

Abstract
Gene disruption via programmable, sequence-specific nucleases represents a promising gene therapy strategy in which the reduction of specific protein levels provides a therapeutic benefit. Proprotein convertase subtilisin/kexin type 9 (PCSK9), an antagonist of the low-density lipoprotein (LDL) receptor, is a suitable target for nuclease-mediated gene disruption as an approach to treat hypercholesterolemia. We sought to determine the long-term durability and safety of PCSK9 knockdown in non-human primate (NHP) liver by adeno-associated virus (AAV)-delivered meganuclease following our initial report on the feasibility of this strategy. Six previously treated NHPs and additional NHPs administered AAV-meganuclease in combination with corticosteroid treatment or an alternative AAV serotype were monitored for a period of up to 3 years. The treated NHPs exhibited a sustained reduction in circulating PCSK9 and LDL cholesterol (LDL-c) through the course of the study concomitant with stable gene editing of the PCSK9 locus. Low-frequency off-target editing remained stable, and no obvious adverse changes in histopathology of the liver were detected. We demonstrate similar on-target nuclease activity in primary human hepatocytes using a chimeric liver-humanized mouse model. These studies demonstrate that targeted in vivo gene disruption exerts a lasting therapeutic effect and provide pivotal data for safety considerations, which support clinical translation.
Keywords: hypercholesterolemia, PCSK9, gene editing, meganuclease, nonhuman primates, AAV, in vivo, genome editing, LDL, gene therapy
Graphical abstract
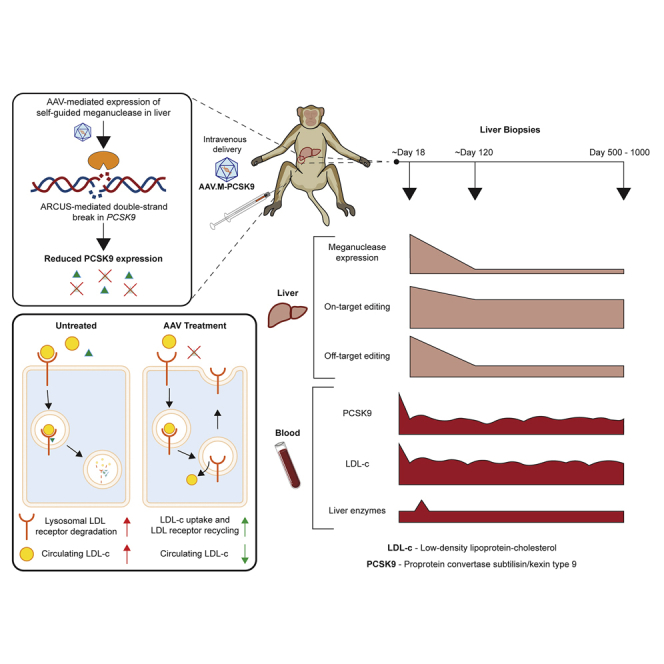
We investigated the long-term durability and safety of proprotein convertase subtilisin/kexin type 9 (PCSK9) knockdown in non-human primate liver by adeno-associated virus-delivered meganuclease to treat hypercholesterolemia. Sustained reductions in circulating PCSK9 and LDL cholesterol, stable PCSK9 locus editing for up to 3 years, and stable, low-frequency off-target editing were achieved.
Introduction
Hypercholesterolemia is one of the leading causes of cardiovascular disease (CVD). The low-density lipoprotein receptor (LDLR) plays a crucial role in the regulation of circulating LDL cholesterol (LDL-c) levels. When proprotein convertase subtilisin/kexin type 9 (PCSK9), a serine protease, binds to hepatic LDLR, it can accelerate the degradation of LDLR and increase LDL-c levels.1 Naturally occurring loss-of-function mutations in PCSK9 are associated with reduced LDL-c levels and a decreased risk of CVD, with no apparent adverse health consequences, rendering PCSK9 a compelling therapeutic target.2, 3, 4 Monoclonal antibodies or RNA interference can successfully reduce PCSK9 levels in humans, with monoclonal antibodies having received approval from the Food and Drug Administration for hypercholesterolemia; however, both approaches require periodic repeated treatments to achieve a sustained reduction in PCSK9.5, 6, 7, 8, 9
Characterizations of germline and liver-specific PCSK9-knockout mice indicate that hepatocytes are the primary source of circulating PCSK9, despite lower levels of PCSK9 expression in the intestine and kidney.10,11 Therefore, liver-directed in vivo genome editing that targets the PCSK9 gene represents a novel approach for treating hypercholesterolemia and preventing cardiovascular events. Several proof-of-concept studies in wild-type mice or chimeric liver-humanized Fah−/−Rag2−/−Il2rg−/− (FRG) mice have employed clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 or zinc finger nuclease (ZFN) technology.12 In these studies, lipid nanoparticles (LNPs), adenovirus, or adeno-associated virus (AAV) vectors deliver Cas9 and guide RNA or ZFN to the liver, achieving disruption of the PCSK9 gene.13, 14, 15, 16, 17, 18 More recently, researchers investigated base editing (BE) technology in mice as a therapeutic alternative to precisely modify the PCSK9 gene.19 Thus far, reports of in vivo genome editing in large animals have focused only on short-term results of dystrophin restoration in the muscle of Duchenne muscular dystrophy dogs and the results of CEP290 editing in the eye of nonhuman primates (NHPs) by EDIT-101, a CRISPR-based experimental medicine that restores CEP290 expression and vision loss in Leber congenital amaurosis type 10.20,21 EDIT-101 recently entered clinics and became the first clinical tool for in vivo genome editing using CRISPR/Cas9 technology.22
Meganucleases are engineered variants of homing endonucleases that can be redirected to different DNA sequences for genome editing applications.23 Because the DNA-binding and cleavage domains of homing endonucleases are closely associated, it is difficult to re-engineer meganucleases to target novel sequences. One attractive feature of meganucleases is their small size, which renders them amenable to all standard gene delivery methods, including AAV vector approaches. We previously reported the development of two generations of meganucleases (M1PCSK9 and M2PCSK9) that target a conserved region in exon 7 of the human and macaque PCSK9 gene.24 We demonstrated the improved specificity of M2PCSK9 and reported that a single infusion of an AAV8 vector expressing M1 or M2PCSK9 in NHPs resulted in a dose-dependent disruption of PCSK9 in the liver, as well as a stable reduction in circulating PCSK9 and serum LDL-c levels for up to 11 months.
Here, we report a long-term follow-up of these NHPs for over 3 years and longitudinal analysis data on the efficiency, durability, and safety of in vivo genome editing in NHPs. We also report the effects of a course of corticosteroid treatment to reduce immune responses to the meganuclease. In vivo genome editing provides a novel approach for (permanently) disrupting the expression of PCSK9 and thus ameliorating hypercholesterolemia.
Results
Long-term follow-up of in vivo genome editing in NHPs
To evaluate the longevity and safety of in vivo genome editing, we followed six NHPs that were previously treated with AAV8-M1PCSK9 or AAV8-M2PCSK9, vectors expressing engineered meganucleases targeting exon 7 of the human and rhesus PCSK9 gene (Table 1).24 The four M1PCSK9-treated NHPs demonstrated dose-correlated, significant, and sustained reductions in serum PCSK9 and LDL-c levels, ranging from 16% to 83% and 44% to 77% of day-0 levels, respectively, for over 3 years (Table 1; Figures 1A, 1D, S1, and S2). RA2343, an AAV8-M2PCSK9-treated animal, also demonstrated a stable reduction in serum PCSK9 (43.7% ± 1.5% of day-0 levels) and LDL-c (60.5% ± 1.3% of day-0 levels) levels for over 3 years (Table 1; Figures 1B, 1E, S1, and S2). All animals maintained stable PCSK9 and LDL-c levels in year 3, without significant differences from year 2. We continuously monitored the health of these animals by daily observations, clinical pathology tests on blood samples collected biweekly or monthly, and semiannual physical examinations and abdominal ultrasound evaluations.
Table 1.
Summary of PCSK9 genome editing in rhesus macaques after systemic AAV vector administration
| Animal No. | Sex | Age at dosing (years) | Endonuclease | AAV serotype | Other treatment | Vector dose (GC/kg) | Levels in steady phasea % of day-0 level (absolute level) |
Editing in the third liver biopsy/necropsy |
|||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PCSK9 (ng/mL) | LDL (mg/dL) | Time | On-target indel %b | Time point | |||||||
| RA1866 | M | 4.9 | M1PCSK9 | AAV8 | 3.0E+13 | 15.5 ± 0.5 (17.9 ± 0.5) | 43.8 ± 1.6 (25.4 ± 0.9) | day 56–1237 (n = 55) | 64.4 | d1070 | |
| RA1857 | M | 4.9 | M1PCSK9 | AAV8 | 6.0E+12 | 54.9 ± 1.8 (36.8 ± 1.2) | 60.1 ± 1.3 (28.8 ± 0.6) | day 56–1237 (n = 55) | 44.4 | d1070 | |
| RA1829 | F | 5.1 | M1PCSK9 | AAV8 | 2.0E+12 | 74.8 ± 2.8 (195.3 ± 7.1) | 67.2 ± 1.2 (49.8 ± 0.9) | day 56–1134 (n = 50) | 24.0 | d989 | |
| RA2334 | M | 4.0 | M1PCSK9 | AAV8 | 2.0E+12 | 83.2 ± 2.9 (193.0 ± 6.6) | 77.4 ± 1.5 (57.8 ± 1.2) | day 56–1134 (n = 50) | 9.5 | d989 | |
| RA2125 | F | 4.2 | M2PCSK9 | AAV8 | 6.0E+12 | 37.5 ± 1.9 (65.5 ± 3.4) | 61.2 ± 2.0 (23.9 ± 0.8) | day 56–280 (n = 16) | 38.8 | d284 (necropsy) | |
| RA2343 | M | 3.9 | M2PCSK9 | AAV8 | 6.0E+12 | 43.7 ± 1.5 (98.7 ± 3.4) | 60.4 ± 1.3 (46.5 ± 1.0) | day 56–1142 (n = 50) | 41.8 | d995 | |
| RA3167 | M | 4.7 | M2PCSK9 | AAV8 | prednisolone (d0-d76) | 6.0E+12 | 77.5 ± 5.0 (103.7 ± 6.7) | 85.7 ± 2.1 (49.7 ± 1.2) | day 56–789 (n = 39) | 19.2 | d642 |
| RA3169 | M | 4.0 | M2PCSK9 | AAV8 | prednisolone (d0-d76) | 6.0E+12 | 20.2 ± 1.2 (36.8 ± 2.1) | 44.4 ± 1.7 (31.1 ± 1.2) | day 56–789 (n = 39) | 57.2 | d642 |
| RA2083 | F | 5.8 | M2PCSK9 | AAV3B | 6.0E+12 | 26.9 ± 1.3 (98.0 ± 4.9) | 57.0 ± 1.7 (37.6 ± 1.1) | day 56–699 (n = 35) | 30.3 | d531 | |
| RA2396 | F | 5.1 | M2PCSK9 | AAV3B | 6.0E+12 | 51.1 ± 2.5 (150.3 ± 7.2) | 61.3 ± 1.8 (74.8 ± 2.2) | day 56–699 (n = 36) | 23.4 | d531 | |
AAV, adeno-associated virus; GC, genome copy; LDL, low-density lipoprotein.
Steady phase is arbitrarily defined as day 56 post vector administration to the most current time point for each animal or to the final time point for RA2125, and the number of time points for LDL is shown in parentheses. Data are presented as the percentage of the day-0 level, and absolute levels are shown in parentheses. The means ± SEM are shown. The reduction compared with pretreatment is significant (p < 0.001) based on a one-sided one-sample t test.
On-target indel % in hepatocytes is based on mean AMP-seq on third biopsy or necropsy (RA2125) samples (Figure 2A) and adjusted 1.4-fold to reflect the indel % in hepatocytes, which represent ~70% of total liver cells.
Figure 1.

Long-term follow-up of in vivo genome editing on rhPCSK9 in rhesus macaques after a single infusion of different AAV-meganuclease vectors
Animals received a single intravenous infusion of AAV8-M1PCSK9, AAV8-M2PCSK9 with or without a course of steroid treatment, or AAV3B-M2PCSK9 vector. (A–C) Serum PCSK9 levels presented as the percentage of levels on day 0. (D–F) Serum low-density lipoprotein cholesterol (LDL-c) levels presented as the percentage of levels on day 0. (G–I) Alanine aminotransferase (ALT) levels during the study. The gray bar indicates the period of prophylactic steroid treatment and tapering course for RA3167 and RA3169. The results from early time points for RA1866, RA1857, RA1829, RA2334, RA2125, and RA2343 have been previously published24 and are included here for completeness of the dataset.
Except for transient elevations during the early phase, as previously reported,24 transaminase levels remained within normal ranges (Figures 1G–1I and S3). We also monitored T cell responses to the AAV8 capsid and meganuclease in peripheral blood mononuclear cells (PBMCs). We found that the magnitude of the T cell response to the meganuclease decreased gradually, as measured by ELISpot, and that none of the animals showed any positive responses after 1 year (Figure S4). Three of the six animals showed low-level, transient T cell responses to the capsid protein during the first 6 months after vector administration.
One of the M2PCSK9-treated animals, RA2125, was mandated for euthanasia on day 284 due to repeated positive reactions during routine semiannual tuberculin skin tests. We harvested tissues from the major organs and lymph nodes at necropsy. Polymerase chain reaction (PCR) analyses for Mycobacterium in the lung tissues and tracheobronchial lymph nodes were negative. Further histopathological analysis of tissues harvested at necropsy gave no evidence of infection with Mycobacterium tuberculosis (TB) in any tissue examined (Table S1). Therefore, we concluded that RA2125 had false-positive results to the TB tests. Before euthanasia on day 284, RA2125 had maintained stable reductions in PCSK9 (37.5% ± 1.9%) and LDL-c (61.2% ± 2.0%) (Table 1; Figures 1B, 1E, S1, and S2). Among the six animals, RA2125 showed the strongest and most persistent T cell response to the meganuclease (Figure S4).
Treating additional animals with M2PCSK9 vectors
Several AAV gene therapy trials have applied short-course corticosteroid treatments to control the acute rise in transaminases that often follows AAV vector administration.25, 26, 27, 28 To determine whether a prophylactic course of corticosteroid treatment can reduce or abolish the magnitude of transaminase elevations in NHPs during in vivo genome editing, we treated two macaques with AAV8-M2PCSk9 (6 × 1012 genome copies [GC]/kg) combined with an 11-week course of oral prednisolone (1 mg/kg/day) followed by tapering. RA3167 had normal liver enzyme levels during the course of prednisolone treatment. During the tapering course, RA3167 had a mild elevation of alanine aminotransferase (ALT) on day 84 and at a few later time points throughout the study (Figures 1H and S3). RA3169 had a transient transaminase elevation that peaked on day 35, similar to the previous six animals, but at a lower magnitude (Figures 1H and S3). RA3169 also showed modest levels of T cell response to the meganuclease which started on day 56 and persisted throughout the study (Figure S4). These data indicate that a corticosteroid treatment can reduce the magnitude of transaminase elevations in NHPs during in vivo genome editing but cannot abolish transaminase elevations or T cell responses to the meganuclease. Due to the low number of subjects in the current study, additional studies involving more animals would be necessary to confirm these observations.
We also treated two macaques with the AAV3B-M2PCSK9 vector. AAV3B is an AAV serotype that is phylogenically distinct from other liver-tropic clade E members of AAV, such as AAV8.29 Similar to the engineered variant of AAV3B called AAVLK03, AAV3B has shown high transduction in NHPs and human hepatocytes and could be used as an alternative serotype in subjects with neutralizing antibodies to other AAVs.30,31 Both macaques, naive for AAV3B but positive for AAV8 neutralizing antibodies (NAbs) (1/10 and 1/5, respectively), showed stable reductions in serum PCSK9 and LDL-c (Table 1; Figures 1C and 1F). RA2396 began to show rising ALT levels as early as day 7, with a peak on day 28 at 1,089 IU/L, higher than that of the other animals in this study (Figure 1I). RA2396 also showed T cell responses to AAV3B capsid on day 28 and to the meganuclease for at least 1 year after vector treatment (Figure S4).
Stable genome editing in the liver of NHPs
To further evaluate the stability of in vivo genome editing following a single infusion of AAV-meganuclease vectors, we performed three consecutive liver biopsies via laparotomy on all remaining nine animals (RA2125 was necropsied on day 284). We evaluate on-target editing by two deep sequencing methods: (1) amplicon-seq, which captures small insertions and deletions (indels); and (2) anchored multiplexed PCR sequencing (AMP-seq), which captures partial sequences of large insertions in addition to small indels.24 Amplicon-seq showed stable editing in all three biopsy samples for all animals except for RA1866, the high-dose AAV8-M1PCSK9-treated animal (Figure 2A). RA2334 and RA3167 showed a reduction in indels after the first liver biopsy but maintained the same level of indels after the second biopsy. AMP-seq showed a reduction in indels after the first liver biopsy in all animals except for RA1866, which showed a modest increase in the second and third biopsy samples (Figure 2B). On average, the on-target indel level measured by AMP-seq was reduced by 32% ± 4% (n = 9) in the second liver biopsies compared with the first biopsies. The indels were well maintained in the third biopsies, without significant changes (97% ± 5% of the second biopsy levels, n = 9). The indel reduction is largely attributed to the reduction in inverted terminal repeat (ITR) insertions, which were more profound in the first liver biopsy samples (63% ± 2% of total indels, n = 10) compared with those in the second (40% ± 3%) and third (34% ± 3%) biopsies. AAV insertions into the double-stranded breaks induced by ZFN, CRISPR, and meganucleases have been previously reported.32, 33, 34, 35 Overall, the on-target editing was well maintained in the liver after the second biopsy (∼4 months) for up to 3 years post-dosing.
Figure 2.

Analyses of on-target and OT editing in consecutive macaque liver biopsy and necropsy samples collected at various time points after vector administration
Each biopsy sample was evaluated by two independent genomic DNA isolations from the biopsy sample followed by next-generation sequencing (NGS). (A and B) On-target editing analyzed by amplicon-seq (A) or AMP-seq (B). Indels in the liver of RA2125 collected at necropsy were analyzed in eight samples, two each from the left, right, middle, and caudate lobes. The means ± SEM are shown. (C and D) Off-target (OT) editing evaluated by ITR-seq on AAV8.M1PCSK9-treated liver biopsy samples (C) or AAV.M2PCSK9-treated liver samples (D). Each dot represents a unique potential OT site detected by ITR-seq and its relative frequency with respect to on-target editing. The number in red above the animal ID indicates the number of OT sites detected in samples from all three time points of the same animal. Detailed OT information can be found in Data S1. UMI, unique molecular identifier.
We also evaluated OT (off-target) editing in the liver samples by two methods: (1) ITR sequencing (ITR-seq), which identifies unique insertion sites in the genome and determines the relative insertion frequency of each site in tissues or cells treated with AAV vectors;35 and (2) amplicon-seq on selective sites predicted by GUIDE-seq.36 OT analyses of the six original animals showed a significant reduction in OT editing in the second liver biopsy samples in published data.24,35 Here, we focused on the third liver biopsy samples to determine whether there were any changes in OT effects in the third biopsy samples. As shown in Figures 2C and 2D and Data S1, the number of OT sites and relative OT site frequencies were similar between the second and third biopsy samples. Further investigation highlighted that some OT sites (2 to 80) were present in all three biopsy samples from the same animal; moreover, a few OTs were present across all animals treated with the same meganuclease (Data S1).
We also evaluated OT editing in each sample by conducting an indel analysis of the top 15 GUIDE-seq-identified OT sites for M1PCSK9 or M2PCSK9. Indel frequencies analyzed by amplicon-seq for each OT site are shown in Figure S5 and Tables S2–S4. Compared to the first biopsy samples, the second biopsy samples showed a significant reduction of indel frequencies in the majority of the OT sites, whereas the third biopsy samples only showed significant changes in a few of the OT sites. Notably, some of the high-rank OT sites identified by in vitro GUIDE-seq were also detected by ITR-seq of the liver samples. In animals treated with the same meganuclease (M1PCSK9 or M2PCSK9), samples with relatively higher on-target editing, such as the RA3169 samples, showed higher OT editing at potential OT sites and more OT sites detected by ITR-seq (Tables S2–S4; Figure 2D).
Evaluation of vector and transgene expression and histopathology in macaque liver
We analyzed vector GC and transgene expression levels in the biopsy samples by quantitative PCR (qPCR) and quantitative reverse-transcriptase PCR (qRT-PCR), respectively. The third biopsy samples obtained at later time points (≥642 days, n = 7) showed an additional loss of vector GCs compared with the second biopsy samples (Figure 3A). qRT-PCR did not detect an additional loss of transgene expression levels in the third biopsy samples compared with those in the second biopsy samples (Figure 3B). In situ hybridization (ISH) of liver sections detected positive signals in only a few hepatocytes scattered in the second and third biopsy samples (Figure 3C; note that the images in the top row show previously published24 long-term residual expression in 3/6 animals for comparison). The loss of vector GC and transgene expression is a desirable feature for in vivo genome editing.
Figure 3.

Analyses of vector genome copies (GCs) and meganuclease expression in consecutive macaque liver biopsy and necropsy samples collected at various time points after vector administration
(A) Vector GCs in the liver, as analyzed by qPCR of genomic DNA. (B) Expression of meganuclease in the liver, as measured by qPCR of total RNA isolated from liver samples followed by reverse transcription and presented as relative expression levels normalized by glyceraldehyde 3-phosphate dehydrogenase levels. RA2125-d284 was analyzed based on eight liver tissue samples collected at necropsy, two each from the left, right, middle, and caudate lobes. The means ± SEM are shown. Data from the first two biopsies of RA1866, RA1857, RA1829, RA2334, RA2125, and RA2343 have been previously published24 and are included here for comparison purposes. (C) Images for ISH using transgene-specific probes (M1PCSK9) for liver biopsy samples at the indicated time points (ISH signal: red; nuclear DAPI stain: blue). Top row: images of third biopsies from animals previously described.24 Middle row: images of sequential biopsies representative of the cohort that received steroid treatment. Bottom row: images of sequential biopsies representative of the AAV3B cohort. Scale bar, 100 μm.
We also performed vector biodistribution analysis on tissue samples collected at the day 284 necropsy of RA2125. Vector GCs in different tissues were 10- to 1,000-fold lower than those in the liver (Figure S6A). In addition, we evaluated on-target editing in these tissues by amplicon-seq. The gall bladder was the only tissue that showed editing above that in pre-PBMC (Figure S6B). However, due to its close proximity to the liver, we cannot rule out the possibility of contamination with liver tissue in the gall bladder sample. In conclusion, the combination of a liver-tropic AAV serotype and a liver-specific promoter to drive the expression of meganuclease prevented editing in non-liver tissues.
In our histopathological analysis of the long-term biopsy samples, the only microscopic findings were mild-to-moderate capsular and subcapsular fibrosis and minimal-to-mild mononuclear cell infiltrates (Table S5; Figure S7). Mononuclear cell infiltrates in the liver have been reported as a common background finding in NHPs.37 We detected capsular and subcapsular fibrosis only in the third liver biopsy samples in all animals, although the fibrosis was not significant and may have been the result of previous liver biopsy procedures, similar to what we have observed in non-nuclease AAV vector-treated NHPs that were subjected to consecutive liver biopsies (L.W. and J.M.W., unpublished data). In summary, the AAV-delivered meganuclease resulted in transient expression of meganuclease in the liver and long-lasting editing effects without major safety concerns.
In vivo genome editing in primary human hepatocytes
To assess whether the AAV meganuclease can also achieve efficient editing in primary human hepatocytes, we treated two FRG mice whose livers had been repopulated with primary human hepatocytes with a tail vein injection of AAV3B-M2PCSK9 at a dose of 1.3 × 1012 GC/mouse. Indel analysis on liver samples harvested on day 49 by AMP-seq showed efficient on-target editing in the hPCSK9 gene in both mice, at 28% (Figure 4A). This level of editing in the macaque would lead to a significant reduction in PCSK9 and LDL. OT editing was evaluated by amplicon-seq for the top 22 OT sites previously identified in vitro by GUIDE-seq.24 DNA isolated from the liver of two FRG mice treated with control AAV vector served as negative controls (ctrl 1 and ctrl 2). The editing of each OT site was significantly higher than that in the control samples (Figure 4B; Table S4). We also evaluated OT editing by ITR-seq. As shown in Figure 4C and Data S2, the numbers of OT sites detected in each sample, ranging from 93 to 233, were in a similar range as those detected in the first liver biopsy samples from AAV.M2PCSK9-treated macaques, although the overall frequencies of the OT sites in the human hepatocytes were higher than those in the macaques (Figure 2D; Data S1). Between the two mice, 29 OT sites were commonly found in both mice (Data S2). We also compared the ITR-seq-identified OT sites in the human hepatocytes in FRG mice with previous GUIDE-seq-identified OT sites in induced pluripotent stem cell (iPSC)-derived human hepatocytes.24 There were 32–54 OT sites, varying between the eight samples from the two FRG mice, also detected by GUIDE-seq in the iPSC-derived human hepatocytes, although the ranking orders of the OT sites were not consistent between the in vivo ITR-seq and in vitro GUIDE-seq data (Data S2). The identity of common OT sites between animals and by different assays warrants further investigation. Histology analyses of the liver sections showed a strong expression of meganuclease in some human hepatocytes (Figures 4E–4H). We performed two-plex immunohistochemistry (IHC) of FAH and I-CreI to assess the transduction efficiency in human hepatocytes. Our analysis showed that 20%–33% of the human hepatocytes were expressing the meganuclease at the end of the study (Figure 4D). One major difference from the NHP study is the lack of immune-mediated toxicity in FRG mice, which may have allowed persistent expression of the meganuclease. Persistent, high expression of meganuclease in cells may have led to high-level OT editing in human hepatocytes. Thus, an anti-meganuclease immune response may be beneficial, as it could help to minimize OT editing.
Figure 4.

In vivo genome editing of the hPCSK9 gene in primary human hepatocytes
Two FRG (Fah−/−Rag2−/−Il2rg−/−) mice engrafted with primary human hepatocytes received a tail vein injection of AAV3B-M2PCSK9 (1.3 × 1012 GC/mouse). Liver samples (four from each mouse) were harvested on day 49 after vector injection for analyses. (A) On-target indel analysis by amplicon-seq and AMP-seq. Individual data points and the means ± SEM are shown (n = 4 from each mouse). (B) Validation of OT editing by amplicon-seq for the top 22 OT sites predicted in vitro by GUIDE-seq. One liver DNA sample from each mouse was analyzed. Details regarding the OT sites and indel % for each mouse are shown in Table S4. (C) OT editing evaluated by ITR-seq. Each dot represents a unique potential OT site detected by ITR-seq and its relative frequency with respect to on-target editing. Four DNA samples (S1–S4, S5–S8) from each mouse are shown. Detailed OT information can be found in Data S2. (D) Quantification of transduction efficiency in human hepatocytes based on two-plex IHC. Individual mouse data points and the mean ± SEM are shown (n = 5). (E) ISH using transgene-specific probes (M1PCSK9) for liver samples (ISH signal: red; nuclear DAPI stain: blue). Representative images of each animal are shown. (F) M1PCSK9 ISH (red) and DAPI staining alone (blue) at higher magnification. Some human hepatocytes, identified by the absence of heterochromatin dots and a smaller size, are marked with “h.” (G) M1PCSK9 ISH (red) combined with immunofluorescence staining for human lamin (white) as a marker for human hepatocytes, which can be identified by ring-like staining surrounding the nucleus. (H) Two-plex IHC staining with antibodies against fumarylacetoacetase as a marker for human hepatocytes (FAH, brown stain) and meganuclease (purple-black nuclear stain). Nuclei were counterstained with methyl green (light green). Scale bars, (E) and (H): 50 μm; (F) and (G): 25 μm.
Discussion
In recent years, preclinical studies have demonstrated applications of genome editing as novel therapies for a broad list of diseases, including CVD. Most preclinical studies have utilized mouse models,13, 14, 15, 16, 17, 18, 19,38,39 with only a few short-term studies using large animal models such as dogs or NHPs.20,21 Encouragingly, in vivo genome editing mediated by ZFNs and CRISPR/Cas9 has entered clinics (ClinicalTrials.gov: NCT02702115, NCT03041324, and NCT03872479). We have shown long-term, stable reductions in PCSK9 and LDL-c as well as the safety profiles in NHPs for more than 3 years by in vivo genome editing of the PCSK9 gene with a single intravenous infusion of AAV meganuclease. On-target and OT editing in the liver were stably maintained from 4 months to almost 3 years, as evidenced by molecular analyses of longitudinal biopsy samples from individual animals—although these analyses cannot reveal all OT editing in the entire liver. Except for a transient elevation of transaminases in the early phase, there were no significant findings from clinical pathology tests or liver histopathology analyses. To our knowledge, this is the first study demonstrating the long-term durability of in vivo genome editing in somatic cells in a large animal model, and it presents valuable data enabling the comparison of off-target effects (1) over time in the same animal, (2) between different nucleases (M1PCSK9 versus M2PCSK9), (3) between different animals treated with the same nuclease, and (4) dose effects.
Compared to widely used CRISPR/Cas9, ZFN, and newly emerging BE and prime editing technologies for in vivo gene editing, meganucleases are relatively difficult to reengineer to target novel sequences. However, they have the unique and attractive feature of being small, which renders them amenable to all standard gene delivery methods, especially the AAV vector approach. Due to the size limitation of the AAV vector, in vivo delivery of ZFN, BE, and Streptococcus pyogenes Cas9 nucleases along with guide RNAs would involve oversized, split, or dual AAV vector systems, which would be less efficient than the conventional single vector system. Smaller Cas9 orthologs, such as SaCas9, CjCas9, and Cas12s, could be packaged by AAV vectors. LNP-formulated messenger RNAs (mRNAs) are the alternative delivery approach, which have no strict size limitations and no pre-existing neutralizing antibodies, elicit transient expression of the nuclease, and can be readministered. Success has been demonstrated in mouse models using LNPs to deliver Cas9 or ZFN for in vivo genome editing;13, 14, 15, 16, 17, 18,40 however, no efficacy and/or safety data in large animals have yet been published.
The durability of therapeutic effects is especially important for gene therapy of genetic diseases, which frequently require life-long efficacy, especially in viral vector-mediated in vivo gene therapy when re-administration is not always possible. In AAV gene therapy trials, long-term follow-up of hemophilia A and B patients treated with AAV vectors showed a gradual decline in hFVIII or hFIX expression in years 2 and 3.41,42 This decline is likely caused by the loss of vector genome in transduced cells during hepatocyte turnover. In the current study, we observed a significant loss of vector GCs (1–2 logs) and transgene expression levels (2–3 logs) in the second biopsies compared with those in the first biopsies (Figure 3). The significant loss during this early phase is likely related to the immune response to the meganuclease, a foreign protein to macaques, which results in profound elimination of episomal vector genomes and the death of some transduced hepatocytes. Most animals exhibited transient transaminitis during this phase; however, most transduced and edited hepatocytes persisted despite vector loss, as demonstrated by the stable on-target indels in the longitudinal liver biopsy samples. We detected a further reduction in vector GCs in 7/10 of the third liver biopsy samples, likely due to hepatocyte turnover, but this reduction had no impact on the editing effects. This finding highlights an important difference between AAV gene therapy and AAV-mediated genome editing: the durability of AAV-mediated in vivo genome editing does not depend on the stability of the vector genome and is not affected by hepatocyte turnover, in contrast to traditional AAV gene therapy approaches. During cell division, edited genomic sequences would be passed to the daughter cells, while episomal vector genomes would be lost. For vector-mediated in vivo genome editing, the loss of the vector genome and its expression are desirable features that can prevent persistent nuclease expression and reduce OT editing.
As a unique challenge in the translational research of in vivo genome editing, this approach is inherently human genome-specific. The engineered meganuclease used in this study targets a sequence conserved between the human and NHP PCSK9 gene, which allows us to evaluate on-target editing and general safety aspects in NHPs. However, human genome-specific OT editing is not accessible in NHPs. Instead, the chimeric liver-humanized FRG mouse model allows us to evaluate on- and off-targeting in primary human hepatocytes in vivo. However, the lack of an immune system in FRG mice leads to persistent meganuclease expression. Sustained overexpression of the nuclease could increase OT activity, as observed in the FRG mouse study. In immune-competent primates, cells overexpressing the meganuclease would harbor high OT activity and are more likely to be targeted by cytotoxic T cells. The asymptomatic and transient immune response to the meganuclease seen was useful in extinguishing OT editing. We are evaluating the potential of further reducing meganuclease expression by a weak promoter to further reduce OT activity without affecting on-target editing.
In summary, we have presented a longitudinal study of in vivo genome editing of PCSK9 in NHPs as well as our strategy and methodology for evaluating long-term in vivo editing efficiency and safety in NHPs. Our data show that in vivo genome editing of PCSK9 mediated by a single infusion of AAV meganuclease is efficient, stable, and safe in NHPs for up to 3 years. This approach may have therapeutic potential for the treatment of familial hypercholesterolemia.
Materials and methods
NHPs
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. The long-term treatments of six rhesus macaques have been previously described,24 and the animals have been continually monitored since vector administration. We acquired blood samples biweekly during the first year and once every 4 weeks after the first year. All blood draws were performed in the morning after an overnight fast. Antech Diagnostics conducted all clinical pathology tests on blood samples, including complete blood counts and differentials, clinical chemistries, and coagulation panels. Antech GLP performed tests on lipid panels, including total cholesterol, high-density lipoprotein, LDL, and triglycerides. Four new rhesus macaques were enrolled in this study. The animals were prescreened for AAV8 or AAV3B neutralizing antibodies as previously described.43 Those with AAV8 or AAV3B NAb titers <1:5 were assigned to the respective groups in this study. The two AAV8 animals also received prednisolone orally (1 mg/kg/day) on study days 0–76, followed by a tapering course that ended on day 102. On day 18 and day 127/130 post-vector infusion, a liver biopsy was performed on each macaque via laparotomy. Animal RA2125 was euthanized on day 284. A third liver biopsy was performed on the remaining nine animals on day 531/642/989/995/1,070 (Table 1).
FRG (Fah−/−Rag2−/−Il2rg−/−) mice
FRG mice with a C57BL/6N background and repopulated with 40%–70% human hepatocytes were purchased from Taconic-Yecuris (Portland, OR, USA). All four mice were repopulated with hepatocytes derived from the same donor. Before and during the study, the FRG mice were maintained on mouse chow with low tyrosine content (0.53%, PicoLab High Energy Mouse Diet 5LJ5) (Lab Supply, Fort Worth, TX, USA), 3% dextrose drinking water cycled with sulfamethoxazole (640 mg/L, Yecuris), trimethoprin (128 mg/L, Yecuris), and nitisinone (NTBC, 8 mg/L, Yecuris) according to the vendor’s recommendations, as previously described.30 On the first day of the NTBC cycle, two mice received a tail vein injection of AAV3B.M2PCSK9 (1.3 × 1012 GCs), and the livers of these mice were harvested on day 49 post-vector administration. Two control mice received an AAV vector expressing VP1, and the livers of these mice were harvested on day 28 post-vector administration.
AAV vectors
AAV8.M1PCSK9 and AA8.M2PCSK9, in which M1PCSK9 or M2PCSK9 was driven by a liver-specific thyroid hormone-binding globulin (TBG) promoter followed by the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) element, have been previously described.24 AAV3B.M2PCSK9 was produced and titered by the Penn Vector Core at the University of Pennsylvania, as previously described.44,45
Enzyme-linked immunosorbent assay (ELISA)
PCSK9 levels in macaque serum samples were measured by ELISA using a PCSK9 ELISA kit (R&D Systems, Minneapolis, MN, USA) following the manufacturer’s protocol.
Interferon (IFN)-γ ELISpot
T cell responses against the AAV capsid and meganuclease were evaluated by an IFN-γ ELISpot assay on PBMCs isolated from macaques, as previously described.24
On-target and OT indel analyses
On-target editing was analyzed by amplicon-seq and anchored multiplexed PCR sequencing (AMP-seq), as described previously,24 with modifications. To measure indels, the target window was defined as a region that is within 10 bp upstream or downstream of the predicted cleavage site. We evaluated OT editing by applying ITR-seq35 or amplicon-seq on potential OT sites previously identified by genome-wide, unbiased identification of double-stranded breaks enabled by sequencing (GUIDE-seq) in monkey or human cell lines. The sequences of PCR primer sets utilized in this work have been previously described.24
Two-plex IHC staining and quantification in FRG mouse liver sections
We performed sequential dual IHC on formalin-fixed, paraffin-embedded liver sections. The sections were deparaffinized with xylene and ethanol, boiled in a microwave for 6 min in 10 mM citrate buffer (pH 6.0) for antigen retrieval, and treated sequentially with 2% H2O2 (15 min; Sigma), avidin/biotin blocking reagents (15 min each; Vector Laboratories), and blocking buffer (1% donkey serum in phosphate-buffered saline [PBS] + 0.2% Triton for 10 min). The sections were then incubated with a rabbit antibody against I-CreI, which cross-reacts with M1PCSK9 and M2PCSK9 (custom-made, 1:2,000, overnight at 4°C) and biotinylated secondary anti-rabbit antibodies (30 min at 37°C; donkey antibodies from Jackson ImmunoResearch) diluted in blocking buffer. We applied a Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA, USA) with the Vector VIP peroxidase substrate according to the manufacturer’s protocol, which revealed bound antibodies as a dark purple precipitate. After the first staining round, the slides were rinsed in distilled water and PBS, and the same staining procedure was repeated, but without the antigen retrieval step, using a rabbit antibody against fumarylacetoacetase (FAH) (1:1,000, Yecuris) as the primary antibody. The sections were then incubated with the FAH antibody for 1 h at 37°C. 3,3′-diaminobenzidine was used as a peroxidase substrate (Vector Laboratories) according to the manufacturer’s protocol, showing the location of bound FAH antibodies as a brown precipitate. Finally, the slides were counterstained with methyl green (Vector Laboratories) to visualize nuclei. Meganuclease-expressing human and mouse hepatocytes were quantified on liver sections stained by two-plex IHC for FAH and I-CreI. We manually analyzed images obtained from scanned sections (Aperio Versa digital slide scanner, Leica Biosystems, Buffalo Grove, IL, USA) by counting I-CreI-positive cells using the counting tool in ImageJ (https://imagej.nih.gov/ij/).
Vector GC, transgene mRNA, histopathology, and ISH analyses
Vector GC, transgene mRNA, histopathology, and ISH analyses were performed on NHP tissues as previously described.24 In some FRG mouse liver sections for ISH, we performed co-staining with an antibody specific for human lamin as a marker for human hepatocytes. After ISH, the sections were boiled in a microwave for 6 min in 10 mM citrate buffer (pH 6.0) for antigen retrieval, blocked in blocking buffer (1% donkey serum in PBS + 0.2% Triton for 10 min), and incubated with a monoclonal rabbit antibody against human lamin (clone EPR4100, Abcam ab108595; 1:300; Cambridge, UK) in blocking buffer overnight at 4°C. Fluorescein isothiocyanate-labeled donkey anti-rabbit IgG was used as a secondary antibody (Jackson ImmunoResearch Labs, West Grove, PA, USA). Finally, the slides were mounted in Fluoromount with 4′,6-diamidino-2-phenylindole (DAPI; Electron Microscopy Sciences, Hatfield, PA, USA).
Statistical analyses
Individual data points and the mean ± the standard error of the mean (SEM) are presented. To determine whether serum PCSK9 and LDL-c levels were significantly reduced during the steady phase (arbitrarily defined as starting at day 56) in the treated macaques compared with pretreatment levels, a one-sided one-sample t test was performed using R Statistical Software (version R.4.0.0). To examine whether serum PCSK9 and LDL levels in the treated macaques changed significantly between years, we performed a two-sided Wilcoxon rank-sum test using R Statistical Software (version R.4.0.0). Statistical significance for OT genome editing, which was evaluated by amplicon-seq of top potential OT sites identified by GUIDE-seq, was assessed using a one-sided Fisher’s exact test (fisher.test, R Statistical Software version R.4.0.0) to compare the indel frequencies before and after vector treatment or for comparison with levels in control animals. A two-sided Fisher’s exact test was used to test differences of OT editing between different time points after vector treatment. We applied the Benjamini-Hochberg procedure to correct for multiple hypothesis testing.46 Statistical significance was assessed at a level of 0.05 after multiple testing adjustments.
Data availability statement
All data and supporting materials are available within the article and Supplemental information. The original deep sequencing data are available at NCBI BioProject under NCBI: PRJNA695573.
Acknowledgments
We thank the Penn Vector Core for supplying AAV vectors, the Program in Comparative Medicine at the Gene Therapy Program (GTP) for animal care and procedures, the Next Generation Technology Core at GTP for performing deep sequencing, Dr. Peter Clark for generating the NGS pipelines, the Immunology Core at GTP for PBMC isolation and immunology assays, the Histology Core at GTP for histology analyses, and Dr. Qiang Wang for providing control FRG mouse DNA. This research was supported by Precision BioSciences.
Author contributions
L.W., D.J., and J.M.W. conceived and designed this study. L.W., C.B., C.C.W., P.B., Z.H., J.W., and Y.Z. performed the experiments. H.Y. conducted the bioinformatics and statistical analyses of the data. M.L. supervised statistical analyses. E.L.B. performed histopathology analysis. L.W. wrote the manuscript, C.B., C.C.W., E.L.B., D.J., and J.M.W. edited the manuscript.
Declaration of interests
J.M.W. is a paid advisor to and holds equity in Scout Bio and Passage Bio; he holds equity in Surmount Bio; he also has sponsored research agreements with Albamunity, Amicus Therapeutics, Biogen, Elaaj Bio, FA212, Janssen, Moderna, Passage Bio, Regeneron, Scout Bio, Surmount Bio, and Ultragenyx, which are licensees of Penn technology. J.M.W. and L.W. are inventors on patents that have been licensed to various biopharmaceutical companies and for which they may receive payments.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.02.020.
Supplemental information
References
- 1.Lambert G., Sjouke B., Choque B., Kastelein J.J., Hovingh G.K. The PCSK9 decade. J. Lipid Res. 2012;53:2515–2524. doi: 10.1194/jlr.R026658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao Z., Tuakli-Wosornu Y., Lagace T.A., Kinch L., Grishin N.V., Horton J.D., Cohen J.C., Hobbs H.H. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am. J. Hum. Genet. 2006;79:514–523. doi: 10.1086/507488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hooper A.J., Marais A.D., Tanyanyiwa D.M., Burnett J.R. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis. 2007;193:445–448. doi: 10.1016/j.atherosclerosis.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 4.Kent S.T., Rosenson R.S., Avery C.L., Chen Y.I., Correa A., Cummings S.R., Cupples L.A., Cushman M., Evans D.S., Gudnason V. PCSK9 Loss-of-Function Variants, Low-Density Lipoprotein Cholesterol, and Risk of Coronary Heart Disease and Stroke: Data From 9 Studies of Blacks and Whites. Circ. Cardiovasc. Genet. 2017;10:e001632. doi: 10.1161/CIRCGENETICS.116.001632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Langslet G., Emery M., Wasserman S.M. Evolocumab (AMG 145) for primary hypercholesterolemia. Expert Rev. Cardiovasc. Ther. 2015;13:477–488. doi: 10.1586/14779072.2015.1030395. [DOI] [PubMed] [Google Scholar]
- 6.Sabatine M.S., Giugliano R.P., Keech A.C., Honarpour N., Wiviott S.D., Murphy S.A., Kuder J.F., Wang H., Liu T., Wasserman S.M., FOURIER Steering Committee and Investigators Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017;376:1713–1722. doi: 10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- 7.Farnier M. An evaluation of alirocumab for the treatment of hypercholesterolemia. Expert Rev. Cardiovasc. Ther. 2015;13:1307–1323. doi: 10.1586/14779072.2015.1111759. [DOI] [PubMed] [Google Scholar]
- 8.Rosenson R.S., Hegele R.A., Fazio S., Cannon C.P. The Evolving Future of PCSK9 Inhibitors. J. Am. Coll. Cardiol. 2018;72:314–329. doi: 10.1016/j.jacc.2018.04.054. [DOI] [PubMed] [Google Scholar]
- 9.Fitzgerald K., White S., Borodovsky A., Bettencourt B.R., Strahs A., Clausen V., Wijngaard P., Horton J.D., Taubel J., Brooks A. A Highly Durable RNAi Therapeutic Inhibitor of PCSK9. N. Engl. J. Med. 2017;376:41–51. doi: 10.1056/NEJMoa1609243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rashid S., Curtis D.E., Garuti R., Anderson N.N., Bashmakov Y., Ho Y.K., Hammer R.E., Moon Y.A., Horton J.D. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc. Natl. Acad. Sci. USA. 2005;102:5374–5379. doi: 10.1073/pnas.0501652102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaid A., Roubtsova A., Essalmani R., Marcinkiewicz J., Chamberland A., Hamelin J., Tremblay M., Jacques H., Jin W., Davignon J. Proprotein convertase subtilisin/kexin type 9 (PCSK9): hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology. 2008;48:646–654. doi: 10.1002/hep.22354. [DOI] [PubMed] [Google Scholar]
- 12.Chadwick A.C., Musunuru K. Treatment of Dyslipidemia Using CRISPR/Cas9 Genome Editing. Curr. Atheroscler. Rep. 2017;19:32. doi: 10.1007/s11883-017-0668-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding Q., Strong A., Patel K.M., Ng S.L., Gosis B.S., Regan S.N., Cowan C.A., Rader D.J., Musunuru K. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ. Res. 2014;115:488–492. doi: 10.1161/CIRCRESAHA.115.304351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X., Raghavan A., Chen T., Qiao L., Zhang Y., Ding Q., Musunuru K. CRISPR-Cas9 Targeting of PCSK9 in Human Hepatocytes In Vivo-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2016;36:783–786. doi: 10.1161/ATVBAHA.116.307227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin H., Song C.Q., Suresh S., Wu Q., Walsh S., Rhym L.H., Mintzer E., Bolukbasi M.F., Zhu L.J., Kauffman K. Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat. Biotechnol. 2017;35:1179–1187. doi: 10.1038/nbt.4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ibraheim R., Song C.Q., Mir A., Amrani N., Xue W., Sontheimer E.J. All-in-one adeno-associated virus delivery and genome editing by Neisseria meningitidis Cas9 in vivo. Genome Biol. 2018;19:137. doi: 10.1186/s13059-018-1515-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conway A., Mendel M., Kim K., McGovern K., Boyko A., Zhang L., Miller J.C., DeKelver R.C., Paschon D.E., Mui B.L. Non-viral Delivery of Zinc Finger Nuclease mRNA Enables Highly Efficient In Vivo Genome Editing of Multiple Therapeutic Gene Targets. Mol. Ther. 2019;27:866–877. doi: 10.1016/j.ymthe.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chadwick A.C., Evitt N.H., Lv W., Musunuru K. Reduced Blood Lipid Levels With In Vivo CRISPR-Cas9 Base Editing of ANGPTL3. Circulation. 2018;137:975–977. doi: 10.1161/CIRCULATIONAHA.117.031335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amoasii L., Hildyard J.C.W., Li H., Sanchez-Ortiz E., Mireault A., Caballero D., Harron R., Stathopoulou T.R., Massey C., Shelton J.M. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362:86–91. doi: 10.1126/science.aau1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maeder M.L., Stefanidakis M., Wilson C.J., Baral R., Barrera L.A., Bounoutas G.S., Bumcrot D., Chao H., Ciulla D.M., DaSilva J.A. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019;25:229–233. doi: 10.1038/s41591-018-0327-9. [DOI] [PubMed] [Google Scholar]
- 22.First CRISPR therapy dosed. Nat. Biotechnol. 2020;38:382. doi: 10.1038/s41587-020-0493-4. [DOI] [PubMed] [Google Scholar]
- 23.Arnould S., Delenda C., Grizot S., Desseaux C., Pâques F., Silva G.H., Smith J. The I-CreI meganuclease and its engineered derivatives: applications from cell modification to gene therapy. Protein Eng. Des. Sel. 2011;24:27–31. doi: 10.1093/protein/gzq083. [DOI] [PubMed] [Google Scholar]
- 24.Wang L., Smith J., Breton C., Clark P., Zhang J., Ying L., Che Y., Lape J., Bell P., Calcedo R. Meganuclease targeting of PCSK9 in macaque liver leads to stable reduction in serum cholesterol. Nat. Biotechnol. 2018;36:717–725. doi: 10.1038/nbt.4182. [DOI] [PubMed] [Google Scholar]
- 25.Nathwani A.C., Rosales C., McIntosh J., Rastegarlari G., Nathwani D., Raj D., Nawathe S., Waddington S.N., Bronson R., Jackson S. Long-term safety and efficacy following systemic administration of a self-complementary AAV vector encoding human FIX pseudotyped with serotype 5 and 8 capsid proteins. Mol. Ther. 2011;19:876–885. doi: 10.1038/mt.2010.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.George L.A., Fogarty P.F. Gene therapy for hemophilia: past, present and future. Semin. Hematol. 2016;53:46–54. doi: 10.1053/j.seminhematol.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 27.Rangarajan S., Walsh L., Lester W., Perry D., Madan B., Laffan M., Yu H., Vettermann C., Pierce G.F., Wong W.Y., Pasi K.J. AAV5-Factor VIII Gene Transfer in Severe Hemophilia A. N. Engl. J. Med. 2017;377:2519–2530. doi: 10.1056/NEJMoa1708483. [DOI] [PubMed] [Google Scholar]
- 28.Miesbach W., Meijer K., Coppens M., Kampmann P., Klamroth R., Schutgens R., Tangelder M., Castaman G., Schwäble J., Bonig H. Gene therapy with adeno-associated virus vector 5-human factor IX in adults with hemophilia B. Blood. 2018;131:1022–1031. doi: 10.1182/blood-2017-09-804419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao G., Vandenberghe L.H., Alvira M.R., Lu Y., Calcedo R., Zhou X., Wilson J.M. Clades of Adeno-associated viruses are widely disseminated in human tissues. J. Virol. 2004;78:6381–6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L., Bell P., Somanathan S., Wang Q., He Z., Yu H., McMenamin D., Goode T., Calcedo R., Wilson J.M. Comparative Study of Liver Gene Transfer With AAV Vectors Based on Natural and Engineered AAV Capsids. Mol. Ther. 2015;23:1877–1887. doi: 10.1038/mt.2015.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lisowski L., Dane A.P., Chu K., Zhang Y., Cunningham S.C., Wilson E.M., Nygaard S., Grompe M., Alexander I.E., Kay M.A. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature. 2014;506:382–386. doi: 10.1038/nature12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anguela X.M., Sharma R., Doyon Y., Miller J.C., Li H., Haurigot V., Rohde M.E., Wong S.Y., Davidson R.J., Zhou S. Robust ZFN-mediated genome editing in adult hemophilic mice. Blood. 2013;122:3283–3287. doi: 10.1182/blood-2013-04-497354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nelson C.E., Wu Y., Gemberling M.P., Oliver M.L., Waller M.A., Bohning J.D., Robinson-Hamm J.N., Bulaklak K., Castellanos Rivera R.M., Collier J.H. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019;25:427–432. doi: 10.1038/s41591-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanlon K.S., Kleinstiver B.P., Garcia S.P., Zaborowski M.P., Volak A., Spirig S.E., Muller A., Sousa A.A., Tsai S.Q., Bengtsson N.E. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019;10:4439. doi: 10.1038/s41467-019-12449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Breton C., Clark P.M., Wang L., Greig J.A., Wilson J.M. ITR-Seq, a next-generation sequencing assay, identifies genome-wide DNA editing sites in vivo following adeno-associated viral vector-mediated genome editing. BMC Genomics. 2020;21:239. doi: 10.1186/s12864-020-6655-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato J., Doi T., Kanno T., Wako Y., Tsuchitani M., Narama I. Histopathology of incidental findings in cynomolgus monkeys ( macaca fascicularis ) used in toxicity studies. J. Toxicol. Pathol. 2012;25:63–101. doi: 10.1293/tox.25.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chadwick A.C., Wang X., Musunuru K. In Vivo Base Editing of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) as a Therapeutic Alternative to Genome Editing. Arterioscler. Thromb. Vasc. Biol. 2017;37:1741–1747. doi: 10.1161/ATVBAHA.117.309881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jarrett K.E., Lee C., De Giorgi M., Hurley A., Gillard B.K., Doerfler A.M., Li A., Pownall H.J., Bao G., Lagor W.R. Somatic Editing of Ldlr With Adeno-Associated Viral-CRISPR Is an Efficient Tool for Atherosclerosis Research. Arterioscler. Thromb. Vasc. Biol. 2018;38:1997–2006. doi: 10.1161/ATVBAHA.118.311221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Finn J.D., Smith A.R., Patel M.C., Shaw L., Youniss M.R., van Heteren J., Dirstine T., Ciullo C., Lescarbeau R., Seitzer J. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018;22:2227–2235. doi: 10.1016/j.celrep.2018.02.014. [DOI] [PubMed] [Google Scholar]
- 41.Nathwani A.C., Reiss U.M., Tuddenham E.G., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pasi K.J., Rangarajan S., Mitchell N., Lester W., Symington E., Madan B., Laffan M., Russell C.B., Li M., Pierce G.F., Wong W.Y. Multiyear Follow-up of AAV5-hFVIII-SQ Gene Therapy for Hemophilia A. N. Engl. J. Med. 2020;382:29–40. doi: 10.1056/NEJMoa1908490. [DOI] [PubMed] [Google Scholar]
- 43.Calcedo R., Vandenberghe L.H., Gao G., Lin J., Wilson J.M. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J. Infect. Dis. 2009;199:381–390. doi: 10.1086/595830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lock M., Alvira M., Vandenberghe L.H., Samanta A., Toelen J., Debyser Z., Wilson J.M. Rapid, simple, and versatile manufacturing of recombinant adeno-associated viral vectors at scale. Hum. Gene Ther. 2010;21:1259–1271. doi: 10.1089/hum.2010.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lock M., Alvira M.R., Chen S.J., Wilson J.M. Absolute determination of single-stranded and self-complementary adeno-associated viral vector genome titers by droplet digital PCR. Hum. Gene Ther. Methods. 2014;25:115–125. doi: 10.1089/hgtb.2013.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benjamini Y., Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J. Roy. Stat. Soc. B Met. 1995;57:289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and supporting materials are available within the article and Supplemental information. The original deep sequencing data are available at NCBI BioProject under NCBI: PRJNA695573.