

Abstract
Despite the availability of drugs that target ERα-positive breast cancer, resistance commonly occurs, resulting in relapse, metastasis, and death. Tamoxifen remains the most commonly-prescribed endocrine therapy worldwide, and “tamoxifen resistance” has been extensively studied. However, little consideration has been given to the role of endoxifen, the most abundant active tamoxifen metabolite detected in patients, in driving resistance mechanisms. Endoxifen functions differently from the parent drug and other primary metabolites, including 4-hydroxy-tamoxifen (4HT). Many studies have shown that patients who extensively metabolize tamoxifen into endoxifen have superior outcomes relative to patients who do not, supporting a primary role for endoxifen in driving tamoxifen responses. Therefore, “tamoxifen resistance” may be better modeled by “endoxifen resistance” for some patients. Here, we report the development of novel endoxifen-resistant breast cancer cell lines and have extensively compared these models to 4HT and fulvestrant (ICI)-resistant models. Endoxifen-resistant cells were phenotypically and molecularly distinct from 4HT-resistant cells and more closely resembled ICI-resistant cells overall. Specifically, endoxifen resistance was associated with ERα and PR loss, estrogen insensitivity, unique gene signatures, and striking resistance to most FDA-approved second- and third-line therapies. Given these findings, and the importance of endoxifen in the efficacy of tamoxifen therapy, our data indicate that endoxifen-resistant models may be more clinically relevant than existing models and suggest that a better understanding of endoxifen resistance could substantially improve patient care.
Implications:
Here we develop and characterize the first endoxifen-resistant models and demonstrate that endoxifen resistance may better model tamoxifen resistance in a subset of patients.
Introduction
Breast cancer is the leading cause of cancer death among women, claiming the lives of over 600,000 individuals every year worldwide (1). Over 70% of all breast tumors express estrogen receptor alpha (ERα), a ligand-activated transcription factor that induces expression of genes involved in tumor growth and survival (2,3). Targeted therapies to treat ERα+ breast cancer have existed for decades and include selective estrogen receptor modulators (SERMs), selective estrogen receptor degraders (SERDs), and aromatase inhibitors (3,4). SERMs, such as tamoxifen and its active primary metabolite 4-hydroxy-tamoxifen (4HT), compete with estrogens for ERα binding, thereby preventing estrogen-induced growth of ERα+ tumor cells. (5,6). SERDs, such as fulvestrant (ICI), prevent nuclear translocation of ERα altogether and target the receptor for degradation (4,7), while aromatase inhibitors block the production of estrogen throughout the body. Although these drugs are initially quite effective, the majority of patients will eventually relapse with distant metastasis, anywhere from 5 to 30+ years after the initial diagnosis (3,4,8). Due to the incidence of ERα+ disease, and the frequency of relapse, ERα+ tumors are responsible for more breast cancer deaths than any other subtype, despite their overall better prognosis (3,8).
Tamoxifen is the oldest and most common therapy worldwide for ERα+ breast cancer. It is a prodrug which must be activated by cytochrome P450 (CYP) enzymes in the liver (9). For decades, the tamoxifen metabolite 4HT has been studied as the clinically-relevant active form of the drug, and nearly every model system of tamoxifen response and resistance has utilized 4HT. However, 4HT concentrations are extremely low in tamoxifen treated patients (≈5 nM) (10,11), and the anti-estrogenic activity elicited at such low concentrations is limited (10,12). Endoxifen is another active metabolite of tamoxifen, with a similar potency to 4HT at equimolar concentrations (13). Endoxifen, however, is found at concentrations between 5–80 nM in the serum of tamoxifen treated patients (10,11,14), a range which corresponds to the IC50 of endoxifen in many ERα+ breast cancer cells (12). Endoxifen is produced primarily via conversion from the tamoxifen metabolite N-desmethyl-tamoxifen, catalyzed by the enzyme CYP2D6 (10,15,16). Variability in circulating endoxifen concentrations is due to the fact that CYP2D6 is a highly polymorphic gene with over 100 variant alleles, the prevalence of which varies widely across different ethnicities (9,14). Interestingly, increased serum levels of endoxifen are associated with better outcomes, while CYP2D6 alterations that result in absent or reduced enzyme activity are associated with reduced tamoxifen response (9,14,17,18). In the laboratory, endoxifen has been characterized as the most potent tamoxifen metabolite at clinically relevant concentrations, (12,19–21) and the molecular mechanisms of endoxifen activity differ strikingly from those of tamoxifen, 4HT, and other anti-estrogens (19). Based on these and other findings, phase I and II clinical trials (NCT01327781 (22); NCT02311933; NCT01273168) investigating the efficacy of endoxifen monotherapy have been conducted with promising results, especially in patients with endocrine resistant disease. For these reasons, there is a pressing need to better understand the molecular mechanisms governing endoxifen activity in ERα+ breast cancer. Given the clinical relevance of endoxifen, and its associations with patient outcomes following tamoxifen therapy, it is also crucial to model “endoxifen resistance” and determine if this condition might better resemble “tamoxifen resistance” in patients. Indeed, models of endoxifen resistance may be more clinically relevant than current models of tamoxifen or 4HT resistance for the large majority of patients.
To this end, we have developed the first cell line models of endoxifen resistance, in parallel with our own models of 4HT and ICI resistance, using MCF7 and T47D cells. Here, we demonstrate that endoxifen-resistant cells differ dramatically from 4HT-resistant cells with regard to their global gene and protein expression profiles, including notable differences in expression of ERα and the progesterone receptor (PGR). Endoxifen-resistant cells, unlike 4HT-resistant cells, are shown to be completely estrogen insensitive and are largely resistant to the majority of FDA-approved second- and third-line therapies used to treat endocrine-resistant disease. These findings further demonstrate that endoxifen’s mechanisms of action are unique and indicate that a better understanding of “endoxifen resistance” is warranted. Finally, endoxifen-resistant models are likely to be clinically relevant and should be included in studies evaluating the efficacy of novel therapies for endocrine resistant breast cancer patients.
Materials and Methods
Cell Culture and Treatments
Parental MCF7 and T47D (RRID:CVCL_0553) cells were purchased in 2008 from American Type Culture Collection (ATCC). Cells were maintained in phenol red-free Dulbecco’s modified Eagle’s medium/F12 (DMEM-F12; Corning Life Sciences, Manassas, VA), containing 10% (v/v) fetal bovine serum (FBS; Gemini Bio-Products, West Sacramento, CA) and 1% (v/v) antibiotic/antimycotic (AA; Invitrogen, Carlsbad, CA), in a humidified 37°C incubator with 5% CO2. Vehicle control and resistant MCF7 cells were developed via 24 months of chronic treatment with 0.1% ethanol or 1 μM concentrations of endoxifen, 4HT, or ICI in regular growth medium. T47D models were developed in an identical manner with 12 months of chronic treatment. Cells were authenticated by IDEXX BioAnalytics (Columbia, MO; last tested 8/2018) and tested for mycoplasma infection every six months using a mycoplasma detection kit from SouthernBiotech (Birmingham, AL; last tested 9/2020). (Z)-endoxifen was provided by the National Cancer Institute (Bethesda, MD), (Z)-4HT was purchased from Sigma Aldrich (St. Louis, MO), and ICI-182,780 was purchased from Tocris Biosciences Inc. (Baldwin, MO). Endoxifen, 4HT, and ICI were solubilized in 100% ethanol.
For all experiments requiring an estrogen treatment, cells were cultured in DMEM/F12 medium containing 10% (v/v) HyClone triple charcoal-stripped FBS (GE Healthcare, Chicago, IL) and 1% (v/v) AA for at least 24 hours prior to treatment. 17-β-estradiol (E2) was purchased from Sigma Aldrich and solubilized in 100% ethanol. Cells were treated with 1 nM E2 for 24 hours unless otherwise noted.
Abemaciclib, palbociclib, ribociclib, everolimus, alpelisib, ipatasertib, venetoclax, and lasofoxifene were purchased from Selleck Chemicals (Houston, TX).
Cell Proliferation and Migration Assays
For proliferation assays, cells were plated at a density of 1,000 cells per well (MCF7) or 1,500 cells per well (T47D) in 96-well plates and treated 24 hours later with the indicated concentrations of each drug. Treatments were performed with four technical replicates per condition. Cells were imaged daily for 5–7 days using an IncuCyte© S3 system (Sartorius, Göttingen, Germany), and confluence was determined using the IncuCyte© base software package.
To create dose response curves and determine IC50 values for second/third-line therapies, cells were plated as above and treated 24 hours later with serial dilutions of each compound for 7–10 days, with four technical replicates per condition. The range of concentrations used for each drug was as follows: abemaciclib (0.008–3125 nM), palbociclib (0.001–65.5 μM), ribociclib (0.001–65.5 μM), everolimus (0.244pM-4.01 μM), alpelisib (0.64 nM-250 μM), ipatasertib (0.25–64 μM), venetoclax (0.5–128 μM), and lasofoxifene (0.25 nM-16.4 μM). At the conclusion of the experiment, cells were imaged and confluence was quantified using the IncuCyte© S3 system.
For migration assays, cells were plated at a density of 32,000 cells per well in 96-well ImageLock plates (Sartorius). After 24 hours, uniform scratches were made in the cell monolayer using the Sartorius WoundMaker tool. Cells were washed once with DMEM/F12 medium, then treated as indicated and imaged every two hours using the IncuCyte© S3 system. Analysis was performed using the IncuCyte© Scratch Wound Analysis Software Module.
Colony Formation Assays
Cells were plated in triplicate at a density of 200 cells per well in 6-well plates and treated for approximately 3 weeks. Colonies were fixed using 5% glutaraldehyde (Sigma-Aldrich) and stained with crystal violet. Colony number and size were quantified using Fiji ImageJ software (imagej.net, RRID:SCR_003070).
Reverse Transcriptase Real-Time Polymerase Chain Reaction
Total RNA was extracted from cells using TRIzol reagent (Invitrogen) and quantified using a DS-11 spectrophotometer (DeNovix, Wilmington, DE). One microgram of cDNA was reverse transcribed using the iScript cDNA synthesis kit (BioRad, Hercules, CA). RT-PCR was performed in triplicate as previously described (23). Quantification of results was based on the threshold cycle and normalized to the TATA binding protein (TBP) control gene. All primers were designed using Primer3 software (http://bioinfo.ut.ee/primer3-0.4.0/primer3, RRID:SCR_003139) and purchased from Integrated DNA Technologies (Coralville, IA). Primer sequences are listed in Supplementary Table S1.
Western Blot Analysis
Cells were lysed and total protein was collected using NETN buffer (20 mM Tris pH 8.0, 150 nM NaCl, 1 mM EDTA and 0.5% NP40) supplemented with an EDTA-free protease inhibitor cocktail tablet (Roche, Indianapolis, IN). Protein concentrations were determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA), and equal amounts of protein lysate were suspended in Laemmli buffer (BioRad), run on 7.5% SDS-PAGE gels, and transferred onto PVDF membranes (BioRad). Membranes were blocked in 5% milk in TBST for 1 hour at room temperature, then probed for ERα (F-10, Santa Cruz Biotechnology, Inc., Dallas, TX), progesterone receptor (HC-190, Santa Cruz Biotechnology), and vinculin (EPR-8185, Abcam, Cambridge, United Kingdom). All primary antibodies were diluted in 5% milk in TBST at 1:500 (ERα), 1:1000 (PR), or 1:2500 (vinculin) and applied to the membranes at 4°C overnight. After washing, membranes were incubated with anti-rabbit or anti-mouse secondary antibodies (Sigma Aldrich, A8275 (RRID:AB_258382) and A4416 (RRID:AB_258167), respectively) diluted 1:2000 in 5% milk in TBST for one hour at room temperature. Visualization was performed using enhanced chemiluminescence on the LI-COR Odyssey Fc system (Lincoln, NE).
Transient Transfection and Luciferase Assays
Cells were plated at a density of 35,000 cells per well in 24-well plates. Following 24 hours, cells were transfected with 100 ng per well of an estrogen response element luciferase reporter construct (ERE-TK-luc) using FuGene6 transfection reagent (Roche Applied Science, Indianapolis, IN) at a ratio of 3 μl of FuGene6 per 1 μg of DNA. Medium was changed after 24 hours, and cells were treated in triplicate for an additional 24 hours, as indicated. Cells were lysed using 1x passive lysis buffer (Promega, Madison, WI), and equal amounts of lysate were assayed using Luciferase Assay Reagent (Promega), and a GloMax-Dual Luminometer (Promega).
RNA Sequencing
Vehicle control and resistant MCF7 cells were plated in triplicate in 100 mm tissue culture dishes and grown to approximately 70% confluence. Total RNA was isolated using TRIzol reagent (Invitrogen), purified using the miRNeasy mini kit (Qiagen, Hilden, Germany), and quantified using a DS-11 spectrophotometer (DeNovix). Paired-end sequencing of total RNA was performed in the Mayo Clinic Gene Expression Core Facility (Rochester, MN) using an Illumina Hi-Seq 4000.
Transcript read quality was assessed using fastqc (v0.11.8) and aligned to hg38 using bowtie-2 (v2.3.3.1). Features were determined using featureCounts (v2.0.1, RRID:SCR_012919) (24). Comparisons between samples were performed using edgeR (v3.28.1, RRID:SCR_012802) (25), and transcripts were filtered using the edgeR filterByExpr() command. P-values were determined by Fisher’s exact test, with the doubletail rejection region and dispersion calculated automatically. Raw and processed datasets are available in the Gene Expression Omnibus (GEO, RRID:SCR_005012, accession GSE164529).
Biological Pathway and Gene Set Enrichment Analysis
Differentially expressed genes (DEGs) identified from our RNA-seq analysis (p≤0.05 and fold change≥2.0) were subjected to pathway analysis using Ingenuity Pathway Analysis software (IPA, RRID:SCR_008653, Ingenuity Systems, Inc., Redwood City, CA) as previously described (23,26). Gene Set Enrichment Analysis (27) (GSEA) was performed using the same set of DEGs and was conducted using standard pathways and significantly enriched pathways (C2.all gene sets and C5.all gene sets).
Reverse Phase Protein Array
MCF7 cells were plated in duplicate in 100 mm tissue culture dishes and grown to approximately 70% confluence. Cell pellets were harvested and washed twice with PBS, then submitted to the Functional Proteomics Reverse Phase Protein Array (RPPA) core facility at M.D. Anderson Cancer Center (Houston, TX) for analysis (28).
Statistical Analysis
For all experiments, reported values indicate the average value of all replicates +/− standard error of the mean. Experiments were performed a minimum of three times, and a representative dataset is shown. Cell proliferation and migration assays were analyzed using a two-way ANOVA with Geisser-Greenhouse correction. All colony formation, gene expression, and luciferase assays were analyzed using a two-tailed Student’s t-test. IC50 values were estimated using GraphPad Prism software (RRID:SCR_002798), by fitting proliferation data from each resistant line to a non-linear curve.
Results
Characterization of endoxifen-resistant cells and comparison to 4HT- and ICI-resistant models
MCF7-derived resistant cell lines were developed by chronic treatment of cells with ethanol control or 1 μM concentrations of endoxifen, 4HT, or ICI for 24 months (Fig. 1A). Resistant cells developed subtle yet distinct morphological changes compared to control cells (Fig. 1A). Specifically, 4HT-resistant cells were smaller and grew in tightly packed clusters. In contrast, endoxifen and ICI-resistant cells were larger, with cytoplasmic hypertrophy, and did not pack together as tightly. In the absence of drug treatment, endoxifen and ICI-resistant cells exhibited slower proliferation rates compared to control cells, while 4HT-resistant cells grew slightly faster (Fig. 1B).
Figure 1. Characterization of endoxifen-resistant MCF7 cells and comparison to 4HT- and ICI-resistant models.

A. Parental MCF7 cells were chronically treated with ethanol vehicle or 1μM doses of endoxifen (ENDX), 4HT, or ICI for 24 months to develop resistant cell lines. Microscopic images of cell lines taken at 10x magnification depicting subtle changes in morphology are shown. B. Proliferation assays showing baseline growth rates of all four cell lines (n=4). C-E. Resistance was confirmed using proliferation, migration, and colony formation assays, respectively. For each assay, control cells and ENDX-R, 4HT-R, and ICI-R cells were treated with 1μM doses of each endocrine therapy for 1 week (proliferation; n=4), 48 hours (migration; n=4), or 3 weeks (colony formation, n=3). Values shown indicate mean +/− SEM. Significance is notated by: ns: p > 0.05; *: p ≤ 0.05; **: p ≤ 0.01; ***: p ≤ 0.001; ****: p ≤ 0.0001.
To confirm resistance, and to assess cross-resistance, proliferation rates of each cell line were determined in response to ethanol vehicle and 1 μM concentrations of endoxifen, 4HT, or ICI. Growth of control cells was, as expected, strongly inhibited by all 3 drugs (Fig. 1C). Endoxifen and ICI-resistant cells were completely resistant to all 3 drugs (Fig. 1C). In 4HT-resistant cells, the anti-proliferative effects of 4HT and endoxifen were greatly diminished, and the cells remained completely sensitive to ICI (Fig. 1C).
Confirmation of resistance was also performed using cell migration and colony formation assays. Endoxifen, 4HT, and ICI all significantly inhibited migration of MCF7 control cells, but did not inhibit migration of their respective resistant cell lines (Fig. 1D). Similarly, all three endocrine therapies significantly inhibited colony formation of control cells, but had no effect on their respective resistant models (Fig. 1E). Endoxifen, 4HT, and ICI decreased both the number and size of colonies formed, exclusively in the control cells (Fig. 1E). In the absence of treatment, however, resistant cells formed significantly fewer colonies compared to control cells.
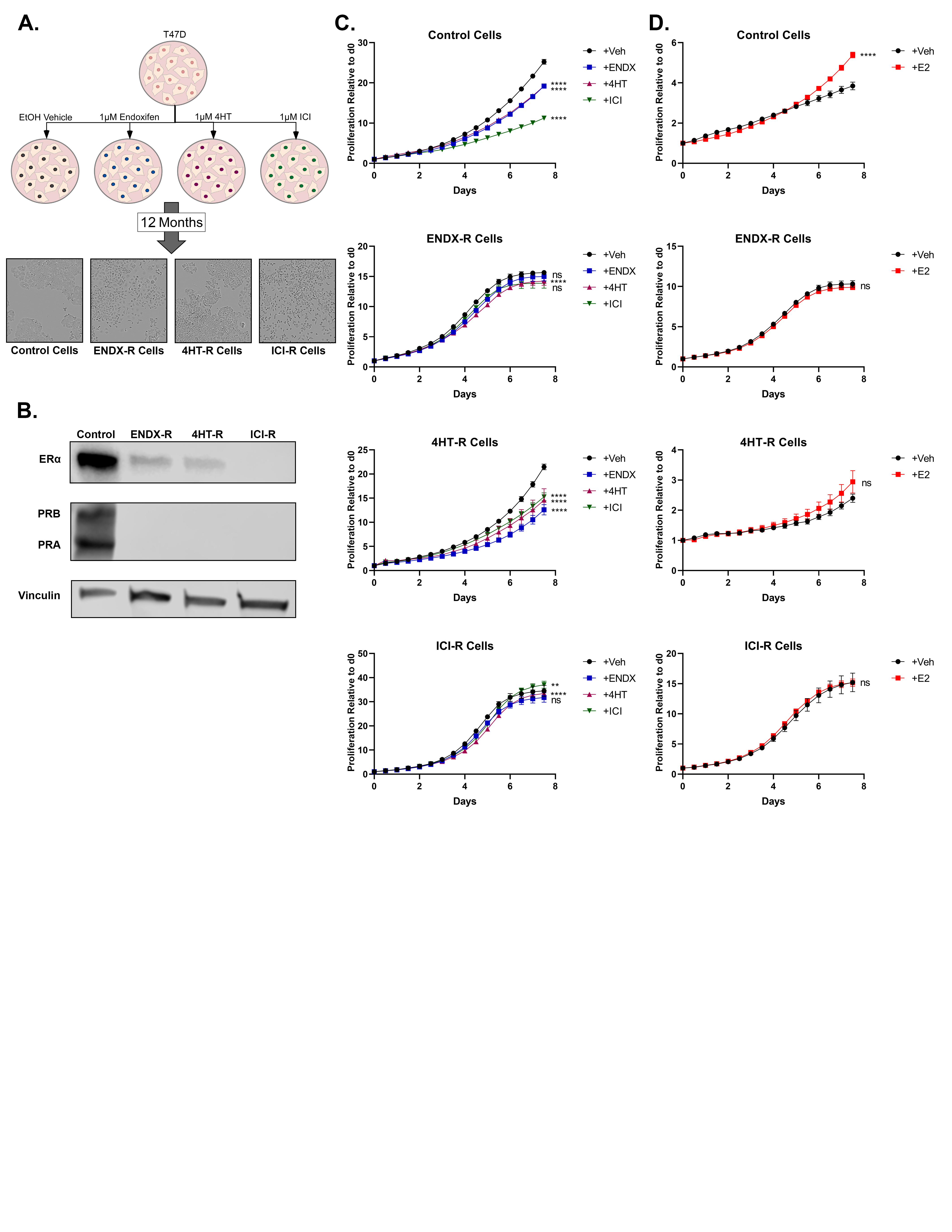
In addition to MCF7 cells, endoxifen, 4HT, and ICI-resistant models were also developed using T47D cells in an identical manner, following 12 months of chronic treatment (Supplementary Figure S1A). Like MCF7 cells, proliferation of control-treated T47D cells was significantly inhibited by all drugs (Supplementary Figure S1C). As observed in the MCF7 resistant lines, both endoxifen-resistant and ICI-resistant T47D cells were essentially resistant to all 3 drugs (Supplementary Figure S1C). However, 4HT-resistant cells remained partially sensitive to all 3 drugs (Supplementary Figure S1C).
ERα expression and pathway activity
The effects of long-term endoxifen treatment on the expression of ERα and its downstream signaling pathways are currently unknown. At the mRNA level, endoxifen-resistant MCF7 cells exhibited substantial downregulation of ERα mRNA with a concomitant loss in progesterone receptor (PGR) expression (Fig. 2A). At the protein level, ERα and PGR were not detected in the endoxifen-resistant model (Fig. 2A). ICI-resistant MCF7 cells were nearly identical to endoxifen-resistant cells in these respects (Fig. 2A). In contrast, 4HT-resistant cells exhibited downregulation of ERα and PGR mRNA levels, albeit to a lesser extent; however, robust levels of ERα and PR protein were maintained (Fig. 2A).
Figure 2. ERα expression and pathway activity in MCF7 resistant cells.

A. RT-PCR (left; n=3) and Western blot (right) analyses of ERα and progesterone receptor (PGR) expression in ENDX-R, 4HT-R, and ICI-R cells. B. Luciferase assays depicting the activity of an estrogen response element reporter construct in indicated cell lines following treatment with 1nM E2 for 24 hours (n=3). C. RT-PCR analysis of estrogen-mediated induction of the ERα target genes PGR, PS2, AREG, and cyclin D1 in each resistant cell line, following 24 hours of 1nM estrogen treatment (n=3). D. Proliferation assay depicting the growth rates of each resistant line in response to 1nM estrogen treatment (n=4). Values shown indicate mean +/− SEM. Significance is notated by: ns: p > 0.05; *: p ≤ 0.05; **: p ≤ 0.01; ***: p ≤ 0.001; ****: p ≤ 0.0001.
Expression of ERα and PGR protein was also assessed in T47D resistant lines. As in MCF7 resistant lines, both ERα and PGR expression was greatly diminished in T47D endoxifen- and ICI-resistant models (Supplementary Figure S1B). 4HT-resistant T47D cells, however, also exhibited decreased ERα expression and undetectable PGR (Supplementary Figure S1B), which represents a striking difference from the 4HT-resistant MCF7 cells.
To further investigate ERα signaling in these models, ERα transcriptional activity was assessed in resistant MCF7 cells via an estrogen response element (ERE) luciferase assay. In agreement with the protein expression profiles, E2 treatment significantly induced ERα signaling in vehicle control and 4HT-resistant MCF7 cells with essentially no impact in endoxifen and ICI-resistant MCF7 cells (Fig. 2B). The effects of E2 on well-known ERα target genes (PGR, trefoil factor 1 (TFF1), amphiregulin (AREG), and cyclin D1 (CCND1)) were also evaluated in MCF7 models and largely paralleled the ERE findings. E2 induced the expression of all four genes in control cells, as well as PGR and TFF1 in 4HT-resistant cells (Fig. 2C). Notably, E2 failed to induce any of these genes in the endoxifen and ICI-resistant models and in fact downregulated many of them (Fig. 2C). With regard to proliferation, E2 stimulated growth of MCF7 control and 4HT-resistant cells, but had no effect on endoxifen- and ICI-resistant MCF7 lines (Fig. 2D). In T47D models, E2 had very little effect on 4HT-, endoxifen-, and ICI-resistant MCF7 lines, despite its robust induction of control cell proliferation (Supplementary Figure S1D).
Global gene expression profiles of MCF7 resistant cell lines
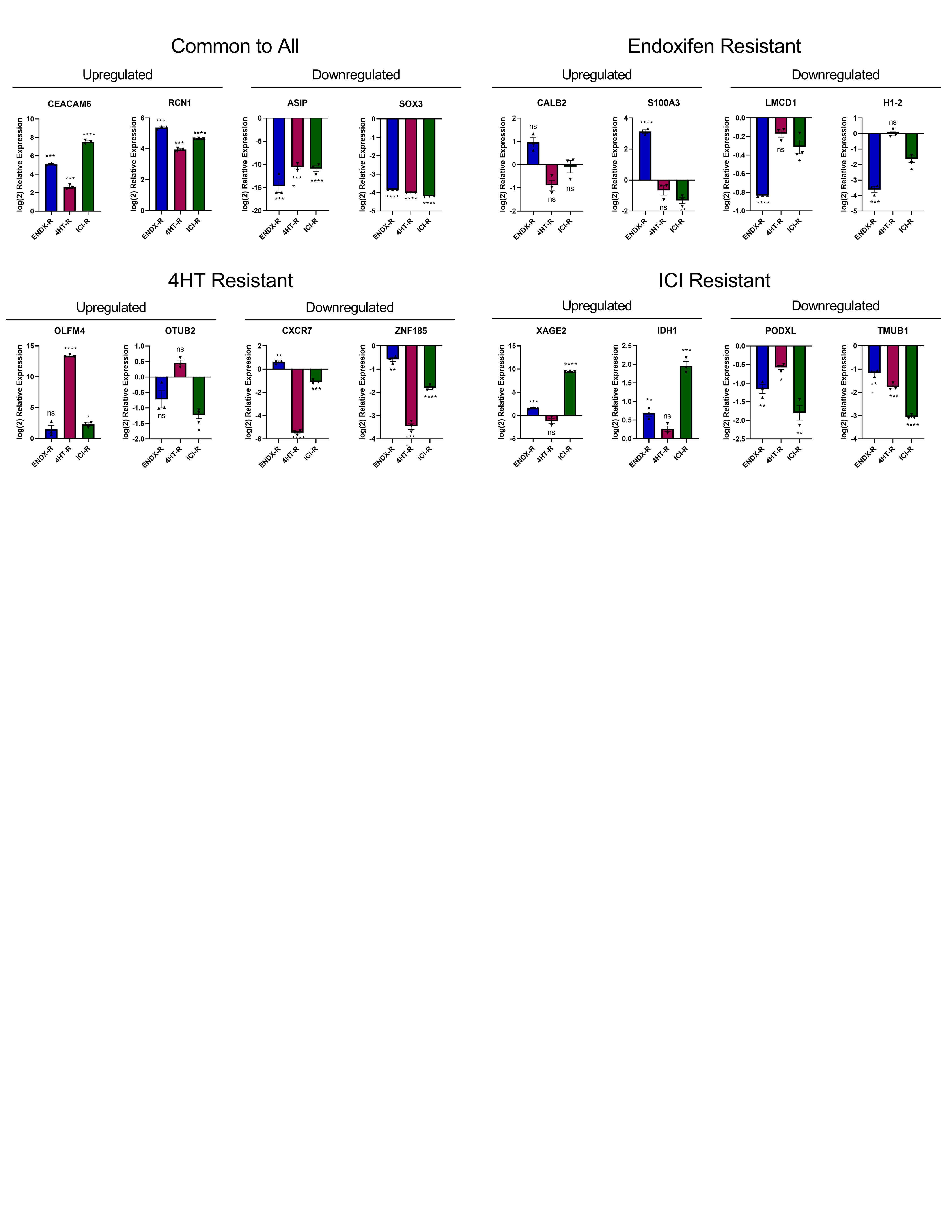
We next investigated the impact of resistance on global gene expression profiles of the MCF7 models using next generation RNA-sequencing (RNA-seq). These analyses revealed substantial differences in the basal gene expression profiles for all three models compared to control cells (Fig. 3A–B). In order to confirm the RNA-seq results, two upregulated genes and two downregulated genes common to all three cell lines, as well as two up- and downregulated genes unique to each cell line, were evaluated by RT-PCR (Supplementary Fig. S2). Results from these studies largely agreed with the RNA-seq findings (Supplementary Fig. S2).
Figure 3. Global gene expression profiles of MCF7 resistant cell lines.

A. Venn diagrams indicating overlap of upregulated (top) and downregulated (bottom; fold change >2; p<0.05) genes in ENDX-R, 4HT-R, and ICI-R cells, relative to vehicle control cells, as detected by RNA-seq. B. Volcano plots depicting gene expression changes in each resistant cell line, with differentially-regulated genes (fold change >2; p<0.05) colored grey. Randomly selected genes for PCR validation are highlighted in orange (differentially regulated in all 3 cell lines), blue (unique to ENDX-R), pink (unique to 4HT-R), or green (unique to ICI-R). Differentially regulated genes were subjected to Ingenuity Pathway Analysis (C) and Gene Set Enrichment Analysis (D). Pathways unique to each cell line are highlighted in blue, red, or green. NES: normalized enrichment score.
DEGs from each cell line were analyzed via Ingenuity Pathway Analysis (IPA) (26) to identify key differences in canonical signaling pathways. Both similarities and differences were observed among the top pathways differentially regulated in each cell line (Fig. 3C). Specific IPA analyses of DEGs common between all three resistant lines, or unique to a given cell line, were also performed and revealed additional pathways of interest (Supplementary Fig. S3).
Gene set enrichment analysis (GSEA) (27) was also performed on DEGs identified in each resistant cell line and revealed largely unique gene sets for each resistant model (Fig. 3D, Supplementary Fig. S4). However, the gene expression profiles of endoxifen- and ICI-resistant cells were more similar to each other and correlated with basal and luminal B signatures, a feature that was not evident in 4HT-resistant cells (Fig. 3D, Supplementary Fig. S4). Unsurprisingly, 4HT and ICI-resistant cells correlated with known gene expression profiles of endocrine and tamoxifen resistance, but interestingly, endoxifen-resistant cells did not (Supplementary Fig. S4), further confirming their uniqueness.
RPPA analysis of resistant cell lines
In addition to RNA-seq, reverse phase protein arrays (RPPA) (28) were utilized to investigate differences in protein expression among the resistant MCF7 cell lines. Each resistant cell line showed a distinct profile of protein expression differences relative to control cells, as demonstrated by independent hierarchical clustering of each cell line (Fig. 4A). Differentially regulated proteins were subjected to IPA analysis to assess differences in activated pathways and, as expected, many differences between cell lines were found (Fig. 4B–C). As with the gene expression profiles, the differentially-activated pathways in endoxifen-resistant cells exhibited more similarities with ICI-resistant cells than with 4HT-resistant cells (Fig. 4C).
Figure 4. RPPA analysis of MCF7 resistant cell lines.

A. Heat map indicating relative expression of all proteins and phosphoproteins analyzed in ENDX-R, 4HT-R, and ICI-R cell lines relative to vehicle control cells. Hierarchical clustering of proteins is shown on the left, while unsupervised clustering of individual replicates is shown on the top. B. Ingenuity Pathway Analysis of differentially expressed proteins, with the top 15 pathways shown for each cell line. Pathways unique to each cell line are highlighted in blue, red, or green. C. Results of IPA analysis comparing top pathways from differentially expressed proteins in each resistant cell line.
Reversibility of resistant cell phenotypes
In order to determine whether the phenotypic changes characteristic of resistance were permanent, all four MCF7 cell lines were withdrawn from their respective treatments for three months. No significant changes in morphology were observed in any cell line following withdrawal (Fig. 5A). Each withdrawn line was treated with vehicle control, endoxifen, 4HT, or ICI to assess proliferation and determine if resistance had been reversed following withdrawal (Fig. 5B). Control cells remained sensitive to all three therapies, while endoxifen and ICI withdrawn cells remained resistant to all three therapies (Fig. 5B). Interestingly, 4HT-resistant cells regained sensitivity to both endoxifen and 4HT and remained potently inhibited by ICI (Fig. 5B). Response to each therapy was also assessed at the migration level (Fig. 5C) where the same patterns were observed.
Figure 5. Reversibility of resistant cell phenotypes in MCF7 cells.

A. Images of resistant MCF7 cells withdrawn from their respective endocrine therapies for 3 months, with non-withdrawn cells shown for comparison, to demonstrate their morphology. Images taken at 10x magnification. Following 3 months of withdrawal, proliferative (B) and migratory (C) responses of ENDX-R, 4HT-R, and ICI-R cells to 1μM doses of each drug were assessed (n=4). D. RT-PCR (top; n=3) and Western blot (bottom) analysis showing expression of ERα and PGR in withdrawn cell lines. E. Proliferative response of withdrawn cells to 1nM estrogen treatment for 7 days (n=4). F. RT-PCR analysis showing expression of selected differentially expressed genes in withdrawn cells (n=3). Values shown indicate mean +/− SEM. Significance is notated by: ns: p > 0.05; *: p ≤ 0.05; **: p ≤ 0.01; ***: p ≤ 0.001; ****: p ≤ 0.0001.
The patterns of ERα and PGR mRNA and protein expression remained largely unchanged by treatment withdrawal, with expression of both receptors remaining high in control and 4HT- resistant cells, and low or undetectable in endoxifen- and ICI-resistant cells (Fig. 5D). It is notable, however, that ERα protein expression was lower, and PGR gene expression higher in 4HT withdrawn cells than in 4HT-resistant cells. The proliferative response of the withdrawn cells to estrogen was also determined. Control and 4HT withdrawn cells responded to estrogen treatment with enhanced proliferation, and interestingly, 4HT withdrawn cells were hypersensitive to estrogen (Fig. 5E). Endoxifen and ICI withdrawn cells remained completely insensitive to estrogen with regard to proliferation (Fig. 5E).
Finally, the effects of drug withdrawal on the expression of differentially up- or downregulated genes in resistant cells were determined. The trends in expression of these genes were mostly unchanged; withdrawn cells still exhibited differential expression of genes common to all 3 resistant cell lines, or unique to their respective resistant cell line (Fig. 5F). Several of the genes (RCN1, SOX3, CALB2, S100A3, H1–2, ZNF185, XAGE2, PODXL, TMUB1) showed a decreased magnitude of differential expression in the withdrawn cells (Fig. 5F), compared to non-withdrawn cells (Supplementary Fig. S2).
Endoxifen-, 4HT-, and ICI-resistant cells exhibit differential sensitivity to clinically-relevant second- and third-line therapies.
Patients whose tumors relapse following endocrine therapy can receive a wide variety of second- and third-line therapies in an attempt to further control disease progression (4,29). We therefore determined the sensitivity of resistant cell lines to 8 clinically-relevant drugs, some of which are already FDA-approved, and some of which are undergoing clinical trials for treatment of endocrine resistant breast cancer (Table 1). Interestingly, endoxifen- and ICI-resistant cells were less responsive than control and 4HT-resistant cells to every therapy except venetoclax, which was equally effective across all models (Fig. 6). With regard to the CDK4/6 inhibitors abemaciclib, palbociclib, and ribociclib, all resistant cells had IC50 values equal to or higher than control. In contrast, 4HT-resistant cells were more responsive to alpelisib and ipatasertib than control cells. These findings represent another stark difference between endoxifen- and 4HT-resistant cells, as endoxifen-resistant cells were the least responsive cell line to both of these drugs.
Table 1.
Mechanism of action and clinical approval status for selected second-line therapies
| Drug | Mechanism of Action | Clinical Status |
|---|---|---|
| Abemaciclib | Cdk 4/6 inhibitor | FDA Approved 2017 |
| Palbociclib | Cdk 4/6 inhibitor | FDA Approved 2015 |
| Ribociclib | Cdk 4/6 inhibitor | FDA Approved 2017 |
| Everolimus | mTOR inhibitor | FDA Approved 2009 Approved for Breast Cancer 2012 |
| Alpelisib | PI3K inhibitor | FDA Approved 2019 for patients with PIK3CA mutations |
| Ipatasertib | Akt inhibitor | Phase III Clinical Trials |
| Venetoclax | BCL2 inhibitor | FDA Approved for chronic lymphocytic leukemia 2016 and acute myeloid leukemia 2018. In Phase II clinical trials for breast cancer. |
| Lasofoxifene | Novel SERM | Phase II Clinical Trials |
Figure 6. Endoxifen, 4HT, and ICI resistant MCF7 cells exhibit differential sensitivity to clinically-relevant second- and third-line therapies.

Control, ENDX-R, 4HT-R, and ICI-R cells were treated with serially-diluted concentrations of drugs commonly used as second- and third-line therapies for endocrine-resistant breast cancer. Drug concentration ranges were chosen individually for each therapy, based on published IC50 values from other breast cancer cell lines. Cells were treated for 7–10 days, after which time, dose response curves were plotted and IC50 values calculated (n=4). N.D.: not defined; an IC50 was unable to be calculated using the chosen drug concentrations. Cmax: maximum concentration reported to be achievable in patient plasma, indicated by a vertical dashed line. Values shown indicate mean +/− SEM.
The most striking contrast, however, existed in response to everolimus and lasofoxifene. For these two drugs, control cells had IC50 values in the picomolar range, or too low to calculate in these experiments, with 4HT-resistant cells showing a similar response (Fig. 6). However, neither endoxifen- nor ICI-resistant cells showed a response until micromolar or higher concentrations were utilized, indicating striking levels of resistance to these two compounds (Fig. 6).
Discussion
Here, we describe the development and characterization of two novel breast cancer cell line models of endoxifen resistance. Results from these studies demonstrate that endoxifen resistance differs substantially from resistance to other previously-characterized forms of “tamoxifen resistance.” In contrast to 4HT-resistant models, endoxifen-resistant cells exhibited loss of ERα and PR expression, estrogen insensitivity, EMT-like signatures, unique gene expression profiles, and striking resistance to many second- and third-line therapies. Interestingly, endoxifen resistance was more similar to ICI resistance, although a number of key differences were observed. Unlike 4HT, resistance to endoxifen was not reversible following drug withdrawal, as cells remained ERα negative, estrogen insensitive and completely resistant to ERα-targeting agents. These findings further highlight the striking differences between endoxifen and other tamoxifen metabolites. Additionally, they provide impetus to further elucidate the molecular mechanisms governing endoxifen resistance, as well as the clinical relevance of such mechanisms in tamoxifen-treated patients.
Given the fact that tamoxifen is still the most widely-prescribed intervention for ERα+ breast cancer worldwide, and the fact that 30–50% of patients on endocrine therapy eventually relapse with metastatic disease (30), Regardless, the molecular mechanisms underlying tamoxifen resistance have been studied extensively in vitro and in the clinic, and a variety of both de novo and acquired resistance mechanisms have been suggested. These include mutation and alternative splicing of ERα, upregulation (i.e., EGF, IGF) or mutation (i.e., PI3K) of other oncogenic signaling pathways, and selection of ERα negative clones from a heterogeneous tumor population (3,30,31). Studies using resistant cell lines, which have existed since the early 1980s (32,33), have been crucial to elucidating these mechanisms. However, as the CYP enzymes which catalyze tamoxifen metabolism are not found in breast tissue, the vast majority of these cell lines have been developed via chronic treatment with 4HT (32–36) and therefore do not reflect the contribution of other active tamoxifen metabolites.
For several decades, 4HT was believed to be the most relevant active tamoxifen metabolite given its higher binding affinity for ERα (37) and its more potent anti-estrogenic activity (38) compared to tamoxifen. However, more recent studies have shown that endoxifen is found at higher concentrations than 4HT in patient serum (39), and that these concentrations correlate with clinical response to tamoxifen (18). Further, at physiologically-relevant concentrations and using pre-menopausal estrogen levels, endoxifen is primarily responsible for suppression of estrogen-mediated growth of ERα+ breast cancer cells under conditions that mimic the pre-menopausal state (12,21).
In tamoxifen-treated patients, endoxifen is produced primarily from CYP2D6-mediated metabolism of N-desmethyl-tamoxifen and 4HT (10,15,16), and circulating endoxifen levels are tightly linked to CYP2D6 genotype (9,14). CYP2D6 is a highly polymorphic gene. A number of variants with absent or decreased CYP2D6 activity have been characterized, with the most common (CYP2D6*4, *5, *10, and *17) found in 15–60% of the general population, varying based on ethnicity (40). These data indicate that up to 50% of all breast cancer patients have two wild-type copies of CYP2D6 and are therefore considered “extensive metabolizers” in regard to endoxifen production. For these patients, response to tamoxifen is likely driven by endoxifen activity. Therefore, “tamoxifen resistance” in extensive metabolizers who adhere to the treatment regimen may not be accurately modeled by 4HT and may be phenotypically and molecularly distinct from resistance in “poor metabolizers,” a possibility that has not yet been explored.
Results presented here support this notion, given that endoxifen-resistant cells exhibited unique gene expression profiles compared to 4HT-resistant cells. Indeed, GSEA analyses performed on differentially regulated genes in 4HT- and ICI-resistant cell lines revealed strong associations with signatures of endocrine resistance. However, no such correlations were identified for endoxifen-resistant cells, indicating their molecular uniqueness from previously reported models. Instead, signatures of endoxifen resistance were associated with a more basal phenotype, stem cell-like features, and activation of epithelial-to-mesenchymal transition (EMT)-related pathways such as tumor necrosis factor and interferon-α. Indeed, 4 of the top 5 proteins identified by RPPA as being up-regulated in endoxifen-resistant cells have known roles in the EMT process, including MUC1 (41), L1CAM (42), LDHA (43), and fibronectin (44). This is of importance given that EMT has been identified as a driving factor of “tamoxifen resistance” (45,46), further implicating the relevance of endoxifen-resistant models in the study of this form of the disease.
Another striking difference between 4HT and endoxifen resistance reported here pertains to ERα expression and estrogen sensitivity. In MCF7-derived models, 4HT-resistant cells maintained ERα expression and estrogen responsiveness, in terms of both cellular proliferation and ERα transcriptional activity. In contrast, endoxifen-resistant cells exhibited estrogen insensitivity and loss of ERα, findings that more closely resembled laboratory and clinical models of ICI resistance (47,48). Interestingly, while these patterns held true for endoxifen-resistant T47D cells, 4HT-resistant T47D cells were marked by a significant decrease in ERα and PGR protein expression as well, which is inconsistent with the observations in MCF7 cells. This difference has been previously observed in long-term estrogen-deprived MCF7 vs T47D cells (49), but the underlying mechanisms remain unknown. Further studies are necessary to elucidate the reasons for this difference in ERα expression between MCF7- and T47D-derived 4HT-resistant models.
Nevertheless, loss of ERα expression has been reported in patients with acquired resistance to tamoxifen (50–54), and is associated with worse outcomes and development of stemness properties (52,55). A 2012 study by Lindstrom et al. found a 48% increased risk of death in patients whose metastatic recurrence was characterized by loss of ERα expression (56), demonstrating a significant need for new strategies to improve prognosis for such patients. Estimates of the percentage of tamoxifen-treated patients in which tumoral ERα expression is lost at the time of recurrence vary from under 20% (53,54) to nearly 50% (50,52). While no studies to date have directly correlated ERα loss in tamoxifen resistance with CYP2D6 genotype, it is interesting to speculate that the patient population characterized by ERα loss at the time of recurrence may be enriched with extensive metabolizers whose disease is more accurately modeled by endoxifen resistance. Further, anti-estrogenic therapies such as aromatase inhibitors or fulvestrant are commonly used in the treatment of tamoxifen resistant patients (3,4,29). However, clinical benefit is only observed in 30–60% of these patients (57,58). Data presented in this study demonstrated that, in a cell line model of endoxifen resistance, other ERα-targeting agents were ineffective, which was not the case for 4HT-resistant models.
In order to determine if changes in ERα expression and estrogen responsiveness were “permanent” in these models, cells were withdrawn from their respective treatments for three months. While estrogen sensitivity was not restored in endoxifen- and ICI-resistant cells, the 4HT-resistant cells experienced a period of estrogen hypersensitivity following withdrawal. This phenomenon has been observed previously in cell line models of tamoxifen resistance (59) and, notably, long term estrogen deprivation (60,61). The lack of a rebound in estrogen sensitivity in endoxifen- and ICI-resistant cells is likely due to the fact that ERα expression did not return in these models following withdrawal. These observations highlight another important difference in resistance mechanisms between the three cell lines and additionally suggest that loss of ERα expression and pathway activity may be permanent following long-term exposure to endoxifen.
Future studies must be performed to further examine the mechanisms underlying these changes. Mutations in regulatory pathways and global epigenetic changes are two possible causes of the ERα silencing observed in these models. While activating ESR1 mutations are a common mechanism of resistance in patients, it is unlikely that they are responsible for the effects observed here. Mutations in ESR1 are incredibly rare in cell lines and have been observed exclusively in the setting of long term estrogen deprivation (62). Given the complete loss of ERα expression and pathway activity in endoxifen- and ICI-resistant models, constitutive ERα activation is unlikely.
This observation is of clinical relevance given that relapse of breast cancer following tamoxifen therapy can occur decades later (63–65). If indeed endoxifen-resistant cells accurately model tamoxifen resistance for a proportion of patients, our findings may provide insight into why additional lines of endocrine therapy may not be effective in some patients after progression on tamoxifen (57,58). These findings further highlight the necessity of evaluating ERα expression and/or pathway activity in patients with disease recurrence or progression when tailoring a treatment plan. Given the cost of long-term endocrine therapy, as well as the adverse side effects experienced by women taking these drugs, eliminating unnecessary and ineffective treatments is of critical importance.
Regardless of whether or not endocrine therapy is continued following resistance, our study has also revealed substantial differences in the responsiveness of endoxifen-resistant cells to second- and third-line therapies. Of the drugs tested here, abemaciclib, palboiclib, ribociclib, everolimus, and alpelisib have already received FDA approval for the treatment of endocrine-resistant breast cancer. Ipatasertib, venetoclax, and lasofoxifene are currently not FDA approved, but are being tested in clinical trials for endocrine resistant patients (NCT03337724, NCT03584009, NCT03781063). While all of these drugs have proven effective in treating endocrine-resistant metastatic disease in some patients, a great deal of variability in response rates has been observed. Little is known about the underlying causes of this variability. However, the data presented here have shown that endoxifen-resistant cells are universally less responsive to these drugs compared to 4HT-resistant models, with the exception of venetoclax. The differential responses to ribociclib, everolimus, and lasofoxifene are particularly striking, as in each case, the IC50 of endoxifen-resistant cells was greater than the maximum concentration achievable in patient serum (Cmax), while the IC50 of 4HT-resistant cells remained well below this threshold. It is therefore possible that patients whose tumors are more accurately modeled by endoxifen resistance (i.e. extensive metabolizers) are also less likely to benefit from these alternative therapies. These findings further highlight the need to better understand endoxifen resistance and to incorporate the use of such resistant models into studies aimed at developing novel therapeutic strategies for endocrine-resistant breast cancer patients. These data raise the crucial question of what therapeutic vulnerabilities exist in endoxifen-resistant cells, or in patients whose tumors are best modeled by endoxifen resistance. Future directions of this project will center on determining causes of endoxifen resistance, as well as identifying and characterizing therapeutic vulnerabilities that can be targeted to effectively inhibit endoxifen-resistant cells.
Though further characterization and studies are needed, this study raises the important point that endoxifen-resistant cell line models are substantially different from tamoxifen-resistant models. This distinction may be especially important when considering a patient’s CYP2D6 genotype. It is possible that clinical tamoxifen resistance differs significantly between extensive, intermediate, and poor metabolizers, and that these differences may explain the wide variability in the efficacy of second- and third-line therapies for tamoxifen-resistant patients. It is also possible that models of endoxifen resistance more accurately reflect “tamoxifen resistance” in a proportion of breast cancer patients. Additionally, endoxifen continues to be developed as an alternative endocrine therapy for both endocrine-sensitive and endocrine-resistant patients. For these reasons, it is imperative to better understand the molecular underpinnings of endoxifen resistance, to elucidate the clinical relevance of endoxifen-resistant models, and to incorporate the use of such models into drug discovery efforts for endocrine-resistant breast cancer. The models presented and described here represent the first steps toward these clinically-relevant goals.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements:
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health through the Mayo Clinic Breast Cancer SPORE Grant P50CA116201 (J. Ingle, M. Goetz, J. Hawse); the Eisenberg Foundation (J. Hawse); and the Mayo Clinic Graduate School of Biomedical Sciences (C. Jones, M. Emch).
Conflicts of Interest: Dr. Goetz reports personal fees from Genomic Health; grants from Pfizer, Lilly and Sermonix; and consulting fees from Lilly, Biovica, Novartis, Sermonix, Context Pharm, Pfizer, and Biotheranostics, outside the submitted work.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018; [DOI] [PubMed] [Google Scholar]
- 2.American Cancer Society. Breast Cancer Facts & Figures 2019–2020. Am Cancer Soc. 2019; [Google Scholar]
- 3.Clarke R, Tyson JJ, Dixon JM. Endocrine resistance in breast cancer - An overview and update. Mol Cell Endocrinol. 2015;418:220–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perez EA. Treatment strategies for advanced hormone receptor-positive and human epidermal growth factor 2-negative breast cancer: The role of treatment order. Drug Resist Updat. 2016; [DOI] [PubMed] [Google Scholar]
- 5.Riggs BL, Hartmann LC. Selective estrogen-receptor modulators - Mechanisms of action and application to clinical practice. N. Engl. J. Med. 2003. [DOI] [PubMed] [Google Scholar]
- 6.Murphy LC, Leygue E, Niu Y, Snell L, Ho SM, Watson PH. Relationship of coregulator and oestrogen receptor isoform expression to de novo tamoxifen resistance in human breast cancer. Br J Cancer. 2002;87:1411–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lai AC, Crews CM. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Demicheli R, Ardoino I, Boracchi P, Coradini D, Agresti R, Ferraris C, et al. Recurrence and mortality according to Estrogen Receptor status for breast cancer patients undergoing conservative surgery. Ipsilateral breast tumour recurrence dynamics provides clues for tumour biology within the residual breast. BMC Cancer. 2010;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goetz MP, Kamal A, Ames MM. Tamoxifen pharmacogenomics: The role of CYP2D6 as a predictor of drug response. Clin. Pharmacol. Ther. 2008. page 160–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mürdter TE, Schroth W, Bacchus-Gerybadze L, Winter S, Heinkele G, Simon W, et al. Activity levels of tamoxifen metabolites at the estrogen receptor and the impact of genetic polymorphisms of phase I and II enzymes on their concentration levels in plasma. Clin Pharmacol Ther. 2011; [DOI] [PubMed] [Google Scholar]
- 11.Jin Y, Desta Z, Stearns V, Ward B, Ho H, Lee KH, et al. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J Natl Cancer Inst. 2005; [DOI] [PubMed] [Google Scholar]
- 12.Wu X, Hawse JR, Subramaniam M, Goetz MP, Ingle JN, Spelsberg TC. The tamoxifen metabolite, endoxifen, is a potent antiestrogen that targets estrogen receptor a for degradation in breast cancer cells. Cancer Res. 2009;69:1722–7. [DOI] [PubMed] [Google Scholar]
- 13.Lim YC, Desta Z, Flockhart DA, Skaar TC. Endoxifen (4-hydroxy-N-desmethyl-tamoxifen) has anti-estrogenic effects in breast cancer cells with potency similar to 4-hydroxy-tamoxifen. Cancer Chemother Pharmacol. 2005; [DOI] [PubMed] [Google Scholar]
- 14.Borges S, Desta Z, Li L, Skaar TC, Ward BA, Nguyen A, et al. Quantitative effect of CYP2D6 genotype and inhibitors on tamoxifen metabolism: Implication for optimization of breast cancer treatment. Clin Pharmacol Ther. 2006; [DOI] [PubMed] [Google Scholar]
- 15.Desta Z, Ward BA, Soukhova NV., Flockhart DA. Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: Prominent roles for CYP3A and CYP2D6. J Pharmacol Exp Ther. 2004; [DOI] [PubMed] [Google Scholar]
- 16.Klein DJ, Thorn CF, Desta Z, Flockhart DA, Altman RB, Klein TE. PharmGKB summary: Tamoxifen pathway, pharmacokinetics. Pharmacogenet Genomics. 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schroth W, Goetz MP, Hamann U, Fasching PA, Schmidt M, Winter S, et al. Association between CYP2D6 polymorphisms and outcomes among women with early stage breast cancer treated with tamoxifen. JAMA. 2009;302:1429–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dezentjé VO, Opdam FL, Gelderblom H, Hartigh den J, Van der Straaten T, Vree R, et al. CYP2D6 genotype- and endoxifen-guided tamoxifen dose escalation increases endoxifen serum concentrations without increasing side effects. Breast Cancer Res Treat. 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hawse JR, Subramaniam M, Cicek M, Wu X, Gingery A, Grygo SB, et al. Endoxifen’s Molecular Mechanisms of Action Are Concentration Dependent and Different than That of Other Anti-Estrogens. PLoS One. 2013;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jayaraman S, Hou X, Kuffel MJ, Suman VJ, Hoskin TL, Reinicke KE, et al. Antitumor activity of Z-endoxifen in aromatase inhibitor-sensitive and aromatase inhibitor-resistant estrogen receptor-positive breast cancer. Breast Cancer Res. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maximov PY, McDaniel RE, Fernandes DJ, Korostyshevskiy VR, Bhatta P, Mürdter TE, et al. Simulation with cells in vitro of tamoxifen treatment in premenopausal breast cancer patients with different CYP2D6 genotypes. Br J Pharmacol. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goetz MP, Suman VJ, Reid JM, Northfelt DW, Mahr MA, Ralya AT, et al. First-in-human phase I Study of the tamoxifen metabolite Z-endoxifen in women with endocrine-refractory metastatic breast cancer. J Clin Oncol. 2017;35:3391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reese JM, Bruinsma ES, Monroe DG, Negron V, Suman VJ, Ingle JN, et al. ERβ inhibits cyclin dependent kinases 1 and 7 in triple negative breast cancer. Oncotarget. 2017;8:96506–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liao Y, Smyth GK, Shi W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014; [DOI] [PubMed] [Google Scholar]
- 25.Robinson MD, McCarthy DJ, Smyth GK. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krämer A, Green J, Pollard J, Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grote T, Siwak DR, Fritsche HA, Joy C, Mills GB, Simeone D, et al. Validation of reverse phase protein array for practical screening of potential biomarkers in serum and plasma: Accurate detection of CA19–9 levels in pancreatic cancer. Proteomics. 2008; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ballinger TJ, Meier JB, Jansen VM. Current landscape of targeted therapies for hormone-receptor positive, HER2 negative metastatic breast cancer. Front. Oncol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ring A, Dowsett M. Mechanisms of tamoxifen resistance. Endocr. Relat. Cancer. 2004. [DOI] [PubMed] [Google Scholar]
- 31.Chang M Tamoxifen resistance in breast cancer. Biomol Ther. 2012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller MA, Lippman ME, Katzenellenbogen BS. Antiestrogen Binding in Antiestrogen Growth-resistant Estrogen-responsive Clonal Variants of MCF-7 Human Breast Cancer Cells. Cancer Res. 1984; [PubMed] [Google Scholar]
- 33.Nawata H, Bronzert D, Lippman ME. Isolation and characterization of a tamoxifen-resistant cell line derived from MCF-7 human breast cancer cells. J Biol Chem. 1981; [PubMed] [Google Scholar]
- 34.Knowlden JM, Hutcheson IR, Jones HE, Madden T, Gee JMW, Harper ME, et al. Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology. 2003; [DOI] [PubMed] [Google Scholar]
- 35.Brünner N, Boysen B, Jirus S, Skaar TC, Holst-Hansen C, Lippman J, et al. MCF7/LCC9: An antiestrogen-resistant MCF-7 variant in which acquired resistance to the steroidal antiestrogen ICI 182,780 confers an early cross- resistance to the nonsteroidal antiestrogen tamoxifen. Cancer Res. 1997; [PubMed] [Google Scholar]
- 36.Lykkesfeldt AE, Madsen MW, Briand P. Altered Expression of Estrogen-regulated Genes in a Tamoxifen-resistant and ICI 164,384 and ICI 182,780 Sensitive Human Breast Cancer Cell Line, MCF-7/TAMR-l. Cancer Res. 1994; [PubMed] [Google Scholar]
- 37.Fabian C, Tilzer L, Sternson L. Comparative binding affinities of tamoxifen, 4-hydroxytamoxifen, and desmethyltamoxifen for estrogen receptors isolated from human breast carcinoma: Correlation with blood levels in patients with metastatic breast cancer. Biopharm Drug Dispos. 1981; [DOI] [PubMed] [Google Scholar]
- 38.Jordan VC, Collins MM, Rowsby L, Prestwich G. A monohydroxylated metabolite of tamoxifen with potent antioestrogenic activity. J Endocrinol. 1977; [DOI] [PubMed] [Google Scholar]
- 39.Jager NGL, Rosing H, Schellens JHM, Linn SC, Beijnen JH. Tamoxifen dose and serum concentrations of tamoxifen and six of its metabolites in routine clinical outpatient care. Breast Cancer Res Treat. 2014; [DOI] [PubMed] [Google Scholar]
- 40.Ingelman-Sundberg M Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): Clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J. 2005. [DOI] [PubMed] [Google Scholar]
- 41.Nath S, Mukherjee P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol. Med. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kiefel H, Bondong S, Hazin J, Ridinger J, Schirmer U, Riedle S, et al. L1CAM: A major driver for tumor cell invasion and motility. Cell Adhes. Migr. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao J, Huang X, Xu Z, Dai J, He H, Zhu Y, et al. LDHA promotes tumor metastasis by facilitating epithelial-mesenchymal transition in renal cell carcinoma. Mol Med Rep. 2017; [DOI] [PubMed] [Google Scholar]
- 44.Jung HY, Fattet L, Yang J. Molecular pathways: Linking tumor microenvironment to Epithelial-mesenchymal transition in metastasis. Clin Cancer Res. 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gupta PB, Pastushenko I, Skibinski A, Blanpain C, Kuperwasser C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jolly MK, Boareto M, Huang B, Jia D, Lu M, Onuchic JN, et al. Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front Oncol. 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaminska KK, Akrap N, Staaf J, Borg Å, Bosch A, Honeth G. Abstract 5907: Fulvestrant resistance in estrogen receptor positive breast cancer models is driven by heterogeneous ER independent transcriptional programs. 2018.
- 48.Paoletti C, Larios JM, Muñiz MC, Aung K, Cannell EM, Darga EP, et al. Heterogeneous estrogen receptor expression in circulating tumor cells suggests diverse mechanisms of fulvestrant resistance. Mol Oncol. 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ribas R, Pancholi S, Guest SK, Marangoni E, Gao Q, Thuleau A, et al. AKT antagonist AZD5363 influences estrogen receptor function in endocrine-resistant breast cancer and synergizes with fulvestrant (ICI182780) in vivo. Mol Cancer Ther. 2015; [DOI] [PubMed] [Google Scholar]
- 50.Encarnación CA, Ciocca DR, McGuire WL, Clark GM, Fuqua SAW, Osborne CK. Measurement of steroid hormone receptors in breast cancer patients on tamoxifen. Breast Cancer Res Treat. 1993; [DOI] [PubMed] [Google Scholar]
- 51.Kuukasjärvi T, Kononen J, Helin H, Holli K, Isola J. Loss of estrogen receptor in recurrent breast cancer is associated with poor response to endocrine therapy. J Clin Oncol. 1996; [DOI] [PubMed] [Google Scholar]
- 52.Johnston SRD, Saccani-Jotti G, Smith IE, Newby J, Dowsett M. Change in oestrogen receptor expression and function in tamoxifen-resistant breast cancer. Endocr Relat Cancer. 1995; [Google Scholar]
- 53.Gutierrez MC, Detre S, Johnston S, Mohsin SK, Shou J, Allred DC, et al. Molecular changes in tamoxifen-resistant breast cancer: Relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J Clin Oncol 2005; [DOI] [PubMed] [Google Scholar]
- 54.Hull DF, Clark GM, Osborne CK, Chamness GC, Knight WA, McGuire WL. Multiple Estrogen Receptor Assays in Human Breast Cancer. Cancer Res. 1983; [PubMed] [Google Scholar]
- 55.D’Assoro AB, Liu T, Quatraro C, Amato A, Opyrchal M, Leontovich A, et al. The mitotic kinase aurora-A promotes distant metastases by inducing epithelial-to-mesenchymal transition in ER + breast cancer cells. Oncogene. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lindström LS, Karlsson E, Wilking UM, Johansson U, Hartman J, Lidbrink EK, et al. Clinically used breast cancer markers such as estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 are unstable throughout tumor progression. J Clin Oncol. 2012; [DOI] [PubMed] [Google Scholar]
- 57.Buzdar A, Jonat W, Howell A, Jones SE, Blomqvist C, Vogel CL, et al. Anastrozole, a potent and selective aromatase inhibitor, versus megestrol acetate in postmenopausal women with advanced breast cancer: Results of overview analysis of two phase III trials. J Clin Oncol. 1996; [DOI] [PubMed] [Google Scholar]
- 58.Dombernowsky P, Smith I, Falkson G, Leonard R, Panasci L, Bellmunt J, et al. Letrozole, a new oral aromatase inhibitor for advanced breast cancer: Double-blind randomized trial showing a dose effect and improved efficacy and tolerability compared with megestrol acetate. J Clin Oncol. 1998; [DOI] [PubMed] [Google Scholar]
- 59.Zhang X, Wang Z-Y. Estrogen Receptor-α Variant, ER-α36, is Involved in Tamoxifen Resistance and Estrogen Hypersensitivity. Endocrinology Oxford Academic; 2013. [cited 2020 Dec 3];154:1990–8. [DOI] [PubMed] [Google Scholar]
- 60.Chan CMW, Martin LA, Johnston SRD, Ali S, Dowsett M. Molecular changes associated with the acquisition of oestrogen hypersensitivity in MCF-7 breast cancer cells on long-term oestrogen deprivation. J Steroid Biochem Mol Biol. 2002; [DOI] [PubMed] [Google Scholar]
- 61.Masamura S, Santner SJ, Heitjan DF, Santen RJ. Estrogen deprivation causes estradiol hypersensitivity in human breast cancer cells. J Clin Endocrinol Metab. 1995; [DOI] [PubMed] [Google Scholar]
- 62.Martin LA, Ribas R, Simigdala N, Schuster E, Pancholi S, Tenev T, et al. Discovery of naturally occurring ESR1 mutations in breast cancer cell lines modelling endocrine resistance. Nat Commun. 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pan H, Gray R, Braybrooke J, Davies C, Taylor C, McGale P, et al. 20-year risks of breast-cancer recurrence after stopping endocrine therapy at 5 years. N Engl J Med. 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ekholm M, Bendahl PO, Fernö M, Nordenskjöld B, Stål O, Rydén L. Two years of adjuvant tamoxifen provides a survival benefit compared with no systemic treatment in premenopausal patients with primary breast cancer: Long-Term follow-up (> 25 years) of the phase III SBII:2pre trial. J Clin Oncol. 2016; [DOI] [PubMed] [Google Scholar]
- 65.Ekholm M, Bendahl PO, Fernö M, Nordenskjöld B, Stål O, Rydén L. Effects of adjuvant tamoxifen over three decades on breast cancer–free and distant recurrence–free interval among premenopausal women with oestrogen receptor–positive breast cancer randomised in the Swedish SBII:2pre trial. Eur J Cancer. 2019; [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.