

Abstract
The transforming growth factor-beta (TGF-β) signaling pathway is the predominant cytokine signaling pathway in the development and progression of hepatocellular carcinoma (HCC). Bone morphogenetic protein (BMP), another member of the TGF-β superfamily, has been frequently found to participate in crosstalk with the TGF-β pathway. However, the complex interaction between the TGF-β and BMP pathways has not been fully elucidated in HCC. We found that the imbalance of TGF-β1/BMP-7 pathways was associated with aggressive pathological features and poor clinical outcomes in HCC. The induction of the imbalance of TGF-β1/BMP-7 pathways in HCC cells could significantly promote HCC cell invasion and stemness by increasing inhibitor of differentiation 1 (ID1) expression. We also found that the microRNA (miR)-17-92 cluster, originating from the extracellular vesicles (EVs) of M2-polarized tumor-associated macrophages (M2-TAMs), stimulated the imbalance of TGF-β1/BMP-7 pathways in HCC cells by inducing TGF-β type II receptor (TGFBR2) post-transcriptional silencing and inhibiting activin A receptor type 1 (ACVR1) post-translational ubiquitylation by targeting Smad ubiquitylation regulatory factor 1 (Smurf1). In vivo, short hairpin (sh)-MIR17HG and ACVR1 inhibitors profoundly attenuated HCC cell growth and metastasis by rectifying the imbalance of TGF-β1/BMP-7 pathways. Therefore, we proposed that the imbalance of TGF-β1/BMP-7 pathways is a feasible prognostic biomarker and recovering the imbalance of TGF-β1/BMP-7 pathways might be a potential therapeutic strategy for HCC.
Graphical abstract
Ning et al. identified that the imbalance of TGF-β1/BMP-7 pathways dominated the interactive communication between cancer cells and M2-polarized tumor-associated macrophages (M2-TAMs) to foster a protumoral microenvironment in hepatocellular carcinoma (HCC), which would shed new light on developing potential therapies for HCC.
Introduction
Liver cancer is the sixth most commonly diagnosed cancer and the fourth leading cause of cancer-related death worldwide, and its incidence and mortality rates have increased in recent years. Hepatocellular carcinoma (HCC) accounts for 75%−85% of primary liver cancers.1 Surgical resection,2 radiofrequency ablation,3 and traditional Chinese medicine4,5 have improved the survival of HCC patients; however, tumor recurrence and metastasis still limit their long-term survival. Thus, efforts should be made to explore the molecular mechanism that governs HCC progression to facilitate the development of novel diagnostic markers and anti-HCC therapies.
HCC is an extraordinarily heterogeneous malignant disease, and studies have illuminated that microenvironment discrepancies can affect the cell plasticity and heterogeneity of HCC,6 which is due to the interaction between cancer cells and their microenvironment components.7 The regulation of HCC induced by the local microenvironment is mainly based on multiple pleiotropic growth factors and cytokine signaling. Dysregulation of ligands, receptors, and their downstream signaling pathway components in the HCC tumor microenvironment has been recognized as an important accelerator of tumor progression with potential therapeutic significance.8
Transforming growth factor-beta (TGF-β) signaling represents the predominant cytokine signaling pathway between tumor cells and the microenvironment involved in the development and progression of HCC. TGF-β, a superfamily of cytokines, controls proliferation, cellular differentiation, migration, and other functions. TGF-β1, a core member of the TGF-β signaling pathway, is known to inhibit hepatocyte proliferation and suppress the progenitor properties of hepatocyte-initiating cells by binding to TGF-β type II receptor (TGFBR2)- and activating TGF-β type I receptor (TGFBR1)-downstream Smad2/3 transcription factors under physiological circumstances.9 However, TGF-β1 signaling aggravates tumor progression by promoting metastasis and angiogenesis under pathological conditions.10 Studies have reported that high TGF-β1 expression (TGF-β1hi) and low TGFBR2 expression (TGFBR2lo) predict a poor disease prognosis in HCC.11,12 Therefore, the disparity in TGF-β1-initiated biological functions between different circumstances indicates the complexity of the TGF-β signaling pathway in HCC development and progression.
Coulouarn et al.13 described two different TGF-β-specific gene signatures in HCC. The first, termed the “early TGF-β signature,” is associated with longer survival and TGFBR2hi, whereas the second, termed the “late TGF-β signature,” is associated with shorter survival and TGFBR2lo.14 Cells with the early TGF-β1 signature retain sensitivity to the antiproliferative effects of TGF-β1 and display an epithelial phenotype, whereas those with a late TGF-β1 signature do not display TGF-β1-dependent cytostasis and present a distinct mesenchymal phenotype.15,16 Studies have shown that the proliferative response to TGF-β1 is TGFBR2 dependent and that TGFBR2 downregulation is beneficial for HCC cell evasion of TGF-β1 antiproliferative control.17 Therefore, the expression of TGFBR2 seems to be a key to shifting from TGF-β1-dependent proliferation inhibition to disease progression.14 Consistently, a previous study showed that compared to patients with high expression, patients with TGFBR2lo in HCC cells had significantly higher recurrence and death rates,12 and this downregulation of TGFBR2 significantly contributed to the acquisition of a more invasive phenotype.12,14 The effects of TGFBR2 expression on tumors are closely related to the tumor environmental background.18,19 However, the effect of TGFBR2 and the mechanisms involving the HCC microenvironment have not yet been fully disclosed.
Dysregulation of growth factor signaling is a common event in HCC patients and can cooperate with TGFBR2 inactivation to promote the progression of some cancers. TGF-α overexpression has been demonstrated to cooperate with TGFBR2 inactivation to promote HCC formation by increasing cell proliferation.20 Moreover, the expression levels of TGFBR2 and human epidermal growth factor receptor-2 (HER2) have a highly significant positive correlation in breast cancer, and the absence of TGFBR2 is an unfavorable prognostic factor in HER2-negative breast tumors.21 A previous study documented the opposing roles of TGFBR2 and bone morphogenetic protein type II receptor (BMPR2) in prostate carcinogenesis:22 TGFBR2 ablation increases the BMP signaling pathway and promotes the stemness of prostate cancer cells.23 BMP proteins are also members of the TGF-β superfamily and have downstream signal transduction pathways similar to those of TGF-β1; these proteins bind to the BMPR2 and BMPR1 on the cell surface and phosphorylate Smad1/5/8 downstream to exert further functions.24 The crosstalk of the TGF-β and BMP pathways has been widely studied in the contexts of kidney disease and bone formation, and the outcomes of these diseases are related to the tuned balance between TGF-β and BMP signaling.25 Therefore, we proposed that the imbalance of TGF-β and BMP pathways is probably involved in the progression of HCC.
In this study, the gene-expression profiles of 359 HCC samples from The Cancer Genome Atlas (TCGA) and 197 primary HCC tissues from Tianjin Medical University Cancer Institute & Hospital (TJMUCH) were included to explore the interactive network between TGF-β and BMP pathways in HCC, and microarray analysis of 64 cases of total RNA samples was used to investigate core factors orchestrating the interaction between the TGF-β and BMP pathways. Finally, the imbalance of TGF-β1/BMP-7 pathways was identified in HCC; it dominated the interactive communication between cancer cells and M2-polarized tumor-associated macrophages (M2-TAMs) to foster a protumoral microenvironment in HCC, and restoring this imbalance fully attenuated M2-TAM-induced HCC progression in vivo and vitro, which sheds new light on developing potential therapies for HCC.
Results
The imbalance of TGF-β1/BMP-7 pathways was detected at the mRNA level in HCC and was associated with poor clinical outcomes
The RNA sequencing (RNA-seq) data of 359 HCC samples were downloaded from TCGA database. We found that either TGF-β1hi or TGFBR2lo correlated with a worse prognosis than TGF-β1lo or TGFBR2hi expression in HCC patients, whereas the other key components of the TGF-β1 signaling pathway, such as TGFBR1, SMAD2, SMAD3, and SMAD4, displayed little influence on the overall survival (OS) of HCC patients (Figure S1A). In addition, the OS of patients with TGF-β1loTGFBR2hi status was apparently longer than that of patients with TGF-β1hiTGFBR2lo, TGF-β1loTGFBR2lo, and TGF-β1hiTGFBR2hi status (p < 0.05) (Figure S1B). The paradoxical effects of TGF-β1 and the corresponding receptor TGFBR2 on clinical outcome indicated the complexity of the TGF-β1 signaling pathway in HCC progression.
Therefore, the differentially expressed genes between TGFBR2hi and TGFBR2lo HCC samples were compared. A total of 3,373 differentially expressed genes were filtered, and the Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology Based Annotation System (KOBAS) tool was used for gene set enrichment analysis (GSEA) of the differentially expressed genes between TGFBR2hi and TGFBR2lo HCC samples.26 Among the top 10 enriched pathways (Figure 1A), the BMP signaling pathway was significantly upregulated; this pathway activates Smad1/5/8 proteins downstream. Among the differentially expressed genes in the BMP signaling pathway with fold change >1.5, BMP-7 and BMP-8B were significantly upregulated, but the antagonists of the BMP pathway—GREM2, NOG, FSTL1, SMAD6, and CHRDL1—were significantly downregulated in TGFBR2lo HCC samples versus TGFBR2hi HCC samples (Figures 1B and S1C), indicating that the inhibition of TGF-β1 signaling was accompanied by the activation of BMP signaling in HCC.
Figure 1.

The imbalance of TGF-β1/BMP-7 pathways was detected at the mRNA level in HCC and was associated with poor clinical outcomes
(A) The KOBAS tool was used for pathway enrichment analysis of the differential gene between TGFBR2hi and TGFBR2lo HCC tissues in TCGA. (B) Differential expression of BMP-8B, BMP-7, GREM2, NOG, FSTL1, and SMAD6 of the BMP pathway between TGFBR2hi and TGFBR2lo HCC tissues in TCGA. (C) Core genes of the TGF-β and BMP pathways were used for unsupervised clustering, and 359 patients from TCGA cohort were divided into two groups: cluster 1 and cluster 2. (D) The consistency of TGF-β and BMP signaling combination groups was compared with clusters 1 and 2. (E) The OS was compared between the imbalanced group and the balanced group for 359 HCC samples. (F) GSEA identified greater enrichment of gene sets related to EMT, liver cancer survival, and proliferation in the imbalanced group compared with the balanced group. For all panels, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Since there is a strong correlation between TGF-β and the BMP pathway, we wondered whether the combination of the two pathways had an effect on the prognosis of HCC. With the use of unsupervised clustering of the expression value of the core genes of the TGF-β and BMP pathways from 359 HCC samples, we identified 2 clusters with different prognoses, indicating that the HCC cohort related to the TGF-β and BMP pathways comprises 2 molecular subtypes with distinct clinical outcomes. Cluster 1 was associated with a shorter OS time, younger age, higher alpha fetoprotein (AFP) levels, higher incidence of cirrhosis-related disease, and more advanced histological grade than cluster 2 (Figures 1C and S1D; log rank p = 0.008). The gene sets related to epithelial-mesenchymal transition (EMT), liver cancer survival, and proliferation were significantly enriched and overexpressed in cluster 1 but not in cluster 2 (Figure S1E). In addition, 18 genes were upregulated, and 20 genes were downregulated in cluster 1 compared with cluster 2 (p < 0.05). Interestingly, genes that participate in activation of the TGF-β pathway, such as TGFBR2, STRAP, MYC, ZFYVE9, UBE2D3, and CDK9, were significantly downregulated, whereas genes involved in inhibition of the TGF-β pathway, such as PMEPA1, CBL, Smad ubiquitylation regulatory factor-2 (SMURF2), and SKIL, were significantly upregulated in cluster 1 compared with cluster 2 (Figure S1F). In contrast to the patterns observed for the TGF-β pathway, genes involved in the activation of the BMP pathway, such as BMP-7, BMP8A, AMH, and BMPR1A, were significantly upregulated, and genes related to inhibition of the BMP pathway, such as SMAD6, FST, and GREM2, were significantly downregulated in cluster 1 compared with cluster 2 (Figure S1G). It revealed that the TGF-β pathway was attenuated, whereas the BMP pathway was enhanced in cluster 1.
To explore the key modulators of poor prognosis in cluster 1, the key genes along either the TGF-β pathway or the BMP pathway were separated into 2 groups by the clustering method. We found that TGFBR2 combined with activin A receptor type 1 (ACVR1) or BMP-7 could classify 359 HCC samples into similar subgroups with high consistency compared with cluster 1 and cluster 2, and the combination of TGFBR2 and ACVR1 had the highest consistency (consistency ratio = 0.73; Figure 1D). ACVR1 is an indispensable type I receptor of the BMP-7 pathway,27,28 which implies that the imbalance of TGF-β1 and BMP-7 pathways induced by TGFBR2 downregulation and ACVR1 upregulation might be the main explanation for the discrepancy between cluster 1 and 2. As we assumed, 2 subgroups clustered by 359 HCC samples, according to the expression of TGFBR2 and ACVR1, presented different prognoses, of which TGFBR2 and ACVR1 showed different expression levels (Figure S1H). Patients in the group with TGFBR2loACVR1hi status were defined as the imbalanced group, and patients in the other group were defined as the balanced group. HCC patients in the imbalanced group suffered from shorter survival than those in the balanced group (Figure 1E). Also, the balanced group was separated into 3 subgroups with different TGFBR2 and ACVR1 expressions: TGFBR2hiACVR1lo, TGFBR2loACVR1lo, and TGFBR2hiACVR1hi. The Kaplan-Meier survival analysis showed that OS in patients with TGFBR2hiACVR1lo was apparently longer than those in patients with TGFBR2loACVR1lo or TGFBR2hiACVR1hi (Figure S1I).
In addition, the imbalanced group showed higher AFP levels, higher histologic grade, higher pathological stage and American Joint Committee on Cancer (AJCC) stage, more distant metastasis and vascular invasion (Table S1), and greater enrichment of EMT-, liver cancer survival-, and proliferation-related genes (Figure 1F). The results indicate that imbalance of TGF-β1/BMP-7 pathways indicates a more aggressive HCC subtype with a poor prognosis.
The imbalance of the TGF-β1/BMP-7 pathways was validated at the protein level in HCC and was associated with aggressive pathological features and poor clinical outcomes
To validate the significance of the imbalance of TGF-β1 and BMP-7 pathways in HCC progression at the protein level, 2 cohorts of patients providing 197 primary HCC tissues from TJMUCH were enrolled. Patients in cohort I provided a total of 64 primary HCC tissues that were used to detect key proteins along the TGF-β1 pathway (including TGF-β1, TGFBR2, and TGFBR1) and BMP-7 pathway (including BMP-7, BMPR2, and ACVR1) by immunohistochemistry (IHC) staining (Figures 2A and 2B), and OS was compared according to the expression of various proteins. The patients with TGFBR2lo, ACVR1hi, TGF-β1hi, and BMP-7 hi expression had shortened OS times compared to those in the alternative expression groups (p < 0.05; Figure 2C). In addition, we found that TGFBR2 expression was significantly, negatively correlated with ACVR1 expression (r = −0.242, p = 0.047; Figures S2A and S2B). Patients with TGFBR2loACVR1hi status were considered the imbalanced group, and other patients were considered the balanced group. Kaplan-Meier survival analysis indicated that HCC patients in the imbalanced group suffered from worse clinical outcomes than those in the balanced group (Figure S2C).
Figure 2.

The imbalance of TGF-β1/BMP-7 pathways was validated at the protein level in HCC and was associated with aggressive pathological features and poor clinical outcomes
(A and B) Representative images of IHC staining for TGF-β1/BMP-7 pathway components in cohort I. Scale bars, 50 μm. (C) Kaplan-Meier survival analysis showing survival rates based on the expression of TGF-β1/BMP-7 pathway components in cohort I. (D) Representative HCC tumor samples showing the expression of 4′,6-diamidino-2-phenylindole (DAPI), hepatocyte paraffin antigen 1 (HepPar-1), TGFBR2, and ACVR1 by multispectral IF staining in cohort II. Scale bars, 50 μm. (E) Scatterplot depicting the correlation between TGFBR2 levels and ACVR1 levels in HepPar-1+ HCC cells in cohort II. (F and G) Kaplan-Meier survival analysis showing OS rates and DFS rates between the imbalanced group and balanced group in cohort II.
Patients in cohort II provided a total of 133 primary HCC tissues that were used to detect the expression pattern of TGFBR2 and ACVR1 in situ by multispectral immunofluorescence (IF) staining (Figure 2D). Spearman’s rank correlation test showed a significant inverse correlation between TGFBR2 levels and ACVR1 expression in HCC cells (r = −0.582, p < 0.001) (Figure 2E). Kaplan-Meier survival analysis showed that the OS rates and disease-free survival (DFS) rates of the imbalanced group were significantly lower than those of the balanced group (Figures 2F and 2G). Furthermore, univariate analysis, followed by a subsequent multivariate Cox proportional hazards analysis, was conducted in cohort II, which indicated that the imbalance of TGF-β1/BMP-7 pathways was a risk factor and an independent prognostic factor for OS (hazard ratio [HR] 1.848; 95% confidence interval [CI] 0.986–3.466; p = 0.055) and DFS (HR 1.506; 95% CI 0.961–2.360; p = 0.074; Table S2). Importantly, larger tumor size, more microvascular invasion, lower differentiation score, and higher advanced Barcelona Clinic Liver Cancer (BCLC) grade were observed in the imbalanced group than in the balanced group (p < 0.05) (Table S3). The above results indicate that the imbalance of TGF-β1/BMP-7 pathways was associated with aggressive pathological features and poor clinical outcomes in HCC.
The imbalance of TGF-β1/BMP-7 pathways dramatically promoted HCC cell invasion by upregulating EMT and stemness via increasing inhibitor of differentiation 1 (ID1)
We compared the expression of both TGFBR2 and ACVR1 among 4 well-known HCC cell lines (Figure S3A) and knocked down TGFBR2 (TGFBR2KD) and overexpressed ACVR1 (ACVR1OE) in HepG2 cells and Hep3B cells separately or simultaneously to interfere with TGF-β1 or BMP-7 signaling and induce the imbalance of TGF-β1/BMP-7 pathways in vitro. The results showed that TGFBR2KD had no effects on the expression of ACVR1 or the phosphorylation of Smad1/5/8 (pSmad1/5/8). Similarly, ACVR1OE did not affect the expression of TGFBR2 or the phosphorylation of Smad2/3. However, the expression of ID1, the core functional target gene along the BMP-7 pathway,29 was sufficiently induced in HepG2 and Hep3B cells with simultaneous ACVR1OE and TGFBR2KD (Figures 3A and S3B), which indicated that the imbalance of TGF-β1/BMP-7 pathways synergistically promotes the switch from the TGF-β1 pathway to the BMP-7 pathway and induces full activation of the BMP-7 pathway in HCC cells.
Figure 3.

The imbalance of TGF-β1/BMP-7 pathways dramatically promoted HCC cell invasion by upregulating EMT and stemness via increasing inhibitor of differentiation (ID1)
(A) Western blotting of the levels of TGF-β1/BMP-7 pathway components in HepG2 cells with separate or simultaneous TGFBR2 knockdown (TGFBR2KD) and ACVR1 overexpression (ACVR1OE). (B) PCR and western blotting of stem cell-related genes, including OCT4, SOX2, and NANOG, in HepG2 cells. (C) PCR and western blotting of EMT-related genes, including E-cadherin, vimentin, slug, and snail, in HepG2 cells. (D) Wound-healing assay in HepG2 cells at 24 and 48 h. Scale bars, 200 μm. (E) Transwell assay after 24 h in HepG2. Scale bars, 200 μm. (F and G) Transwell assay (scale bars, 100 μm) (F) and western blotting (G) of ID1, EMT, and stemness-related genes in TGFBR2KD and ACVR1OE HepG2 cells after transfection with si-ID1. (H) Luciferase activity assays were applied to assess the effect of ID1 on the promoter activity of OCT4, SOX2, NANOG, Snail, and Slug. (I) Transwell assays were performed in TGFBR2KD and ACVR1OE HepG2 cells after transfection with si-SOX2. Scale bars, 100 μm. For all panels, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Next, we detected the mRNA and protein expression of the core EMT genes and stemness-associated genes and found that the expression of mesenchymal markers rather than epithelial markers, such as Snail, Slug, and vimentin, as well as multiple stem cell signature genes, including OCT4, SRY-box 2 (SOX2), and Nanog homeobox (NANOG), increased in HepG2 and Hep3B cells with ACVR1OE or TGFBR2KD alone compared with negative control (NC) cells. Importantly, these genes were significantly increased in HepG2 and Hep3B cells with simultaneous ACVR1OE and TGFBR2KD compared with cells in the other three groups (Figures 3B, C, S3C, and S3D). In addition, the migration capability of TGFBR2KD and ACVR1OE cells was enhanced significantly (Figures 3D and S3E), and more TGFBR2KD and ACVR1OE cells migrated across the Matrigel layer (Figures 3E and S3F). For further validation, we detected EMT-, proliferation-, and invasion-related biomarkers in primary HCC tissues from patients in cohort I (Figure S3G); these include Ki67, E-cadherin, N-cadherin, β-catenin, vimentin, and MMP9. Consistently, the expression of E-cadherin significantly decreased, whereas that of vimentin significantly increased exclusively in the imbalanced group (p < 0.05; Figures S3H and S3I). These results implied that the imbalance of TGF-β1/BMP-7 pathways dramatically stimulated HCC cell invasion by promoting EMT and stemness.
ID1 is well known as a master regulator of cancer stem cells and tumor aggressiveness and can promote tumorigenesis, tumor progression.30 It was reported that ID1 binds to the promoter regions of pluripotent marker genes and leads to transcriptional activation in prostate cancer.23 To investigate whether ID1 was involved in HCC cell invasion induced by the imbalance of TGF-β1/BMP-7 pathways, we transfected NC small interfering (si)RNA (si-NC) and si-ID1 into HepG2 cells with TGFBR2KD and ACVR1OE. The invasion capability and expression of EMT and stemness genes decreased significantly in si-ID1 HepG2 cells compared with si-NC cells (Figures 3F and 3G). Next, luciferase activity assays were used to detected whether ID1 could bind to promoter regions of OCT4, SOX2, NANOG, Snail, and Slug separately. The results revealed that pcDNA3.1-ID1 transfection significantly increased promoter activity with the pGL3-OCT4, pGL3-SOX2, pGL3-NANOG, pGL3-Snail, and pGL3-Slug (Figure 3H). These results indicated that ID1 was potentially responsible for upregulated expression of OCT4, SOX2, NANOG, Snail, and Slug. To prove that the imbalance of TGF-β1/BMP-7 pathways promoted HCC cell invasion by upregulating EMT and stemness, si-SOX2 was transfected into HepG2 cells with TGFBR2KD and ACVR1OE, and the invasion capability of si-SOX2 HepG2 cells decreased significantly compared with that of si-NC cells (Figure 3I). The results above indicated that the imbalance of TGF-β1/BMP-7 pathways dramatically promoted HCC cell invasion by upregulating EMT and stemness induced by ID1.
The imbalance of TGF-β1/BMP-7 pathways was regulated by the microRNA (miR)-17-92 cluster, which promoted cell invasion in HCC
The frequency of TGFBR2 mutations is approximately 0.5%, and that of ACVR1 mutations is approximately 2% in HCC.31 Furthermore, there is rare report about epigenetic modulation of TGFBR2 and ACVR1. Our previous study found that the competing endogenous RNA (ceRNA) network could involve in the invasion and metastasis of HCC.32 MicroRNAs (miRNAs) can inhibit the translation of multiple proteins by interacting with the 3′ untranslated regions (3′ UTRs) of the mRNAs of target genes.33 Thus, we proposed that miRNAs mediate the imbalance of TGF-β1/BMP-7 pathways by simultaneously targeting components or related molecules of these pathways. To examine this possibility, the miRNA expression profiles were compared between the imbalanced group and balanced group of the TJMUCH cohort (64 HCC patients, cohort I). 31 miRNAs were upregulated, and 32 miRNAs were downregulated in the imbalanced group compared to the balanced group (p < 0.05) (Figure 4A). Among them, we found that all members of the miR-17-92 cluster, including miR-17, miR-18a, miR-19a, miR-19b, miR-20a, and miR-92a, were significantly increased in the imbalanced group.
Figure 4.

The imbalance of TGF-β1/BMP-7 pathways was regulated by the miR-17-92 cluster, which promoted cell invasion and stemness in HCC
(A) The miRNA expression was compared in the imbalanced group compared with the balanced group by analyzing the HCC RNA-seq data of cohort I. (B) Networks of miR-17-92 cluster and differentially expressed genes between the imbalanced and balanced groups of 359 HCC patients from TCGA. (C) PCR array of TGF-β1 and BMP-7 pathways in HepG2MIR17HGhi cells compared with HepG2NC and HepG2 cells and in Hep3BMIR17HGlo cells compared with Hep3BNC and Hep3B cells. (D and E) Western blotting for components of the TGFβ1 and BMP-7 pathways and for ID1 and EMT genes. (F) PCR array for stem cell-associated genes in HepG2MIR17HGhi cells compared with HepG2NC and HepG2 cells and in Hep3BMIR17HGlo cells compared with Hep3BNC and Hep3B cells. (G) Wound closure was detected in HepG2MIR17HGhi, HepG2NC, and HepG2 cells at 24 and 48 h (left); wound closure was detected in Hep3BMIR17HGlo, Hep3BNC, and Hep3B cells at 48 and 72 h (right). Scale bars, 200 μm. (H) Cell invasion of HepG2MIR17HGhi and Hep3BMIR17HGlo cells compared with that of their controls. The graphs depict the number of invasive cells after 24 h. Scale bars, 100 μm. For all panels, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
We compared the correlation between the miR-17-92 cluster and ACVR1 in both TCGA cohort and the Tianjin cohort I. There is no significant difference in the expression of the miR-17-92 cluster between the high ACVR1 and low ACVR1 groups (Figures S4A and S4B). However, both miR-17-92 clusters significantly increased in the imbalanced group compared to those in the balanced group (p < 0.05; Figures S4C and S4D). Furthermore, we found that most components of the miR-17-92 cluster had direct interactions with differentially expressed genes related to cell proliferation, EMT, and poor survival in the imbalanced group (Figure 4B). Thus, the results implied that the imbalance of TGF-β1/BMP-7 pathways is correlated with dysregulation of the miR-17-92 cluster in primary HCC tissues.
The levels of MIR17HG and all components of the miR-17-92 cluster were higher in Hep3B cells than in HepG2 cells. Therefore, we overexpressed MIR17HG in HepG2 cells (HepG2MIR17HGhi) and knocked down MIR17HG in Hep3B cells (Hep3BMIR17HGlo) to interfere with miR-17-92 cluster expression in HCC cells (Figures S4E and S4F). The results showed that TGFBR2 mRNA, TGFBR2 protein, and pSmad2/3 protein levels decreased in HepG2MIR17HGhi cells and increased in Hep3BMIR17HGlo cells. Interestingly, although there was no detectable change at the mRNA level in other key functional components along the BMP-7 pathway, the levels of ACVR1, Smad1, Smad5, and pSmad1/5/8 proteins were significantly enhanced in HepG2MIR17HGhi cells and reduced in Hep3BMIR17HGlo cells (Figures 4C and 4D). Moreover, the protein levels of ID1, the expression levels of EMT and stem cell-associated genes, and the migration and invasion capabilities were increased in HepG2MIR17HGhi but attenuated in Hep3BMIR17HGlo cells (Figures 4E−4H). The above results indicated that the miR-17-92 cluster could interfere with the balance of TGF-β1/BMP-7 pathways at both the RNA and protein levels to enhance the aggressive potential of HCC cells.
The miR-17-92 cluster promoted the imbalance of TGF-β1/BMP-7 pathways by interfering with TGFBR2 mRNA expression and enhancing ACVR1 protein expression via Smurf1 silencing
With the consideration that the levels of TGFBR2 mRNA and protein both decreased in HepG2MIR17HGhi cells and increased in Hep3BMIR17HGlo cells, some members of the miR-17-92 cluster might directly bind to TGFBR2 at the 3′ UTR and thus interfere with TGFBR2 RNA expression at the post-transcriptional level. Therefore, a commonly used TargetScan algorithm34 was applied to predict the putative binding sites for each component of miR-17-92 cluster within the 3′ UTR of the TGFBR2 gene. The putative binding sites for miR-17/20a and miR-19a/19b were distinguished, and identical seed sequences were listed. The luciferase reporter assay revealed that miR-17/20a significantly repressed the luciferase activity of the TGFBR2-wild-type (WT) construct compared with the miR-NC construct (Renilla luciferase activity/firefly luciferase activity [Rluc/fluc]: miR-17: 1.000 ± 0.097 versus 0.521 ± 0.108, p = 0.005; miR-20a: 1.000 ± 0.157 versus 0.556 ± 0.053, p = 0.010). However, miR-19a/19b did not affect the luciferase activity of the TGFBR2-WT construct compared with the miR-NC construct (Figure 5A). Thus, these results demonstrate that the miR-17-92 cluster inhibits the TGF-β1 pathway by directly interfering with TGFBR2 RNA expression.
Figure 5.

miR-17-92 cluster promoted the imbalance of TGF-β1/BMP-7 pathways by interfering with TGFBR2 mRNA levels and enhancing ACVR1 protein levels via Smurf1 silencing
(A) Luciferase reporter assays involving different 3′ UTRs were used to identify the targeted relationship of the miR-17-92 cluster with TGFBR2. (B and C) PCR array and western blotting for the mRNA and protein expression of Smurf1 in HepG2MIR17HGhi and Hep3BMIR17HGlo cells compared with their controls. (D) Luciferase reporter assays involving different 3′ UTRs were used to identify the targeting relationship of the miR-17-92 cluster with Smurf1. (E) A PCR array was used to detect the mRNA expression of TGFBR2 and Smurf1 in HepG2 cells transfected with the miR-17-92 cluster. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
However, by contrast, the level of ACVR1 protein rather than mRNA was enhanced in HepG2MIR17HGhi cells and reduced in Hep3BMIR17HGlo cells. The TargetScan algorithm failed to predict the putative binding sites for each member of the miR-17-92 cluster within the 3′ UTR of the ACVR1 gene, which implied that post-translational protein modification rather than post-transcriptional RNA interference is involved in miR-17-92 cluster-mediated regulation of ACVR1 expression in HCC. Smurf1 is a predominant protein regulating the ubiquitylation and degradation of a complex set of functional proteins along the BMP-7 pathway,35 which reduces cellular responses to BMP-7 by triggering proteasomal destruction of ACVR1, Smad1, and Smad5.36 Both the mRNA and protein levels of Smurf1 were significantly decreased in HepG2MIR17HGhi cells and increased in Hep3BMIR17HGlo cells (Figures 5B and 5C), which suggested that RNA interference of Smurf1 was induced by the miR-17-92 cluster. The TargetScan algorithm revealed that miR-17, miR-20a, miR-19a, miR-19b, and miR-92a might directly bind to Smurf1 mRNA, and the luciferase reporter assay demonstrated that miR-17/20a, miR-19a/19b, and miR-92a mimics significantly repressed the luciferase activity of the Smurf1-WT construct compared with the miR-NC construct (Figure 5D).
Furthermore, after transfecting HepG2 cells with miR-17/20 mimics, the mRNA levels of TGFBR2 and Smurf1 were significantly reduced. However, after transfecting HepG2 cells with miR-19a/19b or miR-92a mimics, the mRNA level of Smurf1 but not TGFBR2 was reduced (Figure 5E). The results demonstrate that the miR-17-92 cluster disrupts the balance of TGF-β1/BMP-7 pathways in HCC at both the post-transcriptional and post-translational levels: miR-17 and miR-20a inhibit TGFBR2 mRNA expression, whereas miR-17, miR-20a, miR-19a, miR-19b, and miR-92a silence the Smurf1 gene to inhibit the ubiquitylation and degradation of the ACVR1 protein.
M2-TAMs manipulate the imbalance of TGF-β1/BMP-7 pathways in HCC
To determine whether tumor-infiltrating immune cells were involved in the imbalance of the TGF-β1/BMP-7 pathway, we analyzed the differences in the proportions of infiltrating immune cells between the imbalanced group and balanced group in TCGA cohort using the online tool CIBERSORT;37 we found that the proportions of macrophages, neutrophils, regulatory T cells, and follicular helper T cells were significantly increased, whereas those of monocytes and mast cells were significantly decreased in the imbalanced group versus the balanced group (p < 0.05; Figure 6A). Recent studies indicate that abrogation of TGFBR2 is associated with myeloid cell infiltration in breast cancer.38 Therefore, we compared the distribution of multiple types of myeloid cells (TAMs and neutrophils) in 64 patients from TJMUCH cohort I. The numbers of both CD68+ TAMs and CD163+ M2-TAMs increased in the imbalanced group compared to the balanced group, but no significant difference was detected in the numbers of neutrophils (Figure 6B). The same result was confirmed in 133 patients from TJMUCH cohort II: the number of CD68+CD163+ M2-TAMs dramatically increased in the imbalanced group compared to the balanced group (Figures 6C and 6D). The concordance analysis indicated that the number of M2-TAMs was significantly associated with the expression of TGFBR2 and ACVR1 in HCC tissues; the total amount of M2-TAMs negatively correlated with the mean fluorescent intensity (MFI) of TGFBR2 (r = −0.467, p < 0.001) and positively correlated with that of ACVR1 (r = −0.494 p < 0.001) in HCC cells (Figure 6E). Patients with high M2-TAM infiltration combined with the imbalance of TGF-β1/BMP-7 pathways presented the worst prognosis in terms of both OS and DFS (Figure 6F).
Figure 6.

M2-TAMs manipulate the imbalance of TGF-β1/BMP-7 pathways in HCC
(A) The differences in the proportion of infiltrating immune cells between the imbalanced group and balanced group were analyzed by using CIBERSORT in TCGA cohort. (B) Immunohistochemical (IHC) analysis showed infiltration of CD68+, CD163+, and CD66b+ cells in tumor sections from cohort I. Scale bars, 50 μm. Student’s t test was used to analyze the differences in CD68+, CD163+, and CD66b+ cell infiltration between the imbalanced group and the balanced group. (C) Representative HCC tumor samples of cohort II showing the expression of DAPI, CD68, CD163, HepPar-1, TGFBR2, and ACVR1 via multispectral IF staining. Scale bars, 50 μm. (D) Student’s t test was used to analyze the difference in CD68+CD163+ M2-TAM infiltration between the imbalanced group and balanced group in cohort II. (E) Scatterplot depicting the statistical correlations between CD68+CD163+ M2-TAM infiltration and the expression of TGFBR2 in HepPar-1+ HCC cells and the expression of ACVR1 in HepPar-1+ HCC cells in cohort II. (F) OS curve and DFS curve for M2-TAM infiltration combined with imbalance of TGF-β1/BMP-7 pathways. (G and H) PCR array and western blotting to assess the mRNA and protein expression of TGF-β and BMP-7 pathway components in HepG2 and Hep3B cells cocultured with M2-TAMs compared to cocultured THP-1 cells or THP-1 cells alone. *p < 0.05, **p < 0.01, ***p < 0.001.
Therefore, a Transwell-based indirect coculture system of M2-TAMs and HCC cells was used to investigate whether M2-TAMs could manipulate the imbalance of TGF-β1/BMP-7 pathways in HCC cells. TGFBR2 and Smurf1 mRNA expression was markedly reduced in HepG2 and Hep3B cells cocultured with M2-TAMs compared to those cocultured with THP-1 cells or those not cocultured (p < 0.05; Figure 6G). Consistently, the protein levels of TGFBR2, Smurf1, and pSmad2/3 were remarkably reduced in both HepG2 and Hep3B cells, whereas those of ACVR1, ID1, Smad1, Smad5, and pSmad1/5 were increased with coculture (Figure 6H). The results revealed that M2-TAMs stimulate the imbalance of TGF-β1/BMP-7 pathways in HCC.
M2-TAMs increased the levels of the miR-17-92 cluster in HCC cells via extracellular vesicles (EVs) to exacerbate the imbalance of the TGF-β1/BMP-7 pathway
We compared the levels of the MIR17HG and miR-17-92 cluster in HCC cells cocultured with M2-TAMs and found obvious upregulation of the MIR17HG and miR-17-92 cluster in HepG2 and Hep3B cells cocultured with M2-TAMs compared to those cocultured with THP-1 cells or not cocultured (p < 0.05; Figure 7A). To determine whether the MIR17HG and miR-17-92 cluster in HCC cells was derived from M2-TAMs, we compared the levels of the MIR17HG and miR-17-92 cluster in the cell lysates and EVs of M2-TAMs and found that both expressed higher levels of the MIR17HG and miR-17-92 cluster than THP-1 cells, and the MIR17HG and miR-17-92 cluster was higher in EVs than in M2-TAM cell lysates (p < 0.05; Figure S5). Furthermore, after adding different components of M2-TAM-derived supernatants to HCC cells, EVs in the supernatants exclusively induced increases in the MIR17HG and miR-17-92 cluster in HepG2 cells (p < 0.05; Figure 7B). To confirm that M2-TAM-derived EVs were taken in HCC cells by phagocytosis, M2-TAM-derived EVs were labeled using carboxyfluorescein succinimidyl ester (CFSE) and added to the culture system of HepG2 cells. As predicted, CSFE-labeled EVs were detected in HepG2 cells (Figure 7C), which demonstrated that M2-TAMs indeed transmitted the miR-17-92 cluster to HCC cells via EV release. The EV markers CD63, TSG101, and Alix were detected by western blotting assay; in contrast, calnexin, a negative marker of EVs, was absent in EVs isolated from M2-TAMs (Figure 7D). The morphology and size distribution of the EVs from M2-TAMs were evaluated by transmission electron microscopy (TEM) and nanoparticle tracking analysis (NTA). TEM and NTA analysis showed that the EVs in the isolated fractions were oval or bowl shaped with a size range between 75 nm and 200 nm, which is a typical size range for exosomes (Figures 7E and 7F). Therefore, the EVs we extracted from M2-TAMs were identified as exosomes.
Figure 7.

M2-TAMs increased the levels of the MIR17HG and miR-17-92 cluster in HCC cells via EVs to exacerbate the imbalance of the TGF-β1/BMP-7 pathways
(A) PCR array for the expression of the MIR17HG and miR-17-92 cluster in HepG2 and Hep3B cells cocultured with M2-TAMs compared to THP-1 cells cocultured or cultured alone. (B) The expression of the MIR17HG and miR-17-92 cluster in HepG2 cells was detected by PCR after treatment with EVs from M2-TAMs or EV-free supernatant from M2-TAMs or conditioned media from M2-TAMs. (C) HepG2 cells were treated with CSFE-labeled EVs secreted by M2-TAMs, and HepG2 cells were visualized by staining with DAPI and actin. Scale bars, 50 μm. (D) Western blotting of specific membrane proteins from control cells and EV lysates from M2-TAMs. (E) NTA of EVs from M2-TAMs. (F) TEM for EVs from M2-TAMs. Scale bars, 200 μm. (G−I) HepG2 cells cocultured with M2-TAMs were treated with sh-MIR17HG, and western blotting of TGF-β1 and BMP-7 pathway components and EMT genes was performed. PCR assays of stemness genes and migration (scale bars, 200 μm) and invasion (scale bars, 100 μm) abilities were performed. *p < 0.05, **p < 0.01, ***p < 0.001.
Afterward, we investigated whether the miR-17-92 cluster participated in M2-TAM-induced imbalance of TGF-β1/BMP-7 pathways in HCC. The western blotting results revealed that the imbalance of TGF-β1/BMP-7 pathways in HepG2 cells caused by M2-TAMs was reversed by short hairpin RNA (shRNA) targeting MIR17HG (sh-MIR17HG), which significantly increased the levels of TGFBR2 and pSMAD2/3 but decreased the levels of ACVR1, Smad1, Smad5, pSmad1/5, and ID1. Consistently, after sh-MIR17HG transfection, the levels of multiple EMT-related proteins and stemness-related genes and the migration and invasive abilities of HepG2 cells cocultured with M2-TAMs declined compared to those of cells transfected with nonsense shRNA (Figures 7G−7I). These results implied that M2-TAMs exacerbate the imbalance of the TGF-β1/BMP-7 pathways by increasing the levels of the MIR17HG and miR-17-92 cluster in HCC cells via EV transmission, which further promotes HCC invasion and stemness.
Reversing the imbalance of TGF-β1/BMP-7 pathways effectively attenuated M2-TAM-abundant HCC xenograft growth and metastasis in vivo
We injected Hep3B cells with or without M2-TAMs into nonobese diabetic severe combined immunodeficiency (NOD-SCID) mice to construct M2-TAM-abundant or M2-TAM-scarce HCC xenograft-bearing mouse models. M2-TAM-scarce HCC xenograft-bearing mice were treated with EVs from M2-TAMs at the tumor site after cell transplantation. The results showed that the tumor volume and number of metastatic lung nodules increased significantly in M2-TAM-abundant xenograft-bearing mice compared with M2-TAM-scarce xenograft-bearing mice. Consistent results were obtained in M2-TAM-scarce HCC xenograft-bearing mice treated with EVs from M2-TAMs (Figures 8A and 8B). IHC staining indicated that TGFBR2 decreased and ACVR1 increased both in M2-TAM-abundant xenografts and M2-TAM-scarce xenografts treated with EVs from M2-TAMs. Furthermore, an increase of ID1 and vimentin expression and a decrease of E-cadherin expression were detected in M2-TAMs-scarce xenografts treated with EVs from M2-TAMs. Thus, it revealed that M2-TAMs induced the imbalance of TGF-β1/BMP-7 pathways via EVs and promoted HCC growth and metastasis in vivo (Figure 8C).
Figure 8.

Reversing the imbalance of TGF-β1/BMP-7 pathways effectively attenuated M2-TAM-abundant HCC xenograft growth and metastasis in vivo
(A) The tumor volume, tumor growth curve, tumor weight, and weight curve of the mice are shown. (B) Pulmonary metastatic nodules were detected by hematoxylin and eosin (H&E) staining (scale bars, 100 μm). (C) IHC staining (scale bars, 50 μm) of TGFBR2, ACVR1, ID1, E-cadherin, and vimentin was analyzed under the following conditions: engraftment of Hep3B cells, engraftment of Hep3B cells cocultured with M2-TAMs, and engraftment of Hep3B cells treated with EVs from M2-TAMs. (D) The tumor volume, tumor growth curve, tumor weight, and weight curve of the mice are shown. (E) Pulmonary metastatic nodules were detected by H&E (scale bars, 100 μm). (F) IHC (scale bars, 50 μm) staining of TGFBR2, ACVR1, ID1, E-cadherin, and vimentin was analyzed under the following conditions: engraftment of Hep3B cells cocultured with M2-TAMs + injection of sh-NC lentivirus, engraftment of Hep3B cells cocultured with M2-TAMs + injection of sh-MIR17HG lentivirus, engraftment of Hep3B cells cocultured with M2-TAMs, engraftment of Hep3B cells cocultured with M2-TAMs + injection of DMH1, engraftment of Hep3B cells cocultured with M2-TAMs + injection of NC lentivirus, and engraftment of Hep3B cells cocultured with M2-TAMs + injection of TGFBR2 lentivirus. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
M2-TAM-abundant xenograft-bearing mice were administered an intratumoral injection of sh-MIR17HG lentivirus or TGFBR2 lentivirus or an intraperitoneal injection of the ACVR1 inhibitor DMH1. No significant difference in body weight was detected among variably treated mice and controls, which implied no severe toxicity of the treatments. As predicted, sh-MIR17HG lentivirus reduced the tumor volume and number of lung metastatic nodules in M2-TAM-abundant xenograft-bearing mice (Figures 8D and 8E, d versus e), increased the TGFBR2 expression, and reduced the ACVR1 expression. Similarly, sh-MIR17HG reduced expression of ID1 and vimentin and increased expression of E-cadherin (Figure 8F, d versus e). Consistently, either treatment with TGFBR2 lentivirus (Figures 8D−8F, g versus h) or ACVR1 inhibitor DMH1 (Figures 8D−8F, b versus f) dramatically reduced the tumor volume and number of lung metastatic nodules in M2-TAM-abundant xenograft-bearing mice, as well as the expression of ID1 and vimentin. These results indicate that reversing the imbalance of TGF-β1/BMP-7 pathways effectively attenuates M2-TAM-abundant HCC xenograft growth and metastasis in vivo.
Discussion
In this study, we identified a specific HCC subtype with the imbalance of TGF-β1/BMP-7 pathways, indicated by TGFBR2lo and ACVR1hi; this phenotype significantly promoted HCC invasion and predicted poor clinical outcomes. Systemic bioinformatics analysis of primary HCC samples from TCGA revealed that the imbalanced group showed higher AFP levels, larger tumor sizes, poor differentiation status, and higher metastasis potential than the balanced group. Moreover, expression localization analysis by multispectral IF staining revealed that the imbalance of TGF-β1/BMP-7 pathways was a risk factor and an independent prognostic factor for OS and DFS and was significantly associated with large tumor size, microvascular invasion, portal vein tumor thrombus (PVTT), poor differentiation, and advanced BCLC grade. Thus, the imbalance of TGF-β1/BMP-7 pathways plays a key role in HCC invasion and metastasis, and the specific subtype of HCC with the imbalance of TGF-β1/BMP-7 pathways should be distinguished from other types since patients of the former type have a poor prognosis and a highly aggressive phenotype.
It has been extensively reported that TGF-β1 and BMP-7 signaling can counterbalance the effects of each other. The reciprocal shift of TGF-β1 and BMP-7 signaling is a critical factor in determining the differentiation and development of multiple cell types, such as mucosal Langerhans cells,39 myoblasts,40 and embryonic stem cells.41 The imbalance of TGF-β1/BMP-7 pathways is greatly involved in the progression of kidney disease,42 bone-related disease,43 and some fibrotic-related diseases.44 Moreover, in gastric cancer, the expression of TGFBR2 and ACVR1 indicates different prognoses.45 BMP-7 signaling in TGFBR2-deficient stromal cells provokes epithelial carcinogenesis to elicit squamous cell carcinoma in the forestomach.46 However, whether the dynamic transformation of TGF-β1 signaling to BMP-7 signaling participates in the regulation of HCC development and progression is still unknown. In our study, the interference with the balance of TGF-β1/BMP-7 pathways by simultaneously knocking down TGFBR2 and overexpressing ACVR1 promoted the switch from the TGF-β1 pathway to the BMP-7 pathway and induced full activation of the BMP-7 pathway in HCC cells, significantly exacerbating the aggressiveness and stemness of HCC cells. Therefore, our findings reveal a feasible biomarker for predicting the clinical outcome of HCC patients and demonstrate a novel molecular mechanism for this highly metastatic HCC subtype.
Studies have revealed the antagonistic mechanisms of the TGF-β1 and BMP-7 pathways at different levels of ligand-receptor interaction, pSmad, and target gene transcription in different cell types.25 We demonstrated that TGFBR2KD did not affect downstream BMP-7 at the protein level in HCC cells. It was reported that activation of the TGF-β1 pathway can suppress the expression of ID1 by upregulating SnoN, a transcriptional repressor with no interaction with Smad1/5/8.47 Therefore, we presumed that TGFBR2KD might increase ID1 expression by inhibiting SnoN without affecting the activating step for Smad1/5/8. We next detected SnoN gene transcription in HepG2 and Hep3B cells with TGFBR2KD and confirmed that SnoN significantly decreased, and ID1 accordingly increased compared with those NC-treated cells (Figure S6). Furthermore, it has been reported that miRNA-related post-transcriptional regulation and protein modification-related post-translational regulation also participate in the regulation of the TGF-β1 and BMP-7 pathways.25 In our study, the miR-17-92 cluster was identified to induce the imbalance of TGF-β1/BMP-7 pathways by synergistically and concurrently inhibiting TGFBR2 and stimulating ACVR1 expression by mediating both post-transcriptional regulation and post-translational modification mechanisms. miR-17-92 is an oncogenic miRNA cluster, including 6 mature miR-17-92 miRNAs derived from long noncoding RNA (lncRNA) MIR17HG at 13q31.3, and this cluster is frequently dysregulated in malignancy and plays an important role in the cell cycle, proliferation, apoptosis, and invasion. A previous study reported that the miR-17-92 cluster is highly expressed in HCC tissues compared to nontumorous liver tissues and that forced overexpression of the miR-17-92 cluster in cultured HCC cells enhanced tumor cell colony formation and invasiveness in vitro, but the mechanisms have not yet been clearly elucidated.48 In this study, we found that the miR-17-92 cluster directly decreased TGFBR2 mRNA levels by binding its mRNA 3′ UTR and indirectly increased ACVR1 protein levels by interfering with Smurf1-mediated ubiquitination to induce the imbalance of TGF-β1/BMP-7 pathways and thus to promote cell invasion and stemness in HCC. Therefore, we proposed a novel multilevel regulatory mechanism by which the miR-17-92 cluster promotes the development and progression of HCC.
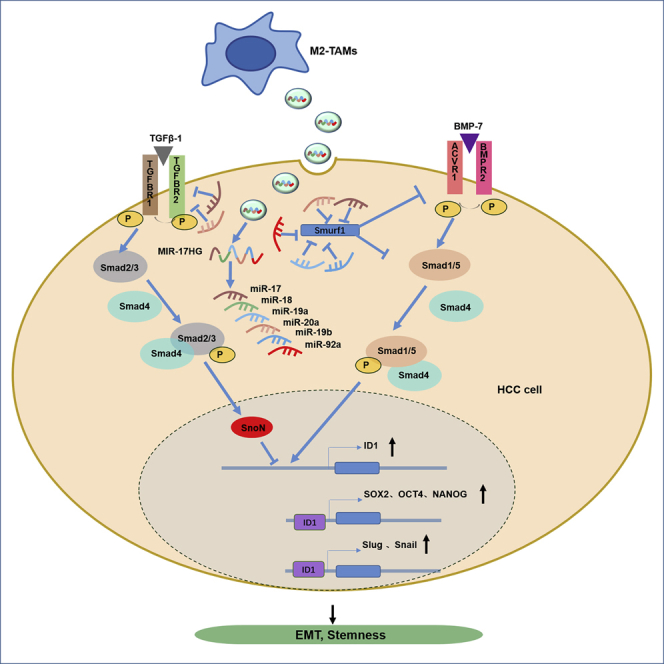
The microenvironment plays a crucial role in carcinogenesis and HCC progression, and multiple inflammatory cells within the microenvironment contribute to the inappropriate activation of a series of oncogenic signaling pathways and aggressive phenotypes of malignant cells. EVs provide an effective transportation method for the shuttling of biomolecules among various cell types in the tumor microenvironment and play a critical role in regulating cancer cell biology. EVs are common membrane-bound nanovesicles that contain diverse biological molecules, such as lipids, proteins, and nucleic acids. EVs are derived from cells through exocytosis, are ingested by target cells, and can transfer biological signals between local or distant cells.49 Some studies have found miR-17-92 cluster members in the EVs of monocytes and macrophages,50 but the mechanism by which the miR-17-92 cluster mediates the interaction between HCC cells and macrophages is not clear. Here, our findings revealed that M2-TAMs in the HCC inflammatory environment could release and transmit EVs containing MIR17HG and miR-17-92 cluster components to HCC cells to induce the imbalance of TGF-β1/BMP-7 pathways and promote HCC progression (Figure S7).
Studies have indicated that M2-TAMs are protumoral inflammatory cells that correlate with a poor prognosis in HCC.51 M2-TAMs could promote HCC progression through transferring the exosome-derived αMβ2 integrin52 and secreting protumoral cytokines interleukin (IL)-653 or growth factor hepatocyte growth factor (HGF).54 Many studies have confirmed that M2-TAMs can promote cancer invasion by stimulating EMT in HCC cells with a late TGF-β1 signature.51,55 Here, we demonstrated an alternative mechanism by which M2-TAMs re-educate HCC cells to switch from the early TGF-β1 signature to the late TGF-β1 signature by inducing the imbalance of TGF-β1/BMP-7 pathways via the miR-17-92 cluster to confer more aggressive phenotypes. Our findings implied that M2-TAMs are the predominant coordinators in the microenvironment that facilitate the communication between oncogenic signaling pathways to mediate the transformation of malignant phenotypes, which finally determines the behavior and fate of HCC in vitro and in vivo. Accordingly, targeting either MIR17HG or ACVR1 effectively inhibited the growth and metastasis of HCC in TAM-abundant tumor-bearing mouse models, which provides new insights for the development of potential therapies for HCC.
We also found that the pathway of amine ligand-binding receptors was significantly upregulated in TGFBR2hi samples compared with TGFBR2lo HCC samples. The amine ligands included acetylcholine, adrenaline, noradrenaline, dopamine, serotonin, and histamine, which biologically act as neurotransmitters in humans, and some amine ligands and receptors have been reported to participate in HCC carcinogenesis.56, 57, 58, 59, 60 However, the cross-talk between amine ligand-binding receptors pathway and the TGF-β pathway has been barely reported. In the future, we would like to further investigate the interaction between these 2 signaling pathways in HCC carcinogenesis.
Moreover, in addition to TAMs and polymorphonuclear leukocytes (PMNs), there are still a large number of immune cells in the microenvironment regulating the development and progression of HCC; in TCGA cohort, we also found that the proportion of T regulatory cells and T follicular helper cells significantly increased, and that of mast cells significantly decreased in the imbalanced group compared to the balanced group. Digital spatial profiling (DSP) technology, a new tool that enables simultaneous and guided detection of multiple proteins and mRNAs from a single tumor tissue, should be used to comprehensively evaluate the interaction between the tumor and the microenvironment61 and generate a panoramic image of core molecular events involved in the imbalance of TGF-β1/BMP-7 pathways in HCC.
However, our study still needs to be further improved since HCC exhibits an inherent high degree of heterogeneity in the tumor and microenvironment and also exhibits complex etiologic diversity. Therefore, patient-derived xenograft (PDX) HCC models62 are warranted to further validate the therapeutic efficacy of the imbalance of TGF-β1/BMP-7 pathways in HCC.
Materials and methods
Patient enrollment and follow-up
Two independent cohorts, together totaling 197 HCC patients, were enrolled in this study and defined as the TJMUCH cohort. One cohort (n = 64 patients who provided paraffin-embedded and snap-frozen tissues) was applied for IHC analysis and whole-genome expression profiling of tumor and normal adjacent tissues from HCC patients treated with partial liver resection surgery at the Department of Hepatology of TJMUCH between 2007 and 2009. A second cohort (n = 133 patients who provided paraffin-embedded tissues), including tumor tissues from HCC patients undergoing curative resection at the same department between 2010 and 2013, was applied for multispectral IF staining. All cases were diagnosed as HCC by 2 experienced pathologists separately, and more than 50% of the cancer cell content was determined for all paraffin-embedded tissues. All patients were followed up for at least 44 months (44.8−79.9 months), and their clinical and pathological information was collected for statistical analysis. This study was approved by the Ethics Committee of Tianjin Medical University (no. TMUHMEC 2012018), and informed consent was obtained from all patients.
Cell lines and animals
The human HCC lines HepG2 and Hep3B and the human leukemia monocytic cell line THP-1 were purchased from Guangzhou Saiku Biotechnology, China. The human HCC lines MHCC97H and HLE were gifted from Professor Hua Guo from the Laboratory of Cancer Cell Biology. All cell lines were authenticated using short tandem repeat (STR) profiling and maintained in a 37°C, 5% CO2 humidified chamber in 10% fetal bovine serum (FBS) + RPMI 1640/Dulbecco’s modified Eagle’s medium (DMEM) (Gibco-BRL, USA). Human TGF-β1 (5 ng/mL; PeproTech, USA) and BMP-7 (100 ng/mL; PeproTech, USA) cytokines were used to activate the TGF-β1 and BMP-7 pathways. THP-1 cells were incubated with human IL-8 (PeproTech, USA) at 37°C for 72 h at 5 ng/mL to induce M2-TAMs, as previously reported.63 Then, the HCC cells were incubated with M2-TAMs for 72 h (HCC cells:M2-TAMs, 1:1). 4- to 6-week-old NOD- SCID male mice were obtained from SPF (Beijing, China) Biotechnology and were maintained under specific pathogen-free conditions. Humane care of the animals conformed with the Guide for the Care and Use of Laboratory Animals criteria of the National Academy of Sciences.
Analysis of HCC data from TCGA
We downloaded data for a total of 418 patients from the Genomic Data Commons (GDC) TCGA Liver Hepatocellular Carcinoma (LIHC) dataset from the University of California, Santa Cruz (UCSC), Xena browser (LIHC: https://xenabrowser.net) and defined this group as TCGA cohort. The gene expression and survival data were retrieved. To exclude the influence of age and complications on survival, we selected samples that met the following criteria: age at operation less than 85 years, no severe inflammation in the adjacent tissue, and OS longer than 80 days. Finally, 359 samples were filtered, and the follow-up time was set to 2,500 days. For the genes mentioned in the article, the median gene expression values were taken as the cutoff to divide the patients into high-expression and low-expression groups. Kaplan-Meier curves were constructed to evaluate the OS of all patients. For univariate survival analysis, log-rank tests were used to explore the differences between two groups.
Functional enrichment analysis of differentially expressed genes in the TGFBR2-high/low subgroup of TCGA
The median expression value of TGFBR2 in 418 HCC cases in TCGA was used as the threshold to divide the samples into two groups: TGFBR2hi and TGFBR2lo. The 359 filtered patient samples contained a total of 177 TGFBR2hi samples and 182 TGFBR2lo samples. These two groups were analyzed for differentially expressed genes (Wilcoxon test). The screening criteria were a p value <0.05 and a FC in both the mean and medium expression values greater than 1.5. A total of 3,373 differentially expressed genes were obtained.
Enrichment analysis with the Reactome pathway database was performed for 3,373 differentially expressed genes using the online functional enrichment tool KOBAS 3.0 (KOBAS: http://kobas.cbi.pku.edu.cn/kobas3/?t=1). All signal transduction pathways were further investigated. The pathways were sorted according to the ratio of the number of foreground genes to the number of background genes, and the top 10 pathways with more than 10 foreground genes were selected for display.
Unsupervised clustering of TCGA data using key genes in the TGF-β and BMP pathways
A total of 49 key genes in the TGF-β and BMP pathways were selected to classify 359 TCGA samples, and log2 (fpkm-uq+1) gene-expression values were retrieved from UCSC Xena and converted to Z scores. The Ward.D2 method was used for hierarchical clustering. Next, all combinations of gene pairs—one in the TGF-β pathway and the other in the BMP pathway—were used to classify the 359 samples into two groups using the unsupervised clustering method (hclust in R, scale = “column” cluster, method = Ward.D2). The consistency ratio was calculated to compare the classification efficiency of the two-gene pattern in subgrouping 359 TCGA samples.
Construction of the miRNA-mRNA regulatory network
The target genes of 6 miRNAs of the miR-17-92 cluster, namely, hsa-miR-17, hsa-miR-18a, hsa-miR-19a, hsa-miR-19b, hsa-miR-20a, and hsa-miR-92a, were downloaded from the database miRTarBase. A total of 181 target genes that were differentially expressed (p < 0.05 and FC > 1.5) in the imbalanced group compared to the balanced group of 359 TCGA HCC samples were visualized in the network. Genes that were up- and downregulated are indicated by different colors. The genes involved in pathways related to EMT, poor survival, and proliferation were retrieved from GSEA’s molecular signature database and are indicated in the network.
Microarray analysis
Sixty-four total RNA samples were obtained from the cancer tissues and adjacent normal tissues, and genome-wide expression profiling and miRNA expression profiling were performed as previously described.32 The profiling data are presented in the Gene Expression Omnibus (GEO) database (GEO: GSE116174 and GSE116182). Raw data were processed using GCOS software and analyzed using R2.9.0 software and Bioconductor 2.4. Student’s t test was performed to evaluate the differential expression of miRNAs between the imbalanced group and the balanced group, and a heatmap was generated with pheatmap software.
IHC
We performed IHC staining using the avidin-biotin-peroxidase complex method, as previously described.63 Primary antibodies and dilutions are listed in Table S4. NCs that omitted the primary antibody were included in all assays. IHC staining was assessed by 2 independent investigators. Cells stained brownish yellow in the cell membrane or in the cytoplasm were regarded as positive. The stained tissue sections were then digitally scanned on an Olympus BX51 microscope for image acquisition at 400× magnification. A standard semiquantitative double-scoring system was applied to evaluate the intensity and proportion of positive cells stained for TGF-β1, TGFBR2, TGFBR1, BMP-7, BMPR2, ACVR1, E-cadherin, N-cadherin, β-catenin, MMP9, and vimentin. Both the intensity and proportion of positive cells were scored from 0 to 3, and for each sample, both scores were added together to obtain a final score. When the final score was less than 4, the samples were included in the negative expression group; otherwise, the samples were included in the positive expression group. For Ki67, CD68, CD163, and CD66b staining, positively stained cells were counted in 5 fields at 200× magnification, and the total number of positive cells was calculated.
Multispectral IF staining
We performed multispectral IF staining as previously described.64 In brief, the slides were heated, deparaffinized using xylene, and rehydrated in graded alcohols. After antigen retrieval and blocking, the primary antibody (Table S5) was applied and incubated at 4°C overnight. Opal polymer horseradish peroxidase (HRP) was used as the secondary antibody. The slides were washed, and tyramide signal amplification (TSA) dye (Opal 7 Color Kit; PerkinElmer, Hopkinton, MA, USA) was applied. The slides were then microwaved to strip the primary and secondary antibodies, washed, and blocked again using blocking solution. Afterward, a second primary antibody and 4′,6-diamidino-2-phenylindole (DAPI) were applied. Finally, slides were coverslipped using ProLong Gold Antifade Reagent (Invitrogen, USA) and inspected using a TissueGnostics automated multispectral microscope. Five fields at 200× magnification from the single-color slides were imaged, and StrataQuest Image Analysis software (v.6.0.1.181) was used to generate a spectral library for unmixing. Index cases stained using the multiplex method were then imaged. Channels were unmixed using the spectral library, and tissues and cells were segmented and scored.
Lentivirus vectors and cell transfection
The following lentivirus vectors were customized by Shanghai GeneChem: ACVR1 expression vector and control vector (Ubi-MCS-3FLAG-CBh-gcGFP-IRES-puromycin), shRNA-TGFBR2 and NC vectors (hU6-MCS-ubiquitin-EGFP-IRES-neomycin), MIR17HG expression vector and control vector (CMV-MCS-IRES-EGFP-SV40-neomycin), and shRNA-MIR17HG and NC vectors (hU6-MCS-CMV-GFP-SV40-neomycin). Transduction was performed as previously described.63
Western blot analysis
Western blotting was performed as previously described.63 Briefly, we generated total protein from each group by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and transferred the proteins to polyvinylidene fluoride membranes. The primary antibodies and dilutions are listed in Table S4. The membranes were incubated with the primary antibody overnight at 4°C and then incubated with secondary antibodies specific to the mouse-, rabbit-, or goat-derived primary antibodies (1:4,000; Santa Cruz Biotechnology, USA) at room temperature for 1 h. Afterward, the membranes were exposed using an enhanced chemiluminescence reagent (Chemicon International, USA).
Wound-healing assay
A wound-healing assay was performed as previously described.63 The cells were allowed to grow to confluence and cultured in serum-free medium for 12 h prior to being scratched with a sterile pipette tip. The cells were washed twice with PBS to remove cell debris and then cultured for the next 24 h (or 48 h) and 48 h (or 72 h). The migration rate was quantified and analyzed by measuring the distance between the wound edges with the IPP 6.0 system at 100× magnification. This assay was independently repeated three times. Independent duplicates were measured in the same manner, and the wound closure rate was calculated from photographs acquired at 24 h (or 48 h) and 48 h (or 72 h).
Transwell assay
Transwell assays were performed as previously described.63 In brief, 2 × 104 cells were harvested and resuspended in 200 mL serum-free DMEM before being plated in the upper chamber. The lower chamber of the Transwell was filled with 500 mL DMEM, supplemented with 10% FBS. The cell suspension was applied to the Matrigel membrane and incubated at 37°C for 48 h. All experiments were repeated three times.
Quantitative real-time PCR analysis
Quantitative real-time PCR was performed using a SYBR Premix Ex Taq Kit (Takara), according to the manufacturer’s instructions. Data were collected and analyzed using a LightCycler 480 instrument (Roche). miRNA was reverse transcribed and relatively quantitated using the TaqMan MicroRNA Reverse Transcription Kit (Thermo Fisher Scientific, USA). All mRNA primers used in this study are shown in Table S6, and TaqMan probes were purchased from Thermo Fisher Scientific. The internal reference gene for mRNA and MIR17HG was β-actin, and that for miRNA was U6. The relative expression levels for each sample were calculated based on the threshold cycle (Ct) value for the target gene normalized to the Ct value of the internal reference gene using the formula 2 − ΔCt (ΔCt = Ct target gene − Ctβ-actin/U6).
3′ UTR luciferase reporter assays
The WT 3′ UTR luciferase reporter vectors TGFBR2 and Smurf1 were constructed and cloned into the XhoI and NotI sites of the psiCHECK-2 vector (Hanbio, China). The mutant-type (Mut) TGFBR2 and Smurf1 3′ UTRs were generated from the WT construct. Then, 293T cells were cotransfected with 0.8 μg 3′ UTR WT/Mut luciferase reporter plasmid and 5 pmol mmu-miRNA mimics/NC (Hanbio, China) using transfection reagents (Lipofectamine 2000 Reagent; Invitrogen, USA), according to the manufacturer’s instructions. After 48 h, the lysates of the 293T cells were harvested, and the luciferase activities were measured using a dual-luciferase reporter assay system (Promega, USA). The data were normalized for transfection efficiency by dividing the fluc activity by the Rluc activity.
Luciferase assay
The OCT4, SOX2, NANOG, Snail, and Slug promoters were cloned into the pGL3-basic vector (Hanbio, China). 293T cells were grown to 60%–80% confluency, after which, the cells were transfected with the pGL3-promoter, pGL3-control, pcDNA3.1-ID1, or pcDNA3.1-NC and Rluc construct (20:1 ratio) using Lipofectamine 2000 Reagent (Invitrogen, USA). The luciferase activities were measured after 48 h (Promega). The luciferase activity was normalized to Rluc activity.
EV experiments
The culture medium was collected, centrifuged at 3,000 rpm for 10 min at 4°C, and then centrifuged at 10,000 × g for 30 min at 4°C to remove cellular debris. Next, the supernatant was filtered using a 0.22-μm filter. EVs were obtained by ultracentrifugation at 100,000 × g for 90 min and then collected and diluted in PBS. For quantitative real-time PCR, the values for EV-packaged lncRNA MIR17HG were normalized against those of a synthesized exogenous reference, λ poly(A)+ RNA (Takara). The values for the EV-packaged miR-17-92 cluster were normalized against those of U6.
NTA
Vesicle suspensions with concentrations between 1 × 107/mL and 1 × 109/mL were examined using a ZetaView PMX 110 (Particle Metrix, Meerbusch, Germany) equipped with a 405-nm laser to determine the size and quantity of the isolated particles. A 60-s video was taken with a frame rate of 30 frames/s, and particle movement was analyzed using NTA software (ZetaView 8.02.28).
TEM
10 μL of exosome solution was placed on a copper mesh grid and incubated at room temperature for 10 min. After washing with sterile distilled water, the exosomes were incubated with uranyl acetate solution (a contrast agent) for 1 min. The sample was then dried for 2 min under incandescent light. The copper mesh was observed and photographed under a transmission electron microscope (H-7650; Hitachi, Tokyo, Japan).
Western blot analysis of EVs
The exosome supernatant was denatured in 5 × SDS buffer and subjected to western blot analysis (10% SDS-polyacrylamide gel electrophoresis; 50 μg protein/lane) using rabbit polyclonal antibodies against CD63 (sc-5275; Santa Cruz, CA, USA), Alix (sc-53540; Santa Cruz, CA, USA), TSG101 (sc-13611; Santa Cruz, CA, USA), and calnexin (10427-2-AP; Promega, Madison, WI, USA). The proteins were visualized on the Tanon 4600 Automatic Chemiluminescence Image Analysis System (Tanon, Shanghai, China).
Xenograft animal model
The animal model was generated according to a previous report.65 Hep3B (1 × 106) and M2-TAMs (1 × 106) were mixed with Matrigel and subcutaneously coinjected into 4- to 6-week-old male NOD-SCID mice. When masses developed after 14 days, the ACVR1 inhibitor DMH1 (S7146; Selleck) (5 mg/kg) was administered to the mice, and the control group was treated with blank PBS containing 10% (v/v) DMSO. Moreover, a total of 2 × 108 TU lentivirus-NC, lentivirus-sh-MIR17HG l, or lentivirus-TGFBR2 lentivirus particles were injected into the tumors at days 14, 21, and 28. Treatment of the mice with EVs (2 μg EVs per injection at the tumor site) or PBS occurred twice a week starting 1 day after cell transplantation and continued until tumor collection. The tumor volume (in cubic millimeters) was estimated every 3 days and was calculated by the following formula: volume = width2 × length/2. After 38 days of implantation, the mice were euthanized. After the tumors were weighed, they were fixed in 10% buffered formalin for further examinations.
Statistical analysis
The statistical analyses were performed with SPSS 16.0 for Windows. The data are presented as the mean ± SD of at least three independent experiments. We used Student’s t test to compare quantitative data between groups and the chi-square test to compare categorical variables. Correlations were analyzed among CD68, CD163, TGFBR2, ACVR1, MIR17HG, and miR-17-92 clusters. The Kaplan-Meier method was used to calculate OS and cumulative recurrence rates, and the log-rank test was used to analyze any differences. For univariate and multivariate analyses, the Cox proportional hazards regression model was employed. p values < 0.05 were considered statistically significant.
Acknowledgments
We thank Dr. Fenglin Zang and Yalei Wang (Department of Pathology, TJMUCH) for giving some pathological suggestions. We thank Cancer Biobank of Tianjin Medical University Cancer Institute & Hospital for its help. This work was supported by National Natural Science Foundation of China (grant numbers 82002601, 82072588, and 81872143), Projects of Science and Technology of Tianjin (18JCQNJC82700), and Key Project of Tianjin Health and Family Planning Commission (16KG126).
Author contributions
J.Y. and Y.Z. conceived and designed the experiments. J.N. and Y.Y. performed the experiments. D.B. was responsible for bioinformatics analysis. J.N., Y.Y., D.B., and J.Y. prepared all of the figures and wrote the manuscript. G.Z. and T.S. collected the HCC samples. P.L. and W.Y. analyzed the data. H.W. and G.Y. offered multispectral immunofluorescent detection equipment. H.L. and X.R. provided the molecular and cellular experimental facility. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.02.016.
Contributor Information
Yi Zhao, Email: biozy@ict.ac.cn.
Jinpu Yu, Email: yujinpu@yahoo.com.
Supplemental information
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Orcutt S.T., Anaya D.A. Liver Resection and Surgical Strategies for Management of Primary Liver Cancer. Cancer Control. 2018;25 doi: 10.1177/1073274817744621. 1073274817744621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Izzo F., Granata V., Grassi R., Fusco R., Palaia R., Delrio P., Carrafiello G., Azoulay D., Petrillo A., Curley S.A. Radiofrequency Ablation and Microwave Ablation in Liver Tumors: An Update. Oncologist. 2019;24:e990–e1005. doi: 10.1634/theoncologist.2018-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ling C.Q., Fan J., Lin H.S., Shen F., Xu Z.Y., Lin L.Z., Qin S.K., Zhou W.P., Zhai X.F., Li B., Zhou Q.H., Chinese Integrative Therapy of Primary Liver Cancer Working Group Clinical practice guidelines for the treatment of primary liver cancer with integrative traditional Chinese and Western medicine. J. Integr. Med. 2018;16:236–248. doi: 10.1016/j.joim.2018.05.002. [DOI] [PubMed] [Google Scholar]
- 5.Zhao H.T., Meng Y.B., Zhai X.F., Cheng B.B., Yu S.S., Yao M., Yin H.X., Wan X.Y., Yang Y.K., Liu H. Comparable effects of Jiedu Granule, a compound Chinese herbal medicine, and sorafenib for advanced hepatocellular carcinoma: A prospective multicenter cohort study. J. Integr. Med. 2020;18:319–325. doi: 10.1016/j.joim.2020.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Li L., Wang H. Heterogeneity of liver cancer and personalized therapy. Cancer Lett. 2016;379:191–197. doi: 10.1016/j.canlet.2015.07.018. [DOI] [PubMed] [Google Scholar]
- 7.Yang J.D., Nakamura I., Roberts L.R. The tumor microenvironment in hepatocellular carcinoma: current status and therapeutic targets. Semin. Cancer Biol. 2011;21:35–43. doi: 10.1016/j.semcancer.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Breuhahn K., Longerich T., Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene. 2006;25:3787–3800. doi: 10.1038/sj.onc.1209556. [DOI] [PubMed] [Google Scholar]
- 9.Seoane J., Gomis R.R. TGF-β Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb. Perspect. Biol. 2017;9:a022277. doi: 10.1101/cshperspect.a022277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J., Han C., Ungerleider N., Chen W., Song K., Wang Y., Kwon H., Ma W., Wu T. A Transforming Growth Factor-β and H19 Signaling Axis in Tumor-Initiating Hepatocytes That Regulates Hepatic Carcinogenesis. Hepatology. 2019;69:1549–1563. doi: 10.1002/hep.30153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng L., Yuan X.Q., Zhang C.Y., Ye F., Zhou H.F., Li W.L., Liu Z.Y., Zhang Y.Q., Pan X., Li G.C. High TGF-β1 expression predicts poor disease prognosis in hepatocellular carcinoma patients. Oncotarget. 2017;8:34387–34397. doi: 10.18632/oncotarget.16166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mamiya T., Yamazaki K., Masugi Y., Mori T., Effendi K., Du W., Hibi T., Tanabe M., Ueda M., Takayama T., Sakamoto M. Reduced transforming growth factor-beta receptor II expression in hepatocellular carcinoma correlates with intrahepatic metastasis. Lab. Invest. 2010;90:1339–1345. doi: 10.1038/labinvest.2010.105. [DOI] [PubMed] [Google Scholar]
- 13.Coulouarn C., Factor V.M., Thorgeirsson S.S. Transforming growth factor-beta gene expression signature in mouse hepatocytes predicts clinical outcome in human cancer. Hepatology. 2008;47:2059–2067. doi: 10.1002/hep.22283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamazaki K., Masugi Y., Sakamoto M. Molecular pathogenesis of hepatocellular carcinoma: altering transforming growth factor-β signaling in hepatocarcinogenesis. Dig. Dis. 2011;29:284–288. doi: 10.1159/000327560. [DOI] [PubMed] [Google Scholar]
- 15.Mu X., Lin S., Yang J., Chen C., Chen Y., Herzig M.C., Washburn K., Halff G.A., Walter C.A., Sun B., Sun L.-Z. TGF-β signaling is often attenuated during hepatotumorigenesis, but is retained for the malignancy of hepatocellular carcinoma cells. PloS One. 2013;8:e63436. doi: 10.1371/journal.pone.0063436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serova M., Tijeras-Raballand A., Dos Santos C., Albuquerque M., Paradis V., Neuzillet C., Benhadji K.A., Raymond E., Faivre S., de Gramont A. Effects of TGF-beta signalling inhibition with galunisertib (LY2157299) in hepatocellular carcinoma models and in ex vivo whole tumor tissue samples from patients. Oncotarget. 2015;6:21614–21627. doi: 10.18632/oncotarget.4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitisin K., Ganesan N., Tang Y., Jogunoori W., Volpe E.A., Kim S.S., Katuri V., Kallakury B., Pishvaian M., Albanese C. Disruption of transforming growth factor-beta signaling through beta-spectrin ELF leads to hepatocellular cancer through cyclin D1 activation. Oncogene. 2007;26:7103–7110. doi: 10.1038/sj.onc.1210513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muñoz N.M., Upton M., Rojas A., Washington M.K., Lin L., Chytil A., Sozmen E.G., Madison B.B., Pozzi A., Moon R.T. Transforming growth factor beta receptor type II inactivation induces the malignant transformation of intestinal neoplasms initiated by Apc mutation. Cancer Res. 2006;66:9837–9844. doi: 10.1158/0008-5472.CAN-06-0890. [DOI] [PubMed] [Google Scholar]
- 19.Tang Y., Kitisin K., Jogunoori W., Li C., Deng C.-X., Mueller S.C., Ressom H.W., Rashid A., He A.R., Mendelson J.S. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc. Natl. Acad. Sci. USA. 2008;105:2445–2450. doi: 10.1073/pnas.0705395105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baek J.Y., Morris S.M., Campbell J., Fausto N., Yeh M.M., Grady W.M. TGF-beta inactivation and TGF-alpha overexpression cooperate in an in vivo mouse model to induce hepatocellular carcinoma that recapitulates molecular features of human liver cancer. Int. J. Cancer. 2010;127:1060–1071. doi: 10.1002/ijc.25127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paiva C.E., Drigo S.A., Rosa F.E., Moraes Neto F.A., Caldeira J.R.F., Soares F.A., Domingues M.A.C., Rogatto S.R. Absence of transforming growth factor-beta type II receptor is associated with poorer prognosis in HER2-negative breast tumours. Ann. Oncol. 2010;21:734–740. doi: 10.1093/annonc/mdp518. [DOI] [PubMed] [Google Scholar]
- 22.Lu X., Jin E.-J., Cheng X., Feng S., Shang X., Deng P., Jiang S., Chang Q., Rahmy S., Chaudhary S. Opposing roles of TGFβ and BMP signaling in prostate cancer development. Genes Dev. 2017;31:2337–2342. doi: 10.1101/gad.307116.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao W., Zhu Q., Tan P., Ajibade A., Long T., Long W., Li Q., Liu P., Ning B., Wang H.Y., Wang R.F. Tgfbr2 inactivation facilitates cellular plasticity and development of Pten-null prostate cancer. J. Mol. Cell Biol. 2018;10:316–330. doi: 10.1093/jmcb/mjx052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miyazono K., Kamiya Y., Morikawa M. Bone morphogenetic protein receptors and signal transduction. J. Biochem. 2010;147:35–51. doi: 10.1093/jb/mvp148. [DOI] [PubMed] [Google Scholar]
- 25.Ning J., Zhao Y., Ye Y., Yu J. Opposing roles and potential antagonistic mechanism between TGF-β and BMP pathways: Implications for cancer progression. EBioMedicine. 2019;41:702–710. doi: 10.1016/j.ebiom.2019.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie C., Mao X., Huang J., Ding Y., Wu J., Dong S., Kong L., Gao G., Li C.-Y., Wei L. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39(Web Server issue):W316–W322. doi: 10.1093/nar/gkr483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li L., Liu Y., Guo Y., Liu B., Zhao Y., Li P., Song F., Zheng H., Yu J., Song T. Regulatory MiR-148a-ACVR1/BMP circuit defines a cancer stem cell-like aggressive subtype of hepatocellular carcinoma. Hepatology. 2015;61:574–584. doi: 10.1002/hep.27543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Macías-Silva M., Hoodless P.A., Tang S.J., Buchwald M., Wrana J.L. Specific activation of Smad1 signaling pathways by the BMP7 type I receptor, ALK2. J. Biol. Chem. 1998;273:25628–25636. doi: 10.1074/jbc.273.40.25628. [DOI] [PubMed] [Google Scholar]
- 29.Korchynskyi O., ten Dijke P. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J. Biol. Chem. 2002;277:4883–4891. doi: 10.1074/jbc.M111023200. [DOI] [PubMed] [Google Scholar]
- 30.Lasorella A., Benezra R., Iavarone A. The ID proteins: master regulators of cancer stem cells and tumour aggressiveness. Nat. Rev. Cancer. 2014;14:77–91. doi: 10.1038/nrc3638. [DOI] [PubMed] [Google Scholar]
- 31.Chen J., Zaidi S., Rao S., Chen J.S., Phan L., Farci P., Su X., Shetty K., White J., Zamboni F. Analysis of Genomes and Transcriptomes of Hepatocellular Carcinomas Identifies Mutations and Gene Expression Changes in the Transforming Growth Factor-β Pathway. Gastroenterology. 2018;154:195–210. doi: 10.1053/j.gastro.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ye Y., Guo J., Xiao P., Ning J., Zhang R., Liu P., Yu W., Xu L., Zhao Y., Yu J. Macrophages-induced long noncoding RNA H19 up-regulation triggers and activates the miR-193b/MAPK1 axis and promotes cell aggressiveness in hepatocellular carcinoma. Cancer Lett. 2020;469:310–322. doi: 10.1016/j.canlet.2019.11.001. [DOI] [PubMed] [Google Scholar]
- 33.Makeyev E.V., Maniatis T. Multilevel regulation of gene expression by microRNAs. Science. 2008;319:1789–1790. doi: 10.1126/science.1152326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grimson A., Farh K.K., Johnston W.K., Garrett-Engele P., Lim L.P., Bartel D.P. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murakami G., Watabe T., Takaoka K., Miyazono K., Imamura T. Cooperative inhibition of bone morphogenetic protein signaling by Smurf1 and inhibitory Smads. Mol. Biol. Cell. 2003;14:2809–2817. doi: 10.1091/mbc.E02-07-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin H., Ying Y., Wang Y.-Y., Wang G., Jiang S.-S., Huang D., Luo L., Chen Y.G., Gerstenfeld L.C., Luo Z. AMPK downregulates ALK2 via increasing the interaction between Smurf1 and Smad6, leading to inhibition of osteogenic differentiation. Biochim. Biophys. Acta Mol. Cell Res. 2017;1864:2369–2377. doi: 10.1016/j.bbamcr.2017.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen B., Khodadoust M.S., Liu C.L., Newman A.M., Alizadeh A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018;1711:243–259. doi: 10.1007/978-1-4939-7493-1_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang L., Huang J., Ren X., Gorska A.E., Chytil A., Aakre M., Carbone D.P., Matrisian L.M., Richmond A., Lin P.C., Moses H.L. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Capucha T., Koren N., Nassar M., Heyman O., Nir T., Levy M., Zilberman-Schapira G., Zelentova K., Eli-Berchoer L., Zenke M. Sequential BMP7/TGF-β1 signaling and microbiota instruct mucosal Langerhans cell differentiation. J. Exp. Med. 2018;215:481–500. doi: 10.1084/jem.20171508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meurer S.K., Esser M., Tihaa L., Weiskirchen R. BMP-7/TGF-β1 signalling in myoblasts: components involved in signalling and BMP-7-dependent blockage of TGF-β-mediated CTGF expression. Eur. J. Cell Biol. 2012;91:450–463. doi: 10.1016/j.ejcb.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 41.Lee P.T., Li W.-J. Chondrogenesis of Embryonic Stem Cell-Derived Mesenchymal Stem Cells Induced by TGFβ1 and BMP7 Through Increased TGFβ Receptor Expression and Endogenous TGFβ1 Production. J. Cell. Biochem. 2017;118:172–181. doi: 10.1002/jcb.25623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meng X.M., Chung A.C., Lan H.Y. Role of the TGF-beta/BMP-7/Smad pathways in renal diseases. Clin. Sci. (Lond.) 2013;124:243–254. doi: 10.1042/CS20120252. [DOI] [PubMed] [Google Scholar]
- 43.Wu M., Chen G., Li Y.P. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016;4:16009. doi: 10.1038/boneres.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeisberg E.M., Tarnavski O., Zeisberg M., Dorfman A.L., McMullen J.R., Gustafsson E., Chandraker A., Yuan X., Pu W.T., Roberts A.B. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 45.Sun Z., Liu C., Jiang W.G., Ye L. Deregulated bone morphogenetic proteins and their receptors are associated with disease progression of gastric cancer. Comput. Struct. Biotechnol. J. 2020;18:177–188. doi: 10.1016/j.csbj.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eikesdal H.P., Becker L.M., Teng Y., Kizu A., Carstens J.L., Kanasaki K., Sugimoto H., LeBleu V.S., Kalluri R. BMP7 Signaling in TGFBR2-Deficient Stromal Cells Provokes Epithelial Carcinogenesis. Mol. Cancer Res. 2018;16:1568–1578. doi: 10.1158/1541-7786.MCR-18-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kawamura I., Maeda S., Imamura K., Setoguchi T., Yokouchi M., Ishidou Y., Komiya S. SnoN suppresses maturation of chondrocytes by mediating signal cross-talk between transforming growth factor-β and bone morphogenetic protein pathways. J. Biol. Chem. 2012;287:29101–29113. doi: 10.1074/jbc.M112.349415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu H., Han C., Wu T. MiR-17-92 cluster promotes hepatocarcinogenesis. Carcinogenesis. 2015;36:1213–1222. doi: 10.1093/carcin/bgv112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He C., Zheng S., Luo Y., Wang B. Exosome Theranostics: Biology and Translational Medicine. Theranostics. 2018;8:237–255. doi: 10.7150/thno.21945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hulsmans M., Holvoet P. MicroRNA-containing microvesicles regulating inflammation in association with atherosclerotic disease. Cardiovasc. Res. 2013;100:7–18. doi: 10.1093/cvr/cvt161. [DOI] [PubMed] [Google Scholar]
- 51.Yeung O.W., Lo C.M., Ling C.C., Qi X., Geng W., Li C.X., Ng K.T., Forbes S.J., Guan X.Y., Poon R.T. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J. Hepatol. 2015;62:607–616. doi: 10.1016/j.jhep.2014.10.029. [DOI] [PubMed] [Google Scholar]
- 52.Wu J., Gao W., Tang Q., Yu Y., You W., Wu Z., Fan Y., Zhang Y., Wu C., Han G. M2 macrophage-derived exosomes facilitate hepatocarcinoma metastasis by transferring α(M) β(2) integrin to tumor cells. Hepatology. 2020 doi: 10.1002/hep.31432. Published online June 28, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.Wan S., Zhao E., Kryczek I., Vatan L., Sadovskaya A., Ludema G., Simeone D.M., Zou W., Welling T.H. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014;147:1393–1404. doi: 10.1053/j.gastro.2014.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dong N., Shi X., Wang S., Gao Y., Kuang Z., Xie Q., Li Y., Deng H., Wu Y., Li M., Li J.L. M2 macrophages mediate sorafenib resistance by secreting HGF in a feed-forward manner in hepatocellular carcinoma. Br. J. Cancer. 2019;121:22–33. doi: 10.1038/s41416-019-0482-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Q., Huang F., Yao Y., Wang J., Wei J., Wu Q., Xiang S., Xu L. Interaction of transforming growth factor-β-Smads/microRNA-362-3p/CD82 mediated by M2 macrophages promotes the process of epithelial-mesenchymal transition in hepatocellular carcinoma cells. Cancer Sci. 2019;110:2507–2519. doi: 10.1111/cas.14101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nie H., Cao Q., Zhu L., Gong Y., Gu J., He Z. Acetylcholine acts on androgen receptor to promote the migration and invasion but inhibit the apoptosis of human hepatocarcinoma. PLoS ONE. 2013;8:e61678. doi: 10.1371/journal.pone.0061678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin X.H., Liu H.H., Hsu S.J., Zhang R., Chen J., Chen J., Gao D.M., Cui J.F., Ren Z.G., Chen R.X. Norepinephrine-stimulated HSCs secrete sFRP1 to promote HCC progression following chronic stress via augmentation of a Wnt16B/β-catenin positive feedback loop. J. Exp. Clin. Cancer Res. 2020;39:64. doi: 10.1186/s13046-020-01568-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liang C., Chen W., Zhi X., Ma T., Xia X., Liu H., Zhang Q., Hu Q., Zhang Y., Bai X., Liang T. Serotonin promotes the proliferation of serum-deprived hepatocellular carcinoma cells via upregulation of FOXO3a. Mol. Cancer. 2013;12:14. doi: 10.1186/1476-4598-12-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yan Y., Pan J., Chen Y., Xing W., Li Q., Wang D., Zhou X., Xie J., Miao C., Yuan Y. Increased dopamine and its receptor dopamine receptor D1 promote tumor growth in human hepatocellular carcinoma. Cancer Commun. (Lond.) 2020;40:694–710. doi: 10.1002/cac2.12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao J., Hou Y., Yin C., Hu J., Gao T., Huang X., Zhang X., Xing J., An J., Wan S., Li J. Upregulation of histamine receptor H1 promotes tumor progression and contributes to poor prognosis in hepatocellular carcinoma. Oncogene. 2020;39:1724–1738. doi: 10.1038/s41388-019-1093-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Decalf J., Albert M.L., Ziai J. New tools for pathology: a user’s review of a highly multiplexed method for in situ analysis of protein and RNA expression in tissue. J. Pathol. 2019;247:650–661. doi: 10.1002/path.5223. [DOI] [PubMed] [Google Scholar]
- 62.Sia D., Moeini A., Labgaa I., Villanueva A. The future of patient-derived tumor xenografts in cancer treatment. Pharmacogenomics. 2015;16:1671–1683. doi: 10.2217/pgs.15.102. [DOI] [PubMed] [Google Scholar]
- 63.Xiao P., Long X., Zhang L., Ye Y., Guo J., Liu P., Zhang R., Ning J., Yu W., Wei F., Yu J. Neurotensin/IL-8 pathway orchestrates local inflammatory response and tumor invasion by inducing M2 polarization of Tumor-Associated macrophages and epithelial-mesenchymal transition of hepatocellular carcinoma cells. OncoImmunology. 2018;7:e1440166. doi: 10.1080/2162402X.2018.1440166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nghiem P.T., Bhatia S., Lipson E.J., Kudchadkar R.R., Miller N.J., Annamalai L., Berry S., Chartash E.K., Daud A., Fling S.P. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N. Engl. J. Med. 2016;374:2542–2552. doi: 10.1056/NEJMoa1603702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang H.C., Chen C.W., Yang C.L., Tsai I.M., Hou Y.C., Chen C.J., Shan Y.S. Tumor-Associated Macrophages Promote Epigenetic Silencing of Gelsolin through DNA Methyltransferase 1 in Gastric Cancer Cells. Cancer Immunol. Res. 2017;5:885–897. doi: 10.1158/2326-6066.CIR-16-0295. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.