

Abstract
Melanoma occurs as a consequence of inherited susceptibility to the disease and exposure to ultraviolet radiation (UVR) and is characterized by uncontrolled cellular proliferation and a high mutational load. The precise mechanisms by which UVR contributes to the development of melanoma remain poorly understood. Here we show that activation of nuclear receptor coactivator 3 (NCOA3) promotes melanomagenesis through regulation of UVR sensitivity, cell cycle progression, and circumvention of the DNA damage response (DDR). Downregulation of NCOA3 expression, either by genetic silencing or small molecule inhibition, significantly suppressed melanoma proliferation in melanoma cell lines and patient-derived xenografts. NCOA3 silencing suppressed expression of xeroderma pigmentosum C and increased melanoma cell sensitivity to UVR. Suppression of NCOA3 expression led to activation of DDR effectors and reduced expression of cyclin B1, resulting in G2/M arrest and mitotic catastrophe. A single nucleotide polymorphism in NCOA3 (T960T) reduced NCOA3 protein expression and was associated with decreased melanoma risk, given a significantly lower prevalence in a familial melanoma cohort than in a control cohort without cancer. Overexpression of wild-type NCOA3 promoted melanocyte survival following UVR and was accompanied by increased levels of UVR-induced DNA damage, both of which were attenuated by overexpression of NCOA3 (T960T). These results describe NCOA3-regulated pathways by which melanoma can develop, with germline NCOA3 polymorphisms in the setting of UVR exposure, enabling enhanced melanocyte survival despite an increased mutational burden. They also identify NCOA3 as a novel therapeutic target for melanoma.
Introduction
Melanoma is the fifth most common malignancy in the United States (1), with a dramatically increasing incidence over the last several decades (2). Ample epidemiological evidence supports the role of UVR in melanoma causation (3–5), supported by whole genome sequencing studies demonstrating a high burden of UV-signature mutations (6–8). Molecular susceptibility to melanoma also plays an important role, and involves high-penetrance loci identified in melanoma-prone families, including CDKN2A and CDK4 (9). However, only a minority of familial melanoma kindreds harbor mutations in known susceptibility genes. Genome-wide association studies have identified low-penetrance susceptibility loci (e.g., MC1R, TYR, TYRP1, and OCA2) (10), but are limited by the small number of single nucleotide polymorphisms (SNPs) that can be tested, and by the caveat that the identified loci may not represent the causal variants. As a result, the precise molecular mechanisms by which melanoma develops following UVR remain poorly understood, necessitating the identification of additional molecular factors that govern both UV and melanoma susceptibility.
NCOA3 (also known as AIB1 or SRC-3) is a member of the nuclear hormone receptor coactivator family, and regulates gene expression through its interaction with various transcription factors (11–13). NCOA3 was initially shown to be amplified in breast cancer, and has a demonstrated oncogenic role in various solid tumors (14–18). We identified NCOA3 as differentially expressed in metastatic melanomas by gene expression profiling (19), and subsequently demonstrated a key prognostic role for NCOA3 in predicting melanoma-specific survival (20,21), including as the first molecular marker to predict melanoma metastasis to the sentinel lymph nodes. However, to date, the precise pathways by which NCOA3 exerts its oncogenic effects in melanoma have not been elucidated. Specifically, a role for NCOA3 in UVR-mediated melanomagenesis has not been previously demonstrated.
In this manuscript, we assess the effects of regulating NCOA3 expression in human melanoma cells as well as in melanocytes, identifying multiple oncogenic pathways regulated by NCOA3 in melanoma progression. These studies describe an important role for NCOA3 in UV susceptibility as well as a rational target for melanoma therapy.
Materials and Methods
Cell culture
C8161.9 human melanoma cells (22) (obtained from Dr. D. Welch, UAB, USA) were grown in DMEM/Ham’s F12 containing 5% FBS and 1X pen/strep. D04 melanoma cells (provided by Dr. B. Bastian, UCSF, USA) were cultured in RPMI medium 1640 containing 5% FBS plus 1X pen/strep. Ma-Mel-12, Ma-Mel-103b, Ma-Mel-66a and Ma-Mel-46 cells (provided by Dr. D. Schadendorf, University of Essen, Germany) were grown in RPMI medium 1640 containing 10% FBS plus 1X pen/strep. Normal human adult epidermal melanocytes (NHEM) (Lonza) were grown in MGM4 medium containing endothelin-3 and 1X pen/strep. Normal human neonatal melanocytes (Mel-F-Neo) (ZenBio) were grown in Mel-2 melanocyte medium containing 1X pen/strep. Patient-derived xenograft (PDX) cells (MM337X, MM341X, MM334X, MM302X, and MM313X) were cultured in ultralow attachment T25 flasks with DMEM/Ham’s F12 medium containing 1X B27 supplement (Gibco), 1X pen/strep, 50ng/ml EGF, 50ng/ml FGF and no serum. These cells were derived from melanoma patients (described in Table S1). All 5 PDX cell lines were authenticated as human, without a match to any cell line in either the ATCC or the DSMZ STR database. Cells were confirmed mycoplasma free by PCR testing, with the last testing performed in summer 2020. Cells were passaged every 2–3 days (37°C and 5% CO2), were kept in culture for the duration of the experiments, and fresh batches of cells were used for repeat experiments. For in vivo experiments, the cells were cultured in their respective media up to the time of tail-vein or subcutaneous injection when they were re-suspended either in RPMI medium 1640 (D04), DMEM/Ham’s F12 (C8161.9) or DMEM/Ham’s F12 with 50% Matrigel (Thermo Fisher Scientific) (MM337X).
Transfections and generation of stable transformants
A shRNA NCOA3 target set (RHS4533-NM_006534) (Dharmacon) and lentiviruses including the anti-luciferase (luc) shRNA were prepared as previously described (23). Subconfluent C8161.9, D04 and Ma-Mel-12 cells were infected with each harvested lentivirus in the presence of 8μg/ml of polybrene, and selected in 1.5μg/ml, 1μg/ml or 0.5μg/ml of puromycin at 48h post-infection, respectively. The consequences of NCOA3 gene silencing were confirmed using a second siRNA targeting a distinct sequence in NCOA3 mRNA. siRNAs were transfected at 100nM with Lipofectamine 2000 and the assays performed at 48h post-transfection. Validated NCOA3 siRNA was purchased from Lifetech (#4427038), and GFP siRNA duplex oligo was purchased from Integrated DNA Technologies:
5’-GCUACGUCCAGGAGCGCACCCUCAA-3’;
3’-CGAUGCAGGUCCUCGCGUGGGAGAA-5’.
To overexpress pcDNA3, human NCOA3 cDNA or T960T NCOA3 polymorphism cDNA in NHEM Mel-F-Neo cells and human XPC cDNA (or a control plasmid expressing enhanced green fluorescent protein, or EGFP), in C8161.9 cells, lipofectamine 2000-mediated transfections (9μL) of (3μg) plasmids expressing control pcDNA3 or EGFP, human XPC cDNA (Addgene#39204), human NCOA3 (Genecopoeia), or T960T were carried out following manufacturer instructions (Thermo Fisher Scientific).
Plasmid construction and site-directed mutagenesis
To introduce the T960T polymorphism on the human NCOA3 plasmid, the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent technologies) and the following simple with the nucleotide change in the center (underlined) were used:
hT960T-mut-acg-F: 5’-GGCTCTATTCCCACGTTGCCTCTTCGGTC-3’
hT960T-mut-acg-R: 5’-GACCGAAGAGGCAACGTGGGAATAGAGCC-3’
The nucleotide change was confirmed by using the following sequencing primers designed with Oligoperfect software (Thermo Fisher Scientific):
Forward pCMV-hNCOA3-seqF: 5’-CTGGGGCTTACCAAACTCAA-3’
Reverse pCMV-hNCOA3-seqR: 5’-CTGCTTGGCCATAGGGATTA-3’
Genotyping of NCOA3 T960T polymorphism
Analyses of patient specimens were performed following approval by the Sutter Health Institutional Review Board, after obtaining written informed consent, and in accordance with the Declaration of Helsinki. The T960T polymorphism (2880A>G) (rs2076546) was investigated in a control cohort without cancer (N=364), in a familial melanoma cohort (from the University of Utah, composed of 97 melanoma patients from independent families that did not carry a CDKN2A gene mutation), and in 23 melanoma cell lines (C8161.9, D04, Ma-Mel-12, LOX, D05, D08, MM415, MM485, WM3211, 1205LU, WM164, WM793, 415LU, C918P, WM1361, WM266.4, CHL, A375, SKMel28, WM115, Ma-Mel-103b, Ma-Mel-66a, Ma-Mel-46) using TaqMan allelic discrimination primers and probes as described (24). The presence of the polymorphism was confirmed by PCR and sequencing. PCR conditions were as follows: 94°C for 2min, followed by 32 cycles of 94°C for 30 seconds, 66°C for 30 seconds and 72°C for 30 seconds and a final extension at 72°C for 5min using the amplification primers T960T-M13F2 (forward: 5’-TGTAAAACGACGGCCAGTCTGGGGCTTACCAAACTCAA-3’) and T960T-M13R2 (reverse: 5’-AGCGGATAACAATTTCACACAGGCTGCTTGGCCATAGGGATTA-3’) with the Platinum PCR SuperMix High Fidelity (Thermo Fisher Scientific), followed by sequencing in both directions using the primers M13F2 (forward: 5-TGTAAAACGACGGCCAGT-3’) and M13R2 (reverse: 5’-AGCGGATAACAATTTCACACAGG-3’).
Quantitative real-time polymerase chain reaction (qRT-PCR) analysis
This assay was performed as described (23). The TaqMan probes for human genes used were as follows (Thermo Fisher Scientific): NCOA3 (Hs00180722_m1), CCNB1 (Hs00259126_m1), CCNB2 (Hs00270424_m1), CCND1 (Hs00277039_m1), CCNA1 (Hs00171105_m1), CHEK2 (Hs00200485_m1), XPC (Hs00190295_m1), HPRT1 (Hs03929098_m1). All reactions were run in triplicate and the expression level of each gene was normalized to the human HPRT1 gene before comparisons. Data was analyzed using the 2−ΔΔCT method.
UV irradiation
UVC (#FG8T5) and UVB (#G8T5E) irradiation were carried out with five UV light bulbs each (Ushio America, Inc.) from a distance of 20cm using Stratalinker UV crosslinker 2400 (Stratagene). For the colony formation assay, C8161.9 and D04 cells were treated with 2.5nM bufalin (#B0261, Sigma) for 6h prior to exposure to 40J/m2 of UVC, and Ma-Mel-12 cells were treated with 5nM bufalin for 6h prior to exposure to 20J/m2 of UVC. For the rescue experiment with the XPC cDNA plasmid (colony formation assay), C8161.9 cells were starved the night before with 0.1% FBS, treated with 50nM bufalin for 6h prior to exposure to 40J/m2 of UVC and immediately followed by transfection of the pcDNA3 control plasmid or the human XPC cDNA plasmid. For γH2AX immunofluorescence, NHEM cells were exposed to 40J/m2 of UVC. For the ELISA assays, NHEM cells were exposed to 40J/m2 of UVC to detect 6–4 photoproducts (6-4PP) and 400J/m2 of UVB to detect cyclopyrimidine dimers (CPD). Mel-F-Neo cells were exposed to 200J/m2 of UVB to detect 6-4PP.
ELISA assay
CPD and 6-4PP photoproducts were detected by using the OxiSelect UV-induced DNA Damage ELISA combo kit (#STA-322-C, Cell Biolabs, Inc.) at 48h post-irradiation and following the manufacturer’s protocol.
Cell survival and colony formation assays
Cell survival was assessed using Cell Counting Kit-8 (Dojindo) following the manufacturer’s instructions on at least triplicate samples. C8161.9, D04 or Ma-Mel-12 stable transformants were plated in 96-well plates at a density of 1,000 cells per well. When using UVC, cells were starved overnight with 0.1% FBS and then plated in 96-well plates at a density of 5,000 cells per well for C8161.9 or D04 and 20,000 cells per well for Ma-Mel-12. When cells were treated with DMSO or bufalin for 6h (2.5nM for C8161.9 and D04 cells, and 5nM for Ma-Mel-12 cells) prior to UVC exposure, parental C8161.9, D04 and Ma-Mel-12 cells were starved the night before with 0.1% FBS and then plated in 96-well plates at a density of 10,000 cells per well. For NHEM cells, 24h post-transfection with pcDNA3 or various human NCOA3 cDNA polymorphism plasmids, cells were plated in 96-well plates at a density of 5,000 cells per well and then exposed to UVC.
For colony formation assays, colonies were stained with crystal violet and counted in triplicate. Five hundred C8161.9 or D04 stable transformants and 1,000 Ma-Mel-12 stable transformants were plated in 60mm dishes and allowed to grow for 7, 10 or 14 days respectively till visible colonies appeared. When cells were treated with DMSO or bufalin for 6h (2.5nM for C8161.9 and D04 cells and 5nM for Ma-Mel-12 cells) prior to UVC exposure, the cells were starved the night before with 0.1% FBS and then 5,000 cells each were plated in 60mm dishes and allowed to grow for 6 days in the case of C8161.9 cells and 10 days for both D04 and Ma-Mel-12 cells.
Cell cycle and apoptosis analyses
Cell cycle and apoptosis were assessed using the Muse Cell Analyzer and the corresponding kits (#MCH100106, Muse Cell Cycle Assay Kit and #MCH100105, Muse Annexin V & Dead Cell Assay Kit) as per the manufacturer’s protocols (EMD Millipore). Ma-Mel-12 and D04 cells were treated with DMSO or 10nM bufalin (40nM for C8161.9 cells) for 2 days before performing the flow cytometric analyses.
Invasion assay
The Matrigel assay for stable transformants was carried out as described (23).
Western analysis
Protein extraction and electrophoresis was performed as described (23). Membranes were first incubated overnight at 4°C with the following antibodies: NCOA3 (#sc-9119 at 1:1000 dil.; Santa Cruz Biotechnology), GAPDH (#MAB374 at 1:1000 dil.; EMD Millipore), CDK1 (#A303–663A at 1:1000 dil., Bethyl Laboratories), CCNB1 (#sc-245 at 1:1000 dil.; Santa Cruz Biotechnology), AURKA (#14475T at 1:1000 dil.; Cell Signaling), PLK1 (#4513T at 1:1000 dil.; Cell Signaling) and then with their respective horseradish peroxidase-labeled secondary antibodies for 1h at room temperature: goat anti-mouse HRP (#170–6516 at 1:1000 dil.; Biorad) and bovine anti-rabbit HRP (#172–1019 at 1:1000 dil.; Biorad). Binding was detected by using luminol reagent (sc-2048; Santa Cruz Biotechnology). For the bufalin experiments, C8161.9 and D04 cells were treated with DMSO or 2.5nM bufalin (5nM for Ma-Mel-12 cells and 40nM for the PDX cells) for 6h before the protein extraction. For the analyses of mitotic catastrophe and centrosome amplification, cells were treated for 6 days with bufalin at 5nM (C8161.9 and D04) or 10nM (Ma-Mel-12) before protein extraction.
Immunofluorescence and FISH
Expression of various proteins was assessed using immunofluorescence performed cultured on coverslips as described (23). Antibodies against NCOA3 (#sc-9119 at 1:1000 dil.; Santa Cruz Biotechnology), XPC (#GTX70294 at 1:500 dil.; Genetex), phospho-Chk2 (Thr68, #2197 at 1:500 dil.; Cell Signaling), p53 (#sc-126 at 1:250 dil.; Santa Cruz Biotechnology), p21 (#sc-6246 at 1:250 dil.; Santa Cruz Biotechnology), CCNB1 (#sc-245 at 1:250 dil.; Santa Cruz Biotechnology), γH2AX (#05–636 at 1:1000 dil.; EMD Millipore), tubulin (#ab6160 at 1:500 dil.; Abcam) and pericentrin (#ab4448 at 1:1000 dil.; Abcam) were followed by secondary antibodies labeled with Alexa Fluor 488, 594 or Cy5 (1:1000 dil.; Lifetech). DAPI staining was used as counterstain. Images were taken at fixed exposures with a Zeiss Axio Image Z2 microscope and the fluorescence intensities of at least 200 cells quantified using Axiovision software. The mean pixel intensities were used for statistical analysis using Microsoft Excel and Data Desk. For the analysis of pericentrin immunopositivity, cells were treated for 6 days with bufalin at 5nM (C8161.9 and D04) or 10nM (Ma-Mel-12) before performing immunofluorescence analysis. For FISH analysis, BAC clones RP11-456N23 (20q13.12), RP5-1049G16 (20q13.12), representing the NCOA3 locus, and RP4–610C12 (20q11.1) for the centromere of chromosome 20, were used (February 2009 freeze of the University of California, Santa Cruz Genome Browser, http://genome.ucsc.edu). All clones were obtained from the Children’s Hospital of Oakland Research Institute (CHORI). BAC DNAs were prepared and labeled as described (25). The quality and mapping of all probes were verified by hybridization to normal metaphase spreads in combination with a commercially available centromeric probe for chromosome 20 (Empire Genomics), before analysis. Hybridization on cell lines, tissue and metaphase preparations was performed as described (25). Images were taken with a Zeiss Axio Imager Z2 controlled by Axiovision software.
Gene expression profiling and SAM analysis
Total RNA was extracted from C8161.9 stable clones expressing shRNA targeting luciferase or NCOA3 using the RNeasy kit (Qiagen). Duplicate samples were submitted to Phalanx Biotech for microarray analysis. After linear normalization, log2 transformation, and supervised hierarchical clustering, the resulting cluster data table was imported into the statistical significance analysis of microarrays software package. Delta was chosen to limit the output gene list so that 5% predicted false positives would be included. Gene ontology analysis of the data was performed using the Panther Classification System (pantherdb.org) (26).
Animal studies
All animal care was in accordance with institutional guidelines and a protocol approved by the CPMCRI Committee on Animal Research. Groups of ten 45-day-old female nude mice were injected i.v. with 100,000 C8161.9 stable transformants. The number of metastatic lung tumors was counted at sacrifice and analyzed using the unpaired, two-sided Student’s t-test as described (25). In subcutaneous studies, 1×106 C8161.9 stable transformants were injected in the right flank of nude mice and tumor growth was followed for 28 days.
In the bufalin experiments, groups of ten 45-day-old female nude mice were injected subcutaneously with 1×106 C8161.9 or D04 cells or with 0.5×106 MM337X cells until the tumors reached a mean tumor volume of at least 75mm3. The mice were then injected i.p. with DMSO (vehicle) or with 1mg/kg of bufalin twice daily for the duration of the experiment. The animals were randomly assigned to treatment groups, and the investigator performing tumor measurements was blinded to the identity of the treatment groups. No samples were excluded from the analysis.
Statistical methods
All quantified data represent an average of at least triplicate samples or as indicated. Statistical significance was determined by the Student’s t-test, randomization test, Chi-square test, Mann-Whitney test, ANOVA, directional Le test or Kolmogorov-Smirnov test, and p values <0.05 were considered significant. PRISM graphing software (GraphPad Software) was used to determine IC50 values. In the in vivo drug efficacy studies, sample sizes were determined prospectively, using a type I error rate of 0.05 and power of 0.8 to detect differences in means of at least 30%. In all experiments, * denotes p<0.05 versus control and error bars represent the Standard Error of the Mean.
Data and materials availability:
Microarray data that support the findings of this study have been deposited in GEO, with the accession code #156062.
Results
Initially, we examined whether NCOA3 is activated in cutaneous melanoma. The NCOA3 gene resides on 20q13, a locus that is amplified in breast cancer (27) and that is frequently gained in melanoma (28,29). We assessed NCOA3 copy number using interphase fluorescence in situ hybridization (FISH) analysis in a cohort of 116 primary melanomas and 86 nevi. All nevi had 2 copies of NCOA3, whereas 29.3% of melanomas had 3 or more copies (Fig. 1A). These results indicate that NCOA3 copy number is significantly elevated in melanoma, providing a potential mechanism for its overexpression at the RNA level observed in cDNA microarray analysis, and identifying it as a candidate diagnostic marker for melanoma.
Fig. 1. NCOA3 copy number and effects of stable expression of anti-NCOA3 shRNA in C8161.9 and Ma-Mel-12 cells.
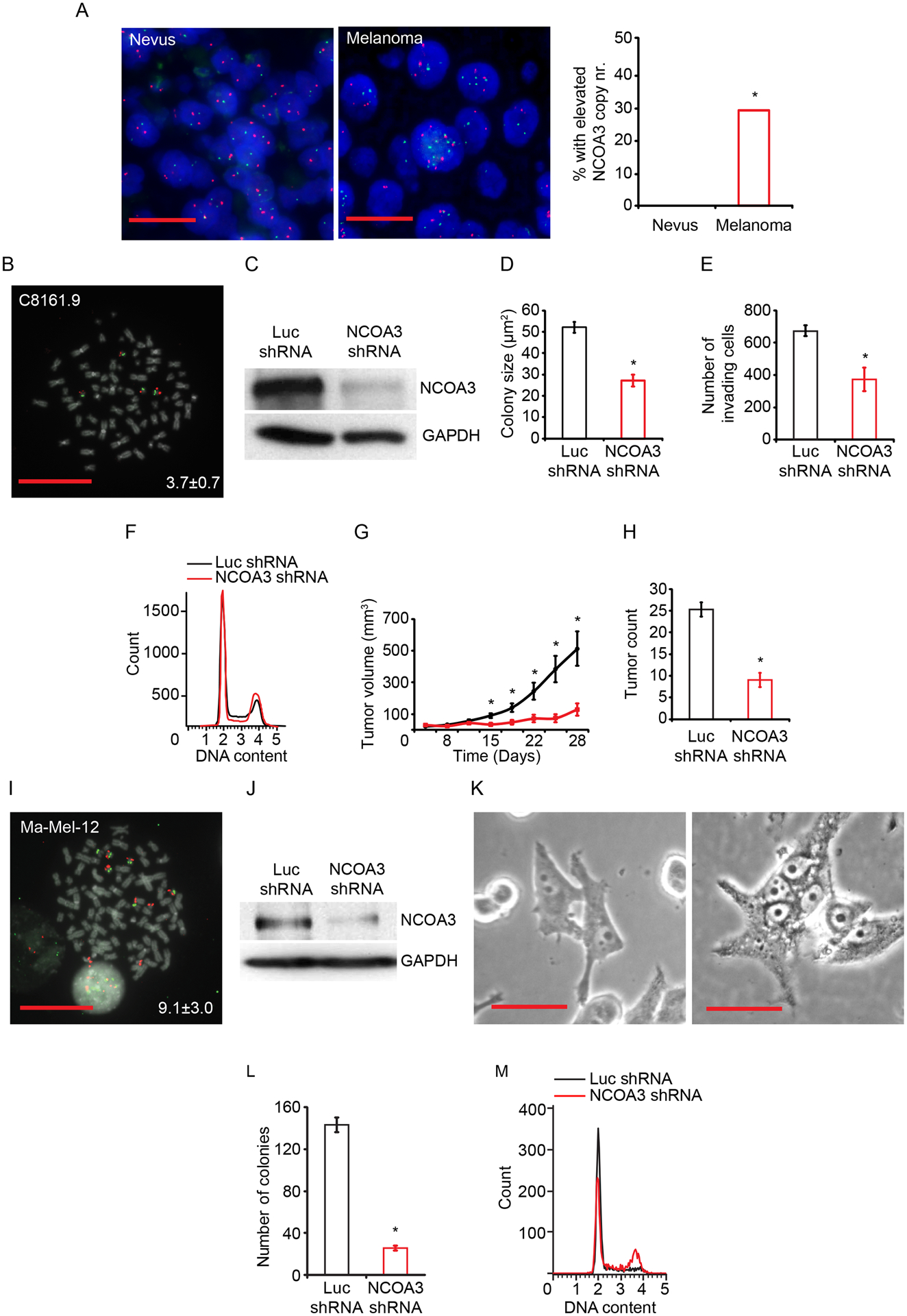
(A) Representative FISH signals for the NCOA3 locus (red) and probes representing the centromere of chromosome 20 (green) in a nevus (left panel) and a melanoma (middle panel). Comparison of mean NCOA3 copy number in nevi (N=86) and melanomas (N=116) (right panel). (B) Representative image of C8161.9 metaphase FISH from NCOA3 locus (red) and probes representing the centromere of chromosome 20 (green). Inset indicates the mean NCOA3 copy number. (C) Western analysis of NCOA3 and GAPDH in C8161.9 cells stably expressing anti-NCOA3 shRNA or anti-luc shRNA. (D) Colony formation assay (in triplicate) of C8161.9 cells stably expressing anti-NCOA3 shRNA or anti-luc shRNA. (E) Invasion into Matrigel (in triplicate) of C8161.9 cells stably expressing anti-NCOA3 shRNA or anti-luc shRNA. (F) Cell cycle analysis (in triplicate) of C8161.9 cells stably expressing anti-NCOA3 shRNA or anti-luc shRNA (statistical analysis provided in Fig. S1B). (G) Tumor volume following subcutaneous injection of C8161.9 cells stably expressing anti-NCOA3 shRNA or anti-luc shRNA. (H) Total tumor counts in the lungs of nude mice i.v. injected with C8161.9 cells stably expressing anti-NCOA3 shRNA or anti-luc shRNA. (I) Representative FISH signals for the NCOA3 locus (red) and probes representing the centromere of chromosome 20 (green) in Ma-Mel-12 cells, with inset indicating the mean NCOA3 copy number. (J) Western analysis of NCOA3 and GAPDH in Ma-Mel-12 cells stably expressing anti-NCOA3 shRNA or anti-luc shRNA. (K) Representative bright-field image of Ma-Mel-12 cells stably expressing anti-NCOA3 shRNA or anti-luc shRNA (quantitation of multinucleation provided in Fig. S1I). (L) Colony formation assay (in triplicate) of Ma-Mel-12 cells stably expressing anti-NCOA3 shRNA or anti-luc shRNA. (M) Cell cycle analysis (in triplicate) of Ma-Mel-12 cells stably expressing anti-NCOA3 shRNA or anti-luc shRNA (quantitation and statistical analysis provided in S1K). *denotes statistically significant differences compared with control. All scale bars are 20 μm except K at 100 μm.
We then determined the consequences of regulating NCOA3 expression in several human melanoma cell lines with elevated NCOA3 copy number (Fig. 1B; Table S1) using both genetic NCOA3 silencing and pharmacological targeting with bufalin, a small molecule inhibitor (30). Elevated NCOA3 copy number was present in melanoma cell lines regardless of BRAF or NRAS mutational status (Table S1). Stable shRNA-mediated downregulation of NCOA3 in C8161.9 cells (Fig. 1C) significantly suppressed melanoma colony formation, viability, and invasion, and was accompanied by G2/M arrest (Fig. 1D–F, S1A–B). NCOA3 silencing resulted in profound reductions both in the subcutaneous growth (Fig. 1G) and metastatic potential (Fig. 1H) of C8161.9 cells in nude mice. The anti-tumor effects produced following NCOA3 silencing were confirmed in D04 melanoma cells, which also harbored elevated NCOA3 copy number (Table S1; Fig. S1C–H).
In addition, we evaluated NCOA3’s role in a short-term melanoma culture, Ma-Mel-12, with a mean of nine copies of NCOA3 (Table S1, Fig. 1I). Stable NCOA3 silencing (Fig. 1J) resulted in profound growth arrest as well as dramatic morphologic changes, characterized by large, multi-nucleated cells, indicative of compromised cell cycle integrity that leads to mitotic catastrophe (31) (Fig. 1K, S1I). Over time, antibiotic treatment resulted in the survival of clones of Ma-Mel-12 cells with sufficient NCOA3 expression to allow propagation in cell culture, enabling analysis of the consequences of NCOA3 silencing. Suppression of NCOA3 expression was accompanied by markedly reduced colony formation (Fig. 1L), viability (S1J), and G2/M arrest (Fig. 1M; Fig. S1K), consistent with the effects observed in C8161.9 and D04 cells. These results indicate that melanoma cells harboring markedly elevated NCOA3 levels are addicted to its oncogenic effects.
Beyond genetic silencing of NCOA3, we assessed the effects of treatment of melanoma cells with the small molecule inhibitor bufalin (30). Administration of nanomolar concentrations of bufalin downregulated NCOA3 expression (Fig. 2A), and produced significant anti-tumor effects in multiple human melanoma cell lines, including patient-derived xenograft lines harboring elevated NCOA3 copy number (Fig. 2B–G; Fig. S2; Table S1). There was a differential sensitivity of melanoma cell lines to bufalin according to NCOA3 copy number status, with cell lines with higher NCOA3 copy number requiring a higher dose of bufalin for cell killing in vitro (Fig. 2C–D, with P<0.001 for each analysis; Table S1). In vivo administration of bufalin resulted in significant anti-tumor effects against C8161.9 (Fig. 2E) and D04 (Fig. 2F) melanoma, as well as against MM337X (Fig. 2G), a xenograft from a patient progressing on sequential treatment with BRAF and MEK inhibitors, and anti-CTLA4 and anti-PD1 antibodies. Bufalin treatment was associated with G2/M arrest and increased apoptosis (Fig. 2H–I; Fig. S2A–E). Thus, pharmacological targeting of NCOA3 resulted in significant anti-tumor activity against multiple melanoma cell lines, similar to NCOA3 gene silencing.
Fig. 2. Effects of bufalin treatment on melanoma cell lines and patient-derived xenografts.
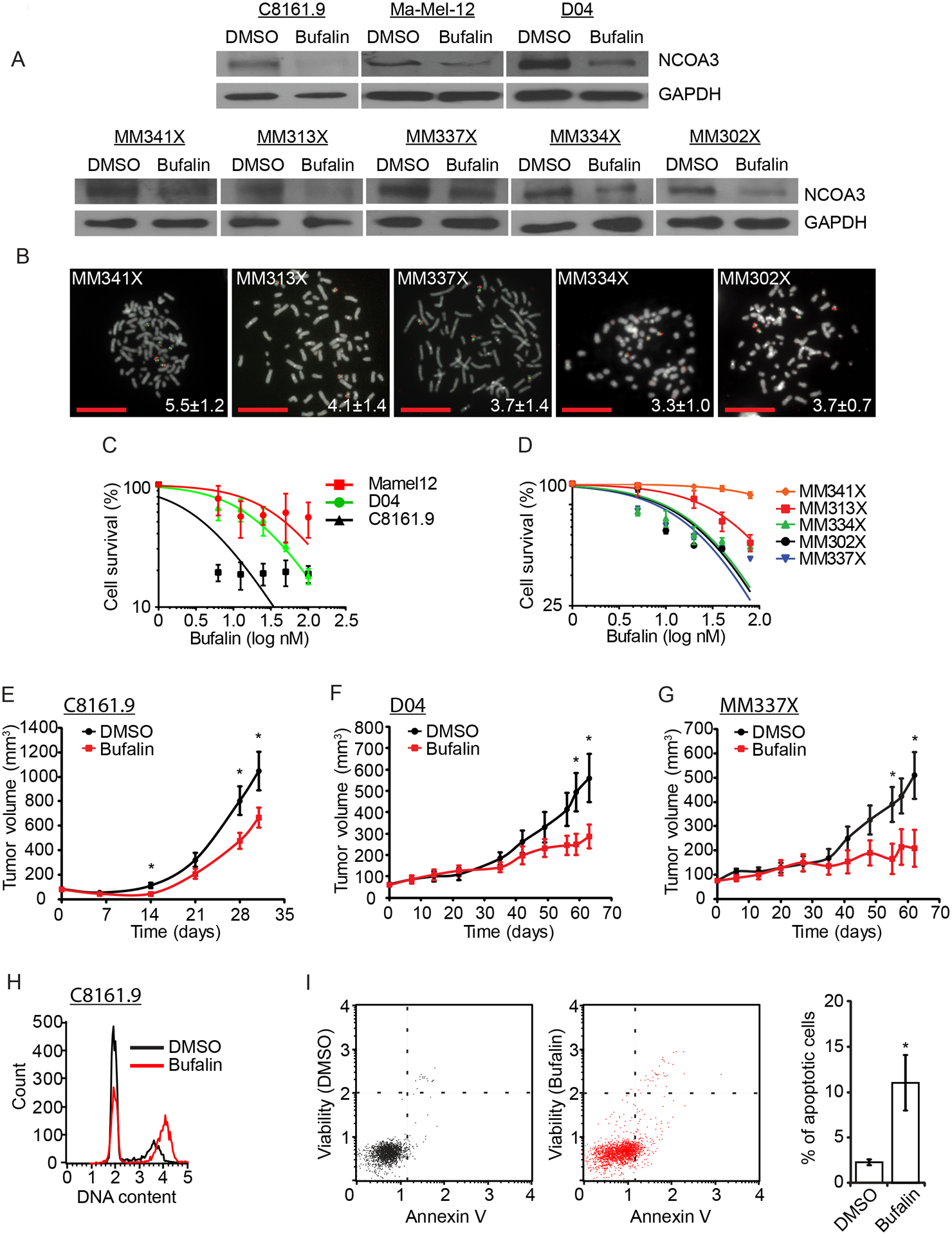
(A) Western analysis of NCOA3 and GAPDH proteins in various melanoma cell lines treated with vehicle (DMSO) or bufalin. (B) Representative metaphase FISH for the NCOA3 locus (red) and probes representing the centromere of chromosome 20 (green) in five melanoma PDX cell lines. Insets indicate the mean NCOA3 copy number. (C) Cell survival analysis (in triplicate) of C8161.9, D04, and Ma-Mel-12 cells treated with bufalin. (D) Cell survival analysis (in triplicate) of five melanoma PDX cell lines treated with bufalin. (E) Tumor volume of C8161.9 cells subcutaneously injected in nude mice treated with vehicle (DMSO) or bufalin. (F) Tumor volume of D04 cells subcutaneously injected in nude mice treated with vehicle (DMSO) or bufalin. (G) Tumor volume of MM337X PDX cells subcutaneously injected in nude mice treated with vehicle (DMSO) or bufalin. (H) Cell cycle analysis (in triplicate) of C8161.9 cells treated with vehicle (DMSO) or bufalin (quantitation and statistical analysis provided in Fig. S2A). (I) Analysis of apoptotic rates (in triplicate), with representative dot plot of the Annexin V versus propidium iodide (PI) assay, indicating percentage of total apoptotic C8161.9 cells treated with vehicle (DMSO) or bufalin. All scale bars are 20 μm.
Having demonstrated both the activation and oncogenic effects of NCOA3 in melanoma, we aimed to identify the pathways by which it promotes melanoma progression. We used transcriptomic analysis to identify the global patterns of gene expression following NCOA3 silencing. Supervised analysis of RNA isolated from C8161.9 clones expressing anti-NCOA3 versus control shRNA, followed by significance analysis of microarrays, identified numerous differentially expressed genes (Fig. 3A). Gene ontology analysis identified several pathways altered following NCOA3 knockdown (Table S2), including regulation of the cell cycle (specifically involving the mitotic phase) and cell division, as well as apoptosis. Prominent among the downregulated genes were cyclins (e.g., CCNB1, CCNB2, CCNA1, and CCND1), and genes that regulate sensitivity to UVR (e.g., XPC) (32). Intriguingly, among the upregulated genes was CHEK2, which plays a key role in the DNA damage response (DDR) activated by a number of DNA-damaging agents, including UVR (32). The differential expression of several of these genes following NCOA3 silencing was confirmed using qRT-PCR (Fig. 3B–C) and immunofluorescence analysis (Fig. 3D–E; Fig. S3A–D). Similar changes were observed following bufalin administration (Fig. S3E–F; S4A–D), and were accompanied by elevated expression of p53 and p21 (Fig. S4E and F), which are activated by CHEK2 (32). This differential gene expression was also validated using RNAi targeting a distinct sequence in human NCOA3 (Fig. S5A–D).
Fig. 3. Microarray analysis and identification of genes in the NCOA3 signal transduction pathway in melanoma cells.
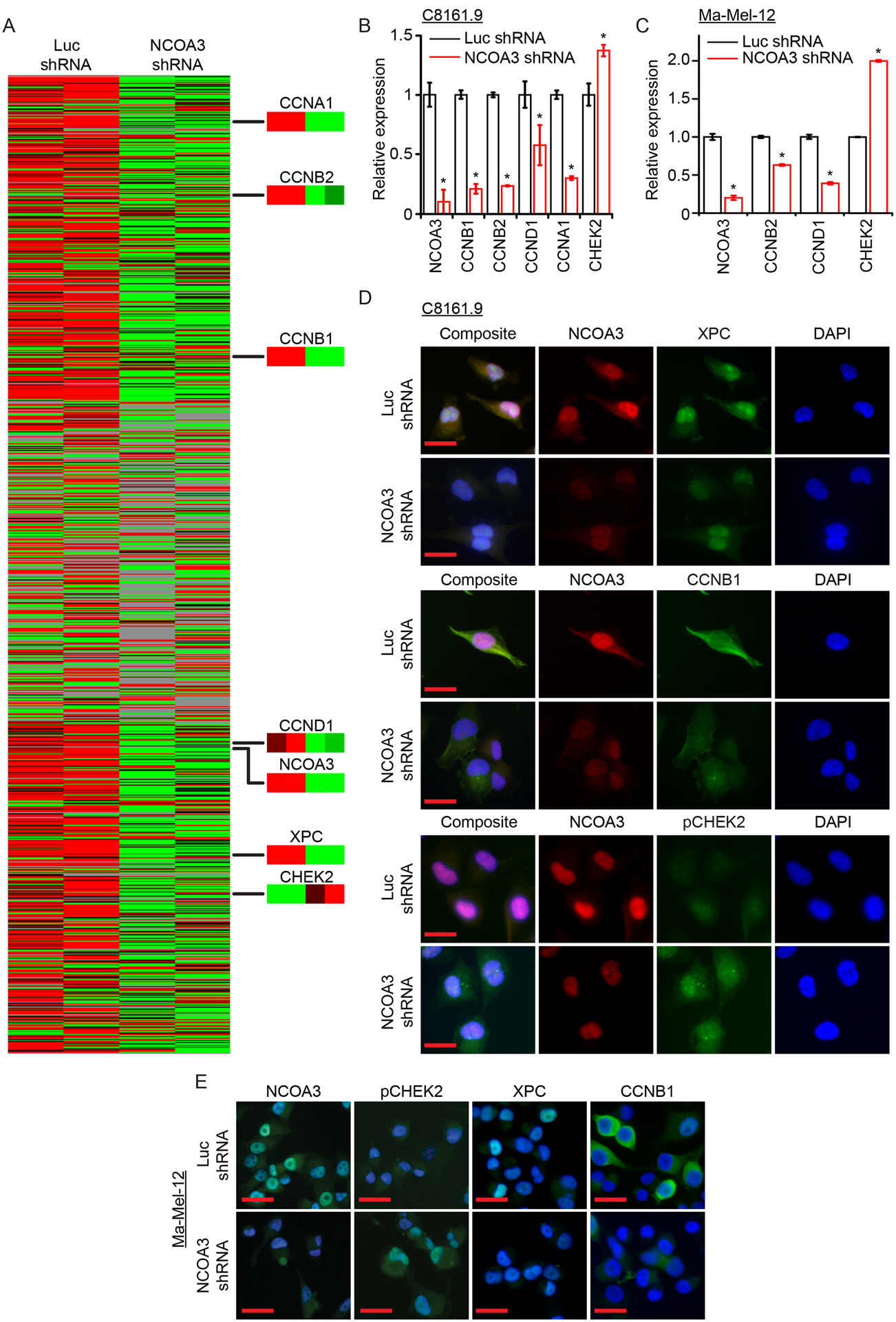
(A) Gene expression profiles using supervised hierarchical analysis of duplicate clones of C8161.9 cells stably expressing anti-luc shRNA (1 and 2) or anti-NCOA3 shRNA (3 and 4). Nodes of gene expression selected demonstrating differential expression of various genes. (B) Quantitative RT-PCR of expression of various genes (in triplicate) in C8161.9 cells stably expressing anti-luc shRNA or anti-NCOA3 shRNA. (C) Quantitative RT-PCR of expression of various genes (in triplicate) in Ma-Mel-12 cells stably expressing anti-luc shRNA or anti-NCOA3 shRNA. (D) Representative images of immunofluorescence detection of various proteins in C8161.9 cells stably expressing anti-luc shRNA or anti-NCOA3 shRNA (quantitation provided in Fig. S3A). (E) Representative images of immunofluorescence detection of various proteins in Ma-Mel-12 stably expressing anti-luc shRNA or anti-NCOA3 shRNA (quantitation provided in Fig. S3B). * denotes statistically significant differences compared with control. All scale bars are 20 μm.
The downregulation of CCNB1 observed following inhibition of NCOA3 was consistent with the G2/M arrest observed in cell cycle analysis, and with the morphologic changes leading to mitotic catastrophe. Intriguingly, bufalin treatment of melanoma cells in culture also promoted morphologic changes of mitotic catastrophe (Fig. 4A; Fig. S6A and B), similar to that observed with stable NCOA3 gene silencing. The phenotypic change induced by bufalin administration was accompanied by reduced expression of other proteins critical to M phase entry and progression (e.g., CDK1, AURKA, and PLK1) (Fig. 4B, C), as well as by centrosome amplification, as evidenced by immunofluorescence analysis of pericentrin immunopositivity (Fig. 4D, E). Beyond the increase of pericentrin numbers, there was a markedly distinct and diffuse pattern of pericentrin immunopositivity in melanoma cells following bufalin treatment (Fig. S6C and D). Taken together, these results indicate that suppression of NCOA3 expression in melanoma cells results in activation of key regulators of DDR, along with downregulation of proteins that promote cell cycle progression, culminating in cell cycle arrest and mitotic catastrophe.
Fig. 4. NCOA3 targeting in melanoma cells results in mitotic catastrophe.
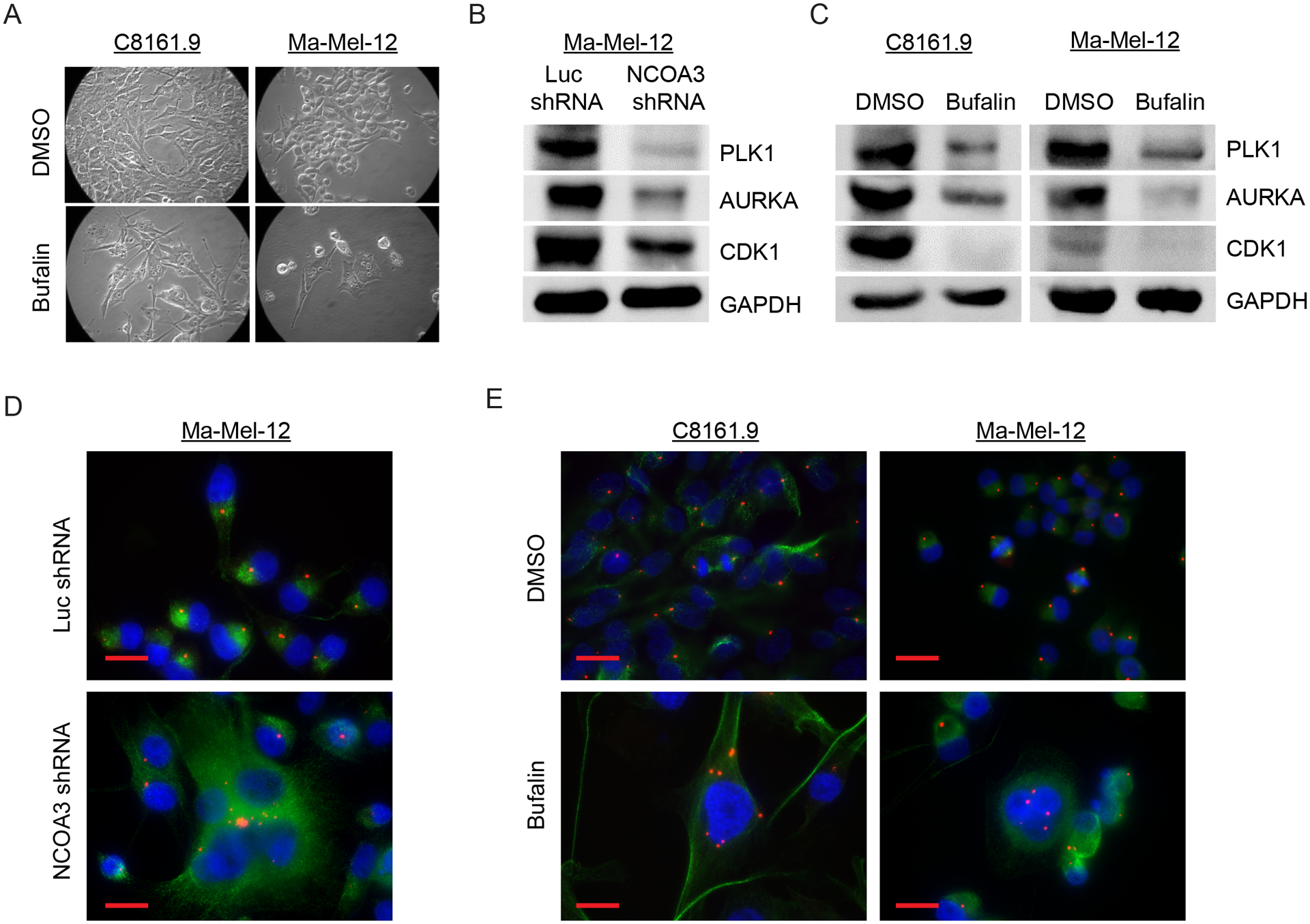
(A) Representative bright field images of C8161.9 and Ma-Mel-12 cells treated with DMSO or bufalin. (B) Western analysis of PLK1, AURKA, CDK1 and GAPDH proteins in Ma-Mel-12 cells stably expressing anti-NCOA3 shRNA or anti-luc shRNA. (C) Western analysis of PLK1, AURKA, CDK1 and GAPDH proteins in C8161.9 and Ma-Mel-12 cells treated with DMSO or bufalin. (D) Representative images of immunofluorescence detecting pericentrin (red) and tubulin (green) in Ma-Mel-12 cells stably expressing anti-NCOA3 shRNA or anti-luc shRNA. (E) Representative images of immunofluorescence detecting pericentrin (red) and tubulin (green) in C8161.9 and Ma-Mel-12 cells treated with DMSO or bufalin. All scale bars are 20 μm.
Given NCOA3’s regulation of XPC expression (Fig. 5A; Fig. S3F), we then determined whether modulation of NCOA3 expression resulted in altered sensitivity to UVR in melanoma cells. Stable NCOA3 silencing resulted in increased sensitivity of melanoma cells to UVR (Fig. 5B–D). Remarkably, brief (6h) exposure to bufalin treatment almost completely abolished the colony formation capacity of melanoma cells following UVR administration (Fig. 5E–G), which was rescued in part by overexpression of XPC cDNA (Fig. 5H; Fig. S6E).
Fig. 5. Effects of NCOA3 levels on UVR sensitivity in human melanoma cell lines.
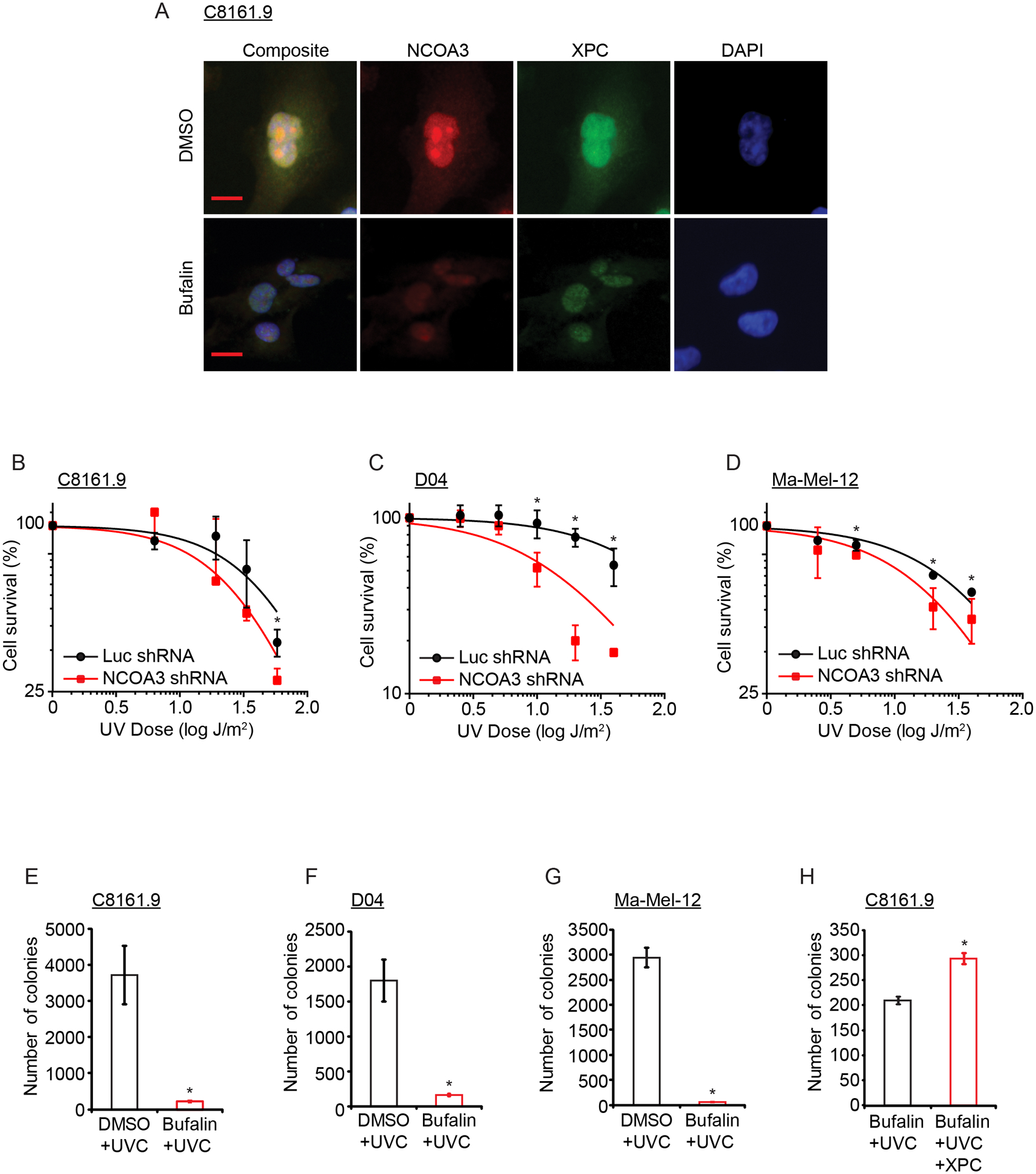
(A) Representative images of immunofluorescence detection of NCOA3 and XPC in C8161.9 cells treated with vehicle (DMSO) or bufalin (quantitation provided in Fig. S3F). (B) Cell survival analysis of UVC-exposed C8161.9 cells (in triplicate). (C) Cell survival analysis of UVC-exposed D04 cells (in triplicate). (D) Cell survival analysis of UVC-exposed Ma-Mel-12 cells (in triplicate). (E) Colony formation assay of UVC-exposed C8161.9 cells previously treated with vehicle (DMSO) or bufalin. (F) Colony formation assay of UVC-exposed D04 cells previously treated with vehicle (DMSO) or bufalin. (G) Colony formation assay of UVC-exposed Ma-Mel-12 cells previously treated with vehicle (DMSO) or bufalin. (H) Effects of XPC cDNA overexpression on colony formation of UVC-exposed C8161.9 cells previously treated with bufalin (all assays performed in triplicate). * denotes statistically significant differences compared with control. All scale bars are 20 μm.
Next, we determined whether regulation of NCOA3 expression in normal melanocytes alters DDR and affects their sensitivity to UV-mediated cytotoxicity. Overexpression of human NCOA3 cDNA into both adult (NHEM) and neonatal (Mel-F-Neo) human melanocyte lines resulted in elevated expression of XPC and CCNB1, while suppressing phospho-CHEK2, p53 and p21 (Fig. 6A; Fig. S7A and B), consistent with the results observed in melanoma cells. In addition, overexpression of NCOA3 cDNA, in the setting of UVR exposure, was accompanied by significantly increased accumulation of the DNA damage marker γH2Ax (Fig. 6B; Fig. S8A and B). Thus, increased NCOA3 expression results in reduced UV sensitivity in both melanoma cells and melanocytes, and is associated with increased accumulation of DNA damage.
Fig. 6. Role of differential NCOA3 levels in UV and melanoma susceptibility.
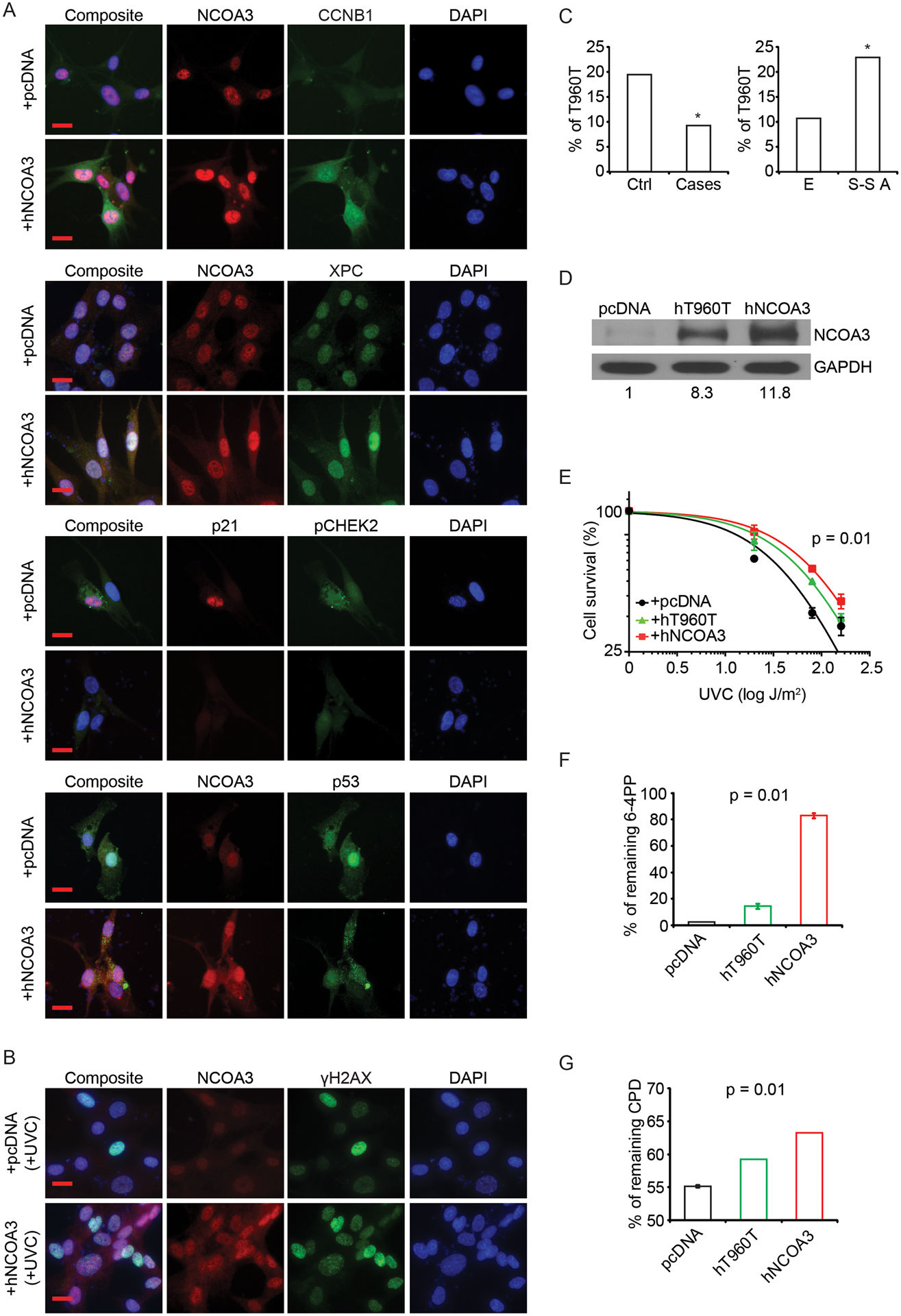
(A) Representative images of immunofluorescence detection of various proteins in NHEM cells transfected with control (pcDNA) or human NCOA3 cDNA plasmids (quantitation provided in Fig. S8A). (B) Representative images of immunofluorescence detection of NCOA3 and γH2AX in NHEM cells transfected with control (pcDNA) or human NCOA3 cDNA plasmids with UVC treatment (quantitation provided in Fig. S8B). (C) Prevalence of the T960T NCOA3 polymorphism in a control population (364 cases) versus a familial melanoma cohort (97 cases) (left panel), and in the European (E; HapMap-CEU) versus Sub-Saharan African populations (S-S A; HapMap-YRI) (right panel). (D) Western analysis of NCOA3 and GAPDH in NHEM cells transfected with control (pcDNA), human T960T NCOA3 polymorphism cDNA (hT960T) or human wild-type NCOA3 cDNA (hNCOA3) plasmids (densitometric values provided were normalized to GAPDH). (E) UVC sensitivity (in triplicate) of NHEM cells transfected with control (pcDNA), human T960T NCOA3 polymorphism cDNA (hT960T) or human wild-type NCOA3 cDNA (hNCOA3) plasmids by cell survival analysis. (F) ELISA assay (in triplicate) of 6-4PP from UVC-exposed NHEM cells transfected with control (pcDNA), human T960T NCOA3 polymorphism cDNA (hT960T) or human wild-type NCOA3 cDNA (hNCOA3) plasmids. (G) ELISA assay (in triplicate) of CPD from UVB-exposed NHEM cells transfected with control (pcDNA), human T960T NCOA3 polymorphism cDNA (hT960T) or human wild-type NCOA3 cDNA (hNCOA3) plasmids. * denotes statistically significant differences compared with control. All scale bars are 20 μm.
Given the regulation of DDR and UV sensitivity by NCOA3, we hypothesized that NCOA3 may be involved in melanoma susceptibility. The NCOA3 T960T (2880A>G or rs2076546) polymorphism is associated with reduced breast cancer risk by virtue of its reduced prevalence in familial breast cancer cases lacking mutations in BRCA1 and BRCA2 when compared with a control cohort (24). Mutations in the CDKN2A gene represent the most commonly identified aberrations in familial melanoma to date (33,34), but are only present in a minority of melanoma-prone families. We compared the prevalence of the T960T polymorphism in a 97-patient familial melanoma cohort without a CDKN2A gene mutation to a control cohort of 364 individuals without evidence of cancer. The 2880A>G variant was present in 9.3% of the familial melanoma cohort versus 19.5% of the control cohort (Fig. 6C; P<0.05). Intriguingly, the 2880A>G variant is present in 10.6% of Europeans (with a higher melanoma risk), compared with 22.6% of a sub-Saharan African population (Fig. 6C; P<0.05, www.ncbi.nlm.nih.gov/SNP). Finally, the T960T polymorphism was present in only two of 23 melanoma cell lines, and none of 53 melanoma specimens examined. Thus, the T960T polymorphism appears to be associated with reduced melanoma risk, and may have a protective role relative to the wild-type construct.
We then assessed the consequences of NCOA3 overexpression following UVR exposure. A cDNA construct encoding the T960T polymorphism was created using site-directed mutagenesis. Intriguingly, overexpression of wild-type NCOA3 cDNA in two melanocyte lines resulted in higher expression of NCOA3 protein when compared with the T960T construct (Fig. 6D; Fig. S8C), even though both cDNA constructs, despite containing different codons, encode the same amino acid. Overexpression of wild-type NCOA3 in both melanocyte lines resulted in resistance to UVR when compared with an empty vector control (Fig. 6E; Fig. S8D). By contrast, overexpression of the T960T cDNA construct produced an intermediate phenotype, with significant differences in cell survival when comparing the three constructs (Fig. 6E; Fig. S8D). UVR generates DNA damage that includes both cyclopyrimidine dimers (CPD) and 6–4 photoproducts (6-4PP) (35), resulting in DNA mutations that promote carcinogenesis. We assessed the proportion of DNA photoproducts remaining following UV treatment of both melanocyte lines expressing an empty vector, wild-type NCOA3 or T960T polymorphism using quantitative ELISA assays. Overexpression of wild-type NCOA3 resulted in a higher level of remaining 6-4PP in NHEM cells following UVC exposure when compared with the control vector. Overexpression of the T960T polymorphism resulted in an intermediate level of 6-4PP, with a significant discrimination between the three groups (Fig. 6F). Comparable results were obtained for remaining CPD levels following UVB treatment of NHEM cells (Fig. 6G), and for 6-4PP levels following UVB exposure in Mel-F-Neo cells (Fig. S8E). Taken together, these results show significant differences in the prevalence of wild-type NCOA3 versus the T960T polymorphism in familial melanoma, as well as differential levels of gene expression, regulation of sensitivity to UVR, and accumulation of UV-mediated DNA damage according to NCOA3 polymorphism status.
Discussion
Our results demonstrate, for the first time, an important role for NCOA3 in melanoma susceptibility through its regulation of UVR sensitivity. NCOA3 activation in melanoma, in part through elevated copy number, directs a transcriptional cascade of gene expression, including activation of genes that promote cell survival following UVR (e.g., XPC) and that promote cell cycle progression (e.g., CCNB1), as well as inactivation of key effectors of DDR (e.g., CHEK2) (32). The reduced photoproduct excision observed upon NCOA3 overexpression in the setting of UVR treatment, despite increased expression of XPC, suggests that the transcriptional cascade of gene expression mitigates the impact of persistent DNA damage on cell survival. Thus, NCOA3 activation drives critical features of melanomagenesis, including molecular susceptibility to melanoma through regulation of sensitivity to UVR, as well as promotion of cell proliferation and cell cycle progression, each of which can be targeted by small molecule inhibition of NCOA3.
A role for NCOA3 in melanoma susceptibility was suggested by the demonstration of a significantly lower prevalence of a NCOA3 polymorphism in a high-risk cohort whose susceptibility to melanoma cannot be accounted for by CDKN2A mutation status. Overexpression of wild-type NCOA3 cDNA resulted in a higher level of protein expression when compared to the T960T construct, despite encoding the same amino acid. These results are consistent with a study in E. coli (36) demonstrating higher protein expression in ACA-containing sequences (such as in wild-type NCOA3) compared with ACG-containing sequences (such as in the T960T polymorphism). This differential expression was associated with enhanced cell survival following UVR exposure and increased accumulation of UV-mediated DNA damage. Thus, these studies advance the association between SNPs and cancer risk. They also provide a mechanistic basis for the differential susceptibility to melanoma by showing that a NCOA3 polymorphism results in a gene dosage effect. Intriguingly, several of the genes regulated by NCOA3 (e.g., p53, CHEK2, and XPC) are themselves established markers of susceptibility to cancer, including melanoma (37–39).
In addition, our results describe a novel role for NCOA3 in controlling DDR, which is recognized to represent an important barrier against carcinogenesis. NCOA3 silencing was shown to upregulate CHEK2 expression, with concomitantly increased phospho-CHEK2 levels. CHEK2 itself activates proteins that promote DNA repair, cell cycle arrest, and apoptosis, in part by activating p53-regulated pathways (40). Accordingly, bufalin-mediated inhibition of NCOA3 resulted in activation of phospho-CHEK2, along with that of p53 and p21. These results indicate that NCOA3 activation drives oncogenesis in part by overriding DDR, thereby removing a critical barrier against carcinogenesis, especially in the setting of UVR exposure.
Taken together, our findings are consistent with a model of melanoma initiation in which elevated NCOA3 expression promotes melanocyte survival following exposure to UVR by overriding DDR and promoting cell cycle progression (Fig. 7). This survival advantage occurs at the expense of increased accumulation of UVR-mediated DNA damage. Over the lifetime of the susceptible individual, significant exposure to UVR can result in both the high mutational burden and uncontrolled cellular proliferation that characterize melanoma. By contrast, these effects are attenuated following expression of the T960T polymorphism, with increased sensitivity to UV-mediated cell death, thereby protecting against the carcinogenic effects of UVR.
Fig. 7.
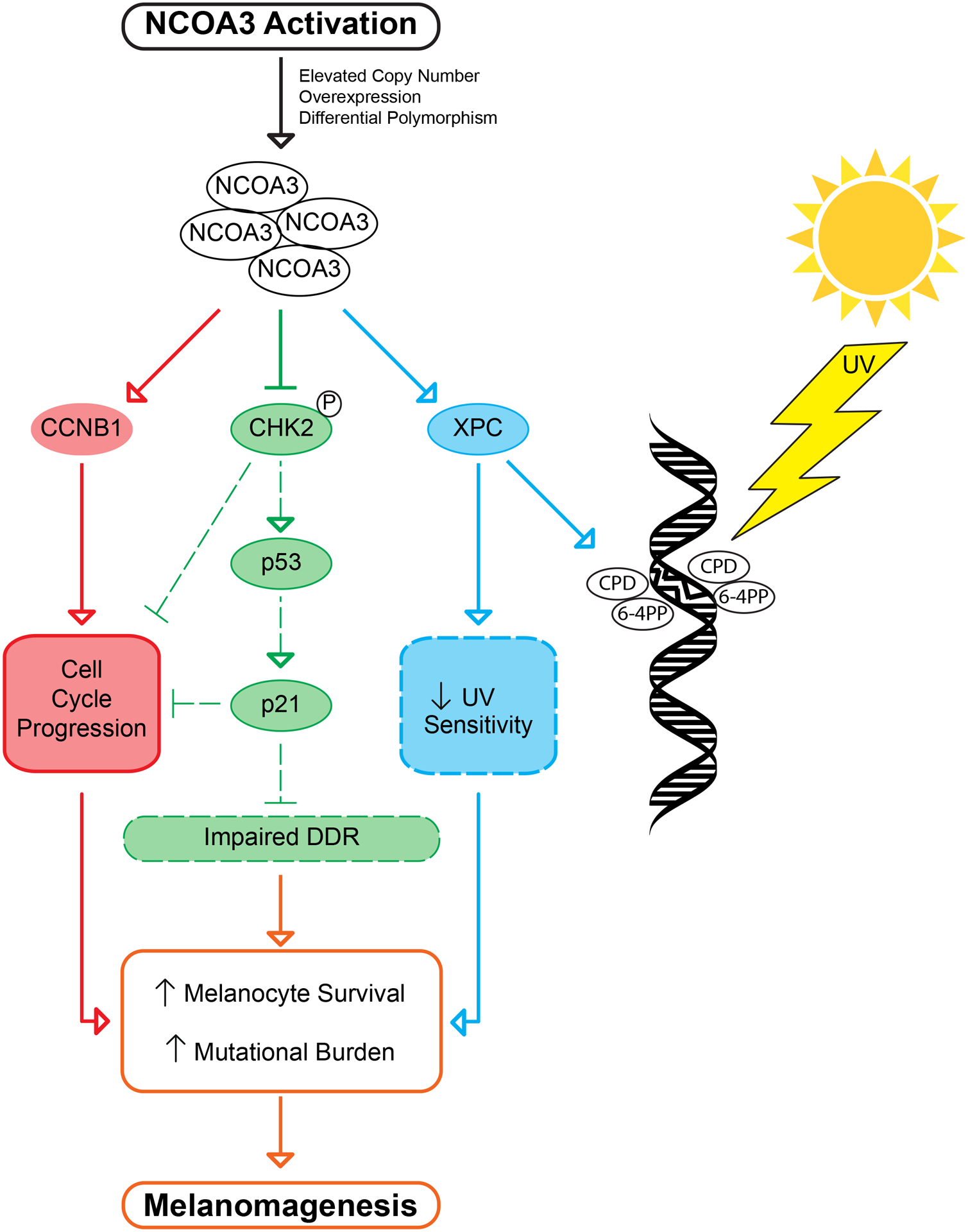
Signal transduction pathways regulated following NCOA3 activation leading to melanomagenesis, including in the setting of UVR exposure.
Beyond its important role in melanoma initiation, our results identify NCOA3 as a novel target for melanoma therapy. Both genetic silencing and pharmacologic targeting of NCOA3 induced profound growth arrest and mitotic catastrophe, resulting in significant anti-tumor effects in vivo. Additional analyses indicated that bufalin administration suppressed expression of key mediators of mitotic progression, including CCNB1, CDK1, AURKA, and PLK1, resulting in centrosome amplification and G2/M arrest. Thus, melanoma cells with elevated NCOA3 copy number are addicted to its oncogenic effects, and are therefore sensitive to NCOA3 targeting, whether by genetic or pharmacological means. Bufalin, which targets both NCOA3 and NCOA1 (30), has been evaluated in early clinical trials of cancer, with good tolerability, but with modest clinical activity in unselected populations (41–44). Our results suggest the potential utility of NCOA3 copy number as a biomarker to identify and enrich patients with melanoma (and potentially other tumors) to undergo treatment with bufalin or other, more specific inhibitors of NCOA3. Intriguingly, NCOA3 was identified as a druggable target in an analysis of the druggable genome (45), further supporting the rationale for NCOA3 targeting.
Taken together, our results have demonstrated an unprecedented role for a molecular marker in distinct stages of tumor progression. These results identify NCOA3 as a candidate susceptibility marker for melanoma, as a potential diagnostic marker (given the presence of copy number elevations in melanomas versus nevi), as a prognostic marker of melanoma survival, and as a target for therapy. Thus, these studies identify a critical role for NCOA3 in UVR-mediated melanomagenesis, and as a rational therapeutic target for melanoma.
Supplementary Material
Statement of Significance:
This manuscript explores NCOA3 as a regulator of the DNA damage response and a therapeutic target in melanoma, where activation of NCOA3 contributes to melanoma development following exposure to ultraviolet light.
Acknowledgments
This work was supported by NIH R01’s CA114337, CA175768, and CA215755 (to M.K.S.), the Cancer Avatar Program Fund of the California Pacific Medical Center Foundation, and the E. A. Dickinson Emeritus Professorship of University California San Francisco (J.E.C.).
Footnotes
Conflict of interest statement: MKS is co-inventor of a patent covering several aspects of the current study.
References:
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7–34 [DOI] [PubMed] [Google Scholar]
- 2.Shaikh WR, Dusza SW, Weinstock MA, Oliveria SA, Geller AC, Halpern AC. Melanoma Thickness and Survival Trends in the United States, 1989 to 2009. J Natl Cancer Inst 2016;108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boniol M, Autier P, Boyle P, Gandini S. Cutaneous melanoma attributable to sunbed use: systematic review and meta-analysis. BMJ 2012;345:e4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lo JA, Fisher DE. The melanoma revolution: from UV carcinogenesis to a new era in therapeutics. Science 2014;346:945–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whiteman DC, Whiteman CA, Green AC. Childhood sun exposure as a risk factor for melanoma: a systematic review of epidemiologic studies. Cancer Causes Control 2001;12:69–82 [DOI] [PubMed] [Google Scholar]
- 6.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A landscape of driver mutations in melanoma. Cell 2012;150:251–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012;44:1006–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010;463:191–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fargnoli MC, Argenziano G, Zalaudek I, Peris K. High- and low-penetrance cutaneous melanoma susceptibility genes. Expert Rev Anticancer Ther 2006;6:657–70 [DOI] [PubMed] [Google Scholar]
- 10.Pho LN, Leachman SA. Genetics of pigmentation and melanoma predisposition. G Ital Dermatol Venereol 2010;145:37–45 [PubMed] [Google Scholar]
- 11.Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, et al. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell 1997;90:569–80 [DOI] [PubMed] [Google Scholar]
- 12.Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, et al. The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature 1997;387:677–84 [DOI] [PubMed] [Google Scholar]
- 13.Xu J, Wu RC, O’Malley BW. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat Rev Cancer 2009;9:615–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, et al. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 1997;277:965–8 [DOI] [PubMed] [Google Scholar]
- 15.Cai D, Shames DS, Raso MG, Xie Y, Kim YH, Pollack JR, et al. Steroid receptor coactivator-3 expression in lung cancer and its role in the regulation of cancer cell survival and proliferation. Cancer Res 2010;70:6477–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tien JC, Liu Z, Liao L, Wang F, Xu Y, Wu YL, et al. The steroid receptor coactivator-3 is required for the development of castration-resistant prostate cancer. Cancer Res 2013;73:3997–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mo P, Zhou Q, Guan L, Wang Y, Wang W, Miao M, et al. Amplified in breast cancer 1 promotes colorectal cancer progression through enhancing notch signaling. Oncogene 2015;34:3935–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dasgupta S, Rajapakshe K, Zhu B, Nikolai BC, Yi P, Putluri N, et al. Metabolic enzyme PFKFB4 activates transcriptional coactivator SRC-3 to drive breast cancer. Nature 2018;556:249–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haqq C, Nosrati M, Sudilovsky D, Crothers J, Khodabakhsh D, Pulliam BL, et al. The gene expression signatures of melanoma progression. Proc Natl Acad Sci U S A 2005;102:6092–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rangel J, Torabian S, Shaikh L, Nosrati M, Baehner FL, Haqq C, et al. Prognostic significance of nuclear receptor coactivator-3 overexpression in primary cutaneous melanoma. J Clin Oncol 2006;24:4565–9 [DOI] [PubMed] [Google Scholar]
- 21.Kashani-Sabet M, Nosrati M, Miller JR 3rd, Sagebiel RW, Leong SPL, Lesniak A, et al. Prospective Validation of Molecular Prognostic Markers in Cutaneous Melanoma: A Correlative Analysis of E1690. Clin Cancer Res 2017;23:6888–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miele ME, Jewett MD, Goldberg SF, Hyatt DL, Morelli C, Gualandi F, et al. A human melanoma metastasis-suppressor locus maps to 6q16.3-q23. Int J Cancer 2000;86:524–8 [DOI] [PubMed] [Google Scholar]
- 23.de Semir D, Bezrookove V, Nosrati M, Dar AA, Wu C, Shen J, et al. PHIP as a therapeutic target for driver-negative subtypes of melanoma, breast, and lung cancer. Proc Natl Acad Sci U S A 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burwinkel B, Wirtenberger M, Klaes R, Schmutzler RK, Grzybowska E, Forsti A, et al. Association of NCOA3 polymorphisms with breast cancer risk. Clin Cancer Res 2005;11:2169–74 [DOI] [PubMed] [Google Scholar]
- 25.De Semir D, Nosrati M, Bezrookove V, Dar AA, Federman S, Bienvenu G, et al. Pleckstrin homology domain-interacting protein (PHIP) as a marker and mediator of melanoma metastasis. Proc Natl Acad Sci U S A 2012;109:7067–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res 2019;47:D419–D26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hodgson JG, Chin K, Collins C, Gray JW. Genome amplification of chromosome 20 in breast cancer. Breast Cancer Res Treat 2003;78:337–45 [DOI] [PubMed] [Google Scholar]
- 28.Barks JH, Thompson FH, Taetle R, Yang JM, Stone JF, Wymer JA, et al. Increased chromosome 20 copy number detected by fluorescence in situ hybridization (FISH) in malignant melanoma. Genes Chromosomes Cancer 1997;19:278–85 [PubMed] [Google Scholar]
- 29.Bastian BC, LeBoit PE, Hamm H, Brocker EB, Pinkel D. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridization. Cancer Res 1998;58:2170–5 [PubMed] [Google Scholar]
- 30.Wang Y, Lonard DM, Yu Y, Chow DC, Palzkill TG, Wang J, et al. Bufalin is a potent small-molecule inhibitor of the steroid receptor coactivators SRC-3 and SRC-1. Cancer Res 2014;74:1506–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene 2004;23:2825–37 [DOI] [PubMed] [Google Scholar]
- 32.Ray A, Milum K, Battu A, Wani G, Wani AA. NER initiation factors, DDB2 and XPC, regulate UV radiation response by recruiting ATR and ATM kinases to DNA damage sites. DNA Repair (Amst) 2013;12:273–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hussussian CJ, Struewing JP, Goldstein AM, Higgins PA, Ally DS, Sheahan MD, et al. Germline p16 mutations in familial melanoma. Nat Genet 1994;8:15–21 [DOI] [PubMed] [Google Scholar]
- 34.Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian SV, et al. A cell cycle regulator potentially involved in genesis of many tumor types. Science 1994;264:436–40 [DOI] [PubMed] [Google Scholar]
- 35.Ravanat JL, Douki T, Cadet J. Direct and indirect effects of UV radiation on DNA and its components. J Photochem Photobiol B 2001;63:88–102 [DOI] [PubMed] [Google Scholar]
- 36.Boel G, Letso R, Neely H, Price WN, Wong KH, Su M, et al. Codon influence on protein expression in E. coli correlates with mRNA levels. Nature 2016;529:358–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bell DW, Varley JM, Szydlo TE, Kang DH, Wahrer DC, Shannon KE, et al. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science 1999;286:2528–31 [DOI] [PubMed] [Google Scholar]
- 38.Li L, Bales ES, Peterson CA, Legerski RJ. Characterization of molecular defects in xeroderma pigmentosum group C. Nat Genet 1993;5:413–7 [DOI] [PubMed] [Google Scholar]
- 39.Malkin D, Li FP, Strong LC, Fraumeni JF Jr., Nelson CE, Kim DH, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990;250:1233–8 [DOI] [PubMed] [Google Scholar]
- 40.Bruno T, De Nicola F, Iezzi S, Lecis D, D’Angelo C, Di Padova M, et al. Che-1 phosphorylation by ATM/ATR and Chk2 kinases activates p53 transcription and the G2/M checkpoint. Cancer Cell 2006;10:473–86 [DOI] [PubMed] [Google Scholar]
- 41.Meng Z, Yang P, Shen Y, Bei W, Zhang Y, Ge Y, et al. Pilot study of huachansu in patients with hepatocellular carcinoma, nonsmall-cell lung cancer, or pancreatic cancer. Cancer 2009;115:5309–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meng Z, Garrett CR, Shen Y, Liu L, Yang P, Huo Y, et al. Prospective randomised evaluation of traditional Chinese medicine combined with chemotherapy: a randomised phase II study of wild toad extract plus gemcitabine in patients with advanced pancreatic adenocarcinomas. Br J Cancer 2012;107:411–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qin TJ, Zhao XH, Yun J, Zhang LX, Ruan ZP, Pan BR. Efficacy and safety of gemcitabine-oxaliplatin combined with huachansu in patients with advanced gallbladder carcinoma. World J Gastroenterol 2008;14:5210–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xie X, Huang X, Li J, Lv X, Huang J, Tang S, et al. Efficacy and safety of Huachansu combined with chemotherapy in advanced gastric cancer: a meta-analysis. Med Hypotheses 2013;81:243–50 [DOI] [PubMed] [Google Scholar]
- 45.Finan C, Gaulton A, Kruger FA, Lumbers RT, Shah T, Engmann J, et al. The druggable genome and support for target identification and validation in drug development. Sci Transl Med 2017;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.