

Abstract
RNA interference (RNAi) offers the potential to treat disease at the earliest onset by selectively turning off the expression of target genes, such as intracellular oncogenes that drive cancer growth. However, the development of RNAi therapeutics as anti-cancer drugs has been limited by both a lack of efficient and target cell-specific delivery systems and the necessity to overcome numerous intracellular barriers, including serum/lysosomal instability, cell membrane impermeability, and limited endosomal escape. Here, we combine two technologies to achieve posttranscriptional gene silencing in tumor cells: Centyrins, alternative scaffold proteins binding plasma membrane receptors for targeted delivery, and small interfering RNAs (siRNAs), chemically modified for high metabolic stability and potency. An EGFR Centyrin known to internalize in EGFR-positive tumor cells was site-specifically conjugated to a beta-catenin (CTNNb1) siRNA and found to drive potent and specific target knockdown by free uptake in cell culture and in mice inoculated with A431 tumor xenografts (EGFR amplified). The generalizability of this approach was further demonstrated with Centyrins targeting multiple receptors (e.g., BCMA, PSMA, and EpCAM) and siRNAs targeting multiple genes (e.g., CD68, KLKb1, and SSB1). Moreover, by installing multiple conjugation handles, two different siRNAs were fused to a single Centyrin, and the conjugate was shown to simultaneously silence two different targets. Finally, by specifically pairing EpCAM-binding Centyrins that exhibited optimized internalization profiles, we present data showing that an EpCAM Centyrin CTNNb1 siRNA conjugate suppressed tumor cell growth of a colorectal cancer cell line containing an APC mutation but not cells with normal CTNNb1 signaling. Overall, these data demonstrate the potential of Centyrin-siRNA conjugates to target cancer cells and silence oncogenes, paving the way to a new class of anticancer drugs.
Graphical abstract
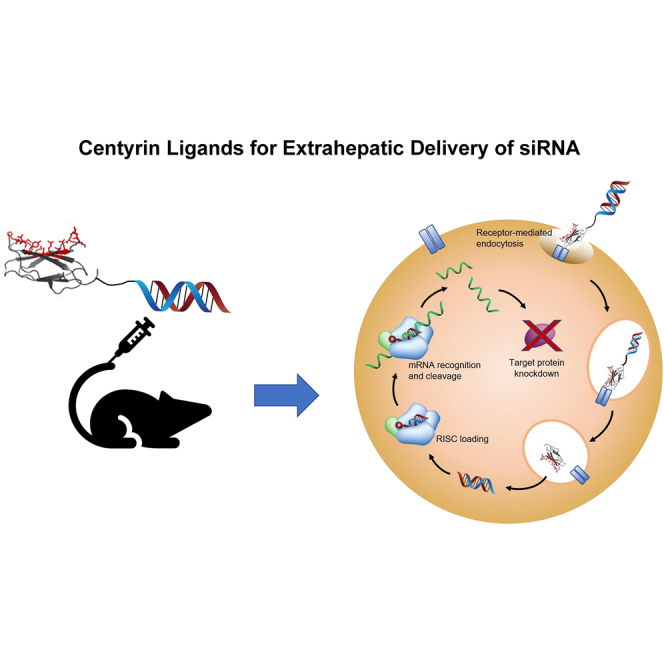
RNA interference offers the potential to silence previously intractable oncogenes but has been limited by inefficient delivery. Here, we combine two technologies to achieve posttranscriptional gene silencing in tumor cells: Centyrins, alternative scaffold proteins binding plasma membrane receptors for targeted delivery, and siRNAs, chemically modified for high metabolic stability and potency.
Introduction
RNAi therapeutics offer tremendous potential to treat diseases driven by previously undruggable targets. They have excellent potency and selectivity by harnessing a natural cellular mechanism that can potentially regulate the expression of any RNA transcript.1 Short interfering RNAs (siRNAs) are generally comprised of double-stranded RNA with 19–21 base pairs and typically with 2 nucleotide overhangs at the 3′ end. Upon entry into cells, the siRNA has to traffic to the cytoplasm and load into the RNA-induced silencing complex (RISC), which mediates binding to a complementary sequence on the target mRNA followed by mRNA cleavage.
Successful translation of this natural mechanism into a therapeutic reality has been challenging and required the development of technologies for safe and efficient delivery of siRNAs to target cells. To date, two platforms for targeted delivery to the liver, lipid nanoparticles and N-acetylgalactosamine (GalNAc) conjugates, have been clinically validated, resulting in the approval of the first RNAi therapeutics, ONPATTRO (patisiran) and GIVLAARI (givosiran).2,3 For the latter, targeted delivery to hepatocytes is mediated by a trivalent GalNAc ligand, which is covalently linked to the siRNA and designed to bind to the asialoglycoprotein receptor (ASGPR).4 However, this approach heavily depends on the siRNA being well protected against a wide variety of extra- and intracellular nucleases during its journey from the subcutaneous site of administration to the cytosol of the hepatocytes in the liver. This is achieved through chemical modifications judiciously placed throughout both strands of the siRNA to achieve high metabolic stability without compromising its ability to functionally load into the RNA-induced silencing complex (RISC).5,6
While clinical successes with liver diseases have been achieved, attempts to deliver siRNA to extrahepatic targets have been more challenging. Different groups have used either antibodies, antibody fragments, or aptamers to deliver siRNA with limited success.7, 8, 9, 10, 11, 12, 13 Using THIOMAB technology, Cuellar et al.7 developed a panel of antibody-siRNA conjugates but detected in vitro silencing at high conjugate concentration with just two of seven conjugates and only very limited silencing detectable in tumor xenografts. Recent data using an anti-CD71 Fab conjugated to modified siRNA dosed either systemically or locally resulted in significant muscle tissue knockdown,8 demonstrating the potential for smaller conjugates, which may penetrate further through tissue. While these data are encouraging and indicate that extrahepatic delivery is plausible, significant efforts are required to identify suitable ligands for cell-specific and functional delivery of siRNAs.
Centyrins are based on a consensus sequence of the FN3 domains in human Tenascin C14,15 and possess biophysical properties that can achieve siRNA delivery to target cells. They are single-domain proteins that can be engineered to bind to targets with high selectivity and affinity, similar to antibodies, although they do not contain any natural cysteine residues, making site-specific conjugation straightforward when a unique cysteine is introduced.16 Moreover, we have developed a high-throughput assay to identify Centyrins that enable maximum payload delivery, using cytotoxin conjugates as a surrogate to predict siRNA delivery potential. In this study, we demonstrate the potential for functional siRNA delivery into multiple cell types utilizing internalizing Centyrins against different cell surface receptors conjugated to highly stabilized siRNA targeting various genes.
Results
EGFR Centyrin-siRNA conjugates potently silence CTNNb1 mRNA in vitro
Centyrins containing a single free cysteine at the C terminus were conjugated to an siRNA duplex modified with a maleimide handle at the 5′ position of the passenger strand. Centyrin-siRNA conjugates were purified and confirmed to contain a single chemical entity with the molecular weight of the desired construct (Figure 1A). An EGFR-binding Centyrin was selected for the first siRNA conjugate, as EGFR binding Centyrins have already been validated for intracellular delivery of cytotoxics.16 EGFR-binding Centyrins were conjugated to a CTNNb1 siRNA (EGFR-1 Cent-siRNA_1 and EGFR-2 Cent-siRNA_1) as described below, with an overall yield of 40%–60% in terms of siRNA mass, and the conjugates were applied to EGFR-expressing A431 cells. Treatment with either of two distinct EGFR-siRNA conjugates for 72 h induced a dose-dependent reduction in CTNNb1 mRNA up to 75% knockdown with an IC50 of 5 nM, in the absence of transfection agent. Neither negative control Centyrin-siRNA conjugate nor siRNA alone reduced CTNNb1 mRNA levels at concentrations at or below 100 nM (Figure 1B). (The slight knockdown observed with non-targeted Centyrin-siRNA conjugates at 1 μM is most likely a result of nonspecific siRNA uptake. No dose-dependent differences in housekeeping gene cycle threshold values were observed, suggesting there was no significant toxicity.) To confirm that gene knockdown induced a reduction in target protein, CTNNb1 protein levels were evaluated by western blot. Approximately 50% decrease in protein level was observed after treatment with EGFR-1-siRNA_1 conjugate at 10 nM for 3 days, confirming that CTNNb1 mRNA knockdown correlates with CTNNb1 protein knockdown (Figure 1C).
Figure 1.

In vitro characterization of EGFR Centyrin-siRNA conjugates.
(A) Isolated siRNA conjugates are fully homogeneous, single chemical entity constructs. siRNA strands separate on the LC and produce defined m/z peaks in MS corresponding to Centyrin-sense strand and free antisense strand (not shown). (B) A431 cells were treated with a range of doses of targeted and non-targeted Centyrin-siRNA conjugates for 72 h, and CTNNB1 mRNA expression was detected by qRT-PCR. All dose levels were evaluated in duplicate. (C) Western blots showing CTNNb1 or GAPDH protein expression after exposure of A431 cells to siRNA targeted to CTNNb1 delivered using lipofectamine or in the EGFR-1 Centyrin-siRNA conjugate in the absence of transfection agent for 72 h. (D) HCC827 cells were pre-treated with or without EGFR-1 Centyrin (227 nM) for 1 h at 4°C followed by treatment with or without 20 nM EGFR-1-Cent-siRNA_1 for 72 h. CTNNb1 mRNA levels were assessed by qRT-PCR and indicate that mRNA knockdown is mediated through EGFR uptake, as conjugate-mediated knockdown is inhibited by EGFR-1-Cent pretreatment (N = 8 per group). (E) EGFR per cell was quantified by flow cytometry using Quantibrite beads (n = 4 for A549 and HCC827 and n = 2 for H3255 cells). (F) HCC827, H3255, or A549 cells were treated with ascending doses of EGFR-1-Centyrin-siRNA conjugate for 72 h, and CTNNb1 mRNA expression was detected by qRT-PCR. All dose levels were evaluated in duplicate. Data represent average ± SEM. ∗p < 0.001 versus control; &p < 0.001.
To explore the relationship between receptor expression level and potency, delivery of siRNA with EGFR Centyrins was evaluated using a panel of cell lines expressing varying levels of EGFR. Significant and potent gene knockdown was evident in two additional tumor cell lines, expressing 500,000 to 1,000,000 copies of the EGFR per cell (Figures 1D and 1E). In contrast, no mRNA knockdown was detected in A549 cells, which exhibit significantly less EGFR per cell (~140,000 receptors per cell) following treatment with conjugates. These data are consistent with our hypothesis that efficient uptake and robust gene silencing activity of EGFR Centyrin-siRNA conjugates requires a threshold level of EGFR on the cell.
The role of the EGFR for uptake of Centyrin-siRNA conjugate was confirmed by a competition assay in the presence of excess free ligand. HCC827 cells were treated with a saturating dose of EGFR-1 Centyrin (227 nM) for 1 h at 4°C and then treated with EGFR-1 Cent-CTNNb1 siRNA _1 conjugate (20 nM) for 72 h at 37°C. Figure 1F shows that 20 nM EGFR-1 Cent-siRNA_1 conjugate results in 57% CTNNb1 mRNA knockdown, while pre-treatment with excess EGFR-1 Centyrin prior to addition of EGFR-1 Cent-siRNA_1 conjugate reduces mRNA knockdown to just 21%, consistent with EGFR-mediated uptake of Centyrin-siRNA conjugate. To understand the temporal profile of the targeted siRNA-mediated knockdown, HCC827 cells were treated with siRNA conjugates for between 24 h and 5 days, and mRNA knockdown was evaluated. CTNNb1 mRNA silencing was evident within 24 h of treatment with EGFR-1-Cent-siRNA_1 conjugates, but more knockdown was observed at 72 h than at 24 h. mRNA knockdown was similar in cells treated with EGFR-targeted siRNA conjugates for 72 h and 120 h (Figure S1).
Robust mRNA knockdown is achieved with Centyrin-siRNA conjugates in tumor xenografts in vivo
To assess in vivo activity, mice bearing A431 tumor xenografts were dosed intravenously (i.v.) on days 1, 3, and 6 with EGFR Centyrin, EGFR Centyrin-siRNA conjugate, or negative Centyrin-siRNA conjugate, where all conjugates were dosed at 10 mg/kg (mpk) siRNA. Tumors were collected 72 h after the final dose. Since Centyrins are small proteins that are cleared rapidly via kidney first-pass clearance, Centyrins that had been genetically fused to the stabilized albumin-binding domain (ABD) ABDCon12,17 which is designed to extend serum half-life, were also evaluated. As expected, addition of the ABD extended serum exposure reduced the siRNA exposure in the kidney and enhanced exposure in the liver, suggesting the clearance mechanism was shifted (Figure S2). Figure 2 shows the relative mRNA levels for tumors from mice treated with EGFR-1 Cent-siRNA_1 conjugates. Significant knockdown of CTNNb1 mRNA is observed in tumors from mice treated with each of the two different EGFR Centyrin-siRNA conjugates compared to a negative control Centyrin-siRNA conjugate, which had no activity. Despite rapid clearance for Centyrins that do not contain a half-life extension domain, up to 50% silencing was detected for EGFR-1 Cent-siRNA conjugates (Figure 2A) and 75% knockdown of CTNNb1 for EGFR-1 Cent-ABD-siRNA_1 conjugates (containing an ABD; Figure 2B). Negative control Centyrin siRNA conjugates containing an ABD showed up to 25% knockdown, which we hypothesize is due to albumin-mediated uptake in tumor cells. Thus, albumin binding may provide an additional mechanism for siRNA uptake into tumor cells, since accumulation of albumin has previously been demonstrated for tumor tissues.18 Reduction of β-catenin was also confirmed at the protein level by western blotting. Nearly 50% loss of β-catenin protein was detected in tumors isolated from mice dosed with EGFR-1-ABD-CTNNb1 siRNA, while negative control Centyrin-siRNA conjugates resulted in minimal protein loss (Figure S3).
Figure 2.

In vivo silencing of CTNNB1 gene by i.v. administration of Centyrin-siRNA conjugate in A431 tumor xenograft model
Mice bearing A431 tumor xenografts were dosed three times every other day with targeted or non-targeted siRNA conjugates containing 10 mpk siRNA or equimolar levels of Centyrin only. Tumors were excised 72 h following the final dose, and CTNNb1 mRNA was assessed by qRT-PCR following tissue lysis (n = 6 mice per group). (A) Silencing with non-half-life extended constructs (red). mRNA silencing is observed with both EGFR-1 and EGFR-2 Centyrin conjugates. Non-binding Centyrin conjugates, Centyrins alone, and vehicle are used as controls. (B) Silencing with conjugates incorporating albumin-binding domain (ABD) fusion for half-life extension (blue). (C) 5′ RACE experiment performed on tumor biopsy from EGFR-1-ABD-siRNA_1-treated animals confirms RNAi mechanism of silencing. PCR products from 5′ RACE assay were run on an agarose gel. Arrow indicates mRNA cleavage product in samples treated with EGFR-1-ABD-siRNA but not vehicle (n = 3 mice per group). Data represent mean ± SEM. ∗p < 0.0001 versus untreated controls, #p < 0.0001.
In order to confirm that an RNAi mechanism was responsible for the observed silencing activity, mRNA cleavage fragments were evaluated from RNA isolated from either vehicle or EGFR-ABD-siRNA-1-treated tumor tissue with a 5′ rapid amplification of cDNA ends (RACE) assay. The PCR products were run on an agarose gel, where an approximately 200 bp band was evident for the EGFR-1 Cent-ABD-siRNA_1-treated samples but not the vehicle controls (Figure 2C). mRNA was extracted from the band and 26 sequences were identified, one of which had an insert that began with the expected sequence derived from the RNA adaptor from the GeneRacer kit and a nearly perfect match with the target cleavage site, as shown in Figure S4.
Since Centyrin-siRNA conjugates (particularly conjugates containing ABDs) were found to accumulate in liver, silencing was also evaluated in liver tissue. In Centyrin-siRNA conjugates that did not contain ABDs, silencing was only observed with the EGFR-siRNA conjugate (EGFR-1 Cent-ABD-siRNA_1) but not the non-binding Centyrin-siRNA conjugate (neg. Cent-ABD-siRNA_1) or the EGFR Centyrin alone. As EGFR is expressed in the liver and EGFR-1 Cent cross-reacts with mouse EGFR, it is likely that the CTNNb1 mRNA silencing observed in the liver is mediated through EGFR-specific uptake (Figure S5A). With the Centyrin-siRNA conjugates fused to an ABD, significant CTNNb1 mRNA silencing was observed for both the non-binding Centyrin-ABD-siRNA conjugate and the EGFR-ABD-siRNA conjugate (Figure S5B). Silencing may be due to albumin-mediated and EGFR-mediated uptake. Silencing was not evaluated in the kidney.
We have previously reported on the critical role of chemical modifications in the sugar-phosphate backbone in stabilizing GalNAc-siRNA conjugates against nucleolytic degradation.5 In our siRNA designs, the ribosugar moieties of each nucleotide are generally modified with either 2′-deoxy-2′-fluoro (2′-F) or 2′-O-methyl (2′-OMe) and two phosphorothioate linkages at each of the free 3′ and 5′ ends of the siRNA. We have also reported that additional enhancement of metabolic stability and in vivo activity can be achieved through further optimization of the siRNA chemistry, specifically by judicious increase in the 2′-OMe and a concomitant decrease in the 2′-F content.4 To address the question whether these findings would translate to Centyrin-conjugated siRNAs and different tissue/cell types, we compared conjugates containing two different siRNA designs, one with a higher and one with an optimized (lower) 2′-F content (Table 1; siRNA_1 and siRNA_2). Further, the impact of the conjugation site was investigated with siRNA_3, which was linked to the Centyrin via the 5′ rather than the 3′ end of the sense strand. Maximum mRNA knockdown and IC50 were modestly improved in vitro with both new constructs with optimized siRNA chemistry (Figure 3A). Moreover, analysis at the protein level showed more than a 5-fold improvement in IC50 for the new modification pattern (siRNA_2: 1.6 nM and siRNA_3: 1.3 nM versus siRNA_1: 9.1 nM; Figure 3B).
Table 1.
Oligonucleotide sequences and chemistries are listed
| CTNNb1 siRNAs | Strand | Sequence |
|---|---|---|
| CTTNNb1 siRNA_1 | sense | U⋅a⋅CuGuUgGAUuGaUuCgAaAL1 |
| antisense | VPu⋅U⋅uCgAaUcAaucCaAcAgUa⋅g⋅c | |
| CTTNNb1 siRNA_2 | sense | u⋅a⋅cuguUgGAUugauucgaaaL1 |
| antisense | VP(Tam)⋅U⋅ucgAaUCaaucCaAcagua⋅g⋅c | |
| CTTNNb1 siRNA_3 | sense | Qu⋅a⋅cuguUgGAUugauucga⋅a⋅a |
| antisense | VP(Tam)⋅U⋅ucgAaUCaaucCaAcagua⋅g⋅c | |
| CTTNNb1 siRNA_4 | sense | u⋅a⋅cuguUgGAUugauucgaaaL2 |
| antisense | VP(Tam)⋅U⋅ucgAaUCaaucCaAcagua⋅g⋅c | |
| KLKB1 siRNA | sense | A⋅a⋅UcCaAaAUAuUcUaCaAaAL1 |
| antisense | VPu⋅U⋅uUgUaGaAuauUuUgGaUu⋅u⋅c | |
| CD68 siRNA | sense | A⋅u⋅GgCgCaGAAuUcAuCuCuAL1 |
| antisense | VPu⋅A⋅gAgAuGaAuucUgCgCcAu⋅g⋅a | |
| SSB siRNA | sense | U⋅a⋅AcAaCaGACuUuAaUgUaAL1 |
| antisense | VPu⋅U⋅aCaUuAaAgucUgUuGuUa⋅g⋅a |
Structures of L1, L2, and Q are in the Supplemental information. Chemistry modification: ⋅, phosphorothioate (PS) linkage; lower case nucleotides, 2′-O-methyl (OMe); upper case nucleotides in italics, 2′-deoxy-2′-fluoro (F); L1, 3′ maleimide; L2, 3′ triglycine; Q, 5′ maleimide; VP, 5′-(E)-vinylphosphonate; (Tam), 2′-O-(N-methylacetamide)thymidine.
Figure 3.

mRNA knockdown (KD) potency of the same oligonucleotide sequence further optimized using changes in siRNA modification chemistry
(A) A431 cells were treated with three different EGFR-1-Cent-ABD-siRNA conjugates containing the same sequence but different chemical modifications or conjugation sites, as indicated in Table 1. (A and B) CTNNb1 mRNA expression after 72 h treatment determined by qRT-PCR (A) and β-catenin protein knockdown after 120 h determined by ELISA (B) are shown. Data represent average of two replicate samples at each dose. (C) Mice bearing A431 tumor xenografts were treated with a single dose of EGFR-1-Cent-siRNA containing various chemistries and conjugation sites (10 mpk by siRNA), and tumors were excised 72 h after dosing. siRNA levels were determined by stem-loop PCR, and (D) CTNNb1 mRNA expression was quantified by qRT-PCR. Chemistries used in siRNA_2 and siRNA_3 result in better tumor knockdown than siRNA_1. n = 6 mice per group. ∗p < 0.0001 versus vehicle. #p < 0.0001. ##p < 0.05.
The trend of improved activity for the two siRNAs with optimized modification pattern observed by in vitro free uptake also translated to improved in vivo activity (Figures 3C and 3D). In tumor xenografts following a single dose of EGFR-1 Cent-ABD-siRNA conjugates (containing 10 mpk siRNA) the original conjugate (siRNA_1) resulted in 51% CTNNb1 mRNA knockdown 72 h after dosing. The conjugates containing the chemically optimized siRNA_2 or siRNA_3 exhibited improved knockdown of CTNNb1 mRNA of 65% and 70%, respectively and a tendency toward higher tumor exposures. This is in agreement with our previously published work on GalNAc mentioned above and indicates that improved metabolic stability through optimized siRNA chemistry plays a similarly important role when targeting tumor tissue (Figure 3D). Comparing the conjugates of CTNNb1_2 and CTNNb1_3, however, the impact of the conjugation site on mRNA knockdown appears to be insignificant, despite the somewhat higher tissue levels observed for siRNA_2.
Generalizability of Centyrin-mediated siRNA delivery
To assess the general utility of Centyrins for targeted delivery of siRNA, Centyrins binding to PSMA, BCMA, or EpCAM were conjugated to CTNNb1 siRNA_1 and evaluated for knockdown in receptor-positive cell lines. Treatment of PSMA Cent-siRNA_1 conjugates in LnCAP cells (200,000 PSMA per cell) resulted in 42% CTNNb1 mRNA knockdown (Figure 4). A431 cells (350,000 EpCAM antigens per cell) treated with EpCAM Cent-siRNA_1 conjugates also induced knockdown of CTNNb1 mRNA by 40%. Impressively, even BCMA Cent-siRNA_1 conjugates were able to significantly silence CTNNb1 mRNA levels in H929 cells, which only express 17,000 BCMA antigens per cell. In all cases, negative control Centyrin-siRNA conjugates had no activity.
Figure 4.

CTNNb1 mRNA expression for cells treated with Centyrin-siRNA conjugates against multiple antigens
(A) LNCAP cells were treated with 200 nM nonbinding Centyrin-siRNA conjugate (neg Cent-siRNA_1) or PSMA-binding Centyrin-siRNA conjugate (PSMA Cent-siRNA_1) against CTNNb1 for 72 h, and mRNA was detected by qRT-PCR. (B) A431 cells were treated with 125 nM nonbinding Centyrin-siRNA conjugate (neg Cent-siRNA_1) or EpCAM-binding Centyrin-siRNA conjugate (EpCAM Cent-siRNA_1) for 72 h. (C) H929 cells were treated with 175 nM non-binding Centyrin-siRNA conjugate or BCMA Centyrin-siRNA conjugate for 72 h, and CTNNb1 mRNA was assessed by qRT-PCR. Significant CTNNb1 mRNA knockdown was observed only with treatment with targeted siRNA conjugates but not non-binding Centyrin-siRNA conjugates. n = 4 per group. Data plotted as mean ± SEM. ∗p < 0.05 versus untreated controls.
To demonstrate that Centyrin-mediated siRNA delivery was generally applicable across different genes, Centyrins were conjugated to siRNAs targeting one of three additional genes: kallikrein B1 (KLKb1), murine CD68, and SSB. Since KLKB1 and murine CD68 are not endogenously expressed by HCC827 cells, cells were first transfected with a luciferase reporter plasmid fused to human KLKb1, murine CD68, human SSB, and human CTNNb1 genes. 24 h after transfection with luciferase reporter, cells were rinsed and treated with Centyrin-siRNA conjugates for 72 h. Figure 5 illustrates significantly improved potency of EGFR-targeted Centyrin-siRNA conjugates compared to non-binding controls for CD68 (Figure 5A), SSB (Figure 5B), KLKb1 (Figure 5C), and CTNNb1 siRNA_1 (Figure 5D)-containing conjugates, where IC50 values were reduced by between 20- and 600-fold. The activity of the non-binding controls appears to be higher than observed previously (as in Figures 1B and 4), which may be an artifact of compromised membrane integrity following transfection of the luciferase reporter plasmids.
Figures 5.

In vitro potency of EGFR Centyrin-siRNA conjugates targeting different genes
HCC827 cells were transfected with plasmid containing luciferase-tagged CD68, KLKb1, SSB1, and CTNNb1 genes. Cells were then treated with a dose range of Centyrin-siRNA conjugates that targeted EGFR (EGFR-1 Cent, red) or were non-binding (neg. Cent, blue) for 72 h. (A–D) Percent luciferase activity is reported as a surrogate for relative gene expression in HCC827 cells transfected with a luciferase reporter gene construct and treated with Centyrin conjugates delivery (A) CD68, (B) KLKb1, (C) SSB1, or (D) CTNNb1 siRNA. Non-binding Centyrin-siRNA conjugates appeared to be more potent than in previous experiments, but this was presumably due to the compromised membrane integrity that occurred following transfection.
Following effective knockdown of a single gene via EGFR-1 Cent-siRNA conjugates, we explored the potential to knock down two genes simultaneously with a single Centyrin conjugated to two different siRNAs. An EGFR Centyrin was first conjugated to an SSB-targeting siRNA at an internal cysteine (E54C) using a thiol maleimide reaction. The Centyrin-SSB conjugate was then conjugated to CTNNb1 siRNA_4 at the C terminus using a sortase transpeptidase reaction,19 as illustrated in Figure 6A. Silencing was compared using an EGFR Centyrin conjugated to either CTNNb1 siRNA, SSB siRNA, or both CTNNb1 and SSB siRNA. Figures 6B and 6C show that the EGFR Centyrin dually conjugated to both SSB and CTNNb1 reduced SSB mRNA to levels equivalent to the EGFR-1 Cent-SSB siRNA mono-conjugate. Similarly, the dual siRNA Centyrin conjugate silenced CTNNb1 to levels comparable to that of the EGFR-1 Cent-CTNNb1 siRNA conjugate. These results indicate that the potency of a dual-function Centyrin conjugate containing two different siRNAs can be equivalent to the individual conjugates. Importantly, these results also demonstrate that no CTNNB1 silencing was observed with a EGFR1-Cent-SSB siRNA, nor was SSB silencing observed with a EGFR1-Cent-CTNNb1 siRNA, further demonstrating the gene specificity of the Centyrin-siRNA conjugates used in these studies.
Figure 6.

Centyrin-siRNA conjugates can be designed to simultaneously inhibit multiple genes
(A) Schematic shows Centyrin dually conjugated to CTNNb1 and SSB siRNA. A431 cells were treated with Centyrin-siRNA conjugates at a range of doses for 72 h, and mRNA expression was assessed by qRT-PCR. Cells were treated at each concentration in triplicate. EGFR1-Cent-dual siRNA (CTNNb1, SSB) contained an EGFR Centyrin conjugated to both CTNNB1 and SSB siRNA, as indicated in schematic in (A). EGFR1-Cent-CTNNb1 siRNA and EGFR1-Cent-SSB siRNA were EGFR Centyrins conjugated only to CTNNb1 or SSB siRNA, respectively. (B) SSB mRNA is shown. Cells treated with Centyrin-siRNA conjugates targeting SSB demonstrate potent knockdown, while that with CTNNb1 siRNA only has no activity. (C) CTNNb1 mRNA is reduced in cells treated with Centyrin-siRNA conjugates against CTNNb1 but not SSB. No difference in potency is observed in Centyrin-siRNA conjugates containing one versus two siRNA, as long as the appropriate siRNA is present.
Centyrin-siRNA conjugates show antitumoral activity in vitro
To assess the potential for using Centyrin-siRNA conjugates to knock down genes that are important for tumor growth, we evaluated cytotoxicity of Centyrin-siRNA conjugates in CTNNb1-dependent cell lines. Mutations in both APC and CTNNb1 are a major driving factor in many colorectal cancers.20, 21, 22 APC is a negative regulator of β-catenin protein levels, and therefore mutations in APC lead to constitutive β-catenin activation, which ultimately activates WNT signaling and drives aberrant growth.23 To assess Centyrin-siRNA conjugate activity in CTNNb1-dependent colorectal cancer cells, we assessed downstream gene signaling and toxicity in a panel of colorectal cell lines with APC mutations using EpCAM-targeted Centyrins conjugated to CTNNb1 siRNA.
To select for the best Centyrin for siRNA delivery, a panel of EpCAM-binding Centyrins was screened using a high-throughput internalization assay. Centyrins were conjugated to a cytotoxic drug, MMAF. Colo205 cells were treated with Centyrin-toxin conjugates at 20 or 2 nM for 72 h and assessed for cytotoxicity. Internalizing Centyrins were selected when they induced more than 50% cytotoxicity when conjugated to vcMMAF. Seven of the EpCAM-binding Centyrin-vcMMAF conjugates were found to induce cytotoxicity in greater than 50% of Colo205 cells at 20 nM (Figure 7), including the positive control EpCAM-binding Centyrin referenced in Figure 4. Of the seven, four were also toxic at 2 nM, while the original positive control was inactive, suggesting improved internalization profiles. EpCAM Centyrin F01 was selected for further evaluation.
Figure 7.

EpCAM Centyrin internalization screen
A panel of 48 EpCAM-binding Centyrins were screened for internalization. Centyrins were rapidly conjugated to MMAF, a cytotoxic drug, and treated on Colo205 cells for 72 h. Each bar represents screening data from a single clone. (A and B) Internalizing Centyrins, as indicated by those reducing viability to less than 50%, are detected at both (A) 20 nM, and (B) 2 nM treatment, as indicated by the red bars. The blue bar indicates the first-generation EpCAM Centyrin, while black indicates the non-binding Centyrins.
Treatment with EpCAM Cent-siRNA_3 for 72 h led to a 50% reduction in CTNNb1 mRNA levels with IC50 values of less than 1 nM in both Colo205 and H358 cells (Figure 8A). Moreover, treatment with EpCAM Cent-siRNA_3 for 9 days potently killed Colo205 cells harboring an APC mutation, while there was no impact on toxicity in H358 cells, which carry wild-type (WT) APC (Figure 8B). To further demonstrate the impact of Centyrin conjugate-mediated CTNNb1 gene knockdowns on downstream signaling, we measured TCF-LEF reporter activity 72 h after EpCAM Cent-siRNA_3 treatment. Three cell lines with mutant APC and significantly enhanced TCF-LEF gene signaling compared to H358 cells expressing WT APC were selected. Treatment with 100 nM EpCAM Cent-siRNA_3 for 3 days inhibited TCF-LEF signaling in all three cell lines to levels similar to or better than lipofectamine-mediated transfection of CTNNb1 siRNA (Figure 8), while negative Centyrin-siRNA_3 conjugates were not significantly changed.
Figure 8.

Efficacy of EpCAM Centyrin-CTNNb1 siRNA conjugates in cells with APC mutations
(A) Colo205 or H358 cells were treated with a dose response of Centyrin-siRNA conjugates that bound to EpCAM (EpCAM Cent-siRNA_3) or that were nonbinding (neg. Cent-siRNA_3) for 72 h. Cells were treated at each concentration in duplicate. EpCAM Centyrin-siRNA conjugates induced potent reduction in CTNNb1 mRNA compared to nonbinding negative control Centyrin-siRNA conjugate in both Colo205 and H358 cells. (B) Colo205 or H358 cells were treated with a dose response of Centyrin-siRNA conjugates for 9 days (with media refreshed with fresh treatment after 5 days), and cell viability was assessed by CellTiterGlo. Cell viability was found to be significantly and selectively reduced in Colo205 cells (containing mtAPC) treated with EpCAM-Centyrin-siRNA conjugates but not in H358 cells, which are not reliant on CTNNb1 for growth as they do not have a mutation in either CTNNb1 or APC. (C) Downstream gene signaling, via a TCF/LEF reporter assay, was assessed in a panel of cells treated with reporters alone, lipofectamine, or transfected with CTNNb1 via lipofectamine or Centyrin-siRNA conjugates for 72 h. EpCAM Centyrin-siRNA conjugates reduced TCF/LEF activity in three cell lines containing mutant APC, to a level similar to that with lipofectamine transfection of free CTNNb1 siRNA. Data are plotted as mean ± SEM. ∗p < 0.05 versus cells + reporters alone.
Discussion
The recent successes in the development of clinically validated technologies for the delivery of RNAi therapeutics to the liver have encouraged and intensified the quest for efficient siRNA delivery to extrahepatic tissues, for which to date there are no clinically proven delivery systems available. Here we explored a delivery system based on Centyrins, a platform based on designed FN3 domain proteins, which can be engineered to target internalizing plasma membrane proteins. Centyrins are particularly amenable to rapid evaluation of intracellular siRNA delivery potential due to the ease of selecting new ligands and introducing functionalities for site-specific conjugation. Centyrins do not contain cysteines, so addition of a defined number of siRNA molecules is easily achieved by introducing cysteines or other conjugation strategies (e.g., peptide ligation). We previously scanned the entire Centyrin scaffold to determine what sites were amenable to conjugation with minimal impact on binding and other biophysical properties.16
Precise tumor targeting and efficient intracellular delivery appear to be essential for successful application of RNAi therapeutics as anticancer agents. Like antibodies, Centyrins can be developed to bind with high affinity and selectivity to numerous antigens, which makes specific tumor targeting across a broad range of tumor antigens feasible. Previously reported approaches for delivery of siRNAs to tumor tissue include encapsulation in cationic nanoparticles. However, these can be toxic, are not selective for tumor cells, and are preferentially sequestered by the reticuloendothelial system.24, 25, 26 Similarly, while cholesterol-siRNA conjugates have demonstrated good potency, they are taken up by numerous cell/tissue types, resulting in lower tumor accumulation.27,28 Previously reported tumor-targeting strategies also include aptamer-siRNA conjugates. However, these can be less selective than traditional biologics and suffer from low metabolic stability and rapid clearance from circulation.9,12,13
In addition to selective cell surface receptor binding, it is essential for the targeting moiety to promote active internalization into the tumor cells, as the highly negative charge of the siRNA prevents passive cell penetration. As the high-throughput internalization screen previously described, Centyrins with good internalization properties can be rapidly identified, conjugated to siRNA, and evaluated for functional siRNA delivery. We selected three Centyrins targeting different antigens (EGFR, EpCAM, and BCMA) using this screening paradigm and demonstrated functional delivery of CTNNb1-siRNA for each of the constructs as evident by target mRNA knockdown. Interestingly, while the degree of mRNA knockdown seems to depend on the target receptor density, the minimum number of receptors required for functional siRNA delivery varied by target and cell type. With EGFR, siRNA delivery required more than 150,000 receptors per cell, while successful knockdown was achieved with BCMA Centyrins binding to as few as 17,000 receptors per cell. The minimal receptor expression is likely determined by a combination of factors, including internalization kinetics, endosomal trafficking, and proliferation rates. The high-throughput screen used to identify Centyrins selects for the best internalizers but does not differentiate for endo/lysosomal trafficking; future efforts should consider screening directly for mRNA knockdown via high-throughput conjugation to siRNA in order to identify the best Centyrins for siRNA delivery across multiple receptors.
Importantly, we found that the mRNA knockdown observed in vitro translated to tumor xenografts as well. Despite rapid clearance of Centyrin-siRNA conjugates in the absence of any half-life extension strategy, we still observed up to 50% mRNA knockdown in A431 tumor xenografts with an EGFR Centyrin-siRNA conjugate. With the addition of an ABD, mRNA knockdown was enhanced to nearly 70%. These data indicate that Centyrin-siRNA conjugates were able to penetrate well through the tumor tissue. Moreover, we show that enhancing stability of the CTNNb1 siRNA (but keeping sequences identical) can improve tumor uptake and knockdown in vivo. Indeed, the EGFR Centyrin-CTNNb1_3 containing the more stabilized siRNA showed enhanced tumor exposure, suggesting that reduced degradation was predominantly responsible for the enhanced activity observed.
Using orthogonal conjugation strategies, we were able to engineer a single Centyrin carrying two different siRNAs. Impressively, there was no loss in potency with the addition of a second siRNA, suggesting that the larger size of the construct does not influence internalization or endosomal escape. Such dually conjugated molecules may be beneficial in order to develop therapies to prevent resistance mechanisms by targeting 2 either horizontal or vertical gene inhibition strategies. Alternatively, two different siRNAs targeting the same gene could be delivered with the potential to achieve enhanced efficacy.29 Future studies should evaluate the feasibility of this strategy in vivo. Centyrins demonstrated equal potency when conjugated to siRNA at either the 5′ (CTNNb1_4) or 3′ end (CTNNb1_2), with no impact on potency. We hypothesize that the small size of Centyrins together with conjugation to the sense strand leaves the unmodified guide strand available and unimpeded for RISC loading.
Overall, we have demonstrated that the combination of tumor-targeting Centyrins with chemically stabilized siRNAs presents a versatile platform for RNAi-mediated gene silencing across multiple tumor types. Combination of Centyrins and chemically stabilized siRNA offers the potential to silence oncogenes that are not druggable with traditional modalities and creating novel therapeutics for cancer and other genetic disorders. For example, CTNNb1 has long been considered an undruggable target, although it is implicated in numerous cancers, especially colorectal cancer where the majority of cases contain aberrant signaling due to mutations in either APC or CTNNb1,30 and more recent data have implicated CTNNb1 signaling in preventing immune cell infiltration into tumors.31,32 Our data show that an optimized EpCAM Centyrin conjugated to a CTNNb1 siRNA can effectively kill tumor cells exhibiting APC mutations but not cells exhibiting normal CTNNb1 signaling. Hence, Centyrin-mediated delivery of CTNNb1 siRNA may provide a novel approach to treat colorectal cancer, with limited off-target toxicity due to the high selectivity of the siRNA target and the tissue-specific delivery approach, which may reduce unwanted effects of gene silencing in healthy tissues.
Materials and methods
General synthesis of siRNA
Oligonucleotide sequences and chemistries are listed in Table 1. Oligo synthesis was performed on either an Applied Biosystems 394, AKTA Oligopilot, or Mermade 12 synthesizer using standard phosphoramidite chemistry on 500–600 Å controlled pore glass (CPG). The phosphoramidites were prepared as 0.15 M solutions in acetonitrile with 15% dimethyl formamide (v/v) added as a cosolvent for 2′ OMe uridine and cytidine. 0.6 M ETT (5-ethylthiotetrazole) was the activation agent during coupling reactions. The oxidizing reagent was 0.02 M I2 in THF/pyridine/water. N,N-dimethyl-N′-(3-thioxo-3H-1,2,4-dithiazol-5-yl)methanimidamide (DDTT), 0.09 M in pyridine, was used as the sulfurizing reagent for the Applied Biosystems 394 and Mermade 12 instruments, while 0.2 M phenylacetyl disulfide (PADS) in 50/50 (v/v) lutidine-acetonitrile was used for the AKTA system. Detritylation was performed using 3% dichloroacetic acid (v/v) in dichloromethane. The single strands were purified by ion exchange chromatography (IEX), with the exception of maleimide-containing strands (see below). The IEX buffers were 20 mM phosphate at pH 8.5 (sense strands) or pH 11 (antisense strands) with 1 M sodium bromide used for the gradient. After IEX purification, the appropriate fractions were pooled, concentrated to a reduced volume, and desalted. Sample volumes were then further reduced by rotary evaporation, filtered through a sterile 0.2 μm filter, and lyophilized to dryness prior to storage of the oligonucleotide powder at −20°C.
Oligos, with the exception of oligos to be included in maleimide-containing duplexes, were annealed using routine methods by dissolving the oligos in annealing buffer, combining the oligos, heating the oligos to 95°C in a water bath, and allowing the water bath to cool to room temperature (RT). Annealed duplexes were stored at −20°C.
Deprotection of antisense strands
Upon completion of synthesis, the support was washed with acetonitrile (ACN) and dried in the column under vacuum or by blowing nitrogen through the column. Subsequently, the support was transferred into a container that could be tightly sealed and shaken with a solution of 5% diethylamine in aqueous ammonia at 35°C for 20 h or 65°C for 5 h; optimization for these conditions has been extensively discussed previously.33 The mixture was filtered into appropriately sized containers according to scale and washed at least twice with water (5× volumes of deprotecting solution). The deprotection of crude oligo was checked by IEX-high-performance liquid chromatography (HPLC) and liquid chromatography-mass spectrometry (LC-MS) and was subsequently purified by IEX-HPLC.
Deprotection of tri-glycine siRNA
The tri-glycine modification for sortase-mediated conjugation was synthesized as a CPG solid support (Supplemental information). The N terminus of the tri-glycine was trifluoroacetic acid (TFA) protected. Upon completion of synthesis, the support was washed with ACN and dried under vacuum or by blowing nitrogen through the synthesis column. Subsequently, the support was transferred into a polypropylene tube that could be tightly sealed and incubated with 50/50 v/v 40% aqueous methyl amine and aqueous ammonia (AMA) at room temperature for 3 h.
Synthesis and deprotection of maleimide-containing oligonucleotides
Precursors for maleimide conjugation were made using either a 3′ amino-modified CPG solid support or a 5′ amino modifier phosphoramidite. In either case, the amine was protected with a TFA group and was deprotected using the same conditions as the tri-glycine oligonucleotides. The single strand was purified by IEX chromatography and desalted under standard conditions prior to maleimide conjugation.
The amine-modified oligomer was dissolved at a concentration of approximately 20 mg/mL in 0.05 M phosphate buffer at pH 7.1. Ten equivalents of the maleimide N-hydroxysuccinimide (NHS) ester were dissolved in ACN at the same volume of the phosphate buffer. The NHS ester solution was added to the aqueous oligonucleotide solution and shaken for 3 h at room temperature. Maleimide-containing oligonucleotides were purified by reverse-phase chromatography (20 mM triethyl ammonium acetate with 80% acetonitrile in Buffer B), as the maleimide hydrolyzes under ion exchange buffer conditions.
After purification, the oligo was dried to a small volume of liquid, not to dryness, at 30°C on a rotary evaporator, and desalted. Due to the sensitive nature of the maleimide, duplexing was performed by freeze-drying using equimolar amounts of each desalted single strand.
Centyrin conjugation to siRNA and conjugate purification
Centyrins were expressed and purified as previously described.16 Centyrins were conjugated to siRNA through either cysteine-specific chemistry via maleimide handles on the siRNA34 or using the sortase reaction.19 For cysteine-maleimide conjugation, cysteine-containing Centyrins in PBS at 50–200 μM were reduced with 5–10 mM tris(2-carboxyethyl)phosphine (TCEP) to yield a free thiol. To remove the TCEP, Centyrin was precipitated by addition of 3 volumes of saturated ammonium sulfate solution (Teknova, Hollister, CA, USA) followed by incubation on ice for 10 min and centrifugation (20 min, 75,600 × g). The supernatant was decanted, and the pellet was resuspended in a 3:1 mixture of saturated ammonium sulfate:PBS, followed by another centrifugation step (20 min, 75,600 × g). The supernatant was decanted, and the final Centyrin pellet was re-dissolved in PBS. The Centyrin was mixed with maleimide-modified siRNA duplex that was dissolved in water immediately prior at a molar ratio of ~1.5:1 Centyrin:siRNA. After 1 h incubation at RT, the reaction was quenched with N-ethyl maleimide.
The conjugate was purified in two steps. Crude reaction was loaded onto an equilibrated QIAGEN Ni-NTA cartridge (Hilden, Germany) and purified using an AKTA AVANT system. Fractions containing conjugate and crude Centyrin were pooled and purified further by ion exchange chromatography (IEX). For IEX, sample was injected onto an AKTA AVANT equipped with a CaptoQ HiTrap column (GE Healthcare, Chicago, IL, USA) equilibrated in Buffer QA (20 mM Tris-HCl [pH 7.5]) and eluted with a 0%–100% gradient of Buffer QB (QA + 2 M NaCl). Fractions containing conjugate were pooled, exchanged into PBS by dialysis or with Zeba desalting columns (Thermo), and concentrated if necessary.
For sortase-catalyzed conjugation, Centyrins with a C-terminal sortase recognition sequence were conjugated to siRNA duplex modified with a tri-glycine peptide. Centyrin (~100 uM), siRNA (~50 uM), and sortase (1 uM) were incubated for 1–2 h at room temperature in buffer containing 50 mM Tris (pH 7.5), 150 mM NaCl, and 10 mM CaCl2. Conjugates were purified essentially as described above.
For dual-siRNA conjugates, a Centyrin with C-terminal sortase recognition site as well as a single cysteine at position 54 (E54C) was used. The first siRNA was conjugated via a maleimide handle to the cysteine and purified as described above. The second siRNA, modified with a tri-glycine handle, was conjugated subsequently using sortase-catalyzed transpeptidation essentially as described above. Purification of the final conjugate proceeded in two steps, using Ni-NTA chromatography to remove the sortase and free siRNA followed by preparative size exclusion chromatography on an AKTA AVANT system equipped with a Superdex75 16/60 column (GE Healthcare) run in PBS.
For conjugates prepared for in vivo studies, endotoxin was removed (if necessary) using an EndoTrapHD column (Hyglos, Munich, Germany). Endotoxin was measured with Endosafe PTS cartridges using an Endosafe device (Charles River, Wilmington, MA).
Characterization of Centyrin-siRNA conjugates
LC-MS was used to confirm identity and purity of the conjugates. Samples were analyzed using an Agilent Model 6230 TOF mass spectrometer system (Agilent, Santa Clara, CA, USA). The instrument was operated in negative electro-spray ionization mode and scanned from m/z 100 to 3,200. Instrument settings included: spray voltage 3,750 V, source temperature 350°C, drying gas flow 12 L/min, nebulizer 35 psi, sheath gas 300°C, sheath gas flow 11 L/min, nozzle voltage 1,200 V, fragmentor 250 V. For the HPLC (Agilent 1290 instrument), the column was a Waters xBridge C8, 2.5 μm particle, 2.1 × 50 mm, and it was run at 75°C with the buffers MSA (25 mM triethylamine, 570 mM 1,1,1,3,3,3-hexafluoro-2-propanol, 10% methanol) and MSB (acetonitrile). 5 μL of sample was injected onto the column, washed for 2 min with 1% MSB, and eluted with a gradient of 1%–50% B over 10 min (flow rate 0.3 mL/min for all).
Target mRNA knockdown after Centyrin-mediated uptake of siRNA
Centyrins were assessed for their ability to mediate uptake of siRNAs into various cell lines in the absence of transfection reagent. Cells were plated in 96-well plates (5,000–10,000 cells per well) for 24 h and then treated with Centyrin-siRNA conjugates in duplicate for 72 h. Cells were lysed with Protein Quant Sample Lysis Reagent (Applied Biosystems, Waltham, MA, USA). Lysates were frozen at −80°C until analysis. Samples were thawed on ice, and RNA in cell lysate was converted to cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) in a ProFlex ThermoCycler (Thermo Scientific, Waltham, MA, USA) according to manufacturer’s instructions. qPCR of cDNA was performed using TaqMan Fast Advanced 2× Master Mix (Applied Biosystems) with TaqMan Gene Expression Assay Primer/Probes (Applied Biosystems) for CTNNb1 and peptidyl-prolyl cis-trans isomerase B (PPIB) on a ViiA 7 Real-Time PCR System (Applied Biosystems). Target gene CT values were normalized by subtracting the PPIB CT value to obtain a deltaCT. The average of untreated deltaCT values was then subtracted from the sample deltaCT. Relative mRNA level was then determined using the equation, % Gene expression = 100 × 2(−deltadeltaCT). Data were log transformed on the x axis, then analyzed using nonlinear regression, applying a 3-parameter model to determine IC50.
Western blot
A431 cells were plated in 6-well plates (100,000 cells per well) and treated with Centyrin-siRNA conjugates or transfected with 50 nM On-Target plus CTNNb1 SMARTpool siRNA (Dharmacon, Lafayette, CO, USA) using Lipofectamine RNAiMAX transfection reagent (Invitrogen, Carlsbad, CA, USA) as a positive control for 3 days. Cells were washed twice with ice-cold PBS and lysed with RIPA buffer (Thermo Scientific) containing PhosSTOP (Sigma, St. Louis, MO, USA) and Complete Mini Protease Inhibitor Cocktail Tablets (Sigma). Total protein concentration was quantified with a Pierce BCA assay (Thermo Scientific), according to manufacturer’s instructions. Samples were prepared using LDS Sample Buffer (Life Technologies) and heated to 95°C for 5 min. For western blots with cell lysates, 20 μg protein was loaded per well into a 4%–12% NuPage Bis-Tris gel (Thermo Scientific) and subjected to electrophoresis at 150 V. Proteins were transferred to 0.45 μm nitrocellulose membranes (Thermo Scientific) by applying 30 V constant for 90 min. Membranes were then blocked for 1 h at room temperature with Odyssey Blocking Buffer (PBS) (Li-Cor, Lincoln, NE, USA), washed with PBS containing 0.1% Tween-20 (Sigma), and stained with rabbit anti-CTNNb1 antibody #9562 (Cell Signaling Technology, Danvers, MA, USA) at 1:1,000 dilution or mouse anti-GAPDH antibody #Ab8245 (Abcam, Cambridge, MA, USA) at 1:10,000 dilution overnight at 4°C. Antibody binding was detected with donkey anti-rabbit IRDye 800CW or donkey anti-mouse IRDye 680RD (Li-Cor) used at 1:20,000 dilution and incubated for 1 h at room temperature. For western blots in tissue lysates, 15 μg of protein was loaded into a 4%–12% NuPage Bis-Tris gel and subject to electrophoresis. After transfer to nitrocellulose membranes, membranes were blocked for 1 h in Odyssey blocking buffer (Li-Cor), and proteins were stained with an anti-β-catenin antibody (Sigma, # HPA029159, 1:1,000) and anti-alpha Tubulin Abs (Abcam, # ab184613, 1:1,000) at 4°C overnight on a shaker. After thorough rinsing, proteins were stained for 1 h with donkey anti-rabbit IRDye 800CW (Li-Cor, 1:5,000) and donkey anti-mouse IRDye 680 (Li-Cor, 1:5,000) in block buffer for 1 h at room temperature. Blots were imaged on an Odyssey Infrared Fluorescence Imaging System (Li-Cor).
β-catenin protein ELISA
A431 cells were plated in 96-well plates at 1,000 cells per well and treated with Centyrin-siRNA conjugates at a range of doses. Five days after treatment, cells were rinsed twice with PBS and lysed with 50 μL RIPA buffer containing HALT protease inhibitors and EDTA. β-catenin protein was assessed with a Human Total Beta-Catenin DuoSet IC ELISA (R&D Systems), according to manufacturer’s instructions with minor modifications. Capture antibody was left on plate overnight at 4°C and horseradish peroxidase (HRP) was detected with BM Chemiluminescence ELISA Substrate (POD) (50 μL/well; Roche). Luminescence was read on a SpectraMax M5 (Molecular Devices), and protein levels were back calculated from a standard curve generated using linear regression.
Xenograft tumor model
Mouse studies were performed at Charles River Laboratories. All animal protocols were approved by Janssen Institutional Animal Care and Use Committee. Ten-week-old female SCID beige mice (Charles River Laboratories) were implanted with 5 × 106 A431 cells suspended in 50% Matrigel (Corning, Corning, NY, USA) in PBS, subcutaneously into the right flank. Tumors were monitored as their volumes approached the target range of 100–150 mm3.
Mice were dosed i.v. three times during the first week (corresponding to days 1, 3, and 6 after tumors reached appropriate size) with EGFR or wild-type Centyrin with or without siRNA. Molecules included both non-half-life extended Centyrins and Centyrins with an ABD fusion. Tumor tissue was collected from all animals 3 days after the final dose. For each mouse, a terminal whole-body perfusion with PBS was performed prior to sample collection. The perfusions were performed by injecting each mouse intraperitoneally (i.p.) with anesthetic (sodium pentabarbitol, 6.5 mg/mL, 500 μL/mouse). Once the perfusion was completed, animals were exsanguinated, and tumors and livers were removed and bisected. Three 3 mm biopsies were collected from the tissue sample using biopsy punches (Sklar, West Chester, PA, USA) and placed in a single RNALater tube (Ambion) and stored at 4°C.
Tissue punches were homogenized in Trizol (Ambion) using a TissueLyser II Bead Homogenizer (QIAGEN), extracted in 1-bromo-2 chloropropane (Sigma), and total RNA was isolated on the KingFisher Flex Nucleic Acid Purification System (Thermo Scientific) using the MagMax RNA Isolation Method (Ambion). CTNNb1 mRNA levels were determined using 50 ng total RNA per sample as described above.
5′ RACE assay
mRNA cleavage fragments were evaluated from 1 μg RNA isolated from either vehicle or EGFR1-Cent-ABD-siRNA_1-treated tumor tissue with a 5′ RACE assay using the Invitrogen GeneRacer Kit with SuperScript III RT and TOPO TA Cloning Kit for Sequencing (L150201), per the manufacturer’s protocol. PCR amplification was done with the Gene Racer 5′ nested primer and gene-specific inner primer (5′-ACCCCCTCCACAAATTGCTGCTGTGT-3′). The PCR product from tumor tissue isolated from mice treated with vehicle or 83v2-ABDcon12-siRNA (5 μL/sample) was run on an agarose gel. Sample was added to 1 μL of 10× Blue Juice (Invitrogen) and loaded on a 1% agarose gel (BioRad) in 1× Tris-borate-EDTA (TBE) (Invitrogen). Track IT 1 kB Plus DNA ladder (Invitrogen) was also run, along with 5 μL of the BioRad ladder. Gel was run at 125 V for 45 min, imaged using a BioRad Gel Doc XR, and analyzed with BioRad Image Lab software. Relevant bands were cut out, and mRNA was extracted from the gel using the Wizard PCR and Gel Cleanup Kit (Promega). Fragments were eluted in 50 μL nuclease-free water and cloned in Invitrogen’s TOPO TA cloning vector, using TOPO TA Cloning Kit for Sequencing (Invitrogen).
Detection of siRNA in tissue samples
Concentrations of siRNA in tissue samples were determined using real-time quantification of microRNAs by stem-loop RT-PCR methods as previously described.35 Tissue samples were weighed, diluted in RIPA buffer (containing HALT protease/phosphatase inhibitor [Thermo Scientific] and Superase RNase inhibitor [Life Technologies]), and lysed in a TissueLyzer (QIAGEN). Samples, siRNA standards, and study test article spike controls were diluted 1:100 or 1:1,000 with TE buffer (pH 8.0) (Thermo Scientific). 25 nM sequence-specific stem-loop Primer (Thermo Scientific; Table 2) was annealed to the siRNA in all assay samples using a ProFlex ThermoCycler (Thermo Scientific), then converted to cDNA using the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems). qPCR of cDNA was performed using TaqMan Fast Advanced 2× Master Mix (Applied Biosystems) with sequence-specific forward and reverse primers and probe (Thermo Scientific; Table 2) and run on a ViiA 7 Real-Time PCR System (Applied Biosystems). CT values for the siRNA standards are transformed to X = log(X) and Y = log(Y), and nonlinear regression curve fit is performed using a sigmoidal dose-response (variable slope) to generate a standard curve. Concentration of siRNA in the unknowns is interpolated from the standard curve. Resultant values are multiplied by the dilution factor to determine nanomolar concentration.
Table 2.
Primer and probe sequences used for stem-loop PCR are shown
| RT stem-loop primer | 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACGCTACT-3′ |
| qPCR probe | (6-FAM)-5′-CTGGATACGACGCTACT-(NFQ)-(MGB)-3′ |
| qPCR forward primer | 5′-TCGTGATTTCGAATCAATCCAAC-3′ |
| qPCR reverse primer | 5′-GTGCAGGGTCCGAGGT-3′ |
Receptor quantitation in cell lines
EGFR, PSMA, EpCAM, and BCMA expression levels were quantified by flow cytometry. Cells were lifted from substrate with Enzyme-Free Cell Dissociation buffer (Thermo Scientific) and then stained with saturating levels of phycoerythrin (PE)-conjugated anti-EGFR antibody (25 μg/mL, #555997, BD Biosciences, Franklin Lakes, NJ, USA), anti-EpCAM antibody (3 μg/mL, #347198, BD Bioscience), anti-PSMA antibody (20 μg/mL; #77228, Abcam), anti-BCMA antibody (300 μg/mL, #357504, BioLegend), or isotype control (BD Biosciences) for 2 h. Excess antibody was rinsed away, and fluorescence was recorded using a FACs Calibur Flow Cytometer (BD Biosciences). Antibody binding was quantified using PE Quantibrite beads (BD Biosciences) as directed by the manufacturer. The number of bound antibodies was then translated to receptor antigen expression by subtracting that bound to isotype control, assuming that each antibody bound to two antigens.
Luciferase assay
To test the activity of Centyrin-siRNA conjugates against targets that are not expressed endogenously in the cell lines used, a luciferase reporter system was used. Cells were plated in 96-well plates (3,000 cells per well) for 4 h, then transfected using Lipofectamine 2000 according to manufacturer’s instructions with the Multi-site psiCheck-2 Dual Luciferase Reporter Plasmid (Promega, Madison, WI, USA) containing the target gene of interest. Cells were thoroughly rinsed 24 h later to remove excess transfection agent and treated with Centyrin-siRNA conjugates for 72 h. Cells were assayed for both Renilla and firefly luciferase expression using the Dual-Glo Luciferase Assay System kit (Promega), essentially according to manufacturer’s instructions (manufacturer’s supplied Luciferase assay Reagent II was diluted 1:1 with complete media prior to use). Luminescence was detected using an Envision multi-mode plate reader (Perkin Elmer, Waltham, MA, USA). Background signal from untransfected cells was subtracted from each assay well, and Renilla expression was normalized to the nonspecific Firefly expression.
Centyrin internalization assay
Centyrins were conjugated to monomethyl auristatin F (MMAF36) using a high-throughput conjugation strategy via a sortase tag. Purified Centyrins in 96-well plates were mixed with the cytotoxic drug payload Gly3-Val-Cit-para-aminobenzyl-MMAF (Levena Biopharma), Sortase A, and sortase buffer (50 mM Tris [pH 7.5], 150 mM sodium chloride, 10 mM calcium chloride final). The conjugation reaction proceeded for 2 h at room temperature, after which proteins were purified using a Ni-NTA multi-trap HP plate (GE Catalog #28-4009-89). Conjugates were filter sterilized and used directly for cell-based cytotoxicity assays. Centyrin-drug conjugates were then diluted to 20 or 2 nM and treated on Colo205 cells for 72 h. Cell viability was assessed using Cell Titer Glo (Promega), according to manufacturer’s instructions. Percent viable cells was determined by normalizing the Cell TiterGlo relative light units (RLU) signal to the average of the untreated cells. The best internalizing Centryins were determined by those that caused more than 50% toxicity at the indicated concentrations.
TCF/LEF assay
Cells were plated in white 96-well tissue-culture-treated plates (5,000 cells per well) and incubated overnight in a humidified incubator at 37°C, 5% CO2. Lentiviral transduction was performed after creating a transduction master mix with CMV-Renilla Control (QIAGEN), TCF/LEF Reporter (QIAGEN), and SureEntry (QIAGEN) in growth medium (RPMI + 10% FBS). Transduction mix and Centyrin-siRNA conjugates were then sequentially added to each well. Transfection samples were prepared with equal volume conjugate to RNAiMAX in serum-free OptiMEM and incubated at room temperature for 20 min prior to sample addition. Following sample addition, the plates were incubated in a humidified incubator at 37°C, 5% CO2. Seventy-two hours later, cells were assessed for firefly/Renilla luminescence using Promega DualGlo, according to manufacturer’s instructions. Firefly luminescence (FL) (TCF/LEF plasmid) is normalized to the constitutively expressed Renilla luciferase (RL) signal. The FL/RL ratio was compared among treatment groups and cell lines.
Viability assay
Cells in growth media (RPMI + 10% FBS) were plated in ultra-low attachment 96-well plates (500 cells per well; Corning 3474) and incubated overnight in a humidified incubator at 37°C, 5% CO2. Cells were then treated with Centyrin-siRNA conjugates and returned to the incubator. After 5 days, cells were replenished with fresh media containing Centyrin-siRNA conjugates in growth media and incubated for an additional 96 h. Cell viability was assessed with CellTiterGlo (Promega), according to the manufacturer’s protocol. Percent viability was determined by normalizing the RLU readings of treated wells to the average of untreated wells.
Statistical analysis
All data were analyzed in GraphPad Prism and presented as mean ± standard error of the mean (SEM). Dose-response curves were all performed in duplicate, concentrations were log transformed, and data were fit to three parameter sigmoidal curves, unless otherwise noted. All single-dose comparisons contain at least four replicates unless noted. Statistically significant differences were evaluated using a one-way ANOVA and compared to untreated controls using a Sidak multiple comparisons test.
Acknowledgments
All work was jointly funded by Janssen and Alnylam. The authors wish to thank Charles River Laboratories for conducting the tumor xenograft studies and the RNA Synthesis team at Alnylam for their help in preparing the siRNAs used for the studies.
Author Contributions
D.K. and S.G. wrote the manuscript. D.K., S.G., V.J., M.A.M., L.S., and V.D. designed experiments for this paper. C.S.T. prepared siRNAs; T.L. and J.M.Z. prepared Centyrin-siRNA conjugates; R.D., E.K., M.R., and M.W. conducted cell-based assays and data analysis. K.H. performed PK/PD on tumor xenografts. M.A.M., V.J., L.S., K.O., and V.D. provided expertise and feedback. K.O., L.S., and M.M. obtained support for this work.
Declaration of interests
D.K., S.G., R.D., K.H., E.K., T.L., M.R., M.W., J.M.Z., K.O., and V.D. are/were employed at Janssen while experiments were conducted. C.S.T., V.J., R.P., M.M., M.A.M., and L.S. are/were employed at Alnylam while experiments were conducted.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.02.015.
Contributor Information
Donna Klein, Email: dklein30@its.jnj.com.
Karyn O’Neil, Email: koneil@arobiotx.com.
Supplemental information
References
- 1.Elbashir S.M., Harborth J., Lendeckel W., Yalcin A., Weber K., Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 2.Sardh E., Harper P., Balwani M., Stein P., Rees D., Bissell D.M., Desnick R., Parker C., Phillips J., Bonkovsky H.L. Phase 1 Trial of an RNA Interference Therapy for Acute Intermittent Porphyria. N. Engl. J. Med. 2019;380:549–558. doi: 10.1056/NEJMoa1807838. [DOI] [PubMed] [Google Scholar]
- 3.Adams D., Gonzalez-Duarte A., O’Riordan W.D., Yang C.C., Ueda M., Kristen A.V., Tournev I., Schmidt H.H., Coelho T., Berk J.L. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018;379:11–21. doi: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- 4.Nair J.K., Willoughby J.L., Chan A., Charisse K., Alam M.R., Wang Q., Hoekstra M., Kandasamy P., Kel’in A.V., Milstein S. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014;136:16958–16961. doi: 10.1021/ja505986a. [DOI] [PubMed] [Google Scholar]
- 5.Foster D.J., Brown C.R., Shaikh S., Trapp C., Schlegel M.K., Qian K., Sehgal A., Rajeev K.G., Jadhav V., Manoharan M. Advanced siRNA Designs Further Improve In Vivo Performance of GalNAc-siRNA Conjugates. Mol. Ther. 2018;26:708–717. doi: 10.1016/j.ymthe.2017.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nair J.K., Attarwala H., Sehgal A., Wang Q., Aluri K., Zhang X., Gao M., Liu J., Indrakanti R., Schofield S. Impact of enhanced metabolic stability on pharmacokinetics and pharmacodynamics of GalNAc-siRNA conjugates. Nucleic Acids Res. 2017;45:10969–10977. doi: 10.1093/nar/gkx818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuellar T.L., Barnes D., Nelson C., Tanguay J., Yu S.F., Wen X., Scales S.J., Gesch J., Davis D., van Brabant Smith A. Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB-siRNA conjugates. Nucleic Acids Res. 2015;43:1189–1203. doi: 10.1093/nar/gku1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sugo T., Terada M., Oikawa T., Miyata K., Nishimura S., Kenjo E., Ogasawara-Shimizu M., Makita Y., Imaichi S., Murata S. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control. Release. 2016;237:1–13. doi: 10.1016/j.jconrel.2016.06.036. [DOI] [PubMed] [Google Scholar]
- 9.Neff C.P., Zhou J., Remling L., Kuruvilla J., Zhang J., Li H., Smith D.D., Swiderski P., Rossi J.J., Akkina R. An aptamer-siRNA chimera suppresses HIV-1 viral loads and protects from helper CD4(+) T cell decline in humanized mice. Sci. Transl. Med. 2011;3:66ra6. doi: 10.1126/scitranslmed.3001581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baumer S., Baumer N., Appel N., Terheyden L., Fremerey J., Schelhaas S., Wardelmann E., Buchholz F., Berdel W.E., Müller-Tidow C. Antibody-mediated delivery of anti-KRAS-siRNA in vivo overcomes therapy resistance in colon cancer. Clin. Cancer Res. 2015;21:1383–1394. doi: 10.1158/1078-0432.CCR-13-2017. [DOI] [PubMed] [Google Scholar]
- 11.Xia C.F., Boado R.J., Pardridge W.M. Antibody-mediated targeting of siRNA via the human insulin receptor using avidin-biotin technology. Mol. Pharm. 2009;6:747–751. doi: 10.1021/mp800194y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dassie J.P., Liu X.Y., Thomas G.S., Whitaker R.M., Thiel K.W., Stockdale K.R., Meyerholz D.K., McCaffrey A.P., McNamara J.O., 2nd, Giangrande P.H. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat. Biotechnol. 2009;27:839–849. doi: 10.1038/nbt.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Esposito C.L., Nuzzo S., Catuogno S., Romano S., de Nigris F., de Franciscis V. STAT3 Gene Silencing by Aptamer-siRNA Chimera as Selective Therapeutic for Glioblastoma. Mol. Ther. Nucleic Acids. 2018;10:398–411. doi: 10.1016/j.omtn.2017.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacobs S.A., Diem M.D., Luo J., Teplyakov A., Obmolova G., Malia T., Gilliland G.L., O’Neil K.T. Design of novel FN3 domains with high stability by a consensus sequence approach. Protein Eng. Des. Sel. 2012;25:107–117. doi: 10.1093/protein/gzr064. [DOI] [PubMed] [Google Scholar]
- 15.Diem M.D., Hyun L., Yi F., Hippensteel R., Kuhar E., Lowenstein C., Swift E.J., O’Neil K.T., Jacobs S.A. Selection of high-affinity Centyrin FN3 domains from a simple library diversified at a combination of strand and loop positions. Protein Eng. Des. Sel. 2014;27:419–429. doi: 10.1093/protein/gzu016. [DOI] [PubMed] [Google Scholar]
- 16.Goldberg S.D., Cardoso R.M., Lin T., Spinka-Doms T., Klein D., Jacobs S.A., Dudkin V., Gilliland G., O’Neil K.T. Engineering a targeted delivery platform using Centyrins. Protein Eng. Des. Sel. 2016;29:563–572. doi: 10.1093/protein/gzw054. [DOI] [PubMed] [Google Scholar]
- 17.Jacobs S.A., Gibbs A.C., Conk M., Yi F., Maguire D., Kane C., O’Neil K.T. Fusion to a highly stable consensus albumin binding domain allows for tunable pharmacokinetics. Protein Eng. Des. Sel. 2015;28:385–393. doi: 10.1093/protein/gzv040. [DOI] [PubMed] [Google Scholar]
- 18.Merlot A.M., Kalinowski D.S., Richardson D.R. Unraveling the mysteries of serum albumin-more than just a serum protein. Front. Physiol. 2014;5:299. doi: 10.3389/fphys.2014.00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Popp M.W., Antos J.M., Grotenbreg G.M., Spooner E., Ploegh H.L. Sortagging: a versatile method for protein labeling. Nat. Chem. Biol. 2007;3:707–708. doi: 10.1038/nchembio.2007.31. [DOI] [PubMed] [Google Scholar]
- 20.Ilyas M., Tomlinson I.P., Rowan A., Pignatelli M., Bodmer W.F. Beta-catenin mutations in cell lines established from human colorectal cancers. Proc. Natl. Acad. Sci. USA. 1997;94:10330–10334. doi: 10.1073/pnas.94.19.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fodde R., Smits R., Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat. Rev. Cancer. 2001;1:55–67. doi: 10.1038/35094067. [DOI] [PubMed] [Google Scholar]
- 22.Sawa M., Masuda M., Yamada T. Targeting the Wnt signaling pathway in colorectal cancer. Expert Opin. Ther. Targets. 2016;20:419–429. doi: 10.1517/14728222.2016.1098619. [DOI] [PubMed] [Google Scholar]
- 23.Kim G., Kurnit K.C., Djordjevic B., Singh C., Munsell M.F., Wang W.L., Lazar A.J., Zhang W., Broaddus R. Nuclear β-catenin localization and mutation of the CTNNB1 gene: a context-dependent association. Mod. Pathol. 2018;31:1553–1559. doi: 10.1038/s41379-018-0080-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mendonça M.C.P., Radaic A., Garcia-Fossa F., da Cruz-Höfling M.A., Vinolo M.A.R., de Jesus M.B. The in vivo toxicological profile of cationic solid lipid nanoparticles. Drug Deliv. Transl. Res. 2020;10:34–42. doi: 10.1007/s13346-019-00657-8. [DOI] [PubMed] [Google Scholar]
- 25.Hattori Y., Nakamura M., Takeuchi N., Tamaki K., Shimizu S., Yoshiike Y., Taguchi M., Ohno H., Ozaki K.I., Onishi H. Effect of cationic lipid in cationic liposomes on siRNA delivery into the lung by intravenous injection of cationic lipoplex. J. Drug Target. 2019;27:217–227. doi: 10.1080/1061186X.2018.1502775. [DOI] [PubMed] [Google Scholar]
- 26.Abrams M.T., Koser M.L., Seitzer J., Williams S.C., DiPietro M.A., Wang W., Shaw A.W., Mao X., Jadhav V., Davide J.P. Evaluation of efficacy, biodistribution, and inflammation for a potent siRNA nanoparticle: effect of dexamethasone co-treatment. Mol. Ther. 2010;18:171–180. doi: 10.1038/mt.2009.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khan T., Weber H., DiMuzio J., Matter A., Dogdas B., Shah T., Thankappan A., Disa J., Jadhav V., Lubbers L. Silencing Myostatin Using Cholesterol-conjugated siRNAs Induces Muscle Growth. Mol. Ther. Nucleic Acids. 2016;5:e342. doi: 10.1038/mtna.2016.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soutschek J., Akinc A., Bramlage B., Charisse K., Constien R., Donoghue M., Elbashir S., Geick A., Hadwiger P., Harborth J. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–178. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 29.Parsons B.D., Schindler A., Evans D.H., Foley E. A direct phenotypic comparison of siRNA pools and multiple individual duplexes in a functional assay. PLoS ONE. 2009;4:e8471. doi: 10.1371/journal.pone.0008471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ting W.C., Chen L.M., Pao J.B., Yang Y.P., You B.J., Chang T.Y., Lan Y.H., Lee H.Z., Bao B.Y. Common genetic variants in Wnt signaling pathway genes as potential prognostic biomarkers for colorectal cancer. PLoS ONE. 2013;8:e56196. doi: 10.1371/journal.pone.0056196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spranger S., Bao R., Gajewski T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–235. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 32.Ganesh S., Shui X., Craig K.P., Park J., Wang W., Brown B.D., Abrams M.T. RNAi-Mediated β-Catenin Inhibition Promotes T Cell Infiltration and Antitumor Activity in Combination with Immune Checkpoint Blockade. Mol. Ther. 2018;26:2567–2579. doi: 10.1016/j.ymthe.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Shea J., Theile C.S., Das R., Babu I.R., Charisse K., Manoharan M., Maier M.A., Zlatev I. An efficient deprotection method for 5′-[O,O-bis(pivaloyloxymethyl)]-(E)-vinylphosphonate containing oligonucleotides. Tetrahedron. 2018;74:6182–6186. [Google Scholar]
- 34.Brinkley M. A brief survey of methods for preparing protein conjugates with dyes, haptens, and cross-linking reagents. Bioconjug. Chem. 1992;3:2–13. doi: 10.1021/bc00013a001. [DOI] [PubMed] [Google Scholar]
- 35.Chen C., Ridzon D.A., Broomer A.J., Zhou Z., Lee D.H., Nguyen J.T., Barbisin M., Xu N.L., Mahuvakar V.R., Andersen M.R. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doronina S.O., Mendelsohn B.A., Bovee T.D., Cerveny C.G., Alley S.C., Meyer D.L., Oflazoglu E., Toki B.E., Sanderson R.J., Zabinski R.F. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: effects of linker technology on efficacy and toxicity. Bioconjug. Chem. 2006;17:114–124. doi: 10.1021/bc0502917. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.