

Abstract
Schlafen11 (SLFN11) inactivation occurs in approximately 50% of cancer cell lines and in a large fraction of patient tumor samples, which leads to chemoresistance. Therefore, new therapeutic approaches are needed to target SLFN11-deficient cancers. To that effect, we conducted a drug screen with the NCATS mechanistic drug library of 1978 compounds in isogenic SLFN11-knockout (KO) and wild-type (WT) leukemia cell lines. Here we report that TAK-243, a first-in-class ubiquitin activating enzyme UBA1 inhibitor in clinical development, causes preferential cytotoxicity in SLFN11-KO cells; this effect is associated with claspin-mediated DNA replication inhibition by CHK1 independently of ATR. Additional analyses showed that SLFN11-KO cells exhibit consistently enhanced global protein ubiquitylation, endoplasmic reticulum (ER) stress, unfolded protein response (UPR), and protein aggregation. TAK-243 suppressed global protein ubiquitylation and activated the UPR transducers PERK, phosphorylated eIF2alpha, phosphorylated IRE1, and ATF6 more effectively in SLFN11-KO cells than WT cells. Proteomic analysis using biotinylated mass spectrometry and RNAi screening also showed physical and functional interactions of SLFN11 with translation initiation complexes and protein folding machinery. These findings uncover a previously unknown function of SLFN11 as a regulator of protein quality control and attenuator of ER stress and UPR. Moreover, they suggest the potential value of TAK-243 in SLFN11-deficient tumors.
Keywords: Schlafen11, TAK-243, unfolded protein response (UPR), DNA replication, CHK1
Introduction
Schlafen11 (SLFN11) drives the cytotoxicity of a wide range of anticancer agents targeting DNA replication: topoisomerase I and II (TOP1 & TOP2) inhibitors, DNA synthesis inhibitors (gemcitabine, hydroxyurea, cytarabine), alkylating agents (cis- and carbo-platin) and poly (ADPribose) polymerase (PARP) inhibitors (talazoparib, olaparib, niraparib, rucaparib) [reviewed in (1)]. SLFN11 is one of the five human Schlafen genes, only found in mammals. SLFN11 is involved in an increasing number of cellular functions including cellular proliferation, DNA replication and native immune response to viral infections (1).
At the molecular level, SLFN11 targets DNA replication by binding to replication forks via replication protein A1 (RPA1) (2,3), blocking the replicative helicase complex (3), inducing the degradation of the replication initiation factor CDT1 (4) and stalled replication forks (5) and causing chromatin opening with activation of the stress response and immediate early response genes (IEGs) (6). Because approximately 50% of cancer cell lines and a large fraction of patient tumors do not express SLFN11 and are resistant to DNA replication-targeted therapies (1,7–15), an unanswered question is how to target SLFN11-negative cancer cells.
The medical importance of SLFN11 beyond anti-cancer therapy includes its reported anti-human immunodeficiency virus (HIV) function due to transfer RNA (tRNA) pool alterations and codon-bias discrimination (16). In non-human primates, SLFN11 has also been shown to reduce protein production from the host genes as well as from viral transcripts without codon optimization (17). Recently SLFN11 has been found to directly bind to the ribosomal protein S4 X-linked (RPS4X) and inhibit its function along with the mTOR pathway resulting in suppression of tumor growth and metastasis (14).
The unfolded protein response (UPR) is a homeostatic mechanism in response to the accumulation of unfolded and aggregated proteins in the endoplasmic reticulum (ER) (18). While at least three branches of the UPR including PERK-eIF2α-ATF4, IRE1-XBP1 and ATF6 are well-known, the relationship between the UPR and DNA replication is unclear. Yet, prolonged and uncontrolled UPR leads to cell cycle arrest and apoptotic cell death (19,20).
Targeting the ubiquitin-proteasome system (UPS) is a recent focus of investigations in preclinical models and clinical trials. The generation of ubiquitin-conjugated proteins in mammals is regulated by two ubiquitin activating (E1) enzymes, UBA1 (also known as UBE1) and UBA6. UBA1 is responsible for >99% of cellular ubiquitin and has a catalytic role in ubiquitin-charging for all E2 ubiquitin-conjugating enzymes (approximately 35) (21).
TAK-243 (initial designation, MLN7243) is a first-in-class inhibitor of the ubiquitin activating enzyme UBA1, in clinical trials as anti-cancer agent. The drug binds free ubiquitin to form a TAK-243–ubiquitin adduct inhibiting UBA1 (22). Consequently, TAK-243 impairs cellular ubiquitin conjugation resulting in proteotoxic stress and ultimately cancer cell death (23–29). Here we report that TAK-243 is more cytotoxic in the absence of SLFN11 in preclinical cancer cell line models. Mechanistically, we found that TAK-243 suppresses DNA replication by activating CHK1 in response to ER stress and UPR, which is related to a new functional role of SLFN11 as protective factor against proteotoxic stress, excessive ubiquitylation and aggregation of cellular proteins, as well as UPR activation.
Materials and methods
Cell lines and reagents
DU145, CCRF-CEM and MOLT4 were obtained from DCTD, DTP (3,30). DMS114, HEK293 and 293T were purchased from ATCC (Virginia, USA). Li-7 was bought from ElabScience (Texas, USA). DU145, HEK293 and 293T cell lines were grown in DMEM medium (11995065; GIBCO) with 10% fetal bovine serum and 1% penicillin-streptomycin. CCRF-CEM, MOLT4, DMS114 and Li-7 cell lines were grown in RPMI medium 1640 (11875093; GIBCO) added with 10% FBS and 1% penicillin-streptomycin at 37°C in 5%CO2. Cell lines in this study were passaged about 15 times; all cell lines were tested by MycoAlert™ Mycoplasma Detection Kit (LT07-418; Lonza). Reagents are listed in the Supplementary Table 1.
Generation of SLFN11-Deleted Cells
SLFN11-deleted DMS114 and Li-7 cells were generated by a CRISPR/Cas9 method previously used in DU145, CCRF-CEM and MOLT4 (30).
Cell viability assay
Cells (2x103 cells) were plated in 96-well white plates (PerkinElmer). After 24-hours incubation, TAK-243 was added and incubated for 72 hours. Cellular viability was determined with ATPlite solution (6016949; PerkinElmer) according to manufacturer’s instructions by using an EnVision™ (PerkinElmer).
Western blot analysis
Cells were lysed with RIPA buffer (150 mM NaCl, 50 mM Tris-HCl (pH 7.5), 1 mM EDTA, 1% NP40 (v/v), 0.1% SDS (v/v) and 0.5% sodium deoxycholate (w/v)), protease inhibitor cocktail (Cell Signaling) and phosphatase inhibitor (ThermoScientific). Cell lysates were loaded into wells of Novex tris-glycine gels (Invitrogen) and transferred into Immun-Blot PVDF membranes (BioRad). Membranes were incubated with the indicated primary antibodies (Supplementary Table 1) in 1% milk/PBS-T at 4 °C and subsequently incubated with the horseradish peroxidase (HRP)-conjugated secondary antibodies (NA931 and NA934; GE Healthcare). Protein signals were visualized by ChemiDoc™ Touch MP. Quantification of band intensity was done using ImageJ software and Image Lab software (BioRad).
EdU (5-ethynyl-2’-deoxyuridine) incorporation assay by flow cytometry
Cells were seeded in 6-well plates and treated by drugs, following incubation with EdU 10 μM for 30 min prior to cell harvest. EdU incorporation was measured by Click-iT™ Plus EdU Alexa Fluor™ 647 Flow Cytometry Assay Kit (Invitrogen) according to the manufacturer’s protocols. For DNA staining, cells were mixed with 500 μL of FACS analysis solution containing DAPI. Analysis of the samples was performed by flow cytometry using a FACS Canto (Becton Dickinson) and FlowJo.
Molecular combing assay
Profiling of replication forks was measured by using DNA combing assay as previously described (31). For the analysis of fork elongation speed, CIdU / ldU fibers were picked up randomly. The length of CIdU and ldU were measured and their ratio was calculated. For the analysis of new origin firing, all DNA fibers in one section of each coverslips were counted from captured images. Green-colored single fibers of IdU were defined as new origin firings. Their percentages were calculated by a relative equation.
Immunofluorescence staining analysis
Cells (5×104 cells) were seeded on chamber slides (Alkali Scientific) and treated with TAK-243. Cells were incubated with 10 μM EdU for 30 min prior to cell harvest. After PBS washing, cells were fixed and permeabilized with 0.3% Triton X-100/PBS for 30 min. Next, cells were incubated with Click-IT Plus reaction cocktail (C10339; Invitrogen) for 30 min followed by washing with PBS and blocking with 3% BSA/PBS-T for 30 min. The cells were incubated overnight with primary antibodies/3% BSA/PBS-T at 4°C. After washing with PBS-T, the cells were incubated with second antibodies/3% BSA/PBST by 1:1000 dilution for 1-2 hours. After washing, cells were mounted with Vectashield with DAPI (H-1200; VECTOR). Images were captured with a Zeiss LSM 780 confocal microscope.
Small interfering RNA transfection
DU145 and its SLFN11-KO cells were seeded in a 6-well plate at a density of 2x105 cells/well. Cells were transfected by the Lipofectamine RNAiMax (13778150; Thermo Fisher Scientific) according to the manufacturer’s instructions for 48 hours. siClaspin was obtained from Dharmacon (siGENOME Human Claspin siRNA SMARTPool; M-00528-00-0005). The siRNA sequences of EIF3B and CCT2 are listed in Supplementary Table 1.
BioID pull down and mass spectrometry and validation
SLFN11-BioID2-HA was cloned by PCR with primers (Supplementary Table 1) into MCS-BioID2-HA was a gift from Kyle Roux (74224; Addgene plasmid) by NheI (R0131; NEB) and BspEI (R0540; NEB) restriction enzyme sites (32). HEK293 cells were transiently transfected with empty vector and SLFN11-BioID2-HA by GeneJuice for 48 hours. Cells were incubated with 40 μM biotin (B4501; Sigma) for 24 hours prior to harvest. After PBS wash, cells were lysed in NETN150 buffer (1% NP40, 150 mM NaCl, 0.1 mM EDTA, and 50 mM Tris [pH 7.5]) supplemented with protease inhibitor cocktail. After centrifugation, supernatant was incubated with 100 μL of Dynabeads™ MyOne™ Streptavidin C1 (65001; Invitrogen) for 24 hours. Beads were washed twice with PBS on a magnetic stand. For mass spectrometry, beads were separated by SDS-PAGE gel and analyzed according to previously described protocols (3).
Flow cytometry analysis with L-homopropargylglycine (HPG) labeling and PROTEOSTAT® staining
In HPG labeling, we used a Click-iT™ HPG Alexa Fluor™ 488 Protein Synthesis Assay Kit (C10428; Invitrogen) to measure HPG incorporation with the method based on the manufacture’s datasheet and a previous literature (33). In brief, cells were seeded at the density of 3.0×105/2 mL with methionine-containing complete RPMI media with FBS. After 24-hours incubation, the cells were washed by pre-warmed PBS and incubated with serum-free RPMI lacking methionine (R7513; Sigma) and HPG 25 μM for 30 min. All cells were harvested with EDTA-trypsin and fixed in PBS with 4% PFA for 15 min on ice. Next, cells were permeabilized in 100 μL of PBS containing 0.1% saponin for 15 min at room temperature and then re-suspended with 250 μL of Click-iT reaction cocktails and incubated for 30 min. The pellets were washed twice and transferred with PBS into 5 mL FACS tubes. FITC fluorescence signal was measured by flow cytometry using a FACS Canto (Becton Dickinson).
In PROTEOSTAT® staining, we used a PROTEOSTAT® Aggresome detection Kit (ENZ-51035; Enzo) to measure misfolded and aggregated proteins using the method provided by the manufacture. In brief, cells were incubated in complete media with FBS for 24 hours and harvested with EDTA-trypsin. Next, cells were mixed with 4% PFA-based fixative solution for 30 min, permeabilized in 0.5% triton X-100-based permeabilization solution for 30 min at room temperature, and then re-suspended with 500 μL of PROTEOSTAT® aggresome red detection reagent using fresh dilution of ENZ-51035-0025 by 1,500 fold. After one-hour incubation, the mixture was transferred into 5 mL FACS tubes. The fluorescence signal was measured by PI channel of the FACS Canto.
Quantification and statistical analysis
All statistical analyses were conducted with GraphPad Prism9.0. Statistical significances were determined by the unpaired t test (Figure 1E, 5I, S5C, S5F, S5H, 6A and 6B), the two-way ANOVA test (Figure S1D, 2B, S2B, 3B, 4B, 4E, S4H and S4J), the ordinary one-way ANOVA test (Figure 2D, 2F, 2G, S2C, S2D, 3F, 3I, 4G, 4H and 5C), Sidak’s multiple comparisons test (Figure S1D, 2D, 4E and S4J) and Tukey’s multiple comparisons test (Figure 2B, 2F, 2G, S2B, S2C, S2D, 3B, 3C, 3F, 3I, 4B, 4G, 4H, S4H and 5C). The threshold for statistical significance was P < 0.05. For the quantitative data, the statistical parameters were shown in the figure legends.
Figure 1. SLFN11-deficient cells are hypersensitive toTAK-243.
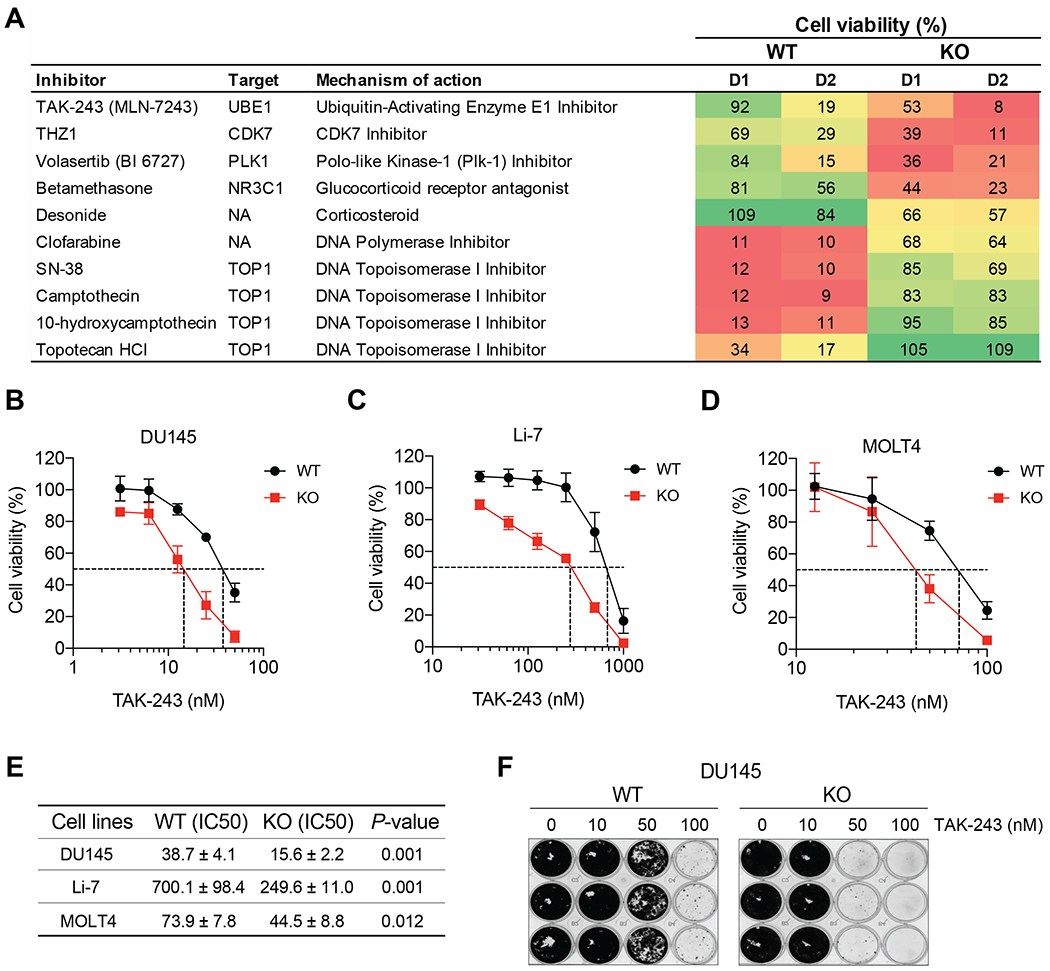
(A) Isogenic leukemic lymphoblasts CCRF-CEM SLFN11-WT and -KO cells were screened with the indicated compounds. D1: 63.2 nM, D2: 189.6 nM. (B-D) TAK-243 cytotoxicity in the indicated SLFN11-WT and -KO isogenic cell lines: prostate cancer DU145, hepatocellular carcinoma Li-7 and leukemic lymphoblasts MOLT4. Cells were treated with the indicated concentrations of TAK-243 for 72 hours. Error bars represent SD for 3 independent experiments. (E) IC50 values of TAK-243 in the different cell lines shown in panels B-D. Mean ± SD and P-values are shown and statistical significance was determined by the unpaired t test. (F) Colony forming assay in DU145 SLFN11-WT and -KO cells treated with the indicated concentrations of TAK-243 for 10 days. The images of colony formation stained with crystal violet.
Figure 5. SLFN11 interactions with translation initiation and protein folding complexes.
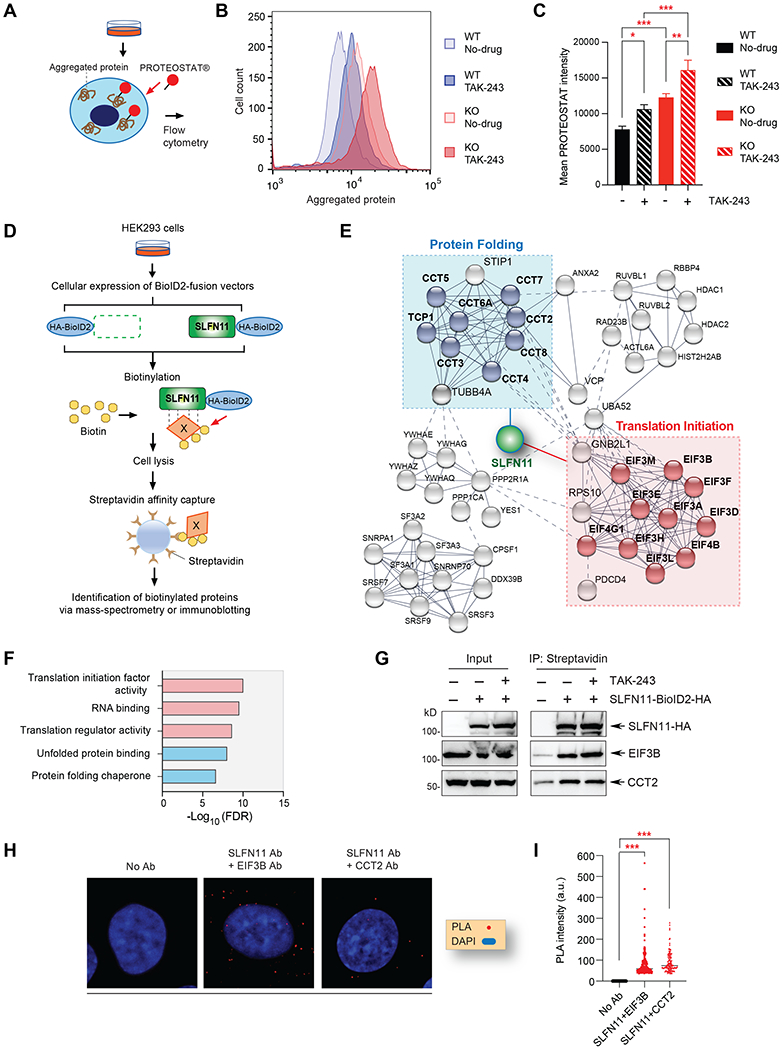
(A) Scheme of the PROTEOSTAT® aggregated protein detection assay. (B) Representative image of aggregated protein in DU145 SLFN11-WT and KO cells with or without TAK-243 (50 nM, 24 hours) measured by flow cytometry. (C) Quantitation of aggregated protein as shown in panel B. The error bars represent SD (N = 5); * P = 0.013, ** P = 0.002, *** P < 0.001. (D) Schematic overview of the biotinylated mass-spectrometry assay and immunoblotting protocol. (E) Protein interaction network of SLFN11 generated by STRING analysis. Line thickness represents the strength of data confidence. (F) Gene ontology (GO) analysis of molecular network associated with SLFN11. (G) Validation of interactions with EIF3B and CCT2. Cells were transfected with SLFN11 vector used in Figure S5E. TAK-243 (0.5 μM) was added for 4 hours prior to cell harvest. After immunoprecipitation with streptavidin, immunoblotting was performed with the indicated antibodies. (H) Proximity ligation assay for SLFN11 interaction with EIF3B and CCT2 in DU145 cells. The red dots indicate the protein interactions. Images were captured by Zeiss LSM 780 ELYRA (63x magnification) and modified by ImageJ software. (I) Quantitation of PLA intensity as shown in panel H by ImageJ software (N>100). *** P < 0.001 (t-test).
Figure 6. Proposed model of SLFN11-mediated protein quality control and connection with TAK-243.
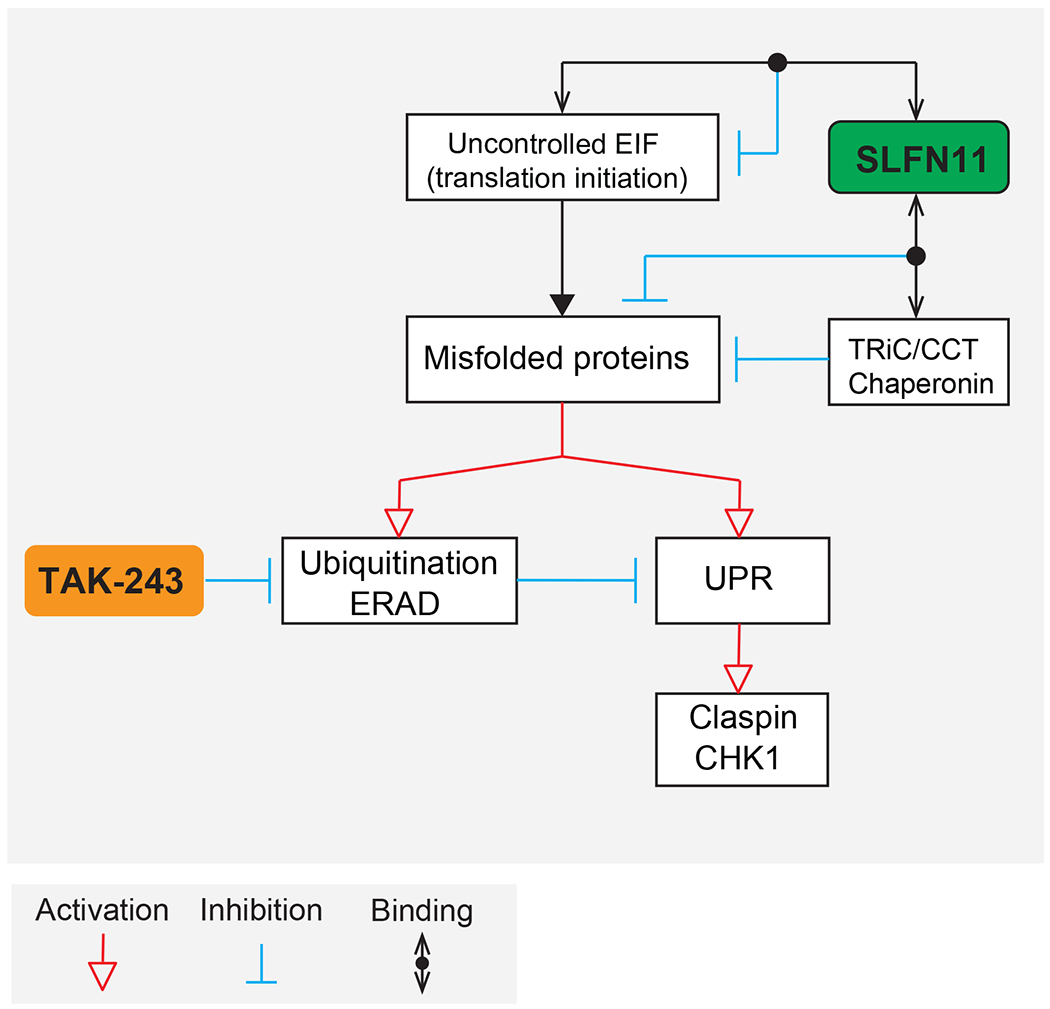
SLFN11 interacts with protein chaperone TRiC/CCT complex and translation initiation complex and reduces protein misfolding. TAK-243 blocks ubiquitination, promotes the accumulation of misfolded proteins and causes ER stress and uncontrolled UPR. In the absence of SLFN11, misfolded proteins accumulate leading to protein ubiquitylation, activation of ER stress and UPR. TAK-243-induced UPR hyperactivation induces CHK1 activation via Claspin.
Figure 2. TAK-243 inhibits DNA replication more effectively in SLFN11-deficient (KO) than proficient (WT) DU145 prostate cancer cells.
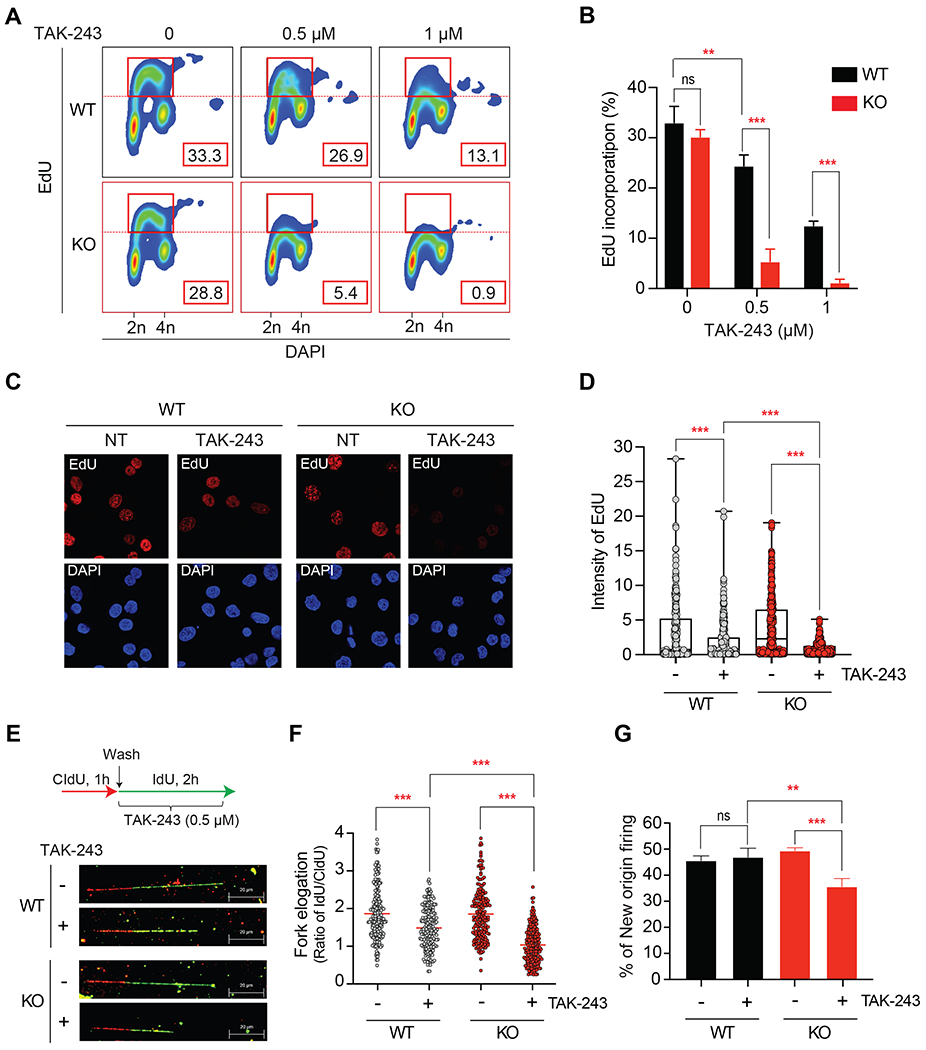
(A) Representative flow cytometry experiment. WT and KO cells were treated with the indicated TAK-243 concentrations for 4 hours, and EdU (10 μM) was added for the last 30 min before harvesting. The percentage of replicating (EdU and DAPI positive) cells measured by FlowJo was indicated in the red boxes. (B) Quantitation of EdU incorporation for 3 independent experiments as shown in panel A. Error bars represent SD; ** P = 0.002, *** P < 0.001. (C) Representative image of EdU incorporation visualized by confocal immunofluorescence microscopy. WT and KO cells were treated with TAK-243 (0.5 μM) for 4 hours. EdU (10 μM) was added for the last 30 min. Nuclei were stained with DAPI (blue). (D) Quantitation of EdU incorporation in WT and KO cells from the immunofluorescence analyses. Error bars represent SD (N = 250-300). *** P < 0.001. (E) Inhibition of replication fork progression analyzed by molecular combing assay. Upper panel: experimental scheme. WT and KO cells were incubated with CldU (100 μM) for 1 hour before adding TAK-243 (0.5 μM) and IdU (100 μM) for 2 hours. Bottom panel: representative images of DNA fibers. The red and green-colored part of fibers represent intake of CldU and IdU, respectively. (F) Ratio of IdU/CIdU (green/red). The length of the green or red-colored section of individual fibers are shown in Figure S2. Red bars represent means (N ≥ 198). *** P < 0.001. (G) New origins defined as ldU-single-labeled fibers measured in the DNA combing assay. Percentage was calculated from the number of green signals divided by the total number of signals (N = 177). Error bars represent SD (N = 3 independent measurements); ** P = 0.003, *** P < 0.001.
Figure 3. TAK-243 inhibits DNA synthesis through CHK1 activation, independently of ATR.
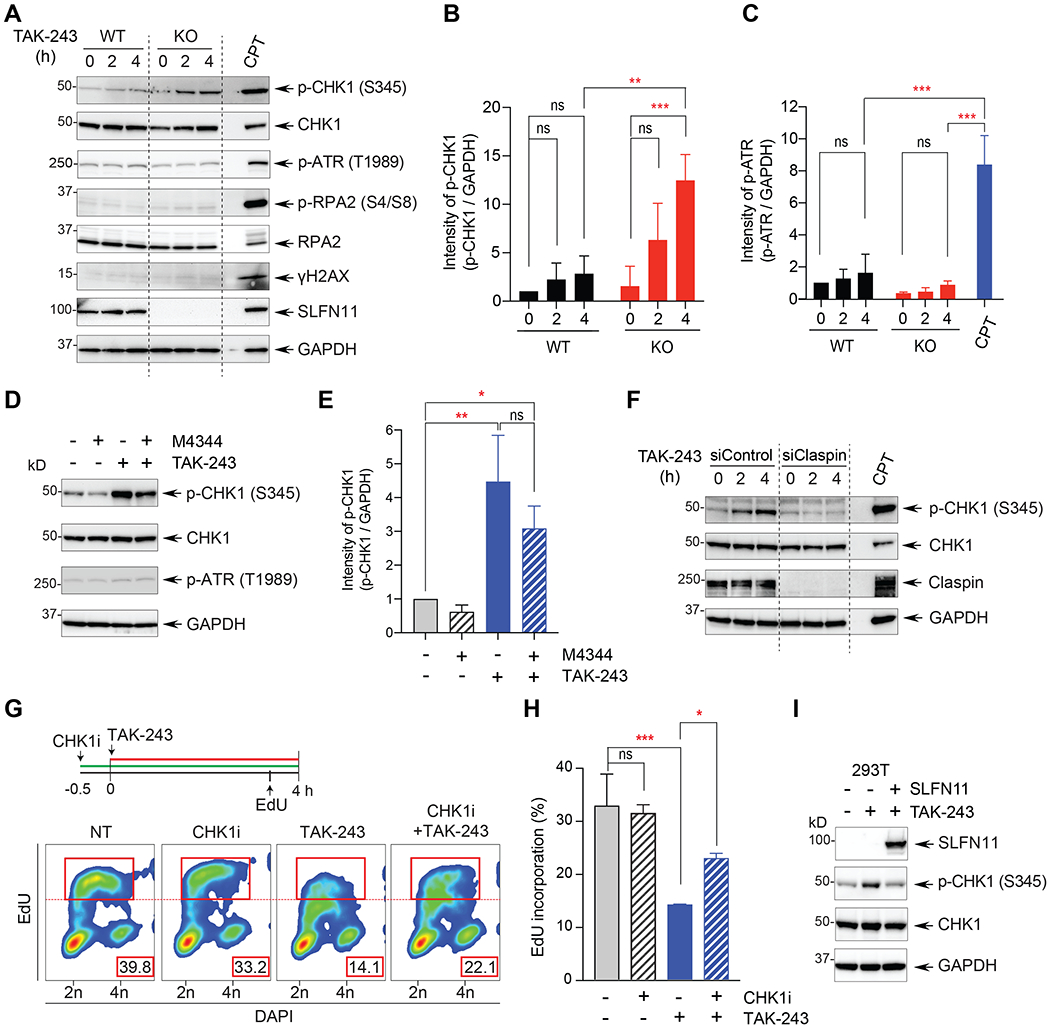
(A) DNA damage response (DDR) proteins detected by Western blotting after TAK-243 treatment. DU145 SLFN11-WT and -KO cells were treated with TAK-243 for the indicated times. WT cells treated with camptothecin (CPT 100 nM) for 4 hours served as positive control. (B, C) Quantitation of p-CHK1 and p-ATR for 3 independent experiments as shown in panel A. Error bars represent SD; ** P = 0.003, *** P < 0.001. (D) CHK1 activation by TAK-243 is independent of ATR. DU145 SLFN11-KO cells were treated with TAK-243 (1 μM) for 4 hours in the absence or presence of the ATR inhibitor (M-4344; 5 nM). (E) Quantitation of p-CHK1 for 3 independent experiments as shown in panel D. Error bars represent SD; * P = 0.042, ** P = 0.002. (F) siClaspin suppresses the CHK1 activation by TAK-243. DU145 SLFN11-KO cells were transfected with siClaspin for 48 hours before TAK-243 treatment (0.5 μM 4 hours).(G) Upper panel shows the experimental scheme used for flow cytometry analysis of DU145 SLFN11-KO cells treated with TAK-243 and the CHK1 inhibitor (LY2606368, 100 nM). EdU (10 μM) was added 30 min prior to cell harvest as indicated. Bottom panel: EdU and DAPI positive cells were quantified by flow cytometry. The percentage of replicating (EdU positive) cells (indicated in the red rectangles) was determined as in Figure 2. (H) Quantitation of EdU incorporation for 3 independent experiments as shown in panel G. Error bars represent SD. * P = 0.039, *** P < 0.001. (I) CHK1 activation by TAK-243 is suppressed by SLFN11. As indicated, SLFN11-negative 293T cells were transfected for 48 hours with SLFN11 construct before TAK-243 treatment (0.5 μM 4 hours).
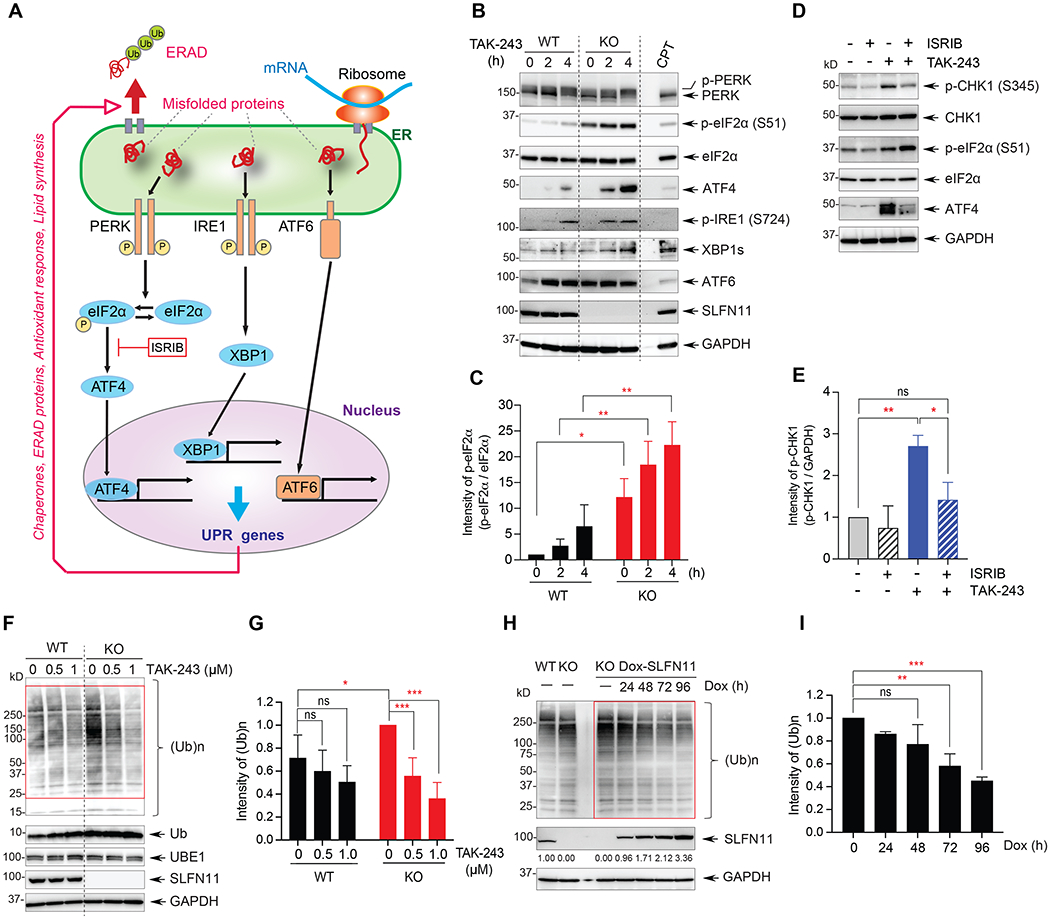
Figure 4. SLFN11 KO cells have high baseline ubiquitylation and TAK-243 further reduces ubiquitin conjugates and eliciting ER stress-mediated UPR in those cells.
(A) Scheme of the unfolded protein response (UPR). ERAD: ER associated degradation of misfolded proteins. (B) Activation of the UPR response by TAK-243 (0.5 μM) was measured by Western blotting in DU145 WT and SLFN11-KO cells as indicated. WT cells treated with CPT (100 nM for 4 hours) served as control. (C) Quantitation of p-eIF2α for 3 independent experiments as shown in panel B. (D) Phosphorylation of eIF2α by combination of TAK-243 and the PERK-ATF4 inhibitor. Cells were treated with TAK-243 (0.5 μM) for 4 hours and the PERK-ATF4 inhibitor (ISRIB; 10 μM) was added 1 hour prior to TAK-243. (E) Quantitation of p-CHK1 for 3 independent experiments as shown in panel D. (F) Ubiquitin conjugates [(Ub)n] determined by Western blotting. DU145 cells were treated with the indicated TAK-243 concentrations for 1 hour. The red rectangle indicates the area quantified in panel G. (G) Quantitation of total ubiquitin conjugates after 1 hour TAK-243 treatment. The error bars represent SD (N = 6); * P = 0.031, *** P < 0.001. (H) Basal ubiquitin conjugates [(Ub)n] changes in doxycycline (Dox)-inducible SLFN11 expressing cells. The cells were treated with Dox 10 μg/ml for the indicated time. The red rectangle indicates the area quantified in panel I. (I) Quantitation of total ubiquitin conjugates after Dox treatment. The error bars represent SD (N = 2); ** P = 0.002, *** P < 0.001.
The rest of Materials and Methods are described in Supplementary Data.
Results
TAK-243 causes selective cytotoxicity in SLFN11-deficient cancer cells.
To detect therapeutic compounds selective for SLFN11-negative cells, we performed a highthroughput drug screening in isogenic leukemic lymphoblast cell lines, CCRF-CEM SLFN11 wild-type (WT) and SLFN11 knockout (KO) (3). As expected, topoisomerase inhibitors, well-known DNA damaging agents, showed increased cytotoxicity in SLFN11-WT cells than SLFN11-KO cells (Figure 1A) (3,15). Conversely, TAK-243 induced greater cytotoxicity towards SLFN11-KO cells (Figure 1A and S1A).
To confirm the selective cytotoxicity of TAK-243 in SLFN11-KO cell lines, we tested three isogenic cancer cell line pairs expressing SLFN11 and their SLFN11-KO counterparts: prostate cancer DU145, hepatocellular carcinoma Li-7 and leukemic lymphoblasts MOLT4. While the DU145 and MOLT4 cell lines were previously reported (30), the Li-7 SLFN11-KO cells were generated by CRISPR/CAS9 for the present study (Figure S1B) and were confirmed to be resistant to CPT (Figure S1C). In all three pairs of cell lines, SLFN11-KO cells were significantly more sensitive to TAK-243 than their WT counterparts (Figure 1B–D). Figure 1E summarizes the differential TAK-243 activity for the three pairs of isogenic SLFN11-expressing and KO cell lines. The sensitivity difference to TAK-243 between the SLFN11-WT and KO cells was confirmed by clonogenic assays. Colony formation in SLFN11-KO cells was significantly reduced at the concentration of 50 nM (p < 0.05) compared to WT cells (Figures 1F and S1D).
TAK-243 also caused more apoptotic cell death in the SLFN11-KO cells compared to SLFN11-WT cells (Figure S1E). In addition, the annexin V-negative and PI-positive population was increased in the SLFN11-KO cells, indicating that TAK-243 caused some necrotic cell death. Apoptotic cell death in SLFN11-KO cells was validated as increased cleaved PARP and cleaved caspase-3 (Figure S1F). These results show that TAK-243 preferentially suppresses cell viability of SLFN11-deficient cancer cells, suggesting that UBA1 activity fosters the survival of SLFN11-KO cells.
TAK-243 inhibits DNA synthesis more effectively in SLFN11-deficient than proficient cancer cells.
We next examined DNA replication in response to TAK-243 treatment because SLFN11 is well established to inhibit DNA replication in response to replication stress (1,3,5,6). We first performed pulse incorporations of the thymidine analog 5-ethynyl-2’-deoxyuridine (EdU) in SLFN11-WT and -KO cells treated with TAK-243 to evaluate DNA synthesis. TAK-243 decreased EdU incorporation within 4 hours significantly more in SLFN11-KO than in SLFN11-WT cells (Figure 2A–B). EdU incorporation was inhibited by more than 80% after 0.5 μM TAK-243 in the SLFN11-KO cells while it was inhibited less than 20% in the SLFN11-WT cells (Figure 2A). The reduced EdU incorporation was observed over 24 hours with a lower concentration of TAK-243 (Figure S2A–B). To confirm the FACS results, immunofluorescence staining was performed after EdU pulse-labeling. SLFN11-KO cells showed enhanced loss of EdU staining after TAK-243 treatment (Figure 2C–D). These results show that TAK-243 suppresses DNA synthesis more effectively in SLFN11-KO cells than SLFN11-WT cells.
To analyze the effects of TAK-243 on DNA replication at the molecular level, we measured fork elongation and replication origin firing using molecular combing (31). SLFN11-WT and -KO cells were incubated with chlorodeoxyuridine (CIdU; red) for 1 hour and then treated with TAK-243 and iododeoxyuridine (IdU; green) for 2 hours (Figures 2E and S2C–D). The ratio of IdU/CIdU represents fork elongation speed with an expected value of 2 in the absence of drug treatment (Figure 2F). The IdU/CldU ratio was significantly decreased in SLFN11-KO cells treated with TAK-243, indicating that TAK-243 reduces replication fork speed more effectively in SLFN11-KO cells (Figure 2E–F and S2C–D). In the same experiments, we also analyzed the ratio of CIdU single-colored fibers versus the total measured fibers to estimate new origin firing (Figure 2G). SLFN11-KO cells treated with TAK-243 showed significantly less origin firing compared to SLFN11-WT cells or untreated cells. These results show that TAK-243 interrupts DNA replication both at the fork elongation and origin firing levels most effectively in SLFN11-KO cells.
TAK-243 blocks DNA synthesis through CHK1 activation via Claspin, but independently of ATR in SLFN11-KO cells.
To determine how TAK-243 suppresses DNA replication, we evaluated the phosphorylation of DNA damage response (DDR) signaling molecules. Under conditions where TAK-243 induced cellular DNA replication inhibition, we observed increased phosphorylation of CHK1 (S345) in the SLFN11-KO cells (Figure 3A and 3B). CHK1 hyperphosphorylation was also detected in the SLFN11-KO cells 10 hours at lower concentrations of TAK-243 (Figure S3A). Notably, in contrast to CPT, which was used as a positive control (31), CHK1 activation by TAK-243 was not accompanied by phosphorylation of RPA2 and γ-H2AX (Figure 3A and S3A), suggesting that TAK-243-induced CHK1 activation is independent of ataxia telangiectasia and RAD3-related (ATR). We confirmed that TAK-243 did not induce detectable ATR activation, which is involved in DDR and is responsive to CPT (Figure 3A, 3C and S3A) (31). Additionally, given that CHK1 is generally activated by ATR (34), we tested the effect of an ATR inhibitor on CHK1 activation by TAK-243. Consistent with the lack of ATR activation by TAK-243, the ATR inhibitor M4344 did not suppress the activation of CHK1 by TAK-243 (Figure 3D and 3E) whereas it blocked ATR activation in the case of CPT (Figure S3B). These data suggest that CHK1 activation by TAK-243 is independent from ATR and different from the classical RPA2-ATR-CHK1 activation by CPT (31).
Because Claspin is a mediator of CHK1 activation (35), and because a recent study showed that thapsigargin, an ER stressor, induces CHK1 activation via Claspin and independently of ATR (36), we hypothesized that CHK1 activation by TAK-243 could be mediated by Claspin independently of ATR. To test this hypothesis, we depleted endogenous Claspin using small interfering RNA (siRNA) prior to TAK-243 treatment. Depletion of Claspin in SLFN11-KO cells completely blocked the phosphorylation of CHK1 by TAK-243 (Figure 3F), consistent with the role of Claspin as a mediator of CHK1 activation by TAK-243.
To examine this possibility further, we tested whether CHK1 inhibition could restore DNA replication in TAK-243-treated cells. SLFN11-KO cells treated with both TAK-243 and the CHK1 inhibitor (LY2606368; prexasertib) showed higher EdU incorporation compared to those treated with TAK-243 alone (Figure 3G–H), indicating that replication inhibition by TAK-243 is mediated by CHK1 activation.
To determine whether CHK1 activation by TAK-243 could be affected by re-expression of SLFN11 in SLFN11-deficient cells, we used 293T SLFN11-deficient cells (16) transiently transfected with SLFN11-WT expressing construct. Re-expression of SLFN11 in SLFN11-deficient cells suppressed TAK-243-induced CHK1 activation (Figure 3I). Together, these results show that TAK-243 suppresses DNA synthesis by activating CHK1 via Claspin independently of ATR.
TAK-243 induces ER stress and UPR more effectively in SLFN11-deficient than proficient cells.
TAK-243 is known to induce ER stress and the UPR (22,23,26–29), and the UPR has been shown to activate CHK1 and inhibit DNA replication (36–38). Also, the activation of PERK-eIF2α pathway by thapsigargin or dithiothreitol has been reported to promote the phosphorylation of CHK1 via Claspin independently of ATR (36).
To test whether SLFN11-KO cells exhibit increased ER stress and uncontrolled UPR, we probed the three classical pathways associated with the UPR (Figure 4A) (18,39) in DU145 WT and SLFN11-KO cells treated with TAK-243 (Figure 4B). Phosphorylated PERK was detected as an electrophoretically upper-shifted band both in SLFN11-WT and -KO cells treated with TAK-243. However, the basal level of phosphorylated eIF2α was higher in SLFN11-KO cells and more effectively induced by TAK-243 in SLFN11-KO than in WT cells (Figure 4B–C). Probing the second IRE1-XBP1 signaling branch of the UPR showed that IRE1 was phosphorylated faster in the SLFN11-KO cells than WT cells. Testing the third UPR pathway also showed that ATF6 was increased in both of SLFN11-WT and KO cells; however, the baseline expression of ATF6 was higher in the SLFN11-KO cells. We confirmed these results in the leukemia CCRF-CEM cells treated for 2 hours (Figure S4A) and in DU145 cells and small cell lung cancer DMS114 cells treated with lower TAK-243 concentrations for 24 hours (Figure S4B–C). Together these experiments show that SLFN11-KO cells have enhanced UPR activation both the absence and presence of TAK-243.
To examine whether phosphorylation of CHK1 (S345) by TAK-243 was dependent on the activation of PERK-ATF4 branch, we treated DU145 SLFN11-KO cells with TAK-243 and the PERK inhibitor ISRIB. ISRIB significantly blocked the phosphorylation of CHK1 in response to TAK-243 (Figure 4D–E). In addition, consistent with the reported accumulation of short-lived proteins including c-Jun and c-Myc upon TAK-243 treatment (22), we found enhanced accumulation of c-Jun and c-Myc in SLFN11-KO cells in response to TAK-243 (Figure S4D), suggesting that SLFN11-KO cells treated with TAK-243 have more global protein accumulation. We also tested the cytotoxicity of the p97 inhibitors, NMS-873 and CB5083 in DU145 SLFN11-WT and -KO cells because these drugs also induce robust UPR (20). SLFN11-KO cells were more sensitive to both p97 inhibitors than WT cells (Figure S4E–F).
Because our data show that SLFN11-KO cells demonstrate enhanced UPR response to TAK-243, which primarily acts as an E1 inhibitor, we measured ubiquitin conjugates in SLFN11-WT and -KO cells. Notably, the overall baseline levels of ubiquitin conjugates were different in the SLFN11-WT and -KO cells. Higher ubiquitin conjugates [(Ub)n] were consistently observed both in the DU145 and CEM SLFN11-KO than SLFN11-WT cells (Figures 4F and S4G), indicating that lack of SLFN11 enhances global ubiquitylation. After TAK-243 treatment, DU145 SLFN11-KO cells lost ubiquitin conjugates in a dose-dependent manner to a greater extent than the SLFN11-WT cells. This effect was not related to different UBE1 protein level. Repeated experiments (N = 6) confirmed high levels of ubiquitin conjugates in SLFN11-KO cells at a steady-state and more robust reduction of ubiquitin conjugates by TAK-243 in SLFN11-KO than WT cells (Figure 4G). Comparable results were obtained in CCRF-CEM SLFN11-WT and -KO cells treated for 1 hour with TAK-243 (Figure S4G–H) and in DU145 SLFN11-WT and -KO cells treated with TAK-243 at lower concentrations for 24 hours (Figure S4I–J). These results demonstrate that TAK-243 depletes ubiquitin conjugates more effectively in the absence of SLFN11. To confirm that the presence of SLFN11 influences the global ubiquitination at the steady-state condition, we generated doxycycline (Dox)-inducible SLFN11-expressing cells. 72-hour exposure to Dox induced robust SLFN11 expression and significantly reduced global cellular ubiquitination by 40-50% (Figure 4H–I). Together, these results show that cells lacking SLFN11 have increased UPR and ubiquitylation, and that TAK-243 suppresses ubiquitin conjugates and increases UPR and ER stress along with CHK1 activation to a greater extent in SLFN11-KO than SLFN11-WT cells.
SLFN11 protects cells from UPR by interacting with protein folding and translation initiation complexes.
To test whether absence of SLFN11 leads to an accumulation of misfolded protein, we evaluated aggregated and newly synthesized proteins by PROTEOSTAT® staining and L-homopropargylglycine (HPG) labelling, respectively (Figure 5A and S5A). SLFN11 WT cells showed less aggregated proteins than KO cells at steady-state condition and TAK-243 increased aggregated proteins in both WT and KO cells (Figure 5B–C). In addition, newly synthesized proteins were less in Li-7 SLFN11 WT cells than KO cells (Figure S5B–C). These data suggest that SLFN11 can decrease aggregated protein and/or newly synthesized protein at steady-state conditions, which is consistent with the lower levels of ubiquitin conjugates in SLFN11-expressing cells.
Next, to determine whether SLFN11 interacts with protein homeostasis and UPR pathways, we performed proteomic analysis with biotinylated mass-spectrometry (Figure 5D) (32). HEK293 SLFN11-proficient cells were transfected with a proximity-dependent biotin identification (BioID)2-fusion empty vector or BioID2-fusion SLFN11. Upon addition of biotin, proteins in close proximity to SLFN11 were biotinylated inside cells. The biotinylated proteins were captured by streptavidin beads and analyzed via mass-spectrometry. As shown in Figure 5E and Supplementary Table 2, TCP1, CCT2, CCT3, CCT4, CCT5, CCT6A, CCT7 and CCT8, which belong to the T-complex protein-1 ring complex (TRiC) and chaperonin containing TCP-1 complex (CCT) in protein folding were identified in SLFN11-BioID transfected cells. In addition, translation initiation complex proteins (EIF3A, EIF3B, EIF3D, EIF3E, EIF3F, FIF3H, EIF3L, EIF3M, EIF4B and EIF4G1) were also enriched in SLFN11-BioID transfected cells. Gene ontology (GO)-term enrichment analysis with the top 100 hits in the list of biotinylated mass-spectrometry proteins showed translation initiation and protein folding-related pathways as prominent protein networks with SLFN11 (Figure 5F and S5D).
To validate the mass spectrometry data, we performed pull-down experiments with the SLFN11-HA-BioID2 construct, and confirmed enrichment of EIF3B, EIF3L and CCT2 in SLFN11-mediated immunoprecipitation (Figure 5G and S5E–F). The interaction of SLFN11 with EIF3B and CCT2 were detected regardless of TAK-243 treatment (Figure 5G). We also validated SLFN11 interaction with EIF3B and CCT2 by proximity ligation assay (PLA) in DU145 cells (Figure 5H–I). Depletion of the TRiC/CCT chaperonin complex genes (CCT6A, CCT4, TCP1, CCT7, CCT3 and CCT2) consistently showed a greater sensitivity of the SLFN11-KO than SLFN11-WT cells (Figure S5G and Supplementary Table 3). Additional experiments confirmed that SLFN11-KO cells were significantly more dependent on CCT2 than SLFN11-WT cells (Figure S5H).
Collectively, these results suggest that SLFN11 plays a role in promoting protein homeostasis and in reducing cellular ubiquitination caused by misfolded proteins.
Discussion
Ubiquitin conjugation and ER stress associated protein degradation (ERAD) by the proteasome are critical for the clearance of misfolded proteins following their cytoplasmic retro-translocation from the ER (40). By blocking the ubiquitin activating enzyme (UBA1), TAK-243 induces irresolvable ER stress and UPR activation (22,26–29). Our data are consistent with the potent and rapid effects of TAK-243 in inhibiting ubiquitin conjugation as visualized by the rapid depletion of cellular polyubiquitylated proteins, especially in SLFN11-KO cancer cells. Our study also shows that the activation of UPR by TAK-243 arrests replication within 2 - 4 hours by activating CHK1 as well as by inducing apoptosis and cell death at later time points. Hence, our data support the mechanism of action of TAK-243 and its preferential antiproliferative effects in SLFN11-KO cancer cells. They are relevant to the reported activity of TAK-243 in patient-derived xenograft (PDx) models (22,24,26–29), and in the context of the ongoing clinical trials of TAK-243 as first-in-class UBA1 inhibitor (NCT03816319 and NCT02045095).
Our flow cytometry, immunofluorescence microscopy and molecular combing analysis show that TAK-243, in addition to targeting UPR, interrupts replicative DNA synthesis within 2 - 4 hours as it depletes global cellular ubiquitylation. We demonstrate that this replication inhibition is related to CHK1 activation via Claspin without detectable ATR-mediated DDR activation (Figure 6). Consistent with our results, two prior studies reported that TAK-243 itself does not cause DNA damage, and that the combination of TAK-243 with UV or gamma irradiation suppresses DNA damage repair (22,26). The lack of γ-H2AX induction by TAK-243 in our study can be reconciled with a previous report showing that diffuse large B-cell lymphoma (DLBCL) cells treated with TAK-243 generate γ-H2AX (27). Our interpretation is that the γ-H2AX induction by TAK-243 in the prior study (27) is related to cell death by apoptosis as γ-H2AX is both a marker of DDR and of apoptotic response (41,42). Our finding that TAK-243 arrests DNA replication by activating CHK1 is consistent with independent reports showing that ER stress and uncontrolled UPR cause non-ATR mediated CHK1 activation (36–38).
An unexpected finding is that SLFN11 attenuates the UPR (Figure 6). Indeed, in two different isogenic cell line models (DU145 prostate cancer and CCRF-CEM leukemia), cells expressing SLFN11 showed significantly lower level of global polyubiquitylation at the steady-state condition, along with attenuated baseline of UPR activity and reduced protein aggregation. Our proteomic analysis identifies potential molecular insights for this novel proteostasis activity of SLFN11, as we observed SLFN11 binding to protein folding and translation initiation factors. Profiling of gene dependencies by a genome-wide RNAi screen also supports that SLFN11 deficient cells are highly dependent on the protein chaperonin and folding genes, implying that absence of SLFN11 leads to protein mis-folding and dependency on cellular ubiquitination, which is inhibited by TAK-243 (Figure 6).
Consistent with the activity of SLFN11 on translation, SLFN11 has been shown to bind type II tRNA leading to its activity as restriction factor against HIV-1 (16) and to cleave type II tRNAs resulting in ATR and ATM inhibition in response to DNA damage (43). SLFN11 has also been reported to limit the infectivity of flaviviruses by targeting host tRNAs (44). Binding and cleavage of tRNAs have also been demonstrated for SLFN13, the close relative SLFN11 (33). Non-human primate SLFN11 also suppresses protein production from viral, non-viral and host transcripts (17). In addition, a recent study showed that SLFN11 suppresses tumor growth and metastasis by inhibiting the RPS4X and mTOR pathway (14).
Our study is also the first to report a potential implication of SLFN11 with protein folding. This finding is plausible with the fact that murine Slfn1 binds to DnaJB6, one of the chaperone proteins of the DnaJ/Hsp (heat-hock protein) family, leading to the stabilization of Hsp70 (45), which plays an important role in protein folding and protein quality control (46). This complex leads to the localization of Slfn1 to the nucleus and induction of cell cycle arrest and quiescence of murine T cells (45). Additional reports showed that mutation of Slfn2 causes a loss of murine immune cell quiescence and chronic ER stress (47,48). Further studies are warranted to explore the detailed molecular connections between SLFN11, proteostasis and control of ER stress and UPR.
Beyond cancer, uncontrolled ER stress in human cells can induce multiple diseases such as atherosclerosis, diabetes, and neurodegenerative disease including Alzheimer’s and Parkinson’s disease (49). In addition, the chaperonin TRiC/CCT has been detected as a key molecule protecting cells from amyloid-like protein misfolding (50). Our mass spectrometry analysis showed TCP1 and CCTs as the SLFN11 interactors. Recently, a detailed immunohistochemistry and RNA expression study showed that SLFN11 is expressed in normal human brain cells and other organic cells (12). Our results that SLFN11-deficient cells are highly dependent on CCT genes, imply abnormality in the protein folding process in the absence of SLFN11. Taken together, these data suggest that SLFN11 might be involved in protecting normal human cells, especially brain and immune cells, from proteotoxicity, and that malfunction of SLFN11 might be associated with various human diseases including neurodegenerative and autoimmune diseases.
Future investigations are warranted to test our results in in vivo xenograft with cell lines and patient-derived models. Our findings may also be relevant for the therapeutic development of TAK-243 with the inclusion of SLFN11 expression as a treatment response predictive biomarker for cancer patient selection. Approximately, 50% of cancer cells do not express SLFN11 (1), therefore TAK-243 may be a therapeutic option for those cancer patients whose tumors are SLFN11-negative and do not respond to DNA damaging agents.
Supplementary Material
Significance.
This study uncovers that SLFN11 deficiency induces proteotoxic stress and sensitizes cancer cells to TAK-243, suggesting that profiling SLFN11 status can serve as a therapeutic biomarker for cancer therapy.
Acknowledgement
This project was supported by the Intramural Program, Center for Cancer Research of the National Cancer Institute, NIH (to Yves Pommier): Z01-BC006150. We appreciated all the staff at NCATs for their contribution in this study.
Footnotes
Conflict of interest disclosure statement: The authors declare no competing interests.
References
- 1.Murai J, Thomas A, Miettinen M, Pommier Y. Schlafen 11 (SLFN11), a restriction factor for replicative stress induced by DNA-targeting anti-cancer therapies. Pharmacol Ther 2019;201:94–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mu Y, Lou J, Srivastava M, Zhao B, Feng XH, Liu T, et al. SLFN11 inhibits checkpoint maintenance and homologous recombination repair. EMBO Rep 2016;17:94–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murai J, Tang SW, Leo E, Baechler SA, Redon CE, Zhang H, et al. SLFN11 Blocks Stressed Replication Forks Independently of ATR. Mol Cell 2018;69:371–84 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jo U, Murai Y, Chakka S, Chen L, Cheng K, Murai J, et al. SLFN11 promotes CDT1 degradation by CUL4 in response to replicative DNA damage, while its absence leads to synthetic lethality with ATR/CHK1 inhibitors. Proc Natl Acad Sci U S A 2021;118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okamoto Y, Abe M, Mu A, Tempaku Y, Rogers CB, Mochizuki AL, et al. SLFN11 promotes stalled fork degradation that underlies the phenotype in Fanconi anemia cells. Blood 2021;137:336–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murai J, Zhang H, Pongor L, Tang SW, Jo U, Moribe F, et al. Chromatin Remodeling and Immediate Early Gene Activation by SLFN11 in Response to Replication Stress. Cell Rep 2020;30:4137–51 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buettner R Awakening of SCHLAFEN 11 by immunohistochemistry: a new biomarker predicting response to chemotherapy. Virchows Archiv 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kagami T, Yamade M, Suzuki T, Uotani T, Tani S, Hamaya Y, et al. The first evidence for SLFN11 expression as an independent prognostic factor for patients with esophageal cancer after chemoradiotherapy. BMC Cancer 2020;20:1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knelson EH, Patel SA, Sands JM. PARP Inhibitors in Small-Cell Lung Cancer: Rational Combinations to Improve Responses. Cancers (Basel) 2021;13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moribe F, Nishikori M, Takashima T, Taniyama D, Onishi N, Arima H, et al. Epigenetic suppression of SLFN11 in germinal center B-cells during B-cell development. PLoS One 2021;16:e0237554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takashima T, Sakamoto N, Murai J, Taniyama D, Honma R, Ukai S, et al. Immunohistochemical analysis of SLFN11 expression uncovers potential non-responders to DNA-damaging agents overlooked by tissue RNA-seq. Virchows Arch 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winkler C, Armenia J, Jones GN, Tobalina L, Sale MJ, Petreus T, et al. SLFN11 informs on standard of care and novel treatments in a wide range of cancer models. Br J Cancer 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou C, Liu C, Liu W, Chen W, Yin Y, Li CW, et al. SLFN11 inhibits hepatocellular carcinoma tumorigenesis and metastasis by targeting RPS4X via mTOR pathway. Theranostics 2020;10:4627–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zoppoli G, Regairaz M, Leo E, Reinhold WC, Varma S, Ballestrero A, et al. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc Natl Acad Sci U S A 2012;109:15030–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li M, Kao E, Gao X, Sandig H, Limmer K, Pavon-Eternod M, et al. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature 2012;491:125–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stabell AC, Hawkins J, Li M, Gao X, David M, Press WH, et al. Non-human Primate Schlafen11 Inhibits Production of Both Host and Viral Proteins. PLoS Pathog 2016;12:e1006066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011;334:1081–6 [DOI] [PubMed] [Google Scholar]
- 19.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 2011;13:184–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou HJ, Wang J, Yao B, Wong S, Djakovic S, Kumar B, et al. Discovery of a First-in-Class, Potent, Selective, and Orally Bioavailable Inhibitor of the p97 AAA ATPase (CB-5083). J Med Chem 2015;58:9480–97 [DOI] [PubMed] [Google Scholar]
- 21.Jin J, Li X, Gygi SP, Harper JW. Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature 2007;447:1135–8 [DOI] [PubMed] [Google Scholar]
- 22.Hyer ML, Milhollen MA, Ciavarri J, Fleming P, Traore T, Sappal D, et al. A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat Med 2018;24:186–93 [DOI] [PubMed] [Google Scholar]
- 23.Barghout SH, Schimmer AD. The ubiquitin-activating enzyme, UBA1, as a novel therapeutic target for AML. Oncotarget 2018;9:34198–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McHugh A, Fernandes K, South AP, Mellerio JE, Salas-Alanis JC, Proby CM, et al. Preclinical comparison of proteasome and ubiquitin E1 enzyme inhibitors in cutaneous squamous cell carcinoma: the identification of mechanisms of differential sensitivity. Oncotarget 2018;9:20265–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Monda JK, Cheeseman IM. Dynamic regulation of dynein localization revealed by small molecule inhibitors of ubiquitination enzymes. Open Biol 2018;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barghout SH, Patel PS, Wang X, Xu GW, Kavanagh S, Halgas O, et al. Preclinical evaluation of the selective small-molecule UBA1 inhibitor, TAK-243, in acute myeloid leukemia. Leukemia 2019;33:37–51 [DOI] [PubMed] [Google Scholar]
- 27.Best S, Hashiguchi T, Kittai A, Bruss N, Paiva C, Okada C, et al. Targeting ubiquitin-activating enzyme induces ER stress-mediated apoptosis in B-cell lymphoma cells. Blood Adv 2019;3:51–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Best S, Liu T, Bruss N, Kittai A, Berger A, Danilov AV. Pharmacologic inhibition of the ubiquitin-activating enzyme induces ER stress and apoptosis in chronic lymphocytic leukemia and ibrutinib-resistant mantle cell lymphoma cells. Leuk Lymphoma 2019;60:2946–50 [DOI] [PubMed] [Google Scholar]
- 29.Zhuang J, Shirazi F, Singh RK, Kuiatse I, Wang H, Lee HC, et al. Ubiquitin-activating enzyme inhibition induces an unfolded protein response and overcomes drug resistance in myeloma. Blood 2019;133:1572–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murai J, Feng Y, Yu GK, Ru Y, Tang SW, Shen Y, et al. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget 2016;7:76534–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seiler JA, Conti C, Syed A, Aladjem MI, Pommier Y. The Intra-S-Phase Checkpoint Affects both DNA Replication Initiation and Elongation: Single-Cell and -DNA Fiber Analyses. Mol Cell Biol 2007;27:5806–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim DI, Jensen SC, Noble KA, Kc B, Roux KH, Motamedchaboki K, et al. An improved smaller biotin ligase for BioID proximity labeling. Mol Biol Cell 2016;27:1188–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang JY, Deng XY, Li YS, Ma XC, Feng JX, Yu B, et al. Structure of Schlafen13 reveals a new class of tRNA/rRNA- targeting RNase engaged in translational control. Nat Commun 2018;9:1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saldivar JC, Cortez D, Cimprich KA. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 2017;18:622–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smits VAJ, Cabrera E, Freire R, Gillespie DA. Claspin - checkpoint adaptor and DNA replication factor. FEBS J 2019;286:441–55 [DOI] [PubMed] [Google Scholar]
- 36.Cabrera E, Hernandez-Perez S, Koundrioukoff S, Debatisse M, Kim D, Smolka MB, et al. PERK inhibits DNA replication during the Unfolded Protein Response via Claspin and Chk1. Oncogene 2017;36:678–86 [DOI] [PubMed] [Google Scholar]
- 37.Malzer E, Daly ML, Moloney A, Sendall TJ, Thomas SE, Ryder E, et al. Impaired tissue growth is mediated by checkpoint kinase 1 (CHK1) in the integrated stress response. J Cell Sci 2010;123:2892–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomas SE, Malzer E, Ordonez A, Dalton LE, van ť Wout EF, Liniker E, et al. p53 and translation attenuation regulate distinct cell cycle checkpoints during endoplasmic reticulum (ER) stress. J Biol Chem 2013;288:7606–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer 2014;14:581–97 [DOI] [PubMed] [Google Scholar]
- 40.Christianson JC, Ye Y. Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat Struct Mol Biol 2014;21:325–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, et al. gammaH2AX and cancer. Nat Rev Cancer 2008;8:957–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Solier S, Pommier Y. The nuclear gamma-H2AX apoptotic ring: implications for cancers and autoimmune diseases. Cell Mol Life Sci 2014;71:2289–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li M, Kao E, Malone D, Gao X, Wang JYJ, David M. DNA damage-induced cell death relies on SLFN11-dependent cleavage of distinct type II tRNAs. Nat Struct Mol Biol 2018;25:1047–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valdez F, Salvador J, Palermo PM, Mohl JE, Hanley KA, Watts D, et al. Schlafen 11 Restricts Flavivirus Replication. J Virol 2019;93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y, Yang Z, Cao Y, Zhang S, Li H, Huang Y, et al. The Hsp40 family chaperone protein DnaJB6 enhances Schlafen1 nuclear localization which is critical for promotion of cell-cycle arrest in T-cells. Biochem J 2008;413:239–50 [DOI] [PubMed] [Google Scholar]
- 46.Benyair R, Ron E, Lederkremer GZ. Protein quality control, retention, and degradation at the endoplasmic reticulum. Int Rev Cell Mol Biol 2011;292:197–280 [DOI] [PubMed] [Google Scholar]
- 47.Berger M, Krebs P, Crozat K, Li X, Croker BA, Siggs OM, et al. An Slfn2 mutation causes lymphoid and myeloid immunodeficiency due to loss of immune cell quiescence. Nat Immunol 2010;11:335–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Omar I, Lapenna A, Cohen-Daniel L, Tirosh B, Berger M. Schlafen2 mutation unravels a role for chronic ER stress in the loss of T cell quiescence. Oncotarget 2016;7:39396–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol 2017;13:477–91 [DOI] [PubMed] [Google Scholar]
- 50.Behrends C, Langer CA, Boteva R, Bottcher UM, Stemp MJ, Schaffar G, et al. Chaperonin TRiC promotes the assembly of polyQ expansion proteins into nontoxic oligomers. Mol Cell 2006;23:887–97 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.