

Abstract
One of the greatest barriers to curative treatment of neuroblastoma (NB) is its frequent metastatic outgrowth prior to diagnosis, especially in cases driven by amplification of the MYCN oncogene. However, only a limited number of regulatory proteins that contribute to this complex MYCN-mediated process have been elucidated. Here we show that the growth arrest-specific 7 (GAS7) gene, located at chromosome band 17p13.1, is preferentially deleted in high-risk MYCN-driven NB. GAS7 expression was also suppressed in MYCN-amplified NB lacking 17p deletion. GAS7 deficiency led to accelerated metastasis in both zebrafish and mammalian models of NB with overexpression or amplification of MYCN. Analysis of expression profiles and the ultrastructure of zebrafish NB tumors with MYCN overexpression identified that GAS7 deficiency led to (i) downregulation of genes involved in cell-cell interaction, (ii) loss of contact among tumor cells as critical determinants of accelerated metastasis, and (iii) increased levels of MYCN protein. These results provide the first genetic evidence that GAS7 depletion is a critical early step in the cascade of events culminating in NB metastasis in the context of MYCN overexpression.
INTRODUCTION
Tumor metastasis -- the translocation of tumor cells from a primary site to a distant site -- is the main cause of most cancer-related deaths and a major obstacle to successful treatment (1-3). Over the past few decades, only modest gains in 5-year survival rates have been achieved in patients with metastatic disease, thus underscoring the urgent need to improve understanding of the mechanisms that underlie metastasis and hold the keys to more effective treatment options for this devastating phenotype (1, 4).
Neuroblastoma (NB), a childhood cancer of the developing peripheral sympathetic nervous system (PSNS), metastasizes at a high rate (5-7). About 50% of patients, especially those older than 18 months of age with amplification of the MYCN oncogene, present with hematogenous dissemination at diagnosis, with tumor cells often found in bone marrow (71%), bone (56%), lymph nodes (31%), liver (30%), and in intracranial or orbital sites (18%) (8). Although a substantial subset of these tumors can spontaneously regress without treatment, high-risk NB with widespread metastasis invariably denotes refractory disease and a poor outcome (9).
Despite the discovery of several recurrent somatic mutations and chromosomal abnormalities that contribute significantly to neuroblastomagenesis (10, 11), the role of such aberrations in NB metastasis remains unclear. However, recent studies with diverse animal models, including mice (12, 13), chickens (14), and zebrafish (15), have begun to reduce this gap. For example, loss of Caspase-8 has been reported to potentiate NB metastasis in chickens (13) as well as in xenografted or TH-MYCN mice (12), while suppression of SEMA3C signaling promotes the detachment and dissemination of xenografted NB cells in chick embryos (14). Using a zebrafish model, we have shown that transgenic coexpression of MYCN and LMO1, a NB susceptibility gene (16, 17), in the PSNS promotes hematogenous metastasis of NB cells in vivo (15). More importantly, we found that genes involved in tumor cell-extracellular matrix interactions were deregulated when MYCN and LMO1 were co-expressed in the transgenic fish, leading to the acceleration of tumor cell dissemination (15).
Recently, the growth arrest-specific 7 (GAS7) gene, encoding for an adaptor protein that coordinates the dynamic activities of the membrane, actin cytoskeleton and microtubules (18-20), has been shown to inhibit migration of lung (21) and breast (22) cancer cells when overexpressed. Methylation of the GAS7 promoter has also been observed in a range of cancers and is associated with a poor survival among patients with oral squamous cell carcinoma (23), colorectal cancer (24, 25), lung cancer (21), and breast cancer (26, 27). However, whether GAS7 is a key negative regulator of NB metastasis, and the impact and mechanisms of deregulated GAS7 activity to metastasis in vivo, remain to be explored.
MATERIALS AND METHODS
Genomic Data Processing and Availability
The copy number profiles from the TARGET whole-genome sequencing project are available at Gene Expression Omnibus (GEO), GSE131189 (RRID: SCR_005012). Whole-genome sequencing data are available from NCBI dbGaP (https://www.ncbi.nlm.nih.gov/gap, NCBI database of Genotypes and Phenotypes, RRID: SCR_002709) with a study ID, phs000218, and an accession number, phs000467. The SNP arrays, Human exon arrays and RNA-seq data were obtained from TARGET NCI pages (https://target-data.nci.nih.gov/). Human Exon arrays were processed with Robust Multichip Average and summarized with the core probe set provided by Affymetrix. SNP arrays were processed with GenomeStudio (RRID: SCR_010973), and copy number were obtained with the Nexus SNPRank algorithm as previously described (28). In addition to the different TARGET datasets, we also used the SEQC RNA-seq dataset at. GEO, GSE62564, for survival analysis.
Vertebrate Animal
All zebrafish were of the wild-type AB strain (ZIRC Cat# ZL1, RRID: ZIRC_ZL1). The zebrafish maintenance and breeding were performed as described previously (29), and studies with both sexes of animals were conducted in accord with Mayo Clinic Institutional Animal Care and Use Committee (IACUC)-approved protocol # A00004637-19. The severe combined immunodeficiency (SCID) beige mice used in the study were maintained under standard conditions of light and temperature with free access to food and water, in accord with IACUC-approved protocol # A00003355-18.
Cell Lines and Cell Culture
The human NB BE(2)-M17 [American Type Culture Collection (ATCC) Cat# CRL-2267, RRID: CVCL_0167) cell line, BE(2)C (ATCC Cat# CRL-2268, RRID: CVCL_0529) and 293T (ATCC Cat# CRL-3216, RRID: CVCL_0063) were obtained from ATCC. The CHP-134 cell line (DSMZ Cat# ACC-653, RRID: CVCL_1124) was obtained from Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures GmbH. The Kelly cell line (ECACC Cat# 92110411, RRID: CVCL_2092) was obtained from sigma Aldrich. The NB SHEP-Tet/N21 cell line (RRID: CVCL_9812) was a generous gift from Dr. M. Schwab at the German Cancer Research Center, Heidelberg, Germany (30). These human NB cell lines were cultured in RPMI 1640 medium (Gibco, 22400) supplemented with 10% (v/v) fetal bovine serum (FBS) (Gibco, 26140079), penicillin (100 U/ml), and streptomycin (100 mg/ml) at 37°C with 5% CO2. Medium was changed once every day. The cell lines were authenticated by ATCC based on STR profiling. The mycoplasma testing was performed at IDEXX BioAnalytics.
Tumor Watch of Zebrafish
MYCN;gas7mut het fish were crossed with heterozygous gas7mut fish. The EGFP-MYCN-positive offspring were sorted at 1 day postfertilization (dpf) and screened every 2 weeks, starting at 5 weeks postfertilization (wpf), for fluorescent EGFP-expressing tumor cell masses. Fish with tumors were separated, genotyped and analyzed further by H&E staining and immunohistochemical assays.
Computation and Statistical Analyses of Human NB Cases
All statistical comparisons of genomic and survival data were done with the Wilcoxon rank-sum test and R programming, respectively. To establish the copy number status of the 17p chromosomal region, we calculated the average copy number of the 12 Mb region from the distal chromosome 17 short arm to the 17p.21 and performed log2 transformation of tumor/blood ratio for the WGS cohort and log R ratio for the SNP cohort. We then used a threshold of 0.05 to determine the status of 17p copy numbers, such as 17p loss (17p−, logR < −0.05), 17p gain (17p+, logR > 0.05), and copy number neutral (17p 2n, −0.05 < logR < 0.05). In our analysis, loss of GAS7 copy number was considered when the copy number of GAS7 regions was less than the median ploidy of the whole tumor. The ploidy estimations were generated using the Complete Genomics Cancer Pipeline 2.0, https://www.completegenomics.com/documents/DataFileFormats_Cancer_Pipeline_2.0.pdf. The output of this pipeline is publicly available in the TARGET site (https://target-data.nci.nih.gov/Public/NBL/WGS/L3/copy_number/CGI/). To estimate the association between GAS7 expression and survival, we applied the Kaplan Scan (KaplanScan) approach, where an optimum survival cut-off is established based on statistical testing (see the illustration of this method at R2: Neuroblastoma Genomics Analysis and Visualization Platform, https://hgserver1.amc.nl/r2/help/r2_tutorials.pdf). Briefly, the KaplanScan approach separates the samples of a dataset into two groups based on the expression of a given gene. It will use every increasing expression value as a cutoff to create two groups and test the p-value in a log-rank test. After obtaining the optimal Kaplan-Meier separation (lower log-rank p-value) from all possible expression thresholds, we used the Benjamini & Hochberg’s procedure (false discovery rate) for multiple testing corrections.
RESULTS
Heterozygous 17p Deletion Harboring the GAS7 Locus in High-Risk NB Confers a Poor Clinical Outcome
A gain of chromosome arm 17q occurs in most high-risk NB cases and is associated with 1p and/or 11q deletions (31-33). In a subset of cases with 17q gain, there is concomitant deletion of the short arm of chromosome 17 (17p), harboring TP53 and GAS7. To assess the contribution of alterations of 17p to NB pathogenesis, we performed whole-genome sequencing (WGS) and single-nucleotide polymorphism (SNP) array analysis on 135 and 914 primary NB samples, respectively. We identified heterozygous 17p deletions in two subsets of high-risk cases, those with amplified MYCN (31.0% WGS and 12.1% SNP) or single-copy MYCN (18.2% WGS and 10.7% SNP), but rarely in low- or intermediate-risk cases, which typically show whole-chromosome 17 gains (Figures 1A and S1A; Table S1), as reported previously (34-36). We attribute the different frequencies of heterozygous 17p deletions detected by WGS versus SNP to the different dynamic detection ranges of these platforms and to the composition of the two cohorts, with the WGS cohort comprising mainly high-risk patients over 18 months of age, while the SNP array cohort more closely reflects the natural distribution of patients with this disease (Figures 1A and S1A; Table S1). We have not detected homozygous 17p deletions in either cohort.
Figure 1. GAS7 is deleted in a subset of high-risk NBs and low levels of its expression are associated with an inferior survival.

(A) Integrative genome viewer visualization of chromosome 17 copy numbers across 135 primary human NBs from a whole-genome sequencing (WGS) cohort (accession number, phs000467, in NCBI dbGaP). Samples are ordered according to overall 17p copy number (CN). COG, Children’s Oncology Group; INSS, International NB Staging System.
(B) Kaplan-Meier survival curves for NB patients in WGS cohort stratified by 17p copy numbers, including 17p deletion (17p−), 17p neutral copy number (17p 2n), and 17p gain (17p+). Differences between the curves are significant by the log-rank test at p=1.95e-4.
(C) Kaplan-Meier survival curves for NB patients stratified by GAS7 expression (Data from GEO: GSE62564). The difference between the curves for GAS7-low versus GAS7-high expression is significant by the log-rank test at p= 6.48e-13.
(D) Box plots comparing expression levels of GAS7 in high-risk tumors with MYCN amplification (HR-AMP) versus those without MYCN amplification (HR-NA) in the context of copy number-neutral 17p (17p 2n) or loss (17p−). P=4.2e-4, by Wilcoxon rank-sum test (Data from GEO: GSE62564).
Further computational analyses showed that heterozygous 17p deletion was associated with poor survival (Figures 1B and S1B), suggesting that genes within the deleted 17p region, such as GAS7 and TP53 (Figures 1A and S1A), might indeed suppress tumor pathogenesis. High frequency of TP53 pathway inactivation has been reported as a critical mechanism underlying NB relapse (37). The support for an indispensable suppressor role of GAS7 came from the comparisons of its expression levels which show significant association of its reduced expression with poorer survival of patients in multiple publicly available datasets (Figures 1C and S1C) and higher risk of NBs by immunohistochemical analysis of human tissue array (Figure S1D-G).
GAS7 Expression is Suppressed by MYCN
In addition to the heterozygous deletion of GAS7 in high-risk NB, the expression levels of GAS7 were also significantly lower in MYCN-amplified versus MYCN single-copy cases (Figures 1D and S2A). Such inverse correlation of expression between GAS7 and MYCN, but not GAS7 and CMYC, was also detected in MYCN non-amplified high-risk NBs with intact chromosome 17p (Figure S2B-D). These suggest that GAS7 expression might be downregulated as a consequence of enhanced MYCN activity in high-risk NBs lacking a 17p deletion. To explore this hypothesis, we first examined the expression levels of GAS7 in the SHEP-Tet/21N human NB cell line, which constitutively expresses MYCN in the absence of doxycycline (30). Upon induction with doxycycline, MYCN expression in SHEP-Tet/21N cells was clearly silenced (Figure 2A, left), while GAS7 expression was significantly upregulated (Figure 2A, right), indicating that such expression is negatively regulated by MYCN. To validate this finding, we used transgenic zebrafish models that overexpress EGFP-MYCN (designated MYCN line) or control EGFP (designated dβh:EGFP line) in the PSNS under control of the dopamine-beta-hydroxylase (dβh) promoter (38). By qRT-PCR analyses of gas7 expression in FACS-sorted EGFP-positive PSNS cells from MYCN or control dβh:EGFP transgenic fish, we detected significantly decreased expression of endogenous gas7 in the MYCN-positive PSNS cells compared with EGFP-positive control PSNS cells (Figure 2B), further demonstrating a negative correlation between GAS7 and MYCN expression.
Figure 2. GAS7 expression is suppressed by high levels of MYCN expression.
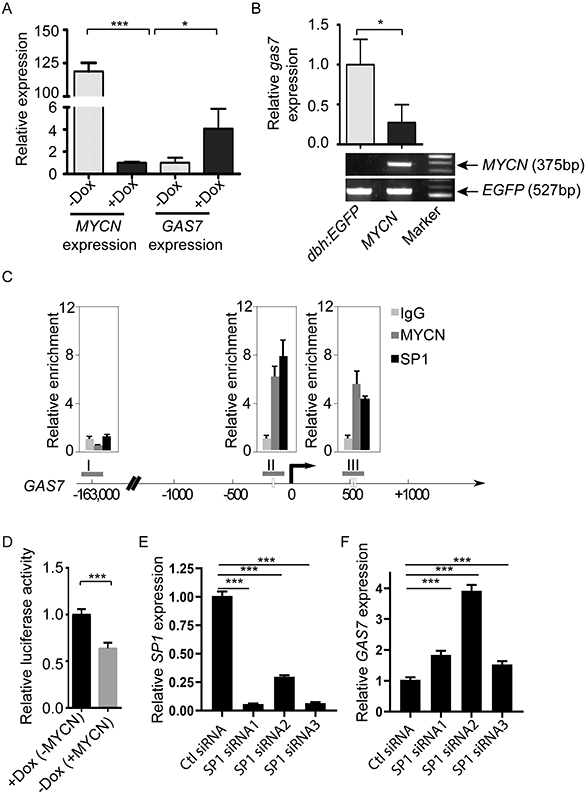
(A) Relative expression of GAS7 versus ACTIN in SHEP-Tet/21N human NB cell line in the presence or absence of doxycycline induction by semiquantitative RT-PCR analysis. The data are presented as means ± SD of triplicate experiments; *p < 0.05 and ***p < 0.001 by two-tailed t test.
(B) Top: Relative expression of gas7 versus elfa in the FACS-sorted EGFP-positive PSNS cells from control dβh-EGFP or dβh-EGFP-MYCN (MYCN) transgenic fish by semiquantitative RT-PCR analysis. The data are presented as means ± SD of triplicate experiments; *p < 0.05 by two-tailed t test. Bottom: Electrophoresis of MYCN and EGFP genotyping confirms the genotypes of transgenic embryos subjected to qRT-PCR analysis.
(C) Dual cross-linking ChIP-PCR with SHEP-Tet/21N human NB cell line showing co-occupancy of SP1 and MYCN at two regions (II and III) of the GAS7 promoter (region I serves as a negative control). Arrow marks the transcription start site. Experiments were performed in duplicate; and error bars represent SEM.
(D) Relative luciferase activity in SHEP-Tet/21N human NB cell line transfected with luciferase reporter vector containing the GAS7 promoter region in the presence or absence of doxycycline (Dox). The data are presented as means ± SEM of duplicate experiments; ***p=0.0009 by two-tailed t test.
(E-F) Relative expression of SP1 (E) or GAS7 (F) versus ACTIN in SHEP-Tet/21N human NB cell line transfected with control siRNA (Ctl siRNA) or three independent SP1 siRNAs (SP1 siRNA1, siRNA2 or siRNA3) by semiquantitative RT-PCR analysis. The data are presented as means ± SD of triplicate experiments; ***p < 0.001 by two-tailed t test.
To determine whether GAS7 is a direct target of MYCN, we analyzed our previously published MYCN chromatin immunoprecipitation (ChIP) sequencing data on a panel of MYCN-amplified NB cell lines, including LAN5, Kelly, NGP, COGN415 and NB-1643 cells (39). Surprisingly, we did not detect an obvious MYCN binding signal at the GAS7 promoter region (Figure S2E), suggesting that MYCN does not bind to the GAS7 promoter region directly. It has been previously reported that MYCN can form a transcription repressor complex with SP1 at the gene promoter region to repress transcription of the gene (40). To determine whether MYCN might bind to GAS7 promoter region indirectly by interacting with SP1, we first performed a dual cross-linking ChIP-PCR assay with the SHEP-Tet/21N human NB cell line, and found that SP1 robustly binds to the GAS7 gene at the regions bound by MYCN (Figure 2C). We detected increased luciferase activity in SHEP-Tet/21N cells transfected with a reporter construct carrying the GAS7 promoter region upon induction with doxycycline (Figure 2D). Additionally, GAS7 expression was significantly upregulated when SP1 was knocked down in the SHEP-Tet/21N cells (Figure 2E and 2F). To further demonstrate that GAS7 can be suppressed by SP1 in the same manner in MYCN-amplified NB cells, we performed the dual cross-linking ChIP-PCR assay with CHP-134 human NB cell line which harbors MYCN amplification and expresses GAS7 at low levels (Figure S2F and S2G). Consistently, we detected robust binding of SP1 to the GAS7 gene at the regions bound by MYCN (Figure S2H). Taken together, these data indicate that MYCN can indirectly repress GAS7 expression in high-risk NB at least partially through forming a transcription repression complex with SP1 at the GAS7 promoter region.
In the analysis of our MYCN ChIP sequencing data, we also identified strong signals of H3K27M3, a repressive mark of transcription, at the transcription start site of the GAS7 gene (Figure S2E). It has been shown that MYCN may prime a subset of genes for epigenetic silencing via activation of polycomb repressive complex 2 (PRC2) components (41). To test whether PRC2 activity might contribute to GAS7 repression, we treated BE(2)C cells with decitabine/5-aza-2'-deoxycytidine (DEC, a DNA methyltransferase inhibitor), or EPZ-6438/tazemetostat (EPZ, a selective EZH2 inhibitor), or both. Interestingly, GAS7 expression was significantly upregulated in the cells treated with either inhibitor alone or in combination of both inhibitors (Figure S2I), suggesting the involvement of PRC2 in the transcriptional repression of GAS7. Together, these findings demonstrate that high levels of MYCN can inhibit GAS7 expression in NB with an intact 17p region, providing a mechanism that could underlie metastasis in a large fraction of high-risk NB cases.
Knockout of gas7 Promotes Hematogenous Metastases in Transgenic Fish Overexpressing MYCN
To establish the role of GAS7 deficiency in NB pathogenesis more firmly, we developed a zebrafish line using transcription activator-like effector nuclease (TALEN)-mediated gas7 knockout. After injecting paired TALEN mRNAs targeting exon 2 of the zebrafish gas7 gene (42) (Figures 3A and S3A) into wild-type zebrafish embryos, we identified a mutant allele with a 7-bp deletion in the F1 generation, resulting in an open reading frame shift and formation of a premature stop codon (Figure 3A and 3B). This mutation would be expected to truncate 85% of the Gas7 protein with loss of majority of the F-BAR (FCH-Bin-Amphiphysin-Rvs) domain (Figure 3B-D), a critical and major domain mediating the interaction of Gas7 with the cell membrane (43-45), and hence would likely generate a function deficient mutant of gas7. To confirm that the targeted allele disrupts the production of a full-length Gas7 protein, we performed immunoblotting with an antibody against the C-terminus of Gas7 on the extracts prepared from wild-type, gas7mut heterozygous (gas7mut het) or homozygous (gas7mut homo) larvae at 5 days postfertilization (dpf). A control Gas7 protein was synthesized by in vitro transcription and translation with the zebrafish gas7 coding region used as a template (Figure 3E). We detected low levels of Gas7 protein expression in the heterozygous mutant extracts, as well as complete loss of Gas7 signal in the homozygous mutant extracts, compared to wild-type extracts or control Gas7 protein (Figure 3E); thus, indicating successful knockout of gas7 in the mutant fish line (designated gas7mut). We did not detect an apparent developmental phenotype or tumor formation in the gas7mut fish. Both heterozygous and homozygous gas7 knockout fish were viable and fertile with a life-span similar to that of their wild-type siblings.
Figure 3. Knockout of gas7 promotes NB metastasis in transgenic fish overexpressing MYCN.

(A) Schematic diagram of the targeted site for TALEN in exon 2 of gas7. TALEN binding sites are indicated by arrows in the nucleotide sequences alignment of wild-type (WT) and mutant gas7 (gas7mut) alleles. Red box marks the 7-base pair (bp) deletion (del) in the gas7 mutant.
(B) Alignment of amino acid sequence of WT and Gas7 mutant protein. The 7-bp deletion leads to a truncated protein product with short regions of altered translation (indicated in red) and pre-mature stop codon (*).
(C) Diagram of the predicted Gas7 mutant protein with loss of most of the F-BAR domain.
(D) Gel electrophoresis of PCR products from WT, gas7mut heterozygous (gas7mut het) or gas7mut homozygous (gas7mut homo) larvae with or without Fok I digestion. Intact bands at 546 bp were detected in gas7mut het or homo larvae after Fok I digestion, suggesting loss of Fok I cut sites in these mutants.
(E) Immunoblot analysis of protein lysates from 5-day old gas7mut het, gas7mut homo or WT larvae zebrafish, as well as in vitro synthesized Gas7 protein (Control). The expression levels of Gas7 are either decreased in the gas7mut het larvae or undetectable in the gas7mut homo larvae. Equal loading was confirmed by stripping the membrane and reprobing with antibody against Gapdh.
(F) Fraction of tumor-bearing fish that developed metastases at 18 weeks of age. Differences in the percentage of MYCN-only versus MYCN;gas7mut het or MYCN;gas7mut homo are significant by Fisher’s exact test at p=0.002 or p=0.0004, respectively.
(G-I) Fluorescence images of MYCN;gas7mut het (G) or MYCN;gas7mut homo compound fish (H), or MYCN-only transgenic fish (MYCN, I) with EGFP-expressing primary tumors in the interrenal gland regions or metastatic tumors (white arrows, G and H) at 18 weeks postfertilization (wpf). Scale bar, 1 mm.
Given the strong association of reduced GAS7 expression with amplified MYCN in high-risk NB (Figure 1D), next we tested whether knockout of gas7 might strengthen or accelerate MYCN overexpression-induced NB pathogenesis in vivo. We first crossed the heterozygous MYCN transgenic fish line with gas7 mutant fish (gas7mut) to generate compound fish with heterozygous overexpression of MYCN and heterozygous loss of gas7 (designated MYCN;gas7mut het). We then crossed the MYCN;gas7mut het fish with heterozygous gas7mut fish and sorted EGFP-MYCN-positive offspring to monitor them for tumor development. There were no substantial differences in tumor onset between MYCN-only transgenic fish and MYCN;gas7mut het or MYCN;gas7mut homo compound fish (Figure S3B), suggesting that knockout of gas7 might not significantly affect MYCN–induced tumorigenesis. Strikingly, among the tumor-bearing fish, both MYCN;gas7mut het (~19%; Figures 3F and 3G) and MYCN;gas7mut homo (~23%; Figures 3F and 3H) compound fish developed widespread EGFP-positive tumor masses distant from the primary tumors that arose from the interrenal gland region (IRG, analogue of the human adrenal gland) at 18 weeks of age. Although we occasionally detected the spread of EGFP-positive tumor masses distant from the IRG region in the MYCN-only transgenic fish over 1 year of age, we did not observe any metastases in MYCN-only fish at 18 weeks of age (Figures 3F and 3I), consistent with our previous observation that metastases were not present in MYCN fish by 6 months of age (15).
To further confirm that the widespread EGFP-positive masses were metastatic tumors, we performed sagittal sections on MYCN-only, MYCN;gas7mut het or MYCN;gas7mut homo compound fish (n=4-6 tumor-bearing fish per group) and examined them in a blinded manner (Figures 4 and S3). All of the primary tumors in these fish arose in the IRG (Figures 4A, 4B, 4J, S3C, and S3I) and consisted of small, undifferentiated round tumor cells with hyperchromatic nuclei, often forming nests that were comparable histologically to the human NBs we described earlier (38). None of the five MYCN-only fish we examined had disseminated tumor masses. This stands in marked contrast to the finding in two of six MYCN;gas7mut het fish and two of four MYCN;gas7mut homo fish, where such masses were found in multiple regions that recapitulated the metastatic sites in children with high-risk NB: for example, the bone (Figures 4A, 4C and 4K); spleen, which functions as lymph nodes in zebrafish (46)(Figures 4D, 4L, S3D and S3J); and liver (Figures 4E and 4M; double arrows point to tumor cells extravasating the blood vesicle in liver, suggesting hematogenous metastasis of tumor cells). Also included were sites rarely seen in humans, such as the gill, analogous to the mammalian lung (47) (Figures 4A, 4F, 4N, S3E and S3K); sclera of the eye (Figures 4A, 4G, 4O, S3F and S3L); pancreas (Figures 4H, 4P, S3G and S3M) and gut (Figures 4I, 4Q, S3H and S3N). These metastatic tumor cells expressed EGFP-MYCN at all sites (Figures 4J-4Q and S3P) and the NB markers tyrosine hydroxylase (TH) (Figures S3C-S3H and S3Q) along with HuC (Figures S3I-S3N), as detected by immunohistochemistry with antibodies against GFP, TH and HuC.
Figure 4. Pathological and immunohistochemical analyses of distant metastases of NB in MYCN transgenic fish with knockout of gas7.

(A-I) H&E-stained sagittal sections of a representative MYCN;gas7mut homo compound fish at 5 months of age. White box in (A) outlines the interrenal gland (IRG), magnified in (B) and (J).
(J–Q) Immunohistochemical analyses with GFP antibody on the sagittal sections of a representative MYCN;gas7mut homo compound fish in magnified views. Disseminated tumor cells were detected in the bone (b, C and K, double black arrowheads), the spleen (D and L, open arrowheads), the liver (E and M, double black arrows), the gill (g, F and N, black arrows), the sclera of the eye (e, G and O, black arrowheads), the pancreas (H and P, open arrows) and the gut (I and Q, double open arrowheads). Scale bars, 2 mm (A) and 50 μm (B–Q).
To rule out the possibility of an off-target effect of TALEN-mediated gas7 knockout on tumor metastasis in our gas7mut fish line, we injected gas7 TALEN mRNAs into the MYCN-expressing embryos and monitored them for tumor metastasis. We consistently detected disseminated tumor cells in organs distant from the IRG, such as the spleen, in MYCN fish with mosaic gas7 knockout at about 5 months of age (Figure S3O-S3Q) but not in their non-injected MYCN fish sibling. Genotyping confirmed the knockout of gas7 in the tumor-bearing fish with metastasis (Figure S3R). These findings indicate that either heterozygous or homozygous knockout of gas7 significantly accelerates hematogenous dissemination of NB in vivo in the context of MYCN overexpression.
Downregulation of GAS7 Expression Promotes the Invasive and Migratory Properties of Human NB Cells
Given the strong correlation between knockout of gas7 and metastasis in our MYCN transgenic zebrafish model, we next asked if this link might be due to an effect on cell invasion and migration. To address this issue, we first infected BE(2)-M17 cells, a MYCN-amplified NB cell line with high levels of endogenous GAS7 expression (Figure S2F and S2G), using lentivirus-based control shRNA, GAS7 shRNA1 or GAS7 shRNA2. By semi-quantitative RT-PCR and immunoblotting analyses, we detected significant downregulation of GAS7 expression at both mRNA (Figure 5A) and protein (Figure 5B) levels in BE(2)-M17 cells infected with either hairpin RNA. We then performed a transwell invasion and migration assay by culturing control or GAS7 shRNAs-infected BE(2)-M17 cells in medium with 1% FBS in the apical chamber over a matrigel-coated transwell permeable membrane. After 24 hours (hrs), the number of BE(2)-M17 cells infected with GAS7 shRNAs that passed through the membrane was about 2- to 8-fold higher than the BE(2)-M17 cells infected with control shRNA, as assessed by crystal violet staining of cells on the lower surface of the membrane (Figure 5C). This suggests a gain in the ability of BE(2)-M17 cells with downregulated GAS7 expression to invade and migrate across a matrigel-coated membrane. In addition, GAS7 shRNA-infected BE(2)-M17 cells also migrated significantly faster than control shRNA infected BE(2)-M17 cells toward a region that had been scraped with a pipette in a wound-healing assay (Figure 5D). A similar effect of faster closure of the wounded area was also observed in GAS7-knockdown Kelly cells, another MYCN-amplified cell line with relatively high levels of endogenous GAS7 expression (Figures S2G and S4A-C). These results indicate that downregulation of GAS7 expression enhances both the invasiveness and migratory capacities of NB cells with MYCN amplification.
Figure 5. Knockdown of GAS7 expression promotes the invasive and migratory properties of human NB cells.
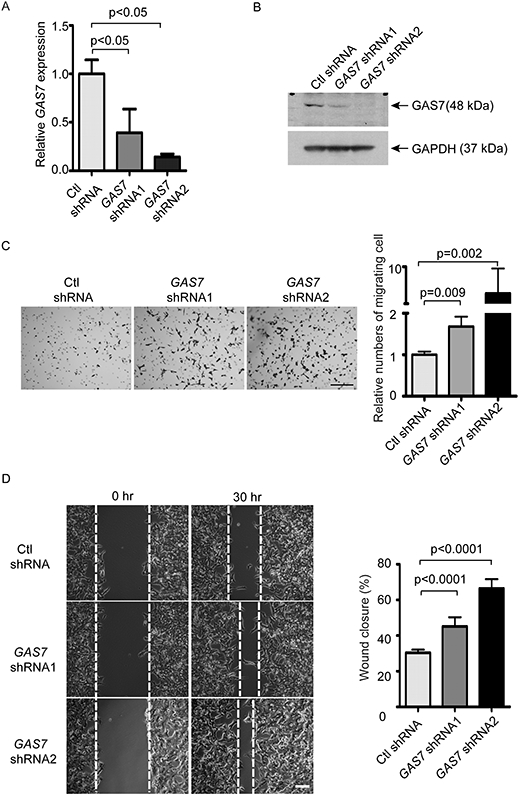
(A) Relative expression of GAS7 versus ACTIN in BE(2)-M17 NB cells after lentivirus infection with scrambled control shRNA (Ctl shRNA) or two independent GAS7 shRNAs (GAS7 shRNA1 or GAS7 shRNA2) by semiquantitative RT-PCR analysis. The data are presented as means ± SD of triplicate experiments; p< 0.05 by two-tailed t test (both comparisons).
(B) Immunoblotting of GAS7 expression in the BE(2)-M17 NB cells after lentivirus infection with control shRNA (Ctl shRNA), GAS7 shRNA1 or GAS7 shRNA2. Levels of GAPDH expression served as a loading control.
(C) Transwell invasion and migration assay of BE(2)-M17 cells infected with lentiviral control shRNA (Ctl shRNA), GAS7 shRNA1 or GAS7 shRNA2. Left: crystal violet-stained migrated cells. Right: relative numbers of cells migrated through the membrane. Scale bar, 200μm. Data are presented as means ± SD of triplicate experiments, differences between groups were analyzed by two-tailed t test.
(D) Wound-healing assay of BE(2)-M17 cells infected with the lentiviral control shRNA (Ctl shRNA) or GAS7 shRNAs (GAS7 shRNA1 or GAS7 shRNA2). Left: representative bright-field pictures at 0- or 30-hr time point. Right: quantification of wound healing in area to which the cells migrated. Scale bar, 200μm. Data are presented as means ± SD of triplicate experiments, differences between groups were analyzed by two-tailed t test.
To test the reverse prediction, that GAS7 overexpression suppresses the invasion and migration of tumor cells, we transduced a CMV promoter-mediated GAS7 expression vector by lentivirus infection of CHP-134 and BE(2)C NB cells, MYCN-amplified lines with low levels of endogenous GAS7 expression (Figure S2G). The increased GAS7 expression levels in the infected cells were detected by immunoblotting (Figure S4D and S4G), as compared to the cells infected with control vector. Stable overexpression of GAS7 significantly slowed down the migration of cells toward the lesion in the wound-healing assay (Figure S4E, S4F, S4H, and S4I) and significantly reduced the numbers of cells that invaded and migrated across the matrigel-coated membrane in the transwell assay (Figure S4J and S4K). Moreover, we did not observe any significant changes in growth rate of cells with knockdown or overexpression of GAS7 over a 4-day culture period (Figure S4L-N), eliminating the possibility that a growth effect had altered the invasion and migration properties of these tumor cells.
Molecular Pathways Altered by Reduced Expression of GAS7
To identify key genes associated with gas7 deficiency in the context of MYCN overexpression that might contribute to tumor metastasis, we first used RNA sequencing (RNA-seq) and gene set enrichment analysis (GSEA) to interrogate global gene expression profiles in tumors from MYCN;gas7mut compound fish with metastasis versus MYCN-only transgenic fish of similar age. We also performed GSEA on a publicly available human NB dataset (GEO: GSE49710) to identify genes and pathways that are significantly related to GAS7 expression in high-risk NBs with MYCN amplification. By comparing genes significantly downregulated in MYCN;gas7mut tumors with those positively associated with GAS7 expression in MYCN-amplified human NBs, we compiled a panel of gene signatures that are enriched and conserved in both fish and human tumors (Figure 6A), including those encoding extracellular matrix proteins, basement membrane proteins, and cell adhesion molecules. Interestingly, genes involved in actin or microtubule-related cellular structures or biological processes, including “contractile fiber”, “focal adhesion”, “actin cytoskeleton”, “cytoskeletal protein binding”, “apical junction” and “actin filament-based process” (Figure 6A), were also significantly altered. In support of this finding, we detected increased acetylation of tubulin in much higher percentage of GAS7-knockdown BE(2)-M17 cells in the leading edge toward the wound lesion by immunofluorescent staining of the acetylated K40 α-tubulin (Figure S5A-C), which is consistent with the known function of GAS7 in the control of cell cytoskeleton dynamics and cell migration (18, 19). To extend this analysis, we further examined expression levels of representative enriched genes, especially those involved in cell junction organization (Figure 6B) and cell adhesion (Figure 6C). Using qRT-PCR analyses of human NB cell lines, we detected downregulation of CDH6, CDH11 and CADM3 in BE(2)-M17 cells infected with GAS7 lentiviral shRNAs (Figure 6D) and decreased expression of their zebrafish homologs in MYCN;gas7mut fish tumors (Figure 6E).
Figure 6. Reduced GAS7 expression downregulates signaling pathways involved in tumor cell-cell interaction.

(A) Enriched and conserved GAS7-associated pathways in both zebrafish and human NBs. Bars on the left represent significantly downregulated pathways in MYCN;gas7mut versus MYCN-only zebrafish tumors by GSEA analysis. Bars on the right represent pathways that are significantly associated with GAS7 expression in MYCN-amplified human NBs (GEO: GSE49710 dataset). The highlighted pathways (in bold-italics) are shown in (B) and (C).
(B, C) GSEA analyses of expression profiles of MYCN;gas7mut versus MYCN-only tumors. Representative enriched gene signatures include the cell junction organization (B, REACTOME cell junction organization) and cell adhesion (C, KEGG cell adhesion molecules cams). Representative enriched genes that were subjected to qRT-PCR analysis are marked in the enrichment plots.
(D) Relative expression values of the representative genes identified from enriched biological processes in BE(2)-M17 cells infected with lentiviral control shRNA (Ctl shRNA), GAS7 shRNA1 or GAS7 shRNA2 by qRT-PCR analyses. All of the values were further normalized to the mean of expression of each given gene in the Ctl shRNA-infected BE(2)-M17 cells. The data are presented as means ± SD of triplicate experiments; ***p < 0.001 by two-tailed t test.
(E) Relative expression of the representative genes in three MYCN;gas7mut or three MYCN-only NBs by qRT-PCR analysis. All values were further normalized to the mean of expression of each given gene in MYCN-only tumors. The data are presented as means ± SD of triplicate experiments; *p < 0.05 and **p < 0.01 by two-tailed t test.
(F) Immunoblotting of MYCN and GAS7 expression in BE(2)-M17 NB cells infected with control shRNA (Ctl shRNA), GAS7 shRNA1 or GAS7 shRNA2. Levels of ACTIN expression served as a loading control.
(G) Immunoblotting of MYCN expression in three MYCN-only, two MYCN;gas7mut het (T1 and T2) and one MYCN;gas7mut homo (T3) NBs. The levels of Actin expression served as a loading control.
(H, I) Electron microscopic analysis of MYCN-only (MYCN) (H) or MYCN;gas7mut het (I) NBs. Enlarged boxed regions are shown on the top right corner within the same figure. Scale bar represents 2μm. Spaces among cells in MYCN;gas7mut het tumor are highlighted by arrows (I).
Since the well-established role of GAS7 is to coordinate the dynamics of actin cytoskeleton and microtubule, we hypothesize that downregulation of the genes mentioned above could be an indirect effect of reduced GAS7 expression. We detected increased MYCN protein levels in both BE(2)-M17 cells infected with GAS7 lentiviral shRNAs (Figure 6F) and MYCN-expressing fish tumors with heterozygous or homozygous loss of gas7 by immunoblotting analyses (Figures 6G and S5D). We also detected upregulation of CDH6, CDH11 and CADM3 in CHP-134 cells with overexpression of GAS7 (Figure S5E), while downregulation of their expression in BE(2)-M17 cells with transient overexpression of MYCN using qRT-PCR analysis (Figure S5F). Together, these data suggest a causative relationship between increased MYCN protein levels and downregulation of signature genes involved in cell junction organization or cell adhesion in NB cells with knockdown of GAS7 or fish tumors with knockout of gas7.
We then questioned whether cell-cell contact might be affected in tumors with gas7 deficiency. Thus, we performed electron microscopic analyses on three MYCN;gas7mut and MYCN-only tumors (Figures 6H, 6I and S5G). We observed non-cohesive tumor cells in all three MYCN;gas7mut fish tumors (Figure 6I and S5G lower panels), while the tumor cells in MYCN-only fish were all tightly connected (Figures 6H and S5G top panels), supporting the idea that reduced expression of cell adhesion genes in the gas7-knockout mutant disrupts the close contact among tumor cells, ultimately leading to widespread metastasis in MYCN;gas7mut compound fish.
Downregulation of GAS7 Promotes Bone Marrow Metastasis of Human NB Cells in Xenografted SCID Mice
To show that the enhanced NB metastasis mediated by knockout of Gas7 can be consistently recapitulated in an in vivo mammalian model, we transplanted the BE(2)-M17 NB cell line infected with a luciferase construct and GAS7 shRNA or control shRNA intraperitoneally into SCID mice. Twelve mice per group were injected with the same number of cells and monitored by Xenogen imaging every 3 weeks for evidence of tumor metastasis. During 11 weeks of observation, six mice injected with GAS7 shRNA-infected BE(2)-M17 cells died, compared to only one injected with control shRNA-infected BE(2)-M17 cells (Table S2), suggesting that the engrafted BE(2)-M17 cells with knockdown of GAS7 may be more aggressive than the engrafted control BE(2)-M17 cells. In addition, the bioluminescent-positive tumor masses expanded faster in mice transplanted with GAS7 shRNA-infected BE(2)-M17 cells than in those transplanted with control shRNA-infected BE(2)-M17 cells (Figures 7A-D and S6A). Additional tumor masses away from the engrafted primary tumors were detected in seven of nine mice transplanted with GAS7 shRNA-infected BE(2)-M17 cells versus only three of twelve mice transplanted with control shRNA-infected BE(2)-M17 cells by 9 weeks post-transplantation (p=0.03, two-tailed probability by Fisher’s exact test; Figures 7A-E and Table S3). These findings indicate that reduced expression of GAS7 in BE(2)-M17 cells promotes tumor cell dissemination in xenografted mice.
Figure 7. Knockdown of GAS7 promotes bone marrow metastasis of NB cells in xenografted SCID mice.

(A-D) Representative bioluminescence imaging of mice injected with luciferase-expressing BE(2)-M17 cells infected with control shRNA- (Ctl, A and C) or GAS7 shRNA (GAS7 KD, B and D) at 2 weeks or 9 weeks post-transplantation (wpt). Colored scale bar represents intensity of bioluminescence in counts. Scale bars, 2cm.
(E) Fraction of xenografted mice developed additional tumor masses away from the engrafted primary tumors at 9 wpt. The difference in the percentage of mice developed additional tumor masses in animals xenografted with control (Ctl) cells versus GAS7 KD cells is significant by Fisher’s exact test at p=0.03.
(F) Gel electrophoresis of PCR product of MYCN genotyping. Left, bone marrow from mice transplanted with control shRNA-infected-BE(2)-M17 NB cells, n=11. Right, bone marrow from mice transplanted with GAS7shRNA-infected-BE(2)-M17 NB cells, n=6. The expression levels of Gapdh serve as a loading control. Positive control (PC) was using gDNA from BE(2)-M17 NB cells as a template. Negative control (NC) was using gDNA from bone marrow of a non-injected SCID mouse as a template. The difference in fraction of mice with detection of MYCN-positive tumor cells in bone marrow of animals xenografted with control (Ctl) cells versus GAS7 KD cells is significant by Fisher’s exact test at p=0.05.
To further validate the metastatic phenotype in this model, we sacrificed all live mice at 11 weeks post-transplantation, including six injected with GAS7 shRNA-infected BE(2)-M17 cells and eleven injected with control shRNA-infected BE(2)-M17 cells (Table S2), and collected the bone marrow cells to detect the presence of MYCN-expressing metastatic tumor cells. By PCR analyses of the DNA extracted from those bone marrow cells, we detected the MYCN oncogene in bone marrow cells from five of six mice transplanted with GAS7-knockdown BE(2)-M17 cells (Figure 7F, right) and three of eleven mice transplanted with control BE(2)-M17 cells (p=0.05, two-tailed probability by Fisher’s exact test; Figure 7F, left). These results support enhanced metastasis of GAS7-knockdown BE(2)-M17 cells to the bone marrow of transplanted SCID mice.
To test the reverse prediction that GAS7 overexpression suppresses metastasis of tumor cells in vivo, we transplanted the same number of BE(2)C cells infected with a luciferase construct and a control vector or a CMV promoter-mediated GAS7 expression vector intraperitoneally into nine SCID mice per group. The injected mice were monitored by Xenogen imaging weekly and sacrificed at 4 weeks post-transplantation. Interestingly, mice transplanted with control BE(2)C cells developed bigger tumors with wider spread in the abdomen than those transplanted with GAS7-overexpressing BE(2)C cells (Figure S6B-E). In addition, we detected significantly increased abundance of human MYCN gene in the DNA extracted from bone marrow cells of five of nine mice transplanted with control BE(2)C cells versus two of nine mice transplanted with GAS7-overexpressing BE(2)C cells by semiquantitative PCR analysis (Figure S6F, p=0.004 by two-tailed Wilcoxon signed-rank test). Therefore, these data support that GAS7 overexpression reduced the metastatic ability of tumor cells in the transplanted SCID mice.
DISCUSSION
MYCN amplification has long been regarded as a hallmark of high-risk NB, since it consistently correlates with advanced-stage, metastatic disease. Although greatly increased expression of this oncogene can drive multiple facets of NB metastasis (48), very little is known about the ancillary proteins that are inhibited or commandeered by MYCN to achieve this feat. We show here, using both zebrafish and mammalian model systems, that reduced expression of the GAS7 gene at chromosome band 17p13.1 contributes importantly to the increased dissemination, invasion, and migration of MYCN-overexpressed or amplified NB cells. Heterozygous deletion of chromosome 17p accounts for the reduced GAS7 expression in a subset of these high-risk cases, while in many others it appears to result from amplified MYCN activity in the context of an intact 17p region. Briefly, our findings support the hypothesis that MYCN represses GAS7 expression by indirectly binding to its promoter via interaction with the SP1 transcription factor or by hypermethylating its promoter via PRC2 activity, and maintains GAS7 expression at low levels during the pathogenesis of high-risk NB without a 17p deletion. In addition to the known function of GAS7 in regulation of cytoskeleton dynamics and neurite formation (18, 19, 43), our RNA-sequencing, RT-PCR, and immunoblotting analyses show that knockdown or knockout of GAS7/gas7 downregulates the expression of a panel of genes involved in tumor cell-cell interaction, including cdh6, cdh11, cadm3, as well as their human homologs, and increases the expression of the MYCN protein, leading in turn to accelerated cell dissemination, migration and metastasis. These results not only provide genetic evidence casting GAS7 as a key suppressor of NB metastasis, but also support a feedback mechanism between GAS7 and MYCN that facilities loss of tumor cell-cell contact leading to enhanced dissemination of NB cells in high-risk cases with high levels of MYCN expression.
It has been shown for two decades that gain of chromosome arm 17q is associated with high-risk NBs (31-33), while the segmental chromosome defect involving loss of 17p was only identified recently in relapsed NB cases (49). Using an integrated WGS of tumor-normal pairs (n=135) and SNP array (n=914) analyses of primary NBs, we found a significant association of heterozygous 17p deletions with primary high-risk cases and poor survival. Although, at most, only about 15% of high-risk NBs would be expected to undergo a 17p deletion with consequent reduced expression of GAS7, we find that GAS7 expression is consistently downregulated in the absence of such deletions, in both human NB cells and PSNS cells from zebrafish, implicating a much broader role for GAS7 suppression in NB metastasis than previously suspected. This conclusion is reinforced by computational analyses of both human exon arrays from the TARGET database and RNA-seq data from the GSE62564 dataset, indicating that expression levels of GAS7 are significantly downregulated in MYCN-amplified high-risk cases with intact 17p regions, in contrast to cases without MYCN amplification. Although the main focus of our study was to clarify the role of GAS7 deficiency in NB metastasis in the context of MYCN overexpression or amplification, we suggest that such loss might also contribute to the pathogenesis of NB harboring a single-copy of MYCN, a prediction warranting further study in the near future.
Early bioinformatics analysis predicated that GAS7 may contain a structural domain resembling the Oct2 transcription factor (43), though no evidence has yet emerged to show that this adaptor protein can regulate gene transcription directly. It has been recently reported that overexpression of GAS7 affects stabilization of β-catenin protein via sequestering hnRNP U and reducing its interaction with proteins involved in ubiquitin-degradation pathway in lung cancer cells (21). Interestingly, hnRNP U is found to be able to stabilize CMYC mRNA in U2OS osteosarcoma cell line and hepatocellular carcinoma cells (50, 51). To test whether reduced GAS7 expression could potentially stabilize MYCN mRNA, we measured the MYCN mRNA half-life in the control- or GAS7-knockdown Kelly cells after Actinomycin D (ActD) treatment. Supporting our hypothesis, we detected an increased half-life of MYCN mRNA from ~77.8 minutes to ~102.8 minutes in the Kelly cells transfected with control siRNA versus GAS7 siRNA (Figure S7A), which also corresponded with increased MYCN expression at protein level (Figure S7B). Consistently, the increased expression levels of MYCN or EGFP-MYCN fusion protein were also detected in BE(2)M17 cells infected with GAS7 lentiviral shRNAs or in MYCN fish tumors with gas7 knockout (Figure 6F and 6G). These findings suggest that overexpression or amplification of MYCN could downregulate GAS7 expression, which in turn would be expected to maintain high levels of the MYCN protein to facilitate dissociation of tumor cells from the primary tumor and subsequent widespread dissemination (Figure S7C). Whether low levels of GAS7 expression are continuously required for the maintenance of metastatic tumor cells at the distant sites would be worth studying in the future.
Given the apparent role of GAS7 as a metastasis suppressor in MYCN-driven high-risk NB, its restoration after remission induction may provide a means to prevent subsequent dispersion and dissemination of tumor cells, especially when MYCN-mediated repression is the underlying mechanism of GAS7 depletion. Thus, we anticipate that the insights provided by this study could stimulate new translational strategies to counter the metastatic potential of high-risk NB with high levels of MYCN expression.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Heterozygous deletion or MYCN-mediated repression of GAS7 in NB releases an important brake on tumor cell dispersion and migration to distant sites, providing a novel mechanism underlying tumor metastasis in MYCN-driven NB.
ACKNOWLEDGMENTS
This work was supported by a grant R01 CA240323 (S.Z.) from the National Cancer Institute; a V Scholar award (D2018-005) from the V Foundation for Cancer Research (S.Z.) and a Platform Grant from the Mayo Center for Biomedical Discovery (S.Z.); support from the Mayo Clinic Cancer Center, and Center for Individualized Medicine (S.Z.); a grant R01 CA1247709, R01 CA180692 and R35 CA220500 (J.M.M.) from the National Cancer Institute.
Footnotes
The authors declare no potential conflicts of interest.
REFERENCES
- 1.Steeg PS. Targeting metastasis. Nature reviews Cancer. 2016;16(4):201–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fidler IJ, Kripke ML. The challenge of targeting metastasis. Cancer metastasis reviews. 2015;34(4):635–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer research. 2010;70(14):5649–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tevaarwerk AJ, Gray RJ, Schneider BP, Smith ML, Wagner LI, Fetting JH, et al. Survival in patients with metastatic recurrent breast cancer after adjuvant chemotherapy: little evidence of improvement over the past 30 years. Cancer. 2013;119(6):1140–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker DL, Schmidt ML, Cohn SL, Maris JM, London WB, Buxton A, et al. Outcome after reduced chemotherapy for intermediate-risk neuroblastoma. The New England journal of medicine. 2010;363(14):1313–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369(9579):2106–20. [DOI] [PubMed] [Google Scholar]
- 7.Park JR, Bagatell R, London WB, Maris JM, Cohn SL, Mattay KK, et al. Children's Oncology Group's 2013 blueprint for research: neuroblastoma. Pediatric blood & cancer. 2013;60(6):985–93. [DOI] [PubMed] [Google Scholar]
- 8.DuBois SG, Kalika Y, Lukens JN, Brodeur GM, Seeger RC, Atkinson JB, et al. Metastatic sites in stage IV and IVS neuroblastoma correlate with age, tumor biology, and survival. Journal of pediatric hematology/oncology. 1999;21(3):181–9. [DOI] [PubMed] [Google Scholar]
- 9.Maris JM. Recent advances in neuroblastoma. The New England journal of medicine. 2010;362(23):2202–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature. 2018;555(7696):321–7. [DOI] [PubMed] [Google Scholar]
- 11.Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teitz T, Inoue M, Valentine MB, Zhu K, Rehg JE, Zhao W, et al. Th-MYCN mice with caspase-8 deficiency develop advanced neuroblastoma with bone marrow metastasis. Cancer research. 2013;73(13):4086–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stupack DG, Teitz T, Potter MD, Mikolon D, Houghton PJ, Kidd VJ, et al. Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature. 2006;439(7072):95–9. [DOI] [PubMed] [Google Scholar]
- 14.Delloye-Bourgeois C, Bertin L, Thoinet K, Jarrosson L, Kindbeiter K, Buffet T, et al. Microenvironment-Driven Shift of Cohesion/Detachment Balance within Tumors Induces a Switch toward Metastasis in Neuroblastoma. Cancer cell. 2017;32(4):427–43 e8. [DOI] [PubMed] [Google Scholar]
- 15.Zhu S, Zhang X, Weichert-Leahey N, Dong Z, Zhang C, Lopez G, et al. LMO1 Synergizes with MYCN to Promote Neuroblastoma Initiation and Metastasis. Cancer cell. 2017;32(3):310–23 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang K, Diskin SJ, Zhang H, Attiyeh EF, Winter C, Hou C, et al. Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature. 2011;469(7329):216–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oldridge DA, Wood AC, Weichert-Leahey N, Crimmins I, Sussman R, Winter C, et al. Genetic predisposition to neuroblastoma mediated by a LMO1 super-enhancer polymorphism. Nature. 2015;528(7582):418–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.She BR, Liou GG, Lin-Chao S. Association of the growth-arrest-specific protein Gas7 with F-actin induces reorganization of microfilaments and promotes membrane outgrowth. Experimental cell research. 2002;273(1):34–44. [DOI] [PubMed] [Google Scholar]
- 19.Gotoh A, Hidaka M, Hirose K, Uchida T. Gas7b (growth arrest specific protein 7b) regulates neuronal cell morphology by enhancing microtubule and actin filament assembly. The Journal of biological chemistry. 2013;288(48):34699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanawa-Suetsugu K, Itoh Y, Ab Fatah M, Nishimura T, Takemura K, Takeshita K, et al. Phagocytosis is mediated by two-dimensional assemblies of the F-BAR protein GAS7. Nature communications. 2019;10(1):4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tseng RC, Chang JW, Mao JS, Tsai CD, Wu PC, Lin CJ, et al. Growth-arrest-specific 7C protein inhibits tumor metastasis via the N-WASP/FAK/F-actin and hnRNP U/beta-TrCP/beta-catenin pathways in lung cancer. Oncotarget. 2015;6(42):44207–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang JW, Kuo WH, Lin CM, Chen WL, Chan SH, Chiu MF, et al. Wild-type p53 upregulates an early onset breast cancer-associated gene GAS7 to suppress metastasis via GAS7-CYFIP1-mediated signaling pathway. Oncogene. 2018;37(30):4137–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li YF, Hsiao YH, Lai YH, Chen YC, Chen YJ, Chou JL, et al. DNA methylation profiles and biomarkers of oral squamous cell carcinoma. Epigenetics. 2015;10(3):229–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim YH, Lee HC, Kim SY, Yeom YI, Ryu KJ, Min BH, et al. Epigenomic analysis of aberrantly methylated genes in colorectal cancer identifies genes commonly affected by epigenetic alterations. Annals of surgical oncology. 2011;18(8):2338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Torabi K, Miro R, Fernandez-Jimenez N, Quintanilla I, Ramos L, Prat E, et al. Patterns of somatic uniparental disomy identify novel tumor suppressor genes in colorectal cancer. Carcinogenesis. 2015;36(10):1103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ronneberg JA, Fleischer T, Solvang HK, Nordgard SH, Edvardsen H, Potapenko I, et al. Methylation profiling with a panel of cancer related genes: association with estrogen receptor, TP53 mutation status and expression subtypes in sporadic breast cancer. Molecular oncology. 2011;5(1):61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conway K, Edmiston SN, May R, Kuan PF, Chu H, Bryant C, et al. DNA methylation profiling in the Carolina Breast Cancer Study defines cancer subclasses differing in clinicopathologic characteristics and survival. Breast cancer research : BCR. 2014;16(5):450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lopez G, Conkrite KL, Doepner M, Rathi KS, Modi A, Vaksman Z, et al. Somatic structural variation targets neurodevelopmental genes and identifies SHANK2 as a tumor suppressor in neuroblastoma. Genome research. 2020;30(9):1228–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists. 1995;203(3):253–310. [DOI] [PubMed] [Google Scholar]
- 30.Lutz W, Stohr M, Schurmann J, Wenzel A, Lohr A, Schwab M. Conditional expression of N-myc in human neuroblastoma cells increases expression of alpha-prothymosin and ornithine decarboxylase and accelerates progression into S-phase early after mitogenic stimulation of quiescent cells. Oncogene. 1996;13(4):803–12. [PubMed] [Google Scholar]
- 31.Bown N, Cotterill S, Lastowska M, O'Neill S, Pearson AD, Plantaz D, et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. The New England journal of medicine. 1999;340(25):1954–61. [DOI] [PubMed] [Google Scholar]
- 32.Caron H. Allelic loss of chromosome 1 and additional chromosome 17 material are both unfavourable prognostic markers in neuroblastoma. Medical and pediatric oncology. 1995;24(4):215–21. [DOI] [PubMed] [Google Scholar]
- 33.Plantaz D, Mohapatra G, Matthay KK, Pellarin M, Seeger RC, Feuerstein BG. Gain of chromosome 17 is the most frequent abnormality detected in neuroblastoma by comparative genomic hybridization. The American journal of pathology. 1997;150(1):81–9. [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Q, Diskin S, Rappaport E, Attiyeh E, Mosse Y, Shue D, et al. Integrative genomics identifies distinct molecular classes of neuroblastoma and shows that multiple genes are targeted by regional alterations in DNA copy number. Cancer Res. 2006;66(12):6050–62. [DOI] [PubMed] [Google Scholar]
- 35.Stallings RL, Nair P, Maris JM, Catchpoole D, McDermott M, O'Meara A, et al. High-resolution analysis of chromosomal breakpoints and genomic instability identifies PTPRD as a candidate tumor suppressor gene in neuroblastoma. Cancer research. 2006;66(7):3673–80. [DOI] [PubMed] [Google Scholar]
- 36.Mosse YP, Greshock J, Margolin A, Naylor T, Cole K, Khazi D, et al. High-resolution detection and mapping of genomic DNA alterations in neuroblastoma. Genes, chromosomes & cancer. 2005;43(4):390–403. [DOI] [PubMed] [Google Scholar]
- 37.Carr-Wilkinson J, O'Toole K, Wood KM, Challen CC, Baker AG, Board JR, et al. High Frequency of p53/MDM2/p14ARF Pathway Abnormalities in Relapsed Neuroblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16(4):1108–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu S, Lee JS, Guo F, Shin J, Perez-Atayde AR, Kutok JL, et al. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer cell. 2012;21(3):362–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bosse KR, Raman P, Zhu Z, Lane M, Martinez D, Heitzeneder S, et al. Identification of GPC2 as an Oncoprotein and Candidate Immunotherapeutic Target in High-Risk Neuroblastoma. Cancer cell. 2017;32(3):295–309 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iraci N, Diolaiti D, Papa A, Porro A, Valli E, Gherardi S, et al. A SP1/MIZ1/MYCN repression complex recruits HDAC1 at the TRKA and p75NTR promoters and affects neuroblastoma malignancy by inhibiting the cell response to NGF. Cancer research. 2011;71(2):404–12. [DOI] [PubMed] [Google Scholar]
- 41.Henrich KO, Bender S, Saadati M, Dreidax D, Gartlgruber M, Shao C, et al. Integrative Genome-Scale Analysis Identifies Epigenetic Mechanisms of Transcriptional Deregulation in Unfavorable Neuroblastomas. Cancer research. 2016;76(18):5523–37. [DOI] [PubMed] [Google Scholar]
- 42.Hung FC, Cheng YC, Sun NK, Chao CC. Identification and functional characterization of zebrafish Gas7 gene in early development. Journal of neuroscience research. 2013;91(1):51–61. [DOI] [PubMed] [Google Scholar]
- 43.Ju YT, Chang AC, She BR, Tsaur ML, Hwang HM, Chao CC, et al. gas7: A gene expressed preferentially in growth-arrested fibroblasts and terminally differentiated Purkinje neurons affects neurite formation. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(19):11423–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frost A, Perera R, Roux A, Spasov K, Destaing O, Egelman EH, et al. Structural basis of membrane invagination by F-BAR domains. Cell. 2008;132(5):807–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu S, Xiong X, Zhao X, Yang X, Wang H. F-BAR family proteins, emerging regulators for cell membrane dynamic changes-from structure to human diseases. Journal of hematology & oncology. 2015;8:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Renshaw SA, Trede NS. A model 450 million years in the making: zebrafish and vertebrate immunity. Disease models & mechanisms. 2012;5(1):38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Menke AL, Spitsbergen JM, Wolterbeek AP, Woutersen RA. Normal anatomy and histology of the adult zebrafish. Toxicologic pathology. 2011;39(5):759–75. [DOI] [PubMed] [Google Scholar]
- 48.Huang M, Weiss WA. Neuroblastoma and MYCN. Cold Spring Harbor perspectives in medicine. 2013;3(10):a014415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eleveld TF, Oldridge DA, Bernard V, Koster J, Colmet Daage L, Diskin SJ, et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet. 2015;47(8):864–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang B, Wang HY, Zhao DX, Wang DX, Zeng Q, Xi JF, et al. The splicing regulatory factor hnRNPU is a novel transcriptional target of c-Myc in hepatocellular carcinoma. FEBS letters. 2020. [DOI] [PubMed] [Google Scholar]
- 51.Weidensdorfer D, Stohr N, Baude A, Lederer M, Kohn M, Schierhorn A, et al. Control of c-myc mRNA stability by IGF2BP1-associated cytoplasmic RNPs. RNA. 2009;15(1):104–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.