

Abstract
Adenosine kinase (ADK) is the key regulator of adenosine and catalyzes the metabolism of adenosine to 5’-adenosine monophosphate. The enzyme exists in two isoforms: a long isoform (ADK-long, ADK-L) and a short isoform (ADK-short, ADK-S). The two isoforms are developmentally regulated and are differentially expressed in distinct subcellular compartments with ADK-L localized in the nucleus and ADK-S localized in the cytoplasm. The nuclear localization of ADK-L and its biochemical link to the transmethylation pathway suggest a specific role for gene regulation via epigenetic mechanisms. Recent evidence reveals an adenosine receptor-independent role of ADK in determining the global methylation status of DNA and thereby contributing to epigenomic regulation. Here we summarize recent progress in understanding the biochemical interactions between adenosine metabolism by ADK-L and epigenetic modifications linked to transmethylation reactions. This review will provide a comprehensive overview of ADK-associated changes in DNA methylation in developmental, as well as in pathological conditions including brain injury, epilepsy, cancer, and diabetes. Challenges in investigating the epigenetic role of ADK for therapeutic gains are briefly discussed.
Keywords: adenosine kinase, epilepsy, development, epigenetic regulator, DNA methylation
1. Introduction
Adenosine kinase (ADK, EC 2.7.1.20) is the key enzyme for the regulation of ambient levels of adenosine (Boison, 2006; Boison and Yegutkin, 2019) and functions by removing adenosine through phosphorylation into 5′-adenosine-monophosphate (AMP) (Park and Gupta, 2008). Adenosine regulates a plethora of biochemical pathways, in addition to the activation of receptor-mediated signaling pathways (Fredholm et al., 2000). As a regulator of adenosine, maladaptive changes in ADK expression have been implicated in a number of pathologies, including epilepsy, brain injury, stroke, diabetes, and cancer (Boison, 2013; Boison and Yegutkin, 2019). Initially, the role of ADK in those pathologies has been linked to its role as regulator of the tissue tone of adenosine, which determines the degree of adenosine receptor activation (Aronica et al., 2011; Giglioni et al., 2008; Li et al., 2008a; Masino et al., 2011; Pawelczyk et al., 2000; Saitoh et al., 2004; Sakowicz-Burkiewicz et al., 2006; Tsuchiya et al., 2012). However, ADK is also a well-characterized key regulator of the transmethylation pathway (Bjursell et al., 2011; Boison et al., 2002; Moffatt et al., 2002; Williams-Karnesky et al., 2013; Xu et al., 2017a; Xu et al., 2017b), which suggests additional functions of ADK beyond its role as a regulator of adenosine receptor activation. Transmethylation is a common biochemical reaction, in which a methyl group derived from the donor S-adenosylmethionine (SAM) is transferred onto an acceptor. Transmethylation reactions include the transfer of methyl groups from SAM to a wide range of acceptors, as varied as lipids, dopamine, and DNA, the latter transmethylation reaction catalyzed by DNA methyltransferases (DNMTs). The resulting product of all transmethylation reactions, S-adenosylhomocysteine (SAH), is then cleaved by SAH hydrolase (SAHH) into adenosine and homocysteine. Importantly, the thermodynamic equilibrium of the SAHH reaction lies on the side of SAH formation. Thus, this reaction, which is critical for regulating the flux of methyl groups through the transmethylation pathway can only proceed, if adenosine levels are kept low (Hoffman et al., 1979), and adenosine is effectively removed through ADK (Boison et al., 2002). Reduced activity of ADK results in the accumulation of SAH (Boison et al., 2002), which itself is a potent inhibitor of DNMT enzyme activity (Rowling et al., 2002) and therefore provides the rationale for the development of SAH analogues as novel DNMT inhibitors (Saavedra et al., 2009). Both ADK knockout mice (Boison et al., 2002) and plants (Moffatt et al., 2002) share the same transmethylation defects, developmental delay and stunted growth; findings also seen in human infants with inborn ADK deficiency (Bjursell et al., 2011). In line with its role as regulator of transmethylation, it was subsequently discovered that ADK also assumes a hitherto unknown role as regulator of DNA methylation, a major epigenetic mechanism (Williams-Karnesky et al., 2013). Those gene mutations support a functional role of ADK as regulator of transmethylation reactions. Mechanistically, ADK thereby regulates transmethylation through mass action by regulating the thermodynamic equilibrium of the SAHH reaction, which in turn determines the concentration of the DNMT inhibitor SAH (Boison, 2013).
Aberrant DNA methylation changes in the CNS have attracted increased attention, with recently identified implications in psychiatric and neurological conditions (Jin and Liu, 2018; Lu et al., 2013). The long-lasting benefits of targeting epigenetic mechanisms such as DNA methylation is one of the primary reasons for this increased attention (Csoka and Szyf, 2009). For instance, recent work demonstrated that transient exposure to low doses of DNA demethylating agents resulted in long-term anti-tumor effects, modulated by genome-wide promoter methylation, which persist well beyond drug withdrawal (Tsai et al., 2012). Similarly, a transient increase in adenosine (Williams-Karnesky et al., 2013) or pharmacological blockade of ADK (Sandau et al., 2019) was sufficient to reverse a global DNA hypermethylation phenotype in epilepsy and to exert long-lasting disease-modifying effects by preventing the progression of epileptogenesis for at least 3 months in an experimental model of TLE (Williams-Karnesky et al., 2013). In addition to epilepsy, rodent studies suggest an association of ADK expression changes with DNA methylation changes in brain injury (Acharya et al., 2017), vascular inflammation (Xu et al., 2017a), angiogenesis (Xu et al., 2017b), atherosclerosis (Wang et al., 2021; Zhang et al., 2018), and cancer (El-Kharrag et al., 2019).
The main purpose of this review is to provide an overview on the contribution of ADK in epigenetic regulation, particularly its ability to modulate DNA methylation in neurodevelopmental and pathological conditions. We will first describe the developmental regulation of ADK isoforms, cellular distribution and function. We then discuss mechanisms of how adenosine metabolism by ADK influences DNA methylation, and review recent efforts linking adenosine perturbation to epigenetic dysregulation in pathological conditions including developmental disorders, brain injury, vascular disorders, cancer, and diabetes. Further, we discuss the effects of targeting ADK and the consequential changes in DNA methylation in these disorders.
2. ADK isoforms: subcellular and cellular localization
The enzyme ADK exists predominantly in two isoforms – ADK long (ADK-L) and ADK short (ADK-S), which are derived through alternative splicing (Singh et al., 1996; Spychala et al., 1996). The two isoforms share similar biochemical and kinetic properties (Singh et al., 1996; Spychala et al., 1996). While expressed in all tissues and organs as documented by Western blot analysis, the expression pattern and levels of the two ADK isoforms vary markedly among different tissues, with the highest expression noted in the liver (Bjursell et al., 2011; Boison et al., 2002; Cui et al., 2011) (Figure. 1). In adult rats, the expression of both isoforms was comparable in liver, kidney, lung and pancreas. In contrast, the expression of the ADK-L isoform dominates in the heart, thymus and skeletal muscle tissues, whereas, the short isoform is prominently expressed in the brain (Cui et al., 2011).
Figure 1. Regulation of ADK expression.

a. Tissue-specific regulation of ADK isoforms in adrenal gland, brain, heart, kidney, liver, pancreas, spleen, muscle and thymus is depicted. The signs ++++, +++, ++, + and – denote, very high, high, moderate, low and no expression, respectively. b. Developmental regulation of ADK isoforms in the brain of mice from embryonic (E5 –E20), postnatal (P4–8), and adult stages. c. Localization of ADK-S in the cytoplasm of astrocytes and localization of ADK-L in the nucleus of both astrocytes and neurons in the adult brain. The ADK expression profiles depicted here are schematic representations based on Western blot analyses from previously reported publications (Bjursell et al., 2011; Boison et al., 2002; Cui et al., 2011).
In addition to the tissue-specific regulation in ADK expression, the two isoforms undergo cell-type specific dynamic changes during brain development (Kiese et al., 2016) (Figure 1) and in response to external stimuli such as brain injury (Gebril et al., 2020). During development, there is a coordinated shift from a predominantly neuronal to a predominantly astrocytic expression pattern (Fedele et al., 2005; Studer et al., 2006). Further, the ADK-L isoform is prominent during development presumably contributing to the increased methylation in the immature brain (Yang et al., 2015). In the adult brain, the expression of ADK-L, although downregulated, is preserved in plastic cells with proliferation potential, such as astrocytes and granular neurons in the dentate gyrus (Fedele et al., 2004; Gebril et al., 2020). In contrast, the cytoplasmic isoform ADK-S is more prominent in the adult phase and is thought to regulate cellular adenosine and thereby control adenosine-receptor activation (Boison, 2013). The developmental downregulation of nuclear ADK-L transcripts in neurons and upregulation of cytoplasmic ADK-S transcripts in astrocytes occurs between P4 and P14 in rodents and is in part facilitated by the binding of transcription factor specificity protein 1 (SP1) to the ADK-L promoter (Cui et al., 2009; Fedele et al., 2005). It is important to note that the developmental regulation of ADK isoforms is based on studies performed in the cortex/cerebrum (Fedele et al., 2005; Studer et al., 2006). Recent investigations in the cerebellum highlight a spatially distinct pattern of ADK expression during neurodevelopment, as well (Gebril et al., 2021). Other non-neuronal cells in the CNS that have reported ADK expression include endothelial cells (Xu et al., 2017a) and oligodendrocytes (Gonzalez-Fernandez et al., 2014), with no detectable ADK expression noted in microglia (Aronica et al., 2011). The tight developmental, tissue and cell-specific regulation of ADK isoforms is indicative of complex transcriptional regulatory mechanisms governing ADK expression (Kiese et al., 2016). However, the physiological relevance of the switch in the expression of the different ADK isoforms remains enigmatic and needs to be investigated.
Inside a cell, the ADK isoforms display distinct subcellular compartmentalization, with ADK-L localized within the nucleus and ADK-S localized in the cytoplasm (Cui et al., 2009). The occurrence of ADK-L in the nucleus suggests an essential role in epigenetic regulation. More specifically, ADK-L facilitates methylation reactions by the removal of adenosine, the obligatory product of all SAM-dependent transmethylation reactions (Boison et al., 2002; Kredich and Martin, 1977; Park and Gupta, 2008) (Figure 2). Co-localization of ADK-L, DNMTs, and SAH in the cell nucleus (Jue et al., 1995; Kloor et al., 2007) suggests that those enzymes are organized as part of an enzymatic pathway, which maintains the metabolic flux of methyl groups. In this review, we discuss the epigenetic mechanisms, mainly DNA methylation influenced by ADK-L in various pathological conditions and how it can be exploited for therapeutic gain.
Figure 2. Molecular, biochemical and epigenetic mechanisms regulated by ADK isoforms.

Schematic shows intracellular adenosine metabolism via ADK-S, equilibration to the extracellular space via equilibrative (ENT) or concentrative (CNT) nucleoside transporters, and associated adenosine receptor-mediated mechanisms. On the other hand, nuclear ADK-L acts as an epigenetic regulator of DNA methylation. ADK-L promotes transmethylation reactions by adenosine conversion to 5’AMP causing a forward shift in S-adenosylmethionine (SAM) to S-adenosylhomocysteine (SAH) transformation, thereby promoting DNA methylation through DNA methyltransferases (DNMT).
3. ADK mediated epigenetic mechanisms in disease
3.1. Neurodevelopmental disorders
As previously mentioned, ADK undergoes coordinated expression changes during early brain development, resulting in a shift from predominant neuronal expression during prenatal and early postnatal brain development to predominant astrocytic expression beyond postnatal day 14 in the mouse (Kiese et al., 2016; Studer et al., 2006). New evidence confirms that ADK-L expressed in the nuclei of neurons and immature precursor cells is involved in embryonic and early postnatal brain development (Gebril et al., 2020). This tight developmental regulation of ADK-L expression combined with the existence of a placental adenosine barrier (von Versen-Hoynck et al., 2009) suggests a critical role for ADK as an epigenetic regulator in brain development. More importantly, imbalance in adenosine homeostasis has been implicated in neuropsychiatric conditions, such as autism and schizophrenia (Boison et al., 2012; Masino et al., 2013; Shen et al., 2012). Further, human mutations in the Adk gene manifests with global developmental delay, cognitive impairment and seizures commencing between the first and third year of life (Bjursell et al., 2011; Staufner et al., 2016). Biochemical analysis in patients with Adk mutations revealed increased plasma levels of methionine, SAM and SAH but normal or mildly elevated homocysteine (Hcy) levels, confirming the link to the transmethylation pathway (Bjursell et al., 2011). In animal studies, adult mice lacking ADK in the brain from fetal development onwards showed significant deficits in social memory in males, contextual learning impairments in both sexes, and a hyper-responsiveness to amphetamine in males. However, mice with a postnatal deletion of ADK in the brain, showed normal social memory and contextual learning but hypo-responsiveness to amphetamine in males (Osborne et al., 2018). The discrepancy in behavioral outcomes in the two transgenic lines, provides an indication for a specific developmental role of ADK. However, evidence linking ADK-mediated epigenetic mechanisms in developmental etiology remains to be elucidated.
3.2. Brain injury
Brain injury can occur as a result of a traumatic or ischemic event causing an insult to the brain and is usually associated with cognitive and behavioral deficits. Dysregulated expression of ADK has been noted in both ischemic as well as traumatic brain injury models. More specifically, a biphasic response in ADK expression is observed, with acute decrease in ADK expression immediately after the onset of injury followed by a progressive increase in ADK expression at chronic time points (Pignataro et al., 2008; Shen et al., 2011). Indeed, there is a link between the ADK expression and the degree of brain injury after ischemic stroke, as demonstrated by three independent experimental models (Shen et al., 2011). First, transgenic mice which have increased ADK expression in striatum (164%) and reduced ADK expression in cortical forebrain (65%) demonstrated increased striatal infarct volume (126%) but almost complete protection of cortex (27%) compared with wild-type controls when subjected to a middle cerebral artery occlusion model of focal cerebral ischemia, suggesting that cerebral injury levels correlate to levels of ADK. Second, transgenic mice with brain-wide ADK overexpression demonstrated increased susceptibility to ischemic insults in a model of LPS-induced stroke tolerance, indicating that ADK activity inversely modulates ischemic tolerance. Third, overexpression or knockdown of ADK in vivo using viral vectors resulted in increased (126%) or decreased (51%) infarct volume, respectively. Together, these data highlight ADK as a promising therapeutic target for modulating the degree of stroke-induced brain injury (Shen et al., 2011).
Until recently, the role of ADK in brain injury was believed to be due to its ability to regulate adenosine tissue tone and the resultant activation of the inhibitory adenosine A1 receptors (Cunha, 2005). However, emerging evidence confirms an epigenetic role for ADK in brain injury (Figure 3). A recent study on neuroepigenetic responses to radiation found that increased levels of 5-methylcytosine and 5-hydroxymethylcytosine in the hippocampus of gamma-irradiated mice coincided with increased levels of both ADK and the DNA methylating enzyme DNMT3a, however a direct link between the enzymes was not investigated (Acharya et al., 2017). Immunofluorescence data showed that ectopic expression of ADK-L co-localized in DAPI stained nuclei in the hippocampus only in irradiated mice (20 cGy exposure) but not in sham exposed mice (0 Gy) (Acharya et al., 2017). The use of the ADK inhibitor 5-iodotubercidin (5-ITU) restored DNA methylation to control levels and improved cognitive performance in mice exposed to 20 cGy radiation injury (Acharya et al., 2017). In a model of controlled cortical impact injury, ectopic expression of ADK-L was observed in the nucleus of dentate granule cell neurons in addition to the increased ADK-S and associated astrogliosis in the hippocampus (Gebril et al., 2020). The use of transgenic mice with reciprocal manipulations to either ablate or overexpress ADK-L specifically in the dentate gyrus and CA1 neurons revealed a novel role for ADK-L in injury induced stem cell proliferation (Gebril et al., 2020). Specifically, inability of mice to express ADK-L in the dentate gyrus and CA1 neurons resulted in a significant increase in neural stem cell proliferation. In contrast, mice, which overexpress ADK-L in dentate and CA1 granule cell neurons had significantly reduced injury-induced neural stem cell proliferation. In line with the genetic deletion of ADK-L, the pharmacological blockade of ADK with 5-ITU also resulted in increased neural stem cell proliferation at 3 days after brain injury. Since changes in DNA methylation play a role in neural stem cell proliferation (Wang et al., 2016), and because the nuclear isoform ADK-L has a specific role in controlling DNA methylation status (Williams-Karnesky et al., 2013), these results imply that ADK-L regulates injury-induced neurogenesis via epigenetic mechanisms. However, whether ADK-L associated global changes in DNA methylation causes gene specific DNA methylation of neurogenic genes remains elusive and warrants further investigation.
Figure 3: ADK and associated epigenetic mechanisms in pathological conditions.
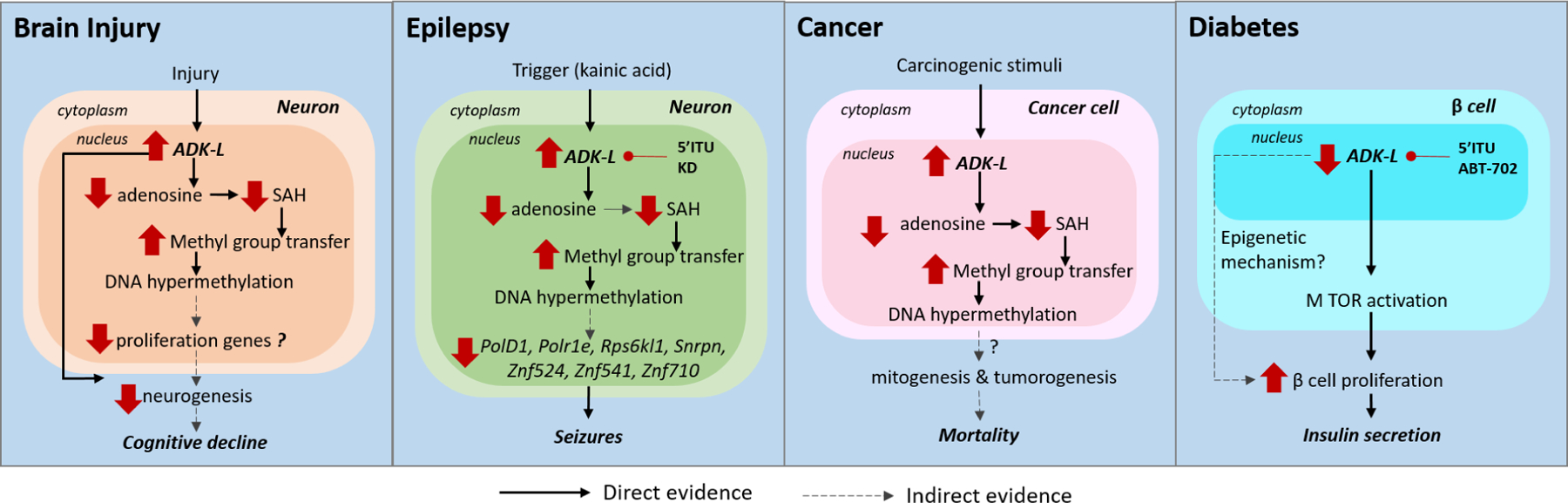
The epigenetic mechanisms mediated by ADK in various pathological conditions are shown. (i) In brain injury, chronic increase in ADK was associated with increased transmethylation leading to DNA hypermethylation and neurogenesis. The links between methylation changes and cognitive decline need to be investigated. (ii) In experimental epilepsy models, a trigger such as kainic acid induces ectopic overexpression of ADK-L and DNA hypermethylation of a network of genes including PolD1, Polr1e, Rps6kl1, Snrpn, Znf524, Znf541, Znf710. ADK inhibition by 5-ITU and ketogenic diet (KD) was able to restore DNA methylation and prevent onset of seizures. (iii) In preclinical cancer models, studies using transgenic mice with ADK deficiency reveals ADK expression changes in tumors may be associated with methylation of mitogenesis and tumorigenesis genes resulting in increased susceptibility to carcinogen responses and associated mortality. (iv) In β-islet cells in the pancreas, ADK inhibition via genetic or pharmacological tools promoted β-cell proliferation and insulin secretion via MTOR activation.
These studies confirm that the expression of ADK, and in particular of ADK-L is dysregulated in various brain injury models suggesting the involvement of ADK-mediated epigenetic regulation. Hence, the pharmacological inhibition of ADK is a promising strategy to restore pathogenic epigenetic modifications, enhance neuroregeneration, and thereby improve injury associated cognitive outcomes. Although triggering neurogenesis is a sought-after therapeutic approach to alleviate brain injury (Mueller et al., 2009), aberrant neurogenesis increases the risk for the development of seizures, especially in TBI survivors (Annegers and Coan, 2000). Notably, the ADK inhibitor 5-ITU has recently been shown to exert antiepileptogenic disease-modifying properties (Sandau et al., 2019), suggesting that ADK inhibition is capable of promoting neurogenesis after a brain injury without the risk of causing seizures. Despite the strong implications for ADK inhibition in alleviating brain injury, whether inhibition of the ADK-L isoform alone or the epigenetic mechanisms modulated by ADK are sufficient to improve the neurocognitive outcomes after brain injury remains to be investigated and would be of significant therapeutic value.
3.3. Epilepsy
Temporal lobe epilepsy (TLE) is the most frequent and most difficult to treat form of epilepsy in adulthood. It is an acquired form of epilepsy, which is triggered by an insult to the brain, such as injury, status epilepticus, stroke, or infection and is often pharmacoresistant. Maladaptive overexpression of both ADK isoforms has been noted in sclerotic hippocampal tissue in a variety of rodent models of epilepsy (Gouder et al., 2004; Li et al., 2008b), as well as in human specimens resected from patients with temporal lobe epilepsy and hippocampal sclerosis (Aronica et al., 2011). Early work on ADK in epilepsy predominantly focused on the adenosine receptor mediated mechanisms that are regulated by the ADK-S isoform (Boison, 2016; Boison and Steinhauser, 2018). However, recent efforts shed light on the important epigenetic influence ADK has on epileptogenesis and epilepsy progression (Williams-Karnesky et al., 2013) (Figure 3).
Epigenetic changes, particularly DNA hypermethylation has been observed in the epileptic brain in both experimental and human epilepsy and may provide novel therapeutic targets to interfere with the epileptogenic process (Debski et al., 2016; Kobow et al., 2013; Younus and Reddy, 2017). Based on the clinical observation that seizure severity is a reliable predictor for the development of pharmacoresistance in epilepsy, it has been hypothesized, that seizures can mediate epigenetic modifications that result in persistent changes in genomic methylation, histone density, and posttranslational modifications; changes that may aggravate the epileptogenic condition (Kobow and Blumcke, 2011) and even confer pharmacoresistance in patients with epilepsy (Kobow et al., 2013). Hence, understanding the epigenetic mechanisms regulated by seizures may be the key to enhance the effectiveness of antiepileptic drugs, development of disease-modifying treatments or even prevent epileptogenesis altogether. In our prior study we used a kainic acid induced TLE model in rats, and demonstrated that pathological overexpression of ADK correlated with increased 5-methylcytidine (5mC) expression and DNA hypermethylation in the epileptic hippocampus, suggesting a functional interaction between ADK and DNA methylation status (Williams-Karnesky et al., 2013). The study used two independent experimental approaches, an ELISA-based assay as well as a rat-specific methylated DNA immunoprecipitation on ChIP (MeDIP-on-ChIP) analysis, to confirm that the global hypermethylation state of hippocampal DNA in epileptic rats is associated with changes in adenosine metabolism. We used adenosine releasing silk-based brain implants and demonstrated that the transient local release of adenosine for only 10 days prevented epilepsy progression with long-lasting benefits through an epigenetic mechanism. The study identified 125 genes with substantially increased methylation in the epileptic brain and 762 genes that showed reduced methylation status during adenosine treatment. Among the two groups, we found 46 genes that were common in the two groups. Interestingly, a large number of the common genes were associated with DNA structural elements and transcription factors (PolD1, Polr1e, Rps6kl1, Snrpn, Znf524, Znf541, Znf710), making them likely candidates as mediators for adenosine-dependent changes in major homeostatic functions in the cell nucleus (Williams-Karnesky et al., 2013). This study suggests that targeting ADK for epilepsy prevention or progression will likely act through a network of gene expression changes caused by site-specific DNA methylation changes of multiple genes.
Apart from adenosine augmentation, inhibition of ADK using 5-iodotubercidin (5-ITU) also reduced hippocampal DNA methylation by 50% (Williams-Karnesky et al., 2013). An independent study showed that transient ADK inhibition with 5-ITU for only 5 days significantly reduced the percent time in seizures by at least 80% in 56% of mice at 6 weeks post-kainic acid (Sandau et al., 2019). Collectively, these findings indicate that 5-ITU restored global DNA methylation levels possibly via ADK-L and prevented epileptogenesis. The prevention of epileptogenesis by ADK inhibition is of significant therapeutic interest, particularly for patients who are at high risk of developing epilepsy, such as patients with severe brain injury (Verellen and Cavazos, 2010). Another high risk situation for secondary epileptogenesis is in patients who undergo surgical resection of an epileptogenic focus. In these patients, seizures recur in about 50% of cases and is a major cause of relapse (de Tisi et al., 2011). Therefore, targeting ADK is a feasible and promising therapeutic strategy for preventing epileptogenesis, especially since transient treatment might be able to provide long-lasting effectiveness. However, the specific role of ADK-L and epigenetic mechanisms underlying epileptogenesis needs to be investigated and there is an urgent need for development of ADK-L specific inhibitors.
An alternative therapeutic option for targeting ADK-mediated epigenetic mechanisms for the treatment of epilepsy is the use of metabolic therapies such as the ketogenic diet. The diet is known to influence epileptogenesis through a combination of adenosine receptor-independent and dependent mechanisms (Murugan M, 2020). For instance, rats fed with ketogenic diet following status epilepticus, demonstrated reduced development of spontaneous seizures and decreased DNA methylation levels both during diet administration and after the return to standard diet (Kobow et al., 2013). Though a direct link between the ketogenic diet, DNA methylation levels and ADK-L must still be demonstrated, taken collectively, these studies indicate that the lasting effects of the ketogenic diet may be conferred via adenosine-mediated epigenetic regulation of the DNA methylome, supporting a key mechanism implicated in epilepsy and epileptogenesis.
3.4. Vascular diseases
Adenosine receptors and extracellular adenosine have been closely associated with several vascular pathologies including atherosclerosis (Koupenova et al., 2012; Wang et al., 2009), restenosis (Yang et al., 2008), and platelet activation (Amisten et al., 2008; Yang et al., 2010). However, the adenosine-mediated epigenetic mechanisms were overlooked. Recent studies using transgenic animal models with conditional deletion of ADK in specific cellular populations reveals a novel role for ADK as an epigenetic regulator in vascular conditions such as hypoxia-induced angiogenesis, atherosclerosis, vascular inflammation and ischemic injury (Wang et al., 2021; Xu et al., 2017a; Xu et al., 2017b; Zhang et al., 2018) (Figure 4).
Figure 4. ADK mediated epigenetic mechanisms in vascular diseases.
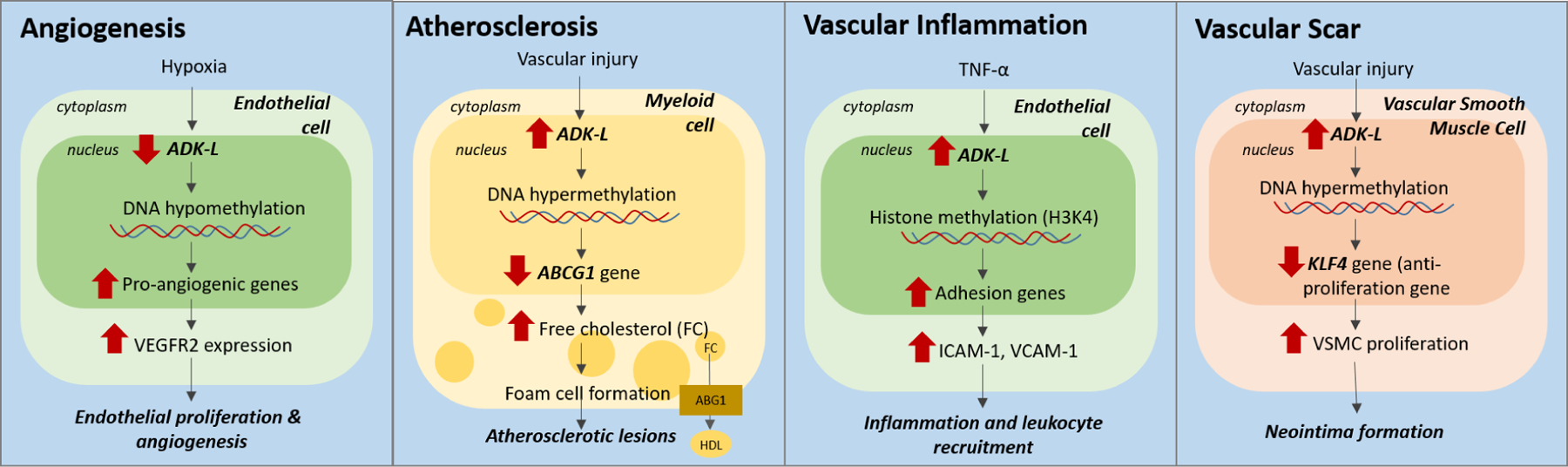
Schematic shows ADK plays an important role as an epigenetic regulator in various vascular pathologies. (i) Hypoxia induces ADK downregulation in endothelial cells, leading to hypomethylation of vascular endothelial growth factor receptor 2 (VEGFR2) and thereby promotes angiogenesis. (ii) In myeloid cells, increase in ADK expression and associated DNA methylation represses the ABCG1 gene, a key regulator of cholesterol trafficking, resulting in the formation of foam cells and atherosclerotic lesions. (iii) In endothelial cells, inflammation (TNF-α)-induced upregulation of ADK promotes histone methylation H3K4 resulting in increased expression of adhesion molecules ICAM-1 and VCAM-1. (iv) In vascular smooth muscle cells (VSMC), ADK expression was associated with DNA hypermethylation and therefore suppression of the KLF4 gene expression, an anti-proliferation gene, resulting in increased VSMC proliferation and neointima formation.
3.4.1. Angiogenesis
Angiogenesis refers to the formation of new blood vessels from pre-existing vessels. It is a common adaptive response to tissue hypoxia and is observed in pathological conditions such as ischemic injury and cancer (Dor and Keshet, 1997; Nishida et al., 2006). Previous work shows that hypoxia is associated with functional inhibition of ADK and prevents the intracellular metabolism of adenosine into AMP (Decking et al., 1997; Morote-Garcia et al., 2009). In line with this, hypoxia induced ADK downregulation in endothelial cells led to hypomethylation of the promoters of a series of pro-angiogenic genes, particularly vascular endothelial growth factor receptor 2 (VEGFR2), promoting angiogenesis and wound healing (Xu et al., 2017b).
3.4.2. Atherosclerosis
Atherosclerosis is a chronic inflammatory vascular disease, characterized by accumulation of foam cells in arterial vessel walls forming plaques. In response to vascular injury, circulating monocytes of myeloid origin are recruited to the injury site, where they differentiate into macrophages and turn into foam cells, a critical step in the initiation of atherosclerosis (Moore et al., 2013). A recent study demonstrated reduced foam cell formation and development of atherosclerotic plaques in mice lacking ADK in myeloid cells (under the LysM promoter) (Zhang et al., 2018). The reduction in foam cell formation was attributed to reduction of methylation of the ABCG1 gene in ADK deficient mice. A reduction of methylation of the ABCG1 gene causes its upregulated expression that results in increased cholesterol efflux and the suppression of pro-inflammatory cytokines (Zhang et al., 2018). The use of A2AR antagonist ZM241385 and A2BR antagonist MRS1754 did not alter ABCG1 gene expression, further confirming that the mechanism is independent of adenosine receptor activation.
3.4.3. Vascular inflammation and neointima formation
Another study showed that ADK deficiency in endothelial cells inhibited inflammation-induced adhesion molecule expression and attenuated leukocyte-endothelial interactions (Xu et al., 2017a). The increased intracellular adenosine from ADK deficiency represses flux through SAM-dependent transmethylation pathways, which leads to reduced histone3 to lysine 4 (H3K4) methylation and thereby the transcription and expression of inflammatory genes encoding E-selectin, ICAM-1, and VCAM-1 (Xu et al., 2017a). A similar study used transgenic mice lacking ADK in vascular smooth muscle cells (VSMCs) to investigate neointima formation, a scar tissue that forms on the inner lining of blood vessels following vascular injury (Wang et al., 2021). The VSMCs migrate into the intima layer after injury, proliferate and produce a large amount of extracellular matrix, resulting in intima formation (Clowes et al., 1983). Genetic and pharmacological inhibition of ADK was sufficient to inhibit VSMC proliferation and thereby attenuate neointima formation (Wang et al., 2021). Mechanistic studies revealed that ADK-deficiency affected the methylation status and increased expression of Krüppel-Like Factor 4 (KLF4), an anti-proliferative gene, causing a reduction in VSMC proliferation. Moreover, increased ADK expression, DNA hypermethylation as well as decreased KLF4 expression were also observed in neointimal VSMCs from stenotic vessels in human femoral arteries suggesting that the preclinical findings are relevant to the human disease and of high translational significance (Wang et al., 2021). Taken together, these studies show that ADK (in myeloid, endothelial or VSMCs) plays an important role as an epigenetic regulator in the development of atherosclerosis and cerebral ischemia/reperfusion injury. Hence ADK-L is a novel and promising therapeutic target for the treatment of vascular diseases.
3.5. Cancer
Cancer is characterized by the occurrence and spread of malignant tumors. Hypermethylation of DNA in cancer has been well established and studied at length (Estecio and Issa, 2011; Huang et al., 2015b; Wolff et al., 2010). In particular, hypermethylation of otherwise non-methylated CpG sites in promoter regions results in silencing relevant tumor suppressor genes. On the other hand, global hypomethylation is also observed in many cancers but is often specific to repetitive elements and influences chromosomal instability as well as transcriptome changes leading to expression of oncogenes (Ehrlich, 2009; Wolff et al., 2010). Aberrant DNA methylation as a hallmark of cancer raises the question of how changes in the transmethylation reaction, and specifically ADK-L-mediated alterations thereof, may influence the maintenance of this disease. The formation, survival and expansion of tumors have strong associations with adenosine metabolism (Boison and Yegutkin, 2019). Recent studies observed a significant increase in the expression of ADK in tumors of patients with glioma and colorectal cancer compared to benign tissue (Giglioni et al., 2008; Huang et al., 2015a). In patients with Grade II and Grade III gliomas, both the isoforms of ADK were increased in the tumor and peritumoral areas (Huang et al., 2015a). In contrast, an early study in rat hepatomas and kidney tumors found an inverse correlation between ADK activity and tumor growth (Jackson et al., 1978). A more recent study noted a global overall reduction in ADK expression in tumors of patients with hepatocellcarcinoma (HCC) with a shift in the relative expression levels of different isoforms of ADK (El-Kharrag et al., 2019). Interestingly, only HCC cases with reduced ADK expression showed recurrence of cancer development, which was not the case in patients with normal ADK levels. Taken together, the correlation of dysregulated ADK expression in cancer tissues suggests a role for adenosine-related mechanisms in carcinogenesis, cancer progression and prognosis. Experimental models have shed some light on the effect ADK has on tumorigenesis. In a preclinical model, transgenic mice with liver ADK deficiency (ADK-Tg, lack endogenous ADK-L and express reduced ADK-S in liver) displayed increased susceptibility to a carcinogen, diethylnitrosamine, and associated mortality (El-Kharrag et al., 2019). On the other hand, augmentation of adenosine inhibited the tumor-initiating potential of breast cancer cells via a receptor-independent mechanism in an in vivo xenograft mouse model (Losenkova et al., 2020). Since angiogenesis is needed for tumorigenesis and survival, the epigenetic regulation of pro-angiogenic genes by ADK is another mechanism by which ADK may be implicated in cancer (Xu et al., 2017b). These findings suggest a potential epigenetic role for ADK in mitogenesis, tumorigenesis, and tumor-associated tissue remodeling and invasion. Whether these mechanisms are mediated by ADK-L and whether there is a direct link between ADK-L and epigenetic changes in the context of tumorigenesis warrants further investigation (Figure 3).
In summary, the association of ADK-L in cell proliferation during development, the correlation of changes in ADK expression in cancerous tissue, and the known epigenetic changes in tumorigenic tissue sets ADK-L up as a potential diagnostic marker as well as a novel therapeutic target for cancer pathologies in which ADK is implicated. The finding that only glioma patients with epilepsy showed upregulated ADK expression in peritumoral tissues which was absent in glioma patients without epilepsy (Huang et al., 2015a), indicates a possible role for ADK in epileptogenesis in these patients. Hence targeting ADK in glioma patients would be beneficial for its anti-tumorigenic properties, as well as, the anti-epileptogenic effects. Further studies should determine the effect of ADK inhibition on DNA methylation in cancer cell lines as well as its effect on cancer cell proliferation and cancer susceptibility in vivo.
3.6. Diabetes
Diabetes is a condition with elevated blood glucose levels or hyperglycemia, resulting from impaired insulin production. It has been proposed that hyperglycemia drives changes in the epigenome resulting in long-term vascular complications, a major cause of morbidity and mortality in type 2 diabetes patients (Rosen et al., 2018). Moreover, genome-wide methylation studies indicate that epigenetic mechanisms can predict nephropathy in type 1 diabetic patients (Bell et al., 2010). Hence, understanding whether and how hyperglycemia-induced changes in chromatin structure is regulated by ADK will be useful in the design of mechanism-based therapeutics, which interfere with long-lasting activating epigenetics and improve patient outcomes in diabetes.
The downregulated expression of ADK has been noted in several experimental models of diabetes (Navarro et al., 2017; Sakowicz-Burkiewicz et al., 2006; Sakowicz and Pawelczyk, 2002). Exogenous insulin administration was able to restore ADK mRNA, protein and enzymatic activity in streptozotocin-induced diabetes mellitus rat, indicating insulin influenced the rate of Adk gene transcription (Sakowicz and Pawelczyk, 2002). Since insulin deficiency and the loss of insulin secreting β-islet cells is the main underlying cause of diabetes (Maedler and Donath, 2004), whether ADK can be targeted to influence insulin production is of therapeutic interest. A small-molecule screening platform identified that ADK inhibitors (5-ITU and ABT-702) were capable of promoting replication of primary β-cells in three species (mouse, rat, and pig) (Annes et al., 2012). However, the ADK inhibitors were only able to induce the proliferation of β-cells but not that of α-cells or fibroblasts. In alignment with this, ADK immunostaining of islet cultures revealed nuclear expression of ADK-L in β-cells, whereas ADK-S was localized to the cytoplasm of fibroblasts and α-cells (Annes et al., 2012). These data suggest that ADK-L regulates β-cell replication following the application of an ADK inhibitor, however whether this process is regulated by epigenetic mechanisms remains to be investigated. Interestingly, a recent study using transgenic mice deficient in ADK specifically in β-cells (under Ins2 promoter) also showed increased pancreatic β-cell proliferation and thereby insulin secretion (Ahmed Abdalhamid Osman et al., 2019). In addition to affecting β-cell proliferation, mice with conditional disruption of ADK expression in β-cells also showed improved glucose tolerance and increased glucose-stimulated insulin secretion in vivo in response to high fat diet, indicating the ability of ADK in regulating insulin tolerance (Navarro et al., 2017). Therefore, modulation of ADK activity is a potential strategy that can not only improve β-cell proliferation and increase insulin secretion, but can also enhance adaptive β-cell response. In summary, the identification of ADK-L as a regulator of β-cell replication and function is an unexpected finding that highlights the therapeutic potential of targeting ADK for the treatment of diabetes.
4. Technical challenges in investigating the epigenetic roles of ADK-L
One of the major challenges in the field is the lack of attention to the roles for specific ADK-isoforms. The isoform specific expression of ADK in tissues can be easily identified using Western blots (Cui et al., 2011; Gebril et al., 2020). With the knowledge on subcellular compartmentalization of the two isoforms, simple immunohistological labeling can be employed to separate the expression of the ADK-L isoform in the nucleus from ADK-S in the cytoplasm (Cui et al., 2009). Currently available ADK inhibitors such as 5-iodotubercidin (5-ITU) and ABT 702 lack isoform-specificity, hence the development of inhibitors with higher selectivity for the nuclear isoform of ADK (ADK-L) is needed. A recent study using a structure-based design approach and molecular dynamics simulation analysis of human ADK, synthesized novel ADK inhibitors (MRS4202, MRS4380 and MRS4203) with higher potency for ADK inhibition (Toti et al., 2016), however their selectivity to specific ADK isoforms remains to be investigated.
The lack of pharmacological tools to dissect the roles of the specific isoforms have led to the reliance on genetic tools to elucidate the functions of the lesser-investigated ADK-L isoform. Most of the experimental studies mentioned in this review use cell-type specific deletion of ADK by crossbreeding ADK flox mice with a cell-type specific Cre, which results in the deletion of both ADK-L and ADK-S isoforms in these cells (Ahmed Abdalhamid Osman et al., 2019; Gebril et al., 2020; Xu et al., 2017a; Xu et al., 2017b). Hence, there is rationale that the observed effects may be due to the indirect effect of ADK-S deficiency. To overcome this drawback, certain studies have taken advantage of the endogenous selectivity in ADK-L expression and combined the use of the ADK flox-cre lines to generate isoform and cell-specific knockout in selective neurogenic neurons (Gebril et al., 2020) and β islet cells(Ahmed Abdalhamid Osman et al., 2019), cells that only express the ADK-L isoform. On the other hand, mice with ADK-L transgene insertion shed light on the gain of function from ADK-L overexpression in addition to the basal endogenous ADK (Gebril et al., 2020). In light of these technical challenges, the use of transgenic tools for investigating ADK-L needs to be carefully considered and the data needs to be cautiously interpreted.
Another important factor to take into consideration when investigating the epigenetic mechanisms regulated by ADK is whether the ADK-S isoform can modulate global DNA methylation. To this end, a study showed that Adk-deficient BHK-AK2 cells expressing ADK-L only showed a robust 400% increase in global DNA methylation, whereas cells with ADK-S showed only a modest 50% increase in global DNA methylation (Williams-Karnesky et al., 2013). These data demonstrate that both isoforms of ADK are involved in the regulation of global DNA methylation. Although the nuclear isoform appears to be more effective in altering the DNA methylation status, the existence of cell-autonomous (nuclear ADK) and non–cell-autonomous (cytoplasmic ADK) effects of ADK is apparent.
5. Conclusion
Based on the body of evidence discussed in this review, the role of ADK-L isoform, as an epigenetic modulator in developmental and pathological conditions is apparent. Dysregulated ADK-L expression and pathogenic DNA methylation seems to be a common feature in many disorders, emphasizing the advantage of targeting ADK for treatment for not just the condition, but also the associated co-morbidities. The link between ADK-L suppression and improved cell proliferation, is another remarkable shared mechanism among conditions such as cancer, TBI, and diabetes. In light of the above, it is possible that epigenetic regulation by ADK forms the very basis of pathologies affecting multiple genes, hence targeting ADK is akin to a globally-acting multi-modal approach that is able to restore complex network function. ADK inhibition although proven beneficial in preclinical studies, has not been translated into clinical success due to concerns associated with liver toxicity (Boison et al., 2002). These toxicity concerns are mainly attributed to the inhibition of ADK-S isoform, which is essential for critical physiological functions (Boison et al., 2002). Hence the development of ADK-L specific inhibitors is not only exciting for its effect on epigenetic regulation but will also overcome the safety concerns, which are currently associated with global ADK inhibition. Further investigations overcoming the technical challenges of investigating the epigenetic functions of ADK-L are needed to confirm the direct link between ADK-L and DNA methylation changes. In conclusion, this review highlights the unprecedented therapeutic potential of targeting ADK and its putative epigenetic functions in various pathological conditions ranging from developmental disorders and epilepsy to vascular diseases and cancer.
Highlights.
ADK is developmentally regulated in a cell-type and isoform specific manner.
The nuclear localization of the ADK-L isoform supports its role as an epigenetic regulator that drives DNA and histone methylation by clearing adenosine.
Epigenetic regulation by ADK is essential for normal neurodevelopment and angiogenesis.
ADK-regulated epigenetic mechanisms are implicated in neurodevelopmental disorders, brain injury, epilepsy, vascular diseases, cancer, and diabetes.
Acknowledgements:
DB gratefully acknowledges research funding support provided by the NIH (NS065957, NS103740) and Citizens United for Research in Epilepsy (DB, CURE Catalyst Award).
Abbreviations:
- 5-ITU
5-iodotubercidin
- 5mC
5-methylcytidine
- ADK
adenosine kinase
- ADK S
adenosine kinase short isoform
- ADK L
adenosine kinase long isoform
- AMP
5′-adenosine-monophosphate
- BHK
baby hamster kidney
- CNT
concentrative nucleoside transporters
- DNMTs
DNA methyltransferases
- ENT
equilibrative nucleoside transporters
- Hcy
homocysteine
- HCC
hepatocellcarcinoma
- KLF4
Krüppel-like factor 4
- SAH
S-adenosylhomocysteine hydrolase
- SAM
S-adenosylmethionine
- SP1
factor specificity protein 1
- TBI
traumatic brain injury
- TLE
temporal lobe epilepsy
- VEGFR2
vascular endothelial growth factor receptor 2
- VSMCs
vascular smooth muscle cells
Footnotes
Conflict of Interest: The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
⊠The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Acharya MM, Baddour AA, Kawashita T, Allen BD, Syage AR, Nguyen TH, Yoon N, Giedzinski E, Yu L, Parihar VK, Baulch JE, 2017. Epigenetic determinants of space radiation-induced cognitive dysfunction. Sci Rep 7, 42885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed Abdalhamid Osman M, Sun YJ, Li RJ, Lin H, Zeng DM, Chen XY, He D, Feng HW, Yang Z, Wang J, Wu C, Cui M, Sun JP, Huo Y, Yu X, 2019. Deletion of pancreatic beta-cell adenosine kinase improves glucose homeostasis in young mice and ameliorates streptozotocin-induced hyperglycaemia. J Cell Mol Med 23, 4653–4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amisten S, Braun OO, Bengtsson A, Erlinge D, 2008. Gene expression profiling for the identification of G-protein coupled receptors in human platelets. Thromb Res 122, 47–57. [DOI] [PubMed] [Google Scholar]
- Annegers JF, Coan SP, 2000. The risks of epilepsy after traumatic brain injury. Seizure 9, 453–457. [DOI] [PubMed] [Google Scholar]
- Annes JP, Ryu JH, Lam K, Carolan PJ, Utz K, Hollister-Lock J, Arvanites AC, Rubin LL, Weir G, Melton DA, 2012. Adenosine kinase inhibition selectively promotes rodent and porcine islet beta-cell replication. Proc Natl Acad Sci U S A 109, 3915–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronica E, Zurolo E, Iyer A, de Groot M, Anink J, Carbonell C, van Vliet EA, Baayen JC, Boison D, Gorter JA, 2011. Upregulation of adenosine kinase in astrocytes in experimental and human temporal lobe epilepsy. Epilepsia 52, 1645–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell CG, Teschendorff AE, Rakyan VK, Maxwell AP, Beck S, Savage DA, 2010. Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med Genomics 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjursell MK, Blom HJ, Cayuela JA, Engvall ML, Lesko N, Balasubramaniam S, Brandberg G, Halldin M, Falkenberg M, Jakobs C, Smith D, Struys E, von Dobeln U, Gustafsson CM, Lundeberg J, Wedell A, 2011. Adenosine kinase deficiency disrupts the methionine cycle and causes hypermethioninemia, encephalopathy, and abnormal liver function. Am J Hum Genet 89, 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, 2006. Adenosine kinase, epilepsy and stroke: mechanisms and therapies. Trends Pharmacol Sci 27, 652–658. [DOI] [PubMed] [Google Scholar]
- Boison D, 2013. Adenosine kinase: exploitation for therapeutic gain. Pharmacol Rev 65, 906–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, 2016. Adenosinergic signaling in epilepsy. Neuropharmacology 104, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Scheurer L, Zumsteg V, Rulicke T, Litynski P, Fowler B, Brandner S, Mohler H, 2002. Neonatal hepatic steatosis by disruption of the adenosine kinase gene. Proc Natl Acad Sci U S A 99, 6985–6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Singer P, Shen HY, Feldon J, Yee BK, 2012. Adenosine hypothesis of schizophrenia--opportunities for pharmacotherapy. Neuropharmacology 62, 1527–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Steinhauser C, 2018. Epilepsy and astrocyte energy metabolism. Glia 66, 1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Yegutkin GG, 2019. Adenosine Metabolism: Emerging Concepts for Cancer Therapy. Cancer Cell 36, 582–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clowes AW, Reidy MA, Clowes MM, 1983. Mechanisms of stenosis after arterial injury. Lab Invest 49, 208–215. [PubMed] [Google Scholar]
- Csoka AB, Szyf M, 2009. Epigenetic side-effects of common pharmaceuticals: a potential new field in medicine and pharmacology. Med Hypotheses 73, 770–780. [DOI] [PubMed] [Google Scholar]
- Cui XA, Agarwal T, Singh B, Gupta RS, 2011. Molecular characterization of Chinese hamster cells mutants affected in adenosine kinase and showing novel genetic and biochemical characteristics. BMC Biochem 12, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui XA, Singh B, Park J, Gupta RS, 2009. Subcellular localization of adenosine kinase in mammalian cells: The long isoform of AdK is localized in the nucleus. Biochem Biophys Res Commun 388, 46–50. [DOI] [PubMed] [Google Scholar]
- Cunha RA, 2005. Neuroprotection by adenosine in the brain: From A(1) receptor activation to A (2A) receptor blockade. Purinergic Signal 1, 111–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Tisi J, Bell GS, Peacock JL, McEvoy AW, Harkness WF, Sander JW, Duncan JS, 2011. The long-term outcome of adult epilepsy surgery, patterns of seizure remission, and relapse: a cohort study. Lancet 378, 1388–1395. [DOI] [PubMed] [Google Scholar]
- Debski KJ, Pitkanen A, Puhakka N, Bot AM, Khurana I, Harikrishnan KN, Ziemann M, Kaspi A, El-Osta A, Lukasiuk K, Kobow K, 2016. Etiology matters - Genomic DNA Methylation Patterns in Three Rat Models of Acquired Epilepsy. Sci Rep 6, 25668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decking UK, Schlieper G, Kroll K, Schrader J, 1997. Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ Res 81, 154–164. [DOI] [PubMed] [Google Scholar]
- Dor Y, Keshet E, 1997. Ischemia-driven angiogenesis. Trends Cardiovasc Med 7, 289–294. [DOI] [PubMed] [Google Scholar]
- Ehrlich M, 2009. DNA hypomethylation in cancer cells. Epigenomics 1, 239–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Kharrag R, Owen R, Boison D, 2019. Adenosine Kinase Deficiency Increases Susceptibility to a Carcinogen. J Caffeine Adenosine Res 9, 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estecio MR, Issa JP, 2011. Dissecting DNA hypermethylation in cancer. FEBS Lett 585, 2078–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedele DE, Gouder N, Guttinger M, Gabernet L, Scheurer L, Rulicke T, Crestani F, Boison D, 2005. Astrogliosis in epilepsy leads to overexpression of adenosine kinase, resulting in seizure aggravation. Brain 128, 2383–2395. [DOI] [PubMed] [Google Scholar]
- Fedele DE, Koch P, Scheurer L, Simpson EM, Mohler H, Brustle O, Boison D, 2004. Engineering embryonic stem cell derived glia for adenosine delivery. Neurosci Lett 370, 160–165. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Arslan G, Halldner L, Kull B, Schulte G, Wasserman W, 2000. Structure and function of adenosine receptors and their genes. Naunyn Schmiedebergs Arch Pharmacol 362, 364–374. [DOI] [PubMed] [Google Scholar]
- Gebril HM, Rose RM, Gesese R, Emond MP, Huo Y, Aronica E, Boison D, 2020. Adenosine kinase inhibition promotes proliferation of neural stem cells after traumatic brain injury. Brain Commun 2, fcaa017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giglioni S, Leoncini R, Aceto E, Chessa A, Civitelli S, Bernini A, Tanzini G, Carraro F, Pucci A, Vannoni D, 2008. Adenosine kinase gene expression in human colorectal cancer. Nucleosides Nucleotides Nucleic Acids 27, 750–754. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Fernandez E, Sanchez-Gomez MV, Perez-Samartin A, Arellano RO, Matute C, 2014. A3 Adenosine receptors mediate oligodendrocyte death and ischemic damage to optic nerve. Glia 62, 199–216. [DOI] [PubMed] [Google Scholar]
- Gouder N, Scheurer L, Fritschy JM, Boison D, 2004. Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis. J Neurosci 24, 692–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DR, Cornatzer WE, Duerre JA, 1979. Relationship between tissue levels of S-adenosylmethionine, S-adenylhomocysteine, and transmethylation reactions. Can J Biochem 57, 56–65. [DOI] [PubMed] [Google Scholar]
- Huang J, He Y, Chen M, Du J, Li G, Li S, Liu W, Long X, 2015a. Adenosine deaminase and adenosine kinase expression in human glioma and their correlation with gliomaassociated epilepsy. Mol Med Rep 12, 6509–6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WY, Hsu SD, Huang HY, Sun YM, Chou CH, Weng SL, Huang HD, 2015b. MethHC: a database of DNA methylation and gene expression in human cancer. Nucleic Acids Res 43, D856–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RC, Morris HP, Weber G, 1978. Adenosine deaminase and adenosine kinase in rat hepatomas and kidney tumours. Br J Cancer 37, 701–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z, Liu Y, 2018. DNA methylation in human diseases. Genes Dis 5, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jue K, Bestor TH, Trasler JM, 1995. Regulated synthesis and localization of DNA methyltransferase during spermatogenesis. Biol Reprod 53, 561–569. [DOI] [PubMed] [Google Scholar]
- Kiese K, Jablonski J, Boison D, Kobow K, 2016. Dynamic Regulation of the Adenosine Kinase Gene during Early Postnatal Brain Development and Maturation. Front Mol Neurosci 9, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloor D, Hermes M, Fink K, Schmid H, Klingel K, Mack A, Grenz A, Osswald H, 2007. Expression and localization of S-adenosylhomocysteine-hydrolase in the rat kidney following carbon monoxide induced hypoxia. Cell Physiol Biochem 19, 57–66. [DOI] [PubMed] [Google Scholar]
- Kobow K, Blumcke I, 2011. The methylation hypothesis: do epigenetic chromatin modifications play a role in epileptogenesis? Epilepsia 52 Suppl 4, 15–19. [DOI] [PubMed] [Google Scholar]
- Kobow K, El-Osta A, Blumcke I, 2013. The methylation hypothesis of pharmacoresistance in epilepsy. Epilepsia 54 Suppl 2, 41–47. [DOI] [PubMed] [Google Scholar]
- Koupenova M, Johnston-Cox H, Vezeridis A, Gavras H, Yang D, Zannis V, Ravid K, 2012. A2b adenosine receptor regulates hyperlipidemia and atherosclerosis. Circulation 125, 354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kredich NM, Martin DV Jr., 1977. Role of S-adenosylhomocysteine in adenosinemediated toxicity in cultured mouse T lymphoma cells. Cell 12, 931–938. [DOI] [PubMed] [Google Scholar]
- Li H, Smolen GA, Beers LF, Xia L, Gerald W, Wang J, Haber DA, Lee SB, 2008a. Adenosine transporter ENT4 is a direct target of EWS/WT1 translocation product and is highly expressed in desmoplastic small round cell tumor. PLoS One 3, e2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Ren G, Lusardi T, Wilz A, Lan JQ, Iwasato T, Itohara S, Simon RP, Boison D, 2008b. Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice. J Clin Invest 118, 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losenkova K, Zuccarini M, Karikoski M, Laurila J, Boison D, Jalkanen S, Yegutkin GG, 2020. Compartmentalization of adenosine metabolism in cancer cells and its modulation during acute hypoxia. J Cell Sci 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Liu X, Deng Y, Qing H, 2013. DNA methylation, a hand behind neurodegenerative diseases. Front Aging Neurosci 5, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maedler K, Donath MY, 2004. Beta-cells in type 2 diabetes: a loss of function and mass. Horm Res 62 Suppl 3, 67–73. [DOI] [PubMed] [Google Scholar]
- Masino SA, Kawamura M Jr., Cote JL, Williams RB, Ruskin DN, 2013. Adenosine and autism: a spectrum of opportunities. Neuropharmacology 68, 116–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino SA, Li T, Theofilas P, Sandau US, Ruskin DN, Fredholm BB, Geiger JD, Aronica E, Boison D, 2011. A ketogenic diet suppresses seizures in mice through adenosine A(1) receptors. J Clin Invest 121, 2679–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffatt BA, Stevens YY, Allen MS, Snider JD, Pereira LA, Todorova MI, Summers PS, Weretilnyk EA, Martin-McCaffrey L, Wagner C, 2002. Adenosine kinase deficiency is associated with developmental abnormalities and reduced transmethylation. Plant Physiol 128, 812–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KJ, Sheedy FJ, Fisher EA, 2013. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol 13, 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morote-Garcia JC, Rosenberger P, Nivillac NM, Coe IR, Eltzschig HK, 2009. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology 136, 607–618. [DOI] [PubMed] [Google Scholar]
- Mueller BK, Mueller R, Schoemaker H, 2009. Stimulating neuroregeneration as a therapeutic drug approach for traumatic brain injury. Br J Pharmacol 157, 675–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugan MBD, 2020. Ketogenic diet, neuroprotection, and antiepileptogenesis. Epilepsy Research. 167: 106444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro G, Abdolazimi Y, Zhao Z, Xu H, Lee S, Armstrong NA, Annes JP, 2017. Genetic Disruption of Adenosine Kinase in Mouse Pancreatic beta-Cells Protects Against High-Fat Diet- Induced Glucose Intolerance. Diabetes 66, 1928–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida N, Yano H, Nishida T, Kamura T, Kojiro M, 2006. Angiogenesis in cancer. Vasc Health Risk Manag 2, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne DM, Sandau US, Jones AT, Vander Velden JW, Weingarten AM, Etesami N, Huo Y, Shen HY, Boison D, 2018. Developmental role of adenosine kinase for the expression of sex-dependent neuropsychiatric behavior. Neuropharmacology 141, 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Gupta RS, 2008. Adenosine kinase and ribokinase--the RK family of proteins. Cell Mol Life Sci 65, 2875–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawelczyk T, Sakowicz M, Szczepanska-Konkel M, Angielski S, 2000. Decreased expression of adenosine kinase in streptozotocin-induced diabetes mellitus rats. Arch Biochem Biophys 375, 1–6. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Maysami S, Studer FE, Wilz A, Simon RP, Boison D, 2008. Downregulation of hippocampal adenosine kinase after focal ischemia as potential endogenous neuroprotective mechanism. J Cereb Blood Flow Metab 28, 17–23. [DOI] [PubMed] [Google Scholar]
- Rosen ED, Kaestner KH, Natarajan R, Patti ME, Sallari R, Sander M, Susztak K, 2018. Epigenetics and Epigenomics: Implications for Diabetes and Obesity. Diabetes 67, 1923–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowling MJ, McMullen MH, Chipman DC, Schalinske KL, 2002. Hepatic glycine N-methyltransferase is up-regulated by excess dietary methionine in rats. J Nutr 132, 2545–2550. [DOI] [PubMed] [Google Scholar]
- Saavedra OM, Isakovic L, Llewellyn DB, Zhan L, Bernstein N, Claridge S, Raeppel F, Vaisburg A, Elowe N, Petschner AJ, Rahil J, Beaulieu N, MacLeod AR, Delorme D, Besterman JM, Wahhab A, 2009. SAR around (l)-S-adenosyl-l-homocysteine, an inhibitor of human DNA methyltransferase (DNMT) enzymes. Bioorg Med Chem Lett 19, 2747–2751. [DOI] [PubMed] [Google Scholar]
- Saitoh M, Nagai K, Nakagawa K, Yamamura T, Yamamoto S, Nishizaki T, 2004. Adenosine induces apoptosis in the human gastric cancer cells via an intrinsic pathway relevant to activation of AMP-activated protein kinase. Biochem Pharmacol 67, 2005–2011. [DOI] [PubMed] [Google Scholar]
- Sakowicz-Burkiewicz M, Kocbuch K, Grden M, Szutowicz A, Pawelczyk T, 2006. Diabetes induced decrease of adenosine kinase expression impairs the proliferation potential of diabetic rat T lymphocytes. Immunology 118, 402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakowicz M, Pawelczyk T, 2002. Insulin restores expression of adenosine kinase in streptozotocin-induced diabetes mellitus rats. Mol Cell Biochem 236, 163–171. [DOI] [PubMed] [Google Scholar]
- Sandau US, Yahya M, Bigej R, Friedman JL, Saleumvong B, Boison D, 2019. Transient use of a systemic adenosine kinase inhibitor attenuates epilepsy development in mice. Epilepsia 60, 615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HY, Lusardi TA, Williams-Karnesky RL, Lan JQ, Poulsen DJ, Boison D, 2011. Adenosine kinase determines the degree of brain injury after ischemic stroke in mice. J Cereb Blood Flow Metab 31, 1648–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HY, Singer P, Lytle N, Wei CJ, Lan JQ, Williams-Karnesky RL, Chen JF, Yee BK, Boison D, 2012. Adenosine augmentation ameliorates psychotic and cognitive endophenotypes of schizophrenia. J Clin Invest 122, 2567–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B, Hao W, Wu Z, Eigl B, Gupta RS, 1996. Cloning and characterization of cDNA for adenosine kinase from mammalian (Chinese hamster, mouse, human and rat) species. High frequency mutants of Chinese hamster ovary cells involve structural alterations in the gene. Eur J Biochem 241, 564–571. [DOI] [PubMed] [Google Scholar]
- Spychala J, Datta NS, Takabayashi K, Datta M, Fox IH, Gribbin T, Mitchell BS, 1996. Cloning of human adenosine kinase cDNA: sequence similarity to microbial ribokinases and fructokinases. Proc Natl Acad Sci U S A 93, 1232–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staufner C, Lindner M, Dionisi-Vici C, Freisinger P, Dobbelaere D, Douillard C, Makhseed N, Straub BK, Kahrizi K, Ballhausen D, la Marca G, Kolker S, Haas D, Hoffmann GF, Grunert SC, Blom HJ, 2016. Adenosine kinase deficiency: expanding the clinical spectrum and evaluating therapeutic options. J Inherit Metab Dis 39, 273–283. [DOI] [PubMed] [Google Scholar]
- Studer FE, Fedele DE, Marowsky A, Schwerdel C, Wernli K, Vogt K, Fritschy JM, Boison D, 2006. Shift of adenosine kinase expression from neurons to astrocytes during postnatal development suggests dual functionality of the enzyme. Neuroscience 142, 125–137. [DOI] [PubMed] [Google Scholar]
- Toti KS, Osborne D, Ciancetta A, Boison D, Jacobson KA, 2016. South (S)- and North (N)- Methanocarba-7-Deazaadenosine Analogues as Inhibitors of Human Adenosine Kinase. J Med Chem 59, 6860–6877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, Shin JJ, Harbom KM, Beaty R, Pappou E, Harris J, Yen RW, Ahuja N, Brock MV, Stearns V, Feller-Kopman D, Yarmus LB, Lin YC, Welm AL, Issa JP, Minn I, Matsui W, Jang YY, Sharkis SJ, Baylin SB, Zahnow CA, 2012. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell 21, 430–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya A, Kanno T, Saito M, Miyoshi Y, Gotoh A, Nakano T, Nishizaki T, 2012. Intracellularly transported adenosine induces apoptosis in [corrected] MCF-7 human breast cancer cells by accumulating AMID in the nucleus. Cancer Lett 321, 65–72. [DOI] [PubMed] [Google Scholar]
- Verellen RM, Cavazos JE, 2010. Post-traumatic epilepsy: an overview. Therapy 7, 527–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Versen-Hoynck F, Rajakumar A, Bainbridge SA, Gallaher MJ, Roberts JM, Powers RW, 2009. Human placental adenosine receptor expression is elevated in preeclampsia and hypoxia increases expression of the A2A receptor. Placenta 30, 434–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zhang W, Zhu C, Bucher C, Blazar BR, Zhang C, Chen JF, Linden J, Wu C, Huo Y, 2009. Inactivation of the adenosine A2A receptor protects apolipoprotein E-deficient mice from atherosclerosis. Arterioscler Thromb Vasc Biol 29, 1046–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xu Y, Yan S, Cao K, Zeng X, Zhou Y, Liu Z, Yang Q, Pan Y, Wang X, Boison D, Su Y, Jiang X, Patel VS, Fulton D, Weintraub NL, Huo Y, 2021. Adenosine kinase is critical for neointima formation after vascular injury by inducing aberrant DNA hypermethylation. Cardiovasc Res. [DOI] [PMC free article] [PubMed]
- Wang Z, Tang B, He Y, Jin P, 2016. DNA methylation dynamics in neurogenesis. Epigenomics 8, 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams-Karnesky RL, Sandau US, Lusardi TA, Lytle NK, Farrell JM, Pritchard EM, Kaplan DL, Boison D, 2013. Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J Clin Invest 123, 3552–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff EM, Byun HM, Han HF, Sharma S, Nichols PW, Siegmund KD, Yang AS, Jones PA, Liang G, 2010. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet 6, e1000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Wang Y, Yan S, Yang Q, Zhou Y, Zeng X, Liu Z, An X, Toque HA, Dong Z, Jiang X, Fulton DJ, Weintraub NL, Li Q, Bagi Z, Hong M, Boison D, Wu C, Huo Y, 2017a. Regulation of endothelial intracellular adenosine via adenosine kinase epigenetically modulates vascular inflammation. Nat Commun 8, 943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Wang Y, Yan S, Zhou Y, Yang Q, Pan Y, Zeng X, An X, Liu Z, Wang L, Xu J, Cao Y, Fulton DJ, Weintraub NL, Bagi Z, Hoda MN, Wang X, Li Q, Hong M, Jiang X, Boison D, Weber C, Wu C, Huo Y, 2017b. Intracellular adenosine regulates epigenetic programming in endothelial cells to promote angiogenesis. EMBO Mol Med 9, 1263–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang AN, Zhang HP, Sun Y, Yang XL, Wang N, Zhu G, Zhang H, Xu H, Ma SC, Zhang Y, Li GZ, Jia YX, Cao J, Jiang YD, 2015. High-methionine diets accelerate atherosclerosis by HHcy-mediated FABP4 gene demethylation pathway via DNMT1 in ApoE(−/−) mice. FEBS Lett 589, 3998–4009. [DOI] [PubMed] [Google Scholar]
- Yang D, Chen H, Koupenova M, Carroll SH, Eliades A, Freedman JE, Toselli P, Ravid K, 2010. A new role for the A2b adenosine receptor in regulating platelet function. J Thromb Haemost 8, 817–827. [DOI] [PubMed] [Google Scholar]
- Yang D, Koupenova M, McCrann DJ, Kopeikina KJ, Kagan HM, Schreiber BM, Ravid K, 2008. The A2b adenosine receptor protects against vascular injury. Proc Natl Acad Sci U S A 105, 792–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younus I, Reddy DS, 2017. Epigenetic interventions for epileptogenesis: A new frontier for curing epilepsy. Pharmacol Ther 177, 108–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Zeng X, Yang Q, Xu J, Liu Z, Zhou Y, Cao Y, Zhang X, An X, Xu Y, Huang L,Han Z, Wang T, Wu C, Fulton DJ, Weintraub NL, Hong M, Huo Y, 2018. Ablation of Myeloid ADK (Adenosine Kinase) Epigenetically Suppresses Atherosclerosis in ApoE(−/−) (Apolipoprotein E Deficient) Mice. Arterioscler Thromb Vasc Biol 38, 2780–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]