, Ji H, Xiao Y, Li K. Global patterns and trends in pancreatic cancer incidence: age, period, and birth cohort analysis. Pancreas 2019;48(2):199–208 doi 10.1097/MPA.0000000000001230. [DOI] [PubMed] [Google Scholar]
- 4.Brune KA, Lau B, Palmisano E, Canto M, Goggins MG, Hruban RH, et al. Importance of age of onset in pancreatic cancer kindreds. Journal of the National Cancer Institute 2010;102(2):119–26 doi 10.1093/jnci/djp466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu C, Hart SN, Polley EC, Gnanaolivu R, Shimelis H, Lee KY, et al. Association Between Inherited Germline Mutations in Cancer Predisposition Genes and Risk of Pancreatic Cancer. JAMA 2018;319(23):2401–9 doi 10.1001/jama.2018.6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yurgelun MB, Chittenden AB, Morales-Oyarvide V, Rubinson DA, Dunne RF, Kozak MM, et al. Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genet Med 2018. doi 10.1038/s41436-018-0009-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen F, Childs EJ, Mocci E, Bracci P, Gallinger S, Li D, et al. Analysis of Heritability and Genetic Architecture of Pancreatic Cancer: A PanC4 Study. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 2019;28(7):1238–45 doi 10.1158/1055-9965.EPI-18-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amundadottir L, Kraft P, Stolzenberg-Solomon RZ, Fuchs CS, Petersen GM, Arslan AA, et al. Genome-wide association study identifies variants in the ABO locus associated with susceptibility to pancreatic cancer. NatGenet 2009;41(9):986–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Childs EJ, Mocci E, Campa D, Bracci PM, Gallinger S, Goggins M, et al. Common variation at 2p13.3, 3q29, 7p13 and 17q25.1 associated with susceptibility to pancreatic cancer. Nat Genet 2015;47(8):911–6 doi 10.1038/ng.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klein A, Wolpin BM, Risch HA, Stolzenberg-Solomon RZ, Mocci E, Zhang M, et al. Genome-wide meta-analysis identifies five new susceptibility loci for pancreatic cancer. Nat Comm 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petersen GM, Amundadottir L, Fuchs CS, Kraft P, Stolzenberg-Solomon RZ, Jacobs KB, et al. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. NatGenet 2010;42(3):224–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolpin BM, Rizzato C, Kraft P, Kooperberg C, Petersen GM, Wang Z, et al. Genome-wide association study identifies multiple susceptibility loci for pancreatic cancer. NatGenet 2014;46(9):994–1000 doi ng.3052 [pii]; 10.1038/ng.3052 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang M, Wang Z, Obazee O, Jia J, Childs EJ, Hoskins J, et al. Three new pancreatic cancer susceptibility signals identified on chromosomes 1q32.1, 5p15.33 and 8q24.21. Oncotarget 2016;7(41):66328–43 doi 10.18632/oncotarget.11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siegel RL, Jacobs EJ, Newton CC, Feskanich D, Freedman ND, Prentice RL, et al. Deaths Due to Cigarette Smoking for 12 Smoking-Related Cancers in the United States. JAMA internal medicine 2015;175(9):1574–6 doi 10.1001/jamainternmed.2015.2398. [DOI] [PubMed] [Google Scholar]
- 15.Lynch SM, Vrieling A, Lubin JH, Kraft P, Mendelsohn JB, Hartge P, et al. Cigarette smoking and pancreatic cancer: a pooled analysis from the pancreatic cancer cohort consortium. AmJEpidemiol 2009;170(4):403–13 doi kwp134 [pii]; 10.1093/aje/kwp134 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bosetti C, Lucenteforte E, Silverman DT, Petersen G, Bracci PM, Ji BT, et al. Cigarette smoking and pancreatic cancer: an analysis from the International Pancreatic Cancer Case-Control Consortium (Panc4). AnnOncol 2012;23(7):1880–8 doi mdr541 [pii]; 10.1093/annonc/mdr541 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang J, Li MD. Converging findings from linkage and association analyses on susceptibility genes for smoking and other addictions. Mol Psychiatry 2016;21(8):992–1008 doi 10.1038/mp.2016.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boardman JD, Blalock CL, Pampel FC. Trends in the genetic influences on smoking. J Health Soc Behav 2010;51(1):108–23 doi 10.1177/0022146509361195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hallfors J, Palviainen T, Surakka I, Gupta R, Buchwald J, Raevuori A, et al. Genome-wide association study in Finnish twins highlights the connection between nicotine addiction and neurotrophin signaling pathway. Addict Biol 2019;24(3):549–61 doi 10.1111/adb.12618.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matoba N, Akiyama M, Ishigaki K, Kanai M, Takahashi A, Momozawa Y, et al. GWAS of smoking behaviour in 165,436 Japanese people reveals seven new loci and shared genetic architecture. Nat Hum Behav 2019;3(5):471–7 doi 10.1038/s41562-019-0557-y. [DOI] [PubMed] [Google Scholar]
- 21.Liu M, Jiang Y, Wedow R, Li Y, Brazel DM, Chen F, et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nature genetics 2019;51(2):237–44 doi 10.1038/s41588-018-0307-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jansen RJ, Tan XL, Petersen GM. Gene-by-Environment Interactions in Pancreatic Cancer: Implications for Prevention. Yale J Biol Med 2015;88(2):115–26. [PMC free article] [PubMed] [Google Scholar]
- 23.Tang H, Wei P, Duell EJ, Risch HA, Olson SH, Bueno-de-Mesquita HB, et al. Axonal guidance signaling pathway interacting with smoking in modifying the risk of pancreatic cancer: a gene- and pathway-based interaction analysis of GWAS data. Carcinogenesis 2014;35(5):1039–45 doi 10.1093/carcin/bgu010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song M, Wheeler W, Caporaso NE, Landi MT, Chatterjee N. Using imputed genotype data in the joint score tests for genetic association and gene-environment interactions in case-control studies. Genet Epidemiol 2018;42(2):146–55 doi 10.1002/gepi.22093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Delaneau O, Marchini J, Genomes Project C, Genomes Project C. Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel. Nat Commun 2014;5:3934 doi 10.1038/ncomms4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet 2010;11(7):499–511 doi 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 27.Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature 2015;526(7571):68–74 doi 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chatterjee N, Carroll R. Semiparametric maximum likelihood estimation exploiting gene-environment independence in case-control studies. Biometrika 2005;92(2):399–418. [Google Scholar]
- 29.Piegorsch WW, Weinberg CR, Taylor JA. Non-hierarchical logistic models and case-only designs for assessing susceptibility in population-based case-control studies. Stat Med 1994;13(2):153–62 doi 10.1002/sim.4780130206. [DOI] [PubMed] [Google Scholar]
- 30.Mukherjee B, Chatterjee N. Exploiting gene-environment independence for analysis of case-control studies: an empirical Bayes-type shrinkage estimator to trade-off between bias and efficiency. Biometrics 2008;64(3):685–94 doi 10.1111/j.1541-0420.2007.00953.x. [DOI] [PubMed] [Google Scholar]
- 31.Mukherjee B, Ahn J, Gruber SB, Chatterjee N. Testing gene-environment interaction in large-scale case-control association studies: possible choices and comparisons. Am J Epidemiol 2012;175(3):177–90 doi 10.1093/aje/kwr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han SS, Rosenberg PS, Garcia-Closas M, Figueroa JD, Silverman D, Chanock SJ, et al. Likelihood ratio test for detecting gene (G)-environment (E) interactions under an additive risk model exploiting G-E independence for case-control data. Am J Epidemiol 2012;176(11):1060–7 doi 10.1093/aje/kws166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kraft P, Yen YC, Stram DO, Morrison J, Gauderman WJ. Exploiting gene-environment interaction to detect genetic associations. Hum Hered 2007;63(2):111–9 doi 10.1159/000099183. [DOI] [PubMed] [Google Scholar]
- 34.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81(3):559–75 doi 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.GTExConsortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015;348(6235):648–60 doi 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun 2017;8(1):1826 doi 10.1038/s41467-017-01261-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hormozdiari F, van de Bunt M, Segre AV, Li X, Joo JWJ, Bilow M, et al. Colocalization of GWAS and eQTL Signals Detects Target Genes. Am J Hum Genet 2016;99(6):1245–60 doi 10.1016/j.ajhg.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 2014;10(5):e1004383 doi 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res 2012;40(Database issue):D930–4 doi 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature 2015;518(7539):317–30 doi 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burre J, Zimmermann H, Volknandt W. Identification and characterization of SV31, a novel synaptic vesicle membrane protein and potential transporter. J Neurochem 2007;103(1):276–87 doi 10.1111/j.1471-4159.2007.04758.x. [DOI] [PubMed] [Google Scholar]
- 42.Cuajungco MP, Kiselyov K. The mucolipin-1 (TRPML1) ion channel, transmembrane-163 (TMEM163) protein, and lysosomal zinc handling. Front Biosci (Landmark Ed) 2017;22:1330–43 doi 10.2741/4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anderson KJ, Cormier RT, Scott PM. Role of ion channels in gastrointestinal cancer. World J Gastroenterol 2019;25(38):5732–72 doi 10.3748/wjg.v25.i38.5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 2014;46(9):989–93 doi 10.1038/ng.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kulminski AM, Huang J, Loika Y, Arbeev KG, Bagley O, Yashkin A, et al. Strong impact of natural-selection-free heterogeneity in genetics of age-related phenotypes. Aging (Albany NY) 2018;10(3):492–514 doi 10.18632/aging.101407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tabassum R, Chauhan G, Dwivedi OP, Mahajan A, Jaiswal A, Kaur I, et al. Genome-wide association study for type 2 diabetes in Indians identifies a new susceptibility locus at 2q21. Diabetes 2013;62(3):977–86 doi 10.2337/db12-0406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bai H, Liu H, Suyalatu S, Guo X, Chu S, Chen Y, et al. Association Analysis of Genetic Variants with Type 2 Diabetes in a Mongolian Population in China. J Diabetes Res 2015;2015:613236 doi 10.1155/2015/613236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heilbronner SR, Hayden BY. Dorsal Anterior Cingulate Cortex: A Bottom-Up View. Annu Rev Neurosci 2016;39:149–70 doi 10.1146/annurev-neuro-070815-013952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Janes AC, Farmer S, Peechatka AL, Frederick Bde B, Lukas SE. Insula-Dorsal Anterior Cingulate Cortex Coupling is Associated with Enhanced Brain Reactivity to Smoking Cues. Neuropsychopharmacology 2015;40(7):1561–8 doi 10.1038/npp.2015.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao MM, Hu F, Zeng XD, Tang HL, Zhang H, Jiang W, et al. Hypothalamic proteome changes in response to nicotine and its withdrawal are potentially associated with alteration in body weight. J Proteomics 2020;214:103633 doi 10.1016/j.jprot.2020.103633. [DOI] [PubMed] [Google Scholar]
- 51.Himeno S, Sumi D, Fujishiro H. Toxicometallomics of Cadmium, Manganese and Arsenic with Special Reference to the Roles of Metal Transporters. Toxicol Res 2019;35(4):311–7 doi 10.5487/TR.2019.35.4.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Madani A, Alack K, Richter MJ, Kruger K. Immune-regulating effects of exercise on cigarette smoke-induced inflammation. J Inflamm Res 2018;11:155–67 doi 10.2147/JIR.S141149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nogueira LM, Newton CC, Pollak M, Silverman DT, Albanes D, Mannisto S, et al. Serum C-peptide, Total and High Molecular Weight Adiponectin, and Pancreatic Cancer: Do Associations Differ by Smoking? Cancer Epidemiol Biomarkers Prev 2017;26(6):914–22 doi 10.1158/1055-9965.EPI-16-0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wittel UA, Hopt UT, Batra SK. Cigarette smoke-induced pancreatic damage: experimental data. Langenbecks Arch Surg 2008;393(4):581–8 doi 10.1007/s00423-007-0273-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ewald N, Bretzel RG. Diabetes mellitus secondary to pancreatic diseases (Type 3c)--are we neglecting an important disease? Eur J Intern Med 2013;24(3):203–6 doi 10.1016/j.ejim.2012.12.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.