

Abstract
Diffuse large B-cell lymphoma (DLBCL) is a biologically and clinically heterogenous disease. Transcriptomic and genetic characterization of DLBCL have increased the understanding of its intrinsic pathogenesis and provided potential therapeutic targets. However, the role of the microenvironment in DLBCL biology remains less understood. Here, we performed a transcriptomic analysis of the microenvironment of 4,655 DLBCLs from multiple independent cohorts and described four major lymphoma microenvironment categories that associate with distinct biological aberrations and clinical behavior. We also found evidence of genetic and epigenetic mechanisms deployed by cancer cells to evade microenvironmental constrains of lymphoma growth; supporting the rationale for implementing DNA hypomethylating agents in selected DLBCL patients. In addition, our work uncovered new therapeutic vulnerabilities in the biochemical composition of the extracellular matrix that were exploited to decrease DLBCL proliferation in pre-clinical models. This novel classification provides a roadmap for the biological characterization and therapeutic exploitation of the DLBCL microenvironment.
INTRODUCTION
DLBCL, the most common lymphoid malignancy in adults worldwide, is curable with anti-CD20-based combination immunochemotherapy in just over 60% of patients. Over the past few years, several novel targeted therapies have been developed but a substantial fraction of patients with relapsed disease continue to die from lymphoma or its complications. Further improvement of treatment outcome relies on elucidating the biology that underlies the clinical behavior of this heterogenous disease. Gene expression analysis based on cell-of-origin (COO) classification originally identified two major subgroups of DLBCL: activated B-cell (ABC) and germinal center B-cell (GCB). These subgroups have distinct clinical behavior and molecular features, reflecting differential pathogenesis(1). More recently, studies of genomic alterations in DLBCL cells including mutations, somatic copy number alterations and structural variants, identified distinct genetic subtypes within the COO subgroups(2–4). ABC-DLBCL patients with co-occurring MYD88 and CD79B mutations or harboring NOTCH1 mutations have poorer prognosis than other ABC-DLBCLs(3). Similarly, lymphomas carrying EZH2 mutations and BCL2 translocations are associated with worse outcome within the GCB-DLBCLs(3). Meanwhile, lymphomas harboring MYC and BCL2 and/or BCL6 rearrangements represent a GCB subgroup with distinctly aggressive biology also referred as high-grade B-cell lymphoma double-hit or triple-hit (HGBL-DH/TH)(5,6).
The biology and clinical behavior of DLBCL result not only from the molecular alternations of DLBCL cells but also from their interaction with the microenvironment. Data from lymphoma patients and animal models indicate that in the lymphoma niche, external stimuli provided by ME cells and the extracellular matrix (ECM) contribute to disease development, progression and response to treatment(7–11). Since the interaction of lymphoma cells and the microenvironment is bidirectional(12,13), it is expected that the DLBCL microenvironment will exhibit molecular and functional complexities that are yet to be defined. Despite the increasingly recognized role of the microenvironment in DLBCL biology(10), the majority of the molecular and therapeutic studies of this disease have been focused on the characterization of the DLBCL cell as an isolated entity(1).
Here, we characterized the DLBCL microenvironment based on the transcriptional footprint of microenvironment cells and processes in a large cohort of patients. We found four distinct microenvironment compositions reflecting unique biological properties and clinical behavior. This newly described categories associate with distinct clinical behavior of genetically similar DLBCLs and provide an array of novel potential targets for innovative therapeutic interventions.
RESULTS:
The lymphoma microenvironment (LME) is characterized by four distinct categories.
We aimed to define the diversity in the composition and functionality of DLBCL microenvironments based on the statistical powered conferred by gene expression profiles of thousands of patients. As a first step towards this end we generated 25 Functional Gene Expression Signatures (FGES) reflecting either distinct cellular subtypes relevant to tumor microenvironment (e.g. cancer associated fibroblasts, tumor infiltrating lymphocytes), non-cellular components of the tumor microenvironment (e.g., immune suppressive cytokines, extracellular matrix), biological processes (e.g., secretion, proliferation) and canonical signaling pathway activation (e.g., PI3K, NF-kB) (Figs. 1a and S1a–c and Table S1). For each FGES we calculated an activity score that directly associates with the magnitude by which a particular effect is present among populations of cells. For example, PI3K and NF-kB FGES are sensitive enough to detect changes in pathway activity upon their specific inhibition in DLBCL cell lines and mice models (Fig. S1d,e). Activity scores of the FGES revealed the expect enrichment for the cell types from which they derive (Figs. 1b and S1b,c), as well as potentially novel functional interdependencies. For example, while the “extracellular matrix” (ECM) FGES was almost exclusively correlated with fibroblasts, the “ECM remodeling” FGES was also correlated with activated macrophages (Fig. 1b), suggesting a potential role for these cells in the ECM microarchitecture. Similarly, they informed on cell states, for example, the biological process “cell proliferation” FGES was almost absent in non-proliferating circulating B cells and progressively higher in B-cell lymphomas ranging from low-proliferating indolent follicular lymphomas (FL) to DLBCLs and the highly proliferative Burkitt lymphomas (BL) (Fig. 1b).
Figure 1. Transcriptomics analysis distinguishes four distinct subtypes of the DLBCL microenvironment.
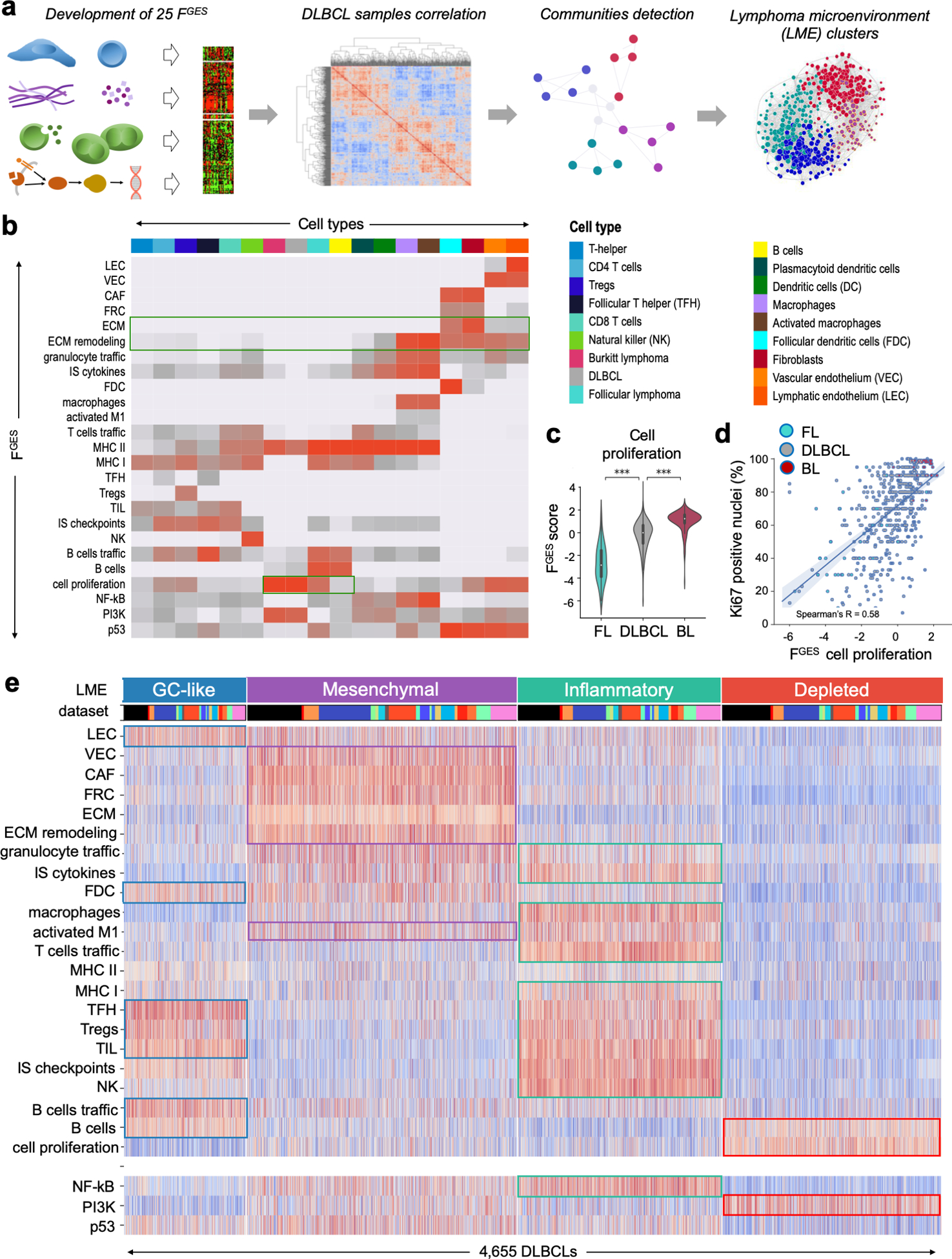
a. Overview of the transcriptomic approach to identify lymphoma microenvironment (LME) signatures in DLBCL. b. Association between functional gene expression signatures (FGES) represented on the y-axis with cell types on the x-axis where different colors represent unique cell types following the palette shown in a. The strength of the association is represented with red intensities with light grey indicating no association. c. Activity score of the “cell proliferation” FGES represented across B-cell lymphomas including 130 follicular lymphomas (FL), 4,655 diffuse large B-cell lymphomas (DLBCL) and 196 Burkitt lymphomas (BL). d. Spearman rank correlation between the activity score of the “cell proliferation” FGES and the percentage of Ki67 positive nuclei determined by immunohistochemistry in 532 B-cell lymphomas. e. Heat map of the activity scores of 25 FGES (x-axis) denoting four major LME clusters termed as GC-like, mesenchymal, inflammatory and depleted. Individual datasets are indicated by different colors. LEC: lymphatic endothelial cells, VEC: vascular endothelial cells, CAF: cancer-associated fibroblasts, FRC: fibroblastic reticular cells, ECM: extracellular matrix, IS/PL: immune suppressive / pro-lymphoma, IA/AL: immune activating / anti-lymphoma, FDC: follicular dendritic cells, MHC: major histocompatibility complex, TFH: follicular T-helper cells, TIL: tumor infiltrating lymphocytes, NK: natural killer cells. ***p < 0.001.
To validate whether the FGES was still informative when applied to gene expression profiles derived from tissues with mixed cell types, we analyzed the activity score of the “cell proliferation” FGES in a cohort of 4,984 B-cell lymphomas gene expression profiles including FL, DLBCL and BL and found that, similar to individual cells, the activity score of these FGES directly correlated with the proliferation rate reported for these diseases (Fig. 1c). Moreover, as an additional independent validation, the “cell proliferation” FGES also correlated with the proliferation rate determined by “gold-standard” Ki67 immunohistochemistry of 532 B-cell lymphoma samples (Fig. 1d). Lastly, we proved that the proportion of “fibroblasts” and “activated macrophages” estimated by FGES correlated with the estimation of these cells obtained from anti-SMA and anti-CD68 immunostaining quantification from matched DLBCL tissues (Fig. S1f).
We then used the 25 FGES to virtually reconstruct the LME of 4,580 DLBCL cases collected from publicly available gene expression databases and included an additional set of 75 DLBCLs profiled by RNA-seq locally (Fig. S2a). We analyzed the correlation between FGES among samples by applying an unsupervised community detection algorithm (Fig. 1a) and obtained four major clusters of LMEs (Fig. 1e). These four LMEs reflect distinct FGES associations or “communities”, and were present in 15% (LME-1), 33% (LME-2), 25% (LME-3) and 27% (LME-4) of the samples, with similar distribution across all tested datasets (Figs. 1e and S2b–f). According to the communities of FGES enriched within each microenvironment, the four distinct LMEs were termed, respectively, as “germinal center-like” due to the presence of FGES from cell types commonly found in germinal centers (GC)(14); “mesenchymal” (MS) for the abundance of FGES from stromal cells and extracellular matrix pathways; “inflammatory” (IN) for the presence of FGES associated with inflammatory cells and pathways; and finally, a “depleted” (DP) form that, contrasting with the other LMEs, was characterized by an overall lower presence of microenvironment derived FGES (Fig. 1e).
LME categories represent a novel gene expression-based DLBCL classification.
We developed a platform-independent algorithm to classify individual lymphomas according to their ABC vs. GCB DLBCL cell of origin subtype (Fig. S3a–c and Table S2). Among the full cohort (n = 4,655), 59% were GCB- and 41% were ABC-DLBCLs (Fig. S3c). These DLBCL subtypes varied in their LME associations. For example, whereas IN-LME sub-type contained a higher proportion of ABC-DLBCLs, the GC-like and MS LMEs were more enriched in GCB-DLBCLs (Fig. 2a, p<0.001, Chi-square test, all comparisons). For the subsequent analysis requiring different molecular or clinical annotations, we used the largest possible subset of the data that had the required information (Fig. S2a). We investigated the association of LME categories with the CCC (Consensus Clustering Classification)(15) gene expression subtypes BCR (B-cell receptor), OxPhos (oxidative phosphorylation) and HR (host response). As might be expected, the DP-LME was not associated with HR cases, whereas BCR and OxPhos did show specific LME patterns (Fig. S3d). In addition, the MS-LME were enriched in the previously described “stromal-1” and “stromal-2” transcriptional signatures(10) (Fig. S3e).
Figure 2. Association of LME categories with genomic alterations in DLBCL cells.
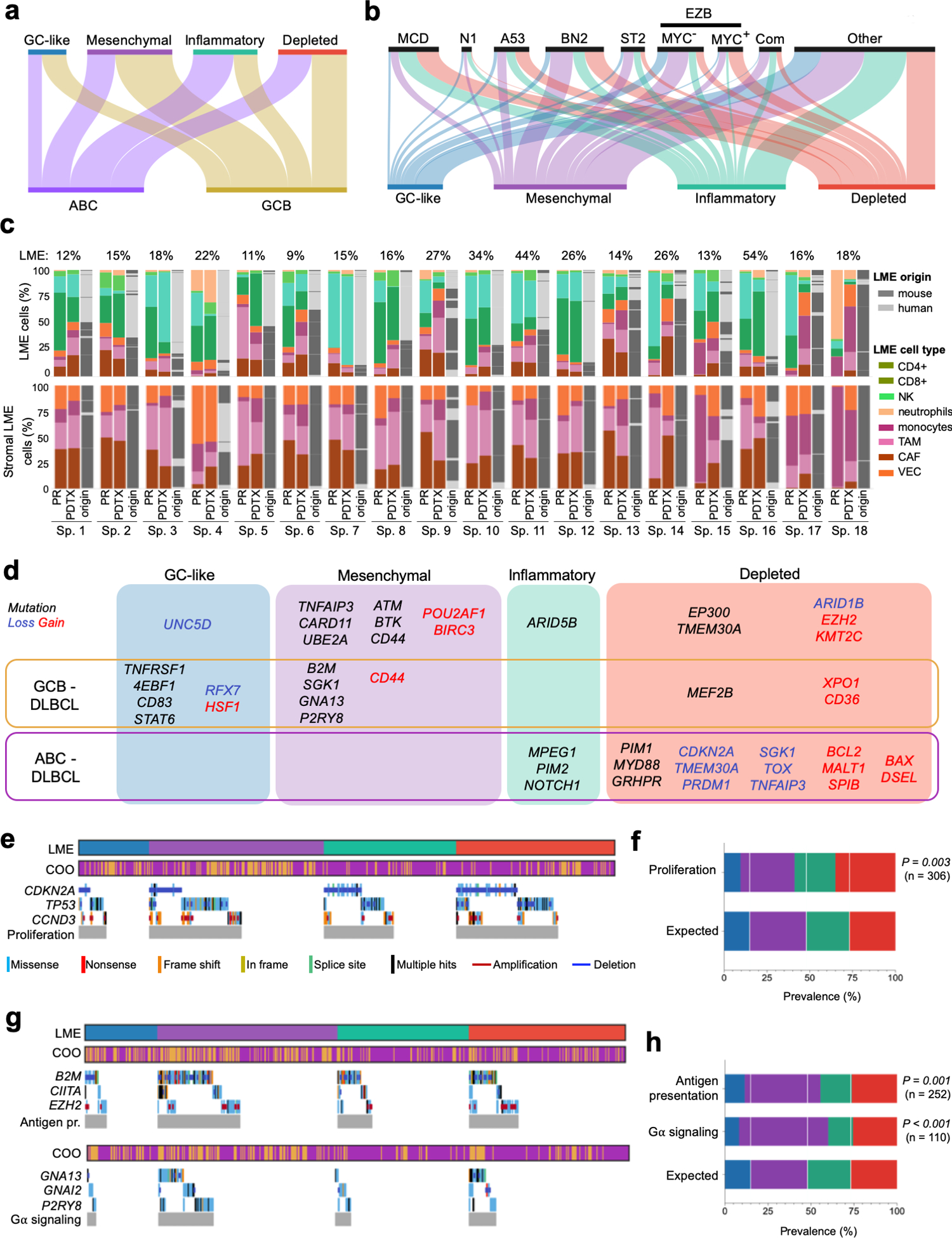
a. Distribution of the DLBCL cell-based COO transcriptomic signature subgroups within the LME categories. b. Distribution of DLBCL genomic subtypes MCD, N1, A53, BN2, ST2, EZB, composite and others within LME categories. c. LME cellular composition (first column for primary samples and second columns for their patient-derived tumor xenograft) and LME origin (mouse vs. human, third column). Upper columns represent percentage of total LME cells and lower columns the percentage of stromal LME cells. The proportion of LME vs. lymphoma cells in primary samples is shown on top of the columns. d. Mutations and copy number alterations significantly enriched (Fisher T-test) in a particular LME category. Alterations also significantly enriched in a COO subgroup are indicated. e. Oncoplot for genomic alterations affecting the proliferation pathway genes CDKN2A, TP53 and CCND3 by LME category. f. Prevalence of proliferation pathway genomic alterations by LME category according to their expected random distribution. The number of cases with these genomic alterations is shown. g. Oncoplot for genomic alterations affecting antigen presentation genes B2M, CIITA and EZH2, and G-protein signaling pathway genes GNAI2, GNA13 and P2RY8 by LME categories. h. Prevalence of antigen presentation and G-protein signaling pathways genomic alterations by LME category according to their expected random distribution. The number of cases with these genomic alterations in each category is shown.
Notably, HGBL-DHs were enriched in DP-LME and secondarily in GC-LME according to the analysis of 1,397 cases with this data available (Fig. S3d). Concurrent rearrangements of MYC and BCL2 are not the only molecular determinants of aggressiveness in DLBCL. Two gene expression signatures, the molecular high-grade (MHG) and the DHL signature (DHITsig)(16), identified additional patients that behave as HGBL-DH without necessarily having these genomic alterations. We developed and applied these classifiers (Fig. S4a–g and Table S3) to our collected dataset in relation to LME categories and found a significant enrichment of MHG and DHITsig patients in the DP-LME category (Fig. S3d). Moreover, the MHG and DHITsig were highly overlapping (Fig. S4d), likely reflecting a similar underlying biology. Lastly, in a mouse model engineered to express myc and bcl2 in germinal center B-cells, the resultant B-cell lymphomas harbor an LME that is similar to DP-LME DLBCLs (p<0.001, Fig. S4h)
DLBCLs were recently classified into genetically defined entities with distinct clinical characteristics(2–4), some of them also associated with microenvironmental features(4). One of these efforts(4) grouped DLBCL into the subtypes MCD (co-occurrence of MYD88 and CD79B mutations), N1 (NOTCH1 mutations), EZB (EZH2 mutations and BCL2 translocations), BN2 (BCL6 fusions and NOTCH2 mutations), ST2 (SGK1 and TET2 mutations) and A53 (aneuploid with TP53 inactivation). The genetic annotation was available from 737 cases from two cohorts (NCICCR and DLC2), and revealed uneven distribution of the four LME categories (Fig. 2b). For example, the poor-prognostic MCD subtype represented 4% of GC-like, 8% of MS, 17.6% of IN and 21% of the DP LME categories (p<0.001, Chi-square test, Fig. 2b). The MCD distribution in LMEs was not in full agreement with the proportion of ABC cases. Although majority of BN2, ST2 and EZB cases had an immune deserted LME (either MS or DP), in EZBs the specific immune deserted LME category associated with their MYC status: EZB-MYC+ had higher proportion of DP-LMEs whereas EZB-MYC− had more MS-LMEs (Fig. 2b). Taken together these data indicates that DLBCL LME categories provide additional orthogonal information that is not captured by previously reported lymphoma classifications.
LME categories associate with specific genomic alterations in lymphoma cells.
The LME is dependent on dynamic interactions between tumor and normal cell types. To better understand the effect of lymphoma cells in driving specific LME patterning, we established 18 patient-derived tumor xenografts (PDTX) by implanting DLBCL tissues from patients into immunosuppressed mice. We then performed RNA-seq in tumors from early passages of each PDTX and used our cell signature deconvolution approach to determine whether murine LME cells were repopulating the PDTX in proportions similar to that from the primary human tissue. Remarkably, the majority of PDTXs maintained similar murine LME categories as were represented in the original primary human lymphoma (Figs. 2c and S5a,b). Since recipient mice are immunosuppressed, the immune components of the LME were largely maintained from the human primary tissues in early passages. In contrast, the LME stromal cells, like cancer-associated fibroblasts (CAFs) and tumor-associated macrophages (TAMs), were recruited from the mouse tissues (Fig. 2c); as has been reported in breast cancer PDTX specimens(17). This suggests an underlying, long-lasting mutual signaling wiring between malignant and LME cells that is maintained in early passages PDTXs.
To explore mechanisms by which lymphomas might induce specific LMEs, we compared their spectra of mutations and copy number alternations (CNA). Mutations were obtained from WES and/or WGS or targeted sequencing from 747 patients whereas whole genome CNA were available for 750 patients (Fig. S2a). The analysis of the 86 most frequent mutations and the 65 most frequent CNAs (Fig. S6a,b, Tables S4 and S5) showed uneven distribution among LME categories with 24 mutations and 22 CNAs significantly enriched in a particular LME (Fig. 2d). Importantly, the distribution of LME categories for 9 out of these unequally distributed 24 mutations and 6 out of the 22 CNAs were not entirely explained by their COO designation (Fig. 2d), and so are potentially linked to direct effects of these mutations. Remarkably, some mutations and CNAs targeting the same gene converged in a particular LME as it was the case for CD44 with the MS-LME and for TMEM30A with the DP-LME (Fig. 2d).
Since CNAs and/or mutations targeting functionally-related genes may result in similar phenotypes, we analyzed such complimentary genomic alterations in relation to LME categories. We considered all genomic aberrations annotated to a pathway independently of their individual statistical significance. We found that genomic alterations resulting in decreased p53 activity and perturbation of cell cycle regulation were more common in DLBCL with DP-LME (Fig. 2e,f). This was validated in an independent dataset of 67 DLBCLs(18) in which 90% of DP-LMEs showed complex CNAs affecting this pathway compared to > 60% for the other LMEs (p < 0.04, Fig. S3d). Accordingly, DP-LMEs also had a higher proportion of A53 lymphomas (Fig. 2b) and strong enrichment for the “cell proliferation” FGES as later characterized. Recent data(19) showed expression of MHC I and MHC II antigen presentation genes and corresponding sparsity of immune infiltrates in murine lymphoma models expressing Ezh2Y641. In agreement with these data, EZH2 genomic alterations as well as mutations in B2M or CIITA that affect MHC I or MHC II, respectively, were enriched in MS-LME (Fig. 2g,h). In addition, MS-LME cases were enriched for mutations in GNA13, GNAI2 and P2RY8 (Fig. 2g,h). Mutations in GNA13/I2 and/or P2RY8 disrupt the Gα signaling pathway that is necessary to transduce homing signaling to lymphoma cells from surrounding immune cells(20), which are depleted from this LME, probably suggesting the acquisition of LME-independent growth mechanisms by lymphoma cells.
LME categories associate with distinct disease courses.
LME-GC-like and LME-DP had, respectively, the highest proportion of low (0–1) and high (4–5) IPI (International Prognostic Index) scores (Fig. S7a). To determine the association of the LME categories with response to R-CHOP chemoimmunotherapy, we compiled and performed RNA-seq of a cohort of 105 newly diagnosed DLBCL patients who were balanced between responders (55%) and non-responders (45%) including refractory (i.e., never achieved complete response) and relapsed (i.e., free from disease < 2 years). The number of responders was highest in GC-like- and lowest in DP-LME pts. (Fig. 3a, Z-tests p = 0.030 for GC-like vs. DP and p = 0.022 for GC-like + MS vs. IN + DP). In contrast, DP-LME had the highest proportion of non-responders and specifically of relapsed cases (Fig. 3a, 40.5% vs. 16.2% for DP-LME vs. all other LMEs, respectively, Z-test p = 0.011). These associations were validated in an independent cohort of unselected 1,349 DLBCL patients who received chemoimmunotherapy and were evaluated according to the RECIL criteria. The proportion of non-responders (SD + PD) was 4.5%, 7.3%, 11.1% and 12.7% for GC-like, MS, IN and DP LMEs, respectively (Fig. S7b, Z-test p = 5×10−4 for GC-like + MS vs. IN + DP LMEs).
Figure 3. Association of DLBCL LME categories with clinical outcome.
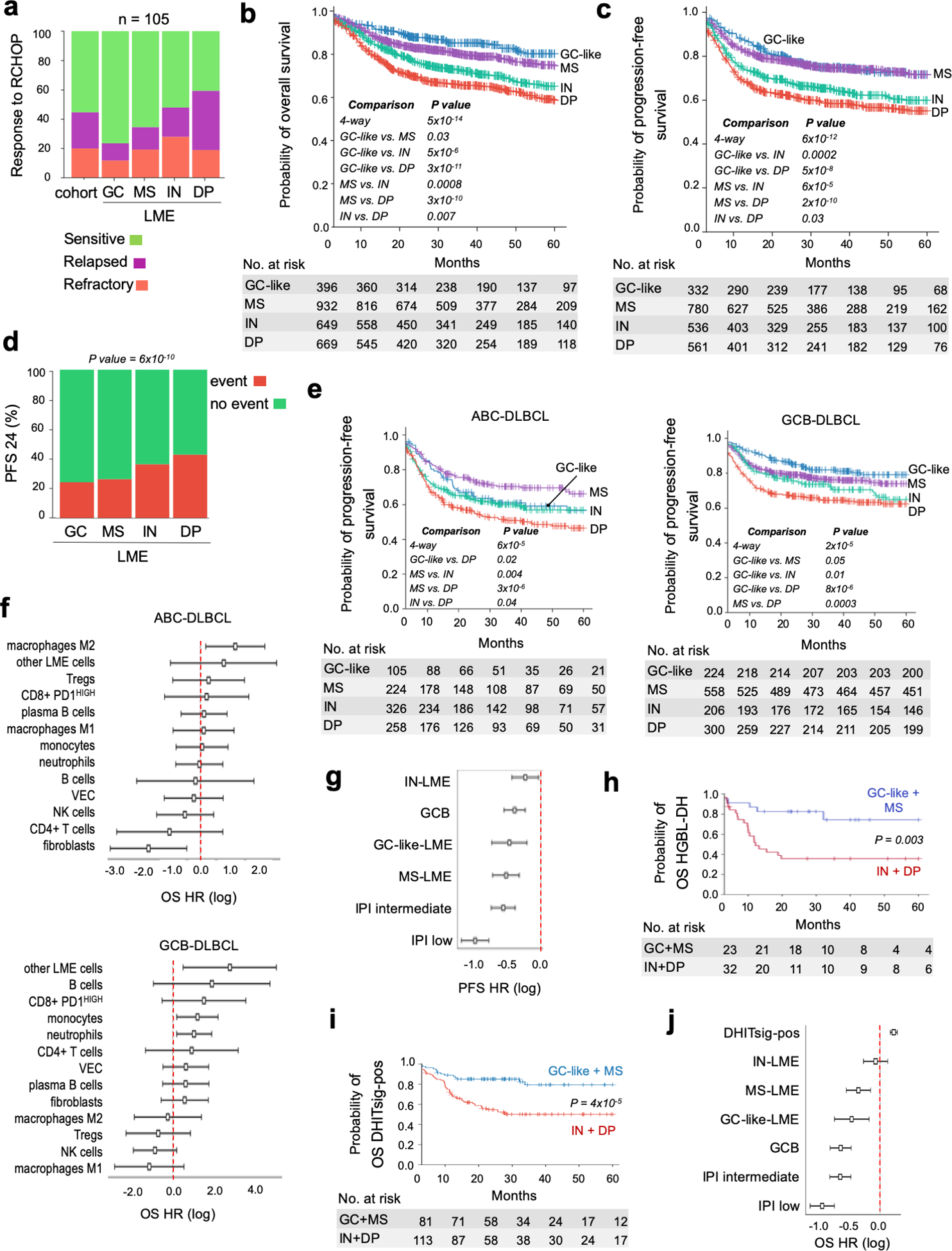
a. Response to chemoimmunotherapy (R-CHOP) in a balanced cohort (n = 105) or responsive and non-responsive (refractory and relapsed) DLBCL patients according to the LME category. b-c. Kaplan-Meier models of overall survival (OS) and progression-free survival (PFS), respectively, according to DLBCL LME category. d. PFS at 24 months (PFS24) in DLBCL patients according to the LME category. Censored patients are not shown. e. Kaplan-Meier models of PFS according to LME category in ABC- and GCB-DLBCL subgroups. Only statistically significant pairwise comparisons are shown. f. Contribution individual LME cellular subtypes to the OS hazard ratio (HR) (log with 95% confidence interval) in ABC- (top) and GCB-DLBCLs (bottom). g. PFS hazard ratio (HR) plots (log with 95% confidence interval) for LME category, COO subgroup and IPI. h. Kaplan-Meier models of OS for HGBL-DH patients segregated into favorable prognosis LMEs (GC-like and MS) and unfavorable prognosis LMEs (IN and DP). i. Kaplan-Meier models of OS for DHITsig-positive patients segregated into favorable prognosis LMEs (GC-like and MS) and unfavorable prognosis LMEs (IN and DP). j. OS hazard ratio (HR) plots (log with 95% confidence interval, n = 2,047) for LME category, COO subgroup, IPI and DHITsig status.
We further analyzed the association of LME categories with overall survival (OS) and progression-free survival (PFS) in patient cohorts of 2,646 and 2,189 DLBCL, respectively. All patients received rituximab-based chemoimmunotherapy resulting in typical OS and PFS Kaplan-Meier curves (Fig. S7c,d). Analysis of OS by LME indicated significant differences in prognosis from better to poor as follows GC-like, MS (p =0.03, vs. GC-like), IN (p = 0.0008, vs. MS) and DP (p = 0.007, vs. IN) LMEs (Fig. 3b); whereas GC-like and MS have similarly favorable PFS curves (p = 0.9) followed by IN (p < 0.001) and DP (p = 0.03, vs. IN), which presented with the poorest PFS (Fig. 3c). Bonferroni’s correction for multiple testing indicates that GC-like and MS are in the same strata for OS and PFS, as well as IN and DP for PFS. LME categories associated with different proportion of cases achieving OS and PFS at 24 months (OS24 and PFS24, respectively) (Figs. 3d and S7e).
When segregated by COO, the DP-LME retained the poorest OS and PFS in both COO subtypes. However, in ABC-DLBCL, the LME with the best PFS and OS was MS, and in GCB-DLBCL, the best LME was GC-like (Fig. 3e and Fig. S7f); suggesting that the biological impact of the microenvironment may be different depending on lymphoma subtype(21). Accordingly, individual LME cells subtypes contributed differently to OS in ABC- and GCB-DLBCLs (Fig. 3f). Overall, the combination of GCB-DLBCL with GC-like LME and ABC-DLBCL with DP-LME resulted in the most favorable and poorest clinical outcome, respectively (Figs. 3e and S7f). In multivariate Cox-proportional hazard analysis for PFS (n = 2,024) and OS (n = 2,052) controlled for COO and IPI, the LME subtypes remained informative with GC-like and MS LMEs associated with better outcome (Figs. 3g and S7g).
Finally, HGBL-DH, that carry significantly lower PFS and OS than DLBCL with neither MYC or BCL2 rearrangements (Fig. S7h), modified their outcome when LME was considered. HGBL-DH with the favorable prognosis LMEs GC-like and MS had significantly better PFS and OS than HGBL-DHs with the unfavorable prognosis LMEs IN and DP (Fig. 3h and Fig. S7h). In multivariate Cox-proportional hazard analysis for OS (n = 486) controlled for COO, IPI and MYC or BCL2 rearrangements, the LME subtypes remained informative with GC-like and MS LMEs associated with better outcome (Fig. S7i). The same was true among the DHITsig-positive DLBCLs; i.e., poor prognostic GCB-DLBCLs with high-grade molecular features including concurrent MYC and BCL2 rearrangements(16) (Fig. 3i,j and Fig. S7j,k). Collectively, these data underline the impact of LME category on outcome regardless of COO subgroup or genetic subtype, suggesting that these host factors make truly critical and independent contributions to lymphoma biology and clinical presentation.
LME categories are composed of distinct cellular communities.
To further understand the nature of each LME category, we expanded the community analysis of FGES signatures by estimating cell percentages in the LME using a RNA-seq deconvolution algorithm(22). This allowed us to more precisely define cell subpopulations and estimate their abundance. We validated this approach to cellular composition analysis by comparing and contrasting the results with mass cytometry performed on reactive tonsillar tissues (Fig. S8a). In addition to cellular composition, we also investigated the enrichment of the LME-dependent signaling and transcriptional pathways HIF (hypoxia inducible factor), TGFB/SMAD (transforming growth factor beta), JAK/STAT (signal transducer and activator of transcription) and TNF (tumor necrosis factor).
The GC-like LME resembled the cellular constitution of the germinal center microenvironment(14) since contained a relatively higher proportion of follicular dendritic cells, lymphatic endothelial cells, total T cells and several CD4+ T cell subpopulations including Tregs and T helper cells (Fig. 4a,b). The ratio of malignant to normal B cells was lower in this subtype. The FGES for cell proliferation activity that primarily captures the activity of malignant cells was lower compared to other LME categories (Fig. S8b). These lymphomas may thus represent transformed variants of indolent, germinal center-derived FLs. Further supporting this notion, 28% of GC-like LME cases harbored the FL hallmark BCL2 translocation and 90% of the LMEs profiled in a cohort of the 132 FLs were also GC-like (Fig. S8c,d). Community analysis of the MS-LME indicated a higher proportion of signatures from vascular endothelial cells, CAFs, fibroblastic reticular cells and ECM (Fig. 4c,d). In addition, there was enrichment for angiogenesis and ECM remodeling process FGES (Fig. S8e) and TGFB/SMAD and HIF pathways (Fig. 4e), which are associated with good prognosis in DLBCL(23–25). Notably, MS-LME was high enriched among primary mediastinal B-cell lymphomas vs. other LME subtypes (n = 287 total cohort, p = 0.003, Fig. S8c).
Figure 4. Distinct cellular communities define DLBCL LME categories.
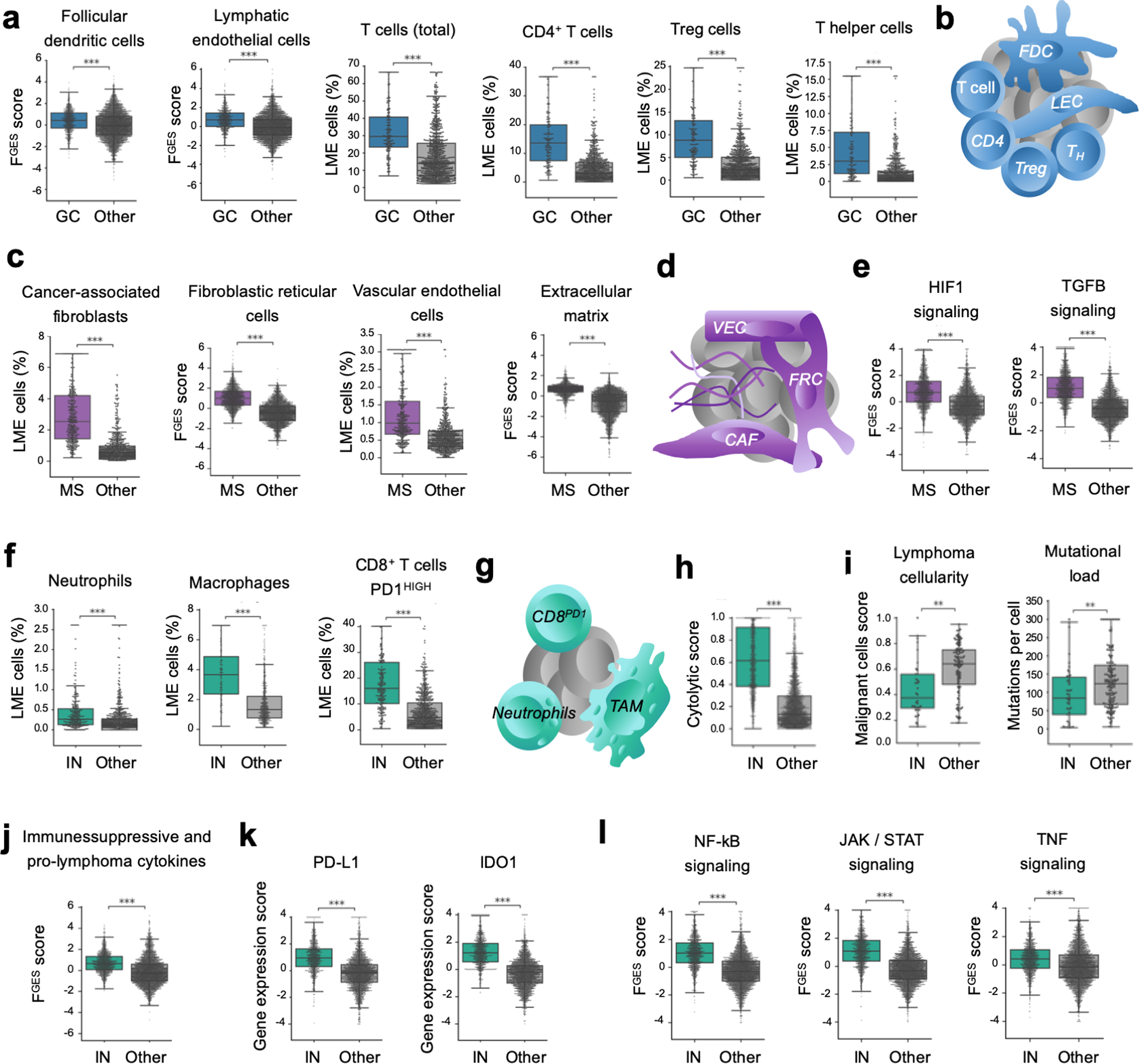
a. Proportion of LME cells significantly enriched in the GC-like LME obtained by cell deconvolution algorithms or functional gene expression signatures. b. Schematic representation of selected features of GC-like-LME. FDC: follicular dendritic cells, LEC: lymphatic endothelial cells. TFH: follicular T helper cells. c. Proportion of LME cells significantly enriched in the MS LME obtained by cell deconvolution algorithms or functional gene expression signatures. d. Schematic representation of selected features of MS-LME. VEC: vascular endothelial cells, FRC: fibroblastic reticular cells, CAF: cancer-associated fibroblasts. e. HIF1 (hypoxia) and TGFB pathways activity. f. Proportion of LME cells significantly enriched in the IN LME obtained by cell deconvolution algorithms. g. Schematic representation of selected features of IN-LME. TAM: tumor-associated macrophages. h. Cytolytic score in the IN-LME vs. other LMEs. i. Lymphoma cellularity (proportion of malignant cells) and mutational load (number of mutations per cell) in DLBCL containing IN-LME vs. other LMEs. j. Immune suppressive / pro-lymphoma cytokines FGES in the IN-LME vs. other LMEs. k. Expression of PD-L1 and IDO1 in lymphomas with IN-LME vs. other LMEs. l. NF-kB, JAK/STAT and TNF-alpha pathways activity. **p<0.01, ***p<0.001
Among the unfavorable LME categories, the IN-LME was enriched in neutrophils FGES associated with poor outcome(26), macrophages (likely M1, Fig. S8f), CD8+ T-cells (over total T-cells or over CD4+ T-cells, Fig. S8f) and the subset of CD8+ T-cells with high PD-1 expression (Fig. 4f,g). The proportion of NK cells was not significantly different among LMEs; however, the NK activity by FGES was significantly higher in IN-LME (Fig. S8f). This finding plus an enrichment of cytotoxicity activity as determined by the cytolytic score(27) (Fig. 4h) and presence of T cell activation markers, suggest certain degree of anti-lymphoma immunity. Indeed, DLBCL with IN-LME presented with the lowest relative number of malignant cells and lower mutational load (Fig. 4i), potentially representing lymphoma cells that may be able to escape tumor immunity(28,29). The IN-LME had features that may contribute to decrease lymphoma immunity such as enrichment of immune suppressive and pro-lymphoma cytokines (including the neutrophils attractant CXCL8(30) as well as IL10(31–34)) (Fig. 4j), expression of the immune checkpoint molecule PD-L1(33,34) and the tryptophan catabolic enzyme IDO1 (indoleamine 2,3-dioxygenase 1)(35,36) (Fig. 4k). As might be expected from this inflammatory milieu given the presence of macrophages and neutrophils FGESs in particular(37), there was a significantly higher activity of the signaling pathways NF-kB(38) (only partially associated with the COO subgroup, Fig S8g), JAK/STAT(34,38) and TNF(39) compared with the other LME categories (Fig. 4l).
DNA methylation patterning of lymphoma cells contribute to a depleted microenvironment.
DLBCLs with a DP-LME were characterized by a minimal presence of FGES derived from microenvironmental cells as well as by higher proportions of proliferating tumor cells (Fig. 5a) and clonal tumor cells (Fig. 5b and Fig. S9a). This category of LME was also common in BL patients (Fig. S8c). DP-LME is also characterized by activation of the PI3K signaling pathway (Fig. S9b); suggesting potential therapeutic vulnerabilities. Higher tumor cell proliferation was insufficient to account for the scarcity of FGES derived from microenvironmental cells. Indeed, majority of the tumor cells stained negative for MHC I and II (by immunohistochemistry) than any other LME (n = 177, p = 0.04, Fig. 5c); although there was no enrichment of MHC mutations in this group. We and others have previously reported(23,24) that aberrant DNA hypermethylation of the SMAD1 promoter allows DLBCL cells to escape from the anti-proliferative effects of LME-derived TGFB ligands. Indeed, analyzing our cohort and the TCGA cohort of 100 DLBCLs, we found that the degree of global DNA hypermethylation and specific SMAD1 promoter methylation were significantly higher among DP-LME (Figs. 5d and S9c). Accordingly, the SMAD1 expression and TGFB pathway activity were also lower (Figs. 5e and Fig. S9d,e). In the full dataset, DLBCL with DP-LME had the lowest SMAD1 expression and TGFB pathway activity than any other LME (Fig. 5f).
Figure 5. Pharmacologically reversible epigenetic mechanisms of ME evasion.
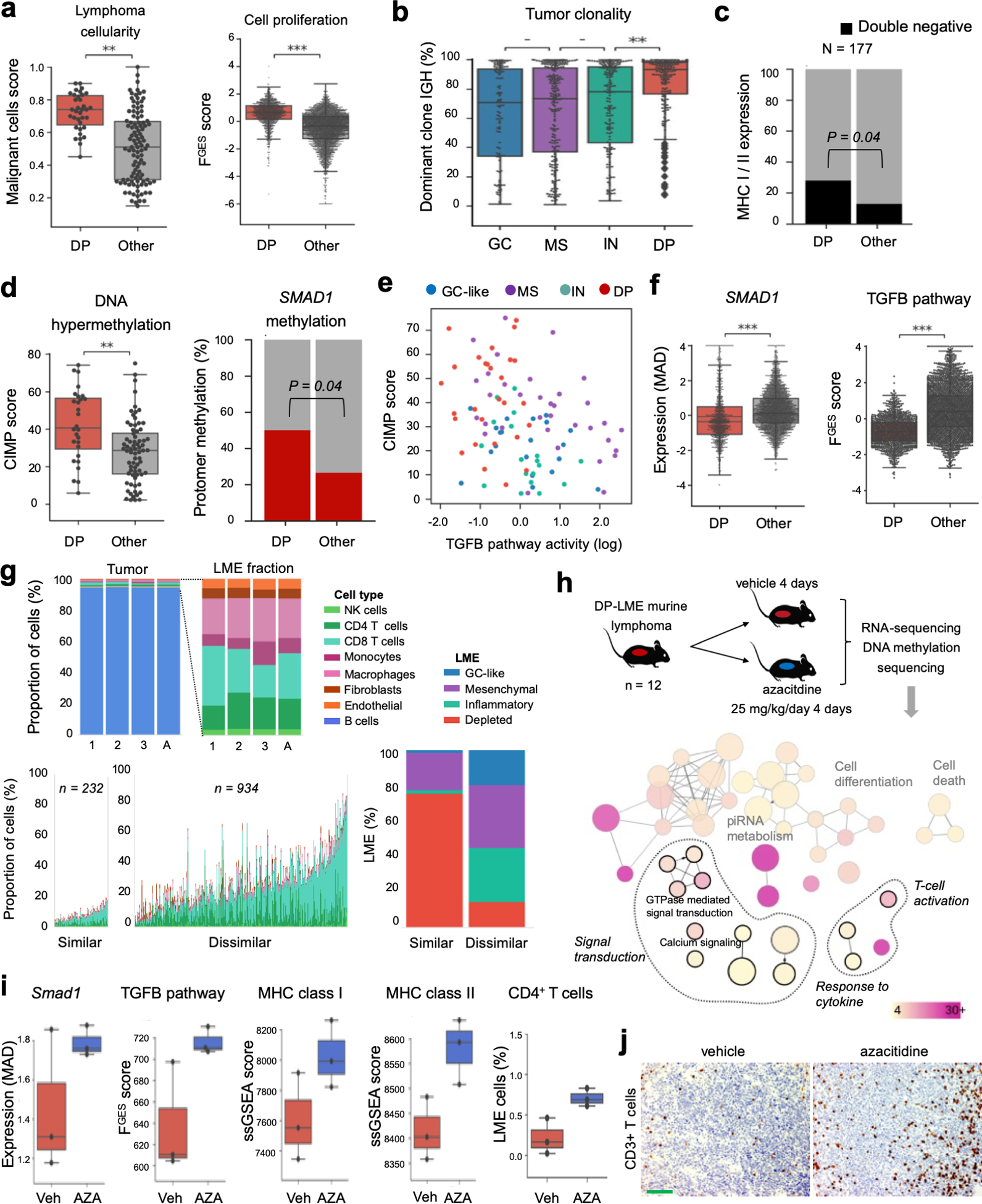
a. Proportion of malignant cells and proliferative activity of DLBCL with DP-LME. b. Tumor clonality (fraction of dominant clone by IGH). c. Proportion in MHC class I and II double negative by immunohistochemistry analysis of 177 DLBCL comparing DP-LME vs. other LMEs patients. d. Genome wide aberrant DNA hypermethylation CIMP score and specific SMAD1 gene promoter methylation in DP-LME vs. other LMEs. e. Correlation plot between CIMP score and TGF pathway activity (PROGEny) in DLBCL according to their respective LMEs. f. SMAD1 gene expression and TGF pathway activity in the full cohort of DLBCL. g. Characterization of the LME in the syngeneic murine B-cell lymphoma model A20 by the LME similarity score. LME human DLBCLs with the highest 20% of similarity score (n = 232) to A20 LME showed enrichment of immune deserted LMEs (96%) with higher proportion of DP-LMEs (78%). h. Schedule of azacytidine administration in A20 mice and tissue analysis (top). Selection of pathways from differentially hypomethylated and overexpressed genes from the A20 mice treated with azacytidine (bottom). The color bar indicates the percent of genes associated with a pathway and the size of the circles the number of genes in a pathway. i. Smad1 expression, TGF pathway activity, MHC class I and II expression and proportion of deconvoluted CD4+ T cells in the A20 lymphomas treated with azacytidine. j. Representative pictures of CD3 immunostaining of A20 lymphomas treated with azacitidine. The bar represents 100 μm. **p<0.01, ***p<0.001
To determine the contribution of aberrant DNA hypermethylation to this microenvironmental scenario we used the A20 syngeneic murine B-cell lymphoma model, that we characterized as having a similar LME constitution as DP-LME DLBCL patients (Fig. 5g). Once tumors developed in the treatment cohort (n = 12), we exposed the mice to the DNA hypomethylating agent azacitidine or vehicle for four consecutive days and then performed RNA-seq and DNA methylation sequencing of the tumors (Fig. 5h). Azacitidine treatment resulted primarily in gene upregulation and hypomethylation (Fig. S9f,g). Pathway analysis of significantly hypomethylated gene promoters and up-regulated genes revealed regaining of “cellular response to cytokines”, “signal transduction” and immune response including “activation of T cells” (Fig. 5h and Table S6). There was upregulation of Smad1 as well as increased TGFB pathway activity (Fig. 5i). In addition, there was induction of MHC class I and II expression and an increased proportion of CD4+ T cells by transcriptome deconvolution (Fig. 5i) and increased in the distribution of tumor infiltrating T cells by immunohistochemistry analysis [4.7% (q25−75: 2.9 – 7.3) vs. 18% (q25−75: 14.65 – 18.9), vehicle vs. azacitidine, P < 0.001 Mann-Whitney test, Fig. 5j]. Overall, these data indicate that cytosine methylation patterning of lymphoma cells may contribute to a depleted LME.
LME heterogeneity decreases with disease progression.
To explore the notion that LME cellular composition changes during evolution of lymphomas, we first established a series of temporally-ordered conditions representing early and late stages of lymphoma development. In each sample we established the LME composition by transcriptome signature deconvolution and measured its complexity by Shannon entropy metric, which has two components: the number of unique cell subtypes (richness) and their equality of distribution (evenness). The Shannon metric directly corresponds with the LME cellular heterogeneity. We obtained paired samples from diagnostic and relapsed DLBCL (n = 1), indolent FL and transformed(t) FL (n = 5), early and late murine lymphoma progression (n = 1) and pre-malignant lymph node and nodal lymphoma from genetically engineered mice models expressing in GCB cells Myc + Bcl2, Ezh2 mutant + Bcl6 or Kmt2d + Bcl2 (n = 3) (Fig. 6a). We observed that the complexity of the LME, measured as the Shannon entropy, decreases with lymphoma development of progression by approximately 40% (95% CI: 20–60%, p = 0.001, paired T-test) (Fig. 6a). This effect is more notorious in the decreasing intratumoral content of T cells, that agrees with recent data demonstrating a significant reduction in tumor T cell infiltration in paired diagnostic compared to relapsed DLBCLs(40).
Figure 6. Changes in LME cellular composition during lymphoma progression.
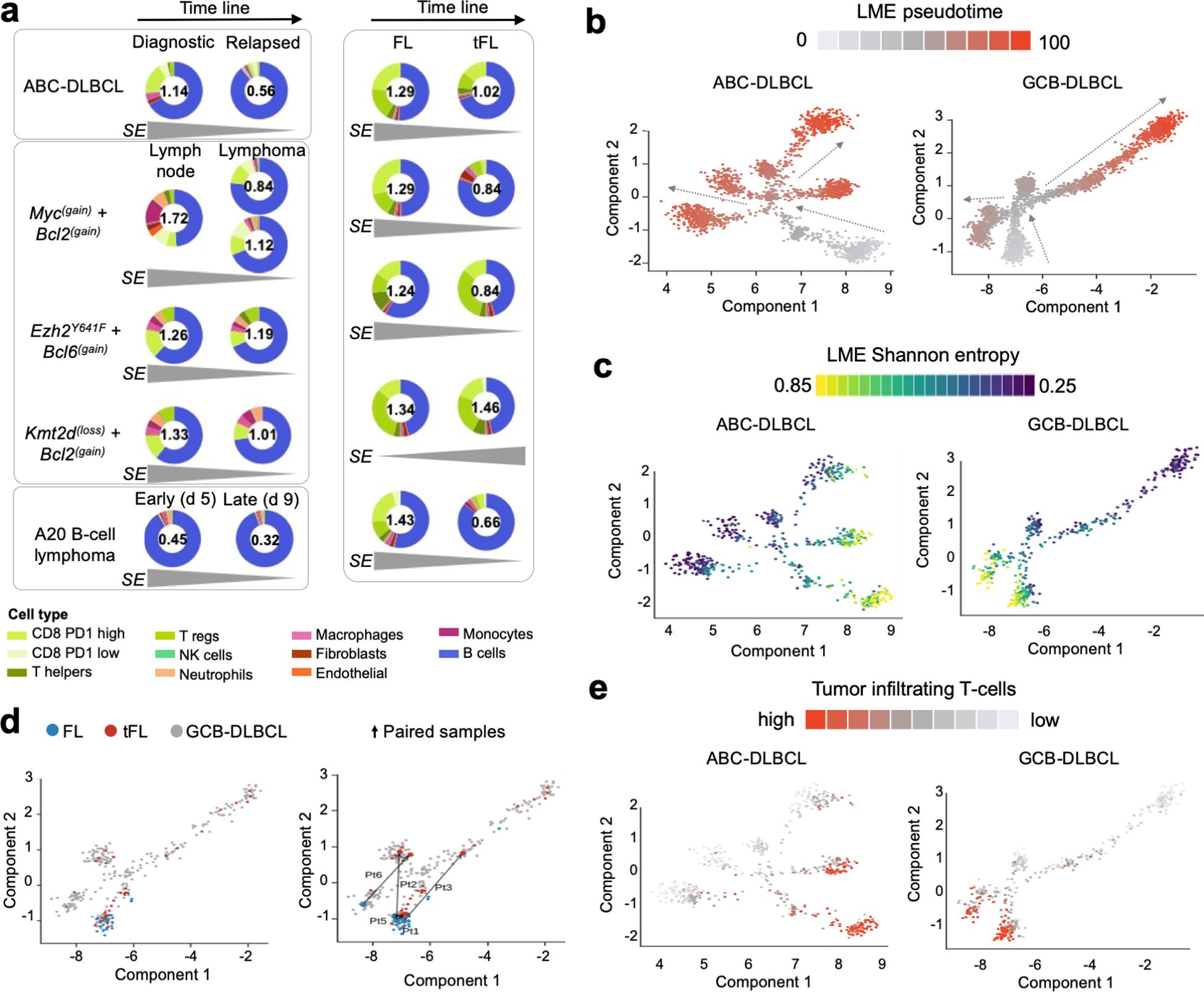
a. LME cellular heterogeneity measured as Shannon entropy (SE) as function of lymphoma progression. Early and late time points include: matched ABC-DLBCL primary and relapsed case, three GEMMs comparing pre-malignant lymph node niches and lymphoma niches, A20 syngeneic B-cell lymphoma model at days 5 and 9 after implantation and five paired indolent FL phase and at the moment of transformation. The SE metric is shown inside every donut plot and the tendency over time is shown at the bottom for each pair. The proportion of cell populations including B-cells are represented in the donut plot. b. Pseudotime analysis of LME progression in ABC and GCB-DLBCLs. c. LME Shannon entropy analysis imposed on the LME pseudotime analysis. d. LME of FL, tFL and de novo GCB-DLBCLs imposed on the LME pseudotime of GCB-DLBCLs (left). Paired FL-tFL samples (n = 5) shown on the LME pseudotime of GCB-DLBCLs (right). e. Analysis of proportion of tumor infiltrating T cells imposed on the pseudotime analysis of BC and GCB-DLBCLs. Density of T cells is shown as color scale.
To characterize the tendency of the LME to immune desertion as function of lymphoma progression in a bigger scale, we applied a pseudotime algorithm to reconstruct LME temporal dynamics using individual samples as points in this trajectory. Using the space of the 25 FGES we projected 2,128 ABC- and 2,867 GCB-DLBCL samples (Fig. 6b). The immune rich GC-LME that resembles the cellular constitution of the germinal center microenvironment(14) was considered as the starting point. The LME Shannon entropy for individual DLBCL samples demonstrated an inversed association with the pseudotime (Figs. 6c and S10a, R = −0.51, p < 0.001) and with the epigenetic age(41,42) of the lymphoma (Fig. S10b, MethylAger R = −0.52, p < 0.001 and epiCMIT R = −0.58, p < 0.001). The projection of FL (n = 37), tFL (n = 33) and de novo DLBCL (n = 267) from two cohorts onto the GCB-DLBCL LME pseudotime also demonstrated an association between lower LME complexity and higher LME pseudotime (Fig. 6d). Furthermore, the projection of five paired FL and tFL samples also showed this tendency (Fig. 6d). Similar to lymphoma paired samples, the LME pseudotime progression was associated with a decrease in the tumor infiltrating T cells (Fig. 6e). This notion could indicate that lymphoma cells may acquire or select for mechanisms decreasing T cell infiltration resulting in immune deserted LMEs.
CAFs produce a lymphoma-restrictive ECM.
In the tumor microenvironment, the ECM is primarily produced and remodeled by the balanced activity of TAMs and CAFs. We thus determined whether the ratio of FGES for TAM vs. CAF impacts prognosis. The TAM/CAF ratio associated with higher risk of death (HR: 9.1), an effect observed to some degree in all LME subtypes but that was particularly strong in DP-LME with a HR: 4.4 (Fig. 7a). To determine which ECM components are more likely to be associated with the activity of CAFs vs. TAMs, we conducted unbiased proteomic analysis of the ECM (matrisome, Fig. 7b) of 18 primary DLBCLs, using the matrisome of inflamed tonsils as a control for normalization. We have enough material in 14 of these DLBCL to conduct RNA-sequencing that we use to estimate TAM/CAF, LME and COO. We identified 131 proteins in the DLBCL matrisome distributed as glycoproteins, ECM regulators, collagens, ECM-affiliated, secreted factors and proteoglycans (Table S7); clustering in three matreotypes (Fig. 7c). Two matreotypes (termed Fm1 and Fm2) associated with higher proportion of CAFs and had higher proportion of collagens and proteoglycans (Fig. 7c); whereas the third matreotype containing a higher proportion of ECM-affiliated and secreted proteins had a higher expression of TAMs (thus termed Mm, Fig. 7c). Reflecting on the low expression of FGES, DP-LME DLBCL had relatively low ECM abundance (Fig. 7c). To dissect cellular contributors to the DLBCL ECM, we correlated the matrisome genes with their median expression in 15 LME cell subtypes and DLBCL cells to obtain their probability of expression according to cell type (Fig. S11a, representative examples). The DLBCL matrisome genes were then ranked from the highest probability of expression in fibroblasts vs. macrophages to obtain their respective contributions to the ECM (Fig. 7d and S11b). Collagens and the proteoglycans decorin (DCN) and biglycan (BGN) were among the strongly associated with the CAFs (Fig. 7d). Conversely, the ECM protein abundance of these collagens and proteoglycans significantly correlated with the estimation of CAFs by FGES (R = 0.88, p = 0.001, n = 9 pts) and fibroblasts proportion by signature deconvolution (R = 0.9, p < 0.001, n = 9 pts) (Fig. S11c). Accordingly, CAF-associated proteins were enriched in the portion of the DLBCL matrisome that correlated with a lower TAM/CAF ratio in 14 patients (Fig. 7e). Lastly and similar to CAF FGES, the expression of the CAF’s ECM proteoglycans DCN and BGN were associated with favorable prognosis, particularly with DLBCL LME-DP patients (Fig. 7f,g).
Figure 7. The DLBCL ECM produced by CAFs and TAMs influences lymphoma biology.
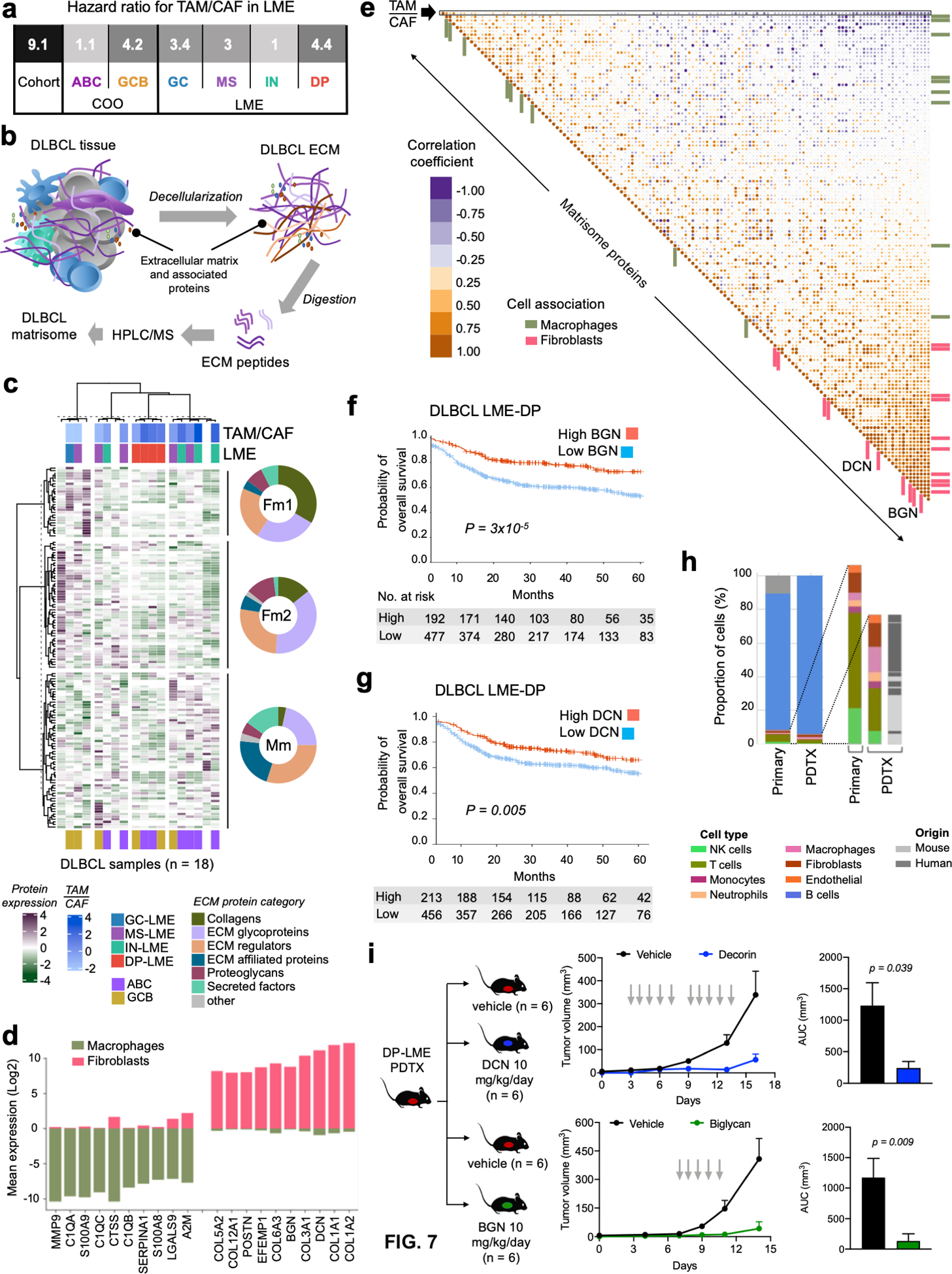
a. Probability of overall survival (OS) for the ratio of CAF/TAM proportions in the LME of DLBCLs for the full cohort, COO classes and LME subtypes. b. Graphical representation of the DLBCL matrisome experiments c. Matrisome heat map of 18 DLBCL samples (x-axis) and 136 proteins (y-axis) by unsupervised hierarchical clustering. Donut plots show the breakdown of the ECM proteins category for each of the three matreotype clusters termed Fm1, Fm2 and Mm. For the 14 samples with available RNA-seq, the TAM/CAF ratio, LME cluster and the COO are shown. d. Top-10 and bottom-10 transcripts of matrisome proteins according to their relative abundance in fibroblasts and macrophages. e. Pearson’s correlation matrix of TAM/CAF ratio and the 136 matrisome proteins. Proteins associated with fibroblasts and macrophages (from d) are depicted with distinct color bars and decorin (DCN) and biglycan (BGN) are shown. f-g. Kaplan-Meier model of OS according to BGN (f) and DCN (g) abundance for DLBCL LME-DP patients. h. Cellular composition by transcript deconvolution of primary DLBCL LME-DP and its PDTX model. Expansion of the LME cellular composition in the primary and PDTX model and indication whether these cells in PDTX are of human or mouse origin. i. DLBCL growth curve (left) and area under the curve (AUC) of tumor volume (right) of a DLBCL LME-DP PDTX treated with vehicle vs. DCN (top) or vehicle vs. BGN (bottom).
By a combination of short and long-range effects(43), microenvironmental proteoglycans have the potential to influence tumor growth. To determine their effects in DLBCL, we first developed two PDTX models from DP-LME ABC-DLBCL patients that conserved these characteristics in the mouse (Fig. 7h and S11d). Once the PDXs were fully established, we injected the mice intraperitoneally with vehicle, DCN or BGN and followed the tumor growth curve. We found that both proteoglycans significantly delayed the progression of the tumor vs. vehicle (p = 0.009 and p = 0.039, for BGN and DCN, respectively in PDTX1 and p = 0.0004 and p = 0.01 for BGN and DCN, respectively in PDTX2, Fig. 7i and S11e). Proteoglycans did not show a statistically significant anti-proliferative effect in isolated PDX-derived lymphoma cells. Although there was no evidence of direct incorporation of the administered proteoglycans into the PDTX ECM, BGN administration resulted, as previously described(44), in 20% increase in DCN content (p = 0.032, vs. vehicle) in the PDTX2 ECM. Overall, this suggest that the anti-proliferative effect of these proteoglycans results from both short- and long-range interactions.
DISCUSSION
We characterized the microenvironment of DLBCL by analyzing a large collection of gene expression profiles and technical innovations, including development of microenvironment-derived functional gene expression signatures, analysis of the ECM composition by proteomics and establishment of PDTX models. Similar to cell-deconvolution algorithms, our methodology reflects the cellular composition of the microenvironment but also provides valuable information on its functionality. This methodology allowed us to describe four basic categories of DLBCL LME with distinct clinical and biological connotations. In addition, by overlaying mutations and epigenetic alterations in lymphoma cells with the temporal ordering of LME changes, we were able to infer potential mechanisms of cancer cell-LME co-evolution. Lastly, we identified novel potential therapeutic targets present in the LME, including ECM proteins exemplified by the proteoglycans DCN and BGN.
Our study represents a comprehensive analysis of the LME in DLBCL that simultaneously integrates characteristics of microenvironmental and malignant cells populations into the prognosis of the disease. Pioneering transcriptomic studies that examined host factors and the stroma in DLBCL identified the microenvironment as a relevant component of disease biology(10,15,45). These initial studies relied on identifying differences in gene expression profiles of tumor samples(10,15,45). Our study refined this analysis be extracting microenvironmental signatures from the confounding transcriptome background of the usually most abundant lymphoma cells, which enabled the identification and quantification of microenvironmental cells within the bulk tissue transcriptome(46). We further optimized this technique to include functional signatures that include the activity of certain cell types such as the T cell and NK cell cytotoxicity as well as pathways not uniquely associated with a cell type such as ECM remodeling and cytokine secretion. The association of these functional signatures suggested the presence of functional cellular ecosystems within the LME and offered a comprehensive understanding of the role of the microenvironment in this disease. This approach provided more biologically relevant evidence than that obtained from the quantification of individual cell populations. Remarkably, the LME mirroring the GC microenvironment confers a better prognosis than the LME depleted of microenvironmental cells, suggesting that analogous to the molecular checkpoints operating in centroblasts(47,48), the microenvironment may provide primordial mechanisms to avoid lymphomagenesis. In turn, lymphoma cells develop and/or select genetic and epigenetic traits that contribute to the evasion of immune microenvironmental constraints(19). Our data suggests that for initially immune-rich DLBCLs, disease progression tends to associate with an immune deserted LME. More broadly, this notion suggests that the co-evolution of lymphoma cells and LME cells (likely also involving non-immune LME cells) progresses towards a state with lower heterogeneity in these two compartments, a concept that remains to be experimentally validated as more LME cell subpopulations are described.
In addition to the restriction imposed by the immune cells to lymphoma development(49), our work suggests that the ECM has also a critical role. The ECM is a network of physically and biochemically distinct macromolecules, comprised of mostly fibrillar proteins, proteoglycans and glycoproteins, that are central to the structural integrity of tissues and the behavioral regulation of malignant and microenvironmental cells. We described that LMEs presenting with a higher proportion of ECM components derived from CAFs (i.e., a CAF matreotype) rather than from TAMs have a favorable prognosis. Some ECM-derived proteins, particularly the small leucine rich proteoglycans(50) BGN and/or DCN, exert short and long-range effects that ultimately impact on lymphoma cell proliferation, as demonstrated in our PDTX experiments. Since the PDTXs were developed in immunocompromised mice, the mechanism appears independent of the establishment of an adaptive lymphoma immune response. Along these lines, previous work in DLBCL has established that CAFs were associated with a possible better outcome(51). In addition, through the secretion into the interstitial matrix of soluble factors like TGFB ligands(52), fibroblasts may contribute to curtailing lymphoma cell progression. A mechanism that can be overcome in DLBCL cells by the epigenetic repression of TGFB transducers such as SMAD1(23,24). The anti-lymphoma role of CAFs, and of TGFB, in DLBCL is in contrast with their pro-tumorigenic role described for the majority of solid tumors(52,53), underlining the need to study the microenvironment in a tumor-specific manner.
In cohort analysis, HGBL-DH patients harboring translocations of MYC and BCL2 as well as DLBCL expressing high molecular grade transcriptional signatures have unfavorable prognosis after frontline therapy, which often justifies the implementation of intensive chemotherapy regimens(16,54). However, at the individual level, long-term responders to standard chemoimmunotherapy do exist, suggesting the presence of a sub-group of less aggressive lymphomas. Although this phenomenon could be explained by the presence of additional genomic and epigenomic alterations(16), our data indicate that distinct biological behavior associates with unique LME categories. HGBL-DH and other molecular high-grade DLBCLs exhibit a significantly better outcome within GC-like and MS LMEs than within IN and DP LMEs. In light of the chemoresistance associated with this lymphoma sub-type, new approaches are considering the addition of targeted therapies such as BCL2 inhibitors(54). The results of our studies suggest that, in clinical trials, targeted agents should be considered not only in the context of particular genetic subtypes but also in consideration of LME categories.
In summary, we defined the LME into four major transcriptionally-defined categories with distinct biological properties and clinical behavior, which complements gene expression subgroups and genetic subtypes of DLBCL in guiding the development of rational therapeutic approaches for these patients.
MATERIALS AND METHODS
Human subjects.
All patients providing samples gave written informed consent. Molecular and clinical data acquisition and analysis and patient-derived tumor xenograft (PDTX) establishment were approved and carried out in accordance with Declaration of Helsinki and were approved by Institutional Review Boards of the New York-Presbyterian Hospital, Weill Cornell Medicine (WCM), New York, USA and Ospedale San Giovanni Battista delle Molinette, Turin, Italy.
Clinical characteristics.
All the DLBCL patients in the WCM cohort were treated with R-CHOP. Response categories were defined as complete response when patients remained free of disease at 5 years, non-responders refractory when complete response was never achieved and non-responders relapsed when they remained free of disease for less than 2 years. All the biopsies for molecular analysis were taken before any treatment at the moment of diagnosis. From the publicly available cohorts, we considered for outcome analysis all gene expression DLBCL datasets with annotated clinical and outcome data including response by RECIL criteria(55), progression-free survival (PFS) and/or overall survival (OS) of patients treated with rituximab-containing chemoimmunotherapy regimens. We excluded data sets in which the OS and/or PFS Kaplan-Meier curves were not representative of unselected DLBCL patients treated with standard of care chemoimmunotherapy and/or datasets with less than 30 samples. Survival analysis was conducted using the Lifelines Python package.
RNA-sequencing.
RNA was extracted from fresh-frozen DLBCL samples maintained in TRIzol (Invitrogen) and libraries (PE 50 or 100 bp) were prepared using the TruSeq RNA sample kit and validated using the Agilent Technologies 2100 Bioanalyzer. Library preparation, sequencing and post-processing of the raw data was conducted at the WCM Epigenomics Core Facility on Illumina HiSeq2500. This dataset (WCM cohort) was combined for normalization and aggregated into training and validation cohorts as described in the specific analysis.
Compilation of a transcriptomic LME dataset.
Publicly available DLBCL gene expression (GE) datasets and our own dataset were compiled into a unique dataset (LME dataset) comprising a total of 4,655 samples (Fig. S2a). Only cohorts with 30 or more lymphoma samples were included. Since our goal was to obtain information on the tumor microenvironment, GE data obtained from selected or isolated lymphoma cells were excluded from this analysis. For comparison, GE cohorts containing HGBL, FL, BL and PMBL samples were also considered. Clinical annotation, pathological information and molecular data were curated and harmonized among all included datasets. Only DLBCL and HGBL patients treated with rituximab-containing regiments were considered for response and outcome comparisons. Our WCM cohort included a balanced number of R-CHOP responsive and non-responsive patients. RNA-sequencing data processing: new and deposited RNA-sequencing reads were processed using a unified pipeline(56). Reads were aligned using Kallisto v0.42.4 to GENCODE v23 transcripts 69 with default parameters. Protein coding, IGH/K/L- and TCR-related transcripts were retained whereas noncoding, histone- and mitochondrial-related transcripts were removed resulting in 20,062 analyzed transcripts. Gene expression was quantified as transcripts per million and log2 transformed. Almost all RNA-sequencing cohorts were sequenced using poly-A enrichment and were combined into the LME dataset. GE microarray data processing: raw and processed data were downloaded from GEO. When possible, expression was reprocessed from raw files. All Affymetrix datasets with available CEL files were re-normalized using the gcRMA package with default parameters. Illumina array probes were converted into genes using one probe with the highest mean values in the cohort per gene. Datasets with more than one platform were spit by platforms. Samples with low Pearson correlation with other samples in the space of all genes (< 0.8 for Affymetrix platforms and < 0.7 for Illumina platforms) for each cohort were excluded as well as outliers according to the principal component analysis (PCA) projection. Microarray GE datasets were combined into the LME dataset if they were obtained in the same institution, on the same platform, had no significant batch effects on PCA projection in the LME signature space and had no significant differences in outcomes.
Purified cell type compendium collection.
We collected 5,069 RNA-sequencing gene expression datasets from purified cell populations including normal and lymphoma cells (DLBCL, FL and BL) from public data sources (NCBI GEO(57), SRA(58), ENA, Array Express, Protein Atlas(59), BluePrint, ImmPort) to create a cell gene expression compendium. We included datasets using the following criteria: isolated from human tissue, poly-A or total RNA-sequencing performed with read length higher than 31 bp, having at least 4M of coding read counts, passed quality control by FASTQC and no contamination with other cell types detected (< 2%).
Development of LME gene expression signatures.
We developed functional gene expression signatures (FGES) of tumor microenvironmental cells, cellular states, physiological and pathological processes and signaling pathways using a combination of gene expression signatures and literature curation. FGES for cell types were obtained from purified gene expression cell population signatures manually curated to include only those genes that are exclusively expressed in the defined cell type or specifically associated with particular biological process using data from more than 500 publications. The association of gene expression profiles from 5,009 cell populations with the curated signatures was determined by tSNE projections and Mann-Whitney tests. Signature scores were calculated using in-house python implementation of the ssGSEA(60). Some cellular FGES were also validated using publicly available single cell RNA-sequencing data(61). The activity scores of biological pathways were calculated using PROGENy (Pathway RespOnsive GENes)(62). As result, 25 FGES (including 3 signaling pathways) were developed (Table S1). LME clustering: FGES signatures were used to identify microenvironmental patterns among DLBCL gene expression samples by unsupervised dense clustering using the Louvain method for community detection(63). FGES intensities were median-transformed within each cohort. Non-DLBCL samples were also transformed using DLBCL samples’ median and MAD values. Inter-sample similarity was calculated using Pearson correlation. The resulting distance matrix was converted into a graph where each sample formed a node and two nodes formed an edge with weight equal to the pair’s Pearson correlation. Edges with weight lower than specified thresholds were removed and the Louvain community detection algorithm was applied to calculate graph partitioning into clusters. To mathematically determine the optimum threshold for observed clusters we employed minimum David Bolduin, maximum Calinski Harabasz and Silhouette scores excluding separations with low-populated clusters (< 5% of samples).
Development of a COO classifier.
Training samples (n = 1,968) were obtained from the datasets GSE117556, Leipzig Lymphoma(64), GSE31312, GSE10846, GSE87371, GSE11318, GSE32918, GSE23501, LLMPP and GSE93984 data sets. For each data set, we selected samples having COO labeling followed by another round of random selection to obtain a balanced COO ABC:GCB ratio of 40:60 per dataset. Validation samples (n = 928) with COO labelling were selected from the datasets GSE34171 (GPL96+GPL97), BCA (DCL1 + DCL2)(19), GSE22898, GSE64555, WCM (GSE145043), GSE19246 and the NCICCR data sets. A final data set of samples originally considered COO unknown or unclassified (n = 1,169) was aggregated from COO unlabeled samples included in the previous data sets plus the GSE69051, GSE69049, E-TABM-346, GSE68895, GSE38202, GSE12195, ICGC_MALY_DE and NCICGCI data sets. To eliminate platform and dataset batch effects without introducing scaling batch effects we applied rank transformation to each sample independently as follows: Expression Geneset A = [ x1,x2,x3…xN-1, xN ] sorted ascending [x2, x15,xN-1…x1, xN] [1, 2, 3 … N-1, N]. Missing genes were omitted in the rank transformation and genes with equal values were assigned with the average rank. To perform binary classification, we used a gradient boosting decision tree classifier in LightGBM. To perform feature selection, we estimated the feature importance to the model using Shap package (https://github.com/slundberg/shap). To develop a classifier the initial geneset was curated from(65). We conducted recursive feature elimination until 32 features were left and a set of genes with the best cross-validation was chosen (Table S2).
Development of DHIT-signature classifier.
The initial geneset of 104 genes was curated from(16). The same procedure applied to the COO classifier was performed with slight modifications. The BCA (DLC1+DLC2) cohort (n = 157 GCB-DLBCL samples) was split into training and validation samples. Due to class disbalance we applied a weighted f1-score during all procedures. Feature selection resulted in 20 genes (Table S3). The classifier was applied to all the GCB-DLBCL samples from the final dataset. For comparison, the original DHITsig score was calculated in each data set using coefficients from the original publication(16) followed by median transformation of the score for each data set and combined.
Other gene expression signatures.
The CYT score for cytolytic activity was calculated as previously described(27), values were then clipped using 5% lower and upper quantiles, projected to [0, 1] and combined. The stromal-1 and stromal-2 signatures were calculated using ssGSEA and then median transformed within each cohort.
LME heterogeneity and pseudotime analysis.
Shannon diversity indexes were calculated from the LME cell deconvolution profiles. To calculate the LME heterogeneity index for each tumor, we performed cell deconvolution on the bulk RNA-sequencing samples from tumor tissue. From this prediction, we calculated the estimated proportion (p) of cells that belong to each distinct cell type. The subpopulation diversity index was then calculated as Shannon Index: DI = −Σi(pi × lnpi), with larger values representing higher LME heterogeneity within the lymphoma. The monocle 2.0 R package(66) was implemented for dimensionality reduction and the construction of pseudotime. All samples were assigned COO subgroups and LME categories. Dimension reduction was run with DDRTree method(67) and the pseudotime was set with GC-like LME as zero point. This analysis was performed thrice with same hyperparameters for samples classified as ABC-DLBCL (n = 2,128) and GCB-DLBCL (n = 2,867). Then, the projection was used to display Shannon entropy and tumor infiltrating T cells. Paired FL-tFL RNA-seq samples (n = 6) were obtained from GSE142334 and only samples with matched HLA within pairs were included.
Deconvolution of cell percentages from bulk DLBCL transcriptomes.
A recently described algorithm was used for cell deconvolution(22). Briefly, the sorted cell population compendium was used to develop a machine learning based cell deconvolution algorithm to calculate the percentage of different cell types from bulk RNA-sequencing mixtures based on the minor difference between cell subpopulations. We used a two-stage hierarchical learning procedure for gradual boosting a LightGBM model that included training on an artificial RNA-sequencing mixtures of different cell types including immune and stromal cell populations. Artificial RNA-sequencing mixtures were created by admixing different datasets of sorted cells together in various cell proportions and the LightGBM model was trained to predict the admixed cell percentage. Then, the model was used to reconstruct proportions of cell subpopulations using the information from the proportion of the major cell populations and subpopulations. The algorithm estimates the RNA proportion of a cell type in bulk RNA-sequencing mix of a sample, which could be converted into cell percentage if the RNA concentration of a cell type was known(68). Total RNA abundance in isolated cells was quantified using Qubic.
Mass cytometry (CyTOF) and immunohistochemistry analysis.
Inflamed tonsils were procured from the WCM-New York Presbyterian Hospital as pathology discards. Tonsils were mechanically dissociated and incubated in a digestion buffer (25 mg/mL collagenase A, 25 mg/mL dispase II, 250 mg/mL DNAse in a solution of 140 nM NaCl, 5 mM KCl, 2.5 mM phosphate buffer saline pH 7.4, 10 mM HEPES, 2 mM CaCl2 and 1.3 mM MgCl2) until a single cell suspension was obtained. Homogenous fresh cell suspensions were divided for RNA-sequencing and CyTOF analyses. Briefly, cells for CyTOF analysis were barcoded and pooled for incubation with FcR blocking reagent (Miltenyi Biotech), stained with 300 μl of the antibody panel per 107 cells and resuspended in nucleic acid Ir-intercalator (Fluidigm). Samples were analyzed at the Mass Cytometry Core of WCM. Data was processed and analyzed as previously described(69). Clustering analysis was performed using the Python implementation of PhenoGraph run on all samples simultaneously and then manually curated into cell populations. CAFs and TAMs were identified by immunohistochemistry using anti-SMA and anti-CD68 antibodies, respectively, and quantified using QuPath(70).
Genetic alterations analysis.
Whole exome sequencing (WES) and whole genome sequencing (WGS) next generation sequencing (NGS) quality control analysis was performed using FastQC v0.11.5, FastQ Screen v0.11.1 and MultiQC v1.6. Sample correspondence was checked using HLA comparison and the Conpair algorithm(71). For WES low quality reads were filtered using FilterByTile/BBMap v37.90 and aligned to human reference genome GRCh38 (GRCh38.d1.vd1 assembly) using BWA v0.7.17. Duplicate reads were removed using MarkDuplicates v2.6.0, indels were realigned by IndelRealigner and recalibrated by BaseRecalibrator (both of GATK v3.8.1). Variant calling: germline and somatic single nucleotide variations, small insertions and deletions were detected using Strelka v2.9 and annotated using Variant Effect Predictor v92.1. Copy number alterations (CNA) were evaluated by customized version of Sequenza v2.1.2. Translocations from WGS samples were called using DELLY (v0.8.1)(72) and annotated with AnnotSV (v2.2)(73). Public genomic data: whole genome mutations were available for NCICCR, WCM and GSE98588/phs000573 cohorts. Targeted mutations were available for the GSE117556 (70 genes), GSE22898 (322 genes), BCA/DLC2 (144 genes) and GSE87371 (34 genes) cohorts. Whole genome CNA calls were available for NCICCR, WCM, GSE34171_1 and GSE98588 cohorts. Association of LME with specific genetic alterations: NCICCR, BCA and WCM cohorts were used to analyze disbalances in the proportion of mutations in relation to LME categories. NCICCR, WCM and GSE87371 cohorts were used to analyze disbalances in the proportion of CNAs in relation to LME categories. Ploidy as accessed using weighted-mean and reported as decimal number. The gene-level CNAs were obtained intersecting segments with hg19 gene coordinates using BEDTools(74). Gene CNAs were analyzed relative to ploidy levels. Additionally, we curated mutations and CNAs for selected genes including MYD88, CD79B, EZH2, CREBBP, EP300, KMT2D, TP53, B2M, CIITA, GNA13, CCND3, GNAI2, P2RY8, CD70 and CDKN2A, across all datasets and platforms.
Tumor clonality.
To process immunome data from RNA sequences into quantitated clonotypes we applied MiXCR v2.1.7(75). Single clonotypes were grouped into clones with unique VDJ combination and identical CDR3 nucleotide sequences. For B cells the clones were further aggregated into clone groups if the VDJ combination was the same and CDR3 nucleotide sequences differed no more than 1 nucleotide. The biggest clone group was assigned as tumor if the absolute clonotypes counts > 20; the relative clonotypes counts > 5%; the ratio of the second biggest group to the first < 0.6 and the group contains an enriched clone > 25%. The tumor light chain was called if there was an enriched clonotype in one of the light chains. In cases with an enriched clone in both chains the biggest by absolute counts was selected.
DNA methylation analysis.
Library preparation, sequencing and post-processing of the raw data were performed at the WCM Epigenomics Core Facility following the enhanced reduced representation bisulfite sequencing method (eRRBS)(76). Briefly, DNA was digested and then ligated with 5-methylcytosine-containing indexed Illumina adapters. Adaptor ligated DNA fragments were size selected and bisulfite converted using the EZ DNA methylation Kit (Zymo Research, Irvine, CA). Amplification was performed on size selected DNA fractions (150 to 250 bp and 250 to 400 bp) and purification steps were done using Agencourt AMPure XP (Beckman Coulter, Brea, CA) beads. Libraries were sequenced on Illumina HiSeq2500. Bisulfite reads were aligned to the bisulfite-converted hg19 reference genome using Bismark(77). In addition to the WCM cohort (GSE145043), two publicly available datasets were used in this analysis (TCGA and GSE23967). To estimate CIMP values we selected probes located in CpG islands in gene promoters (i.e. regions TSS200, TSS1500, 5’UTR and first exon) excluding the genes located in chromosomes X and Y. For CIMP scores we selected unmethylated probes (i.e. 95th percentile < 0.2) from 60 normal immune cells subtypes from dataset GSE35069. Probes that were methylated (beta-value > 30%) in less than one third of tumor samples were also excluded, yielding 8,137 informative probes that were used to compute CIMP scores as their mean methylation values for samples included in the datasets TCGA and WCM. The datasets GSE23501 (gene expression) and GSE23967 (HELP methylation assay) were used to validate SMAD1 promoter methylation on one available probe and TGFB pathway activity.
Mice experiments.
All the experiments were conducted under approval of the WCM Institutional Animal Care and Use Committee at the Research Animal Resource Center. Patient derived tumor xenografts (PDTX) were established in female and male NSG B2M mice (NOD.Cg-B2mtm1Unc Prkdcscid Il2rgtm1Wjl/SzJ) and then serially propagated into NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, Jackson Laboratory). PDTX were analyzed by RNA-sequencing to determine whether molecular features from the original lymphoma were maintained in the xenografted tissue. As part of the PDTX characterization, PDTX implanted in NSG mice were treated with CHOP chemotherapy to determine whether the original patient response was reproduced in the mice. Clonal correspondence was proven via BRC rearrangement analyses. Molecular and therapeutically characterized PDTX were subsequently implanted in NSG mice to assess the anti-tumor effect of proteoglycans. Once tumors reached a palpable size, mice were randomized to receive vehicle (PBS), decorin (R&D Biosystems) 0.4 mg/kg intraperitoneally or biglycan (R&D Biosystems) 0.4 mg/kg intraperitoneally according to the schedule shown in the figures. Mice were treated daily in a weekend-off schedule and tumor sizes were recorded every other day. Body weight was used as surrogate for drug toxicity. PDTX establishment, characterization and treatment were conducted at the WCM, Mayer Cancer Center, PDTX Core Facility. Murine lymphoma model: mouse B-cell lymphoma A20 cells were subcutaneously implanted in the inguinal lymph node pad into female and male BALB/c mice (at least 8-week old) and once tumors were fully established and palpable, mice were randomized to receive either vehicle (saline solution) or azacitidine 12.5 mg/kg/day by subcutaneous injection for four consecutive days. Authenticated A20 cells were purchased from the ATCC repository (TIB-208). We routinely conducted analysis of Mycoplasm sp. in cellular cultures by PCR. At the end of the treatment, mice were sacrificed and tumors extracted for analysis. Transcription and DNA methylation analysis of tumors: RNA was isolated according to the kit manufacturer’s instructions (Zymo Quick-RNA Miniprep) from cryopreserved A20 tumors and from bio-banked cryopreserved whole tissues (spleen and tumors) for genetically engineered lymphoma mice models. The Illumina TruSeq Stranded polyA Library Prep Kit was used for cDNA preparation. Sequencing was performed using an Illumina NovaSeq 6000 SP Flowcell at WCM Genomics Resources Core Facility. Transcripts were quantified against Gencode mm38 genome using Salmon(78). Transcript abundances were summarized with tximport in R version 3.6.2. DESeq2(79) was used for differential expression analyses. DNA was isolated according to the kit manufacturer’s instructions (Zymo Quick-DNA Miniprep). Library preparation, sequencing and post-processing of the raw data were performed at the WCM Epigenomics Core Facility following the eRRBS method. Libraries were sequenced on Illumina NovaSeq 6000 SP Flowcell. Bisulfite reads were aligned to the bisulfite-converted mm10 reference genome using Bismark(77) and differential methylation was assessed using MethylKit(80). Briefly, promoter regions were defined as 2.5 kb upstream and 0.5 kb downstream of the TSS, and differential methylation was calculated on those regions, with a minimum number of covered bases of 3. Tumor tissues were processed for paraffin embedding and CD3 immunohistochemistry analysis at the WCM Comparative Pathology Core Facility. CD3 stained cells were quantified using ImageJ software.
Analysis of murine lymphoma microenvironment signatures.
We used Kallisto v.0.42.4(81) to align RNA-sequencing reads to the transcriptome reference GRCm38.p6. Transcript annotation (protein-coding and non-coding), transcript to gene mapping and annotation to human homologs for murine genes were retrieved from the Ensembl database (release 96)(82). Only protein-coding genes with human homologs were used in subsequent analysis. To measure the similarity between the human lymphoma sample microenvironment and a particular murine lymphoma phenotype, we developed the Microenvironment Similarity (MES) metric which is a reversed Euclidean distance between estimated percentages of different cell types in murine and human samples as follows: MES (P, s) = 1i(phumani- pmousei)2, where P is a murine lymphoma phenotype; s is a human lymphoma sample; phumani is the estimated cell percentage of cell type i in the sample s; pmousei is the median estimated cell percentage of cell type i among murine samples with phenotype P.
Proteomics and matrisome analysis.
Lymphoma tissues were processed based on a modified decellularization protocol(83) using 3% peracetic acid and incubated in a 1% Triton X-100, 2 mM EDTA solution for 27 h. Then, tissues were successively incubated in distilled water for 24 h followed by 600 U/ml DNAse in PBS for 24 h and followed by distilled water for 72 h. Tissue fragments were then dried using a vacuum filtration system and digested and prepared for tandem mass tag (TMT) mass spectrometry analysis using a modify protocol(84). Briefly, samples were resuspended in 8 M urea and subjected to reduction with DTT and alkylation with iodoacetamide. After that, samples were diluted to 2 M urea with 25 mM ammonium bicarbonate and incubated with PNGase F for 2 h at 37°C. Lys-C were added for 2 h incubation at 37°C after deglycosylation. Trypsin was added for overnight digestion at 37°C. The resulting peptides were desalted by C18 Stage-tips. Lyophilized peptides were then labeled with the TMT reagents (Thermo Fisher Scientific) according to manufacturer’s protocol. Labeled peptides were mixed, lyophilized, desalted prior to LC-MS analysis. An EASY-nLC 1200 coupled on-line to a Fusion Lumos mass spectrometer was used (Thermo Fisher Scientific). Buffer A (0.1% formic acid in water) and buffer B (0.1% formic acid in 80 % acetonitrile) were used as mobile phases for gradient separation. A 75 μm I.D. column (ReproSil-Pur C18-AQ, 3 μm, Dr. Maisch GmbH, German) was packed in-house for peptides separation. Peptides were separated with a gradient of 5–10% buffer B over 1 min, 10%−42% buffer B over 231 min, and 42%−100% B over 5 min at a flow rate of 300 nL/min. Full MS scans were acquired in the Orbitrap mass analyzer over a range of 400–1500 m/z with resolution 60,000 at m/z 200. Top 15 most abundant precursors were selected with an isolation window of 0.7 Thomson and fragmented by higher-energy collisional dissociation with normalized collision energy of 40. MS/MS scans were acquired in the Orbitrap mass analyzer. For protein identification, raw files were processed using the MaxQuant computational proteomics platform version 1.5.5.1 (Max Planck Institute, Munich, Germany) and the fragmentation spectra were used to search the UniProt human protein database. Oxidation of methionine and protein N-terminal acetylation were used as variable modifications for database searching. Both peptide and protein identifications were filtered at 1% false discovery rate based on decoy search using a database with the protein sequences reversed. To identify the abundance of ECM proteins, we used the matrisome database to find the overlapping proteins between the ones identified from the tissues and the ones present in the database of identified human ECM proteins (http://matrisomeproject.mit.edu). As a control to normalize between subsequent mass spectrometry runs, we used decellularized human tonsils from one individual and computed the ratios of the abundance of proteins found in the DLBCL tissues to the abundance found in the tonsils. Experiments were conducted at the WCM Metabolomics and Proteomics Core Facility. For samples with matched matrisome-transcriptome data, the RNA was extracted using Qiagen RNeasy kit following the manufacturer’s instructions. The sequencing was performed using an exome capture protocol and libraries were constructed using Illumina RNA-Prep with enrichment (tagmentation kit with dual indexes) according to manufacturer’s protocol and sequenced on Novaseq 6000 SP Flowcell.
Data sharing:
WCM transcriptomics and DNA methylation samples (n = 184) are deposited in Gene Expression Omnibus GSE145043.
Supplementary Material
Statement of Significance:
In a translational relevant transcriptomic-based classification we characterized the microenvironment as a critical component of the B-cell lymphoma biology and associated it with the DLBCL clinical behavior establishing a novel opportunity for targeting therapies.
Acknowledgements
Funding for this work was provided by the Leukemia and Lymphoma Society (LLS-SCOR 7012-16 and LLS TRP R6510-19), Italian Association for Cancer Research (AIRC 20198) and the National Institutes of Health - National Cancer Institute (R01CA242069).
Footnotes
Conflict of interest:
J.P.L. served as a consultant for Sutro, Miltenyi, AstraZeneca, Epizyme, Roche/Genentech, BMS/Celgene, Regeneron, ADC Therapeutics, MEI Pharma, Bayer and Gilead/Kite. N.K., A.B., Z.A., V.S., E.T., N.K., N.M., F.F., M.T., N.A. and N.F. hold shares in BostonGene. A.M.M. received research funding from Sanofi and Janssen, served as consultant for Epizyme, Constellation and Jubiland, and served as scientific advisor for KDAC. L.C. received research funding from BMS/Celgene. All other authors declare no competing interests.
REFERENCES
- 1.Frontzek F, Lenz G. Novel insights into the pathogenesis of molecular subtypes of diffuse large B-cell lymphoma and their clinical implications. Expert Rev Clin Pharmacol 2019;12(11):1059–67 doi 10.1080/17512433.2019.1683447. [DOI] [PubMed] [Google Scholar]
- 2.Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 2018;24(5):679–90 doi 10.1038/s41591-018-0016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med 2018;378(15):1396–407 doi 10.1056/NEJMoa1801445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 2020;37(4):551–68 e14 doi 10.1016/j.ccell.2020.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson NA, Savage KJ, Ludkovski O, Ben-Neriah S, Woods R, Steidl C, et al. Lymphomas with concurrent BCL2 and MYC translocations: the critical factors associated with survival. Blood 2009;114(11):2273–9 doi 10.1182/blood-2009-03-212191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scott DW, King RL, Staiger AM, Ben-Neriah S, Jiang A, Horn H, et al. High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements with diffuse large B-cell lymphoma morphology. Blood 2018;131(18):2060–4 doi 10.1182/blood-2017-12-820605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kant S, Kumar A, Singh SM. Tumor growth retardation and chemosensitizing action of fatty acid synthase inhibitor orlistat on T cell lymphoma: implication of reconstituted tumor microenvironment and multidrug resistance phenotype. Biochim Biophys Acta 2014;1840(1):294–302 doi 10.1016/j.bbagen.2013.09.020. [DOI] [PubMed] [Google Scholar]
- 8.Cayrol F, Diaz Flaque MC, Fernando T, Yang SN, Sterle HA, Bolontrade M, et al. Integrin alphavbeta3 acting as membrane receptor for thyroid hormones mediates angiogenesis in malignant T cells. Blood 2015;125(5):841–51 doi 10.1182/blood-2014-07-587337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mourcin F, Pangault C, Amin-Ali R, Ame-Thomas P, Tarte K. Stromal cell contribution to human follicular lymphoma pathogenesis. Front Immunol 2012;3:280 doi 10.3389/fimmu.2012.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med 2008;359(22):2313–23 doi 10.1056/NEJMoa0802885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ciavarella S, Vegliante MC, Fabbri M, De Summa S, Melle F, Motta G, et al. Dissection of DLBCL microenvironment provides a gene expression-based predictor of survival applicable to formalin-fixed paraffin-embedded tissue. Ann Oncol 2018;29(12):2363–70 doi 10.1093/annonc/mdy450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Charette M, Houot R. Hide or defend, the two strategies of lymphoma immune evasion: potential implications for immunotherapy. Haematologica 2018;103(8):1256–68 doi 10.3324/haematol.2017.184192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pandey S, Mourcin F, Marchand T, Nayar S, Guirriec M, Pangault C, et al. IL-4/CXCL12 loop is a key regulator of lymphoid stroma function in follicular lymphoma. Blood 2017;129(18):2507–18 doi 10.1182/blood-2016-08-737239. [DOI] [PubMed] [Google Scholar]
- 14.Allen CD, Okada T, Cyster JG. Germinal-center organization and cellular dynamics. Immunity 2007;27(2):190–202 doi 10.1016/j.immuni.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monti S, Savage KJ, Kutok JL, Feuerhake F, Kurtin P, Mihm M, et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 2005;105(5):1851–61 doi 10.1182/blood-2004-07-2947. [DOI] [PubMed] [Google Scholar]
- 16.Ennishi D, Jiang A, Boyle M, Collinge B, Grande BM, Ben-Neriah S, et al. Double-Hit Gene Expression Signature Defines a Distinct Subgroup of Germinal Center B-Cell-Like Diffuse Large B-Cell Lymphoma. J Clin Oncol 2019;37(3):190–201 doi 10.1200/JCO.18.01583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med 2011;17(11):1514–20 doi 10.1038/nm.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Monti S, Chapuy B, Takeyama K, Rodig SJ, Hao Y, Yeda KT, et al. Integrative analysis reveals an outcome-associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B cell lymphoma. Cancer Cell 2012;22(3):359–72 doi 10.1016/j.ccr.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ennishi D, Takata K, Beguelin W, Duns G, Mottok A, Farinha P, et al. Molecular and Genetic Characterization of MHC Deficiency Identifies EZH2 as Therapeutic Target for Enhancing Immune Recognition. Cancer Discov 2019;9(4):546–63 doi 10.1158/2159-8290.CD-18-1090. [DOI] [PubMed] [Google Scholar]
- 20.Muppidi JR, Schmitz R, Green JA, Xiao W, Larsen AB, Braun SE, et al. Loss of signalling via Galpha13 in germinal centre B-cell-derived lymphoma. Nature 2014;516(7530):254–8 doi 10.1038/nature13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Young RM, Shaffer AL 3rd, Phelan JD, Staudt LM. B-cell receptor signaling in diffuse large B-cell lymphoma. Semin Hematol 2015;52(2):77–85 doi 10.1053/j.seminhematol.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zaytcev A, Chelushkin M, Nuzhdina K, Bagaev A, Dyykanov D, Zyrin V, et al. Abstract 853: Novel machine learning based deconvolution algorithm results in accurate description of tumor microenvironment from bulk RNAseq. Cancer Research 2020;80(16 Supplement):853- doi 10.1158/1538-7445.Am2020-853. [DOI] [Google Scholar]
- 23.Clozel T, Yang S, Elstrom RL, Tam W, Martin P, Kormaksson M, et al. Mechanism-based epigenetic chemosensitization therapy of diffuse large B-cell lymphoma. Cancer Discov 2013;3(9):1002–19 doi 10.1158/2159-8290.CD-13-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stelling A, Wu CT, Bertram K, Hashwah H, Theocharides APA, Manz MG, et al. Pharmacological DNA demethylation restores SMAD1 expression and tumor suppressive signaling in diffuse large B-cell lymphoma. Blood Adv 2019;3(20):3020–32 doi 10.1182/bloodadvances.2019000210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evens AM, Sehn LH, Farinha P, Nelson BP, Raji A, Lu Y, et al. Hypoxia-inducible factor-1 {alpha} expression predicts superior survival in patients with diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol 2010;28(6):1017–24 doi 10.1200/JCO.2009.24.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manfroi B, Moreaux J, Righini C, Ghiringhelli F, Sturm N, Huard B. Tumor-associated neutrophils correlate with poor prognosis in diffuse large B-cell lymphoma patients. Blood Cancer J 2018;8(7):66 doi 10.1038/s41408-018-0099-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160(1–2):48–61 doi 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vesely MD, Schreiber RD. Cancer immunoediting: antigens, mechanisms, and implications to cancer immunotherapy. Ann N Y Acad Sci 2013;1284:1–5 doi 10.1111/nyas.12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smyth MJ, Dunn GP, Schreiber RD. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol 2006;90:1–50 doi 10.1016/S0065-2776(06)90001-7. [DOI] [PubMed] [Google Scholar]
- 30.Manfroi B, McKee T, Mayol JF, Tabruyn S, Moret S, Villiers C, et al. CXCL-8/IL8 Produced by Diffuse Large B-cell Lymphomas Recruits Neutrophils Expressing a Proliferation-Inducing Ligand APRIL. Cancer Res 2017;77(5):1097–107 doi 10.1158/0008-5472.CAN-16-0786. [DOI] [PubMed] [Google Scholar]
- 31.Gupta M, Han JJ, Stenson M, Maurer M, Wellik L, Hu G, et al. Elevated serum IL-10 levels in diffuse large B-cell lymphoma: a mechanism of aberrant JAK2 activation. Blood 2012;119(12):2844–53 doi 10.1182/blood-2011-10-388538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beguelin W, Sawh S, Chambwe N, Chan FC, Jiang Y, Choo JW, et al. IL10 receptor is a novel therapeutic target in DLBCLs. Leukemia 2015;29(8):1684–94 doi 10.1038/leu.2015.57. [DOI] [PubMed] [Google Scholar]
- 33.Azzaoui I, Uhel F, Rossille D, Pangault C, Dulong J, Le Priol J, et al. T-cell defect in diffuse large B-cell lymphomas involves expansion of myeloid-derived suppressor cells. Blood 2016;128(8):1081–92 doi 10.1182/blood-2015-08-662783. [DOI] [PubMed] [Google Scholar]
- 34.Li L, Zhang J, Chen J, Xu-Monette ZY, Miao Y, Xiao M, et al. B-cell receptor-mediated NFATc1 activation induces IL-10/STAT3/PD-L1 signaling in diffuse large B-cell lymphoma. Blood 2018;132(17):1805–17 doi 10.1182/blood-2018-03-841015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu-Monette ZY, Xiao M, Au Q, Padmanabhan R, Xu B, Hoe N, et al. Immune Profiling and Quantitative Analysis Decipher the Clinical Role of Immune-Checkpoint Expression in the Tumor Immune Microenvironment of DLBCL. Cancer Immunol Res 2019;7(4):644–57 doi 10.1158/2326-6066.CIR-18-0439. [DOI] [PubMed] [Google Scholar]
- 36.Liu XQ, Lu K, Feng LL, Ding M, Gao JM, Ge XL, et al. Up-regulated expression of indoleamine 2,3-dioxygenase 1 in non-Hodgkin lymphoma correlates with increased regulatory T-cell infiltration. Leuk Lymphoma 2014;55(2):405–14 doi 10.3109/10428194.2013.804917. [DOI] [PubMed] [Google Scholar]
- 37.Nie M, Yang L, Bi X, Wang Y, Sun P, Yang H, et al. Neutrophil Extracellular Traps Induced by IL8 Promote Diffuse Large B-cell Lymphoma Progression via the TLR9 Signaling. Clin Cancer Res 2019;25(6):1867–79 doi 10.1158/1078-0432.CCR-18-1226. [DOI] [PubMed] [Google Scholar]
- 38.Feist M, Schwarzfischer P, Heinrich P, Sun X, Kemper J, von Bonin F, et al. Cooperative STAT/NF-kappaB signaling regulates lymphoma metabolic reprogramming and aberrant GOT2 expression. Nat Commun 2018;9(1):1514 doi 10.1038/s41467-018-03803-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakayama S, Yokote T, Hirata Y, Akioka T, Miyoshi T, Hiraoka N, et al. TNF-alpha expression in tumor cells as a novel prognostic marker for diffuse large B-cell lymphoma, not otherwise specified. Am J Surg Pathol 2014;38(2):228–34 doi 10.1097/PAS.0000000000000094. [DOI] [PubMed] [Google Scholar]
- 40.Santiago R, Johnson N, Dmitrienko S, Papadakis A, Benlimame N, Mercier F, et al. Changes in Tumor Immune Micro-Environment in Diffuse Large B-Cell Lymphoma (DLBCL): A Comparative Study of Relapsed Versus Diagnostic DLBCL. Blood 2019;134(Supplement_1):3968- doi 10.1182/blood-2019-122207. [DOI] [Google Scholar]
- 41.Horvath S DNA methylation age of human tissues and cell types. Genome Biol 2013;14(10):R115 doi 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duran-Ferrer M, Clot G, Nadeu F, Beekman R, Baumann T, Nordlund J, et al. The proliferative history shapes the DNA methylome of B-cell tumors and predicts clinical outcome. Nature Cancer 2020;1(11):1066–81 doi 10.1038/s43018-020-00131-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neill T, Schaefer L, Iozzo RV. Decorin: a guardian from the matrix. Am J Pathol 2012;181(2):380–7 doi 10.1016/j.ajpath.2012.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Subbarayan K, Leisz S, Wickenhauser C, Bethmann D, Massa C, Steven A, et al. Biglycan-mediated upregulation of MHC class I expression in HER-2/neu-transformed cells. Oncoimmunology 2018;7(4):e1373233 doi 10.1080/2162402X.2017.1373233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 2002;346(25):1937–47 doi 10.1056/NEJMoa012914. [DOI] [PubMed] [Google Scholar]
- 46.Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol 2016;17(1):218 doi 10.1186/s13059-016-1070-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017;171(2):481–94 e15 doi 10.1016/j.cell.2017.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pasqualucci L, Dalla-Favera R. The genetic landscape of diffuse large B-cell lymphoma. Semin Hematol 2015;52(2):67–76 doi 10.1053/j.seminhematol.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nicholas NS, Apollonio B, Ramsay AG. Tumor microenvironment (TME)-driven immune suppression in B cell malignancy. Biochim Biophys Acta 2016;1863(3):471–82 doi 10.1016/j.bbamcr.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 50.Appunni S, Anand V, Khandelwal M, Gupta N, Rubens M, Sharma A. Small Leucine Rich Proteoglycans (decorin, biglycan and lumican) in cancer. Clin Chim Acta 2019;491:1–7 doi 10.1016/j.cca.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 51.Haro M, Orsulic S. A Paradoxical Correlation of Cancer-Associated Fibroblasts With Survival Outcomes in B-Cell Lymphomas and Carcinomas. Front Cell Dev Biol 2018;6:98 doi 10.3389/fcell.2018.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Calon A, Tauriello DV, Batlle E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin Cancer Biol 2014;25:15–22 doi 10.1016/j.semcancer.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 53.Fiori ME, Di Franco S, Villanova L, Bianca P, Stassi G, De Maria R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol Cancer 2019;18(1):70 doi 10.1186/s12943-019-0994-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marullo R, Rutherford SC, Leonard JP, Cerchietti L. Therapeutic implication of concomitant chromosomal aberrations in patients with aggressive B-cell lymphomas. Cell Cycle 2016;15(17):2241–7 doi 10.1080/15384101.2016.1207839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Younes A, Hilden P, Coiffier B, Hagenbeek A, Salles G, Wilson W, et al. International Working Group consensus response evaluation criteria in lymphoma (RECIL 2017). Ann Oncol 2017;28(7):1436–47 doi 10.1093/annonc/mdx097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vivian J, Rao AA, Nothaft FA, Ketchum C, Armstrong J, Novak A, et al. Toil enables reproducible, open source, big biomedical data analyses. Nat Biotechnol 2017;35(4):314–6 doi 10.1038/nbt.3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res 2013;41(Database issue):D991–5 doi 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leinonen R, Sugawara H, Shumway M, International Nucleotide Sequence Database C. The sequence read archive. Nucleic Acids Res 2011;39(Database issue):D19–21 doi 10.1093/nar/gkq1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Uhlen M, Karlsson MJ, Zhong W, Tebani A, Pou C, Mikes J, et al. A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science 2019;366(6472) doi 10.1126/science.aax9198. [DOI] [PubMed] [Google Scholar]
- 60.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013;14:7 doi 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.James KR, Gomes T, Elmentaite R, Kumar N, Gulliver EL, King HW, et al. Distinct microbial and immune niches of the human colon. Nat Immunol 2020;21(3):343–53 doi 10.1038/s41590-020-0602-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schubert M, Klinger B, Klunemann M, Sieber A, Uhlitz F, Sauer S, et al. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat Commun 2018;9(1):20 doi 10.1038/s41467-017-02391-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blondel A, Guillaume JL, Lambiotte R, Lefebvre E Fast unfolding of communities in large networks. J Stat Mech 2008;2008(10):P10008 doi 10.1088/1742-5468/2008/10/P10008. [DOI] [Google Scholar]
- 64.Loeffler-Wirth H, Kreuz M, Hopp L, Arakelyan A, Haake A, Cogliatti SB, et al. A modular transcriptome map of mature B cell lymphomas. Genome Med 2019;11(1):27 doi 10.1186/s13073-019-0637-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, Staudt LM. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci U S A 2003;100(17):9991–6 doi 10.1073/pnas.1732008100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 2014;32(4):381–6 doi 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 2017;14(10):979–82 doi 10.1038/nmeth.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Racle J, de Jonge K, Baumgaertner P, Speiser DE, Gfeller D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife 2017;6 doi 10.7554/eLife.26476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chevrier S, Levine JH, Zanotelli VRT, Silina K, Schulz D, Bacac M, et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 2017;169(4):736–49 e18 doi 10.1016/j.cell.2017.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep 2017;7(1):16878 doi 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bergmann EA, Chen BJ, Arora K, Vacic V, Zody MC. Conpair: concordance and contamination estimator for matched tumor-normal pairs. Bioinformatics 2016;32(20):3196–8 doi 10.1093/bioinformatics/btw389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012;28(18):i333–i9 doi 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Geoffroy V, Herenger Y, Kress A, Stoetzel C, Piton A, Dollfus H, et al. AnnotSV: an integrated tool for structural variations annotation. Bioinformatics 2018;34(20):3572–4 doi 10.1093/bioinformatics/bty304. [DOI] [PubMed] [Google Scholar]
- 74.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26(6):841–2 doi 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bolotin DA, Poslavsky S, Davydov AN, Frenkel FE, Fanchi L, Zolotareva OI, et al. Antigen receptor repertoire profiling from RNA-seq data. Nat Biotechnol 2017;35(10):908–11 doi 10.1038/nbt.3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Garrett-Bakelman FE, Sheridan CK, Kacmarczyk TJ, Ishii J, Betel D, Alonso A, et al. Enhanced reduced representation bisulfite sequencing for assessment of DNA methylation at base pair resolution. J Vis Exp 2015(96):e52246 doi 10.3791/52246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011;27(11):1571–2 doi 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 2017;14(4):417–9 doi 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol 2012;13(10):R87 doi 10.1186/gb-2012-13-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 2016;34(5):525–7 doi 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 82.Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, et al. Ensembl 2018. Nucleic Acids Res 2018;46(D1):D754–D61 doi 10.1093/nar/gkx1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Beachley VZ, Wolf MT, Sadtler K, Manda SS, Jacobs H, Blatchley MR, et al. Tissue matrix arrays for high-throughput screening and systems analysis of cell function. Nat Methods 2015;12(12):1197–204 doi 10.1038/nmeth.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Naba A, Clauser KR, Hynes RO. Enrichment of Extracellular Matrix Proteins from Tissues and Digestion into Peptides for Mass Spectrometry Analysis. J Vis Exp 2015(101):e53057 doi 10.3791/53057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.