

Abstract
The Src family of kinases (SFKs) are homologs of retroviral oncogenes, earning them the label of proto-oncogenes. Their functions are influenced by positive and negative regulatory tyrosine phosphorylation events and inhibitory and activating intramolecular and extramolecular interactions. This regulation is disrupted in their viral oncogene counterparts. However, in contrast to most other proto-oncogenes, the genetic alteration of these genes does not seem to occur in human tumors and how and if their functions are altered in human cancers remains to be determined. To look for proteomic level alterations, we took a more granular look at the activation states of SFKs based on their two known regulatory tyrosine phosphorylations but find no significant differences in their activity states when comparing immortalized epithelial cells to cancer cells. SFKs are known to have other less well studied phosphorylations, particularly within their unstructured N-terminal unique domains (UD), although their role in cancers has not been explored. In comparing panels of epithelial cells to cancer cells we find a decrease in S17 phosphorylation in the UD of Src in cancer cells. Dephosphorylated S17 favors the dimerization of Src that is mediated through the UD and suggests increased Src dimerization in cancers. These data highlight the important role of the UD of Src and suggest that a deeper understanding of proteomic level alterations of the unstructured UD of SFKs may provide considerable insights into how SFKs are deregulated in cancers.
Implications
This work highlights the role of the N-terminal unique domain of Src kinases in regulating their signaling functions and possibly in their deregulation in human cancers.
Introduction
Src belongs to a family of closely related non-receptor tyrosine kinases that phosphorylate a plethora of substrates and participate in many different cellular signaling pathways (1–3). All Src family kinases (SFKs) share a simple modular domain structure consisting of SH3, SH2, and catalytic kinase domains (KD) followed by a short C-terminal tail, all of which are highly homologous within the family. However N-terminal sequences of approximately 70–80 residues are entirely divergent imparting a unique identity and possibly unique and non-redundant function to individual members. Src and other closely related SFK members undergo N-terminal myristoylation by N-myristoyl transferase (NMT) (4, 5). The lipid moiety along with charged amino acids within the first 10–17 residues account for their localization to membranes (4, 6). The N-terminal unique domain and its myristoyl moiety are also involved in Src dimerization (7).
The activities of Src are regulated in a complex fashion by intramolecular interactions that are often in competition with extramolecular interactions and which are modulated by post-translational modifications (extensively reviewed (8–11)). Phosphorylation at Tyr 527 (using chicken c-src numbering) by c-src kinase (CSK) is inhibitory while phosphorylation at Tyr 416 is activating, although neither of these phosphorylations by themselves exert full positive or negative regulatory control. Crystallographic studies reveal that Src is held in an autoinhibited state by intramolecular interactions wherein the SH2 domain is bound to its phosphorylated C-terminal Tyr 527 and the SH3 domain engages a proline-rich SH2-KD linker region and the kinase N-lobe. In this state the active site of the kinase domain is disrupted by displacement of the αC helix. These intramolecular interactions are weak in nature, and can be destabilized by solicitation of the SH2 and SH3 domains by higher affinity ligands (12) or by dephosphorylation of Tyr 527 by a number of phosphatases (13). Auto-phosphorylation at Tyr 416 stabilizes the activation loop, an event that in most kinases is required for catalytic activity. Thus, in the most simple paradigm, Src has been thought to be regulated negatively and positively by phosphorylations at Y527 and Y416 (11).
The Src family of non-receptor tyrosine kinases have been implicated in cancer pathogenesis for more than 4 decades, yet their role has remained difficult to define. The fact that the homologous v-src oncogene imparts a tumorigenic attribute to the Rous Sarcoma Virus forms the basis for the belief that the c-src protooncogene must somehow be involved in human cancer pathogenesis. However after massive efforts in the characterization of human tumor genomes, it is evident that the genetic alteration of c-src or its family members is not seen in human cancers. There are rare amplifications across the c-src locus seen in some tumors, but there is little evidence that c-src is the driver gene in these amplicons, particularly since over-expression of c-src is not transforming or tumorigenic without mutational alteration (14, 15). Exhaustive clinical testing of several Src tyrosine kinase inhibitors involving a wide variety of epithelial cancers showed almost no antitumor activity worthy of pursuit(16–27).
Therefore it remains to be defined if and how the functions of Src are perturbed in human cancer pathogenesis. In lieu of genetic alterations, it remains possible that Src is deregulated in other ways. In this work we undertook an in-depth analysis of the phosphorylation-state cycles of Src in an effort to explore how Src may be deregulated in cancer cells.
Methods
Cell culture and reagents
A total of forty-three cell lines were used in this study including six immortalized epithelial cell lines, thirty six cancer cell lines, and a fibroblast cell line. The source of each cell line is described in Supplementary table 1. Cell lines were used within six months of purchase or receipt from the gifting laboratory. With the exception of MCF-10A and MDA-MB-468, no additional authentication was performed. All cells tested negative for mycoplasma contamination by polymerase chain reaction (PCR) and were subsequently grown in fresh medium (RPMI1640, DMEM/F12, Myco’s 5A, MEM) supplemented with 10% fetal bovine serum (Gemini Bio-Products) and penicillin and streptomycin (ThermoFisherScientific). Some HMECs including MCF10A cells were cultured in DMEM/HamF12 media supplemented with 5% Horse serum, bovine insulin, recombinant EGF, hydrocortisone. Other HMECs were cultured in Clonetics MEGM media containing EGF, hydrocortisone, insulin, and bovine pituitary extract.
Western blotting and antibodies
Western blot analysis was performed by SDS-PAGE separation of cell lysates. Briefly, cell lysates were made in modified RIPA buffer (containing 150 mmol/L NaCl, 0.1% SDS, 1% Nonidet P40, 1% sodium deoxycholate and 10 mmol/L sodium phosphate, pH 7.2), which was supplemented with leupeptin, aprotinin, phenylmethylsulfonyl fluoride (PMSF), sodium vanadate, and phosphatase inhibitor cocktail (Roche # 04906845001). The protein concentrations were estimated by the Pierce BCA assay (ThermoFisherScientific). Equal amounts of protein (30 ugs) were resolved on 8–10% denaturing SDS polyacrylamide gel and transferred onto a PVDF membrane. The membranes were blocked with 3% bovine serum albumin (BSA), stained with primary antibodies overnight at 4°C, washed 3 times with TBST (Tris-buffered saline containing 1% Triton-X-100), stained with secondary antibodies for 1 hour at room temperature and detected using chemiluminescence. Antibodies for the following proteins were from these vendors: SFK (SantaCruz Bitoech sc-18), pY416-SFK (Cell Signaling cs-2101 and cs-6943), pY527-SFK (Cell Signaling cs-2105), non-pY416-SFK (Cell Signaling cs-2102), non-pY527-SFK (Cell Signaling cs-2107), pS17-Src (Cell Signaling 12432s), Src (Cell Signaling 2110s), HA (SantaCruz Biotech sc-7392 and Cell Signaling cs-3724), Flag (Cell Signaling cs-2368), actin (SantaCruz Biotech sc-1616), GAPDH (Cell Signaling cs-2118), control mouse IgG (SantaCruz Biotech sc-2025), control rabbit IgG (SantaCruz Biotech sc-2027).
Immunoprecipitation and SFK states determination
Cells were trypsinized and seeded onto regular tissue culture treated plates or in Corning CoStar ultra-low attachment plates where they remain in suspension. After 24 hours of culture in the adherent or suspended state, cells were collected and washed once with PBS. Cell lysates were harvested in 0.5 ml ice-cold modified RIPA buffer as indicated above and cleared by centrifugation at 16,000g. 400ug of lysates were subjected to two rounds of immunoprecipitation (IP). The first IP was done using 1ug of anti-pY416-SFK antibodies (cs-6943 or control IgG sc-2027) and the second IP was done from the unbound fraction using 1ug of anti-non-pY416-SFK antibodies (cs-2102 or control IgG Ms sc-2025). The immune complexes were pelleted using 30ul of protein G sepharose beads (GE Healthcare) at 4°C with constant rotation overnight and washed 3 times in mRIPA buffer resuspended to a volume of 350ul. The resuspended (350ul) bound proteins from the 1st and 2nd immune complexes and the unbound fraction remaining in the 350ul of final supernatant were all heated in sample buffer and equal volumes run on SDS-PAGE and immunoblotted using antibodies to detect pY416-SFK, pY527-SFK, and non-pY527-SFK. The anti-pY416 immunoblots verify that IPs with anti-pY416-SFK antibodies are able to immunodeplete the pY416-SFK fraction to completion, thus allowing clean separation of pY416 and non-pY416 fractions of SFKs on different lanes of the immunoblot. The quantitative analysis of the pY527-SFK and non-pY527-SFK immunoblots of these fractions allows the determination of the four possible states of SFKs. Since a pY527-SFK immunoblot cannot be directly and quantitatively compared with a non-pY527-SFK immunoblot, a separate determination of the ratio of total cellular pY527-SFK to non-pY527-SFK was necessary and this was separately obtained for each cell and condition by dasatinib treatment. Dasatinib inhibits the SFK family as well as CSK and thus inhibits both the Y416 autophosphorylation and the Y527 phosphorylation by CSK (Supplementary Figure 1A)(28–30). Dasatinib treatment at 1uM for 30 minutes completely dephosphorylates Y527 of SFKs and allows direct quantitative analysis of the ratio of pY527-SFK to non-pY527-SFK by immunoblotting (Supplementary Figure 1B).
Proteomic analysis and mass spectrometry
24 hours before transfection, cells were seeded in media without antibiotics. Plasmids containing Src followed by a PreScission protease cleavage site and GST were transfected in non-cancer and cancer cells (typically 20 μg plasmid per 15 cm dish; 2 dishes per cell lines) using Lipofectamine 2000 according to the manufacturer’s instructions (Invitrogen). 24h after transfection, cells were collected and seeded into 2 15 cm dish (adherent state) and 2 ultra low attachment 10 cm dishes (non-adherent state). 48 hours after transfection, cells were lysed using modified RIPA buffer as described above. The lysates were incubated at 4°C for 30 minutes with constant rotation, and then centrifuged at 16,000 g for 5 min. The supernatant was collected and 4000 ug were incubated with previously washed Glutathione Sepharose 4B beads (GE Healthcare) overnight at 4°C with constant rotation. Next day, the beads were washed once with modified RIPA buffer, twice with modified RIPA buffer without protease inhibitors and 3 times with ice-cold PBS. RapiGest SF Surfactant (Waters) was added to a final concentration of 0.01%. Typically, around 5% of the sample was run in a gel for Coomassie staining. The rest of the sample was used for tryptic digestion. Beads were resuspended in 5 mM DTT in 100mM NH4HCO3 and incubated for 30 min at room temperature. After this, iodoacetamide was added to a final concentration of 7.5 mM and samples incubated for 30 additional minutes. 0.5ug of sequencing grade trypsin (Promega) was added to each sample and incubated at 37C overnight. Supernatants of the beads were recovered, and beads digested again using 0.5ug trypsin in 100mM NH4HCO3 for 2 hrs. Peptides from both consecutive digestions were recovered by solid phase extraction using C18 ZipTips (Millipore), and resuspended in 0.1% formic acid for analysis by LC-MS/MS. Peptides resulting from trypsinization were analyzed on an Orbitrap Fusion Lumos tribrid mass spectrometer (Thermo Scientific), connected to a NanoAcquity™ Ultra Performance UPLC system (Waters). A 15-cm EasySpray C18 column (Thermo Scientific) was used to resolve peptides (90-min 2–30% gradient with 0.1% formic acid in water as mobile phase A and 0.1% formic acid in acetonitrile as mobile phase B). MS was operated in data-dependent mode to automatically switch between MS and MS/MS. The top 10 precursor ions with a charge state of 2+ or higher were fragmented by EThcD. Peak lists were generated using PAVA in-house software (31). All generated peak lists were searched against the translated sequence of the Src-GST construct expressed, plus the human subset of the SwissProt database (SwissProt.2016.9,6) using Protein Prospector (32) A randomized version of all entries was concatenated to the database for estimation of false discovery rates in the searches. The database search was performed with the following parameters: Peptide tolerance in searches was 20 ppm for precursor and 30 ppm for product ions, respectively. Cysteine carbamidomethylation was include as a fixed modification, and acetylation of the N terminus of the protein, pyroglutamate formation from N terminal glutamine, oxidation of methionine and phosphorylation of serine, threonine and tyrosine as variable modifications. For relative quantitation of the phosphopeptide abundances between samples, the total intensity for precursor ions on the MS traces was integrated during the elution time using Skyline (33).
Immunofluorescence microscopy
Cells were seeded onto glass cover slips in 12-well plates that had been coated with 0.1% gelatin/0.0025% poly-L-lysine and dried. The next day cells were rinsed with cold PBS, fixed in 4% paraformaldehyde for 20 minutes, permeabilized with 0.2% Triton X-100 for 10 minutes and blocked in 2% BSA for 30 minutes. Cells were then stained at room temperature for 30 minutes with primary antibody in 2% BSA and detected with fluorophore conjugated secondary antibodies. Primary antibodies were anti-src rabbit polyclonal and monoclonal antibodies (Cell signaling # 2108, 2109) and secondary antibodies were anti-rabbit IgG Alexa Fluor 647 (Invitrogen A21244). The cells were washed with PBS, incubated in 5.0 μM Hoechst 33342 (as a nuclear counterstain; Thermo Scientific #62249) for 2 minutes, and washed again in PBS. The coverslips were gently dried and mounted onto glass microscope slides (Fisher Scientific #12-550-15) using ProLong Gold antifade reagent (without DAPI; Life Technologies Corporation #P36930). The slides were protected from light and allowed to cure for 24 hours prior to imaging. Imaging was performed using a Zeiss Confocal Laser-Scanning upright Microscope 780-LSM. Equipment used were a Plan-Apochromat 63x/1.40 Oil DIC M27 objective and two laser lines (405nm, 633nm). The image acquisition, adjustment, and exportation were performed using the ZEN 3.1 lite software from Carl Zeiss Microscopy GmbH.
Quantitative analysis of protein expression
For quantitative comparison of protein bands, the individual bands were quantified by densitometry using ImageJ (NIH). Corrected values were obtained by subtracting background densitometry reading from the raw band values. Each gel had a common lane of MCF10A lysates to allow comparison between gels. For each gel the values were normalized relative to the MCF10A lane on that gel. These MCF10A-normalized values were then stratified into non-cancer (6 cell lines) and cancer (37 cell lines) groups and the data points, distribution and means were plotted using GraphPad Prism software. The statistical significance of the difference was determined using the Mann Whitney test and the one-tailed p-value calculated using GraphPad Prism software.
Site-directed mutation
The serine 17 of Src was mutated to alanine using the QuickChange site-directed mutagenesis kit (Agilent Technologies), PfuUltra II Fusion High-fidelity DNA Polymerase (Agilant Technologies), a PCR cycler (C1000 Touch, Bio-Rad), and using following primers (Integrated DNA Technologies Inc): 5’-GCGCCGCGCCCTGGAGCCCGCCGAGAA-3’ and 5’-TTCTCGGCGGGCTCCAGGGCGCGGCGC-3’. The product was treated with DpnI endonuclease to digest the parental DNA template and to select for mutation-containing-synthetic DNA. The mutated DNA mutation was transformed into DH5a elctrocompetent cells (ThermoFisherScientific), and the purified plasmid was verified by sequencing the entire Src cDNA insert.
Results
A panel of immortalized human epithelial cell lines and human cancer cell lines were used to evaluate the expression and phosphorylation state of SFKs at the major regulatory Y419 and Y530 sites (using human Src numbering). The antibody reagents used for this analysis target epitopes largely conserved among the SFK family and this analysis reflects the combined expression and phosphorylation of the ubiquitously expressed members Src, Yes, and Fyn in these cells. The SFKs show activity and substrate preferences that are adhesion state dependent as shown by us and others (34–38). Thus we assayed these cells both in the adherent and suspended states so that our analyses also encompass the dynamic changes in SFKs that correlate with anchorage state. The expression of SFKs varies among the cell panel, both within the non-cancer and cancer cells (Figure 1A). SFK Y419 phosphorylation does not differentiate cancer from non-cancer cells but interestingly it is significantly induced in the unanchored state in most cells (Figure 1B,C). SFK Y530 phosphorylation is variable across these cells and shows only a slight decrease in the unanchored state in cancer and non-cancer cells (Figure 1B,C).
Figure 1.
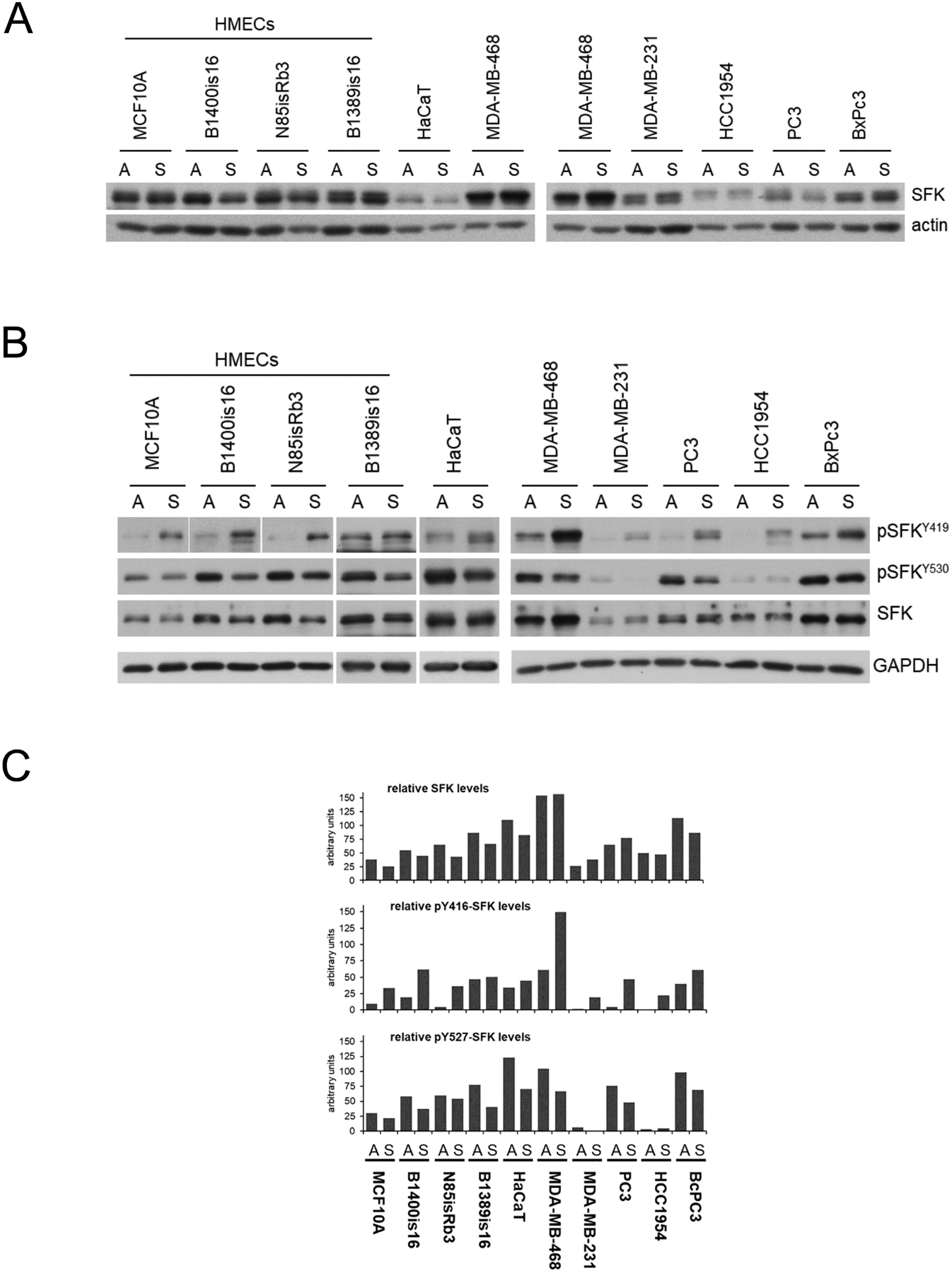
A) Cell lysates from the indicated cancer and non-cancer cell lines were immunoblotted as shown. The SFK antibodies recognize the c-terminal region of this family highly homologous among the family members. B) Cell lysates were immunoblotted against the indicated phospho-specific antibodies marking the highly homologous positive and negative regulatory tyrosines of the SFK family. The Y416 and Y527 residue numbering refers to the commercial names of the antibodies; the actual residue number is different for each SFK family member. C) The immunoblots of part B were quantified, normalized to GAPDH expression, and graphically displayed as shown.
These immunoblots assay the total phosphorylation of Y419 and Y530 as distinct variables. However the activity of each SFK molecule may depend on the combined input from these regulatory residues. To look at the activity state of SFKs in more depth we undertook to characterize them according to the 4 possible Y419/Y530 phosphorylation states in order to describe what fraction of the SFKs are in each specific state. This was done by developing methodologies, using antibody reagents specific for the phosphorylated and for the non-phosphorylated states of these two tyrosine residues and designing an immunodepletion and immunoblotting strategy to detect the 4 states of Y419/Y530 phosphorylation. This data yields a number of observations. First, it provides a glimpse of the overall distribution of regulatory states adopted by Src (Figure 2). Only a small fraction of the total cellular Src is in the fully active state (pY419 / Y530). By far the majority of cellular Src is in the fully inactive auto-inhibited mode (Y419/pY530) or the partially active mode (Y419/Y530). In some circumstances a small fraction is also detectable in a dual phosphorylated pY419/pY530 mode that is not well accounted for by the current paradigms for the activation of Src and likely represents a transient state. Second, comparing non-cancer to cancer cell types finds no significant distinction between them in the overall distribution of Src activity states. Third, comparing adherent to suspended states, it is evident that there is a significant shift towards dephosphorylation of Y530 and phosphorylation of Y419 with the loss of anchorage. This is more pronounced in non-cancer cells, and less evident in cancer cells.
Figure 2.
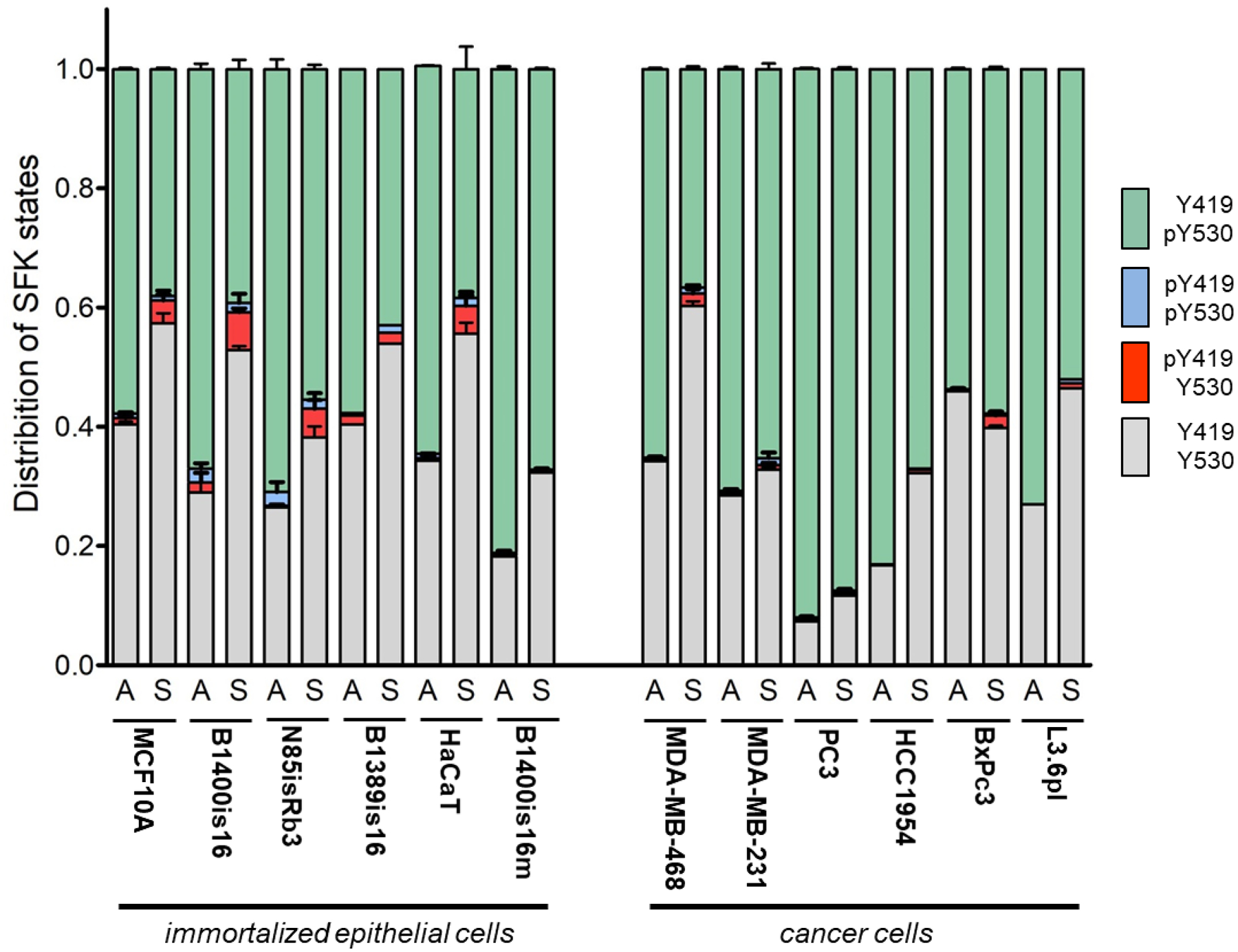
A) Schematic depiction of the fractionation strategy used to determine the fraction of SFK molecules in each of the four possible Y416 and Y527 phosphorylated and non-phosphorylated regulatory states. B) The distribution of SFKs in the four possible regulatory states is shown for the indicated cell lines.
The localization of SFKs was compared among several cancer cell lines and immortalized epithelial cell lines. Two different antibodies including monoclonal and polyclonal antibodies were used to account for non-specific background staining patterns. Both antibodies are directed to epitopes at the kinase domain and c-terminus of Src and thus target many members of the SFK family due to the high degree of homology in this region. This was done to identify the localization of SFK family members as a whole. The staining patterns are consistent between the two different antibodies and reveal predominantly plasma membrane staining as well as a punctate intracellular pool that is consistent with endosomal localization (Figure 3 and Supplementary Figure 2). This localization pattern is consistent with previous reports (39–44). There are no clear differences in subcellular localization that emerge when comparing these cancer cells to the immortalized epithelial cells (Figure 3 and Supplementary Figure 2).
Figure 3.
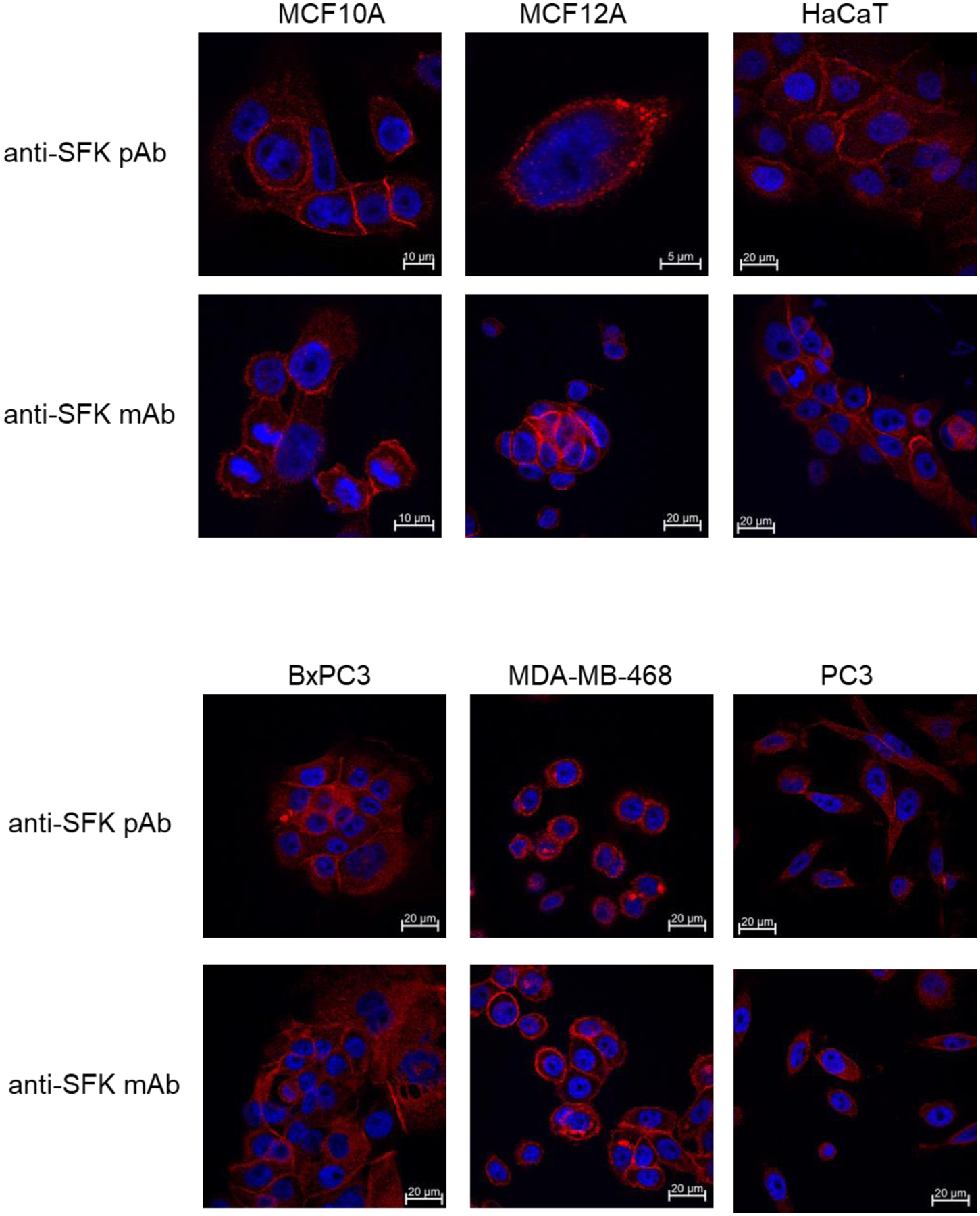
The indicated cell types were grown on cover slips, fixed in paraformaldehyde and stained using either anti-SFK polyclonal or monoclonal antibodies. These antibodies target the c-terminal homologous region of SFKs and react with Src, Yes, and Fyn. After application of secondary antibodies conjugated with Alexa Fluor 647 the images were acquired using confocal microscopy with the appropriate laser excitation. We used two different antibodies in order to best account for background staining and we used a far-red fluorophore which exhibits much less endogenous fluorescence in most cells. All images were acquired using a 63X objective. Some images are cropped. The scale bars indicate the field size for each individual image. A more comprehensive set of images is provided in Supplementary Figure 2.
While Y419 and Y530 phosphorylation are the best understood regulatory mechanisms of Src, there are other phosphorylations that are described but less understood. To begin to explore whether there are distinct phosphorylation events that characterize the Src protein in cancer cells we undertook a phosphoproteomic analysis of Src. A full length Src construct fused with C-terminal GST was expressed in cells for 48 hours and subsequently purified from cell lysates using glutathione beads. This experimental design was expanded in parallel to two breast cancer cell lines and two human mammary epithelial cell (HMEC) lines. Furthermore, Src expression in each cell type was studied in the adherent and in the non-adherent state, with the split occurring 24 hours following transfection. The Src-GST protein was purified from all experimental arms and studied by mass spectroscopy.
Multiple phosphorylation events were detected in Src in these cells encompassing 10 different tyrosine and serine residues. Most of these phosphorylations showed no quantitative differences between the cancer cells and the HMECs except for phosphorylation of S17 which seemed to be lower in cancer cells. The analysis of S17 phosphorylation was then pursued by western blotting of a panel of cancer cell lines and non-cancer cell lines in anchored and unanchored states. S17 phosphorylation does not appear to be regulated by the state of anchorage. However in comparing different cell lines, this analysis confirms that S17 phosphorylation is decreased in some cancer cells compared with non-cancer cells (Figure 4A). This is partly due to a reduced expression of Src in some cells and partly due to a reduced S17 phosphorylation of Src in other cells. The analysis was extended to a larger panel of cancer cell lines revealing that there are other cancer cells with reduced S17 phosphorylation compared to non-malignant epithelial cell lines (Figure 4B,C,D). On average the S17 phosphorylation in cancer cells is significantly reduced compared with non-cancer cells (Figure 4E).
Figure 4.
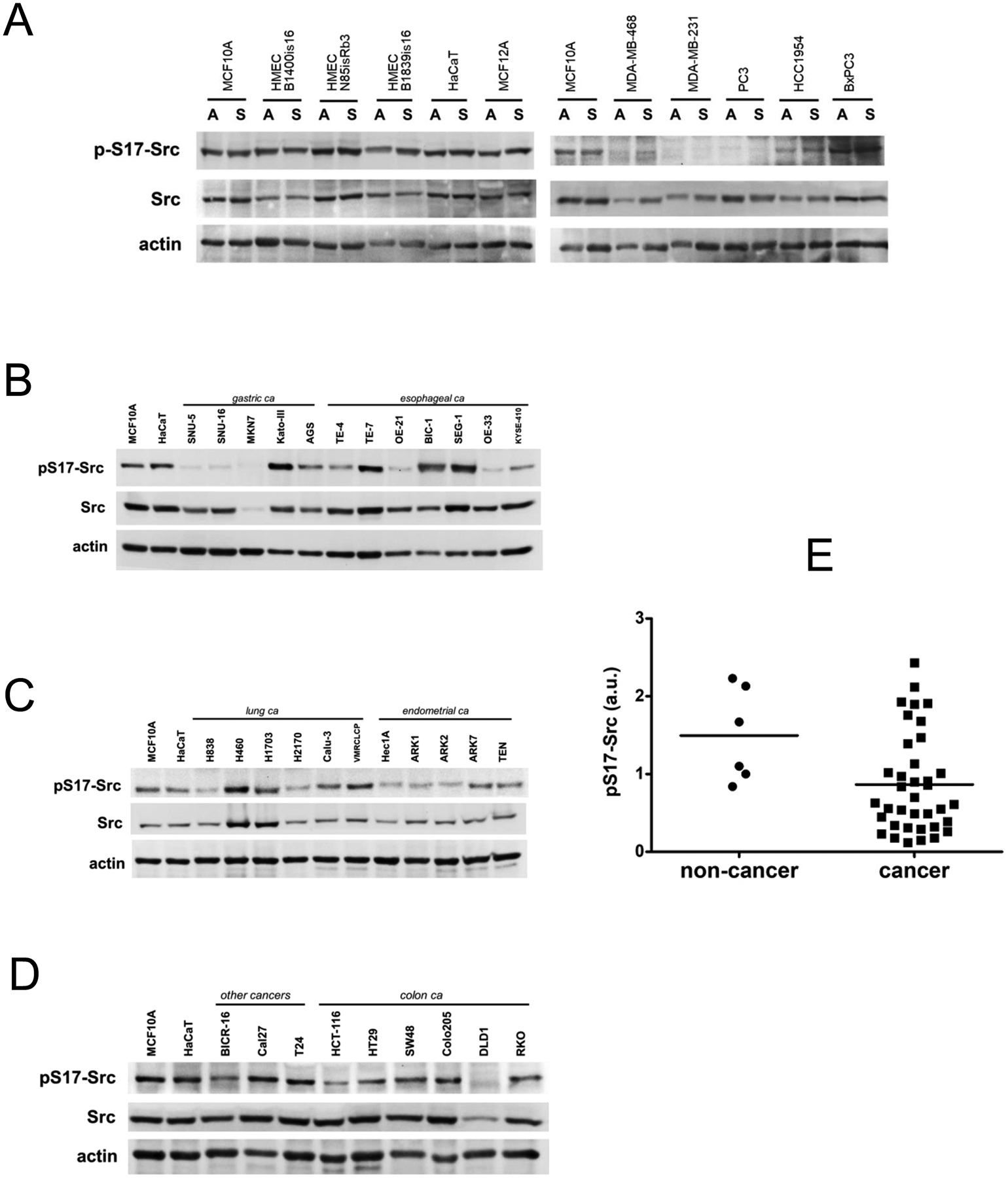
A) Cell lysates from the indicated cancer and non-cancer cell lines were immunoblotted as shown. Cell lysates were harvested in the adherent state (A) or in the suspended (S) state. S17 is in the unique N-terminal domain of Src and is specific for Src. B-D) Cell lysates from the indicated cell lines in the adherent state were collected and immunoblotted as indicated. The same lysates were loaded in the first two lanes of each gel to allow comparisons across the entire cell panel. E) The level of S17 phosphorylation in each cell line was quantitatively determined by densitometry analysis, normalized to actin, normalized relative to MCF10A on the first lane of each blot, and graphically depicted on the grouped scatter plot.
Although the S17 phosphorylation of Src has been described and associated with certain signaling and biologic activities (45, 46), its relevance to the biochemistry of Src function is not well known. S17 resides within the intrinsically disordered N-terminal unique domain (UD) of Src, a domain that has no homology between individual members of the Src family. Because of its unstructured attribute, the UD has evaded analysis by crystallographic studies. We recently described the important role of the UD and its N-terminal myristoyl modification in mediating dimerization of Src (7). This dimerization is mediated through interactions between the UD and the kinase domain (KD) of a dimerization partner. The phosphorylation of S17 may interfere with this interaction and be disruptive to dimerization. We assayed the effects of S17 phosphorylation on Src dimerization through comparison with the S17A mutant of Src. This analysis shows that the S17A mutant dimerizes much more than the wildtype (Figure 5A). This has been repeated in several independent experiments (Supplementary Figure 3). The increased propensity of the S17A mutant to dimerize suggests that S17 phosphorylation is disruptive to dimerization. This would be consistent with our prior work showing that dimerization involves the interaction of the N-terminal myristoylated region with the kinase domain of a dimerization partner (7). Thus the finding of reduced S17 phosphorylation in some cancers suggests that increased Src dimerization is an attribute of some cancers. S17A mutation also affects Y419 autophosphorylation, but not the phosphorylation of the unique Src substrate CDCP1 (Figure 5B).
Figure 5.
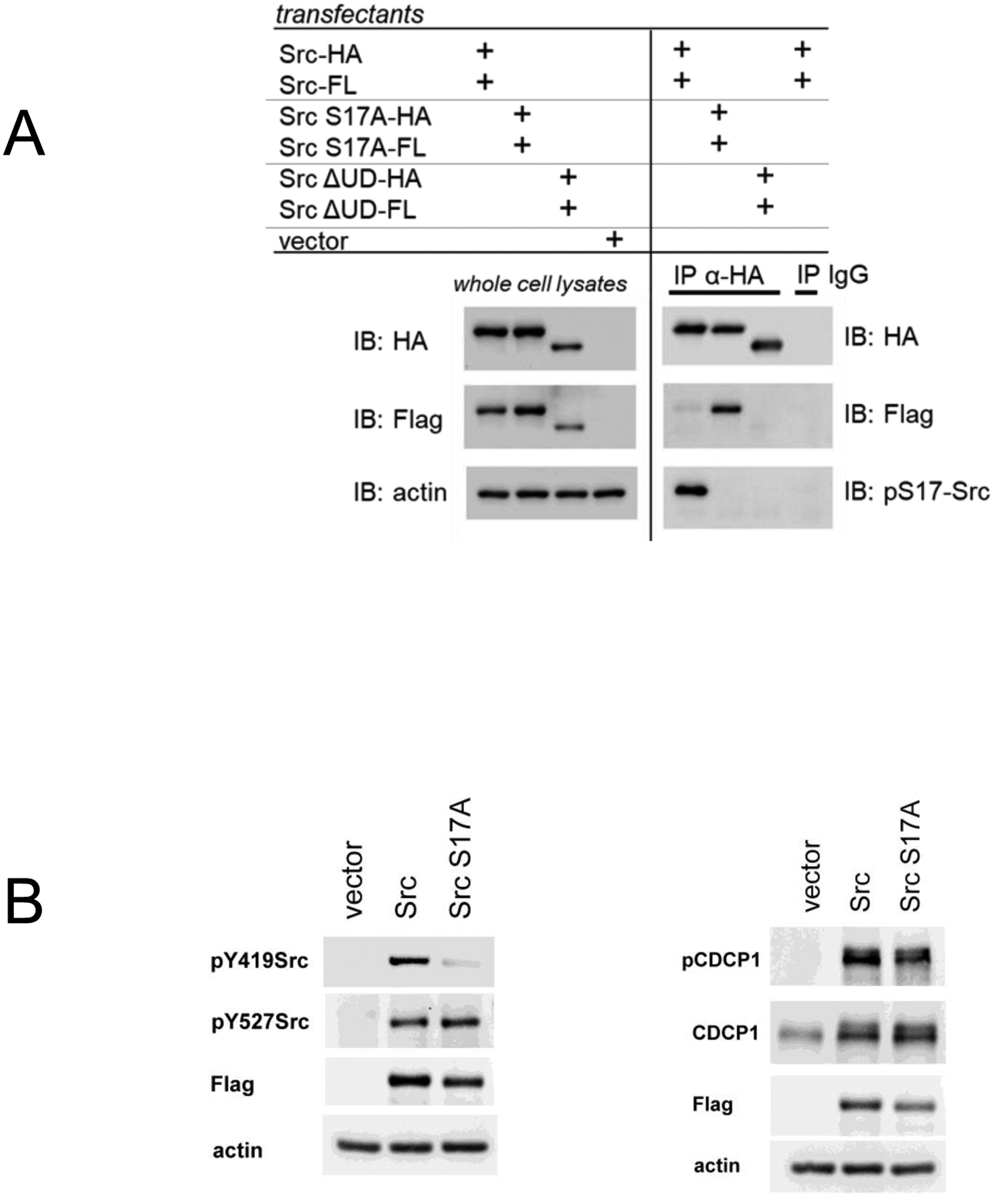
A) SYF cells were transfected with both HA and Flag tagged Src constructs and their level of dimerization determined by co-immunoprecipitation. This is shown for the wildtype Src, the S17A mutant, and the ΔUD deletion mutant that lacks the entire N-terminal unique domain. The wildtype Src does also dimerize, however a lower exposure is shown here to best demonstrate the increased dimerization seen with the S17A mutant. Other experiments are shown in Supplementary Figure 3. B) SYF cells were transfected as in part A and whole cell lysates immunoblotted as indicated.
Src is localized to the membrane through its N-terminal myristoyl modification and this can potentially be affected by UD phosphorylations near the N-terminal such as S17. To directly assess this we looked at the subcellular localization of the S17A mutant in comparison with wildtype Src. Although both constructs have membrane and intracellular fractions, the S17A mutant has an increase in the intracellular pool (Figure 6)
Figure 6.
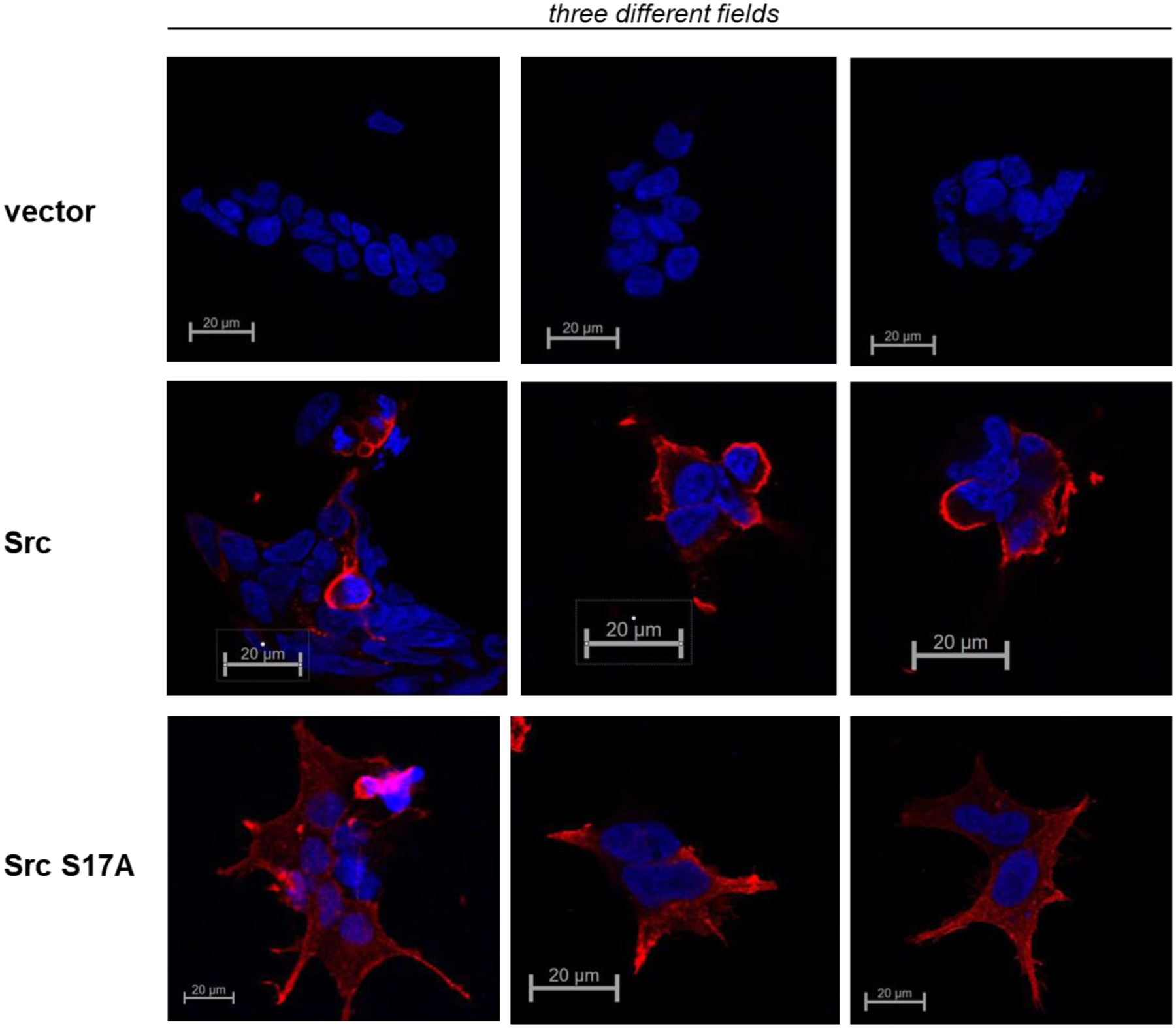
HEK293 cells were transiently transfected to express wildtype Src or the Src S17A mutant, both with c-terminal Flag tags. 24 hours after transfection the cells were stained with Flag antibodies and secondary antibodies conjugated to Alexa Fluor 647.
Discussion
Many of the oncogenes identified in tumor viruses were subsequently found to have protooncogene homologs in normal mammalian genomes, and many of these protooncogenes were subsequently found to be genetically altered and activated in human cancers. Many of these cancers are now effectively treated by inhibiting these activated oncoproteins, completing the full validation of the oncogene hypothesis of human cancer pathogenesis. However this paradigm has not panned out for Src. Despite decades of efforts, it is apparent that there is much more to be learned about the normal and abnormal functions of Src kinases. At the most basic level, it remains unclear if and how Src function is altered in cancers and how this is relevant to cancer biology. In this work we took a fresh look at Src activity states based on its known mechanisms of regulation. Our exploratory analysis of Src regulation in cancer cells led us away from its structured regions and highlight the S17 phosphorylation in the unstructured UD as a clue to how Src may be deregulated in cancers. This calls for much more understanding of the functions of the UD and its phosphorylation as our understanding of this region remains primitive. Indeed the biggest gap in our understanding of this family of kinases lies in the N-terminal UD. This unstructured region has evaded analysis by crystallographic studies which has informed much of our current understanding of Src regulation. However the importance of intrinsically disordered proteins (IDPs) or intrinsically disordered regions (IDRs) of proteins has become apparent in recent years by the sheer abundance of such regions in the eukaryotic proteome (47–49). These regions often mediate multiple interactions with modest affinities that offer a substantial degree if regulation through post-translational modifications and can function as signaling hubs (50–55). Considerable lines of evidence suggest that the UD of SFKs are critically involved in the regulation of SFKs. Although divergent among the SFK family, these regions are conserved across species suggesting that they harbor specific functions. These functions are lost by UD deletion or transferred by UD swapping experiments (56–59). Further consistent with a regulatory function, the UDs of SFKs are phosphorylated and dephosphorylated on several sites, affecting their signaling functions (46, 60–63).
The UD is known to be important in transformation by v-src as deletion mutants are defective in transformation (64). Therefore biologically relevant events involving this region in cancers would not be surprising. S17 has been reported to be phosphorylated by PKA (45, 65), although other kinases may also be able to phosphorylate this residue. We were unable to produce the dephosphorylation of Src S17 in our SYF cell transfection assays despite using either H89 or KT5720 PKA inhibitors, suggesting redundancy with other kinases.
There is only limited data regarding the role and relevance of S17 phosphorylation. S17 phosphorylation has been implicated in Src-mediated regulation of Rap1 and of ERK (65, 66). Whether S17 phosphorylation positively or negatively affects potential tumorigenic functions of Src remain unknown, since we don’t currently have a good understanding of how Src promotes human cancer pathogenesis. However S17 phosphorylation is dispensable for the transforming function of v-src (67), suggesting that it is more likely to have a negative effect on tumorigenic functions. There is some data suggesting that S17 phosphorylation of Src mediates antiproliferative signals although this has not been studied that extensively (65). Our data showing a decrease in S17 phosphorylation in some cancers is consistent with the existing limited evidence favoring a suppressive role linked with this phosphorylation event. Clearly much more work is needed to better understand how S17 phosphorylation affects the biochemical, signaling, and biological functions of Src. Although the S17 residue is unique to Src, there are other phosphorylated residues in the other members of SFKs which may play analogous or non-redundant roles.
We have been studying the functions of the Src UD region and previously reported that it mediates Src dimerization in a biologically relevant manner, affecting the phosphorylation of substrates. In the current study, we find that S17 phosphorylation negatively affects dimerization and that S17 phosphorylation is reduced in some cancers. The implication of these findings is that an increase in Src dimerization may somehow be relevant to cancer pathogenesis. Src is known to exist in dimeric or monomeric forms (68, 69) although there is little known about whether dimers and monomers have different signaling functions, or whether the monomer/dimer ratios have any relevance to their signaling function. It is clear that despite decades of efforts, there remains much more to be learned about the Src UD, the phosphorylation of the UD, Src dimerization, and Src biology.
Supplementary Material
Acknowledgements
We thank Nathaniel Gulizia, Courtney A. Dreyer and Jennifer Merino for technical assistance. We acknowledge the use of the UCSF core facilities including the Cancer Center Laboratory for Cell Analysis core and the UCSF Biomedical Mass Spectrometry and Proteomics Resource Center. UCSF Mass Spectrometry Facility (A.L. Burlingame, Director) is supported by the Dr. Miriam And Sheldon G. Adelson Medical Research Foundation (AMRF) and NIH P41GM103481 and 1S10OD016229. ARS was supported by a grant for postgraduate studies in Life and Matter Sciences of the Fundación Ramón Areces. DSS is currently receiving funding by the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 882247.
Footnotes
Conflicts: The authors have no conflicts to disclose.
References
- 1.Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23:7906–9. [DOI] [PubMed] [Google Scholar]
- 2.Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. [Review] [621 refs]. Annual Review of Cell & Developmental Biology. 1997;13:513–609. [DOI] [PubMed] [Google Scholar]
- 3.Erpel T, Courtneidge SA. Src family protein tyrosine kinases and cellular signal transduction pathways. [Review] [76 refs]. Current Opinion in Cell Biology. 1995;7:176–82. [DOI] [PubMed] [Google Scholar]
- 4.Resh MD. Myristylation and palmitylation of Src family members: the fats of the matter. Cell. 1994;76:411–3. [DOI] [PubMed] [Google Scholar]
- 5.Resh MD. Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochimica et biophysica acta. 1999;1451:1–16. [DOI] [PubMed] [Google Scholar]
- 6.McCabe JB, Berthiaume LG. Functional Roles for Fatty Acylated Amino-terminal Domains in Subcellular Localization. Molecular biology of the cell. 1999;10:3771–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spassov DS, Ruiz-Saenz A, Piple A, Moasser MM. A Dimerization Function in the Intrinsically Disordered N-Terminal Region of Src. Cell reports. 2018;25:449–63 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sicheri F, Kuriyan J. Structures of Src-family tyrosine kinases. Current opinion in structural biology. 1997;7:777–85. [DOI] [PubMed] [Google Scholar]
- 9.Boggon TJ, Eck MJ. Structure and regulation of Src family kinases. Oncogene. 2004;23:7918–27. [DOI] [PubMed] [Google Scholar]
- 10.Xu W, Doshi A, Lei M, Eck MJ, Harrison SC. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol Cell. 1999;3:629–38. [DOI] [PubMed] [Google Scholar]
- 11.Roskoski R Jr. Src protein-tyrosine kinase structure and regulation. Biochemical and biophysical research communications. 2004;324:1155–64. [DOI] [PubMed] [Google Scholar]
- 12.Moarefi I, LaFevre-Bernt M, Sicheri F, Huse M, Lee CH, Kuriyan J, et al. Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature. 1997;385:650–3. [DOI] [PubMed] [Google Scholar]
- 13.Roskoski R Jr. Src kinase regulation by phosphorylation and dephosphorylation. Biochemical and biophysical research communications. 2005;331:1–14. [DOI] [PubMed] [Google Scholar]
- 14.Iba H, Takeya T, Cross FR, Hanafusa T, Hanafusa H. Rous sarcoma virus variants that carry the cellular src gene instead of the viral src gene cannot transform chicken embryo fibroblasts. Proceedings of the National Academy of Sciences of the United States of America. 1984;81:4424–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shalloway D, Coussens PM, Yaciuk P. Overexpression of the c-src protein does not induce transformation of NIH 3T3 cells. Proceedings of the National Academy of Sciences of the United States of America. 1984;81:7071–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campone M, Bondarenko I, Brincat S, Hotko Y, Munster PN, Chmielowska E, et al. Phase II study of single-agent bosutinib, a Src/Abl tyrosine kinase inhibitor, in patients with locally advanced or metastatic breast cancer pretreated with chemotherapy. Ann Oncol. 2011. [DOI] [PubMed] [Google Scholar]
- 17.Gucalp A, Sparano JA, Caravelli J, Santamauro J, Patil S, Abbruzzi A, et al. Phase II Trial of Saracatinib (AZD0530), an Oral SRC-inhibitor for the Treatment of Patients with Hormone Receptor-negative Metastatic Breast Cancer. Clin Breast Cancer. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herold CI, Chadaram V, Peterson BL, Marcom PK, Hopkins J, Kimmick G, et al. Phase II Trial of Dasatinib in Patients with Metastatic Breast Cancer Using Real-Time Pharmacodynamic Tissue Biomarkers of Src Inhibition to Escalate Dosing. Clin Cancer Res. 2011. [DOI] [PubMed] [Google Scholar]
- 19.Lara PN, Longmate J, Evans CP, Quinn DI, Twardowski P, Chatta G, et al. A phase II trial of the Src-kinase inhibitor AZD0530 in patients with advanced castration-resistant prostate cancer: a California Cancer Consortium study. Anticancer Drugs. 2009;20:179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayer E, Baurain J-F, Sparano JA, Strauss LC, Campone M, Fumoleau P, et al. A phase 2 trial of dasatinib in patients with advanced HER2-positive and/or hormone receptor-positive breast cancer. Clinical Cancer Research. 2011. [DOI] [PubMed] [Google Scholar]
- 21.Reddy SM, Kopetz S, Morris J, Parikh N, Qiao W, Overman MJ, et al. Phase II study of saracatinib (AZD0530) in patients with previously treated metastatic colorectal cancer. Investigational new drugs. 2015;33:977–84. [DOI] [PubMed] [Google Scholar]
- 22.Arcaroli J, Quackenbush K, Dasari A, Powell R, McManus M, Tan AC, et al. Biomarker-driven trial in metastatic pancreas cancer: feasibility in a multicenter study of saracatinib, an oral Src inhibitor, in previously treated pancreatic cancer. Cancer medicine. 2012;1:207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fury MG, Baxi S, Shen R, Kelly KW, Lipson BL, Carlson D, et al. Phase II study of saracatinib (AZD0530) for patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC). Anticancer research. 2011;31:249–53. [PMC free article] [PubMed] [Google Scholar]
- 24.Gangadhar TC, Clark JI, Karrison T, Gajewski TF. Phase II study of the Src kinase inhibitor saracatinib (AZD0530) in metastatic melanoma. Investigational new drugs. 2013;31:769–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gubens MA, Burns M, Perkins SM, Pedro-Salcedo MS, Althouse SK, Loehrer PJ, et al. A phase II study of saracatinib (AZD0530), a Src inhibitor, administered orally daily to patients with advanced thymic malignancies. Lung cancer (Amsterdam, Netherlands). 2015;89:57–60. [DOI] [PubMed] [Google Scholar]
- 26.Mackay HJ, Au HJ, McWhirter E, Alcindor T, Jarvi A, MacAlpine K, et al. A phase II trial of the Src kinase inhibitor saracatinib (AZD0530) in patients with metastatic or locally advanced gastric or gastro esophageal junction (GEJ) adenocarcinoma: a trial of the PMH phase II consortium. Investigational new drugs. 2012;30:1158–63. [DOI] [PubMed] [Google Scholar]
- 27.Molina JR, Foster NR, Reungwetwattana T, Nelson GD, Grainger AV, Steen PD, et al. A phase II trial of the Src-kinase inhibitor saracatinib after four cycles of chemotherapy for patients with extensive stage small cell lung cancer: NCCTG trial N-0621. Lung cancer (Amsterdam, Netherlands). 2014;85:245–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–51. [DOI] [PubMed] [Google Scholar]
- 29.Guerrouahen BS, Futami M, Vaklavas C, Kanerva J, Whichard ZL, Nwawka K, et al. Dasatinib inhibits the growth of molecularly heterogeneous myeloid leukemias. Clin Cancer Res. 2010;16:1149–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kurebayashi J, Kanomata N, Moriya T, Kozuka Y, Watanabe M, Sonoo H. Preferential antitumor effect of the Src inhibitor dasatinib associated with a decreased proportion of aldehyde dehydrogenase 1-positive cells in breast cancer cells of the basal B subtype. BMC cancer. 2010;10:568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guan S, Price JC, Prusiner SB, Ghaemmaghami S, Burlingame AL. A data processing pipeline for mammalian proteome dynamics studies using stable isotope metabolic labeling. Molecular & cellular proteomics : MCP. 2011;10:M111 010728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clauser KR, Baker P, Burlingame AL. Role of accurate mass measurement (+/− 10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal Chem. 1999;71:2871–82. [DOI] [PubMed] [Google Scholar]
- 33.Schilling B, Rardin MJ, MacLean BX, Zawadzka AM, Frewen BE, Cusack MP, et al. Platform-independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in skyline: application to protein acetylation and phosphorylation. Molecular & cellular proteomics : MCP. 2012;11:202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spassov DS, Baehner FL, Wong CH, McDonough S, Moasser MM. The transmembrane src substrate Trask is an epithelial protein that signals during anchorage deprivation. Am J Pathol. 2009;174:1756–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei L, Yang Y, Zhang X, Yu Q. Altered regulation of Src upon cell detachment protects human lung adenocarcinoma cells from anoikis. Oncogene. 2004;23:9052–61. [DOI] [PubMed] [Google Scholar]
- 36.Petch LA, Bockholt SM, Bouton A, Parsons JT, Burridge K. Adhesion-induced tyrosine phosphorylation of the p130 src substrate. Journal of cell science. 1995;108 (Pt 4):1371–9. [DOI] [PubMed] [Google Scholar]
- 37.Shattil SJ. Integrins and Src: dynamic duo of adhesion signaling. Trends in cell biology. 2005;15:399–403. [DOI] [PubMed] [Google Scholar]
- 38.Loza-Coll MA, Perera S, Shi W, Filmus J. A transient increase in the activity of Src-family kinases induced by cell detachment delays anoikis of intestinal epithelial cells. Oncogene. 2005;24:1727–37. [DOI] [PubMed] [Google Scholar]
- 39.David-Pfeuty T, Bagrodia S, Shalloway D. Differential localization patterns of myristoylated and nonmyristoylated c-Src proteins in interphase and mitotic c-Src overexpresser cells. Journal of cell science. 1993;105 (Pt 3):613–28. [DOI] [PubMed] [Google Scholar]
- 40.de Diesbach P, Medts T, Carpentier S, D’Auria L, Van Der Smissen P, Platek A, et al. Differential subcellular membrane recruitment of Src may specify its downstream signalling. Experimental cell research. 2008;314:1465–79. [DOI] [PubMed] [Google Scholar]
- 41.Kaplan KB, Swedlow JR, Varmus HE, Morgan DO. Association of p60c-src with endosomal membranes in mammalian fibroblasts. The Journal of cell biology. 1992;118:321–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kasahara K, Nakayama Y, Kihara A, Matsuda D, Ikeda K, Kuga T, et al. Rapid trafficking of c-Src, a non-palmitoylated Src-family kinase, between the plasma membrane and late endosomes/lysosomes. Experimental cell research. 2007;313:2651–66. [DOI] [PubMed] [Google Scholar]
- 43.Reinecke J, Caplan S. Endocytosis and the Src family of non-receptor tyrosine kinases. Biomolecular concepts. 2014;5:143–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sandilands E, Frame MC. Endosomal trafficking of Src tyrosine kinase. Trends in cell biology. 2008;18:322–9. [DOI] [PubMed] [Google Scholar]
- 45.Obara Y, Labudda K, Dillon TJ, Stork PJ. PKA phosphorylation of Src mediates Rap1 activation in NGF and cAMP signaling in PC12 cells. Journal of cell science. 2004;117:6085–94. [DOI] [PubMed] [Google Scholar]
- 46.Amata I, Maffei M, Igea A, Gay M, Vilaseca M, Nebreda AR, et al. Multi-phosphorylation of the Intrinsically Disordered Unique Domain of c-Src Studied by In-Cell and Real-Time NMR Spectroscopy. ChemBioChem. 2013;14:1820–7. [DOI] [PubMed] [Google Scholar]
- 47.Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nature reviews Molecular cell biology. 2015;16:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. Journal of molecular biology. 1999;293:321–31. [DOI] [PubMed] [Google Scholar]
- 49.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–82. [DOI] [PubMed] [Google Scholar]
- 50.Gsponer J, Babu MM. The rules of disorder or why disorder rules. Progress in biophysics and molecular biology. 2009;99:94–103. [DOI] [PubMed] [Google Scholar]
- 51.Borg M, Mittag T, Pawson T, Tyers M, Forman-Kay JD, Chan HS. Polyelectrostatic interactions of disordered ligands suggest a physical basis for ultrasensitivity. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:9650–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van Roey K, Dinkel H, Weatheritt RJ, Gibson TJ, Davey NE. The switches.ELM resource: a compendium of conditional regulatory interaction interfaces. Science signaling. 2013;6:rs7. [DOI] [PubMed] [Google Scholar]
- 53.Van Roey K, Gibson TJ, Davey NE. Motif switches: decision-making in cell regulation. Current opinion in structural biology. 2012;22:378–85. [DOI] [PubMed] [Google Scholar]
- 54.Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. The FEBS journal. 2005;272:5129–48. [DOI] [PubMed] [Google Scholar]
- 55.Kim PM, Sboner A, Xia Y, Gerstein M. The role of disorder in interaction networks: a structural analysis. Molecular systems biology. 2008;4:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carrera AC, Paradis H, Borlado LR, Roberts TM, Martinez C. Lck unique domain influences Lck specificity and biological function. The Journal of biological chemistry. 1995;270:3385–91. [DOI] [PubMed] [Google Scholar]
- 57.Hoey JG, Summy J, Flynn DC. Chimeric constructs containing the SH4/Unique domains of cYes can restrict the ability of Src(527F) to upregulate heme oxygenase-1 expression efficiently. Cellular signalling. 2000;12:691–701. [DOI] [PubMed] [Google Scholar]
- 58.Summy JM, Qian Y, Jiang BH, Guappone-Koay A, Gatesman A, Shi X, et al. The SH4-Unique-SH3-SH2 domains dictate specificity in signaling that differentiate c-Yes from c-Src. Journal of cell science. 2003;116:2585–98. [DOI] [PubMed] [Google Scholar]
- 59.Werdich XQ, Penn JS. Src, Fyn and Yes play differential roles in VEGF-mediated endothelial cell events. Angiogenesis. 2005;8:315–26. [DOI] [PubMed] [Google Scholar]
- 60.Joung I, Kim T, Stolz LA, Payne G, Winkler DG, Walsh CT, et al. Modification of Ser59 in the unique N-terminal region of tyrosine kinase p56lck regulates specificity of its Src homology 2 domain. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:5778–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hansen K, Alonso G, Courtneidge SA, Ronnstrand L, Heldin CH. PDGF-induced phosphorylation of Tyr28 in the N-terminus of Fyn affects Fyn activation. Biochemical and biophysical research communications. 1997;241:355–62. [DOI] [PubMed] [Google Scholar]
- 62.Johnson TM, Williamson NA, Scholz G, Jaworowski A, Wettenhall RE, Dunn AR, et al. Modulation of the catalytic activity of the Src family tyrosine kinase Hck by autophosphorylation at a novel site in the unique domain. The Journal of biological chemistry. 2000;275:33353–64. [DOI] [PubMed] [Google Scholar]
- 63.Amata I, Maffei M, Pons M. Phosphorylation of unique domains of Src family kinases. Frontiers in genetics. 2014;5:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Calothy G, Laugier D, Cross FR, Jove R, Hanafusa T, Hanafusa H. The membrane-binding domain and myristylation of p60v-src are not essential for stimulation of cell proliferation. Journal of virology. 1987;61:1678–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmitt JM, Stork PJ. PKA phosphorylation of Src mediates cAMP’s inhibition of cell growth via Rap1. Mol Cell. 2002;9:85–94. [DOI] [PubMed] [Google Scholar]
- 66.Klinger M, Kudlacek O, Seidel MG, Freissmuth M, Sexl V. MAP kinase stimulation by cAMP does not require RAP1 but SRC family kinases. The Journal of biological chemistry. 2002;277:32490–7. [DOI] [PubMed] [Google Scholar]
- 67.Cross FR, Hanafusa H. Local mutagenesis of Rous sarcoma virus: the major sites of tyrosine and serine phosphorylation of pp60src are dispensable for transformation. Cell. 1983;34:597–607. [DOI] [PubMed] [Google Scholar]
- 68.Le Roux A-L, Castro B, Garbacik ET, Garcia Parajo MF, Pons M. Single molecule fluorescence reveals dimerization of myristoylated Src N-terminal region on supported lipid bilayers. ChemistrySelect. 2016;1:642–7. [Google Scholar]
- 69.Irtegun S, Wood RJ, Ormsby AR, Mulhern TD, Hatters DM. Tyrosine 416 is phosphorylated in the closed, repressed conformation of c-Src. PloS one. 2013;8:e71035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.