

Abstract
Genome-editing technologies that enable the introduction of precise changes in DNA sequences have the potential to lead to a new class of treatments for genetic diseases. Epidermolysis bullosa (EB) is a group of rare genetic disorders characterized by extreme skin fragility. The recessive dystrophic subtype of EB (RDEB), which has one of the most severe phenotypes, is caused by mutations in COL7A1. In this study, we report a gene-editing approach for ex vivo homology-directed repair (HDR)-based gene correction that uses the CRISPR-Cas9 system delivered as a ribonucleoprotein (RNP) complex in combination with donor DNA templates delivered by adeno-associated viral vectors (AAVs). We demonstrate sufficient mutation correction frequencies to achieve therapeutic benefit in primary RDEB keratinocytes containing different COL7A1 mutations as well as efficient HDR-mediated COL7A1 modification in healthy cord blood-derived CD34+ cells and mesenchymal stem cells (MSCs). These results are a proof of concept for HDR-mediated gene correction in different cell types with therapeutic potential for RDEB.
Keywords: epidermolysis, genome editing, CRISPR, AAV, skin, HDR, RDEB, gene therapy
Graphical abstract
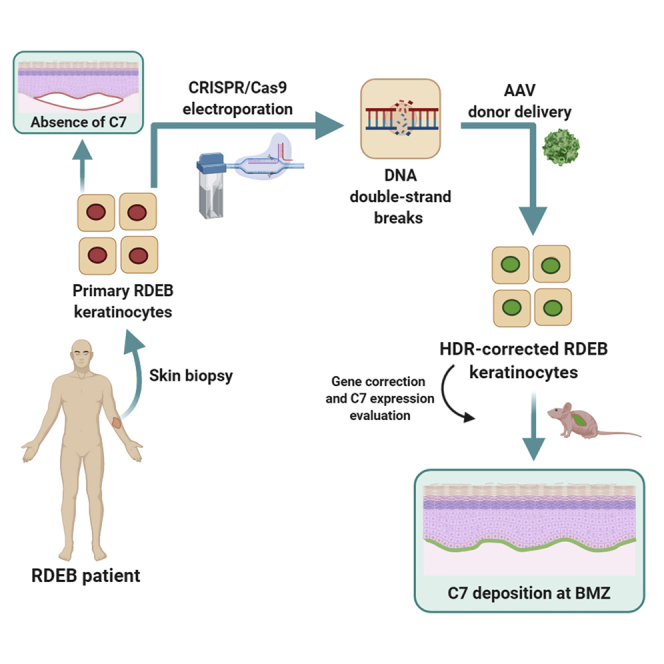
By using CRISPR-Cas9 delivered as a ribonucleoprotein complex in combination with donor DNA templates delivered by adeno-associated viral vectors, Bonafont and colleagues describe highly efficient HDR-mediated gene correction in recessive dystrophic epidermolysis bullosa patient cells. This advanced therapy method opens the way to genome-editing treatments for genetic skin disease.
Introduction
Epidermolysis bullosa (EB) is a group of rare genetic diseases characterized by skin fragility. The generalized severe recessive dystrophic EB subtype (RDEB-sev gen) displays an extremely severe phenotype, with clinical manifestations including blistering of the skin and mucosa, pseudosyndactyly, and a high risk of developing metastatic squamous cell carcinoma. Mutations throughout COL7A1, the gene encoding type VII collagen (C7), are present in all RDEB patients, making this gene an important target for precision medicine therapies.1
In recent years, different site-specific nucleases able to perform double-stranded DNA breaks (DSBs), such as meganucleases, zinc-finger nucleases, transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated protein 9 (Cas9), have been used as tools to induce gene sequence modification. Genome-editing approaches take advantage of cellular DNA repair mechanisms triggered by nuclease-induced DSBs to introduce insertions or deletions (indels) in the gene sequence when DNA repair occurs through the non-homologous end joining (NHEJ) pathway or to precisely correct it by means of a DNA donor repair template when DSBs are repaired via the homology-directed repair (HDR) pathway. NHEJ is the most common DNA repair mechanism, but recent advances in molecular tools have increased the efficiency of HDR-based gene correction strategies, allowing for more precise modifications.2, 3, 4
Most cellular and molecular therapy protocols for EB have focused on keratinocytes and fibroblasts, the main skin cell types. HDR-based protocols using TALENs and DNA oligonucleotide donors demonstrated gene correction in fibroblasts derived from RDEB patients, albeit with limited efficacy (2%).5 Similar approaches using meganucleases6 or minicircle-based CRISPR-Cas9 delivery7 achieved comparable success. Higher gene-editing rates have been reported using integration-deficient lentiviral vectors both for the delivery of CRISPR-Cas9 and donor DNA to correct a mutation in exon 2 of COL7A1 in keratinocytes and fibroblasts.8 Recently, efficient HDR-based COL7A1 correction has been achieved in induced pluripotent stem cells derived from RDEB patient cells, showing bi-allelic correction in about 60% of treated cells.9 HDR-based strategies have also been deployed to restore the expression of LAMB3, one of the genes whose loss of function causes junctional EB (JEB), by using adenoviral vectors for expression of CRISPR-Cas9 in immortalized keratinocytes.10 Although these approaches represent significant contributions to the establishment of protocols for the therapy of different genodermatoses by HDR-based gene editing, gene correction has not yet been reported within useful therapeutic ranges in somatic stem cells derived from patients.
Our group recently demonstrated the highly efficient NHEJ-based restoration of C7 expression after administration of dual guide CRISPR-Cas9 ribonucleoproteins (RNPs) in primary RDEB keratinocytes to delete an exon of COL7A1 that contains a pathogenic mutation.11 Although this approach has clear clinical translation potential for RDEB patients carrying mutations in small exons within the COL7A1 collagenous domain, HDR-based protocols offer more accurate gene sequence correction. Furthermore, a single set of HDR gene-editing tools could allow correction of many different mutations, provided they are within the length of the designed donor template, which could be useful for a larger cohort of RDEB patients. In this context, adeno-associated viral vectors (AAVs), which mediate highly efficient delivery of single-stranded DNA (ssDNA) and promote higher HDR rates than integration-deficient lentiviral vectors (IDLVs) without compromising biosafety, have taken the lead as vectors for donor template delivery. Hence, the combination of AAVs with CRISPR-Cas9 is a promising genome-editing toolkit for HDR-mediated correction in primary patient cells.
Due to the extensive multi-organ involvement in patients with EB, complementary clinical protocols to skin gene therapy have been attempted.12, 13, 14, 15, 16, 17 Recently, hematopoietic stem cell transplantation (HSCT) with subsequent systemic infusions of mesenchymal stem cells (MSCs) ameliorated the RDEB phenotype.12 Thus, given the potential therapeutic value of CD34+ cells and MSCs in EB management, we have also explored HDR-based gene editing in these cell types, achieving high modification rates comparable to those reached in primary keratinocytes.
Altogether, our data prove the feasibility of efficient ex vivo marker-free HDR-based gene modification of different cell types relevant to RDEB treatment.
Results
Highly efficient DNA nuclease activity in the vicinity of the c.6527insC mutation using a CRISPR-Cas9 RNP complex delivered in primary RDEB cells
Since HDR is greatly enhanced by generating DSBs near target sites, we tested the DNA cutting efficiency of CRISPR-Cas9 RNPs with an RNA guide, sg2, which had been previously validated.11 For this purpose, we used a chemically modified, single-molecule guide RNA (sgRNA) rather than the dual component CRISPR RNA (crRNA):trans-activating crRNA (tracrRNA) guide used in our previous study. This guide targets a sequence in COL7A1 intron 79 and directs cleavage 7 bp upstream of exon 80 (Figure 1A). Sg2-RNPs were delivered by electroporation into primary RDEB keratinocytes derived from three different donors (patient 1 [P1] and P3 were homozygous for the c.6527insC mutation in exon 80, and P2 was homozygous for the c.6501G>T mutation in exon 79) and NHEJ-mediated indel generation was assessed by Tracking of Indels by Decomposition (TIDE) analysis of Sanger sequencing chromatograms of PCR amplicons spanning the target region (Figure 1B). This analysis showed that up to 94% of COL7A1 alleles carried indels in the predicted position in RNP-nucleofected primary keratinocytes. The most frequent indel variant was a T insertion at the CRISPR cutting site (c.6502-7insT) (Figure 1C).
Figure 1.

HDR-based COL7A1 gene-editing strategy
(A) HDR-based strategy using AAV6 for donor template delivery and electroporation of CRISPR-Cas9 RNPs. CRISPR-Cas9-induced double-stranded (ds) breaks in the proximity of pathogenic mutations trigger HDR repair. The sg2 ds break is indicated (ray). LHA, left homology arm; RHA, right homology arm. (B) TIDE analysis of indel generation within intron 79 of COL7A1 in primary keratinocytes nucleofected with CRISPR-Cas9 RNPs from three RDEB donors. Estimated percentage of sequences carrying insertions or deletions is shown. Mean ± SD (bars) are shown. (C) Frequencies of most common indel variants generated by sg2 CRISPR-Cas9 RNP.
AAV transduction optimization and HDR-based gene correction estimation in primary RDEB keratinocytes
To assess the most suitable AAV serotype for patient keratinocyte transduction with donor template DNA, we first evaluated the efficiency of a wide array of AAV serotypes (AAVs 1, 2, 5, 6, 7, 8, 9, and DJ; Figure 2A) to transduce human keratinocytes with a construct for GFP expression. Flow cytometry analysis revealed that AAV6 was the most effective serotype and was therefore used to package constructs for donor DNA delivery.
Figure 2.

AAV transduction and genotyping of RDEB keratinocytes after AAV and RNP treatment
(A) Transduction efficacy of eight different AAV serotypes in primary keratinocytes. Percentage of GFP+ cells determined by flow cytometry for each serotype is shown. AAV6 donor vector was used at a an MOI of 30,000. Mean ± SD (bars) are shown. (B) Common mutations in COL7A1 region contained in AAV donor template constructs; exons are represented as red boxes. Symmetrical (Sym) and asymmetrical (Asym) vector designs are shown; exons are represented as blue boxes. Primers used for PCR genotyping were F (out) and R (in) (purple arrows). (C) PCR genotyping in primary RDEB keratinocytes treated with the gene-editing protocol. Duplicate experiments for each condition were performed. Upper (filled arrowhead) and lower (open arrowhead) bands correspond to non-recombinant and intron 79-lacking recombinant alleles, respectively; recombinants were found only in cells treated with both AAV-donor template and CRISPR-Cas9 RNP. Frequencies of HDR are shown below. R1, R2, experimental replicates 1 and 2.
After optimization of the electroporation and transduction conditions (Figure S1), RDEB primary keratinocytes were electroporated with the sg2 RNP and subsequently transduced with AAV6 vectors carrying the HDR donor templates. Two different donor template designs covering different exons were tested: one with symmetric (Sym) and the other with asymmetric (Asym) homology arms, comprising COL7A1 exons 74–84 and 77–88, respectively (Figure 2B). The genomic region composed of both vectors together covers 35% of the mutations described in the DEB registry.18 For both designs, homology arms flanked a fusion of exons 79 and 80, so that the recombinant allele would lack intron 79, thus avoiding the appearance of indels in the recombinant allele and providing a simple genotyping strategy. The experiments were performed in duplicate. The DNAs of the edited keratinocytes were analyzed by PCR amplification of a region comprising exons 79 and 80. A smaller molecular weight band of a size consistent with the recombinant allele was detected only in samples from keratinocytes treated with both AAV-donor templates and the CRISPR-Cas9 RNP (Figure 2C). Cloning and Sanger sequencing of the PCR products showed a correction frequency of between 35% and 50% for the c.6527insC mutation in exon 80. No significant difference in gene correction efficacy was observed between the two donor templates (Figure 2C).
To further characterize the HDR-based correction of the epidermal stem cell compartment, we isolated 22 clones with proliferative ability. PCR genotyping showed that 5 out of 21 clones analyzed had been biallelically corrected (Figure S2).
C7 expression after HDR-based correction in RDEB primary keratinocytes
Highly efficient COL7A1 gene correction should lead to C7 expression in a significant proportion of patient cells within the bulk population of edited keratinocytes. To verify C7 restoration after HDR gene editing, we analyzed C7 expression by immunofluorescence and western blot in RDEB keratinocytes treated with the sg2 RNP and transduced with the two different donor templates. The number of C7-expressing cells detected by immunofluorescence analysis (Figure 3A) was consistent with the high frequency of HDR-mediated gene correction shown by PCR and Sanger sequencing (approximately 40%) (Figure 2C). To verify that the highly proliferative cell pool within the treated keratinocyte population had been targeted, we performed anti-C7 and Ki-67 co-staining in treated P1 keratinocytes, and found abundant co-localization demonstrating the growth potential of corrected cells (Figure S3).
Figure 3.

C7 expression in gene-corrected RDEB polyclonal keratinocytes population
RDEB primary keratinocytes were treated with CRISPR-Cas9 and donor template containing AAV6, and C7 expression restoration was assessed by immunofluorescence and western blot from cellular extracts. (A) C7 immunofluorescence analysis. Left, upper panel: positive control, healthy donor keratinocytes (Co). Right upper panel: untreated RDEB P1 keratinocytes, showing lack of C7 expression. Left, lower panel: AAV6-symmetrical donor plus RNP-treated RDEB keratinocytes. Right lower panel: AAV6-asymmetrical donor template plus RNP-treated RDEB keratinocytes. Scale bars, 50 μm. (B) Western blot analysis of C7 restoration in untreated (P1), healthy (Co), and treated RDEB patient cells (two experimental replicates). Graphs show C7 expression normalized to vinculin and relative to control as mean ± SD (n = 3). ∗∗p < 0.01. (C) NGS analysis of COL7A1 transcripts in gene-edited keratinocyte populations. Precisely corrected transcripts (green) are the most abundant in all of the treated samples. Transcripts lacking E80 (blue) are also present. Non-corrected transcripts are shown in red.
Consequently, western blot analysis of extracts from patient cells treated with AAVs and Cas9 RNPs showed that C7 expression after correction with both donor templates was similar to that found in the keratinocyte sample from healthy donors (Figure 3B). This shows that both tested donor template designs are useful for correcting primary RDEB cells, thereby potentially increasing the population of RDEB patients who could benefit from this gene therapy strategy.
To further study COL7A1 gene expression after HDR correction, we analyzed the COL7A1 transcripts by RT-PCR and next-generation sequencing (NGS) and found that the accurately corrected transcripts in the treated samples ranged from 60% to 80% (Figure 3C). This frequency is higher than the DNA modification determined by PCR (Figure 2C), probably because corrected transcripts are more stable than un-corrected ones, which are degraded by the nonsense-mediated mRNA decay (NMD) cellular mechanism. Our analysis also revealed the presence of transcripts lacking exon 80 (ΔE80), most likely as a consequence of indel generation by the sg2 guide, which can potentially affect the splice acceptor site at the 5′ of the exon. Transcripts lacking mutant exon 80 would thus contribute to the detected total C7 protein. Taken together, in all of the treated samples, at least 85% of the COL7A1 mRNA expression could lead to functional C7, taking into account both internally truncated and full-length variants (Figure 3C). However, in a fraction of treated cells that co-express both variants, C7 secretion might be hampered.
Long-term skin engraftment of gene-corrected RDEB keratinocytes and expression of C7 in regenerated skin
To determine whether the observed high frequency of gene correction is sufficient to achieve skin adhesion restoration, in vivo skin regeneration assays were performed. Healthy, non-treated, and bulk populations of gene-edited patient keratinocytes, combined with COL7A1-null RDEB fibroblasts from the same RDEB patient, were used to generate skin equivalents that were transplanted onto immunodeficient mice. Histological analysis showed normal skin architecture in grafts from healthy and gene-edited keratinocytes 12 weeks after transplantation (Figures 4C and 4E), while blisters were observed in grafts from untreated P1 keratinocytes (Figure 4A). Immunohistochemical analysis did not detect C7 in regenerated tissue from untreated RDEB P1 keratinocytes (Figure 4B). In contrast, grafts from edited RDEB keratinocytes (P1 HDR) displayed C7 continuous deposition along the basement membrane zone (BMZ) of the regenerated skin (Figure 4D; Figure S4), similar to grafts regenerated from healthy donor keratinocytes (Figure 4F). All of the tissue samples showed expression of human involucrin, thus demonstrating human skin regeneration in mice (insets in Figures 4A, 4C, and 4E).
Figure 4.

Histological appearance and C7 expression in gene-corrected grafts
(A–F) Sections from representative grafts generated from (A and B) uncorrected RDEB (P1), (C and D) HDR-corrected RDEB (P1 HDR), and (E and F) healthy donor control (Co) cells. (A, C, and E) Hematoxylin and eosin (H&E) staining; insets show immunohistochemistry (IHC) detection of human involucrin. (B, D, and F) Anti-C7 IHC staining. The asterisks in (A) and (B) and the inset in (A) indicate the epidermal-dermal separation in uncorrected grafts. Scale bars, 100 μm.
HDR-based correction in RDEB patient cells with a mutation in exon 79 of COL7A1
The different donor templates tested covered a large number of exons within the COL7A1 gene, which potentially enables gene correction for different RDEB-causing mutations. Therefore, after demonstrating high correction efficacy in cells from P1, who has a mutation in exon 80,19 we tested the exon 79–80 fusion strategy to correct cells from a patient carrying a biallelic mutation in E79 (P2) that results in near complete absence of C7 expression (Figure S5). PCR genotyping showed correction rates similar to those achieved with P1 cells (Figure 5A). Consistently, a high proportion of these treated P2 keratinocytes were C7-positive, as shown by immunofluorescence analysis (Figure 5B). Detection of extracellular and intracellular C7 by western blot analysis indicated that the restored C7 protein was properly secreted (Figure 5C).
Figure 5.

Gene-editing frequencies and evaluation of C7 expression and proliferation status in P2 keratinocytes
(A) PCR genotyping of P2 and P1 RDEB cells treated with RNP plus donor template-detected uncorrected (upper band, filled arrowhead) and corrected (lower band, open arrowhead) alleles. Similar gene correction rates (shown below) were observed in cells from both patients. C+ HK P1, HDR-corrected P1. IX and λ are molecular weight markers. (B) C7 immunofluorescence (red) detection after genome-editing treatment (Sym and Asym). Ki-67 immunostaining (green) shows proliferative status. Nuclei visualized by DAPI (blue). Scale bars, 100 μm. (C) Intracellular and extracelullar (s-C7) C7 expression determined by western blot analysis in treated cells.
HDR-based gene editing in CB-CD34+ cells and BM-MSCs as potential sources for systemic infusion of autologous stem cells
Allogenic HSCT has been considered as a therapeutic option for EB treatment. Although HSCT could offer benefits for symptom amelioration, it is a high-risk procedure with a high probability of severe complications. However, autologous HSCT using gene-corrected cells could offer a safer therapeutic alternative to treat EB systemically. Therefore, we decided to treat cord blood (CB)-isolated CD34+ cells and bone marrow-derived MSCs (BM-MSCs) from three healthy donors with the same HDR toolkit successfully tested in keratinocytes. Five days after treatment, HDR-mediated gene modification in CD34+ cells was assessed by PCR. Forty-eight percent of COL7A1 alleles were found to be modified, a frequency of gene modification similar to that found in treated keratinocytes. Two different AAV6 MOIs were tested, 5,000 and 10,000, in cells from three healthy donors, showing similar modification rates in all treated samples (Figure 6A).
Figure 6.

Gene editing-based correction in CB-CD34+ cells and BM-MSCs
(A) CD34+ HDR-based gene-editing frequencies in CD34+ cells from three healthy donors (HD 1–3), showing HDR percentages in all samples. (B) MSC HDR-based gene-editing frequencies in three healthy donors (HD 1–3). Treated P1 keratinocytes (C+ HK P1) were included as reference. MOIs of 5,000, 10,000, and 30,000 were used.
To prove that gene editing-based modification is maintained in clonogenic progenitors, gene-edited CD34+ bulk cells were cultured in methylcellulose and burst-forming unit erythroid (BFU-E) cells and colony-forming unit granulocyte-macrophage (CFU-GM) cells were analyzed (Figure S6). PCR genotyping showed the presence of the HDR-introduced modification in most of the colonies analyzed.
In addition, MSCs from three healthy donors were treated with the same reagents, and all treated samples showed genetic modification in close to 50% of COL7A1 alleles (Figure 6B). Comparable gene-editing frequencies for different donors were also observed, showing the reproducibility of the approach. Therefore, this study provides a proof of concept that BM-derived stem cells are susceptible to correction by following the same strategy proposed for keratinocytes.
Discussion
Genetically modified stem cells offer new therapeutic solutions for currently untreatable genetic diseases. Ex vivo genetic manipulation and subsequent grafting of CD34+ cells from patients to regenerate their hematopoietic system has shown great benefits for the clinical treatment of severe blood disorders.20, 21, 22 Additional strategies for genetic disease correction based on gene-editing techniques are currently entering clinical trials (ClinicalTrials.gov: NCT03745287), which could presage a revolution in modern medicine. In addition, MSC-based cell therapy shows clinical benefits for wound healing defects and treatment of inflammatory disorders, which offers new cell therapy approaches for a wide array of conditions.23,24
Keratinocytes and fibroblasts are the main skin cell types and have been the focus of many ex vivo gene and cell therapy approaches for cutaneous genetic disease. EB is one of the most devastating rare skin diseases, and the RDEB subtype, with complete absence of C7 expression, is considered the most severe form. A large number of COL7A1 gene mutations have been described in these patients, making this gene the most prominent target for EB gene therapy.1 Recently, a phase I clinical trial showed benefits in patients transplanted with skin equivalents containing autologous epidermal stem cells treated with gamma-retroviral vectors for COL7A1 expression, a classical gene replacement approach. Patients showed wound healing amelioration and C7 deposition and anchoring fibril formation.25, 26, 27 Although the addition of functional copies of COL7A1 is the simplest and most proven gene therapy approach, several shortcomings of this strategy, including possible insertional mutagenesis and inappropriate transgene expression resulting from random integration, warrant the development of strategies for precise modification of pathogenic gene mutations. Altering the genome sequence accurately and efficiently has only recently been achieved thanks to the advancement of gene editing techniques that have made it possible to address gene correction in cell populations without the need for drug selection, with ease and cost comparable to gene replacement protocols. In fact, previous work from our group11 described a highly efficient NHEJ-based gene-editing approach for COL7A1 correction in RDEB patient cells. This work established CRISPR-Cas9 delivered as RNP as the most efficient tool for genome editing in primary keratinocytes. The therapy proposed was specific for exon 80 of COL7A1, which contains a highly prevalent mutation in the Spanish RDEB patient cohort.19 Although similar approaches based on exon removal are transferable to other exons within the collagenous domain of COL7A1, it would be desirable to have a unique set of tools capable of genetically correcting more exons. In this context, the HDR-based strategy presented in this study constitutes an interesting therapeutic option to potentially treat a large number of mutations within COL7A1, regardless of the location of the exon in the gene. The approach, which consists of an AAV vector to deliver the donor template DNA along with a CRISPR-Cas9 system delivered by electroporation as RNP, has been tried before in other cell types.2,3,28 Previous studies showed that AAVs based on engineered serotype 2 capsids work efficiently for transduction of human skin keratinocytes.29,30 In the present study, we tested a large collection of AAV serotypes to determine the most efficient transduction conditions in primary keratinocytes and found that the AAV6 serotype performed best. We found similar HDR-based correction ratios for the two different homology donor designs tested in primary keratinocytes. In our study, the symmetrical donor could correct mutations in any exons ranging from E74 to E84 and asymmetrical donor template from E77 to E88. Therefore, these reagents may be used to treat patients affected by a wide number of mutations within COL7A1. Furthermore, a large collection of AAVs spanning different gene regions of COL7A1 could be developed, aiming to precisely correct any mutation throughout the gene. This is an important advantage over the previously proposed NHEJ-based approach11 because some exons are not amenable to exon removal strategies, including at least exons 1, 2, 3, 24, 27, or 113,31 and correcting mutations in these will require precise HDR-mediated gene modification. Beyond EB treatment, this approach could be transferred to any genome-editing application in primary keratinocytes to treat mutations causing other skin diseases or knock-in genes (reporters, therapeutic molecules) to create models of skin pathologies.
Previous work from our group demonstrated the feasibility of achieving HDR correction in a patient-derived keratinocyte cell line by using tropism-modified AAV2 vectors to deliver targeting constructions in which selection cassettes were flanked by genomic DNA sequences homologous to the COL7A1 exon 80 region, along with TALEN nucleases to trigger recombination. After selection treatment, most isolated keratinocyte clones were genetically corrected.29 However, although we demonstrated the feasibility of C7 restoration by HDR in a RDEB keratinocyte cell line, drug selection and clone isolation were necessary, which makes translation into the clinic very difficult. Izmiryan et al.8 used integration-deficient lentiviral vectors to deliver CRISPR-Cas9 and a donor template DNA sequence for correction of a mutation in exon 2 of COL7A1 in RDEB keratinocytes and achieved 11% and 15% of the normal level of COL7A1 mRNA expression in keratinocytes and fibroblasts, respectively. When skin equivalents made with these cells were transplanted onto immunodeficient mice, C7 deposited at the dermal-epidermal junction was sufficient to prevent blistering. In addition, a recent study in induced pluripotent stem cells (iPSCs) derived from RDEB patients showed biallelic correction efficiencies close to 60% using an HDR-based protocol.9 Although this approach achieves a very high gene-editing correction efficacy, it needs reprogramming of iPSCs to keratinocytes, which still represents a big hurdle since there is as yet no definitive evidence that epidermal stem cells capable of supporting long-term skin regeneration can be derived from iPSCs, and risk of tumorigenesis from iPSC-derived keratinocytes has to be considered. Thus, primary keratinocytes are still the most suitable cell type for skin regeneration in terms of clinical translation.
In this study, we overcame the limitations of previously reported protocols and implemented a selection marker-free, highly efficient approach, with a DNA correction frequency of between 35% and 50% in RDEB primary keratinocytes. Accurately corrected transcripts ranged from 60% to 80% of total COL7A1 mRNA. A significant percentage of transcription, 13%–31%, corresponded to ΔE80 transcripts. These are most likely originating from COL7A1 alleles carrying NHEJ-induced sequence alterations at the splice acceptor site of exon 80, which is in the vicinity of the cutting site of the sg2 guide. This ΔE80 transcript population could probably be reduced by using a guide RNA farther apart from exon 80, where most NHEJ-induced indels would not affect splicing of COL7A1, so the frequency of internally truncated variants would be negligible relative to full-length COL7A1 mRNA expression.
The co-expression of truncated and full-length C7 variants in a fraction of treated keratinocytes could potentially hinder triple-helix formation and hence proper C7 secretion. However, we have found that grafts from treated patient keratinocytes displayed C7 deposition at the BMZ and extracellular, secreted C7 was detected by western blot analysis of bulk-edited patient cells, which proves that our polyclonal HDR-based protocol can provide enough secreted C7 to sustain dermal-epidermal attachment.
Bulk populations of keratinocytes edited with this protocol were able to achieve normal human skin regeneration with restoration of dermal-epidermal adhesion when transplanted onto nude mice. Using a polyclonal population greatly facilitates translation into the clinics, since it avoids the technically challenging and labor-intensive epidermal stem cell clone isolation step. Therefore, we propose CRISPR-Cas9 RNP electroporation in combination with AAV6-mediated delivery of donor DNA templates as the optimal toolkit for keratinocyte genome editing.
Although the guide RNAs that direct Cas9 to specific genomic sequences can tolerate some mismatches to the DNA target and thus potentially promote unwanted off-target mutagenesis, our previous study of putative off-target activity directed by the guide RNA used to promote HDR in our protocol did not identify any off-target indels in multiple genomic loci bearing different combinations of mismatches with the guide RNA, including sites with up to three mismatches, 1 or 2 bp bulges, and coding region sites with four mismatches.11 Paired ssDNA breaks elicited by Cas9 mutants have been shown to reduce unwanted repair outcomes while still promoting high HDR correction efficiencies.32, 33, 34 Although the use of recombinant mutant Cas9 “nickases” in CRISPR-Cas9 RNP electroporation experiments has yet to be demonstrated in keratinocytes, these reagents could provide additional safety in HDR correction protocols.
Although AAV vectors remain in transduced cells primarily as concatemeric episomal DNA, they are known to integrate with low frequency into host mammalian genomes, and some studies have suggested that integration of vector fragments could potentially represent a genotoxicity risk.35 However, vector integration studies in human gene therapy clinical contexts have shown that AAV vector integration events occurred at very low frequencies with random distributions, and no serious side effects or cancer development have been described in any patient treated with rAAV gene therapy.36,37
Cell types other than keratinocytes might also be therapeutic targets. In the last decade HSCT has been considered for EB treatment,17 although the mechanisms underlying the benefits of this intervention have yet to be elucidated and BM ablation in patients with severe skin fragility pathology can be lethal.38 Recently, Ebens et al.12 showed that HSCT followed by MSC infusions could ameliorate the RDEB phenotype. MSCs can potentially express C7 and have an immunoregulatory role in wound healing, which could contribute to decreased inflammation and dermal-epidermal adhesion restoration.24,39 Grafting of autologous gene-edited CD34+ cells and MSCs could be considered to improve safety in future trials. Herein, we show efficient gene editing in both CD34+ cells and MSCs from three different healthy donors, demonstrating that efficient HDR-based correction is attainable in these cell types, which could be useful for EB or other pathologies treated with HSCT.
Overall, our study offers an effective ex vivo genome-editing platform that enables genetic correction of various cell types relevant for EB therapy, which opens the way for developing new therapeutic approaches.
Materials and methods
Keratinocyte cell culture and isolation of clones
Patient keratinocytes were originally obtained from skin biopsies from three RDEB (RDEB-sev gen) patients bearing mutations in the COL7A1 gene. Skin biopsies were obtained from patients after approval from the Ethics Committee of the collaborating hospital upon informed consent. P1 and P3 are homozygous carriers for the c.6527insC frameshift mutation in exon 80, and P2 is a homozygous carrier for the c.6501G>T mutation in exon 79 (ClinVar: VCV000977652).
Primary human RDEB and healthy donor keratinocytes were cultured as previously described.10 Human primary RDEB keratinocytes were plated onto lethally irradiated 3T3-J2 cells and cultured in keratinocyte growth cFAD medium (KCa), a 3:1 mix of Dulbecco’s modified Eagle’s medium and Ham’s F12 medium (Gibco-BRL, Barcelona, Spain) containing fetal bovine calf serum (HyClone, GE Healthcare, Logan, UT, USA) (10%), penicillin-streptomycin (1%), glutamine (2%), insulin (5 μg/mL; Sigma-Aldrich), adenine (0.18 mmol/L; Sigma-Aldrich), hydrocortisone (0.4 μg/mL; Sigma-Aldrich), cholera toxin (0.1 nmol/L; Sigma-Aldrich), triiodothyronine (2 nmol/L; Sigma-Aldrich), epidermal growth factor (EGF) (10 ng/mL; Sigma-Aldrich), and Y-27632 ROCK inhibitor (Sigma-Aldrich) at 10 μM. P1 and P3 are homozygous for the c.6527insC mutation. P2 is homozygous for the c.6501G>T mutation.
Production of AAV vectors for delivery of donor template DNA
Homology arms were amplified by PCR from wild-type genomic DNA. For the symmetric donor construct, the left homology arm (LHA) comprises 1,008 bp including exon 79 and the upstream sequence, and the right homology arm (RHA) comprises 798 bp including exon 80 and the downstream sequence. For the asymmetric design, LHA is 556 bp and RHA is 1,461 bp. For each construct, both arms were assembled with an AAV backbone plasmid containing AAV2 inverted terminal repeats (ITRs) using the Gibson assembly method (New England Biolabs). For vector production, 293 cells were transfected in five 15-cm2 plates with 6 μg of AAV vector plasmids and 22 μg of pDGM6 plasmid (an expression vector for AAV6 capsid [cap], AAV2 replication [rep], and adenoviral helper genes, a gift from D. Russell) using 120 μL of 1 mg/mL polyethylenimine (PEI) per plate (molecular weight [MW] of 25 kDa) (Polysciences). 72 h after transfection, vectors were purified using a Takara AAVpro purification kit following the manufacturer’s protocol. Vector titers were assessed by digital drop PCR (ddPCR) using probes on the ITR region of the AAV vector.
CRISPR-Cas9 delivery and AAV6 transduction in primary keratinocytes
Electroporation conditions were optimized by electroporating 1 μg of GFP mRNA with the 4D-Nucleofector system (Lonza, Switzerland) using established Amaxa electroporation codes for human keratinocytes and the Neon system (Invitrogen) with a 1,700-V/20-ms/1-pulse configuration. Sg2 gRNA1 is a chemically modified sgRNA (Synthego, Redwood City, CA, USA) complementary to the DNA sequence 5′-CCTGCAGACCCTACATAGAG-3′.11 1.6 μg of sgRNA mixed with 6 μg of Cas9 protein (Integrated DNA Technologies, IA, USA) was delivered to 1 × 105 primary keratinocytes in Opti-MEM medium (Thermo Fisher Scientific). The electroporation platform used for delivery of the RNPs was the 4D-Nucleofector system (Lonza, Switzerland), electroporation code CM137.
After electroporation, cells were transduced in suspension for 1 h with the AAV vectors at an MOI of 30,000 in a final volume of 50 μL of Opti-MEM medium (Thermo Fisher Scientific). Cells were then plated onto feeder layer-containing plates. For MSCs and CD34+ cells, 3.2 μg of sgRNA and 6 μg of Cas9 were used. The Amaxa electroporation codes were CM119 and DZ100 for MSCs and CD34+ cells, respectively. For viral transduction, MSCs were incubated with the AAV6 vectors for 15 min in suspension and then plated in culture medium. For CD34+ cell transduction, AAV6 was added directly to the well.
Genotyping of gene-targeted keratinocytes
Six days post-treatment, genomic DNA was isolated by isopropanol precipitation of keratinocyte lysates (lysis buffer was 100 mM Tris [pH 8], 5 mM EDTA, 0.2% SDS, 200 mM NaCl, 1 mg/mL Proteinase K; Roche Diagnostics, Mannheim, Germany) and resuspended in Tris-EDTA (TE) buffer. Approximately 50 ng of genomic DNA was used for PCR amplification. PCR primers used for out-in genotyping were forward (F) 5′-CACCAGCATTCTCTCTTCC-3′ and reverse (R) 5′-GTTCTTGGGTACTCACCA C-3′ (F is located outside and R is within the homology arms). The PCR program was as follows: 98°C for 1 min, 5 cycles of 98°C for 30 s, 68°C for 30 s, 72°C for 45 s, decreasing annealing temperature 1°C every cycle, followed by 30 cycles of 94°C for 30 s, 63°C for 30 s, 72°C for 45 s, and then 72°C for 10 min. PCR products were analyzed in 1.5% agarose gel. The molecular weight marker was IX (Sigma-Aldrich). For sequencing, PCR products were treated with illustra ExoProStar (GE Healthcare, UK), sequenced using a BigDye Terminator v1.1 cycle sequencing kit (Thermo Fisher Scientific, Waltham, MA, USA), and examined on a 3730 DNA analyzer (Life Technologies, Carlsbad, CA, USA). Chromatograms were analyzed using Sequencher (Gene Codes, Ann Arbor, MI, USA). Bio-Rad Image Lab software 6.0 was used for PCR band densitometry.
Western blot analysis
Keratinocytes were lysed in protein extraction buffer (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, 1% Nonidet P-40, 4 mM EDTA) containing proteinase inhibitors cocktail (cOmplete Mini, EDTA-free; Roche Diagnostics, Mannheim, Germany). Lysates were incubated for 30 min on ice and centrifμged at 15,000 × g for 30 min at 4°C. Supernatants were collected and protein concentrations were measured using the Bradford assay (Bio-Rad, Hercules, CA, USA). For each sample, 40 μg of total protein was resolved on NuPAGE Novex 3%–8% Tris-acetate electrophoresis gels (Invitrogen, Carlsbad, CA, USA) and electro-transferred to nitrocellulose membranes (Invitrogen, Carlsbad, CA, USA). For type VII collagen analysis, blots were probed with a monospecific polyclonal anti C7 antibody (a generous gift from Dr. A. Nystrom, University of Freiburg). An antibody against vinculin was used as a loading control. Visualization was performed by incubating with horseradish peroxidase (HRP)-conjugated anti-rabbit antibody (Amersham, Burlington, MA, USA) and West Pico chemiluminescent substrate (Pierce, Rockford, IL, USA). Blots from three different cell lysates were densitometrically quantified using ImageJ (NIH, Bethesda, MD, USA).40 Vinculin was used for normalization, and mean values were expressed relative to control. An unpaired t test with Welch’s correction was performed using GraphPad Prism version 9.0.1 (GraphPad, La Jolla, CA, USA).
Immunofluorescence and immunohistochemical staining
For immunofluorescence detection of C7 in keratinocytes, cells grown on glass coverslips were fixed in methanol/acetone (1:1) for 10 min at −20°C. After washing three times in phosphate-buffered saline (PBS) and once in PBS with 3% bovine serum albumin (BSA) (Sigma-Aldrich, St. Louis, MO, USA) for 30 min, cells were incubated with monospecific polyclonal anti C7 antibody at 1:5,000 dilution. Secondary antibody (Alexa Fluor 488, Invitrogen, Carlsbad, CA, USA) was used at 1:1,000 dilution. After the final washing step in PBS, preparations were mounted using Mowiol (Hoechst, Somerville, NJ, USA) mounting medium and 20 μg/mL DAPI (Sigma-Aldrich, St. Louis, MO, USA) for nuclei visualization. Immunoperoxidase detection of C7 in paraffin-embedded, formalin-fixed sections was carried out with Proteinase K antigen retrieval as described.41 Immunoperoxidase staining for human involucrin was performed using rabbit SY5 monoclonal antibody (Sigma) and 4A4 monoclonal antibody, respectively, on paraffin sections without antigen retrieval. The ABC peroxidase kit (Vector Laboratories) with diaminobenzidine as a substrate was used.
NGS COL7A1 transcriptional analysis
Primers with NGS adapters used for the PCR amplification were as follows: F, 5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCTAGGGGTCAGGACGGCAAC-3 and R, 5′-GACTGGAGTTCAGACGTGTGCTCTTCCGATCTCAGCTCCAGTAGGTCCAGTCAG-3′. Amplicons were measured by Qubit technology. NGS was performed on an Illumina 2 × 250-bp configuration, with coverage of 50,000×. Data analysis was conducted with NGS Genotyper v1.4.0. From the data obtained, aberrant sequences also observed in the non-treated sample were removed from the treated samples.
Generation of skin equivalents, grafting onto mice, and graft analysis
Animal studies were approved by our Institutional Animal Care and Use Committee according to national and European legal regulations. Polyclonal keratinocytes corrected by HDR-based gene editing were seeded on fibrin dermal equivalents containing P1 fibroblasts null for C7 expression, prepared as previously described.42 Bioengineered skin equivalents were grafted onto the back of 7-week-old female immunodeficient mice (nu/nu, NMRI background) purchased from Elevage Janvier (France) as previously described.43 Grafting was performed under sterile conditions, and mice were housed in pathogen-free conditions for the duration of the experiment at the CIEMAT Laboratory Animal Facility (Spanish registration no. 28079-21 A). All handling was carried out under sterile conditions, and all experimental procedures were according to European and Spanish laws and regulations. Mice were sacrificed after 12 weeks post-grafting, and grafts were harvested for skin histology and immunohistochemistry analyses.
CD34+ cell purification, culture, and modification
CD34+ cells from healthy donors were obtained from umbilical CB samples provided by the Centro de Transfusiones de la Comunidad de Madrid after informed consent. Hematopoietic stem and progenitor cells (HSPCs) from CB samples were obtained by fractionation in Ficoll-Hypaque according to the manufacturer’s instructions (GE Healthcare) and purified by a magnetic-activated cell sorting (MACS) CD34 MicroBead kit (Miltenyi Biotec). Human CD34+ cells were grown under normoxic conditions in StemSpan medium (STEMCELL Technologies) supplemented with 1% GlutaMAX (Gibco), 1% penicillin/streptomycin solution (Gibco), 100 ng/mL human stem cell factor (hSCF, EuroBiosciences), human FMS-like tyrosine kinase 3 ligand (hFlt3-L, EuroBiosciences), human thrombopoietin (hTPO, R&D Systems), 20 ng/mL human interleukin-3 (hIL-3, Novus Biologicals), and 35 nM UM171 (STEMCELL Technologies).
Cells were electroporated with the DZ-100 4D-Nucleofector system code (Lonza, Switzerland). 1.6 μg of sgRNA mixed with 4.5 μg of Cas9 protein (Integrated DNA Technologies, IA, USA) was delivered to 1 × 105 CD34+ cells. After electroporation, cells were transduced with the AAV6 vectors at MOIs of 5,000 and 10,000.
900 CD34+ cells were resuspended in 3 mL of enriched methylcellulose medium (StemMACS HSC-CFU complete with erythropoietin [Epo], Miltenyi Biotec). Each mL of the triplicate was seeded in a M35 plate and incubated under normoxic conditions. After 14 days, colonies were counted using an inverted microscope (Nikon Diaphot), and CFU-GM and BFU-E cells were identified and counted. PCR genotyping of the colonies was performed with the following primers: F, 5′-GGTCTACCAGGAGAGCGTGGTA-3′ and R, 5′-ACCCCACCAAGGAAACTGA-3′.
Generation, expansion, and modification of BM-MSCs
BM samples were obtained from three volunteer donors. Five to 10 mL of BM was obtained by iliac crest aspiration under local anesthesia. All samples were collected after informed consent was obtained. All procedures were approved by the Clinical Research Ethics Committee of the Health Area of Salamanca and were in accordance with the Helsinki Declaration of 1975. To obtain BM-MSCs, low-density mononuclear cells (MNCs) were isolated by Ficoll-Paque Plus (GE Healthcare, Sweden) density gradient centrifugation and cultured in α-minimum essential medium (α-MEM) (Gibco, USA) supplemented with 5% platelet lysate (Cook Medical, USA), 1% penicillin/streptomycin (Gibco), and 1 ng/mL human basic fibroblast growth factor (bFGF, PeproTech, USA). Cells were seeded at a concentration of 10,000 cells/cm2 in culture flasks (Corning Life Sciences, USA) and cultured at 37°C. For the expansion of BM-MSCs, cell medium was changed every 2–4 days and adherent cells were serially passaged using 0.25% trypsin/EDTA (Sigma-Aldrich, USA) upon reaching near confluence (70%–90%).
Cells were electroporated with the CM-119 4D-Nucleofector system code (Lonza, Switzerland). 3.2 μg of sgRNA mixed with 6 μg of Cas9 protein (Integrated DNA Technologies, IA, USA) was delivered to 1 × 105 MSCs. After electroporation, cells were transduced with AAV6 at an MOI of 10,000.
Acknowledgments
This work was supported by Spanish grants PI17/01747, PI20/00615, AC17/00054 (MutaEB-E-rare), and CIBERER ER18TRL714 from the Instituto de Salud Carlos III and grant SAF2017-86810-R from the Ministry of Economy and Competitiveness, all co-funded with European Regional Development Funds, and Avancell-CM grant (S2017/BMD-3692). Authors are indebted to Almudena Holguín and Nuria Illera for grafting experiments, and to Jesus Martínez and Edilia De Almeida for animal maintenance and care.
Author contributions
J.B., A.M., W.S., S.V., M.H.P., F.L., and R.M. designed the experiments. J.B., A.M., W.S., R.R., and B.D. performed molecular and cellular studies. M.D.R. contributed to experimental results discussion and provided reagents and ethical approval for the studies. J.B., M.G., B.D., E.C.-S., and F.L. performed animal experiments and histological analysis. J.B., R.H.-S., and L.U. performed cell isolation, culture, and targeting experiments in HSCs and MSCs. J.B., F.L., and R.M. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.02.019.
Contributor Information
Fernando Larcher, Email: fernando.larcher@ciemat.es.
Rodolfo Murillas, Email: rodolfo.murillas@ciemat.es.
Supplemental information
References
- 1.Has C., Bauer J.W., Bodemer C., Bolling M.C., Bruckner-Tuderman L., Diem A., Fine J.D., Heagerty A., Hovnanian A., Marinkovich M.P. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. 2020;183:614–627. doi: 10.1111/bjd.18921. [DOI] [PubMed] [Google Scholar]
- 2.Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gomez-Ospina N., Scharenberg S.G., Mostrel N., Bak R.O., Mantri S., Quadros R.M., Gurumurthy C.B., Lee C., Bao G., Suarez C.J. Human genome-edited hematopoietic stem cells phenotypically correct mucopolysaccharidosis type I. Nat. Commun. 2019;10:4045. doi: 10.1038/s41467-019-11962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin R.M., Ikeda K., Cromer M.K., Uchida N., Nishimura T., Romano R., Tong A.J., Lemgart V.T., Camarena J., Pavel-Dinu M. Highly efficient and marker-free genome editing of human pluripotent stem cells by CRISPR-Cas9 RNP and AAV6 donor-mediated homologous recombination. Cell Stem Cell. 2019;24:821–828.e5. doi: 10.1016/j.stem.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Osborn M.J., Starker C.G., McElroy A.N., Webber B.R., Riddle M.J., Xia L., DeFeo A.P., Gabriel R., Schmidt M., von Kalle C. TALEN-based gene correction for epidermolysis bullosa. Mol. Ther. 2013;21:1151–1159. doi: 10.1038/mt.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Izmiryan A., Danos O., Hovnanian A. Meganuclease-mediated COL7A1 gene correction for recessive dystrophic epidermolysis bullosa. J. Invest. Dermatol. 2016;136:872–875. doi: 10.1016/j.jid.2015.11.028. [DOI] [PubMed] [Google Scholar]
- 7.Hainzl S., Peking P., Kocher T., Murauer E.M., Larcher F., Del Rio M., Duarte B., Steiner M., Klausegger A., Bauer J.W. COL7A1 editing via CRISPR/Cas9 in recessive dystrophic epidermolysis bullosa. Mol. Ther. 2017;25:2573–2584. doi: 10.1016/j.ymthe.2017.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Izmiryan A., Ganier C., Bovolenta M., Schmitt A., Mavilio F., Hovnanian A. Ex vivo COL7A1 Correction for recessive dystrophic epidermolysis bullosa using CRISPR/Cas9 and homology-directed repair. Mol. Ther. Nucleic Acids. 2018;12:554–567. doi: 10.1016/j.omtn.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackow J., Guo Z., Hansen C., Abaci H.E., Doucet Y.S., Shin J.U., Hayashi R., DeLorenzo D., Kabata Y., Shinkuma S. CRISPR/Cas9-based targeted genome editing for correction of recessive dystrophic epidermolysis bullosa using iPS cells. Proc. Natl. Acad. Sci. USA. 2019;116:26846–26852. doi: 10.1073/pnas.1907081116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benati D., Miselli F., Cocchiarella F., Patrizi C., Carretero M., Baldassarri S., Ammendola V., Has C., Colloca S., Del Rio M. CRISPR/Cas9-mediated in situ correction of LAMB3 gene in keratinocytes derived from a junctional epidermolysis bullosa patient. Mol. Ther. 2018;26:2592–2603. doi: 10.1016/j.ymthe.2018.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonafont J., Mencía Á., García M., Torres R., Rodríguez S., Carretero M., Chacón-Solano E., Modamio-Høybjør S., Marinas L., León C. Clinically relevant correction of recessive dystrophic epidermolysis bullosa by dual sgRNA CRISPR/Cas9-mediated gene editing. Mol. Ther. 2019;27:986–998. doi: 10.1016/j.ymthe.2019.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ebens C.L., McGrath J.A., Tamai K., Hovnanian A., Wagner J.E., Riddle M.J., Keene D.R., DeFor T.E., Tryon R., Chen M. Bone marrow transplant with post-transplant cyclophosphamide for recessive dystrophic epidermolysis bullosa expands the related donor pool and permits tolerance of nonhaematopoietic cellular grafts. Br. J. Dermatol. 2019;181:1238–1246. doi: 10.1111/bjd.17858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujita Y., Abe R., Inokuma D., Sasaki M., Hoshina D., Natsuga K., Nishie W., McMillan J.R., Nakamura H., Shimizu T. Bone marrow transplantation restores epidermal basement membrane protein expression and rescues epidermolysis bullosa model mice. Proc. Natl. Acad. Sci. USA. 2010;107:14345–14350. doi: 10.1073/pnas.1000044107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iinuma S., Aikawa E., Tamai K., Fujita R., Kikuchi Y., Chino T., Kikuta J., McGrath J.A., Uitto J., Ishii M. Transplanted bone marrow-derived circulating PDGFRα+ cells restore type VII collagen in recessive dystrophic epidermolysis bullosa mouse skin graft. J. Immunol. 2015;194:1996–2003. doi: 10.4049/jimmunol.1400914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petrof G., Lwin S.M., Martinez-Queipo M., Abdul-Wahab A., Tso S., Mellerio J.E., Slaper-Cortenbach I., Boelens J.J., Tolar J., Veys P. Potential of systemic allogeneic mesenchymal stromal cell therapy for children with recessive dystrophic epidermolysis bullosa. J. Invest. Dermatol. 2015;135:2319–2321. doi: 10.1038/jid.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tamai K., Uitto J. Stem cell therapy for epidermolysis bullosa—does it work? J. Invest. Dermatol. 2016;136:2119–2121. doi: 10.1016/j.jid.2016.07.004. [DOI] [PubMed] [Google Scholar]
- 17.Tolar J., Wagner J.E. Allogeneic blood and bone marrow cells for the treatment of severe epidermolysis bullosa: repair of the extracellular matrix. Lancet. 2013;382:1214–1223. doi: 10.1016/S0140-6736(13)61897-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van den Akker P.C., Jonkman M.F., Rengaw T., Bruckner-Tuderman L., Has C., Bauer J.W., Klausegger A., Zambruno G., Castiglia D., Mellerio J.E. The international dystrophic epidermolysis bullosa patient registry: an online database of dystrophic epidermolysis bullosa patients and their COL7A1 mutations. Hum. Mutat. 2011;32:1100–1107. doi: 10.1002/humu.21551. [DOI] [PubMed] [Google Scholar]
- 19.Escámez M.J., García M., Cuadrado-Corrales N., Llames S.G., Charlesworth A., De Luca N., Illera N., Sánchez-Jimeno C., Holguín A., Duarte B. The first COL7A1 mutation survey in a large Spanish dystrophic epidermolysis bullosa cohort: c.6527insC disclosed as an unusually recurrent mutation. Br. J. Dermatol. 2010;163:155–161. doi: 10.1111/j.1365-2133.2010.09713.x. [DOI] [PubMed] [Google Scholar]
- 20.Ferrua F., Cicalese M.P., Galimberti S., Giannelli S., Dionisio F., Barzaghi F., Migliavacca M., Bernardo M.E., Calbi V., Assanelli A.A. Lentiviral haemopoietic stem/progenitor cell gene therapy for treatment of Wiskott-Aldrich syndrome: interim results of a non-randomised, open-label, phase 1/2 clinical study. Lancet Haematol. 2019;6:e239–e253. doi: 10.1016/S2352-3026(19)30021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kohn D.B., Booth C., Kang E.M., Pai S.Y., Shaw K.L., Santilli G., Armant M., Buckland K.F., Choi U., De Ravin S.S., Net4CGD consortium Lentiviral gene therapy for X-linked chronic granulomatous disease. Nat. Med. 2020;26:200–206. doi: 10.1038/s41591-019-0735-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Río P., Navarro S., Wang W., Sánchez-Domínguez R., Pujol R.M., Segovia J.C., Bogliolo M., Merino E., Wu N., Salgado R. Successful engraftment of gene-corrected hematopoietic stem cells in non-conditioned patients with Fanconi anemia. Nat. Med. 2019;25:1396–1401. doi: 10.1038/s41591-019-0550-z. [DOI] [PubMed] [Google Scholar]
- 23.Lamo-Espinosa J.M., Mora G., Blanco J.F., Granero-Moltó F., Nuñez-Córdoba J.M., Sánchez-Echenique C., Bondía J.M., Aquerreta J.D., Andreu E.J., Ornilla E. Intra-articular injection of two different doses of autologous bone marrow mesenchymal stem cells versus hyaluronic acid in the treatment of knee osteoarthritis: multicenter randomized controlled clinical trial (phase I/II) J. Transl. Med. 2016;14:246. doi: 10.1186/s12967-016-0998-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rashidghamat E., Kadiyirire T., Ayis S., Petrof G., Liu L., Pullabhatla V., Ainali C., Guy A., Aristodemou S., McMillan J.R. Phase I/II open-label trial of intravenous allogeneic mesenchymal stromal cell therapy in adults with recessive dystrophic epidermolysis bullosa. J. Am. Acad. Dermatol. 2020;83:447–454. doi: 10.1016/j.jaad.2019.11.038. [DOI] [PubMed] [Google Scholar]
- 25.Eichstadt S., Barriga M., Ponakala A., Teng C., Nguyen N.T., Siprashvili Z., Nazaroff J., Gorell E.S., Chiou A.S., Taylor L. Phase 1/2a clinical trial of gene-corrected autologous cell therapy for recessive dystrophic epidermolysis bullosa. JCI Insight. 2019;4:e130554. doi: 10.1172/jci.insight.130554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marinkovich M.P., Tang J.Y. Gene therapy for epidermolysis bullosa. J. Invest. Dermatol. 2019;139:1221–1226. doi: 10.1016/j.jid.2018.11.036. [DOI] [PubMed] [Google Scholar]
- 27.Siprashvili Z., Nguyen N.T., Gorell E.S., Loutit K., Khuu P., Furukawa L.K., Lorenz H.P., Leung T.H., Keene D.R., Rieger K.E. Safety and wound outcomes following genetically corrected autologous epidermal grafts in patients with recessive dystrophic epidermolysis bullosa. JAMA. 2016;316:1808–1817. doi: 10.1001/jama.2016.15588. [DOI] [PubMed] [Google Scholar]
- 28.Gaj T., Staahl B.T., Rodrigues G.M.C., Limsirichai P., Ekman F.K., Doudna J.A., Schaffer D.V. Targeted gene knock-in by homology-directed genome editing using Cas9 ribonucleoprotein and AAV donor delivery. Nucleic Acids Res. 2017;45:e98. doi: 10.1093/nar/gkx154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chamorro C., Mencía A., Almarza D., Duarte B., Büning H., Sallach J., Hausser I., Del Río M., Larcher F., Murillas R. Gene editing for the efficient correction of a recurrent COL7A1 mutation in recessive dystrophic epidermolysis bullosa keratinocytes. Mol. Ther. Nucleic Acids. 2016;5:e307. doi: 10.1038/mtna.2016.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sallach J., Di Pasquale G., Larcher F., Niehoff N., Rübsam M., Huber A., Chiorini J., Almarza D., Eming S.A., Ulus H. Tropism-modified AAV vectors overcome barriers to successful cutaneous therapy. Mol. Ther. 2014;22:929–939. doi: 10.1038/mt.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bornert O., Kühl T., Bremer J., van den Akker P.C., Pasmooij A.M., Nyström A. Analysis of the functional consequences of targeted exon deletion in COL7A1 reveals prospects for dystrophic epidermolysis bullosa therapy. Mol. Ther. 2016;24:1302–1311. doi: 10.1038/mt.2016.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kocher T., Wagner R.N., Klausegger A., Guttmann-Gruber C., Hainzl S., Bauer J.W., Reichelt J., Koller U. Improved double-nicking strategies for COL7A1-editing by homologous recombination. Mol. Ther. Nucleic Acids. 2019;18:496–507. doi: 10.1016/j.omtn.2019.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ran F.A., Hsu P.D., Lin C.Y., Gootenberg J.S., Konermann S., Trevino A.E., Scott D.A., Inoue A., Matoba S., Zhang Y., Zhang F. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen X., Janssen J.M., Liu J., Maggio I., ’t Jong A.E.J., Mikkers H.M.M., Gonçalves M.A.F.V. In trans paired nicking triggers seamless genome editing without double-stranded DNA cutting. Nat. Commun. 2017;8:657. doi: 10.1038/s41467-017-00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donsante A., Miller D.G., Li Y., Vogler C., Brunt E.M., Russell D.W., Sands M.S. AAV vector integration sites in mouse hepatocellular carcinoma. Science. 2007;317:477. doi: 10.1126/science.1142658. [DOI] [PubMed] [Google Scholar]
- 36.Kaeppel C., Beattie S.G., Fronza R., van Logtenstein R., Salmon F., Schmidt S., Wolf S., Nowrouzi A., Glimm H., von Kalle C. A largely random AAV integration profile after LPLD gene therapy. Nat. Med. 2013;19:889–891. doi: 10.1038/nm.3230. [DOI] [PubMed] [Google Scholar]
- 37.Gil-Farina I., Fronza R., Kaeppel C., Lopez-Franco E., Ferreira V., D’Avola D., Benito A., Prieto J., Petry H., Gonzalez-Aseguinolaza G., Schmidt M. Recombinant AAV integration is not associated with hepatic genotoxicity in nonhuman primates and patients. Mol. Ther. 2016;24:1100–1105. doi: 10.1038/mt.2016.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gostyńska K.B., Yenamandra V.K., Lindemans C., Duipmans J., Gostyński A., Jonkman M.F., Boelens J.J. Allogeneic haematopoietic cell transplantation for epidermolysis bullosa: the Dutch experience. Acta Derm. Venereol. 2019;99:347–348. doi: 10.2340/00015555-3097. [DOI] [PubMed] [Google Scholar]
- 39.Petrova A., Georgiadis C., Fleck R.A., Allison L., McGrath J.A., Dazzi F., Di W.L., Qasim W. Human mesenchymal stromal cells engineered to express collagen VII can restore anchoring fibrils in recessive dystrophic epidermolysis bullosa skin graft chimeras. J. Invest. Dermatol. 2020;140:121–131.e6. doi: 10.1016/j.jid.2019.05.031. [DOI] [PubMed] [Google Scholar]
- 40.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Niskanen J., Dillard K., Arumilli M., Salmela E., Anttila M., Lohi H., Hytönen M.K. Nonsense variant in COL7A1 causes recessive dystrophic epidermolysis bullosa in Central Asian Shepherd dogs. PLoS ONE. 2017;12:e0177527. doi: 10.1371/journal.pone.0177527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Llames S.G., Del Rio M., Larcher F., García E., García M., Escamez M.J., Jorcano J.L., Holguín P., Meana A. Human plasma as a dermal scaffold for the generation of a completely autologous bioengineered skin. Transplantation. 2004;77:350–355. doi: 10.1097/01.TP.0000112381.80964.85. [DOI] [PubMed] [Google Scholar]
- 43.Duarte B., Miselli F., Murillas R., Espinosa-Hevia L., Cigudosa J.C., Recchia A., Del Río M., Larcher F. Long-term skin regeneration from a gene-targeted human epidermal stem cell clone. Mol. Ther. 2014;22:1878–1880. doi: 10.1038/mt.2014.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.