

Abstract
Pancreatic Ductal Adenocarcinoma (PDAC) displays a dismal prognosis due to late diagnosis and high chemoresistance incidence. For advanced disease stages or patients with comorbidities, treatment options are limited to gemcitabine alone or in combination with other drugs. While gemcitabine resistance has been widely attributed to the levels of one of its targets, RRM1, the molecular consequences of gemcitabine resistance in PDAC remain largely elusive. Here we sought to identify genomic, epigenomic, and transcriptomic events associated with gemcitabine resistance in PDAC and their potential clinical relevance. We found that gemcitabine-resistant cells displayed a co-amplification of the adjacent RRM1 and STIM1 genes. Interestingly, RRM1, but not STIM1, was required for gemcitabine resistance, while high STIM1 levels caused an increase in cytosolic calcium concentration. Higher STIM1-dependent calcium influx led to an impaired ER stress response and a heightened NFAT activity. Importantly, these findings were confirmed in patient and patient-derived xenograft samples. Taken together, our study uncovers previously unknown biologically relevant molecular properties of gemcitabine-resistant tumors, revealing an undescribed function of STIM1 as a rheostat directing the effects of calcium signaling and controlling epigenetic cell fate determination. It further reveals the potential benefit of targeting STIM1-controlled calcium signaling and its downstream effectors in PDAC.
Keywords: RRM1, STIM1, SOCE, ER stress, NFAT
Introduction
Pancreatic ductal adenocarcinoma (PDAC) patients display a dismal 7-9% 5-year survival rate due to late diagnosis and therapeutic resistance (1). The current first-line treatment includes FOLFIRINOX (5-fluorouracil-oxaliplatin-irinotecan) with the combination of gemcitabine and nab-paclitaxel as an alternative. Still, patients displaying a more advanced disease or comorbidities that preclude intensive therapy generally receive either gemcitabine alone or in combination with either capecitabine or S-1 (tegafur-gimeracil-oteracil) (2,3). Thus, inevitably, with disease progression, gemcitabine-based treatment is administered to most patients. Unfortunately, the response to such therapy is low and variable, establishing gemcitabine resistance as a major hurdle in PDAC treatment (4-6). Therefore, it is of utmost importance to understand the effects of gemcitabine treatment on PDAC.
Many studies attribute gemcitabine resistance to the upregulation of one of its main targets, ribonucleotide reductase (7-12), while also identifying differential expression of gemcitabine metabolizing enzymes as predictive of treatment response (13,14). Still, the broader effects of gemcitabine on tumors remain elusive. Previous studies suggested that gemcitabine sensitivity highly depends on genetic changes in the tumor and the cellular response to chemotherapeutic-induced stress (15). In addition to or as a result from acting on its primary target, many chemotherapies induce apoptosis through cell stress. The integrated stress response pathway is activated upon ER stress, amino acid deprivation, heme deprivation or viral infection and elicits two responses. First, cells attempt to resolve the stress source by inducing pro-survival genes and, if failing to do so, activating apoptotic genes (16). Consequently, cells heavily rely on the transcription factor ATF4, which is translated and translocates to the nucleus upon stress mediating the activation of stress-induced genes (17,18). Furthermore, stress conditions, such as ER or oxidative stress, are tightly coupled to calcium signaling. This stress-calcium interplay controls the transcription of apoptotic, invasive, or proliferative genes and can thus dramatically alter cellular phenotype (19,20). The cellular stress response is highly variable and depends on the molecular and epigenetic context of the cell. Therefore, chemotherapy-resistant cells may present an altered dependency on the integrated stress response, rendering the targeting of the latter potentially useful in certain contexts. Thus, a better understanding of the molecular mechanisms affected by gemcitabine resistance and their response to cellular stress is of great importance.
Here we investigated gemcitabine resistance in PDAC by characterizing gemcitabine-resistant cells and validated our results in patient samples as well as in naïve and gemcitabine-treated patient-derived xenografts. We identified an amplification in chromosome 11 harboring genes involved in resistance and genes whose functions were elusive in this context. Among them is STIM1, whose overexpression provokes an aberrant calcium signaling program, eliciting ER stress-resistance, a rewiring of several transcription factors and widespread epigenetic reprogramming in resistant cells. Taken together, our data provide new insights into mechanisms accompanying gemcitabine-resistance in PDAC and reveal a novel alteration of calcium signaling which may influence tumor progression.
Materials and Methods
Cell culture:
CFPAC-1 (RRID:CVCL_1119) were purchased from the American Tissue Culture Collection (ATCC). L3.6pl (Par) (RRID:CVCL_0384), Panc1 (RRID:CVCL_0480) and BxPC-3 (RRID:CVCL_0186) cells were provided by Dr. Elisabeth Hessmann, (University Medical Center Göttingen, Germany), HCT116 (RRID:CVCL_0291), DLD-1 (RRID:CVCL_0248), SJSA (RRID:CVCL_1697), MG63 (RRID:CVCL_0426) and HEK293T (RRID:CVCL_0063) cells were kindly provided by Prof. Dr. Matthias Dobbelstein (University Medical Center Göttingen, Germany). All cells were obtained after 2014 at which time numerous parental cell stocks were cryopreserved. Cells were maintained in culture for a maximum of 2-3 months on average before thawing new stock and their identity was regularly confirmed. All cells tested negative for Mycoplasma using the MycoAlert Mycoplasma Detection Kit (Lonza #LT07-318). L3.6pl (Par) and GemR were cultured in Minimum Essential Medium Eagle (Gibco, life technologies); Panc1, CFPAC-1, SJSA, MG63 and HEK293T in Dulbecco’s Modified Eagle’s Medium (Gibco, life technologies); BxPC-3 and DLD-1 in Roswell Park Memorial Institute Medium (Gibco, life technologies); and HCT116 in McCoy’s 5A Medium (Gibco, life technologies). All media were supplemented with 10% FBS (Sigma-Aldrich) and 1% penicillin and streptomycin (Sigma-Aldrich); Minimum Essential Medium Eagle was supplemented with 1% L-glutamine (Sigma-Aldrich). Further cell culture experiments are described in Supplemental Materials and Methods, including siRNA sequences (Table S1).
Protein harvesting and western blot:
Protein was harvested and western blot performed as described (21,22) using the following antibodies: RRM1 (Cell Signaling #8637, RRID:AB_11217623), STIM1 (Sigma-Aldrich S6197, RRID:AB_1079007), ATF4 (Cell Signaling #11815, RRID:AB_2616025), HA (Roche #3F10, RRID:AB_2314622), GAPDH (Origene #TA802519, RRID:AB_2626378), HSC70 (Santa Cruz #sc-7298, RRID:AB_627761), anti-rabbit IgG (Jackson ImmunoResearch Labs #211-032-171, RRID:AB_2339149) and anti-mouse IgG (Jackson ImmunoResearch Labs #115-035-174, RRID:AB_2338512). Further details are in Supplemental Material and Methods.
RNA extraction and quantitative PCR:
RNA was extracted and qPCR run as described earlier (21,23). Gene expression levels were normalized to the housekeeping gene GAPDH. Further details are in Supplemental Material and Methods, including gene expression primer sequences (Table S2).
Chromatin immunoprecipitation:
ChIP was performed as previously described (21,24). qPCR was used to probe IP and the enrichment at each site was calculated by normalizing the IP values to their respective inputs. The following antibodies were used: H3K27ac (Diagenode #C15410196, RRID:AB_2637079) and ATF4 (Cell Signaling #11815, RRID:AB_2616025). Further details are in Supplemental Material and Methods, including primer sequences (Table S3).
Next generation sequencing:
Sequencing libraries for ChIP-seq and RNA-seq were prepared using the KAPA HyperPrep (Roche) or the Microplex Library Preparation V2 (Diagenode) and the TruSeq RNA Library Prep V2 (Illumina) kits, respectively. Library quality was assessed using a Bioanalyzer 2100 (Agilent). The samples were sequenced on a HiSeq4000 (Illumina) at the NGS Integrative Genomics Core Unit (NIG) at the UMG or at the Genome Analysis Core at the Mayo Clinic. CASAVA 1.8.2 was used to demultiplex the bcl files to fastq files. Further analyses are in Supplemental Materials and Methods.
Transcript Profiling:
The high throughput sequencing data in this publication has been deposited in NCBI's Gene Expression Omnibus(25) and are accessible through GEO Series accession number GSE152124 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE152124).
Patient-derived xenografts:
For patient-derived xenograft (PDX) model generation, pieces of bulk primary PDAC tissue from patients who underwent tumor resection at the UMG were subcutaneously transplanted in both flanks of NMRInu/nu mice. Tumors grew until their volume exceeded 1cm3 (F1 generation). Upon harvesting of tumors, one portion of the tissue was embedded in paraffin as described previously (26), while the other half was subcutaneously transplanted into both flanks of another NMRInu/nu mouse for further tumor expansion (F2 generation). For gemcitabine/nab-paclitaxel co-treatment, F3-generation PDX-material from GöPDX13 was transplanted into both flanks of four NMRInu/nu mice. When tumor volumes reached 200mm3, mice were randomized into vehicle (0.9% saline) and chemotherapy arms. Gemcitabine (Sigma-Aldrich; 100mg/kg) was administered intraperitoneally 2x/week, nab-paclitaxel (Abraxane, Celgene; 30mg/kg) was given weekly by tail vain injection. For gemcitabine treatment alone, F4 generation GöPDX13 material was transplanted into seven NMRInu/nu mice, which were randomized into vehicle and gemcitabine (100mg/kg) arms. Here, gemcitabine was administered 3x/week. Mice were sacrificed when endpoint criteria (e.g. weight loss ≥ 20%) were reached (evident upon 3x nab-paclitaxel injections for the gemcitabine/nab-paclitaxel study and upon 6 injections in the gemcitabine solo arm) and PDX tumors were paraffin-embedded for histological assessment. Animal procedures were conducted in accordance with the protocols approved by the Institutional Animal Care and Use Committee (33.9-42502-04-17/2407). The generation and utilization of PDX models have been approved by the ethical review board of the UMG (70112108). Immunohistochemistry details are in Supplemental Materials and Methods.
Statistics:
GraphPad Prism v5.04 (RRID:SCR_002798) was used for statistical analyses. One-way ANOVA followed by Newman-Keuls multiple comparison test was used for comparisons of more than two conditions. A non-linear regression with a variable slope and a bottom constrain between 0 and 2 was used to determine IC50 values, which were analyzed using unpaired two-tailed student’s t-test. Linear regressions were analyzed using Spearman’s correlation. P<=0.05 were considered statistically significant. *P≤0.05, **P≤0.01, ***P≤0.001, ns=not significant.
Results
Amplification in chromosome 11 confers gemcitabine resistance
To study chemoresistance in PDAC, a gemcitabine-resistant human cell line (GemR) was established by treating parental L3.6pl (Par) cells with increasing gemcitabine concentrations (Fig. 1A). Cells were considered resistant once the half maximal inhibitory concentration (IC50) was 50-fold higher in GemR (IC50:223.70 nM ± 26.45 nM) compared to Par (IC50:3.70 nM ± 0.11 nM) (Fig. 1B and Fig. S1A).
Fig. 1.
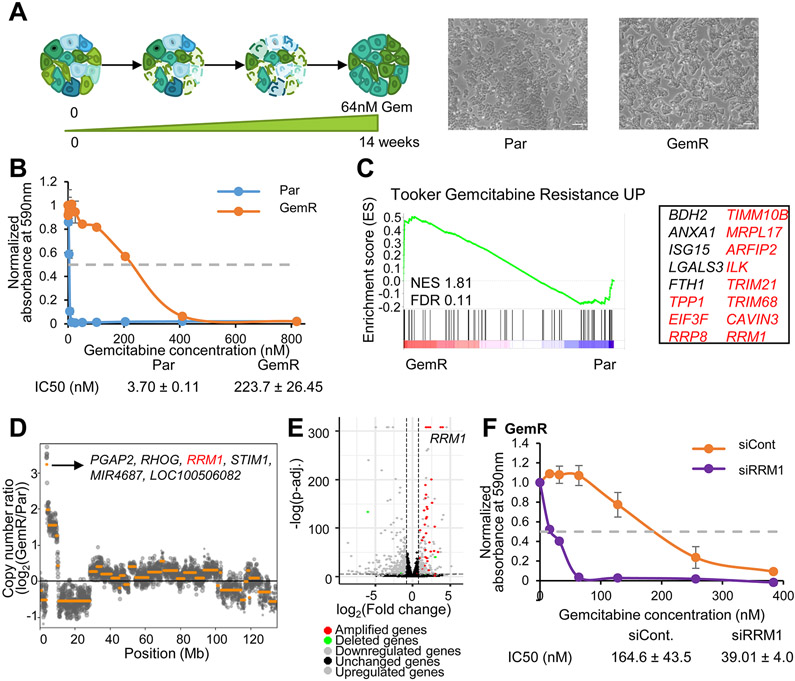
Amplification of a portion of chromosome 11 elicits gemcitabine resistance. (A) Scheme depicting the establishment of GemR. Images of Par and GemR. Scale=1.36mm. (B) Proliferation assay of Par and GemR treated with gemcitabine for 7 days. The absorbance of cell titer blue was normalized to the respective vehicle absorbance. Mean ±SD, n=2. IC50 values ±SD, n=2. (C) GSEA showing an enrichment of the gemcitabine resistance signature in GemR. Significantly enriched genes are listed and classified into amplified genes on chr11 (in red). (D) Copy number variation analysis of GemR compared to Par in chr11 and highest amplified genes. (E) Volcano plot of differentially regulated, amplified and deleted genes in GemR compared to Par. (F) GemR proliferation assay upon RRM1 knockdown and treatment with gemcitabine for 7 days. The absorbance of solubilized crystal violet was normalized to the respective vehicle absorbance. Mean ±SD, n=2. IC50 values ±SD, n=2.
Transcriptome-wide mRNA sequencing and low coverage whole genome sequencing was performed on GemR and Par to identify acquired traits upon resistance. Gene Set Enrichment Analysis (GSEA) showed an enrichment for the “Gemcitabine Resistance UP" signature in GemR. Surprisingly, 11 out of 16 of the significantly enriched genes identified were located on chromosome 11 (Fig. 1C and Table S4). Copy number variation analysis revealed that a region of chromosome 11 (chr11: 3,810,838-10,012,224), encompassing most of the genes contained within this signature, was amplified in GemR compared to Par. RRM1 was identified as highly amplified and upregulated in GemR along with other genes whose association with gemcitabine resistance are unknown (Fig. 1D-E, Fig. S1B, and Table S5). RRM1 is a ribonucleotide reductase subunit and one of the main targets of gemcitabine (27). Its upregulation has also been tightly associated with this chemotherapeutic agent, being reported to drive gemcitabine resistance in vitro and in vivo (8-12). Consistently, we found that RRM1 levels correlate with gemcitabine resistance in vitro and that RRM1 depletion restores gemcitabine sensitivity in GemR (Fig. 1F and Fig. S1C-D). Interestingly, the amplified region not only includes RRM1, but extends for over 6 Mb. While the role of RRM1 in gemcitabine resistance has been established, the effects of the co-amplification of the various other genes remain elusive. It is therefore plausible that co-amplified genes confer additional advantageous molecular properties to tumor cells.
GemR display attenuated ATF4 activity and diminished ER-stress response
We hypothesized that additional genes co-amplified on chromosome 11 may influence the cellular phenotype. As the epigenetic landscape can shape the cellular response to external stimuli and provides an excellent readout for transcription factor and upstream signaling activity, we compared the epigenomic profiles of Par and GemR. For this, we performed chromatin immunoprecipitation followed by next-generation sequencing (ChIP-seq) for the active transcription mark H3K27ac. Despite the identified amplification, about an equal number of acetylated regions were lost and gained in GemR compared to Par (Fig. 2A). Bioinformatic characterization of these regions revealed that sequence motifs for AP1 transcription factors were enriched in both gained and lost regions. Motifs for the transcription factor ATF3 were also enriched in the gained regions, likely due to the sequence similarity to AP1 motifs. Interestingly, regions displaying decreased H3K27ac levels showed an enrichment for the motifs of the stress-responsive transcription factor ATF4 and its downstream target CHOP (Fig. 2B and Fig. S2A). Given the reported importance of ATF4 in mediating stress response, we performed ChIP-seq for ATF4 in Par following induction of ER stress by thapsigargin. Interestingly, 24% of the lost H3K27ac regions in GemR overlapped with ATF4 peaks. Consistently, lower H3K27ac signal intensity was observed at those sites in GemR compared to Par (Fig. 2C). Accordingly, ATF4 target genes displayed decreased H3K27ac occupancy near their transcriptional start site (TSS) in GemR (Fig. 2D and Fig. S2B).
Fig. 2.
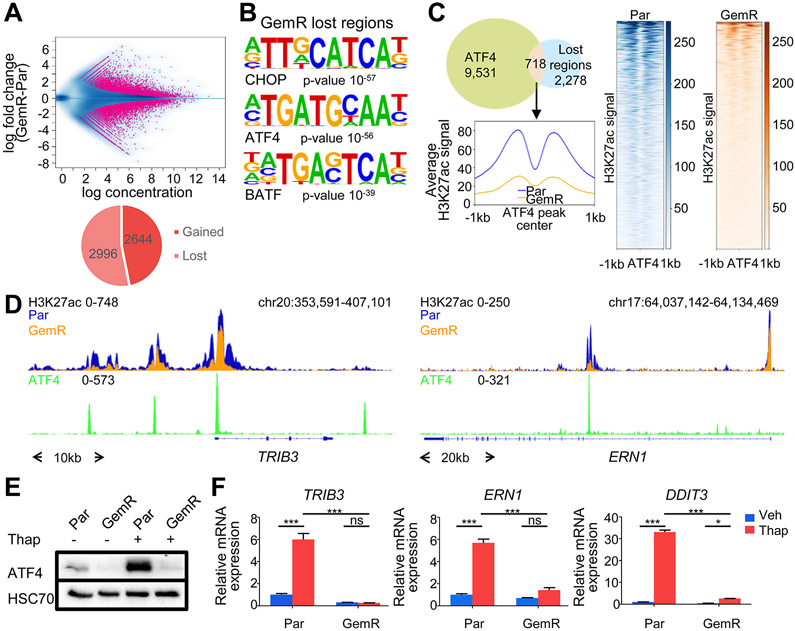
ATF4 activity and ER stress response are dampened in GemR. (A) MA plot and pie chart of differentially occupied H3K27ac regions in GemR and Par. (B) Top most significantly enriched motifs in H3K27ac lost regions in GemR. (C) Venn diagram of ATF4 peaks in Par after thapsigargin (Thap) treatment and H3K27ac lost regions in GemR. Aggregate plot and heatmaps of H3K27ac on ATF4 summits of overlapping regions. (D) ATF4 and H3K27ac profiles around the TSS of stress responsive genes. (E) Western blot of ATF4 in Par and GemR treated with thapsigargin (Thap). (F) Expression of stress responsive genes upon thapsigargin (Thap) treatment in Par and GemR. Mean ±SD, n=3. *P≤0.05, **P≤0.01, ***P≤0.001, ns=not significant.
To investigate whether ATF4 activity and the stress response were affected in GemR compared to Par, we induced ER stress in Par and GemR. Strikingly, upon ER stress, ATF4 protein levels, which dramatically increased in Par, were not detectable in GemR (Fig. 2E and Fig. S2C). Consistently, GemR failed to activate ATF4 target genes, such as TRIB3, ERN1 and DDIT3 (encoding CHOP) (Fig. 2F). In conclusion, GemR are unable to activate ATF4 translation and induce downstream ER stress responsive genes.
STIM1 amplification elicits a higher store-operated calcium entry driving ER stress resistance
Long-term thapsigargin treatment inhibits cell proliferation via induction of the ER stress response pathway. Therefore, we examined whether GemR displayed differential responsiveness to thapsigargin compared to Par. Indeed, GemR were significantly more resistant to the anti-proliferative effects of thapsigargin (IC50: >819.2 nM) compared to Par (IC50: 5.09 nM ± 0.20 nM) (Fig. 3A and Fig. S3A). Consistently, analysis of DepMap data revealed that thapsigargin sensitivity highly correlated with gemcitabine sensitivity in pancreatic cancer cell lines (Fig. S3B). ER stress is triggered by the accumulation of unfolded proteins or changes in redox, calcium, or nutrient levels in the ER (16,28). Furthermore, thapsigargin is a SERCA-pump inhibitor, which affects ER calcium storage. Therefore, we examined whether protein-coding genes involved in these processes were aberrantly regulated and amplified in GemR. Interestingly, STIM1, an ER calcium sensor coding gene, was among the most amplified and highly upregulated genes in GemR, being co-amplified with RRM1 in a focal amplification within the larger amplified region on chr11. Previous studies have also reported the upregulation of STIM1 and RRM1 upon gemcitabine treatment and gemcitabine resistance in pancreatic cancer cells (8,29). Notably, analysis of DepMap data revealed that RRM1 and STIM1 amplifications are highly correlated in cancer cell lines, including pancreatic cancer. Additionally, analysis of TCGA data revealed that 5% of pancreatic cancer patients display a gain of both genes irrespective of treatment modality (Fig. 3B). Consistently, we were able to identify several established cell lines that displayed an amplification and an increased expression of STIM1. For example, the pancreatic and colorectal cancer cell lines Panc1 and DLD1, respectively, highly co-expressed RRM1 and STIM1, while the osteosarcoma cell line SJSA only expressed high levels of STIM1 (Fig. S3C).
Fig. 3.
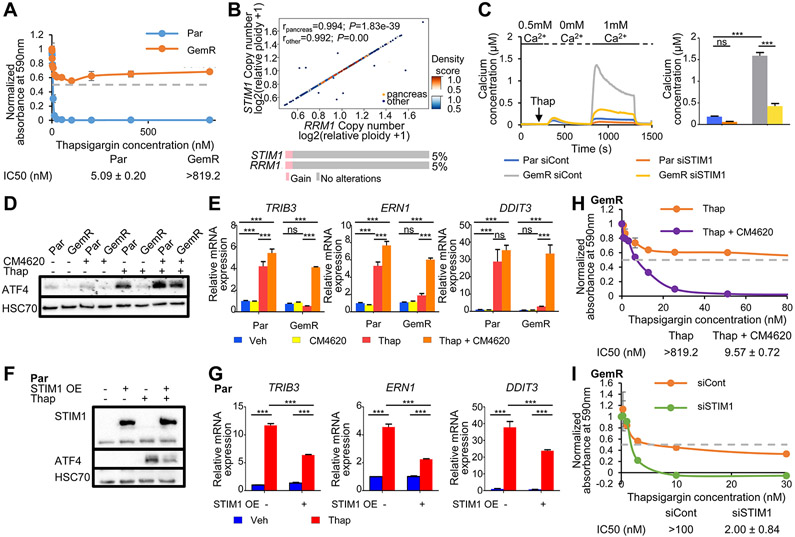
Amplification of STIM1 leads to increased SOCE and ER stress resistance in GemR. (A) Proliferation assay of Par and GemR treated with thapsigargin for 7 days. The absorbance of cell titer blue was normalized to the respective vehicle absorbance. Mean ±SD, n=2. IC50 values ±SD, n=2. (B) Density scatter plot showing the Spearman correlation of the copy number of RRM1 and STIM1 in pancreatic and other cancer cell lines obtained from DepMap. rpancreas=0.994, P=1.83e-39; rother=0.992, P=0.00. Oncoprint and percentage of pancreatic cancer patients displaying a gain of STIM1 and RRM1 from TCGA PanCancer Atlas Studies data (cBioportal). (C) Fura-2 based cytosolic calcium imaging and quantification of ΔSOCEmax. Mean ±SEM, n=334 (Par siCont), 143 (Par siSTIM1), 347 (GemR siCont), 243 (GemR siSTIM1). (D) Western blot showing ATF4 levels upon SOCE inhibition by CM4620 and thapsigargin (Thap) treatment in Par and GemR. (E) Expression of stress responsive genes upon SOCE inhibition by CM4620 and thapsigargin (Thap) treatment in Par and GemR. Mean ±SD, n=3. (F) Western blot of STIM1 and ATF4 levels upon STIM1 overexpression and thapsigargin (Thap) treatment in Par. (G) Expression of stress responsive genes upon STIM1 overexpression and thapsigargin (Thap) treatment in Par. Mean ±SD, n=3. (H) Proliferation assay of GemR treated with thapsigargin and the SOCE inhibitor CM4620 for 7 days. The absorbance of cell titer blue was normalized to the respective vehicle absorbance. Mean ±SD, n=2. IC50 values ±SD, n=2. The profile of GemR treated with thapsigargin (Thap) only was previously shown in Fig. 3A. (I) Proliferation assay of GemR upon STIM1 knockdown and thapsigargin treatment (Thap). The absorbance of solubilized crystal violet was normalized to the respective vehicle absorbance. Mean ±SD, n=2. IC50 values ±SD, n=2. *P≤0.05, **P≤0.01, ***P≤0.001, ns=not significant.
STIM1 is an ER calcium sensor that interacts with and activates ORAI calcium channels in the plasma membrane following ER calcium store depletion. This leads to ORAI channel opening, allowing extracellular calcium to enter the cytosol in a process termed store-operated calcium entry (SOCE) (30,31). Fluorescence calcium measurements revealed comparable calcium levels at resting conditions and upon thapsigargin-induced ER calcium store depletion in Par and GemR. However, GemR displayed a highly increased SOCE compared to Par, which could be reversed by STIM1 depletion (Fig. 3C). While recent studies have pointed at the effects of STIM1 on ER stress response (32,33), no such correlation has been reported in cancer. Moreover, the possible effects elicited by increased SOCE on ER stress response remain elusive. Thus, we investigated whether higher STIM1 levels, and consequently increased SOCE, could lead to ER stress resistance in GemR. To address this, SOCE was prevented by either treating with the SOCE inhibitor, CM4620, or by chelating extracellular calcium from the media with EGTA before the induction of ER stress by thapsigargin. Notably, as assessed via ATF4 accumulation, treatment with either CM4620 or EGTA restored the stress response to thapsigargin in GemR to levels comparable to thapsigargin treatment alone in Par (Fig. 3D and Fig. S4A-B). This confirms that ER stress resistance in GemR is conferred by elevated SOCE elicited by STIM1. This conclusion was further supported by the ability of combined CM4620 or EGTA and thapsigargin treatment to rescue the expression of ER stress responsive genes in GemR (Fig. 3E and Fig. S4C). Moreover, overexpression of STIM1 in Par cells was sufficient to lower ATF4 levels and impair the induction of stress responsive genes upon thapsigargin treatment (Fig. 3F-G and Fig. S4D). Similarly, STIM1 overexpression in other pancreatic cancer cell lines, namely BxPC-3 and CFPAC-1, decreased ATF4 accumulation and dampened the induction of stress-responsive genes following thapsigargin treatment (Fig. S4E-J). Consistent with these effects, inhibition of SOCE with CM4620 (Fig. 3H and Fig. S5A-C) or STIM1 depletion (Fig. 3I and Fig. S5D-H) restored the anti-proliferative effects of thapsigargin in GemR. This was further validated in the colorectal cancer cell line DLD1, which expressed higher levels of both STIM1 and RRM1 and was more resistant to the anti-proliferative effects of thapsigargin compared to HCT116. Consistently, SOCE inhibition restored the sensitivity of DLD1 to thapsigargin to levels similar to HCT116 (Fig. S6A-D). Thus, higher levels of STIM1, and thereby SOCE, in GemR as well as other tumor cell lines provide a survival advantage under ER stress conditions.
Since STIM1 and RRM1 are commonly co-amplified and have important physiological functions, we tested whether they act synergistically. For this, we monitored cell proliferation upon STIM1 and/or RRM1 depletion and thapsigargin or gemcitabine treatment. RRM1 levels did not influence cell growth upon thapsigargin treatment and the depletion of both RRM1 and STIM1 was not synergistic (Fig. S5D-E, H). Similarly, while RRM1 depletion restored gemcitabine responsiveness, STIM1 knockdown did not appreciably influence GemR growth upon gemcitabine treatment nor did it synergize with RRM1 depletion (Fig. S1C, Fig. S5H and Fig. S7A-B). Furthermore, SOCE inhibition did not influence the effects of gemcitabine treatment on cell proliferation in either GemR or Par cells (Fig. 1B, Fig. S1A and Fig. S7C-D). Together, these findings confirm that while STIM1 and RRM1 are co-amplified in human tumors and cancer cell lines, they independently affect calcium-associated ER stress and gemcitabine responsiveness, respectively.
STIM1 depletion restores ER stress-induced transcriptomic and epigenomic changes
To further characterize the role of STIM1 in ER stress resistance, we performed mRNA sequencing in Par, GemR, and STIM1-depleted GemR treated with thapsigargin. Consistent with GemR being resistant to ER stress, gene set enrichment analysis (GSEA) displayed an enrichment of the “Unfolded Protein Response” in Par compared to GemR following treatment with thapsigargin (Fig. 4A and Table S6). Hierarchical clustering revealed two gene clusters whose expression was influenced by STIM1 (Fig. 4B). Genes within cluster 1 were upregulated in thapsigargin-treated Par, but failed to be activated in GemR. Importantly, their induction was rescued by STIM1 depletion in GemR, and were thus referred to as “down (DN-)reversed” genes. Cluster 2 genes were not induced in Par, but upregulated in GemR in response to thapsigargin. Notably, STIM1 depletion in GemR reversed their induction by thapsigargin and were therefore referred to as “UP-reversed” genes (Table S7). Consistent with our observations, the DN-reversed cluster includes the ER-stress responsive genes TRIB3, ERN1 and DDIT3, whose induction by thapsigargin was rescued upon STIM1 depletion in GemR (Fig. 4C). Moreover, STIM1-depletion restored ATF4 accumulation in response to thapsigargin treatment in GemR (Fig. 4D and Fig. S8A). To validate our findings in another pancreatic cancer cell line, we assessed the induction of DN-reversed genes in STIM1-amplified Panc1 cells. Here we observed low levels of induction of DN-reversed genes and ATF4 upon thapsigargin treatment, which were rescued by STIM1 depletion (Fig. 4E-F and Fig. S8B).
Fig. 4.
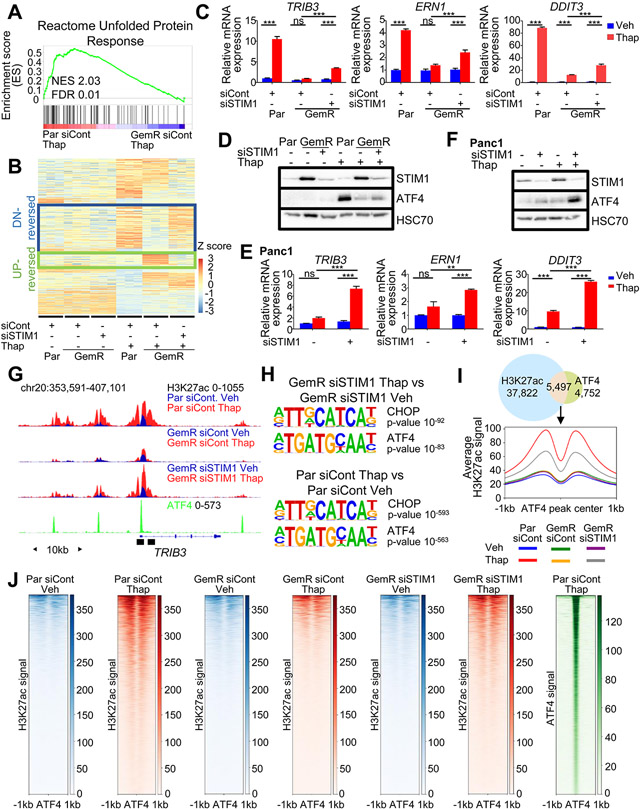
STIM1 depletion sensitizes GemR to ER stress and partially rescues H3K27ac around ATF4-occupied regions. (A) GSEA showing an enrichment for the unfolded protein response upon thapsigargin (Thap) treatment in Par compared to GemR. (B) Heatmap showing the Z-score of each gene ordered into 4 clusters identified by hierarchical clustering highlighting gene clusters: DN-reversed and UP-reversed. (C) Expression of DN-reversed genes upon thapsigargin (Thap) treatment in Par, GemR and STIM1-depleted GemR. Mean ±SD, n=3. (D) Western Blot of ATF4 and STIM1 levels upon a STIM1 knockdown and thapsigargin (Thap) treatment in Par and GemR. (E) Expression of DN-reversed genes upon thapsigargin (Thap) treatment in Panc1 and STIM1-depleted Panc1. Mean ±SD, n=3. (F) Western Blot of ATF4 and STIM1 levels upon a STIM1 knockdown and thapsigargin (Thap) treatment in Panc1. (G) ATF4 profile in Par treated with thapsigargin (Thap) and H3K27ac profile in Par and GemR upon thapsigargin (Thap) treatment and STIM1 depletion. Black boxes indicate the regions used for ChIP qPCR. (H) Top most significantly enriched motifs on gained H3K27ac regions in thapsigargin-treated (Thap) STIM1-depleted GemR compared to vehicle-treated STIM1-depleted GemR (top) and on gained H3K27ac regions in Par treated with thapsigargin (Thap) compared to vehicle-treated Par (bottom). (I) Venn diagram of ATF4 peaks in Par treated with thapsigargin (Thap) and gained H3K27ac regions in Par treated with thapsigargin (Thap) compared to vehicle-treated Par. Aggregate plot of H3K27ac on ATF4 summits of overlapping regions. (J) Heatmaps of H3K27ac and ATF4 on ATF4 summits of overlapping regions from Fig. 4I. *P≤0.05, **P≤0.01, ***P≤0.001, ns=not significant.
We next sought to uncover the molecular and transcriptional mechanisms responsible for the differential gene regulation observed in GemR. Based on our initial epigenome mapping studies, we rationalized that ER stress-induced gene expression changes may be coupled to epigenetic reprogramming. Indeed, in accordance with the gene expression data, H3K27ac occupancy increased near the TSS of DN-reversed genes in Par, but not in GemR upon thapsigargin treatment. STIM1 depletion as well as SOCE inhibition by CM4620 in GemR partially rescued the H3K27ac gain on the TSS of these genes with thapsigargin (Fig. 4G and Fig. S8C-F). Consistent with our earlier findings, STIM1-depletion in GemR restored an enrichment of ATF4 and CHOP motifs in H3K27ac gained regions upon thapsigargin treatment in a manner similar to what we observed following thapsigargin treatment in Par cells (Fig. 4H), where 53% of ATF4 peaks overlapped with H3K27ac gained regions in Par (thapsigargin vs vehicle). On these regions, a significant increase in H3K27ac was only observed in Par and STIM1-depleted GemR, but not in GemR upon thapsigargin treatment (Fig. 4I-J and Fig. S8G). This confirms that GemR cells fail to recruit epigenetic factors to DN-reversed genes in a STIM1-dependent manner, indicating that STIM1-dependent SOCE rewires the cellular epigenome and transcriptome, attenuating the activation of stress-specific genes.
NFAT is aberrantly activated in STIM1-amplified cells
After characterizing the effects of STIM1 amplification on ER stress-induced gene expression, we examined genes that were specifically induced by thapsigargin in the presence of STIM1 amplification (UP-reversed cluster), which included KDM7A, KRT14 and KLF4. These genes were upregulated upon thapsigargin treatment in GemR, but less induced in Par and STIM1-depleted GemR (Fig. 5A). Similarly, SOCE inhibition diminished the induction of these genes by thapsigargin in GemR (Fig. S9A). These effects were not limited to GemR since STIM1-amplified Panc1 cells also displayed an upregulation of KDM7A and KLF4 upon thapsigargin treatment in a STIM1-dependent manner (Fig. 5B). Moreover, H3K27ac signal intensity on the TSS of UP-reversed genes displayed a significant increase in GemR compared to Par and STIM1-depleted GemR upon thapsigargin treatment (Fig. 5C and Fig. S9B). To uncover the underlying mechanisms by which this subset of genes was specifically induced in response to ER stress in GemR cells, we employed EnrichR and GSEA. NFAT-related pathways were identified by EnrichR, while GSEA displayed an enrichment for the “NFAT transcription factor pathway” in thapsigargin-treated GemR cells (siCont vs siSTIM1) (Fig. 5D, Fig. S9C and Table S8). Consistently, NFAT and NFAT-AP1 motifs were enriched in genomic regions displaying increased H3K27ac in the same comparison (Fig. 5E).
Fig. 5.
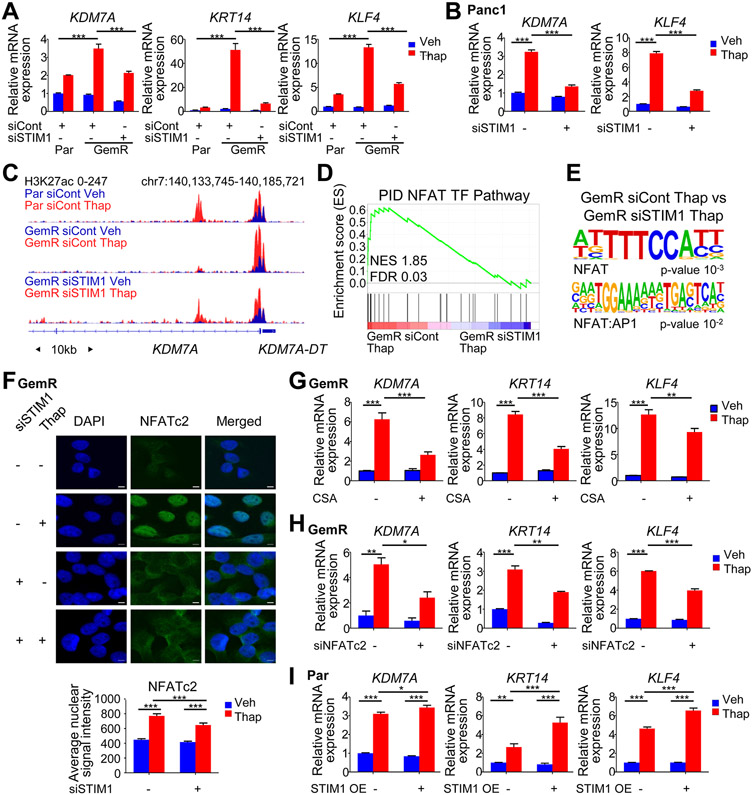
STIM1 amplified cells aberrantly activate NFAT. (A) Gene expression of UP-reversed genes upon thapsigargin (Thap) treatment and STIM1 depletion in Par and GemR. Mean ±SD, n=3. (B) Gene expression of UP-reversed genes upon thapsigargin (Thap) treatment and STIM1 depletion in Panc1. Mean ±SD, n=3. (C) H3K27ac profile around the TSS of KDM7A in Par, GemR and STIM1-depleted GemR treated with thapsigargin (Thap). (D) GSEA showing an enrichment for the NFAT TF pathway in GemR compared to STIM1-depleted GemR both treated with thapsigargin (Thap). (E) Motif analysis showing a significant enrichment for NFAT motifs on gained H3K27ac regions in GemR compared to STIM1-depleted GemR both treated with thapsigargin (Thap). (F) NFATc2 immunofluorescence and average nuclear signal intensity in GemR. Scale=5μm. Mean ±SEM, n=68 (GemR siCont Veh), 57 (GemR siCont Thap), 30 (GemR siSTIM1 Veh), 82 (GemR siSTIM1 Thap). (G) Expression of UP-reversed genes upon cyclosporine A (CSA) and thapsigargin (Thap) treatments in GemR. Mean ±SD, n=3. (H) Expression of UP-reversed genes upon NFATc2 knockdown and thapsigargin (Thap) treatment in GemR. Mean ±SD, n=2. (I) Expression of UP-reversed genes upon STIM1 overexpression and thapsigargin (Thap) treatment in Par. Mean ±SD, n=3. *P≤0.05, **P≤0.01, ***P≤0.001, ns=not significant.
NFAT activation by calcium signaling promotes its translocation to the nucleus, thereby enabling target gene activation. Consistent with our findings that GemR cells display pronounced SOCE, NFAT nuclear translocation was increased in thapsigargin-treated GemR and decreased by STIM1 depletion (Fig. 5F and Fig. S9D). To confirm the importance of NFAT in driving the expression of UP-reversed genes, we treated GemR with the calcineurin inhibitor cyclosporine A (CSA) to attenuate NFAT activation. We observed that the induction of UP-reversed genes by thapsigargin was dampened upon CSA treatment (Fig. 5G). Among the various NFAT proteins, NFATc2 is more tightly linked to STIM1 and SOCE (34,35). NFATc2 is also the only NFAT family member contained in the UP-reversed gene cluster. Consistent with a critical role in mediating the effects of altered calcium signaling in STIM1-amplified cells, NFATc2 depletion significantly dampened the induction of UP-reversed genes in thapsigargin-treated GemR (Fig. 5H and Fig. S9E). Overexpression of STIM1 in Par, BxPC-3 and CFPAC-1 further confirmed that the upregulation of these genes by thapsigargin was due to increased SOCE elicited by higher STIM1 (Fig. 5I and Fig. S9F-G). Notably, the overexpression of NFATc2 in Par did not affect ATF4 levels and the ER stress response (Fig. S9H). This suggests that heightened SOCE independently leads to a dampened ER stress response and an aberrant NFATc2 activation. In conclusion, STIM1 amplification facilitates and increased SOCE, thereby promoting the upregulation of NFATc2 and calcium-mediated activation of NFATc2-dependent gene expression.
STIM1 levels correlate with ATF4 and NFAT activity in primary PDAC and patient-derived xenografts
To examine the in vivo relevance of our findings, we performed immunohistochemistry for STIM1, KRT14, and ATF4 in naïve primary tumor tissue derived from resected PDAC patients and in corresponding PDX-models derived from these specimens (Fig. S10A). Remarkably, in one patient tumor (GöPat15: Fig. 6A) and its corresponding PDX (GöPDX15; Fig. 6B-C), where STIM1 expression was low, we observed readily detectable nuclear ATF4, but only low levels of KRT14 expression. In contrast, another patient tumor (GöPat4; Fig. 6A) and its corresponding PDX sample (GöPDX4; Fig. 6B-C) displayed higher STIM1 and correspondingly high KRT14 levels, but only cytoplasmic ATF4 expression. Next, we tested the effects of chemotherapy by treating PDXs with gemcitabine alone or in combination with nab-paclitaxel (Fig. S10A) and subsequently explored the expression of the aforementioned proteins. Notably, treatment of GöPDX13 with gemcitabine alone, or co-treatment with gemcitabine and nab-paclitaxel, resulted in increased STIM1 and KRT14 expression and lower nuclear ATF4 levels compared to the vehicle-treated GöPDX13 (Fig. 6C-E). Furthermore, mRNA-seq and GSEA analysis revealed an enrichment for UP-reversed genes in GöPDX13 co-treated with gemcitabine and nab-paclitaxel compared to untreated (Fig. 6F). Taken together, STIM1 is not only positively and negatively correlated with KRT14 expression and ATF4 nuclear localization, respectively, in naïve patient tumors, but is also altered in response to treatment both in vitro and in vivo. Thus, STIM1 levels could be exploited as a potential biomarker and/or therapeutic target for naïve and treated patients presenting a priori and acquired ER stress, and possibly gemcitabine resistance.
Fig. 6.
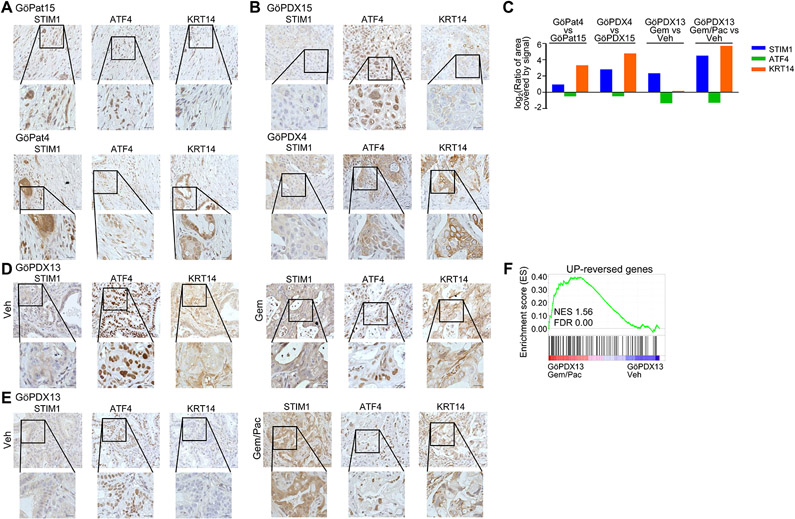
ATF4 and KRT14 expression correlate with STIM1 levels in PDAC patients and PDXs. (A & B) Immunohistochemistry for STIM1, ATF4 and KRT14 in naïve patient tumor material (Pat) (B) and in the respective naïve PDX. (C) Quantification of STIM1, ATF4 and KRT14 staining in naïve patient samples, as well as in naïve PDXs and gemcitabine and gemcitabine and nab-paclitaxel co-treated PDXs. (D) STIM1, ATF4 and KRT14 staining in vehicle (Veh) as well as in gemcitabine (Gem) treated PDXs. (E) Immunohistochemistry for STIM1, ATF4 and KRT14 in vehicle (Veh) and gemcitabine and nab-paclitaxel (Gem/Pac) co-treated PDXs. (F) GSEA showing an enrichment for the UP-reversed genes in GöPDX13 co-treated with gemcitabine and nab-paclitaxel (Gem/Pac) compared to vehicle-treated GöPDX13. For all immunohistochemistry images: scale=20 μm (zoomed out) and 50 μm (zoomed in).
Discussion
In this study, we examined molecular alterations resulting from prolonged gemcitabine treatment of PDAC and identified the co-amplification of RRM1 and STIM1 as responsible for gemcitabine resistance and for altered calcium signaling, downstream transcriptomic and epigenomic alterations, respectively. While STIM1 amplification does not augment RRM1-driven gemcitabine resistance, it shifts calcium signaling via increased SOCE, thereby reciprocally dampening the ER stress response and increasing NFAT activity (Fig. 7A).
Fig. 7.
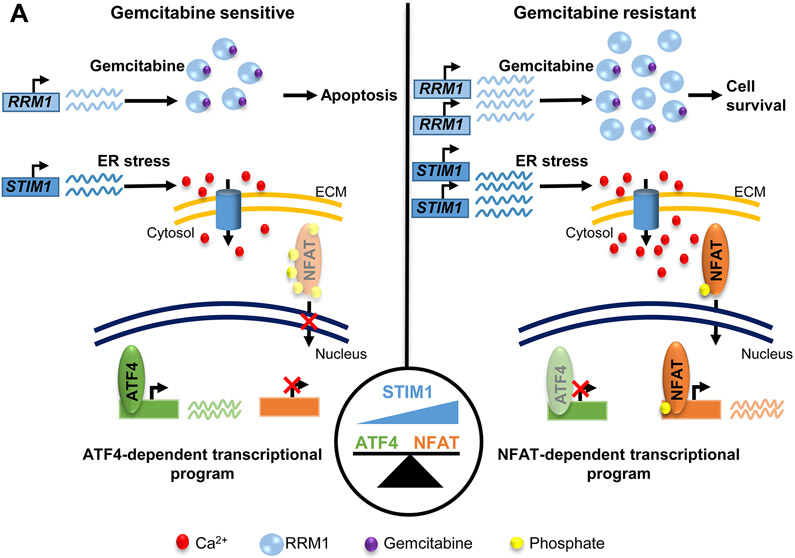
STIM1 acts as rheostat balancing between ATF4 and NFAT-dependent transcriptional programs. (A) Scheme depicting the amplification of RRM1 and STIM1 upon gemcitabine resistance. While the upregulation of RRM1 drives gemcitabine resistance, increased STIM1 levels elicit a calcium signaling shift, leading to a dampened ER stress response and an aberrant NFAT activation.
RRM1, one of the main targets of gemcitabine, was found to be amplified in GemR and to drive gemcitabine resistance. To date, studies have failed to show that RRM1 levels are prognostic since its expression in naïve patients did not correlate with therapeutic response to gemcitabine (14,36). We postulate that RRM1 levels and copy number might correlate with gemcitabine response only in patient tumors after selective pressure caused by treatment. Thus, examining patient samples after treatment would help address this.
Gene amplifications are common in tumors, and their overexpression is known to drive cancer progression. Recently, studies have revealed the importance of co-amplified neighboring genes in tumorigenesis. For example, in HER2-positive breast cancer, the amplified region encompasses not only the oncogenic driver ERBB2, but also GRB7, MIEN1, PNMT, PGAP3, and TCAP (37). While HER2 overexpression drives HER2-positive breast cancer, GRB7 and MIEN1 affect tumorigenesis downstream and independent of HER-2, respectively (38-40). Similarly, the co-amplification of RRM1 and STIM1 elicits independent effects, where RRM1 does not affect ER stress resistance and NFAT activation, while STIM1 does not influence gemcitabine resistance. This suggests that the co-amplification of these genes endows tumor cells with distinct molecular properties, thereby potentially providing multiple survival advantages. Moreover, it is plausible that persistent ER stress or perturbed SOCE stimulation may elicit a selective pressure to amplify STIM1, which could result in the co-amplification of RRM1 and elicit gemcitabine resistance. This is supported by our finding that many treatment-naïve tumors display a co-amplification of STIM1 and RRM1. It is also possible that the upregulation of STIM1 may help promote or facilitate the emergence gemcitabine resistance by promoting cell survival upon gemcitabine treatment during resistance acquisition. In support of this, STIM1 depletion was shown to promote the pro-apoptotic effects of gemcitabine in pancreatic cancer cells (29). Furthermore, STIM1, and thereby SOCE, are known to regulate various metabolic processes (41,42), and could thereby help tumor cells adapt to metabolic changes which could arise as a consequence of RRM1 upregulation during the acquisition of resistance.
ER stress activates ATF4 and elicits an initial pro-survival and secondary pro-apoptotic response, where the former is suggested to be hijacked by many tumors (43). One such example is the hijacking of the pro-survival pathway upon hypoxia, where ATF4 promotes the transcription of VEGF, while activating antioxidant genes (43-45). Thus, the prevailing view is that rather than leading to apoptosis, ER stress is used by tumors to adapt to stressful environments. Still, some PDAC tumors have been characterized to express higher levels of factors controlling ER homeostasis and conferring ER stress resistance (46). Our data supports this alternative mechanism whereby increased SOCE in STIM1-amplified tumors leads to ER stress resistance and NFAT activation. Interestingly, NFAT promotes the transcription of HIF1A in a STIM1-dependent manner in T cells (41), while STIM1 itself has been associated with hypoxic-driven tumorigenesis in hepatocarcinoma (47). STIM1 and thereby SOCE are important regulators of melanoma aggressive behavior, controlling cellular oxidative stress through redox regulation of NFATc2 (20,48). Moreover, STIM and ORAI are important regulators of the pathobiology of several cancers (49). In PDAC, NFATs have been extensively characterized and shown to drive pancreatic cancer development and growth. NFATs are central in inflammation-driven pancreatic cancer development (50,51) and promote the silencing of CDKN2B in late-stage pancreatic intraepithelial neoplasia lesions (52). Furthermore, NFATs have been described to promote cell proliferation and tumor growth by fostering MYC expression in pancreatic cancer (53-55). Taken together, we suggest that rather than hijacking the pro-survival pathway of the ER stress response, STIM1-overexpressing tumors profit from an alternative STIM1-dependent/ATF4-independent pro-survival mechanism. In this case, STIM1 may act as a rheostat balancing between ER stress and NFAT activation, making STIM1 an attractive potential therapeutic target. Thus, STIM1 may also serve as a potential indicator of NFAT activation and ER stress resistance.
While very little is known about calcium homeostasis in PDAC, calcium signaling is key in the development of acute pancreatitis (56) where its therapeutic utility has been recently studied. In fact, the ORAI1 inhibitor CM4620 is currently being tested in a phase II clinical trial in acute pancreatitis patients (NCT04195347) (57-59). Notably, chronic pancreatitis is a known risk factor for the development of pancreatic cancer and is characterized by increased inflammation (60). While the role of ORAI and SOCE has been described specifically in acute pancreatitis, it is worth noting that heightened NFAT activity promotes acinar to ductal metaplasia and fosters the progression of chronic pancreatitis to pancreatic cancer in mouse models (26,61). Consequently, SOCE inhibitors may prevent progression from chronic pancreatitis to PDAC. Hence, it is possible that some PDACs display aberrant calcium signaling obtained during previous chronic pancreatitis or due to other selective pressures. Therefore, analyzing STIM1 levels in PDAC could potentially predict tumor sensitivity to stress, while tumors with high STIM1 expression might benefit from STIM and ORAI inhibitors.
In conclusion, this study unravels novel independent molecular properties of gemcitabine-resistant tumors in PDAC. Through the amplification of RRM1, tumors become resistant to gemcitabine, while STIM1 acts as a rheostat balancing ER stress and NFAT activity in a SOCE-dependent manner. Furthermore, the co-amplification can occur spontaneously in treatment-naïve cancer cells, making STIM1 a potential mediator of aberrant NFAT activation and SOCE inhibitors potential novel therapeutic agents for PDAC patients.
Supplementary Material
Statement of significance.
Gemcitabine-resistant and some naïve tumors co-amplify RRM1 and STIM1, which elicit gemcitabine resistance and induce a calcium signaling shift, promoting ER stress resistance and activation of NFAT signaling.
Acknowledgments:
The authors thank M. Dobbelstein, J. Choo, F. Wegwitz, E. Prokakis and J. Henck for fruitful discussions. F. Wegwitz and E. Prokakis for assistance in microscopy. This project was funded by grants from the Deutsche Krebshilfe (70112505; PIPAC consortium) to S.A.J., Z.N., J.G., V.E. and E.H., the Deutsche Forschungsgemeinschaft (SFB1190 and SFB1027) to I.B., the National Institute of Diabetes and Digestive and Kidney Diseases (T32 DK07198) to A.Q.W., and the National Cancer Institute (CA 102701) to S.A.J.
Abbreviations:
- PDAC
Pancreatic Ductal Adenocarcinoma
- RRM1
Ribonucleotide Reductase Catalytic Subunit M1
- STIM1
Stromal Interaction Molecule 1
- ER
Endoplasmic Reticulum
- ATF4
Activating Transcription Factor 4
- ChIP
Chromatin Immunoprecipitation
- Thap
Thapsigargin
- SERCA
Sarco/endoplasmic Reticulum Calcium-ATPase
- NFAT
Nuclear Factor of Activated T-cells
- PDX
Patient-derived xenograft
Footnotes
Conflict of interest: The authors declare no potential conflicts of interest.
References
- 1.American Cancer Society. Cancer Facts & Figures 2020. 2020;
- 2.Sohal DPS, Kennedy EB, Khorana A, Copur MS, Crane CH, Garrido-Laguna I, et al. Metastatic pancreatic cancer: ASCO clinical practice guideline update. J Clin Oncol. 2018;36:2545–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ryan DP. Chemotherapy for advanced exocrine pancreatic cancer. In: Goldberg RM, Savarese DMF, editors. UpToDate. UpToDate in Waltham, MA; 2020. [Google Scholar]
- 4.Cunningham D, Chau I, Stocken DD, Valle JW, Smith D, Steward W, et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol. 2009;27:5513–8. [DOI] [PubMed] [Google Scholar]
- 5.Scheithauer W, Schüll B, Ulrich-Pur H, Schmid K, Raderer M, Haider K, et al. Biweekly high-dose gemcitabine alone or in combination with capecitabine in patients with metastatic pancreatic adenocarcinoma: A randomized phase II trial. Ann Oncol. Elsevier Masson SAS; 2003;14:97–104. [DOI] [PubMed] [Google Scholar]
- 6.Hamada C, Okusaka T, Ikari T, Isayama H, Furuse J, Ishii H, et al. Efficacy and safety of gemcitabine plus S-1 in pancreatic cancer: A pooled analysis of individual patient data. Br J Cancer. Nature Publishing Group; 2017;116:1544–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akita H, Zheng Z, Takeda Y, Kim C, Kittaka N, Kobayashi S, et al. Significance of RRM1 and ERCC1 expression in resectable pancreatic adenocarcinoma. Oncogene. Nature Publishing Group; 2009;28:2903–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou J, Zhang L, Zheng H, Ge W, Huang Y, Yan Y, et al. Identification of chemoresistance-related mRNAs based on gemcitabine-resistant pancreatic cancer cell lines. Cancer Med. 2019;1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakano Y, Tanno S, Koizumi K, Nishikawa T, Nakamura K, Minoguchi M, et al. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br J Cancer. 2007;96:457–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakahira S, Nakamori S, Tsujie M, Takahashi Y, Okami J, Yoshioka S, et al. Involvement of ribonucleotide reductase M1 subunit overexpression in gemcitabine resistance of human pancreatic cancer. Int J Cancer. 2007;120:1355–63. [DOI] [PubMed] [Google Scholar]
- 11.Wang C, Zhang W, Juan Fu M, Yang A, Huang H, Xie J. Establishment of human pancreatic cancer gemcitabine-resistant cell line with ribonucleotide reductase overexpression. Oncol Rep. 2015;33:383–90. [DOI] [PubMed] [Google Scholar]
- 12.Bergman AM, Eijk PP, Ruiz Van Haperen VWT, Smid K, Veerman G, Hubeek I, et al. In vivo induction of resistance to gemcitabine results in increased expression of ribonucleotide reductase subunit M1 as the major determinant. Cancer Res. 2005;65:9510–6. [DOI] [PubMed] [Google Scholar]
- 13.Farrell JJ, Elsaleh H, Garcia M, Lai R, Ammar A, Regine WF, et al. Human Equilibrative Nucleoside Transporter 1 Levels Predict Response to Gemcitabine in Patients With Pancreatic Cancer. Gastroenterology. AGA Institute American Gastroenterological Association; 2009;136:187–95. [DOI] [PubMed] [Google Scholar]
- 14.Maréchal R, Bachet JB, MacKey JR, Dalban C, Demetter P, Graham K, et al. Levels of gemcitabine transport and metabolism proteins predict survival times of patients treated with gemcitabine for pancreatic adenocarcinoma. Gastroenterology. Elsevier Inc.; 2012;143:664–674.e6. [DOI] [PubMed] [Google Scholar]
- 15.Tiriac H, Belleau P, Engle DD, Plenker D, Deschênes A, Somerville TDD, et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov. 2018;8:1112–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. 2016;17:1374–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu PD, Harding HP, Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol. 2004;167:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–108. [DOI] [PubMed] [Google Scholar]
- 19.Monteith GR, Prevarskaya N, Roberts-Thomson SJ. The calcium-cancer signalling nexus. Nat Rev Cancer. Nature Publishing Group; 2017;17:367–80. [DOI] [PubMed] [Google Scholar]
- 20.Zhang X, Gibhardt CS, Will T, Stanisz H, Körbel C, Mitkovski M, et al. Redox signals at the ER –mitochondria interface control melanoma progression . EMBO J. 2019;38:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamdan FH, Johnsen SA. DeltaNp63-dependent super enhancers define molecular identity in pancreatic cancer by an interconnected transcription factor network. Proc Natl Acad Sci. National Academy of Sciences; 2018;115:E12343–E12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagarajan S, Hossan T, Alawi M, Najafova Z, Indenbirken D, Bedi U, et al. Bromodomain Protein BRD4 Is Required for Estrogen Receptor-Dependent Enhancer Activation and Gene Transcription. Cell Rep. 2014;8:460–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mishra VK, Wegwitz F, Kosinsky RL, Sen M, Baumgartner R, Wulff T, et al. Histone deacetylase class-I inhibition promotes epithelial gene expression in pancreatic cancer cells in a BRD4-and MYC-dependent manner. Nucleic Acids Res. 2017;45:6334–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Najafova Z, Tirado-Magallanes R, Subramaniam M, Hossan T, Schmidt G, Nagarajan S, et al. BRD4 localization to lineage-specific enhancers is associated with a distinct transcription factor repertoire. Nucleic Acids Res. 2017;45:127–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen NM, Neesse A, Dyck ML, Steuber B, Koenig AO, Lubeseder-Martellato C, et al. Context-Dependent Epigenetic Regulation of Nuclear Factor of Activated T Cells 1 in Pancreatic Plasticity. Gastroenterology. Elsevier, Inc; 2017;152:1507–1520.e15. [DOI] [PubMed] [Google Scholar]
- 27.De Sousa Cavalcante L, Monteiro G. Gemcitabine: Metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur J Pharmacol. Elsevier; 2014;741:8–16. [DOI] [PubMed] [Google Scholar]
- 28.Carreras-Sureda A, Pihán P, Hetz C. Calcium signaling at the endoplasmic reticulum: fine-tuning stress responses. Cell Calcium. 2018;70:24–31. [DOI] [PubMed] [Google Scholar]
- 29.Kondratska K, Kondratskyi A, Yassine M, Lemonnier L, Lepage G, Morabito A, et al. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochim Biophys Acta - Mol Cell Res. Elsevier B.V.; 2014;1843:2263–9. [DOI] [PubMed] [Google Scholar]
- 30.Prakriya M, Lewis RS. Store-operated calcium channels. Physiol Rev. 2015;95:1383–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: Dynamic calcium signal transducers. Nat Rev Mol Cell Biol. Nature Publishing Group; 2012;13:549–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gilon P, Roe MW, Evans-Molina C, Kono T, Tong X, Taleb S, et al. Impaired store-operated calcium entry and STIM1 loss lead to reduced insulin secretion and increased endoplasmic reticulum stress in the diabetic B-cell. Diabetes. 2018;67:2293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Da Conceicao VN, Sun Y, Zboril EK, De la Chapa JJ, Singh BB. Loss of Ca2+ entry via Orai-TRPC1 induces ER stress, initiating immune activation in macrophages. J Cell Sci. 2020;133:jcs237610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kar P, Parekh AB. Distinct Spatial Ca2+ Signatures Selectively Activate Different NFAT Transcription Factor Isoforms. Mol Cell. The Authors; 2015;58:232–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kar P, Nelson C, Parekh AB. Selective activation of the transcription factor NFAT1 by calcium microdomains near Ca2+ release-activated Ca2+ (CRAC) channels. J Biol Chem. 2011;286:14795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ashida R, Nakata B, Shigekawa M, Mizuno N, Sawaki A, Hirakawa K, et al. Gemcitabine sensitivity-related mRNA expression in endoscopic ultrasound-guided fine-needle aspiration biopsy of unresectable pancreatic cancer. J Exp Clin Cancer Res. 2009;28:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferrari A, Vincent-Salomon A, Pivot X, Sertier AS, Thomas E, Tonon L, et al. A whole-genome sequence and transcriptome perspective on HER2-positive breast cancers. Nat Commun. 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chu PY, Li TK, Ding ST, Lai IR, Shen TL. EGF-induced Grb7 recruits and promotes ras activity essential for the tumorigenicity of Sk-Br3 breast cancer cells. J Biol Chem. 2010;285:29279–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Janes PW, Lackmann M, Church WB, Sanderson GM, Sutherland RL, Daly RJ. Structural determinants of the interaction between the erbB2 receptor and the Src homology 2 domain of Grb7. J Biol Chem. 1997;272:8490–7. [DOI] [PubMed] [Google Scholar]
- 40.Katz E, Dubois-Marshall S, Sims AH, Faratian D, Li J, Smith ES, et al. A gene on the HER2 amplicon, C35, is an oncogene in breast cancer whose actions are prevented by inhibition of Syk. Br J Cancer. 2010;103:401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vaeth M, Maus M, Klein-Hessling S, Freinkman E, Yang J, Eckstein M, et al. Store-Operated Ca2+ Entry Controls Clonal Expansion of T Cells through Metabolic Reprogramming. Immunity. Elsevier Inc.; 2017;47:664–679.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maus M, Cuk M, Patel B, Lian J, Ouimet M, Kaufmann U, et al. Store-Operated Ca2+ Entry Controls Induction of Lipolysis and the Transcriptional Reprogramming to Lipid Metabolism. Cell Metab. Elsevier Inc.; 2017;25:698–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Urra H, Dufey E, Avril T, Chevet E, Hetz C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends in Cancer. Elsevier Inc.; 2016;2:252–62. [DOI] [PubMed] [Google Scholar]
- 44.Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, et al. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 2005;24:3470–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rouschop KM, Dubois LJ, Keulers TG, Van Den Beucken T, Lambin P, Bussink J, et al. PERK/eIF2α signaling protects therapy resistant hypoxic cells through induction of glutathione synthesis and protection against ROS. Proc Natl Acad Sci U S A. 2013;110:4622–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Milan M, Balestrieri C, Alfarano G, Polletti S, Prosperini E, Nicoli P, et al. Pancreatic Cancer Cells Require the Transcription Factor MYRF to Maintain ER Homeostasis. Dev Cell. Elsevier Inc.; 2020;55:398–412.e7. [DOI] [PubMed] [Google Scholar]
- 47.Li Y, Guo B, Xie Q, Ye D, Zhang D, Zhu Y, et al. STIM1 Mediates Hypoxia-Driven Hepatocarcinogenesis via Interaction with HIF-1. Cell Rep. The Authors; 2015;12:388–95. [DOI] [PubMed] [Google Scholar]
- 48.Stanisz H, Saul S, Müller CSL, Kappl R, Niemeyer BA, Vogt T, et al. Inverse regulation of melanoma growth and migration by Orai1/STIM2-dependent calcium entry. Pigment Cell Melanoma Res. 2014;27:442–53. [DOI] [PubMed] [Google Scholar]
- 49.Prevarskaya N, Skryma R, Shuba Y. Calcium in tumour metastasis: New roles for known actors. Nat Rev Cancer. Nature Publishing Group; 2011;11:609–18. [DOI] [PubMed] [Google Scholar]
- 50.Baumgart S, Chen NM, Zhang JS, Billadeau DD, Gaisina IN, Kozikowski AP, et al. GSK-3β governs inflammation-induced NFATc2 signaling hubs to promote pancreatic cancer progression. Mol Cancer Ther. 2016;15:491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baumgart S, Chen NM, Siveke JT, König A, Zhang JS, Singh SK, et al. Inflammation-Induced NFATc1-STAT3 transcription complex promotes pancreatic cancer initiation by KrasG12D. Cancer Discov. 2014;4:688–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baumgart S, Glesel E, Singh G, Chen N, Reutlinger K, Zhang J, et al. Restricted heterochromatin formation links NFATc2 repressor activity with growth promotion in pancreatic cancer. Gastroenterology. 2012;142:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buchholz M, Schatz A, Wagner M, Michl P, Linhart T, Adler G, et al. Overexpression of c-myc in pancreatic cancer caused by ectopic activation of NFATc1 and the Ca2+/calcineurin signaling pathway. EMBO J. 2006;25:3714–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.König A, Linhart T, Schlengemann K, Reutlinger K, Wegele J, Adler G, et al. NFAT-Induced Histone Acetylation Relay Switch Promotes c-Myc-Dependent Growth in Pancreatic Cancer Cells. Gastroenterology. 2010;138:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Singh G, Singh SK, König A, Reutlinger K, Nye MD, Adhikary T, et al. Sequential activation of NFAT and c-Myc transcription factors mediates the TGF-β switch from a suppressor to a promoter of cancer cell proliferation. J Biol Chem. 2010;285:27241–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Raraty M, Ward J, Erdemli G, Vaillant C, Neoptolemos JP, Sutton R, et al. Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc Natl Acad Sci U S A. 2000;97:13126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.NCT04195347. Study of CM4620 to Reduce the Severity of Pancreatitis Due to Asparaginase [Internet]. 2019. [cited 2020 Aug 3]. Available from: https://clinicaltrials.gov/ct2/show/NCT04195347
- 58.NCT03401190. CM4620 Injectable Emulsion Versus Supportive Care in Patients With Acute Pancreatitis and SIRS [Internet]. 2018. [cited 2020 Aug 3]. Available from: https://clinicaltrials.gov/ct2/show/NCT03401190
- 59.NCT03709342. A PK/PD Study of CM4620-IE in Patients With Acute Pancreatitis [Internet]. 2018. [cited 2020 Aug 3]. Available from: https://clinicaltrials.gov/ct2/show/NCT03709342
- 60.Saluja A, Dudeja V, Dawra R, Sah RP. Early Intra-Acinar Events in Pathogenesis of Pancreatitis. Gastroenterology. Elsevier, Inc; 2019;156:1979–93. [DOI] [PubMed] [Google Scholar]
- 61.Chen NM, Singh G, Koenig A, Liou GY, Storz P, Zhang JS, et al. NFATc1 links EGFR signaling to induction of sox9 transcription and acinar-ductal transdifferentiation in the pancreas. Gastroenterology. Elsevier, Inc; 2015;148:1024–1034.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.