

Abstract
Ferroptosis is a new form of regulated cell death resulting from the accumulation of lipid-reactive oxygen species (ROS). A growing number of studies indicate ferroptosis as an important tumor suppressor mechanism having therapeutic potential in cancers. Previously, we identified TAZ, a Hippo pathway effector, regulates ferroptosis in renal and ovarian cancer cells. Since YAP (Yes-associated protein 1) is the one and only paralog of TAZ, sharing high sequence similarity and functional redundancy with TAZ, we tested the potential roles of YAP in regulating ferroptosis. Here, we provide experimental evidence that YAP removal confers ferroptosis resistance, while overexpression of YAP sensitizes cancer cells to ferroptosis. Furthermore, integrative analysis of transcriptome reveals S-phase kinase-associated protein 2 (SKP2), an E3 ubiquitin ligase, as a YAP direct target gene that regulates ferroptosis. We found that the YAP knockdown represses the expression of SKP2. Importantly, the genetic and chemical inhibitions of SKP2 robustly protect cells from ferroptosis. In addition, knockdown of YAP or SKP2 abolishes the lipid peroxidation during erastin-induced ferroptosis. Collectively, our results indicate that YAP, similar to TAZ, is a determinant of ferroptosis through regulating the expression of SKP2. Therefore, our results support the connection between Hippo pathway effectors and ferroptosis with significant therapeutic implications.
Keywords: Ferroptosis, Hippo pathway, Yes-associated protein 1 (YAP), S-phase kinase-associated protein 2 (SKP2), cell death
Introduction
Ferroptosis
Ferroptosis is a newly recognized form of regulated cell death characterized by iron dependency and lipid peroxidation (1). Ferroptosis was first discovered while investigating the death mechanisms of erastin, a compound developed to eliminate cancer cells bearing RAS mutation. Since the initial description of ferroptosis, significant advances have been made in the genetic determinants and biological processes of ferroptosis. For example, other than iron, zinc is also essential for ferroptosis (2). Furthermore, the oxidative radicals generated by NADPH oxidases (NOXs) are important sources of oxidative stresses that lead to lipid peroxidation and ferroptosis. Furthermore, mammalian cells fight against ferroptosis using glutathione peroxidase 4 (GPX4) using glutathione as a co-factor (1,3). Therefore, ferroptosis can be triggered when the GPX4 function is compromised by either: 1) depletion of glutathione, caused by xCT inhibitors (erastin, sulfasalazine) or 2) direct inhibition of GPX4 by RSL. 3) In addition, ferroptosis can be also triggered by cystine deprivation (the limiting component for glutathione synthesis) or excessive glutamate that limits the efficacy of xCT and cystine import (3).
Regulators and genetic determinants of ferroptosis
Many genetic determinants have been identified to regulate ferroptosis by affecting different biological processes, including cystine metabolism, anti-oxidant capacity, lipid metabolism, and the level of labile iron, as reviewed in previous literatures (4-7). For example, a genome-wide CRISPR-based screen identified acyl-CoA synthetase long-chain family member 4 (ACSL4) and lipid reprogramming as essential for ferroptosis (8). MESH1 was recently found to be essential for ferroptosis as the first cytosolic NADPH phosphatase and its induction contributes to the NADPH depletion during ferroptosis (9). Recently, a kinome screen identified ATM and DNA damage response as a relevant determinant of ferroptosis by regulating iron metabolism (10,11) providing the rationales of using ionizing radiation to enhance ferroptosis efficacy. However, much remains unknown whether non-genetic factors may regulate ferroptosis sensitivity.
Hippo Pathway effectors: YAP/TAZ
The Hippo pathway is an evolutionarily conserved tumor suppressor pathway that senses the cell density and mechanic cues to regulate cell proliferation and organ sizes (12). The final biological function of the Hippo pathway is mediated through two paralogous transcriptional coactivators: Yes-associated protein 1 (YAP1; hereafter referred to as YAP) and WW-domain-containing transcription regulator 1 (WWTR1; also known as and hereafter referred to as TAZ). Both proteins are transcriptional co-activators that interact with the members of the transcriptional enhancer factor (TEA)-domain (TEAD) family of transcriptional factors to regulate the transcription of many genes in the proliferation, self-renewal, and cell death (13). YAP and TAZ are often overexpressed or amplified in many human tumors to mediate the proliferation, self-renewal, metastasis, and chemo-resistance of multiple cancer types. Both YAP and TAZ have overlapping functions and the expression of one protein often recapitulates the phenotypic effects seen by the other protein (14). YAP and TAZ proteins are also regulated similarly by the phosphorylation of a cascade of kinases in the upstream Hippo pathway to dictate their subcellular locations, degradation, and activation. Notably, evidence of the YAP-specific or TAZ-specific functions starts to emerge (15). It was therefore with great interest to investigate the shared and distinct functions of YAP and TAZ in different biological contexts.
Recently, we observed that ferroptosis sensitivity of cancer cells is highly influenced by cellular density and confluency (16,17). In addition, nutrient-dropping screens have identified the cystine addiction of renal and breast cancer cells (18,19). As summarized in recent literature (7), we found that the Hippo pathway effector, TAZ, is an important determinant of ferroptosis by regulating cell-type specific NOXs. Since YAP is the one and only paralog of TAZ, we tested for the potential roles of YAP to ferroptosis in cancer cells. Our results indicate that YAP regulates ferroptosis through SKP2, supporting the connection between Hippo pathway effectors and ferroptosis which may be evolutionarily conserved. Furthermore, we found that SKP2 knockdown repressed the expression of TTK (Threonine Tyrosine Kinase) and TFRC (Transferrin Receptor) mRNA, which contributes to the ferroptosis protection phenotypes. SKP2 is often over-expressed in tumors, associates with poor outcome, and plays critical functions in oncogenesis, cell cycle progression, and senescence. Our findings that link SKP2, as a target gene of YAP, to ferroptosis provide new insights into SKP2 as an attractive therapeutic target for cancers.
Materials and Methods
Cell lines, plasmids, and reagents
MDA-MB-231, H1975, MCF7, T47D, and HEK293T cell lines were obtained from American Type Culture Collection (ATCC) through Duke Cell Culture Facility. The RCC4 cell line was a kind gift from Dr. Denise Chan (University of California, San Francisco) and was further authenticated by DNA Diagnostics Center Medical with the short tandem repeat method. CAOV2 cell line was provided by Dr. Zhiqing Huang and Dr. Susan Murphy (Duke University), which was genetically authenticated at the Duke University Analysis Facility. Short tandem repeat profiling was performed at the Duke University DNA Analysis Facility on June 5, 2018; Mycoplasma testing was conducted at the Cell Culture Facility, Duke University on June 5, 2018. The cell lines were cultured in Dulbecco’s modified eagle medium (Thermo Fisher Scientific #11995) with 10% heat-inactivated fetal bovine serum (GE Healthcare Life Sciences #SH30070.03HI) in a humidified incubator at 37°C and 5% CO2. The YAPS127A plasmid (Addgene #33092) was a kind gift from Dr. Corinne Linardic (Duke University). pLKO.1-shYAP1 and pLKO.1-shYAP2 were purchased from Addgene (Addgene #27368 and #27369). pCMV-YAP-5SA, pCMV-YAP-5SA/S94A, FLAG-Skp2 (Addgene #27371, #33103 and #89142). TTK and TFRC cDNA (DNASU: HsCD00436189 and HsCD00940270) siYAPpool (M-012200-00-0005), siSKP2pool (M-003324-04-0005), and individual SKP2-targeting siRNAs (D-003324-05-0005 and D-003324-07-0005) were purchased from Dharmacon. siNT (Qiagen SI03650318) and individual YAP-targeting siRNAs were purchased from Qiagen: siYAP#1 target sequence: CAG GTG ATA CTA TCA ACC AAA; siYAP#2 target sequence: CAC ATC GAT CAG ACA ACA ACA. Erastin was obtained from the Duke University Small Molecule Synthesis Facility. SKP2 inhibitor II (Calbiochem-500517) and SKP2 inhibitor IV (Calbiochem-530236) were purchased from Sigma-Aldrich.
Overexpression and knockdown lentivirus infections
For stable overexpression and shRNA-mediated knockdown, the lentivirus infections were performed as previously reported(20). In short, HEK293T cells were transfected with TransIT-LT1 (Mirus Bio) with plasmids to generate lentivirus. After adding the lentivirus with 8 μg/ml polybrene in media, the target cells were selected with 1 μg/ml puromycin or 200 μg/ml hygromycin. For siRNA-mediated gene knockdown, cells were seeded and transfected with siRNAs with RNAiMax transfection reagent (ThermoFisher Scientific) for 2 days, which were followed by downstream applications.
Cell viability assays
The cells were seeded at the ratio of 2500 to 4000 cells per 96 well or 100,000 to 300,000 cells per 6 well and transfected with siRNAs for 2 days. When the cells reached around 70% confluence, erastin alone or co-treatment of erastin with SKP2 inhibitors, Ferrostatin-1 or Z-VAD-FMK were applied to the media for 24 hours. The cell viability was measured by the CellTiter-Glo assay (Promega #G7570) according to the manufacturer’s instructions or by crystal violet staining, which was further quantified by determining the absorbance at 570 nm after dissolving the staining in 10% acetic acid.
Lipid peroxidation measurement
The lipid peroxidation levels were determined by C11-BODIPY staining (Thermo Fisher Scientific D3861) as previously described (16). In short, cells were stained with 10 µM of C11-BODIPY dye for 1 hour after the cells were transfected with siRNAs for 2 days and erastin treatment overnight. The negative control: cells without C11-BODIPY staining and the positive control: cells treated with cumene hydroperoxide were included to adjust the PMT voltages of flow cytometry analysis (FACSCantoTM II, BD Biosciences).
RT-qPCR
The steps of RNA extraction and RT-qPCR were described previously(2). In short, the total RNAs were extracted by the RNeasy mini kit (Qiagen #74104) with the treatment of DNase I (Qiagen #79254). 1μg of total RNAs were reverse transcripted into cDNAs by SuperScriptTM II Reverse Transcriptase (ThermoFisher Scientific #18064) with random hexamers. The gene expressions were measured by StepOnePlus qPCR System with Power SYBR Green PCR Mix (Applied Biosystems, ThermoFisher Scientific #4367659). The primers used are listed in Table S1.
Western blots
Total cell lysates were prepared after washing cells with ice-cold phosphate-buffered saline (PBS), lysed in RIPA buffer (Sigma R0278) containing freshly added protease inhibitor (Roche #04693159001) and PhosSTOP phosphatase inhibitor cocktail (Roche #04906837001). The protein amount in cell lysates was quantified by BCA protein assay (ThermoFisher Scientific #23228). Equal amounts of protein extracts were boiled with 4x laemmli buffer, loaded in SDS-PAGE, and transferred to the PVDF membrane (Millipore #IPVH00010). The membranes were then blocked with 5% non-fat milk or BSA for 1 h at room temperature and incubated overnight at 4 °C with 1:1000 dilution of primary antibodies (YAP/TAZ: Cell Signaling Technology (CST) #8418; YAP: CST #14074; beta-tubulin: CST #86298; SKP2: Thermo Fisher Scientific #323300; vinculin: Santa Cruz Biotechnology #sc-73614). The membranes were later incubated with HRP-conjugated goat anti-rabbit or anti-mouse IgG antibodies (CST #7074 or CST #7072) for 1h at room temperature. After washing, the membranes were developed and captured by Amersham ECL prime western blotting detection reagent (GE Healthcare Life Sciences RPN2232) and Bio-Rad ChemiDocTM Imaging System.
Transcriptome analysis
The analyses of microarray (17) and RNA-seq (16) were previously described and the data were deposited into NCBI Genome Expression Omnibus (GEO) as GSE146354 (microarray of YAP knockdown in RCC4 cells) and GSE146353 (RNA-seq of YAP knockdown in CAOV2 cells). Briefly, RCC4 cells were treated with siYAP for two days and then treated with 1 μM erastin for 7 hours. Total RNAs were extracted by RNeasy mini kit, labeled, and hybridized by Affymetrix U133A 2.0 arrays. RMA (robust multi-array average) method was used to analyze the intensities of probes and then zero transformation (Δlog2) was applied compared to the control group, siNT, non-targeting. Later, those probesets with 20.8-fold changes in at least two samples were selected for hierarchical clustering. The RNA-Seqwas performed on the Illumina HiSeq4000 50bp SR system at Sequencing and Genomic Technologies Shared Resource at Duke Center for Genome and Computational Biology.
Datamining
Human breast invasive carcinoma samples from the TCGA dataset were plotted by GEPIA (21) for pair-wise gene expression correlation analysis with Pearson correlation coefficient in Fig S2I.
Statistical analyses and data availability
The data were analyzed by Student’s t-test or ANOVA (one- or two-way) using GraphPad Prism version 8.0.1 (GraphPad Software). Error bars represent SEM, and p-value < 0.05 was considered significantly different (*p < 0.05; **p < 0.01; and ***p < 0.001). All data and reagents in this study are available from the authors upon request.
Results
YAP regulates ferroptosis
MDA-MB-231 cell is a basal-type breast cancer cell line known to be sensitive to ferroptosis and a well-established cellular model for studying YAP/TAZ (22). To ensure the specificity of cell death phenotype, we validated that the erastin-induced reduction in cell viability of MDA-MB-231 was rescued by ferroptosis inhibitor (Ferrostatin-1), but not the caspase inhibitor (Z-VAD-FMK) (Fig. S1A-B). To determine the role of YAP in the ferroptosis of MDA-MB-231, we knockdowned YAP by siRNAs and confirmed the knockdown efficiency by RT-qPCR (Fig. 1A). To exclude the possibility of potential off-target effects from the pooled siRNAs (siYAP pool), we further validated the results using two additional individual siRNAs (siYAP #1 and siYAP #2). Cell viability assay revealed that YAP knockdown by multiple siRNAs all significantly rescued the erastin-induced ferroptosis (Fig. 1B). To further validate the results, we performed stable YAP knockdown by plasmid-encoded short-hairpin RNAs (shRNAs) that target different regions of YAP transcripts from the siRNAs. The knockdown efficiency of shRNAs for YAP at RNA and protein levels was confirmed by RT-qPCR and western blot, respectively (Fig. 1C-D). We found that the stable knockdown of YAP by shRNAs also significantly rescued erastin-induced ferroptosis in MDA-MB-231 cells as well as a luminal breast cancer cell line (MCF7) (Fig. 1E, Fig S1C). We then extended our study to another cell line, H1975, a non-small cell lung cancer cells found to highly sensitive to ferroptosis. The two YAP shRNAs reduced the YAP mRNA and protein levels in H1975 (Fig. 1F-G). Interestingly, while TAZ knockdown led to a compensatory overexpression of YAP (16,17), the knockdown of YAP in multiple cell lines did not significantly increase TAZ protein levels in MDA-MB-231 (Fig. 1D), H1975 (Fig. 1G), MCF7 (Fig. S1D) and T47D cells (Fig. S1E). Importantly, YAP knockdown in H1975 also conferred resistance to erastin-induced ferroptosis using CellTiter-Glo cell viability assay (Fig. 1H) and phase-contrast microscopy (Fig. 1I). On the other hand, the expression of a phosphorylation-defective and constitutively active form of YAP (YAPS127A) (23) in RCC4 cell (Fig. 1J), significantly sensitized to ferroptosis (Fig. 1K). Consistently, another constitutively active and phosphorylation-defective form of YAP (YAP-5SA) also sensitized ferroptosis in H1975 cells (Fig. S1F). We also tested the wild-type YAP and found it also sensitized ferroptosis YAP, but the effects seemed milder than the phosphorylation-defective YAP mutant (Fig S1F). To confirm that the transcriptional activity of YAP was essential for promoting ferroptosis, we introduced a transcriptional inactive S94A mutant on YAP-5SA and found this additional mutation eliminated the ferroptosis-enhancing capacity in RCC4 cells (Fig. S1G). Collectively, these results indicate the ferroptosis phenotype of YAP depend on transcriptional activity of YAP. This is consistent with a previous report showing that NF2-YAP, regulated by cell contact, mediates ferroptosis in mesothelioma cells (24). To compare the ferroptosis resistance of YAP, TAZ or combined YAP/TAZ in MDA-MB-231 cells, we compared the ferroptosis protection of both YAP and TAZ knockdown, or in combination (Fig. S1H). We found that a sub-optimal knockdown of either YAP or YAZ still protected erastin-induced ferroptosis with siYAP had a stronger effect (Fig. S1H). However, the combined knockdown of both YAP and TAZ had the most robust protection effects against erastin (Fig. S1H), indicating the role of both YAP and TAZ in MDA-MB-231 cells. Collectively, these data strongly support that YAP, like TAZ, is a robust regulator of ferroptosis in multiple cancer cell types.
Figure 1. YAP regulates ferroptosis.
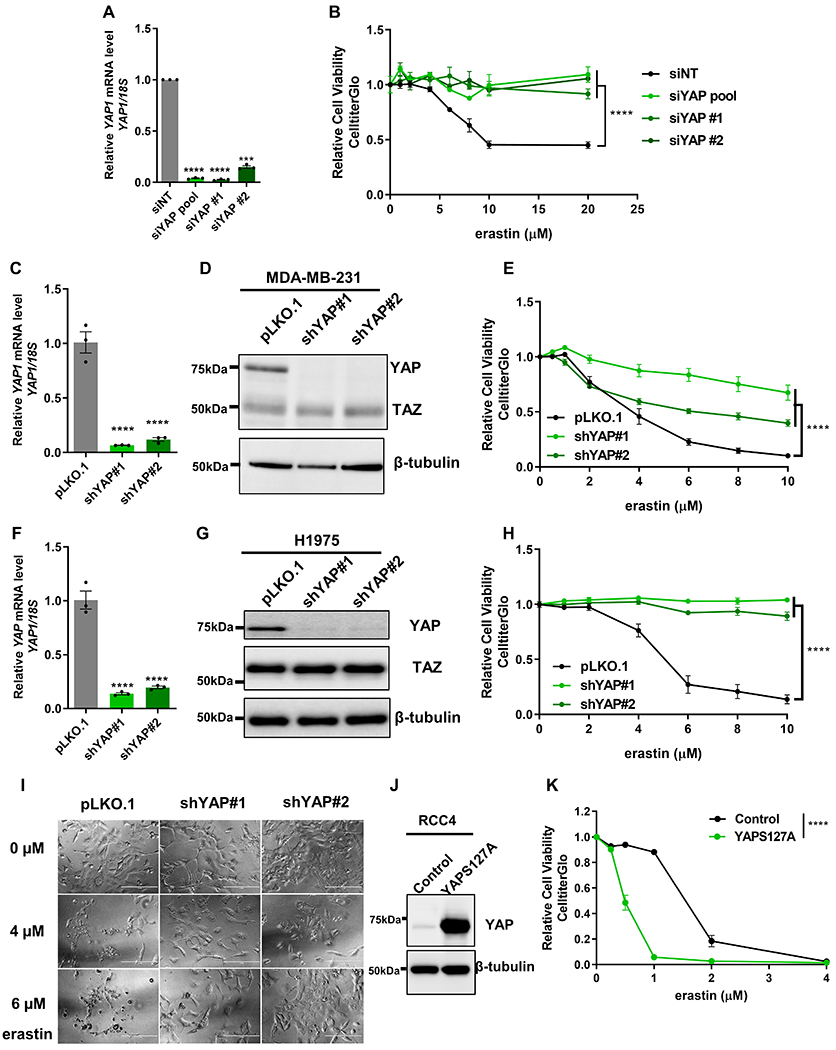
(A) The knockdown efficiency of siRNAs targeting YAP in MDA-MB-231 cells was confirmed by RT-qPCR. n=3; mean ± SEM; one-way ANOVA; ***p < 0.001; ****p < 0.0001.
(B) Knockdown of YAP by pooled or individual siRNAs rendered MDA-MB-231 cells more resistant to erastin-induced ferroptosis. n=2; mean ± SEM; two-way ANOVA; ****p < 0.0001.
(C) The knockdown efficiency of shRNAs targeting YAP in MDA-MB-231 cells was confirmed by RT-qPCR. n=3; mean ± SEM; one-way ANOVA; ****p < 0.0001.
(D) The knockdown efficiency of shRNAs targeting YAP was confirmed by western blots in MDA-MB-231 cells.
(E) The knockdown of YAP by shRNAs rendered MDA-MB-231 cells more resistant to erastin. n=4; mean ± SEM; two-way ANOVA; ****p < 0.0001.
(F) The knockdown efficiency of shRNAs targeting YAP in H1975 cells was confirmed by RT-qPCR. n=3; mean ± SEM; one-way ANOVA; ****p < 0.0001.
(G) The knockdown efficiency of shRNAs targeting YAP in H1975 cells was confirmed by western blots.
(H) Knockdown of YAP by shRNAs rendered H1975 cells more resistant to erastin-induced ferroptosis using Cell Titer Glo. n=4; mean ± SEM; two-way ANOVA; ****p < 0.0001.
(I) Knockdown YAP by shRNAs lets H1975 more resistant to erastin-induced death when observed under light microscopy. Scale bar: 200 µm.
(J) Western blot of RCC4 with transduced with either control vector or constitutively activated form of YAP, YAPS127A.
(K) The constitutively activated form of YAP, YAPS127A, sensitized RCC4 cells to erastin-induced ferroptosis. n=3; mean ± SEM; two-way ANOVA; ****p < 0.0001.
Identify YAP target genes relevant for ferroptosis through an integrated genomic approach
Because YAP is a transcriptional coactivator that mediates various phenotypes through affecting gene expression, we hypothesized that a subset of YAP target mRNAs was essential for ferroptosis and their reduced expression upon YAP knockdown protected ferroptosis. To identify such YAP target genes that may regulate ferroptosis, we took an integrative genomic approach by comparing the transcriptome response to YAP knockdown and previously completed genome-wide siRNA screen under ferroptosis (2). First, we knocked down YAP expression (Fig. 2A) and used microarrays to define the transcriptional response (Fig. 2B). RNA was isolated from RCC4 cells transfected with either control (siNT) or YAP-targeting siRNAs (siYAP) in triplicates (Figure 2A-B), with or without erastin treatments (GSE146354). We found that the knockdown of YAP specifically reduced the expression of YAP, but not to its paralog gene TAZ. Instead, there was a modest increase of WWTR1 (TAZ) mRNA (Fig. 2A). Gene expression analysis revealed YAP knockdown repressed the expression of several known YAP target genes such as the BCL2 family gene (MCL1) (25) and cell cycle-related genes (AURKA/B, CDC20, CCNA2) (26), consistent with previous studies (Fig. 2B). To identify YAP target genes that are essential for ferroptosis, we integrated these downregulated genes upon YAP knockdown with a genome-wide RNAi screen of cystine deprivation-induced ferroptosis (2,17) (Fig. 2C). Among the 27 candidate genes identified in this analysis (Fig. 2C), some have been reported as ferroptosis markers or regulators such as ACSL4 (Acyl-CoA Synthetase Long-Chain Family Member 4) (8), supporting the value of the approach and the list of candidates. In addition, we noted that the YAP knockdown repressed the levels of TTK (Fig. S2A), the top kinase hit essential for ferroptosis in our kinome screen (11). Among the candidates, we were especially interested in SKP2 (S-Phase Kinase-Associated Protein 2), encoding the substrate recognition component of the SCF E3 ubiquitin ligase complex that is important for cell cycle progression (27), because of the following reasons. First, we used RT-qPCR to validate the mRNA downregulation of some candidate genes after YAP removal and found SKP2 mRNA was significantly repressed (Fig. 2D and Fig. S2A-E). Similar SKP2 repression upon YAP knockdown was further supported in an ovarian cancer cell line, CAOV2, by RNA-seq (GSE146353; Fig. 2E and Fig. S2F) as well as at the protein level in MDA-MB-231 breast cancer cells (Fig. 2F). Interestingly, TAZ knockdown in MDA-MB-231 cells (Fig. S2G) also downregulated the SKP2 level (Fig. S2H). Second, gene set enrichment analysis (GSEA) of YAP-knockdown microarray data revealed a significant depletion of multiple cell cycle gene sets (Fig. 2G), consistent with the expected SKP2 depletion phenotypes. Third, we analyzed the expression levels of YAP and SKP2 and found a significant positive correlation (p=1.3e-15) in the breast invasive carcinoma dataset by GEPIA (Gene Expression Profiling Interactive Analysis) database (21) (Fig. S2I). Fourth, previous chromatin immunoprecipitation (ChIP)-seq studies have indicated that the regulatory regions of SKP2 gene were physically associated with YAP-TEAD transcriptional complex (28,29), suggesting that SKP2 is a direct target gene of YAP.
Figure 2. Identify potential YAP target genes that regulate ferroptosis through integrated genomics.
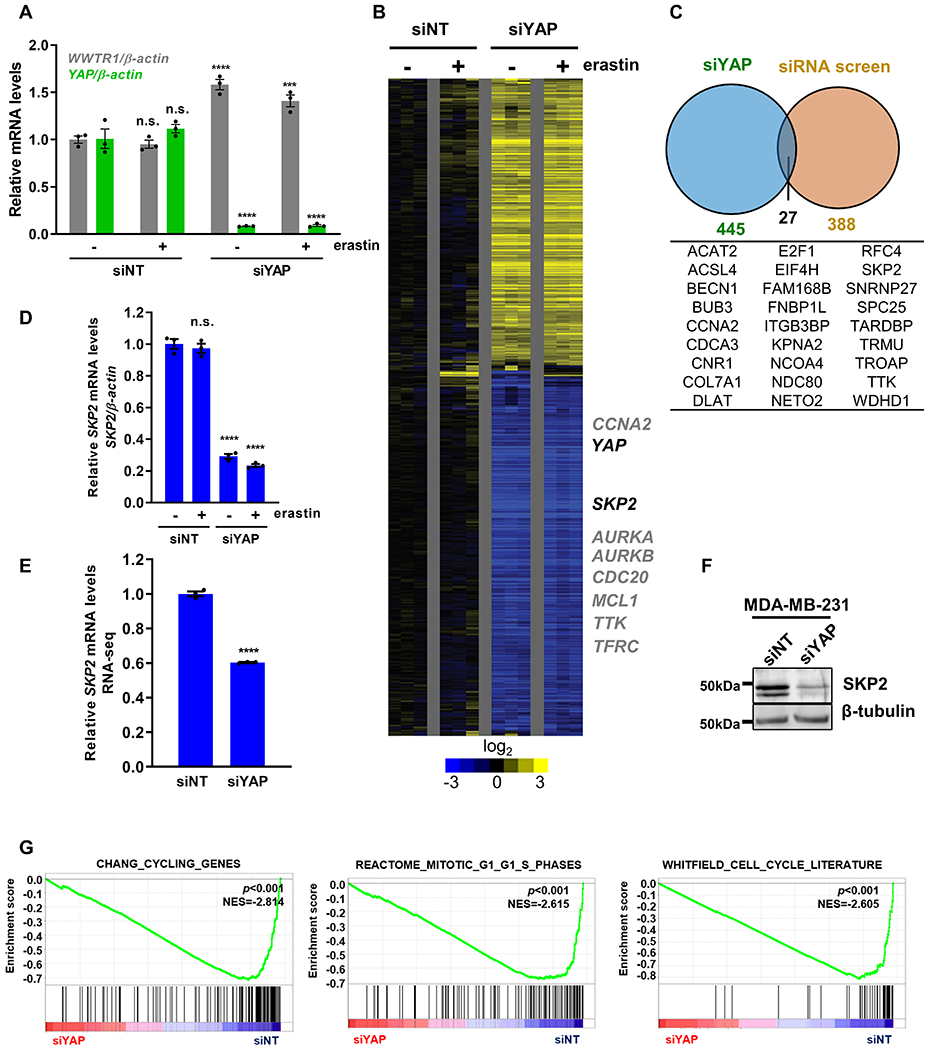
(A) The mRNA expression levels of YAP and WWTR1 (TAZ) were determined by RT-qPCR after siRNA-mediated YAP knockdown in RCC4 cells. n=3; mean ± SEM; one-way ANOVA; n.s.: not significant; ***p < 0.001; ****p < 0.0001.
(B) The heatmap of the transcriptome response of RCC4 cells to YAP knockdown without (-) or with (+) erastin treatment. n=3 per group.
(C) Venn diagram showing genes that were both downregulated upon YAP knockdown and identified as hits in the siRNA ferroptosis screen.
(D) RT-qPCR was used to validate the downregulation of SKP2 mRNA level upon YAP knockdown in RCC4 cells. n=3; mean ± SEM; one-way ANOVA; n.s.: not significant; ****p < 0.0001.
(E) RNA-seq revealed that SKP2 mRNA level was downregulated when YAP was knockdown in CAOV2 cells. n=3; mean ± SEM; Student’s t-test; ****p < 0.0001.
(F) Western blots validated that YAP knockdown decreased SKP2 protein level in MDA-MB-231 cells.
(G) GSEA analyses show the YAP knockdown in RCC4 cells led to the depletion of several cell cycle-related genesets.
SKP2 is essential for ferroptosis
To investigate if SKP2 plays a role in regulating ferroptosis, we manipulated SKP2 expression and activities by both genetic knockdown and pharmacological inhibition. First, we knock downed SKP2 by pooled and individual siRNAs in MDA-MB-231 cells (Fig. 3A) and found that SKP2 silencing by all tested siRNAs robustly protected the cells from erastin-induced cell death (Fig 3B). SKP2 knockdown also conferred ferroptosis resistance in RCC4 cells (Fig. 3C-D). Next, we treated MDA-MB-231 cells with two different cell-permeable SKP2 inhibitors, SKP2 inhibitor II (30) and SKP2 inhibitor IV (31). We found both SKP2 inhibitors also significantly protected MDA-MB-231 cells from ferroptosis using crystal violet staining or CellTiter-Glo assay (Fig. 3E-H). Similarly, the ferroptosis protection effect of these SKP2 inhibitors was also observed in RCC4 cells based on crystal violet staining (Fig. 3I-K) or CellTiter-Glo assay (Fig. 3L-M). Taken together, these data provide compelling evidence that SKP2 is essential for the ferroptosis sensitivity of multiple cancer cells.
Figure 3. SKP2 is essential for ferroptosis.
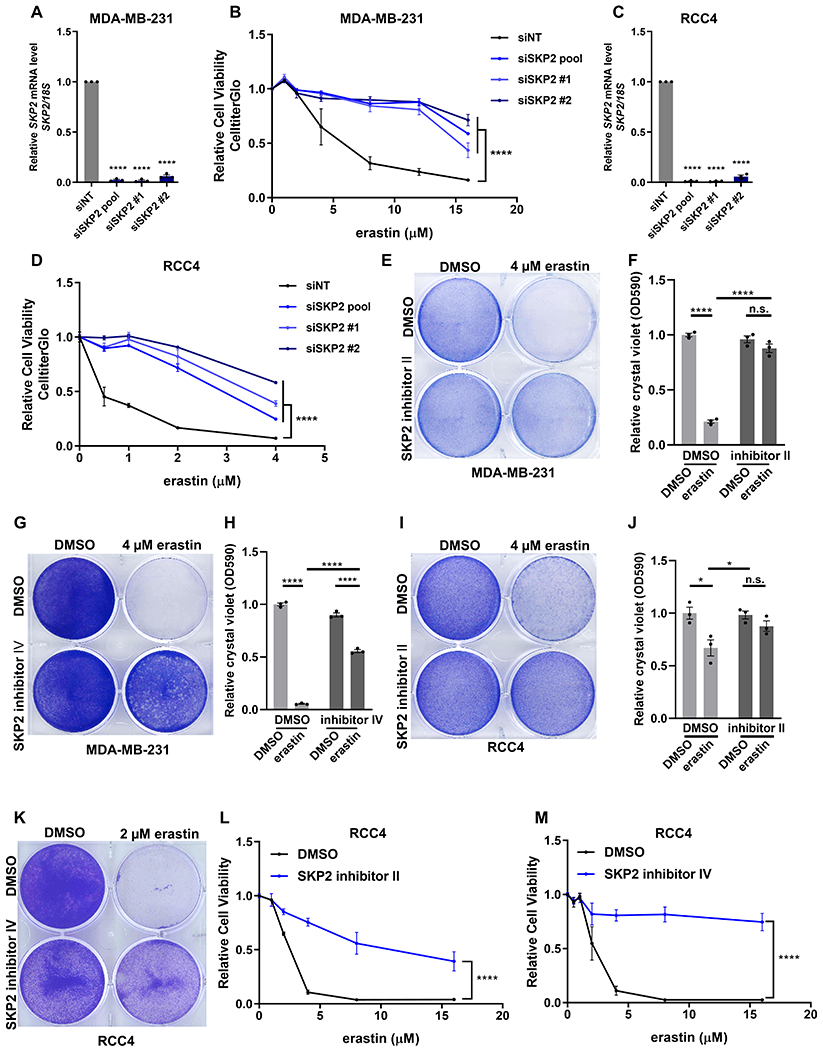
(A) The knockdown efficiency of siRNAs targeting SKP2 was confirmed by RT-qPCR in MDA-MB-231 cells. n=3; mean ± SEM; one-way ANOVA; ****p < 0.0001.
(B) The relative cell viabilities of MDA-MB-231 cells after transfection with control (siNT) or SKP2-targeting siRNAs by pooled siRNAs (siSKP2 pool) or individual siRNAs (siSKP2 #1 and siSKP2 #2) for 2 days. The transfected cells were then treated with indicated dosages of erastin for 1 day and evaluated for their viability. n=3; mean ± SEM; two-way ANOVA; ****p < 0.0001.
(C) The knockdown efficiency of siRNAs targeting SKP2 in RCC4 cells was confirmed by RT-qPCR. n=3; mean ± SEM; one-way ANOVA; ****p < 0.0001.
(D) The relative cell viabilities of RCC4 cells after SKP2 knockdown using indicated siRNAs followed by erastin treatment. n=3; mean ± SEM; two-way ANOVA; ****p < 0.0001.
(E-H) Relative cell viability of MDA-MB-231 cells after 1-day treatment of erastin with 40 μM SKP2 inhibitor II (E-F) or 10 μM SKP2 inhibitor IV (G-H). The viabilities were evaluated by either crystal violet staining (E, G) or CellTiter-Glo assays (F, H). n=3; mean ± SEM; two-way ANOVA; n.s.: not significant; ****p < 0.0001.
(I-M) Cell viability assays of RCC4 cells after 1-day treatment of erastin with 40 μM SKP2 inhibitor II (I-J, L) or 10 μM SKP2 inhibitor IV (K, M) by crystal violet staining (I, K) and CellTiter-Glo assays (J, L, M). n=3; mean ± SEM; two-way ANOVA; n.s.: not significant; *p < 0.05; ****p < 0.0001.
SKP2 is essential for the YAP-enhanced ferroptosis and lipid peroxidation
To investigate the mechanistic link of ferroptosis by YAP and SKP2, we first confirmed that YAPS127A overexpression upregulated SKP2 protein expression in RCC4 cells (Fig. 4A). By knocking down SKP2, the ferroptosis-enhancing effect of YAPS127A overexpression was almost completely abolished as measured by CellTiter-Glo assay (Fig. 4B). Therefore, the downregulation of SKP2 is a major contributes to the ferroptosis resistance conferred by the YAP silencing. Since the hallmark of ferroptosis is lipid ROS, we determined the features of YAP or SKP2 on the lipid peroxidation levels by flow cytometric analysis with C11-BODIPY581/591 staining. Compared to the control group, we observed that knockdown of YAP or SKP2 significantly reduced the erastin-induced increase of lipid peroxidation (Fig. 4C). Consistently, we found that the overexpression of YAP S127A increased the level of lipid peroxidation under erastin treatment (Fig. 4D). Furthermore, this increase was abolished by SKP2 knockdown (Fig. 4D), consistent with the role of SKP2 in the ferroptosis-enhancing capacity of YAP S127A. Reciprocally, the over-expression SKP2 cDNA overrides the ferroptosis resistance by YAP knockdown (Fig. 4E). Taken together, these data suggest that SKP2 is the downstream target of YAP to regulate ferroptosis.
Figure 4. SKP2 is essential for the YAP-enhanced ferroptosis and lipid peroxidation.
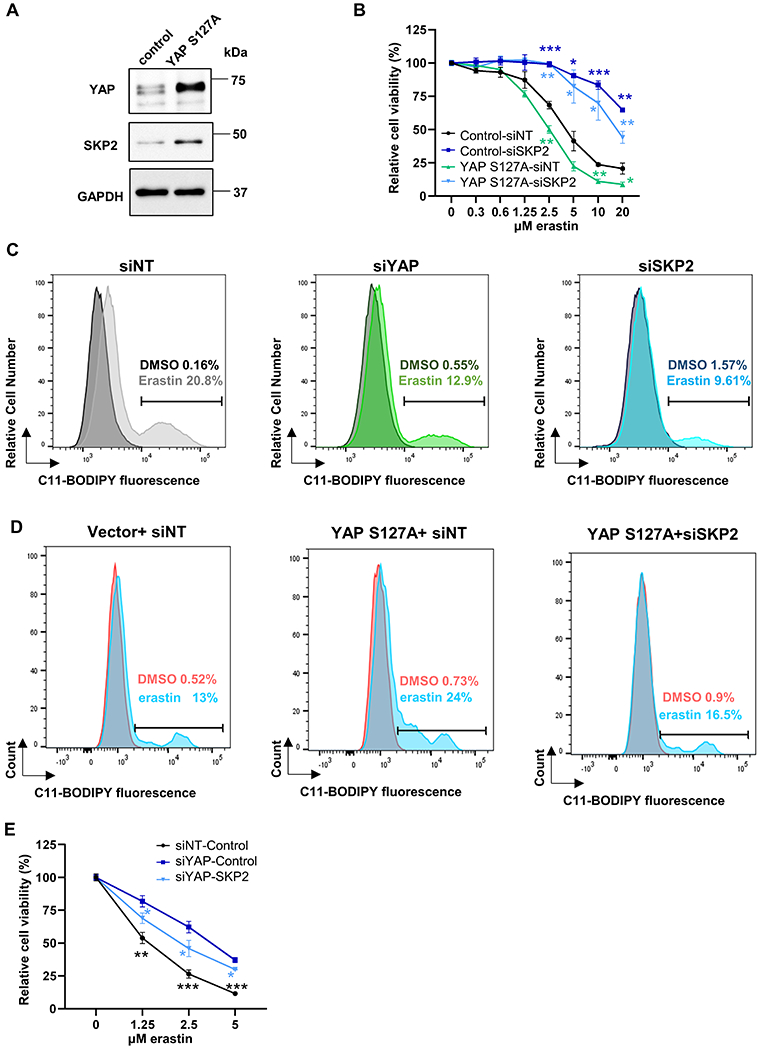
(A) The overexpression of YAP S127A in RCC4 cells upregulated SKP2 protein as determined by western blots.
(B) Genetic interaction between YAP and SKP2. RCC4 cells were treated with siSKP2 after YAP was stably overexpressed. The data of cell viability determined by CellTiter-Glo were normalized to the DMSO controls. n=3; mean± SEM, two-way ANOVA; n.s.: not significant; ****p < 0.0001.
(C) Inhibition of YAP or SKP2 decreases the elevated lipid peroxidation induced by erastin treatment. Representative data from one of three independent experiments in MDA-MB-231 cells are shown.
(D) The increase in lipid peroxidation triggered by YAP-S127A upon erastin treatment was abolished by SKP2 knockdown. Representative data from one of three independent experiments in RCC4 cells are shown.
(E) Overexpression of SKP2 cDNA abolished the ferroptosis resistance triggered by YAP knockdown in RCC4 cells. n=3; mean ± SEM; two-way ANOVA; *p < 0.05, **p < 0.01, ***p < 0.001.
TTK and TFRC are potential SKP2 target genes that regulate ferroptosis.
Although p21 is a well-known direct target of SKP2 (32) shown to regulate ferroptosis (33,34), we did not observe a significant effect of p21 manipulation on SKP2 knockdown-mediated ferroptosis resistance. Therefore, we investigate the genes downstream of SKP2 whose expression was affected by YAP knockdown and may affect ferroptosis (Fig. 2B). Interestingly, we found that both TTK (Threonine Tyrosine Kinase) and TFRC (Transferrin Receptor) mRNAs were both downregulated by SKP2 knockdown (Fig. 5A-C). As noted earlier, TTK was also repressed by YAP knockdown (Fig. S2A) and the top hit in a kinome screen of ferroptosis (11). Furthermore, TFRC is involved in iron uptake and is recognized as a marker of ferroptosis (35). TFRC was also found to be repressed by YAP removal (24). Given the downregulation of TFRC and TTK upon SKP2 knockdown, we determined their contribution to the ferroptosis protection phenotypes. The restoration of either TTK or TFRC significantly mitigated the ferroptosis protection of SKP2 knockdown (Fig. 5D). Therefore, TTK and TFRC downregulation contributed significantly to the ferroptosis protections of SKP2 knockdown. Together, we have provided data to support that YAP regulates ferroptosis by affecting the expression of SKP2, which in turn regulates the expression of TTK and TFRC, resulting in the ferroptosis protection phenotypes.
Figure 5. The repression of TTK and TFRC contributed to the ferroptosis resistance by SKP2 knockdown.
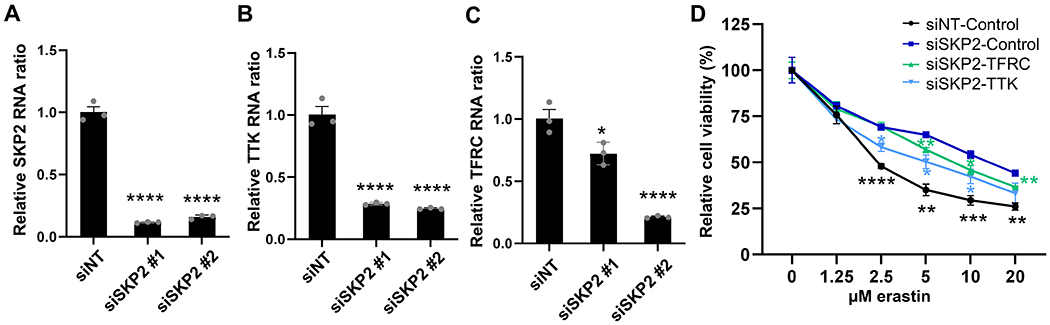
(A) Validation of SKP2 knockdown by siRNA in MDA-MB-231 cells by qRT-PCR. n=3; mean ± SEM; one-way ANOVA; ****p < 0.0001.
(B-C) TTK (B) and TFRC (C) RNAs were downregulated by SKP2 knockdown in MDA-MB-231 cells by qRT-PCR. n=3; mean ± SEM; one-way ANOVA; ****p < 0.0001.
(D) Overexpression of TTK and TFRC resensitized RCC4 cells to ferroptosis protected by SKP2 knockdown. n=3; mean ± SEM; two-way ANOVA; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Discussion
Here, we present compelling evidence that YAP, same as TAZ, is critical for ferroptosis in cancer cells. A similar observation has been reported in the contexts of the cell contact in mesothelioma cells by another research group (24). We extended the finding of YAP-regulated ferroptosis to additional cancer cells and identified SKP2 as a YAP target gene that regulates ferroptosis. To the best of our knowledge, this is the first report showing the regulation of ferroptosis by SKP2. Since SKP2 is a key regulator of the cell cycle regulation, senescence, and oncogenesis (36), our results reveal the potential connection between ferroptosis with these SKP2-regulated processes. Together with our previous findings on the role of TAZ in regulating ferroptosis, these results have shown the role of both Hippo pathway effectors in regulating ferroptosis in a wide variety of cancer cells.
The components of the Hippo pathway are highly conserved during evolution. Upstream of YAP/TAZ, the Hippo pathway comprises a kinase cascade that regulates YAP and TAZ across multiple organisms. These kinases integrate the upstream inputs from a wide variety of external stimuli and environmental cues to regulate the activity of YAP/TAZ and downstream cellular proliferation and organ sizes (12,13). These environmental factors that affect the YAP and TAZ activities include cell density, cellular contact, mechanical properties, pressures, and metabolic status (12). Therefore, the regulation of ferroptosis susceptibility by YAP/TAZ suggests that the potential role of various mechanic stresses, metabolic cues, and osmotic pressures in solid tumors to regulate ferroptosis of tumor cells. For example, both epithelial-mesenchymal transition (EMT) and fibrosis are prominent features associated with enhanced ferroptosis sensitivity (37). While the basis of the EMT-associated ferroptosis remains largely unknown, these EMT-associated mechanical “stiff” environments may activate the YAP/TAZ and promote the ferroptosis of cancer cells. Furthermore, the dynamic and spatial changes in the YAP/TAZ activity during development may modulate the ferroptosis sensitivity of different tissues or distinct regions and shape the organ development and tissue regeneration, similar to the role of other regulated cell death in the proper development. As the evolutionary conservation of the Hippo pathway, ferroptosis-like processes have been described in several model organisms, including C. elegans (38), D. melanogaster (39), and A. thaliana (40). Therefore, it will be important to determine whether the connection between the Hippo pathway and ferroptosis is also evolutionary conserved across diverse model organisms.
Overexpression of SKP2 in tumors is associated with poor clinical outcomes and confer treatment resistance. As a putative oncoprotein, SKP2 promotes proliferation, preventing senescence and apoptosis, maintaining self-renewal and metastasis; therefore, SKP2 has emerged as an attractive therapeutic target for cancers (36). Here, we showed that YAP regulates the transcription of SKP2 mRNA, which contributes to the proliferative program and ferroptosis susceptibility of YAP activation. we also showed that genetic and chemical inhibition of SKP2 robustly protected multiple cancer cells from ferroptosis. The functional properties of SKP2 associate with YAP/TAZ activation. That is, a high level of YAP/TAZ protects cells from apoptosis but promotes ferroptosis. Reciprocally, the inactivation of YAP/TAZ sensitizes tumor cells to apoptosis but inhibits ferroptosis (17,24,41). Like YAP, SKP2 expression is associated with mesenchymal state and tumorigenic potential (42). Consistent with this concept, many ferroptotic proteins also mediate oncogenic processes, including the loss of VHL (18) and DDR2 and RIPK3 activation (20,43). In addition, YAP has also been found to regulate SKP2 acetylation and promote polyploid conversion and growth (44). Reciprocally, YAP is also regulated by SCF(SKP2) E3 ligase complex-mediated non-proteolytic, K63-linked polyubiquitination, that enhances YAP-TEAD interaction and their transcriptional activity, and growth-promoting function (45). Therefore, there may be a positive feedback loop between YAP and SKP2 in the propagation of the proliferative gene expression program and potential ferroptosis susceptibility.
SKP2 is the substrate recognition component of the SCF-SKP2 complex that targets cycle control elements, including p27 and p21 for proteome degradation, to allow cell cycle entry and G1/S transition (27,46). p21 is recently shown to associate with proliferation arrest and ferroptosis resistance (33,34). However, the role of p21 remains unclear in our system. Instead, we found that the repression of the TTK and TFRC mRNAs upon SKP2 knockdown contributes to the ferroptosis protection phenotypes. TFRC encodes transferrin receptor, which mediates the iron uptake and serves as a ferroptosis biomarker (35). Therefore, the downregulation of TFRC upon the YAP/SKP2 knockdown may reduce cellular iron and mitigate ferroptosis. In contrast, much remains unknown about the potential role of TTK in ferroptosis. TTK encodes Monopolar spindle1 (MPS1), a kinase essential for chromosome alignment and duplication during mitosis (47). Interestingly, TTK expression is particularly high in the ferroptosis-sensitive basal-type breast cancer cell lines and inhibition of TTK sensitized breast tumors to ionization radiation by impairing DNA damage repair (48). This is reminiscent of the ferroptosis protection upon the inhibition of ATM which also impairs DNA damage repair and sensitizes tumor cells to radiation(49). In the future, it will be important to identify the direct SKP2 target proteins responsible for the TTK/TFRC downregulation and the mechanism by which their downregulation contribute to the ferroptosis protection phenotypes of SKP2 inhibition.
Our results have therapeutic implications for cancers and other human diseases involving ferroptosis. While ferroptosis-inducing agents have significant anti-tumor potential, it is not clear how to select tumors that are most likely to respond to these ferroptosis-inducing treatments. Our results indicated that YAP or SKP2-activated tumors may be particularly sensitive to ferroptosis and respond to ferroptosis-based therapeutics. Since a high level of YAP or SKP2 is known to cause chemoresistance (36,44,50), our results also suggest that a potential mechanistic link between chemoresistance and enhanced ferroptosis sensitivity, justifying the efforts to eliminate these treatment-resistant tumors by ferroptosis. For neurodegenerations and other human diseases that involve ferroptosis, our results suggest that inhibitors of YAP or SKP2 may interfere with ferroptosis and improve the disease progression and clinical outcomes.
Supplementary Material
Implications:
This study reveals that YAP promotes ferroptosis by regulating SKP2, suggesting novel therapeutic options for YAP-driven tumors.
Acknowledgments
We acknowledge the financial support in part by DOD grants (W81XWH-17-1-0143, W81XWH-15-1-0486, W81XWH-19-1-0842), NIH grants (GM124062, 1R01NS111588-01A1), Duke Bridge Fund, and Duke Cancer Institute (DCI) pilot fund to J.T. Chi.
Footnotes
Conflict of interest statement: The authors have declared that no conflict of interest exists.
References
- 1.Scott J Dixon KML, Lamprecht Michael R., Skouta Rachid, Zaitsev Eleina M., Gleason Caroline E., Patel Darpan N., Bauer Andras J., Cantley Alexandra M., Yang Wan Seok, Morrison Barclay III, Stockwell Brent R.. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012;149(5):1060–72 doi 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen P-H, Wu J, Xu Y, Ding C-KC, Mestre AA, Lin C-C, et al. Zinc transporter ZIP7 is a novel determinant of ferroptosis. Cell Death & Disease 2021;12(2):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wan Seok Yang RS, Matthew E. Welsch, Kenichi Shimada, Rachid Skouta, Viswanathan Vasanthi S., Cheah Jaime H., Clemons Paul A., Shamji Alykhan F., Clish Clary B., Brown Lewis M., Girotti Albert W., Cornish Virginia W., Schreiber Stuart L., Stockwell Brent R.. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014;156(1-2):317–31 doi 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017;171(2):273–85 doi 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ 2016;23(3):369–79 doi 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin C-C, Chi J-T. Ferroptosis of epithelial ovarian cancer: genetic determinants and therapeutic potential. Oncotarget 2020;11(39):3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang W-H, Chi J-T. Hippo pathway effectors YAP/TAZ as novel determinants of ferroptosis. Molecular & Cellular Oncology 2019:1699375 doi 10.1080/23723556.2019.1699375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nature Chemical Biology 2017;13(1):91–8 doi 10.1038/nchembio.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding C-KC, Rose J, Sun T, Wu J, Chen P-H, Lin C-C, et al. MESH1 is a cytosolic NADPH phosphatase that regulates ferroptosis. Nature Metabolism 2020;2:270–7 doi 10.1038/s42255-020-0181-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen PH, Tseng WH, Chi JT. The Intersection of DNA Damage Response and Ferroptosis-A Rationale for Combination Therapeutics. Biology (Basel) 2020;9(8) doi 10.3390/biology9080187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen PH, Wu J, Ding CC, Lin CC, Pan S, Bossa N, et al. Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism. Cell Death Differ 2019;27(27):1008–22 doi 10.1038/s41418-019-0393-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016;29(6):783–803 doi 10.1016/j.ccell.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dey A, Varelas X, Guan K-L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nature Reviews Drug Discovery 2020;19(7):480–94 doi 10.1038/s41573-020-0070-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev 2014;94(4):1287–312 doi 10.1152/physrev.00005.2014. [DOI] [PubMed] [Google Scholar]
- 15.Plouffe SW, Lin KC, Moore JL, Tan FE, Ma S, Ye Z, et al. The Hippo pathway effector proteins YAP and TAZ have both distinct and overlapping functions in the cell. Journal of Biological Chemistry 2018;293(28):11230–40 doi 10.1074/jbc.RA118.002715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang WH, Huang Z, Wu J, Ding CC, Murphy SK, Chi JT. A TAZ-ANGPTL4-NOX2 Axis Regulates Ferroptotic Cell Death and Chemoresistance in Epithelial Ovarian Cancer. Mol Cancer Res 2020;18(1):79–90 doi 10.1158/1541-7786.Mcr-19-0691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang WH, Ding CC, Sun T, Rupprecht G, Lin CC, Hsu D, et al. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep 2019;28(10):2501–8 e4. doi 10.1016/j.celrep.2019.07.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang X, Wu J, Ding CK, Lu M, Keenan MM, Lin CC, et al. Cystine Deprivation Triggers Programmed Necrosis in VHL-Deficient Renal Cell Carcinomas. Cancer Res 2016;76(7):1892–903 doi 10.1158/0008-5472.CAN-15-2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang X, Ding CK, Wu J, Sjol J, Wardell S, Spasojevic I, et al. Cystine addiction of triple-negative breast cancer associated with EMT augmented death signaling. Oncogene 2017;36(30):4235–42 doi 10.1038/onc.2016.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin CC, Mabe NW, Lin YT, Yang WH, Tang X, Hong L, et al. RIPK3 upregulation confers robust proliferation and collateral cystine-dependence on breast cancer recurrence. Cell Death Differ 2020;27(7):2234–47 doi 10.1038/s41418-020-0499-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res 2017;45(W1):W98–W102 doi 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zanconato F, Battilana G, Forcato M, Filippi L, Azzolin L, Manfrin A, et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nature Medicine 2018;24(10):1599–610 doi 10.1038/s41591-018-0158-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes & Development 2007;21(21):2747–61 doi 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 2019;572(7769):402–6 doi 10.1038/s41586-019-1426-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, et al. Elucidation of a Universal Size-Control Mechanism in Drosophila and Mammals. Cell 2007;130(6):1120–33 doi 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.von Gise A, Lin Z, Schlegelmilch K, Honor LB, Pan GM, Buck JN, et al. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proceedings of the National Academy of Sciences 2012;109(7):2394–9 doi 10.1073/pnas.1116136109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nature Cell Biology 1999;1(4):193–9 doi 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 28.Stein C, Bardet AF, Roma G, Bergling S, Clay I, Ruchti A, et al. YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLOS Genetics 2015;11(8):e1005465 doi 10.1371/journal.pgen.1005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jang W, Kim T, Koo JS, Kim SK, Lim DS. Mechanical cue-induced YAP instructs Skp2-dependent cell cycle exit and oncogenic signaling. EMBO J 2017;36(17):2510–28 doi 10.15252/embj.201696089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rico-Bautista E, Yang C-C, Lu L, Roth GP, Wolf DA. Chemical genetics approach to restoring p27Kip1 reveals novel compounds with antiproliferative activity in prostate cancer cells. BMC Biology 2010;8(1):153 doi 10.1186/1741-7007-8-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Q, Xie W, Kuhn DJ, Voorhees PM, Lopez-Girona A, Mendy D, et al. Targeting the p27 E3 ligase SCF(Skp2) results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood 2008;111(9):4690–9 doi 10.1182/blood-2007-09-112904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang W, Nacusi L, Sheaff RJ, Liu X. Ubiquitination of p21Cip1/WAF1 by SCFSkp2: substrate requirement and ubiquitination site selection. Biochemistry 2005;44(44):14553–64 doi 10.1021/bi051071j. [DOI] [PubMed] [Google Scholar]
- 33.Venkatesh D, Stockwell BR, Prives C. p21 can be a barrier to ferroptosis independent of p53. Aging (Albany NY) 2020;12(18):17800–14 doi 10.18632/aging.103961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, et al. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell reports 2018;22(3):569–75 doi 10.1016/j.celrep.2017.12.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feng H, Schorpp K, Jin J, Yozwiak CE, Hoffstrom BG, Decker AM, et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep 2020;30(10):3411–23 e7 doi 10.1016/j.celrep.2020.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cai Z, Moten A, Peng D, Hsu CC, Pan BS, Manne R, et al. The Skp2 Pathway: A Critical Target for Cancer Therapy. Semin Cancer Biol 2020. doi 10.1016/j.semcancer.2020.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017;547(7664):453–7 doi 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perez MA, Magtanong L, Dixon SJ, Watts JL. Dietary Lipids Induce Ferroptosis in Caenorhabditiselegans and Human Cancer Cells. Developmental Cell 2020;54(4):447–54.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hernández-Gallardo AK, Missirlis F. Loss of ferritin in developing wing cells: Apoptosis and ferroptosis coincide. PLOS Genetics 2020;16(1):e1008503 doi 10.1371/journal.pgen.1008503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Distefano AM, Martin MV, Cordoba JP, Bellido AM, D’Ippolito S, Colman SL, et al. Heat stress induces ferroptosis-like cell death in plants. J Cell Biol 2017;216(2):463–76 doi 10.1083/jcb.201605110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Danovi S, Rossi M, Gudmundsdottir K, Yuan M, Melino G, Basu S. Yes-associated protein (YAP) is a critical mediator of c-Jun-dependent apoptosis. Cell Death & Differentiation 2008;15(1):217–9. [DOI] [PubMed] [Google Scholar]
- 42.Šimečková Š, Kahounová Z, Fedr R, Remšík J, Slabáková E, Suchánková T, et al. High Skp2 expression is associated with a mesenchymal phenotype and increased tumorigenic potential of prostate cancer cells. Scientific Reports 2019;9(1):5695 doi 10.1038/s41598-019-42131-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin C-C, Yang W-H, Lin Y-T, Tang X, Chen P-H, Ding C-KC, et al. DDR2 upregulation confers ferroptosis susceptibility of recurrent breast tumors through the Hippo pathway. Oncogene 2021:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang S, Chen Q, Liu Q, Li Y, Sun X, Hong L, et al. Hippo Signaling Suppresses Cell Ploidy and Tumorigenesis through Skp2. Cancer Cell 2017;31(5):669–84 e7 doi 10.1016/j.ccell.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yao F, Zhou Z, Kim J, Hang Q, Xiao Z, Ton BN, et al. SKP2-and OTUD1-regulated non-proteolytic ubiquitination of YAP promotes YAP nuclear localization and activity. Nature communications 2018;9(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bornstein G, Bloom J, Sitry-Shevah D, Nakayama K, Pagano M, Hershko A. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J Biol Chem 2003;278(28):25752–7 doi 10.1074/jbc.M301774200. [DOI] [PubMed] [Google Scholar]
- 47.Pachis ST, Kops G. Leader of the SAC: molecular mechanisms of Mps1/TTK regulation in mitosis. Open Biol 2018;8(8) doi 10.1098/rsob.180109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chandler BC, Moubadder L, Ritter CL, Liu M, Cameron M, Wilder-Romans K, et al. TTK inhibition radiosensitizes basal-like breast cancer through impaired homologous recombination. J Clin Invest 2020;130(2):958–73 doi 10.1172/jci130435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Durant ST, Zheng L, Wang Y, Chen K, Zhang L, Zhang T, et al. The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci Adv 2018;4(6):eaat1719 doi 10.1126/sciadv.aat1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marti P, Stein C, Blumer T, Abraham Y, Dill MT, Pikiolek M, et al. YAP promotes proliferation, chemoresistance, and angiogenesis in human cholangiocarcinoma through TEAD transcription factors. Hepatology 2015;62(5):1497–510. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.