

Abstract
Immune checkpoint blockade (ICB) has improved cancer care, but ICB is only effective in some patients. The molecular mechanisms that influence ICB therapy response are not completely understood. The non-classical major histocompatibility complex (MHC) class I molecule HLA-E and its mouse ortholog, Qa-1b, present a limited set of peptides in a TAP1-dependent manner to the NKG2A/CD94 heterodimer to transduce an inhibitory signal to natural killer (NK) and CD8+ T cells. However, deficiency of TAP1 allows Qa-1b to present an alternative peptidome to Qa-1b-restricted T cell receptors of cytotoxic T cells. In this study, we used CRISPR-Cas9 to study the relationship between TAP1, Qa-1b, and response to anti-PD1 therapy. We hypothesized that immunotherapy response in TAP1 deficient tumors would be influenced by Qa-1b. Strikingly, using a syngeneic orthotopic mouse model, we found that while TAP1 deficient tumors were resistant to anti-PD1 treatment, anti-PD1 response was significantly enhanced in tumors lacking both TAP1 and Qa-1b. This increased sensitivity is partially dependent on NK cells. TAP1-deficient tumors were associated with an increase of intratumoral regulatory T cells (Treg) and neutrophils, while tumors lacking both TAP1 and Qa-1b exhibited an increased CD8+ T cell to Treg ratio. These data suggest that direct inhibition of Qa-1b may alter the immune microenvironment to reverse resistance to anti-PD1 therapy, particularly in the context of antigen processing defects.
Keywords: HLA-E, Qa-1b, TAP1, regulatory T cells, melanoma, immunotherapy, checkpoint blockade, CRISPR-Cas9, NK depletion
Introduction
In adaptive anti-tumor immunity, T cell receptors (TCR) recognize and bind to major histocompatibility complex (MHC) – antigen peptide complexes to generate a primary activation signal in T cells[1]. In intact immune signaling, the non-classical MHCI molecule Qa-1b, the mouse ortholog of human HLA-E, is generally thought to present a very small number of highly conserved peptides. The most commonly presented of these peptides is known as the Qa-1 determinant modifier (Qdm) and is derived from the leader sequence of classical MHCI. The Qa-1b/Qdm complex transduces a regulatory signal through the NKG2/CD94 heterodimeric receptor found on the cell surface of NK cells and a subset of T lymphocytes[2]. It is well established that Qa-1b/Qdm signals predominantly through the NKG2A/CD94 complex. Ligation of Qa-1b/Qdm to NKG2A/CD94 induces phosphorylation of the intracellular immunoreceptor tyrosine-based inhibitory motif (ITIM) domain of the NKG2A molecule, thereby inhibiting NK cell or T cell function. Qa-1b/Qdm can also bind NKG2C/CD94, albeit with lower affinity, to transmit an activation signal. Proper expression of Qdm is required to maintain immune tolerance to self-tissue[3]. Under some circumstances, including viral infection and tumor-related antigen processing defects, Qa-1b/Qdm:NKG2/CD94 signaling is abrogated.[4] In such situations, an alternative set of neopeptides binds Qa-1b in a TAP1-independent manner and is presented to CD8+ T cells by Qa-1b [4–6]. This alternative Qa-1b-presented peptidome can trigger cytotoxic T cell activation and induce immune elimination of infected cells or tumor cells [7]. Previous studies showed that TAP1 expression and function is essential for Qdm generation and proper loading onto the Qa-1b molecule [8]. TAP1 deficiency results in instability of MHC-peptide complexes, causing both classical and non-classical MHCs to be digested via endocytosis [9].
Complete and persistent activation of CD8+ cytotoxic T cells requires a secondary activation signal via binding of the co-stimulatory factors CD80 (also known as B7-1) and CD86 (also known as B7-2) on professional antigen presenting cells to the co-stimulatory receptor CD28 on the surface of T cells. Following activation, T cells upregulate co-inhibitory receptors such as cytotoxic T lymphocyte antigen 4 (CTLA-4) and programmed cell death 1 (PD1). CTLA-4 binds to CD80 and CD86 with high affinity and competitively inhibits T cell activation by removing the secondary activation signal. Tumor and stromal cells also upregulate PD-1 ligand (PD-L1), which induces apoptosis of T cells and inhibits T cell functioning through interaction with PD1. Therefore, use of anti-CTLA-4 and anti-PD-1 antibodies prevents these co-inhibitory ligands from impairing T cell function[1].
While the general mechanism of ICB on lymphocytes is relatively clear, only a small proportion of patients benefit from ICB treatment. Factors such as PD-L1 levels, mutation burden and mutation signatures can impact sensitivity and outcome of ICB treatment[1]. Some specific genes are believed to be predictive factors. Previous studies have shown that inhibition of NKG2A or deletion of TAP1 or Qa-1b can influence anti-tumor immunity[10–13].
In this study, we examined the effects of the TAP1-independent Qa-1b-presented alternative peptidome on antitumor immunity in TAP1-impaired tumors. One possible outcome was that tumors lacking both TAP1 and Qa-1b would respond poorly to anti-PD1 therapy due to a lack of both classical and non-classical MHC peptide presentation. Surprisingly, we found that anti-PD1-mediated tumor killing was greatest in the double knock-out (KO) tumors, which could be partially dampened by NK cell depletion. TAP1 KO was associated with an increase of infiltrated CD4+ Tregs, while the double KO was associated with a marked increase in CD8+ cytotoxic T lymphocyte (CTL) to CD4+ Treg ratio. These results suggest that TAP1-independent Qa-1b function supports the accumulation of regulatory T cells, thereby impairing response to checkpoint blockade therapy. These data reveal important functional crosstalk between classical TAP-dependent MHC complexes and Qa-1b/HLAE, particularly in tumors with impaired antigen processing machinery, that can dramatically influence immunotherapy efficacy.
Materials and methods
Antibodies
We used antibodies for flow cytometry and other immunologic assays as listed in Table 1. All antibodies were purchased from the manufacturers as listed.
Table 1.
Antibodies used for flow cytometry staining.
| Target antigen | Supplier | Clone | Catalog number |
|---|---|---|---|
| Ly6G | Biolegend | 1A8 | 127643 |
| Ly6C | Biolegend | HK1.4 | 128043 |
| IA/IE | Biolegend | M5/114.15.2 | 107622 |
| PD-L1 | Biolegend | 10F.9G2 | 124312 |
| CD11b | Biolegend | M1/70 | 101225 |
| CD11c | Biolegend | N418 | 117318 |
| CSF1R | Biolegend | AFS98 | 135513 |
| CD45 | BD | 30-F11 | 565079 |
| F4/80 | BD | T45-2342 | 565614 |
| CD103 | BD | M290 | 741739 |
| Ki67 | eBioscience | SolA15 | 56-5698-82 |
| PD-1 | Biolegend | 29F.1A12 | 135221 |
| Foxp3 | Invitrogen | FJK-16s | 11-5773-82 |
| CD3 | Biolegend | 145-2C11 | 100351 |
| IFNγ | Biolegend | XMG1.2 | 505821 |
| Perforin | Biolegend | S16009A | 154304 |
| Granzyme B | Biolegend | QA16A02 | 372210 |
| CD25 | BD | PC61 | 564022 |
| CD4 | BD | GK1.5 | 564298 |
| CD8 | Biolegend | 53–6.7 | 100762 |
| NK1.1 | Biolegend | PK136 | 108713 |
| Tigit | BD | R19-760 | 744217 |
| H2-Kb | eBioscience | AF6-88.5.5.3 | 11-5958-82 |
| H2-Db | eBioscience | 28-14-8 | 17-5999-82 |
| Qa-1b | BD | 6A8.6F10.1A6 | 566640 |
| CD16/32 | Biolegend | 93 | 101320 |
| NK1.1 | Biolegend | PK136 | 108708 |
| CD45 | Biolegend | 30-F11 | 103108 |
| CD3 | Biolegend | 145-2C11 | 100312 |
Qa-1b and TAP1 knockout in B16F10 cells
pSpCas9(BB)-2A-GFP (PX458) plasmid was a gift from F. Zhang (Addgene plasmid #48138)[14]. Guide RNA sequences (5’-GGCTATGTCATTCGCGGTCC-3’) targeting mouse H2-T23 gene exon 3 and (5’-CTAGGATGGCCCAGCGACTC-3’) targeting mouse TAP1 gene exon 1 were cloned into PX458 plasmid respectively. The plasmids were then amplified using One Shot™ Stbl3™ (Thermo Fisher #C737303) and were validated by Sanger Sequencing at Genewiz. Parental B16F10 mouse melanoma cells (ATCC® CRL-6475™) were sorted one cell per well into a 96-well plate and cultured to generate B16F10 parental single cell clones. The plasmids were then transfected into two B16F10 parental single cell clones using GenJet™ In Vitro DNA Transfection Reagent for B16-F10 Cells (SignaGen® SL100489-B16). 48 hours after transfection, GFP+ cells were sorted by the Flow Cytometry Core Facility (FCCF) at Memorial Sloan Kettering Cancer Center (MSKCC) into 15ml conical tubes and were cultured in DMEM+10%FBS+1%Pen/Strep culture media. The cells were cultured in culture medium with mouse IFNγ (100 IU/ml) for 48 hours. Qa-1b knockout cells were gated as PE-Qa-1b negative cells. TAP1 knockout cells were gated as APC-H2-Db reduced cells. The sorted cells were seeded at one cell per well into 96 well plates. DNA of the single cell clones was extracted using DNeasy Blood & Tissue Kit (QIAGEN #69506). Primers F(5’-GCCTACGATGGCCAGGATT-3’)-R(5’-GATTTCCCCCAAACCGCAGA-3’) targeting the Qa-1b gene editing region and primers F(5’-GGTTCCCGGGACTTTACGC-3’)-R(5’-CAGCTCTCGGAACAAGGCAA-3’) targeting the TAP1 gene editing region were used in PCR cloning. QIAquick Gel Extraction Kit (QIAGEN #28706) was used to purify the PCR product and samples were sent to Genewiz for next generation sequencing (amplicon seq). PX458 plasmid carrying the TAP1 guide oligo was transfected into confirmed Qa-1b knockout single cell clones. The same sorting and validation strategy was followed to generate TAP1/Qa-1b double knockout clones. Cell proliferation assay was performed on the validated single cell clones using CellTiter-Glo® 2.0 (Promega #G9241). Clones with similar growth rates were used in subsequent mouse experiments. Cells of passage number five or lower were used for all experiments and were not routinely tested for mycoplasma. For each mouse experiment, the set of cell lines used originated from the same B16F10 parental clone. A second set of cell lines, originating from a different independent B16F10 parental clone, was used for validation.
Animal studies
Animal studies were conducted in compliance with a protocol approved by MSKCC’s IACUC committee and Cleveland Clinic’s IACUC committee. Female wild-type C57BL/6J mouse were purchased from Jackson Laboratory. Validated and selected cell lines were thawed, expanded and injected simultaneously into mice in the same experiment to prevent batch effects. 0.2 million cells in 100 ul PBS were injected into left flanks of each 7 to 8-week-old mice. Mice were randomized every 3 days since day 10 after injection once the tumor size reached about 50 mm3 (5 mm in diameter). Tumor sizes were calculated by the formula: (length*width*width)/2.
For NK cell depletion, 200 ug of monoclonal anti-NK1.1 antibody (Clone PK136, Bio X Cell) were injected i.p. on days −1 and 1 relative to tumor injection, and then twice a week until euthanization. Mouse IgG2a isotype control (Bio X Cell) was used in the control groups.
Treatment with immune checkpoint blockade was initiated when mice were randomized. 100 ug of anti-PD-1 antibody (Clone RMP1-14, Bio X Cell) were injected i.p. twice a week for two weeks. Rat IgG2a isotype control (Bio X cell) was injected in the control groups. All mice were euthanized after the two-week treatment.
Flow cytometry analysis
B16F10 parental, Qa-1b knockout, TAP1 Qa-1b double knockout, and TAP1 knockout cells were cultured in medium with IFNγ (100 IU/ml) for 48 hours to induce murine MHC class I surface expression. The cells were then washed with PBS+1% FBS and stained with anti-murine Qa-1b, H2-Kb, H2-Db antibodies and DAPI.
Mice between day 7 and day 10 post-randomization were examined. Fresh tumors and control mouse spleens were dissociated into single cell suspensions through 100um and 70um strainers consecutively. Cell suspension was washed with PBS 3 times. Red blood cell lysis buffer (MACS #130-094-183) was used following the protocol of the supplier. Cells were then washed twice with PBS, stained with LIVE/DEAD® Fixable Aqua Dead Cell Stain Kit (Thermo Fisher #L34957) at 4°C in the dark for 30 minutes. We then incubated cells with anti-mouse CD16/32 antibody,1ul per million cells, in 100ul of PBS+1%FBS at 4°C in the dark for 10 minutes. Cells were divided into two parts for different flow panels, then washed twice with PBS+1% FBS before surface staining with different antibody cocktails respectively at 4°C in the dark for 30 minutes. Cells were washed twice with PBS+1% FBS after surface staining. For myeloid panel, cells were fixed by incubation in BD Cytofix™ Fixation Buffer (BD 554655) at 4°C for 20 minutes. For T/NK panel, cells were fixed by incubation in BD Cytofix/Cytoperm™ Fixation/Permeabilization solution (BD 554722), followed by washing with 1X BD Perm/Wash™ Buffer (BD 554723). Then cells were stained with intracellular antibody cocktail at 4°C in the dark for 30 minutes. Samples were then analyzed using the LSR Fortessa (BD) flow cytometric analyzer. Data files were analyzed using FCS Express 7 (De Novo Software).
For validation of NK cell depletion, mice were euthanized and splenocytes were collected on days 1, 7, 11 and 25 relative to tumor injection. Cell suspensions were washed and stained according to the same procedures described above.
Histology
Mice were euthanized between day 10 and day 14 post randomization. Tumors were resected and fixed in 10% neutral buffered formalin (Sigma HT501128) for 24 hours at room temperature, and then washed with 70% ethanol for three times and stored in 70% ethanol at 4°C in the dark. Tumor tissues were embedded in paraffin by the molecular Cytology Core Facility (MCCF) at Memorial Sloan Kettering Cancer Center. Samples were sectioned at 4μm. Subsequent immunohistochemistry was performed by Histowiz Inc. on a Bond Rx autostainer (Leica Biosystems) with enzyme treatment (1:1000) using standard protocols. Antibodies used were rat monoclonal CD45, CD3, CD4, CD8, foxp3 primary antibody (provided by Histowiz) and rabbit anti-rat secondary (Vector, 1:100). Bond Polymer Refine Detection (Leica Biosystems) was used according to manufacturer’s protocol. After staining, sections were dehydrated and film coverslipped using a TissueTek-Prisma and Coverslipper (Sakura). Whole slide scanning (40x) was performed on an Aperio AT2 (Leica Biosystems).
Statistical analysis
Cell proliferation assay data were presented as mean ± standard deviation (SD), and comparisons were performed using two-way ANOVA. All mouse and flow cytometry data were presented as mean ± SEM unless otherwise specified. Tumor volumes were compared between two treatment groups of the same cell line injected using two-way ANOVA. Gated cell population from flow data was compared using one-way ANOVA and Tukey’s post-hoc test. P < 0.05 was considered to be statistically significant (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Results
Absence of TAP1 abrogates surface expression of classical MHCI but not Qa-1b
We designed CRISPR guide RNAs targeting exon 1 of the mouse TAP1 gene and exon 3 of the mouse H2-T23 gene which encodes Qa-1b. A clonal population of B16F10 cells was transfected with Cas9 and either guide RNA. Qa-1b-negative cells were selected for use for single cell sorting. An H2-Db_low gate was used to select TAP1 deficient cells for single cell sorting. H2-Db is a surface murine classical MHCI molecule whose stable surface expression is dependent on TAP1. Single cell clones were expanded and further confirmed by next generation sequencing-based amplicon sequencing (Fig. S1). Cell lines with frameshifts were selected and validated by flow cytometry. We found that the TAP1 deficient cells expressed very low levels of H2-Kb and H2-Db molecules (both are mouse classical MHCIa molecules), but retained intermediate levels of Qa-1b expression, whereas the Qa-1b deficient cells showed only a slight reduction of H2-Kb and H2-Db surface expression (Fig. 1A, B). Based on the literature, we presume that Qdm is expressed in wild-type (WT) B16F10 cells [13, 15]. In the parental lines, Qa-1b-Qdm presentation and classical MHC Ia-peptide presentation are intact, whereas the Qa-1b-alternative peptidome is predicted not to be employed. In Qa-1b KO cells, all Qa-1b mediated peptide presentation is abolished, while classical MHC Ia-peptide presentation remains intact [7]. In TAP KO cells, both Qdm loading and classical MHC Ia-peptide presentation are inhibited, while presentation of the alternative Qa-1b-peptidome to Qa-1b-restricted CD8 T cells is activated [7]. In double KO cells, both Qa-1b mediated and classical MHC Ia mediated antigen peptide presentation are abolished. By comparing (i) parental versus Qa-1b KO and (ii) TAP1 KO versus TAP1/Qa-1b double KO, we can dissect the impact of Qa-1b-Qdm and the alternative Qa-1b peptidome, respectively, on anti-PD1 therapeutic response.
Figure 1. Validation of clonal CRISPR knock-out (KO) cell lines.
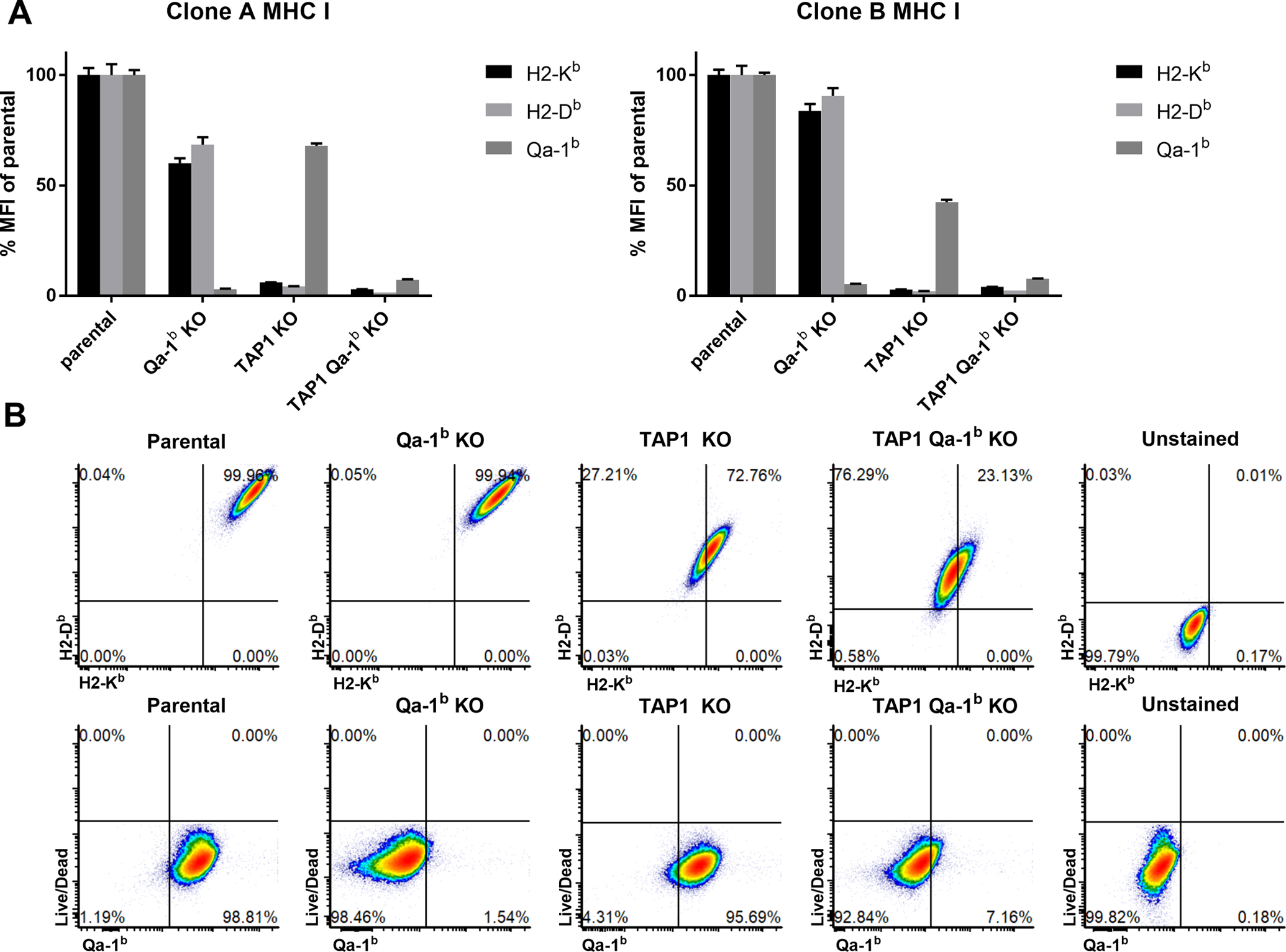
Single cell clone phenotypes were confirmed by flow analysis of H2-Kb, H2-Db and Qa-1b cell surface expression. Two sets of single cell clones were selected and used in mouse experiments. Values are shown as mean ± SEM. (A) Flow cytometry of independent clones after 48 h incubation with IFNγ (100 IU/mL). (B) Representative flow cytometry plots.
Loss of Qa-1b sensitizes TAP1/MHCIa deficient tumors to anti-PD-1 treatment
We first tested the in vitro growth rate of our CRISPR-engineered cell lines. Cells lacking Qa-1b and/or TAP1 grew equivalently compared with the clonal B16F10 parental control, suggesting that neither gene has a cell autonomous influence on proliferation or survival (Fig. 2A). To study the effects of Qa-1b in a TAP1 deficient setting, we injected the B16F10 parental, Qa-1b KO, TAP1 KO and TAP1/Qa-1b double KO B16F10 tumor cells into syngeneic WT C57BL/6J mice subcutaneously. Mice were randomized into treatment and control groups once tumors reached 50 – 150 mm3. The mice were treated with either anti-PD-1 or rat IgG2a isotype control twice a week, four times in total following randomization (Fig. 2B). The parental tumors showed little sensitivity to anti-PD1 treatment. Response was improved in Qa-1b deficient tumors (Fig. 2C,S2), although the percent tumor reduction relative to IgG control at Day 14 was not significantly different from parental tumors (Fig. 2D). Surprisingly, while TAP1 deficient tumors were deficient in cell surface classical MHCIa (Fig. 1A,B) and essentially resistant to anti-PD1 therapy, TAP1/Qa-1b double knockout tumors showed marked response (Fig. 2C). Notably, the percent tumor reduction relative to IgG at Day 14 was significantly improved in Qa-1b/TAP1 double KO tumors compared to tumors lacking only TAP1. Conversely, there was no significant improvement in percent tumor reduction between parental and Qa-1b single KO tumors (Fig. 2D). We found similar results using another set of B16F10 single cell clones engineered with the same CRISPR guides (Fig. 2E,F,S2). These results suggest that Qa-1b plays an immunosuppressive function in the context of TAP1/MHCIa deficient tumors that can be reversed by targeting Qa-1b.
Figure 2. Deletion of Qa-1b in TAP1 deficient tumors restores sensitivity to anti-PD-1 treatment.
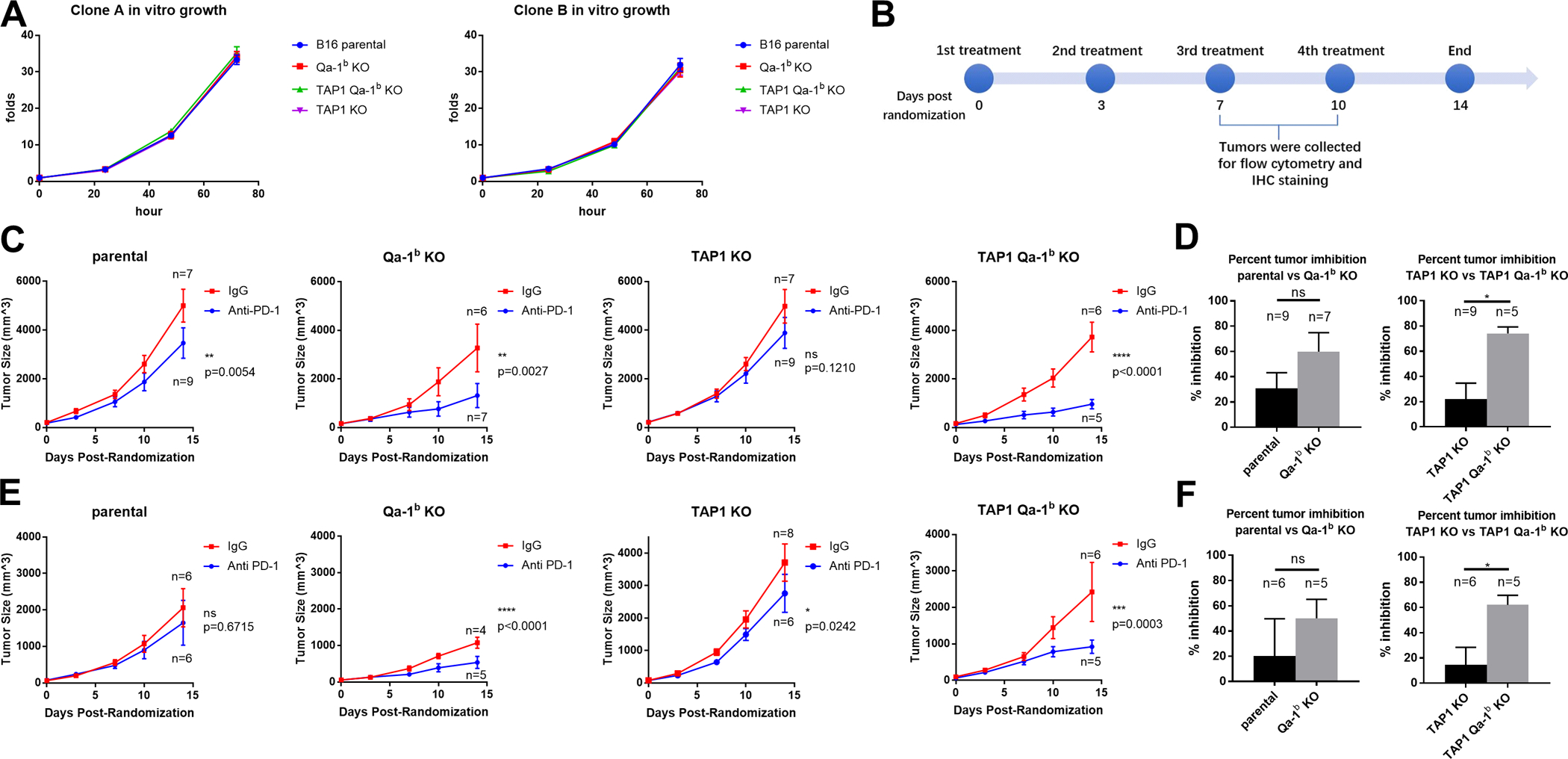
B16F10 parental, Qa-1b KO, TAP1 KO and TAP1/Qa-1b double KO clones were injected subcutaneously into syngeneic C57BL6/J mice. Mice were treated with either anti-PD-1 or IgG2a isotype control every three days after randomization. Graphs show tumor growth for two weeks post randomization. Values are shown as mean ± SEM. (A) In vitro growth of single cell clones used in the subsequent animal experiments. (N=3) (B) Timeline of animal experiments. 30 mice per cell line were injected and randomly divided into anti-PD-1 treated group and IgG2a treated control group. (C) Clone A derived KO lines were treated as labeled and tumor volume is shown. (D) Comparison of percent tumor inhibition at day 14 for Clone A derived KOs. Y axis is calculated using formula: (mean of IgG treated tumors minus size of anti-PD-1 treated tumor) / mean of IgG treated tumors * 100%. (E, F) Results for Clone B derived KOs. Tumor volumes are shown over time for each treatment. Statistical significance was determined by two-way ANOVA and Tukey’s post hoc test for tumor volume comparisons. Student’s t-test was used for percent tumor inhibition. *p<0.05, **P<0.01, ***p<0.001, ****p<0.0001
Qa-1b is required for the accumulation and activity of Tregs in TAP1/MHCIa deficient tumors
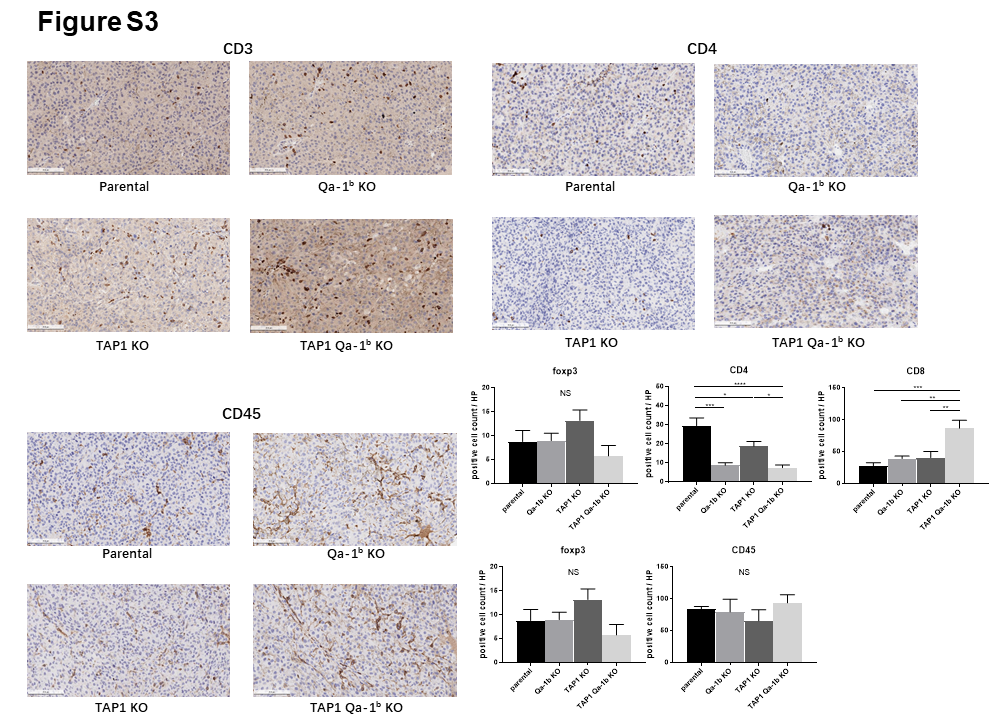
To examine the enhanced response to anti-PD1 therapy in TAP1/Qa-1b double KO tumors, we collected, dissociated, and analyzed tumors by flow cytometry at day 7–10 post-randomization. There was no statistically significant difference in total CD45+ immune cell infiltration among the tumors; however, there was a trend toward decreased infiltration in anti-PD1-treated TAP1 deficient tumors (Fig. 3A). We next analyzed the composition of the immune infiltrates from each tumor group. CD4+ T cells were enriched in anti-PD1-treated TAP1 deficient tumors. Co-deletion of Qa-1b abrogated this effect (Fig. 3B). Notably, these changes in CD4+ T cell composition were driven by primarily by the FoxP3+CD25+ Treg compartment and less so by IFNγ+ or FoxP3− CD4+ T cells (Fig. 3B). Conversely, we found that TAP1 deficient tumors showed decreased CD8+ T cell infiltration compared to parental tumors. This effect was abrogated by co-deletion of Qa-1b (Fig. 3C). We also analyzed cytolytic activity and proliferation markers to determine T cell functional status. Enhanced T cell activation was not correlated with anti-PD1 response in TAP1/Qa-1b double KO relative to parental tumors; however, it is striking that even with significantly diminished MHC Class Ia and Ib expression (Fig. 1A,B), TAP1/Qa-1b double KO tumors still exhibited significant immune infiltration and T cell activation (Fig. 3C, S3, S4). Considering the opposing effects we observed of TAP1 and Qa-1b depletion on Tregs and CD8+ T cells, we also examined the CD8+ T cell to Treg ratio (CD8:Treg). We found that CD8:Treg is significantly enhanced in anti-PD1-treated TAP1/Qa-1b double KO tumors relative to tumors lacking only TAP1 (Fig. 3D). This result was confirmed by IHC staining of tumor sections (Fig. 4). These results suggest that TAP1/MHCIa deficient tumors become resistant to anti-PD1 therapy at least in part through enhanced accumulation of Tregs leading to inhibition of CD8+ T cell proliferation and activation. Importantly, this effect is, at least in part, dependent upon Qa-1b and can be reversed through Qa-1b deletion (Fig. 3D).
Figure 3. Flow cytometry analysis of tumor-infiltrating adaptive immune cell populations from anti-PD-1 treated tumors.
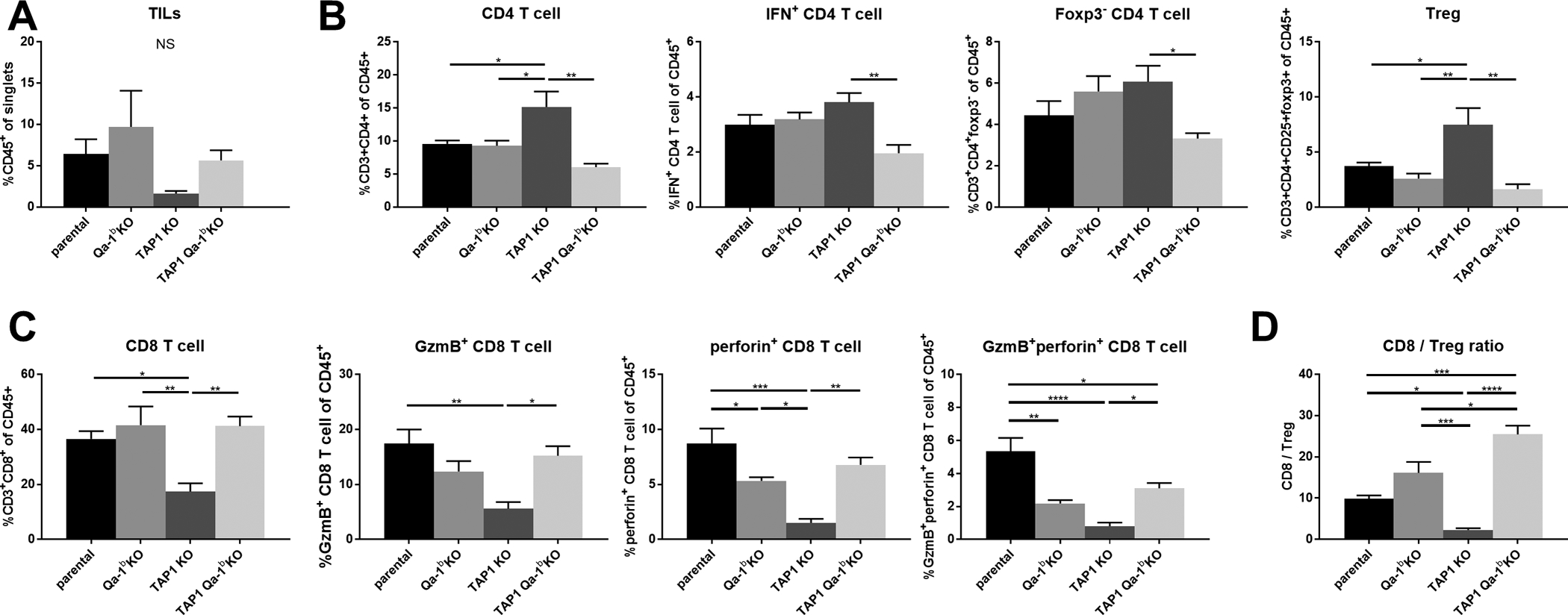
Mice were euthanized between days seven and ten post-randomization. Tumors were excised, dissociated, and stained with a pre-optimized adaptive immune marker panel. Relative numbers of the following cells are shown for each KO: (A) overall tumor-infiltrating immune cells, (b) tumor-infiltrating CD4+ T cells, (C) tumor-infiltrating CD8+ T cells, (D) tumor-infiltrating lymphocyte ratios. Values represent mean ± SEM (n=4). Statistical significance was determined using one-way ANOVA and Tukey’s post-hoc test. *p<0.05, **P<0.01, ***p<0.001, ****P<0.0001.
Figure 4. Immunohistochemical analysis of tumor infiltrating immune cells.
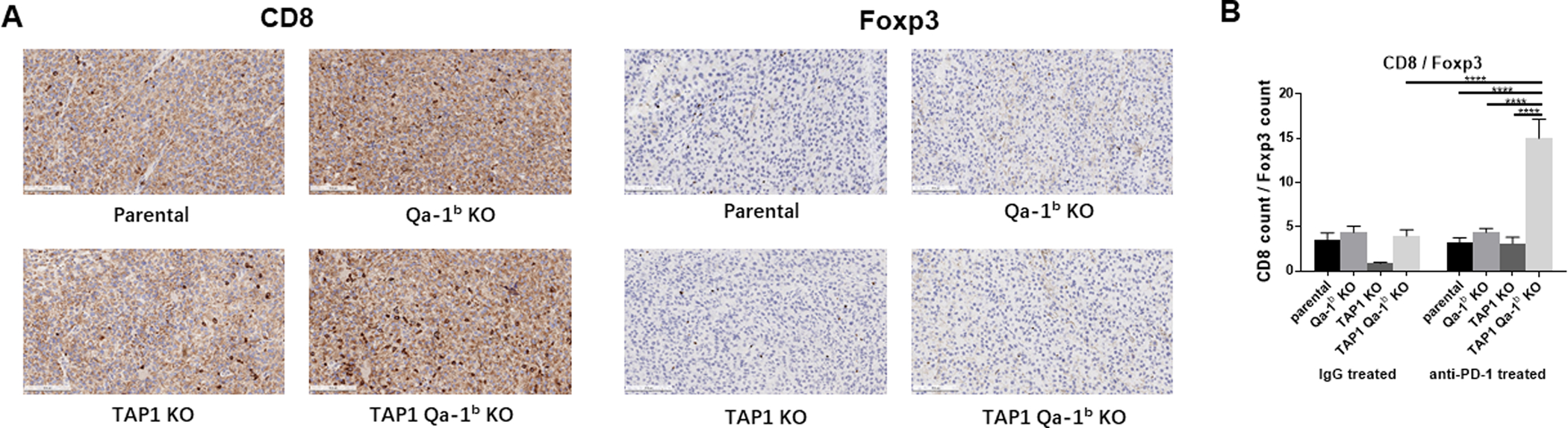
(A) Representative examples of CD8, foxp3 positive cells in paraffin sections from B16F10 parental, Qa-1b KO, TAP1 KO, and TAP1/Qa-1b double KO tumors treated with anti-PD-1. Scale bars represent 200 um. (B) Quantification of positive cells is shown. Data were collected from five images per slide. Values represent mean ± SEM. *p<0.05, **P<0.01, ***p<0.001, ****p<0.0001
TAP1/MHCIa and Qa-1b influence the tumor neutrophil and NK cell compartments
We also analyzed innate immune cell populations and their functional markers by flow cytometry. Interestingly, we observed an increase of neutrophils in the anti-PD-1-treated TAP1 KO tumors compared with parental and Qa-1b KO tumors. However, co-deletion of Qa-1b significantly decreased this effect (Fig. 5A). Neutrophils may be a pro-tumor immune cell population [16], as a subpopulation of neutrophils has been found to play a direct immune inhibitory role through secretion of inhibitory cytokines [16–18]. We did not find any significant differences in monocytes, dendritic cells, or macrophages among the tumor groups (Figs. 5B–D, S3). Although NK cells and T cells that express the NKG2A/CD94 receptor complex are known to be inhibited by Qa-1b–Qdm peptide signaling, we did not detect significant differences in NK cell abundance among the different tumor groups (Fig. 5E). However, the NK cell to Treg ratio was significantly increased in the anti-PD-1 treated TAP1/Qa-1b double KO tumors compared with all other tumor groups (Fig. 5F).
Figure 5. Flow cytometry analysis of tumor-infiltrating innate immune cell populations of anti-PD-1 treated tumors.
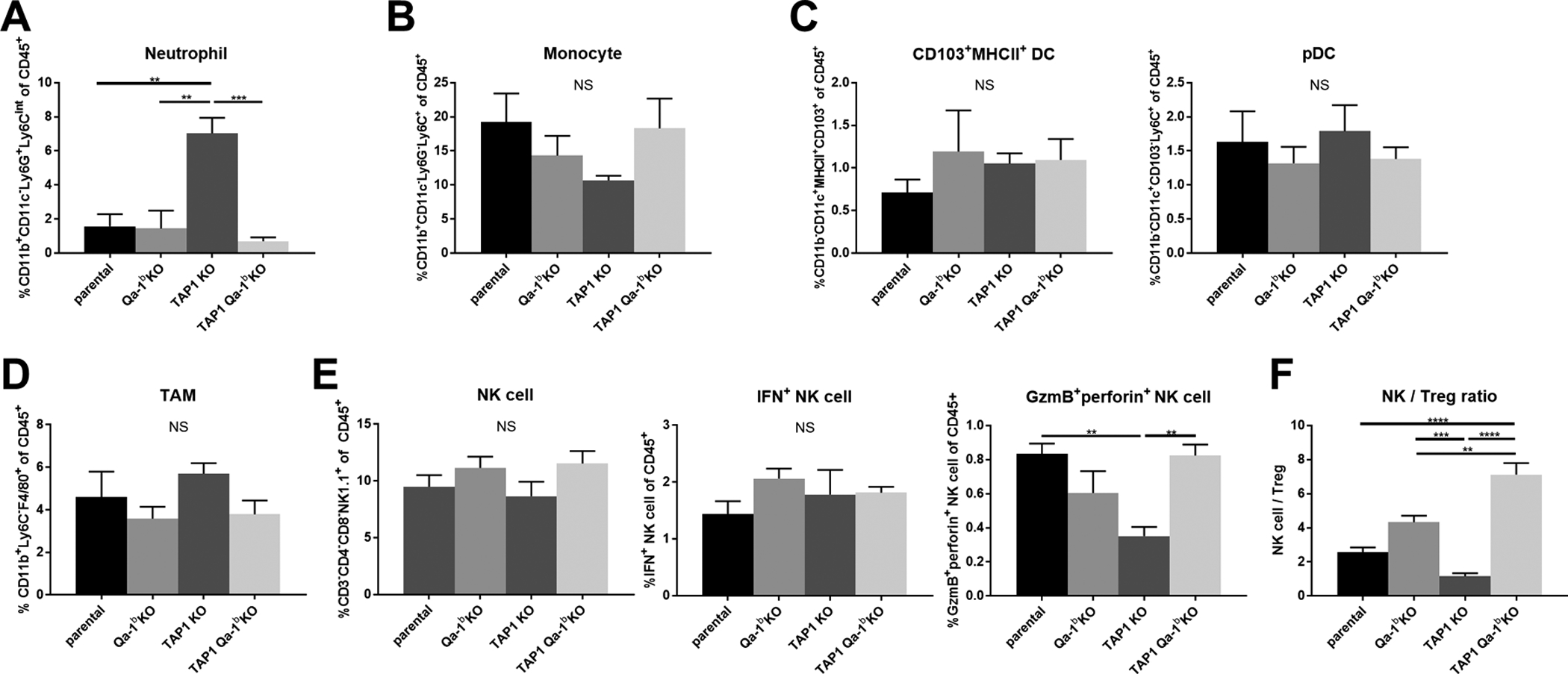
Mice were euthanized between days seven and ten post-randomization. Tumors were excised, dissociated, and stained with a pre-optimized adaptive immune marker panel. Frequency for each KO is shown: (A) neutrophils (B) monocytes (C) dendric cells (D) macrophages. (E) natural killer cells and their functional status. Data are shown in mean ± SEM (n=4). (F) NK cell / Treg ratios. Values represent mean ± SEM (n=4). Statistical significance was determined using one-way ANOVA and Tukey’s post-hoc test. *p<0.05, **P<0.01, ***p<0.001, ****P<0.0001.
Sensitivity of TAP1/Qa-1b double KO tumors to immune checkpoint blockade is partially dependent on NK cells
To elucidate the role of NK cells in the response of double KO tumors to anti-PD-1 treatment, NK depletion was performed using double KO cell lines. To deplete NK cells, mice were treated with either anti-NK1.1. Controls were treated with mouse IgG2a. Mice were randomized and treated with either anti-PD-1 or rat IgG2a isotype control in the same way as described above. Splenocytes were collected from the NK cell depleted and isotype control group at multiple time points to validate efficacy of NK depletion by anti-NK1.1 antibody (Fig. 6A,B). The rat IgG2a treated tumors showed no significance with each other, but both showed higher growth rate than the anti-PD-1 treated tumors, regardless of NK cell depletion. Notably, response to anti-PD-1 was somewhat dampened in the NK cell depleted mice compared with NK intact mice, whereas the growth rates of non-anti-PD-1 treated tumors were not significantly different when NK cells were depleted (Fig. 6C).
Figure 6. NK cell depletion partially impairs sensitivity of TAP1/Qa-1b double KO tumors to anti-PD-1 treatment.
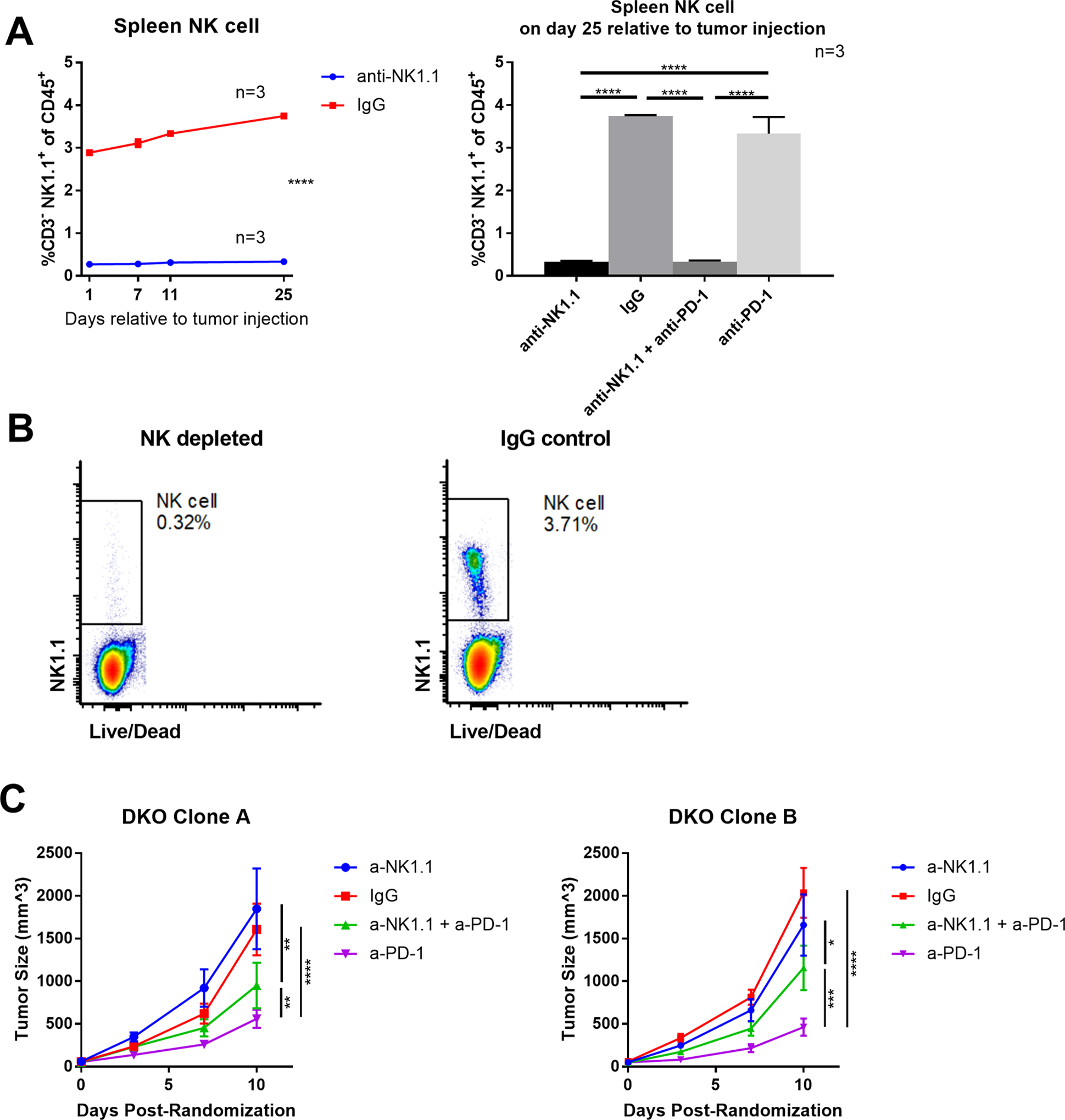
TAP1/Qa-1b double KO clones were injected subcutaneously into syngeneic C57BL6/J mice, 65 mice per clone. Mice were treated with either anti-NK1.1 or IgG2a isotype control at day −1, 1, and then two times per week relative to tumor injection until euthanization. Mice from each treatment arm were euthanized at day 1, 7, 11, and 25 relative to tumor injection and splenocytes were collected to validate NK cell depletion by flow cytometry. Mice were treated with either anti-PD-1 or IgG2a isotype control every three days after randomization. Graphs show NK cell percentage of splenocytes at multiple timepoints, and tumor growth for 10 days post randomization. Values are shown as mean ± SEM. (A) Summary of spleen NK cell flow cytometry analysis. (B) Representative flow cytometry analysis and summary of spleen NK cell. (C) Double KO clones were treated as labeled and tumor volume is shown. Statistical significance was determined using one-way ANOVA and Tukey’s post-hoc test. *p<0.05, **P<0.01, ***p<0.001, ****P<0.0001.
Discussion
Anti-tumor immunity is a complex biological process in which the functional status of various tumor-associated genes may enable increased sensitivity of tumors to the immune system by making them more visible and prone to be attacked by cytotoxic cells. H2-T23 encodes murine Qa-1b (human HLA-E ortholog), which transmits inhibitory signals to NK cells and a subset of T cells by presenting a limited set of highly conserved peptides. The most common of these peptides, AMAPRTLLL, is referred to as the Qa-1b determinant modifier (Qdm) [15]. Qdm is derived from the leader sequence of H2-Db, a murine MHC Ia molecule. Qa-1b-mediated presentation of Qdm is highly depended on TAP1 [4]. Loss of TAP1 prevents loading of Qdm onto Qa-1b and enables Qa-1b to present a more diverse alternative set of neopeptides to Qa-1b-restricted CD8+ cytotoxic T cells to facilitate anti-tumor immunity [19]. Notably, depletion of TAP1 also results in a sharp decrease of classical MHC class Ia cell surface expression, thereby removing almost all conventional MHC Ia - peptide presentation [20].
Therefore, based on this mechanism and a recent CRISPR screening reporting a relationship between ICB treatment and TAP1 or Qa-1b [10], we first hypothesized that TAP1 depleted tumors should respond well to anti-PD1 therapy via recognition of the altered Qa-1b peptidome, whereas TAP1/Qa-1b double knock-out tumors would have the worst response among the groups due to a lack of both Class Ia and Ib MHC-antigen peptide presentation to CTLs. We found that Qa-1b KO tumors grew slowly prior to randomization and were sensitive to anti-PD1 therapy, consistent with previously reported results [10, 11]. This may be explained by enhanced NK cell-mediated tumor killing. We found that TAP KO tumors were essentially resistant to anti-PD1 therapy, while, surprisingly, TAP1/Qa-1b double KO tumors displayed the greatest anti-PD1-mediated tumor inhibition.
We found that TAP1 KO was accompanied by an increase of infiltrated CD4+ Tregs, while the double KO was associated with a marked increase in CD8+ cytotoxic T lymphocyte (CTL) to CD4+ Treg ratio. Previous studies have shown that foxp3+ Tregs including CD4+ Tregs and CD8+ Tregs could suppress anti-tumor immunity [21, 22]. Treg targeting can be a potential strategy to increase efficacy of immunotherapy [23]. Although the proportion of total NK cells among CD45+ cells was not altered, perforin and granzyme B double positive NK cells as well as their ratio to Tregs were significantly upregulated in the double knock-out tumors compared to tumors lacking only TAP1. We also found that in our NK depletion study, double knock-out tumors are still sensitive to anti-PD-1 treatment, although the response was slightly weakened. Because both Class Ia and Ib MHC-mediated antigen peptide presentation were diminished in the double KO tumors, we suspect that either the remaining Class Ia MHC is sufficient to mediate T cell recognition or there may be an alternative antigen peptide presentation-mechanism mediating T cell killing in these tumors. Future studies of the infiltrating TCR repertoire may help address this issue.
Most innate immune cell populations examined in our flow panel - including monocytes, macrophages, and dendritic cells - weren’t significantly altered in the tumor groups. Tumor associated macrophages (TAMs) are one of the predominant regulatory cell populations in anti-tumor immunity. They not only express PD-L1 to inhibit CD8 T cell activity, but can inhibit CTL proliferation depending on metabolism of L-arginine. Additionally, TAMs secrete IL-10, which recruits Tregs and creates an inhibitory tumor microenvironment for T cells. [24, 25] These data suggest that regulation of these innate immune cells is not dependent upon Qa-1b-Qdm or the altered Qa-1b peptidome.
Notably, the neutrophil percentage in the anti-PD-1 treated TAP1 deficient tumors was increased in a Qa-1b-dependent manner relative to parental tumors. Generally, neutrophils are thought to be inhibitory to anti-tumor immunity. The mechanism that drove the change of neutrophils in TAP1 KO tumors in this study is unclear. One possible explanation is that the survival time and function of neutrophils may be dependent upon the levels of various cytokines in the tumor microenvironment [18]. For example, tumor associated neutrophils (TANs) recruited by IFN-g exhibit an anti-tumor effect, while TANs recruited by TGF-beta are immunosuppressive [16, 17, 26]. It is possible that TAP1 depletion-mediated loss of classical MHCIa-peptide presentation may disrupt the balance of cytokines in the tumor microenvironment by altering T cell activation.
In summary, we found that Qa-1b can support the accumulation and function of regulatory T cells in TAP1 deficient tumors, thereby impairing response to checkpoint blockade therapy. Co-deletion of Qa-1b results in a dramatic increase in the CD8+ T cell to Treg ratio and reverses this resistance to anti-PD1 therapy. As such, targeting of Qa-1b/HLA-E, particularly in tumors with impaired antigen processing machinery, may improve patient responses to anti-PD1 therapy.
Summary
We found that resistance to anti-PD1 therapy in TAP1 deficient tumors is reversed by co-deletion of Qa-1b. This phenotype is associated with increased CD8+ T cells and decreased tumor associated neutrophils and CD4+ regulatory T cells.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Implications:
This study reveals important functional crosstalk between classical TAP-dependent MHC complexes and Qa-1b/HLA-E, particularly in tumors with impaired antigen processing machinery. This can dramatically influence immunotherapy efficacy.
Acknowledgements
We thank members of the Chan lab for their suggestions and critical reading of the manuscript. We acknowledge funding sources including Pershing Square Sohn Cancer Research grant (TAC), the PaineWebber Chair (TAC), NIH R01 CA205426 (TAC), and NIH R35 CA232097 (TAC).
Funding: This study was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748. We acknowledge funding sources including Pershing Square Sohn Cancer Research grant (TAC), the PaineWebber Chair (TAC), NIH R01 CA205426, NIH R35 CA232097 (TAC), and the STARR Cancer Consortium (TAC).
Footnotes
Conflict of Interest: TAC is a co-founder of Gritstone Oncology and holds equity. TAC holds equity in An2H. TAC acknowledges grant funding from Bristol-Myers Squibb, AstraZeneca, Illumina, Pfizer, An2H, and Eisai. TAC has served as an advisor for Bristol-Myers Squibb, Illumina, Eisai, NysnoBio, and An2H. MSK has licensed the use of TMB for the identification of patients that benefit from immune checkpoint therapy to PGDx.
References
- 1.Havel JJ, Chowell D, and Chan TA, The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer, 2019. 19(3): p. 133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leone P, et al. , MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst, 2013. 105(16): p. 1172–87. [DOI] [PubMed] [Google Scholar]
- 3.Daniel C, Nolting J, and von Boehmer HJI, Mechanisms of self–nonself discrimination and possible clinical relevance. Innumotherapy, 2009. 1(4): p. 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oliveira CC and van Hall T, Alternative Antigen Processing for MHC Class I: Multiple Roads Lead to Rome. Front Immunol, 2015. 6: p. 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolpert EZ, et al. , Generation of CD8+ T cells specific for transporter associated with antigen processing deficient cells. PNAS, 1997. 94(21): p. 11496–11501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sabapathy K, Nam SJCD, and Differentiation, Defective MHC class I antigen surface expression promotes cellular survival through elevated ER stress and modulation of p53 function. Cell Death & Differentiation, 2008. 15(9): p. 1364–1374. [DOI] [PubMed] [Google Scholar]
- 7.Oliveira CC, et al. , The nonpolymorphic MHC Qa-1b mediates CD8+ T cell surveillance of antigen-processing defects. J Exp Med, 2010. 207(1): p. 207–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aldrich CJ, et al. , Identification of a Tap-dependent leader peptide recognized by alloreactive T cells specific for a class Ib antigen. Cell, 1994. 79(4): p. 649–658. [DOI] [PubMed] [Google Scholar]
- 9.Blum JS, Wearsch PA, and Cresswell P.J.A.r.o.i., Pathways of antigen processing. Annu Rev Immunol, 2013. 31: p. 443–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manguso RT, et al. , In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature, 2017. 547(7664): p. 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andre P, et al. , Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell, 2018. 175(7): p. 1731–1743 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Godfrey DI, et al. , Unconventional T cell targets for cancer immunotherapy. 2018. 48(3): p. 453–473. [DOI] [PubMed] [Google Scholar]
- 13.van Montfoort N, et al. , NKG2A blockade potentiates CD8 T cell immunity induced by cancer vaccines. Cell, 2018. 175(7): p. 1744–1755. e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ran FA, et al. , Genome engineering using the CRISPR-Cas9 system. Nat Protoc, 2013. 8(11): p. 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurepa Z, Hasemann CA, and Forman J.J.T.J.o.e.m., Qa-1b binds conserved class I leader peptides derived from several mammalian species. J Exp Med, 1998. 188(5): p. 973–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fridlender ZG, et al. , Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell, 2009. 16(3): p. 183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jablonska J, et al. , Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest, 2010. 120(4): p. 1151–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu L, et al. , Tumor-Associated Neutrophils in Cancer: Going Pro. Cancers (Basel), 2019. 11(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Hall T, et al. , The other Janus face of Qa-1 and HLA-E: diverse peptide repertoires in times of stress. Microbes and Infection, 2010. 12(12–13): p. 910–918. [DOI] [PubMed] [Google Scholar]
- 20.de la Salle H, et al. , HLA class I deficiencies due to mutations in subunit 1 of the peptide transporter TAP1. 1999. 103(5): p. R9–R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arias DAA, et al. , Disruption of CD8+ Treg activity results in expansion of T follicular helper cells and enhanced antitumor immunity. Cancer Immunol Res, 2014. 2(3): p. 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanaka A and Sakaguchi S.J.C.r., Regulatory T cells in cancer immunotherapy. Cell Research, 2017. 27(1): p. 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanaka A and Sakaguchi S.J.E.j.o.i., Targeting Treg cells in cancer immunotherapy. Eur J Immunol, 2019. 49(8): p. 1140–1146. [DOI] [PubMed] [Google Scholar]
- 24.Yang L and Zhang Y, Tumor-associated macrophages: from basic research to clinical application. J Hematol Oncol, 2017. 10(1): p. 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gordon SR, et al. , PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. 2017. 545(7655): p. 495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hey YY, Tan JK, and O’Neill HC, Redefining Myeloid Cell Subsets in Murine Spleen. Front Immunol, 2015. 6: p. 652. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.