

Abstract
Regulatory T cells (Tregs) are known to inhibit anti-tumor immunity, yet the specific mechanism by which intratumoral Tregs promote tumor growth remains unclear. To better understand the roles of intratumoral Tregs, we selectively depleted tumor-infiltrating Tregs using anti-CD25-F(ab′)2 near-infrared photoimmunotherapy (NIR-PIT). Depletion of tumor-infiltrating Tregs induced transient but synchronized IFN-γ expression in CD8 T and natural killer (NK) cells. Despite the small fraction of CD8 T and NK cells contained within examined tumors, IFN-γ produced by these CD8 T and NK cells led to efficient and rapid tumor vessel regression, intratumoral ischemia, and tumor necrosis/apoptosis and growth suppression. IFN-γ receptor expression on vascular endothelial cells was required for these effects. Similar findings were observed in the early phase of systemic Treg depletion in tumor-bearing Foxp3DTR mice; combination with IL-15 therapy further inhibited tumor growth and achieved increased complete regression. These results indicate the pivotal roles of intratumoral Tregs in maintaining tumor vessels and tumor growth by suppressing CD8 T and NK cells from producing IFN-γ, providing insight into the mechanism of Treg-targeting therapies.
Keywords: Regulatory T cell, Near-infrared photoimmunotherapy, IFN-γ, Endothelial cell, Tumor ischemia
Introduction
Regulatory T cells (Tregs) inhibit anti-tumor immunity at different phases and locations (e.g. priming phase in lymph nodes and effector phase in tumor tissue) by suppressing the function of T, NK, and antigen-presenting cells (1–3). Tumors recruit and accumulate Tregs in tumor-microenvironment to evade anti-tumor immune activities (4,5). Nevertheless, our knowledge regarding roles of Tregs in tumor immunity is largely derived from analyses after systemic depletion of Tregs (6–9) or systemic manipulation of Treg function (10). The specific roles that intra- and extra-tumoral Tregs play remain unclear due to technical difficulties in depleting or manipulating Tregs in a spatially specific manner.
Near-infrared photoimmunotherapy (NIR-PIT) is a novel technology to selectively target and deplete cells using antibodies, against surface antigens, conjugated with a phthalocyanine dye, IR700. Once the antibody-IR700 conjugate is exposed to NIR-light, the targeted cells rapidly undergo necrosis (11,12). Currently, EGFR-targeting NIR-PIT is in phase 3 clinical trial in head and neck cancer (ClinicalTrials.gov Identifier: NCT02422979). In order to achieve spatially specific depletion of Tregs, we have previously developed anti-CD25-F(ab′)2 NIR-PIT (13). An injection of anti-CD25-F(ab′)2-IR700 conjugate followed by NIR irradiation on the tumor induces rapid, selective depletion of pre-existing intratumoral Tregs, without depleting T cells that upregulated CD25 after the NIR-PIT, and suppresses tumor growth (13). This therapeutic effect depends on the presence of CD8 T and NK cells and their production of IFN-γ (13). However, the exact target of IFN-γ and the mechanisms of the consequent tumor regression remain to be elucidated.
In this study, we sought to understand the roles played by intratumoral Tregs by selectively depleting these Tregs utilizing anti-CD25-F(ab′)2 NIR-PIT. We found that depletion of tumor-infiltrating Tregs caused rapid regression of tumor vessels, intratumoral ischemia, and necrosis/apoptosis of tumor cells in an IFN-γ-dependent manner. IFN-γ receptor expression on vascular endothelial cells was required for these changes. We further examined a combination therapy with IL-15 administration to induce complete eradication of the tumors. Our study sheds light on the understanding of the roles that intratumoral Tregs play in maintaining tumor microenvironment that supports tumor growth.
Materials and Methods
Mice
C57BL/6 (expressing either Ly5.1 or Ly5.2) mice, BALB/c mice, Ifngr1−/−, Ifngr1flox/flox, Tie2Cre, Foxp3DTR, and albino mice on a C57BL/6 background were purchased from Jackson Laboratory. Il15ra−/− mice (Jackson Laboratory) were backcrossed to C57BL/6 mice for 10 generation. All mouse experiments were performed in accordance with animal protocols approved by National Institutes of Health Animal Care and Use Committee. Males and females at 6-10-week-old were used.
Tumor cell lines and subcutaneous and lung metastasis tumor models
MC38, LL/2, and 4T1 tumor cell lines [American Type Culture Collection (ATCC)] and EO771 tumor cell line (CH3 BioSystems) were maintained in RPMI 1640 medium supplemented with 10 % fetal calf serum, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 0.05 mM 2-mercaptoethanol. All cell lines were tested negative for Mycoplasma (PCR at Frederick National Laboratory for Cancer Research) and authenticated (ATCC) before use. Subcutaneous tumors were generated by inoculating 4.0×105 MC38 cells, 8.0×105 EO771 cells, or 2.0×105 LL/2 cells in 100 μl PBS in right dorsum of C57BL/6-background mice, and 1.0×105 4T1 cells in BALB/c mice. Tumor size (V), calculated as V = (major axis) x (minor axis)2 × 1/2, was followed up until it reached 2500 mm3. Mice with approximately 100 mm3-tumor were randomly grouped for experiments unless otherwise stated. For lung metastases generation, 1.0×106 MC38 or EO771 cells in 300 μl PBS were intravenously injected.
Near-infrared photoimmunotherapy (NIR-PIT)
IR700-conjugated anti-CD25-F(ab′)2 and isotype matched control-F(ab′)2 were prepared as described in Supplementary Information. Subcutaneous tumor-bearing mice received 50 μg IR700-conjugated F(ab′)2 in 200 μl PBS intravenously, were shaved for irradiation, and, on the next day, underwent NIR-PIT (λ = 690 ± 5 nm) on tumor at 50 J/cm2 (ML7710 Laser System, Modulight Inc, Tanbere, Finland).
Flow cytometry analysis
Single cell suspensions of tissues were incubated with anti-CD16/32 antibody, followed by fluorophore-conjugated antibodies. For intracellular-staining, minced tumors were incubated with 3 μg/ml brefeldin A (4 hours) without exogenous stimulation, made into single cell suspensions, fixed and permeabilized, and stained with antibodies. Where indicated, ex vivo stimulation with 50 ng/ml phorbol 12-myristate 13-acetate (PMA) and 1 μM ionomycin (4 hours) was performed. See Supplementary Information for details.
Histological analysis
Lectin perfusion assay by DyLight 594-conjugated Tomato-lectin intravenous injection, vessel leakiness assay by FITC-conjugated dextran sulfate intravenous injection, pimonidazole-binding assay by pimonidazole intravenous injection, multiplex immunohistochemistry including phopho-STAT1 staining, propidium-iodide intravenous injection assay, TUNEL assay using Click-iT TUNEL Alexa Fluor Imaging Assay, and transmission electron microscopy studies are described in detail, along with quantification methods, in Supplemental Information.
Statistical analysis
P-values between unpaired variables were calculated using Student’s t test. Where indicated, paired t-test and χ2 test were used. P-values less than 0.05 were considered significant. The results were shown as mean ± SEM.
Additional materials and methods
Additional details and product catalog numbers are described in the Supplementary Materials Methods and Supplementary Table.
Results
Selective depletion of intratumoral Tregs by anti-CD25-F(ab′)2 NIR-PIT suppresses tumor growth.
In order to selectively deplete intratumoral Tregs, we employed anti-CD25-F(ab′)2 NIR-PIT (13). CD25 was highly and selectively expressed in CD3+CD4+Foxp3+ Tregs residing in tumor (Figure S1A and B), and anti-CD25-F(ab′)2 NIR-PIT selectively depleted intratumoral CD3+CD4+CD25+Foxp3+ Tregs by more than 90 % within 30 minutes of NIR-light exposure (Figure 1A), while Tregs in the lymph nodes and spleen and CD3+CD4+Foxp3- non-Tregs in tumor remained intact (Figures 1A and B). We further confirmed that immune cells other than Tregs were not depleted, through a comprehensive analysis of lymphoid and myeloid cell populations in tumor tissues (Figure S1A–F and Figure S2A and B). IR-700-conjugated anti-CD25-F(ab′)2 does not bind to tumor cells nor does it induce non-specific cell death upon NIR-light irradiation (13). The fraction of Tregs among CD4+ T cells began to recover from Day 2 and reached near pre-treatment levels by Day 4 (Figure 1C). This recovery was much quicker than that after systemic depletion of Tregs in Foxp3DTR mice by intravenous injection of diphtheria toxin (DT) (Figure S3A and B), indicating that recruitment of circulating Tregs, not conversion from non-Treg CD4 T cells, mainly contributes to the replenishment of intratumoral Tregs after anti-CD25-F(ab′)2 NIR-PIT.
Figure 1. Selective depletion of intratumoral Tregs suppresses tumor growth.

(A) Selective depletion of intratumoral Tregs analyzed 30 minutes after anti-CD25-F(ab′)2 NIR-PIT on MC38 tumor (approximately 100 mm3). Representative flow cytometry data showing frequency of CD25+Foxp3+ Tregs in CD3+CD4+ T cells in indicated tissues (n = 3, each). (B) Frequency and cell number of CD3+CD4+CD25+Foxp3+ Tregs and CD3+CD4+Foxp3- non-Treg cells in total cells in MC38 tumor 30 minutes after control-F(ab′)2 or anti-CD25-F(ab′)2 NIR-PIT, analyzed by flow cytometry (n = 3, each). (C) Changes in the frequency of Foxp3+CD25+ Tregs among CD3+CD4+ T cells in MC38 and EO771 tumors over 4 days after anti-CD25-F(ab′)2 NIR-PIT, analyzed by flow cytometry (n = 3 at each time point). (D) Frequency of indicated subsets of lymphocytes in total cells in untreated MC38, EO771, LL/2, and 4T1 tumors, analyzed by flow cytometry (n ≥ 5, each). *: compared to MC38 tumor, †: compared to EO771 tumor. (E) Tumor growth after control-F(ab′)2 or anti-CD25-F(ab′)2 NIR-PIT on MC38, EO771, LL/2, and 4T1 tumors.
* and †: p < 0.05, ** and ††: p < 0.01.
Next, in order to analyze how CD8 T and NK cell infiltration levels affect the anti-tumor effect of intratumoral Treg depletion, we performed anti-CD25-F(ab′)2 NIR-PIT on different murine tumors that showed similar Treg frequencies (Figure 1D). Interestingly, the growth of tumors with higher CD8 T and NK cell infiltration (MC38 and EO771) was significantly suppressed by intratumoral Treg depletion, while minimal effects were observed in tumors with lower T and NK cell infiltration (LL/2 and 4T1) (Figure 1E). In responsive tumor models (MC38 and EO771), the tumor shrunk from Day 1 to Day 3 after anti-CD25-F(ab′)2 NIR-PIT but re-grew after Day 5, coinciding with the recovery of Tregs in the tumors (Figure 1C). When intratumoral Treg depletion was repeated on MC38 tumor, the period of tumor regression/suppression extended but all tumors re-grew after Day 8 (Figure S4A and B). We used MC38 and EO771 tumors in subsequent experiments to examine the mechanism of the anti-tumor effect of intratumoral Treg depletion.
Anti-tumor effect of intratumoral Treg depletion depends on IFN-γ signaling in non-bone-marrow-derived stromal cells.
Our previous study demonstrates that the anti-tumor effect of intratumoral Treg depletion by anti-CD25-F(ab′)2 NIR-PIT requires IFN-γ production by CD8 T and NK cells (13). Similarly, the anti-tumor effect observed in systemic depletion of Tregs in tumor-bearing Foxp3DTR mice is IFN-γ-dependent (7,14). However, the exact target of IFN-γ after intratumoral Treg depletion remains unknown.
We first analyzed kinetics of IFN-γ expression in CD8 T and NK cells after anti-CD25-F(ab′)2 NIR-PIT intratumoral Treg depletion. IFN-γ was produced within 30 minutes of intratumoral Treg depletion, without exogenous stimulations such PMA and ionomycin, but began to decrease between 2 and 6 hours after treatment on MC38 and EO771 tumors (Figure 2A and B). This rapid IFN-γ production was associated with increased expression of perforin (Figure 2A) and activation markers including CD69 (13), but expression of TNF-α and granzyme B did not change (Figure 2A). Ex vivo stimulation of tumor-infiltrating CD8 T and NK cells with PMA and ionomycin enabled production of IFN-γ and perforin regardless of prior intratumoral Treg depletion (Figure S5A and B). These results indicate that although tumor-infiltrating CD8 T and NK cells possessed the ability to express IFN-γ and perforin, intratumoral Tregs actively inhibited the expression of these effector molecules, and that the short but synchronized induction of IFN-γ up to 6 hours of treatment accounted for the anti-tumor effect of anti-CD25-F(ab′)2 NIR-PIT. Of note, anti-CD25-F(ab′)2 NIR-PIT did not significantly change the frequencies of tumor-infiltrating CD8 T and NK cells (Figure S1D), contrasting to systemic depletion of Tregs in Foxp3DTR mice model that induces tumor-infiltration of activated CD8 and CD4 T cells (6–9).
Figure 2. IFN-γR expression in non-bone-marrow-derived stromal cells is required for the anti-tumor effect of intratumoral Treg depletion.

(A) Induction of IFN-γ and perforin, but not TNF-α and granzyme B, in CD3+CD8+ T and CD45+NK1.1+ NK cells 30 minutes after anti-CD25-F(ab′)2 on MC38 tumor, analyzed by flow cytometry (n ≥ 4, each). Tumor tissues were minced and incubated with brefeldin A (4 hours) without exogenous stimulation before flow cytometry analysis. Refer to Figure S5 for results after PMA/ionomycin stimulation. (B) Changes in the frequency of IFN-γ or perforin-expressing cells in CD3+CD8+ T cells and CD45+NK1.1+ NK cells, using the method depicted in (A), in MC38 and EO771 tumors following anti-CD25-F(ab′)2 NIR-PIT (n ≥ 4 at each time point). (C and D) Anti-tumor effect of anti-CD25-F(ab′)2 NIR-PIT on MC38 tumor was abrogated by intravenous injection of anti-IFN-γ antibody (100 μg, one day before NIR-PIT) (C) and in Ifngr1−/− mice (D). (E and F) Anti-tumor effect of anti-CD25-F(ab′)2 NIR-PIT on MC38 tumor was abrogated in Ifngr1−/− mice reconstituted with WT bone-marrow (WT→Ifngr1−/−) (E), while that was maintained in WT mice reconstituted with Ifngr1−/− bone-marrow (Ifngr1−/−→WT ) (F).
*: p < 0.05, **: p < 0.01.
Next, to determine the target and role of IFN-γ after intratumoral Treg depletion, we neutralized IFN-γ or used various Ifngr1-deficient mouse models. The anti-tumor effect of intratumoral Treg depletion by anti-CD25-F(ab′)2 NIR-PIT was abrogated in wild-type (WT) mice injected with anti-IFN-γ antibody one day before NIR-PIT (Figure 2C), in Ifngr1−/− mice (Figure 2D), and in Ifngr1−/− mice reconstituted with wild-type bone-marrow cells (WT→Ifngr1−/−) (Figure 2E), suggesting that both IFNγ production and IFNγR1 expression on non-bone-marrow-derived cells were required for the efficacy. By contrast, anti-CD25-F(ab′)2 NIR-PIT remained effective in WT mice reconstituted with Ifngr1−/− bone-marrow cells (Ifngr1−/−→WT) (Figure 2F), indicating that IFNγR1 expression on bone-marrow-derived cells was dispensable. In both Ifngr1−/−→WT and WT→Ifngr1−/− mice, more than 95 % of peripheral blood and intratumoral leukocytes were derived from reconstituted bone-marrow cells (Figures S5C and D). The frequency of CD8 T and NK cells in tumor and their IFN-γ-expression was compatible between the tumors in Ifngr1−/− and WT mice (Figure S5E). These results collectively indicate that, after selective depletion of intratumoral Tregs by anti-CD25-F(ab′)2 NIR-PIT, IFN-γ produced from activated CD8 T and NK cells directly acted on non-bone-marrow-derived host stromal cells in tumor tissue.
Selective depletion of intratumoral Tregs causes IFN-γ-dependent acute tumor vessel regression.
Among the stromal cells in tumor tissue, fibroblasts and pericytes can differentiate from bone-marrow-derived cells (15–17), whereas little evidence suggest that endothelial cells are bone-marrow derived (18). A recent study demonstrated that forced expression of IFN-γ in tumor cells directly acts on endothelial cells to cause rapid vessel regression and intratumoral ischemia (19). However, the magnitude of IFN-γ forced to be produced from all the tumor cells is considered to be much higher than that of IFN-γ physiologically produced from tumor-infiltrating CD8 T and NK cells that usually consist of small fractions in the tumor (Figure 1D). Therefore, it is still undetermined whether more physiological, short-lasting induction of IFN-γ from CD8 T and NK cells observed upon depletion of intratumoral Treg (Figure 2B) is sufficient to cause similar vascular changes. Thus, we next analyzed the changes in tumor vasculature after intratumoral Treg depletion.
Immunohistochemical analysis of tumor vessels using an endothelial cell marker CD31 showed narrowed vessel lumens, occurring within 4 hours of anti-CD25-F(ab′)2 NIR-PIT on MC38 tumors (Figures 3A, and B). Blood perfusion was decreased in these vessels (demonstrated by the lack of endothelial cell labeling by intravenously injected-DyLight 594-conjugated Tomato-lectin) in both MC38 and EO771 tumors (Figures 3A, C and S6A–C). Empty structures of collagen IV+ basement membrane sleeves without CD31+ endothelial cell lining remained after vessel regression (Figure 3A). These changes were clearly observed up to 48 hours after depletion of intratumoral Tregs (Figure 3B and C), leading to decreased tumor vessel density (Figure 3D). Anti-CD25-F(ab’)2 NIR-PIT registrant 4T1 tumor showed poor vessel perfusion, which did not change by intratumoral Treg depletion (Figures S6B and C). These findings suggest a role for Tregs to maintain tumor vessels and blood perfusion. Of note, we did not detect CD25 expression in endothelial cells nor were tumor vessels rapidly depleted by anti-CD25-F(ab′)2 NIR-PIT (Figures 3D and E). Contrasting to the tumor vessels, blood vessel structure and perfusion remained unchanged in adjacent non-tumor tissue, where lymphocytes were scarce (Figure S6D). Importantly, the acute tumor vessel regression upon anti-CD25-F(ab′)2 NIR-PIT was observed in Ifngr1−/−→WT mice but not in Ifngr1−/− and WT→Ifngr1−/− mice bearing MC38 tumors (Figures 3F and S6E), indicating that this process required the expression of IFN-γR1 in non-bone-marrow-derived stromal cells in tumor tissue.
Figure 3. Selective depletion of intratumoral Tregs causes IFN-γ-dependent rapid vessel regression.

(A) Representative immunohistochemistry of CD31 (Top), DyLight 594-conjugated Tomato-lectin and CD31 (Tomato-lectin perfusion assay, Middle), and Collagen IV and CD31 (Bottom), 4 hours after control-F(ab′)2 or anti-CD25-F(ab′)2 NIR-PIT on MC38 tumor (n ≥ 3, each). Bar: 100 μm. (B and C) Quantitation of regressed vessels (B) and vessel perfusion (C) on immunohistochemistry, as in (A), at indicated time points following control-F(ab′)2 or anti-CD25-F(ab′)2 NIR-PIT on MC38 tumor (n ≥ 3 at each time point). (D) Quantitation of vessel density on CD31 immunohistochemistry at indicated time points after anti-CD25-F(ab′)2 NIR-PIT on MC38 tumor (n ≥ 3 at each time point). (E) CD25 was not expressed in endothelial cells (CD45-CD31+) in MC38 tumor, analyzed by flow cytometry (n = 3, representative data). (F) Vessel perfusion in MC38 tumors, analyzed by Tomato-lectin perfusion assay, was maintained in Ifngr1−/− and WT→Ifngr1−/− mice, but was impaired in Ifngr1−/−→WT bone-marrow chimera mice, 4 hours after anti-CD25-F(ab′)2 NIR-PIT (n = 3, each). (G and H) Impaired tumor vessel perfusion, analyzed by Tomato-lectin perfusion assay, in early phase of Treg depletion in MC38 tumor-bearing Foxp3DTR mice. Diphtheria toxin (DT, 25 ng/g) was intraperitoneally injected on Day −3, −2, and −1 of analysis. Anti-IFN-γ antibody injection (100 μg on Day −3 and −1) abrogated impaired perfusion. Representative immunohistochemistry (G) and quantitation summary (H) (n = 3, each). Bar: 100 μm. (I) Transmission electron microscope images show degeneration of endothelial cells (white arrowheads: obscured intracellular organelle with or without cytoplasmic vacuolation, black arrowheads: swollen mitochondria and increased electron densities in cytoplasm) 4 hours after anti-CD25-F(ab′)2 NIR-PIT on MC38 tumor. Bar: 2 μm. (J) Quantitation of degenerated vessels in (I). In each group, 22 vessels from 2 tumors were analyzed with χ2 test.
*: compared to no treatment, †: compared to control-F(ab′)2 NIR-PIT, * and †: p < 0.05, ** and ††: p < 0.01.
We next examined the effects of Treg depletion on tumor vessels in another model, Foxp3DTR mice. Previous studies using B16 melanoma-bearing Foxp3DTR mice have shown that Treg depletion by intravenous DT injection causes tumor vessel normalization, characterized by decreased vessel density and improved vessel perfusion, 3 to 4 days after DT treatment (9). We hypothesized that this decreased vessel density was preceded by vessel regression, and thus performed Tomato-lectin perfusion assay 1 day after completion of DT treatment, when more than 95 % of intratumoral and systemic Tregs were depleted (Figure S3A and B). We found that vessel perfusion in MC38 tumor was significantly impaired in DT-treated mice, but not when combined with an IFN-γ-neutralizing antibody injection (Figures 3G and H). These results indicate that systemic Treg depletion induced the vessel regression in an IFN-γ-dependent manner, similar to the findings with intratumoral Treg depletion by anti-CD25-F(ab′)2 NIR-PIT.
To further characterize tumor vessel regression induced by anti-CD25-F(ab′)2 NIR-PIT intratumoral Treg depletion, we performed transmission electron microscopy (TEM) analysis (Figure 3I). The most characteristic changes observed were various degrees of shrinkage or swelling of endothelial cells, associated with obscured intracellular organelle with or without cytoplasmic vacuolation (Figure 3I, white arrowheads) or associated with swollen mitochondria and increased electron densities in cytoplasm (Figure 3I, black arrowheads). These changes were rarely observed in tumors after control-F(ab′)2 NIR-PIT (Figures 3I and J).
To this end, we performed in vivo live perfusion imaging of the tumor by injecting IR800-conjugated bovine serum albumin 18 hours after anti-CD25-F(ab′)2 NIR-PIT and observed decreased tumor vessel perfusion (IR800 optical signal) extending whole tumor both in vivo and in harvested tumors (Figure S7A–D).
Vessel function is partially normalized after intratumoral Treg depletion.
Increased vessel leakiness is an important factor causing uneven and poor blood perfusion in tumor vessels, which can be normalized by IFN-γ (20–23). To assess the changes in vessel leakiness after anti-CD25-F(ab′)2 NIR-PIT intratumoral Treg depletion, a dextran leakage assay was performed. MC38 tumor showed area with dextran leakage, which decreased after intratumoral Treg depletion (Day 3), even with adjustment for the number of tumor vessels (Figures S8A–C), suggesting a partial normalization of vessel function following the acute vessel regression. Tumor vessel coverage by NG2+ pericytes did not change after anti-CD25-F(ab′)2 NIR-PIT (Figure S8D). However, collagen IV+ basement membrane structure, that was maintained in the early phase of vessel regression (4 hours, Figure 3A), degraded over time (18 hours, Figure S8D). Endothelial cells of tumor vessels are characterized by decreased VCAM-1 expression and increased FasL expression, both of which negatively affect the influx of CD8 T cells to the tumor tissue (24), but the expression levels of these molecules did not change by intratumoral Treg depletion (Figure S8E).
Tumor vessel regression by intratumoral Treg depletion leads to tumor ischemia and necrosis/apoptosis.
We next asked if tumor vessel regression induced by intratumoral Treg depletion caused ischemia in the tumor. We first performed pimonidazole-binding assay at various timepoints after anti-CD25-F(ab′)2 NIR-PIT and mice in control groups. Pimonidazole binding to both tumor (MC38 and EO771) and immune cells increased as early as 4 hours after intratumoral Treg depletion and persisted for 48 hours (Figures 4A–D). Similarly, expression of carbonic anhydrase IX (CAIX), another ischemic/hypoxic marker, increased after intratumoral Treg depletion (Figures S9A and B). Systemic Treg depletion in Foxp3DTR mice also demonstrated increased pimonidazole-positive ischemic areas in MC38 tumors on Day 1 of DT treatment completion, which was diminished by IFN-γ-neutralization (Figures S9C and D). Moreover, development of intratumoral ischemia was dependent on IFN-γR1 expression in non-bone-marrow-derived stromal cells as was seen in Ifngr1−/−→WT but not in WT→Ifngr1−/− bone-marrow chimera mice (Figures 4E and F). The acute intratumoral ischemia resulted in a significant increase in necrosis and apoptosis as demonstrated by propidium iodide injection assay (Figures 4G and H) and TUNEL assay (Figures 4I and J), respectively, both of which were abrogated in Ifngr1−/− mice (Figure 4G–J). Altogether, these data indicate that tumor vessel regression induced by intratumoral Treg depletion led to tumor ischemia, apoptosis and necrosis in a manner dependent on physiologically produced IFN-γ that acted on non-bone-marrow-derived host stromal cells.
Figure 4. IFN-γ-dependent rapid vessel regression is followed by tumor ischemia and necrosis/apoptosis of tumor.

(A and B) Flow cytometry indicates increased pimonidazole positivity in CD45- tumor cells and CD45+ immune cells in pimonidazole-binding assay following anti-CD25-F(ab′)2 NIR-PIT. Representative histograms at 18 hours after anti-CD25-F(ab′)2 NIR-PIT (A) and changes of pimonidazole+ hypoxic tumor and immune cell frequencies in MC38 and EO771 tumors (B, n = 3 at each time point). (C and D) Increased pimonidazole+ areas in pimonidazole-binding assay in MC38 tumors 18 hours after anti-CD25-F(ab′)2 NIR-PIT. Representative immunohistochemistry (C) and quantitation summary (D) (n = 3, each). Bar: 250 μm. (E and F) Flow cytometry analysis of pimonidazole positivity in CD45- tumor cells and CD45+ immune cells in pimonidazole-binding assay, 18 hours after anti-CD25-F(ab′)2 NIR-PIT on MC38 tumor in Ifngr1−/−, and Ifngr1−/−→WT and WT→Ifngr1−/− bone-marrow chimera mice. Representative histograms (E) and quantitation summery in CD45- tumor cells (F) (n ≥ 3, each). (G and H) Increased necrotic areas positive for propidium iodide in propidium iodide injection assay, observed 18 hours after anti-CD25-F(ab′)2 NIR-PIT on MC38 tumor, which were abrogated in Ifngr1−/− mice. Representative histological images (G) and quantitation summary (H) (n = 3, each). Bar: 250 μm. (I and J) Increased TUNEL-positive apoptotic cells 18 hours after anti-CD25-F(ab′)2 NIR-PIT on MC38, which was abrogated in Ifngr1−/− mice. Representative histological images (I) and quantitation summary (J) (n = 3, each). Bar: 250 μm.
*: p < 0.05, **: p < 0.01.
Compatible with ischemic reaction, we observed induction of cytokines and chemokines related to acute inflammation (Figure S10), induction of VEGF expression (Figure S10), influx of neutrophils (Figure S1D), and polarization of intratumoral macrophages and monocytes into regulatory phenotypes, exhibiting CD206highMHCIIlow and Ly6Clow, respectively (Figures S1D–F), in the tumor.
IFN-γ directly acts on endothelial cells to cause acute tumor vessel regression.
To determine if IFN-γ directly acted on endothelial cells to cause tumor vessel regression, we generated Tie2CreIfngr1flox/flox mice. In these mice, IFN-γR1 was selectively depleted in endothelial cells, among non-bone-marrow-derived stromal cells in the tumor tissue (Figure 5A). Although Tie2Cre is also expressed in hematopoietic-lineage cells (25) and thus these cells in Tie2CreIfngr1flox/flox mice lack IFN-γR1, our bone-marrow reconstitution experiments using WT and Ifngr−/− mice excluded the contribution of IFN-γR1 expressed on bone-marrow-derived cells to the anti-tumor effect of anti-CD25-F(ab’)2 NIR-PIT (Figures 2 and 3). In Tie2CreIfngr1flox/flox mice, the therapeutic effect on MC38 tumors was abrogated, while it was maintained in control Ifngr1flox/flox mice (Figure 5B). We repeated the experiment using Tie2CreIfngr1flox/flox mice reconstituted with WT bone-marrow (WT→Tie2CreIfngr1flox/flox) and control mice (WT→Ifngr1flox/flox). The therapeutic effect was abrogated in WT→Tie2CreIfngr1flox/flox mice, but was maintained in control WT→Ifngr1flox/flox mice (Figure 5C). Of note, in both WT→Ifngr1flox/flox and WT→Tie2CreIfngr1flox/flox mice, more than 95 % of intratumoral Tregs and Tie2-expressing macrophages were derived from reconstituted bone-marrow (Figure S11A–C). Importantly, induction of acute vessel regression and ischemia were also abrogated in Tie2CreIfngr1flox/flox mice, while they were observed in control Ifngr1flox/flox mice (Figures 5D–G). These results indicate that IFN-γR1 expression on tumor vascular endothelial cells was necessary for the anti-tumor effect of intratumoral Treg depletion and that IFN-γ produced by CD8 T and NK cells directly acted on endothelial cells to cause acute vessel regression and intratumoral ischemia.
Figure 5. IFN-γ directly targets endothelial cells to cause vessel regression.

(A) IFN-γRI expression in endothelial cells (CD45-panCK-CD31+), pericytes (CD45-panCK-CD31-NG2+), and fibroblasts (CD45-panCK-CD31-PDFGRα+) in MC38 tumors in WT, Ifngr1−/−, or Tie2CreIfngr1flox/flox mice, analyzed by flow cytometry (n = 2, each). (B) Anti-tumor effect of anti-CD25-F(ab′)2 NIR-PIT on MC38 tumors was abrogated in Tie2CreIfngr1flox/flox mice. Ifngr1flox/flox mice were used as control. (C) Anti-tumor effect of anti-CD25-F(ab′)2 NIR-PIT on MC38 tumors was abrogated in WT→Tie2CreIfngr1flox/flox mice. WT→Ifngr1flox/flox mice were used as control. (D and E) Immunohistochemistry of DyLight 594-conjugated Tomato-lectin and CD31 (endothelial cells) in Tomato-lectin perfusion assay, 18 hour after anti-CD25-F(ab′)2 NIR-PIT on MC38 tumors in Ifngr1flox/flox or Tie2CreIfngr1flox/flox mice. Representative images (D, Bar: 250 μm) and quantitation summary (E) (n = 3, each). (F and G) Immunohistochemistry of pimonidazole and CD31 in pimonidazole-binding assay, 18 hour after anti-CD25-F(ab′)2 NIR-PIT on MC38 tumors in Ifngr1flox/flox or Tie2CreIfngr1flox/flox mice. Representative images (F, Bar: 500 μm) and quantitation summary (G). (n = 3, each).
**: p < 0.01.
CD8 T and NK cells induce IFN-γ/STAT1 signaling around tumor vessels and in endothelial cells.
It is intriguing how a small number of intratumoral CD8 T and NK cells can induce tumor vessel regression in such an effective manner (Figures 3A–C). Interestingly, CD8 T and NK cells and Tregs were found, histologically, near tumor vessels in both MC38 and EO771 tumors (Figures 6A and B). Thus, we next hypothesized that intratumoral Treg depletion could induce IFN-γ-mediated STAT1 signaling in cells around tumor vessels. Without treatment, only weak phosphorylation of STAT1 Tyr701, the main residue phosphorylated by IFN-γ (26), was observed in scattered areas in MC38 tumors (Figure 6C). Upon intratumoral Treg depletion, cells nearby vessels and endothelial cells readily showed STAT1 Tyr701 phosphorylation (Figures 6C and D). These findings suggest that IFN-γ produced by CD8 T and NK cells around tumor vessels effectively targeted endothelial cells.
Figure 6. Depletion of intratumoral Tregs causes IFN-γ/phospho-STAT1 signaling around tumor vessels.

(A and B) Perivascular localization of CD8+ T cells, NKp46+ NK cells, and CD4+Foxp3+ Tregs analyzed by immunohistochemistry in untreated MC38 tumors. Representative images (A, Bar: 100 μm) and frequency of indicated lymphocytes located in indicated distances from tumor vessels (B) (n = 3, each). (C) Increased phospho-STAT1 (Tyr701) positivity around tumor vessels analyzed by immunohistochemistry 60 minutes after anti-CD25-F(ab′)2 NIR-PIT (n = 3, each, representative images). For negative control, frozen sections were treated with lambda protein phosphatase (λ-PP) before the phospho-STAT1 staining. Inset: phospho-STAT1 (Tyr701) positivity CD31+ endothelial cells. Bar: 500 μm. (D) Percentage of phospho-STAT1-positive areas analyzed by distance from tumor vessels (Top) and phospho-STAT1-positive tumor vessels (Bottom) in (C) (n = 3, each).
*: p < 0.05, **: p < 0.01.
Increased complete eradication is achieved by anti-CD25-F(ab′)2 NIR-PIT combined with rhIL-15 therapy, not PD-1 blockade.
Finally, we aimed to boost the anti-tumor effect of CD8 T and NK cells to eradicate established tumors by combining anti-CD25-F(ab′)2 NIR-PIT with another treatment. A combination with anti-PD-1 antibody therapy failed to enhance effector activity of CD8 T and NK cells after intratumoral Treg depletion, showing only marginal additional tumor control (Figures S12A and B). PD-L1 expression levels on tumor cells, monocytes, macrophages, and dendritic cells in MC38 tumors did not change after intratumoral Treg depletion (Figure S12C).
We then evaluated another combination with recombinant human IL-15 (rhIL-15) treatment (Figure 7A). This combination therapy significantly decreased tumor volume, leading to complete regression of some MC38 and EO771 tumors (Figure 7B) and significantly improved survival (Figure S13A). Anti-CD25-F(ab′)2 NIR-PIT and rhIL-15 treatment synergistically increased the frequency of tumor-infiltrating CD8 T and NK cells, and their granzyme B expression was increased by rhIL-15 (Figures 7C–E). However, expression of IFN-γ and perforin did not increase. Histological analysis revealed that rhIL-15 treatment alone increased CD8 T cell infiltration to the periphery of the tumor but not to the center, which did not change by the combination therapy (Figures 7F and G). By contrast, anti-CD25-F(ab′)2 NIR-PIT preferentially targeted more centrally located tumor cells through induction of ischemic necrosis/apoptosis (Figure 5). The combination of anti-CD25-F(ab′)2 NIR-PIT and rhIL-15 therapy, therefore, targeted both the center and periphery of the tumor, while increasing the cytotoxic activity of lymphocytes by rhIL-15. This likely explains the increased tumor control and complete tumor eradication.
Figure 7. Combination of rhIL-15 treatment with anti-CD25-F(ab’)2 NIR-PIT synergize to increase complete eradication of tumor.
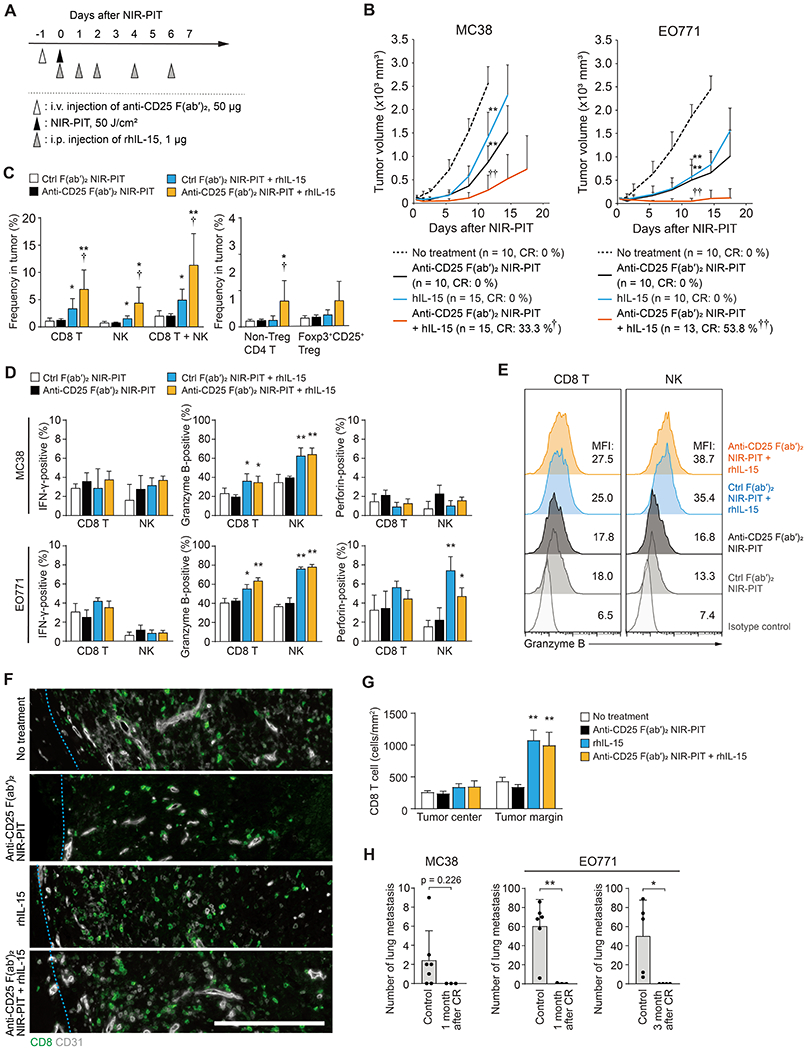
(A) Regimen of anti-CD25-F(ab′)2 NIR-PIT and rhIL-15 combination therapy. (B) Anti-CD25-F(ab′)2 NIR-PIT and rhIL-15 synergistically impaired growth of MC38 and EO771 tumors. Sample size and complete regression (CR) rate in each group are indicated. χ2 test was used to calculate p-values of CR ratio. *: compared to no treatment group, †: compared to other three groups. See Supplemental Figure 13A for the survival curves. (C) Frequency of indicated lymphocyte subsets 5 days after indicated treatments on MC38 tumor, analyzed by flow cytometry (n ≥ 5, each). *: compared to no treatment group and anti-CD25-F(ab′)2 NIR-PIT group, †: compared to rhIL-15 group. (D) Positivity of IFN-γ, granzyme B, and perforin in CD3+CD8+ T cells or CD45+NK1.1+ NK cells, analyzed by flow cytometry 3 days after indicated treatments on MC38 or EO771 tumor (Day 3, n ≥ 5, each) *: compared to no treatment group and anti-CD25-F(ab′)2 NIR-PIT group, †: compared to rhIL-15 group. (E) Granzyme B expression in CD3+CD8+ T cells or CD45+CD3-NK1.1+ NK cells analyzed by flow cytometry 3 days after indicated treatments on MC38 tumor (n ≥ 5). (F and G) rhIL-15 treatment induced infiltration of CD8 T cells to the periphery of EO771 tumors (Day 3). Representative immunohistochemistry (F, Bar: 250 μm) and Quantitation summary (G). (n = 3, each). (H) Prevention of lung metastasis formation after re-challenge by intravenous injection of 1×106 MC38 or EO771 tumor cells at 1 month or 3 months of complete regression by anti-CD25-F(ab′)2 NIR-PIT and rhIL-15 combination therapy (n ≥ 3, each).
* and †: p < 0.05, ** and ††: p < 0.01.
Trans-presentation of IL-15/IL-15Rα complex by myeloid and epithelial cells to lymphocytes is important for IL-15 efficacy (27,28). We analyzed changes in IL-15Rα expression in tumor and immune cells and found increased IL-15Rα expression in Ly6Chigh inflammatory-monocytes after anti-CD25-F(ab′)2 NIR-PIT (Figures S13B and C). However, since the frequency of Ly6Chigh inflammatory-monocytes decreased after the NIR-PIT (Figure S1F), the contribution of these cells in trans-presenting IL-15 may be limited.
After complete remission of local tumor, systemic antitumor immunity lasted for at least several months and prevented the formation of lung metastasis when the mice were re-challenged with intravenous administration of the same tumors (Figure 7H).
Discussion
Although the pivotal roles of Tregs to inhibit anti-tumor immunity is well-established (6–9), the specific roles that intra- and extra-tumor Tregs play remain unclear due to technical difficulties in selectively depleting or manipulating Tregs in a spatially specific manner. In the current study, we took advantage of a highly selective method of depleting cells from the tumor microenvironment, NIR-PIT (11–13), which enabled us to observe the effects of selective removal of intratumoral Tregs without employing systemic depletion strategies.
Our previous study of anti-CD25-F(ab′)2 NIR-PIT demonstrated that depletion of intratumoral Tregs caused tumor regression in a manner dependent on IFN-γ-production from CD8 T and NK cells (13). IFN-γ can exhibit anti-tumor effects directly on tumor cells and also indirectly through T cells, macrophages, and stromal cells, including tumor vessels, depending on the context of IFN-γ production (26). Previous investigations have shown that doxycycline-mediated induction of IFN-γ in tumor cells directly acts on endothelial cells to cause rapid tumor vessel regression and intratumoral ischemia (19,29), however, magnitude of IFN-γ induced from all tumor cells would be significantly higher than that induced upon intratumoral Treg depletion. Therefore, the mechanism by which IFN-γ causes tumor regression after intratumoral Treg depletion remained to be elucidated.
In this study, we utilized anti-CD25-F(ab′)2 NIR-PIT (13) and various Ifngr1 knockout models and identified that IFN-γ produced by tumor-infiltrating CD8 T and NK cells upon intratumoral Treg depletion directly acted on endothelial cells through IFN-γR1, inducing acute vascular regression and intratumoral ischemia, leading to impaired tumor growth. It was surprising how effectively the physiological, short-lasting (up to 6 hours) production of IFN-γ from limited numbers of tumor-infiltrating CD8 T and NK cells after intratumoral Treg depletion induced these changes. Because of the perivascular distribution of CD8 T and NK cells, IFN-γ/phospho-STAT1 signaling was preferentially induced around tumor vessels and in the endothelial cells, suggesting effective IFN-γ targeting of vascular endothelial cells. As a source for IFN-γ, CD8 T and NK cells may equally contribute, since they show similar IFN-γ production kinetics upon intratumoral Treg depletion and similar perivascular distribution patterns. This seems consistent with our previous finding showing similar contributions by CD8 T and NK cells to the anti-tumor effect of anti-CD25-F(ab′)2 NIR-PIT (13). In addition to IFN-γ induction, simultaneous deprivation of Treg-derived VEGF (30) upon intratumoral Treg depletion may also have additional effects on the regression of tumor vasculature and following vessel recovery.
Interestingly, intratumoral Treg depletion alone did not increase the number and effector activity of tumor-infiltrating CD8 T and NK cells, unlike systemic Treg depletion in Foxp3DTR mouse model and by administration Treg-depleting antibodies (6–9). IFN-γ can also modulate tumor vasculature by polarizing vasculature-supporting M2 macrophages into pro-inflammatory M1 macrophages, and by inducing CXCL9/10 in tumors (9, 26). However, we neither observed macrophage polarization into M1 phenotype nor induction of CXCL9/10 after intratumoral Treg depletion, which may be due to the limited amount and duration of IFN-γ production and developing ischemia upon intratumoral Treg depletion.
There is an interesting relationship between IFN-γ-dependent vessel regression observed in our study and tumor vessel normalization, which is also reported to be IFN-γ-dependent (20,22,23) but was only partially observed in this study. Tumor vessel normalization is observed during pruning and reorganization after some therapies and is associated with improved tumor oxygenation and lymphocyte infiltration into tumor (20). Studies have shown that IFN-γ expressed from CD4 and CD8 T cells by immune checkpoint inhibitors (22,23) induces vessel normalization. On the other hand, doxycycline-mediated IFN-γ induction in tumor cells causes acute regression of tumor vessels as discussed above (19,29). The question of whether IFN-γ can cause both tumor vessel regression and normalization may be reconciled by considering differences in the amount and duration of IFN-γ expressed in tumors (31); High levels of IFN-γ expression (19,29) or short-lasting but synchronized expression of IFN-γ, as observed in this study, may preferentially cause rapid vessel regression, while low level but long-lasting expression of IFN-γ from tumor-infiltrating CD4 and CD8 T cells induced by immune checkpoint inhibitors (22,23) may shift towards tumor vessel normalization.
Although development of intratumoral ischemia after intratumoral Treg depletion could control tumor growth, it failed to achieve complete regression. Re-growth of the tumors observed is likely due to the relative resistance to ischemia at the periphery of tumors and to the limited increase in number and cytotoxic activity of CD8 T and NK cells after intratumoral Treg depletion. In order to augment therapeutic efficacy, we added rhIL-15 administrations (1 μg x 5 doses) to anti-CD25-F(ab′)2 NIR-PIT (28,32). IL-15 was chosen over IL-2 because, while IL-15 preferentially expand and activate cytotoxic lymphocytes, IL-2 also expand Tregs systemically (27), which can accelerate recovery of intratumoral Tregs after their depletion. Consequently, rhIL-15 combination achieved increased complete eradication of MC38 and EO771 tumors. While depletion of intratumoral Tregs preferentially targets the center of tumors by inducing tumor vessel regression and intratumoral ischemia, rhIL-15 treatment increased infiltration of cytotoxic lymphocytes at the periphery of tumors and their cytotoxic action, eradicating residual tumor cells. It is also intriguing that the combination of IL-15 may enhance and prolong anti-tumor memory T cell development and survival (27). We observed strong anti-tumor memory at 3 months after achieving complete tumor regression by the combination of IL-15 therapy, as shown by the rejection of a tumor re-challenge.
There are some limitations in this study. First, anti-CD25-F(ab′)2 used in this study is derived from clone PC-61.5.3 that blocks IL-2 binding to CD25 and can potentially inhibit the function of IL-2 produced in initial activation of lymphocytes after intratumoral Treg depletion (33). Thus, we plan to use an anti-CD25-F(ab′)2 derived from a clone that has minimal effects on IL-2 activity (34). It would also be prudent to use a non-IL-2 blocking antibody when we clinically translate anti-CD25-F(ab′)2 NIR-PIT intertumoral Treg depletion therapy in the future, possibly combined with tumor cell-targeting NIR-PIT.
Another limitation is that we could not perform experiments using human tumor samples because of technical difficulties in using human tumors for selective intratumoral Treg depletion, requiring further studies in the future. The structure of tumor vessels vary more significantly in human tumors compared to mouse experimental models (35) and these vessels may exhibit different sensitivities to IFN-γ. Also intriguingly, some types of human tumors are more heavily infiltrated by cytotoxic lymphocytes compared to murine tumors (36–39). The results of current study suggest that these lymphocyte-rich human tumors would be more sensitive to intratumoral Treg depletion by anti-CD25-F(ab′)2 NIR-PIT than murine tumors.
In summary, we demonstrated that IFN-γ induced in CD8 T and NK cells upon selective depletion of intratumoral Tregs directly targeted endothelial cells to cause rapid vessel regression, intratumoral ischemia, and necrosis/apoptosis of tumor cells. Combination with rhIL-15 treatment further increased complete tumor eradication. These data shed light on the role that Tregs play in the tumor microenvironment and help advance clinical translation of anti-Treg cancer therapies such as anti-CD25-F(ab′)2 NIR-PIT.
Supplementary Material
Statement of significance.
Intratumoral Treg depletion induces synchronized intratumoral CD8 T and NK cell activation, IFN-γ-dependent tumor vessel regression, and ischemic tumor necrosis/apoptosis, indicating the roles of intratumoral Tregs to support the tumor vasculature.
Acknowledgements
We thank Mr. Kunio Nagashima for great assistance for electron microscopy studies and Ms. Erina He for her assistance in the creation of the graphical abstract.
Footnotes
Conflict of interest statement
P.L.C., H.K., and N.S. have filed U.S. national and international patent applications for near infrared photoimmunotherapy of suppressor cells to treat cancer used in this study.
Regarding C.O.: This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E and 75N910D00024. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
References
- 1.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol 2008;8:523–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burzyn D, Benoist C, Mathis D. Regulatory T cells in nonlymphoid tissues. Nature immunology 2013;14:1007–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Speiser DE, Ho PC, Verdeil G. Regulatory circuits of T cell function in cancer. Nat Rev Immunol 2016;16:599–611 [DOI] [PubMed] [Google Scholar]
- 4.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nature medicine 2004;10:942–9 [DOI] [PubMed] [Google Scholar]
- 5.Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nature reviews Cancer 2012;12:298–306 [DOI] [PubMed] [Google Scholar]
- 6.Quezada SA, Peggs KS, Simpson TR, Shen Y, Littman DR, Allison JP. Limited tumor infiltration by activated T effector cells restricts the therapeutic activity of regulatory T cell depletion against established melanoma. J Exp Med 2008;205:2125–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bos PD, Plitas G, Rudra D, Lee SY, Rudensky AY. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J Exp Med 2013;210:2435–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, et al. Fc-Optimized Anti-CD25 Depletes Tumor-Infiltrating Regulatory T Cells and Synergizes with PD-1 Blockade to Eradicate Established Tumors. Immunity 2017;46:577–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carretero R, Sektioglu IM, Garbi N, Salgado OC, Beckhove P, Hammerling GJ. Eosinophils orchestrate cancer rejection by normalizing tumor vessels and enhancing infiltration of CD8+ T cells. Nat Immunol 2015;16:609–17 [DOI] [PubMed] [Google Scholar]
- 10.Sawant DV, Yano H, Chikina M, Zhang Q, Liao M, Liu C, et al. Adaptive plasticity of IL-10+ and IL-35+ Treg cells cooperatively promotes tumor T cell exhaustion. Nat Immunol 2019;20:724–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kobayashi H, Choyke PL. Near-Infrared Photoimmunotherapy of Cancer. Acc Chem Res 2019;52:2332–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitsunaga M, Ogawa M, Kosaka N, Rosenblum LT, Choyke PL, Kobayashi H. Cancer cell-selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nature medicine 2011;17:1685–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato K, Sato N, Xu B, Nakamura Y, Nagaya T, Choyke PL, et al. Spatially selective depletion of tumor-associated regulatory T cells with near-infrared photoimmunotherapy. Sci Transl Med 2016;8:352ra110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jang JE, Hajdu CH, Liot C, Miller G, Dustin ML, Bar-Sagi D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep 2017;20:558–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rajantie I, Ilmonen M, Alminaite A, Ozerdem U, Alitalo K, Salven P. Adult bone marrow-derived cells recruited during angiogenesis comprise precursors for periendothelial vascular mural cells. Blood 2004;104:2084–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song S, Ewald AJ, Stallcup W, Werb Z, Bergers G. PDGFRβ+ perivascular progenitor cells in tumours regulate pericyte differentiation and vascular survival. Nat Cell Biol 2005;7:870–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raz Y, Cohen N, Shani O, Bell RE, Novitskiy SV, Abramovitz L, et al. Bone marrow-derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J Exp Med 2018;215:3075–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Purhonen S, Palm J, Rossi D, Kaskenpaa N, Rajantie I, Yla-Herttuala S, et al. Bone marrow-derived circulating endothelial precursors do not contribute to vascular endothelium and are not needed for tumor growth. Proc Natl Acad Sci U S A 2008;105:6620–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kammertoens T, Friese C, Arina A, Idel C, Briesemeister D, Rothe M, et al. Tumour ischaemia by interferon-γ resembles physiological blood vessel regression. Nature 2017;545:98–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer cell 2014;26:605–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer 2017;17:457–74 [DOI] [PubMed] [Google Scholar]
- 22.Tian L, Goldstein A, Wang H, Ching Lo H, Sun Kim I, Welte T, et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 2017;544:250–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng X, Fang Z, Liu X, Deng S, Zhou P, Wang X, et al. Increased vessel perfusion predicts the efficacy of immune checkpoint blockade. The Journal of clinical investigation 2018;128:2104–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nature medicine 2014;20:607–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang Y, Harrington A, Yang X, Friesel RE, Liaw L. The contribution of the Tie2+ lineage to primitive and definitive hematopoietic cells. Genesis 2010;48:563–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ivashkiv LB. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol 2018;18:545–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol 2006;6:595–601 [DOI] [PubMed] [Google Scholar]
- 28.Jabri B, Abadie V. IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat Rev Immunol 2015;15:771–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Briesemeister D, Sommermeyer D, Loddenkemper C, Loew R, Uckert W, Blankenstein T, et al. Tumor rejection by local interferon gamma induction in established tumors is associated with blood vessel destruction and necrosis. Int J Cancer 2011;128:371–8 [DOI] [PubMed] [Google Scholar]
- 30.Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011;475:226–30 [DOI] [PubMed] [Google Scholar]
- 31.De Palma M, Jain RK. CD4+ T Cell Activation and Vascular Normalization: Two Sides of the Same Coin? Immunity 2017;46:773–5 [DOI] [PubMed] [Google Scholar]
- 32.Waldmann TA, Miljkovic MD, Conlon KC. Interleukin-15 (dys)regulation of lymphoid homeostasis: Implications for therapy of autoimmunity and cancer. J Exp Med 2020;217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lowenthal JW, Corthesy P, Tougne C, Lees R, MacDonald HR, Nabholz M. High and low affinity IL 2 receptors: analysis by IL 2 dissociation rate and reactivity with monoclonal anti-receptor antibody PC61. J Immunol 1985;135:3988–94 [PubMed] [Google Scholar]
- 34.Ortega G, Robb RJ, Shevach EM, Malek TR. The murine IL 2 receptor. I. Monoclonal antibodies that define distinct functional epitopes on activated T cells and react with activated B cells. J Immunol 1984;133:1970–5 [PubMed] [Google Scholar]
- 35.Nagy JA, Chang SH, Shih SC, Dvorak AM, Dvorak HF. Heterogeneity of the tumor vasculature. Semin Thromb Hemost 2010;36:321–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kurebayashi Y, Ojima H, Tsujikawa H, Kubota N, Maehara J, Abe Y, et al. Landscape of immune microenvironment in hepatocellular carcinoma and its additional impact on histological and molecular classification. Hepatology 2018;68:1025–41 [DOI] [PubMed] [Google Scholar]
- 37.Kurebayashi Y, Kubota N, Sakamoto M. Immune microenvironment of hepatocellular carcinoma, intrahepatic cholangiocarcinoma and liver metastasis of colorectal adenocarcinoma: Relationship with histopathological and molecular classifications. Hepatol Res 2021;51:5–18. [DOI] [PubMed] [Google Scholar]
- 38.Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov 2019;18:197–218 [DOI] [PubMed] [Google Scholar]
- 39.Galon J, Angell HK, Bedognetti D, Marincola FM. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity 2013;39:11–26 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.