

Abstract
Although prime editors are a powerful tool for genome editing, which can generate various types of mutations such as nucleotide substitutions, insertions, and deletions in the genome without double-strand breaks or donor DNA, the conventional prime editors are still limited to their target scopes because of the PAM preference of the Streptococcus pyogenes Cas9 (spCas9) protein. Here, we describe the engineered prime editors to expand the range of their target sites using various PAM-flexible Cas9 variants. Using the engineered prime editors, we could successfully generate more than 50 types of mutations with up to 51.7% prime-editing activity in HEK293T cells. In addition, we successfully introduced the BRAF V600E mutation, which could not be induced by conventional prime editors. These variants of prime editors will broaden the applicability of CRISPR-based prime editing technologies in biological research.
Keywords: CRISPR-Cas, genome editing, prime editing, PAM variants
Graphical abstract
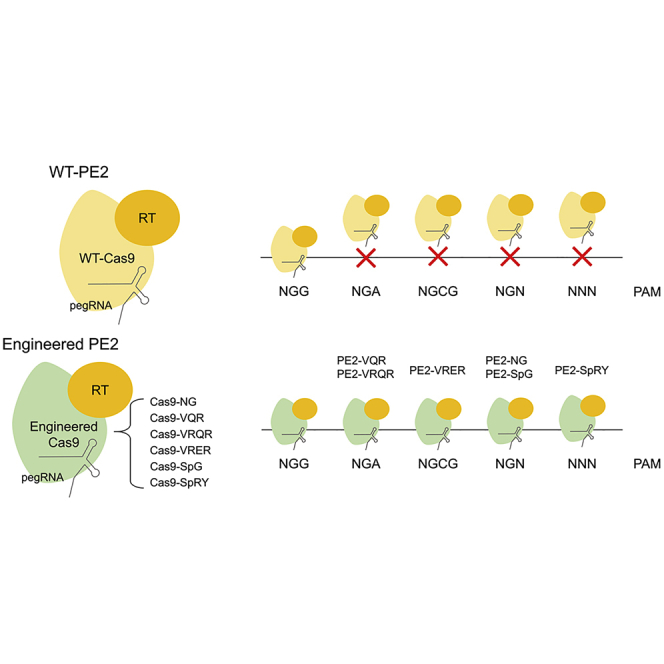
Although prime editors can generate various types of mutations without donor DNA, they are still limited to their target scopes because of the PAM preference of the spCas9 protein. Kim and colleagues engineered the prime editors to expand their target range using various PAM-flexible Cas9 variants.
Introduction
CRISPR-Cas systems have been widely repurposed for targeted genome engineering in living cells and organisms.1 Although Cas nucleases such as Cas9 and Cas12a are widely used for gene knockout and gene correction, they induce DNA double-strand breaks (DSBs),2, 3, 4, 5, 6 which are associated with undesired outcomes such as unwanted genomic mutations7 and activation of p53 in response to DNA damage.8,9 To overcome these limitations, cytosine and adenine base editors have been developed for targeted C:G to T:A and A:T to G:C conversion without DNA DSBs.10, 11, 12, 13, 14 Recently, Anzalone et al.15 developed prime editors (PEs), which can write new genetic information without DSBs or donor DNA. PE2 utilizes an engineered Moloney murine leukemia virus (M-MLV) reverse transcriptase (RT) fused to a Streptococcus pyogenes Cas9 (spCas9)-H840A nickase and prime editing guide RNA (pegRNA) to manipulate the target site. PEs allow the introduction of various types of mutations such as transition, transversion, insertion, and deletion in the various organisms;16,17 however, the NGG protospacer adjacent motif (PAM) preference of spCas9 restricts the targetable loci of PEs in the genome.
Previously, the PAM limitation of spCas9 was overcome by engineering spCas9. Joung et al.18,19 developed the VQR and VRQR variants for NGA PAM recognition and the VRER variant for NGCG PAM recognition by structure-based directed evolution. Hu et al.20 developed the xCas9 variant for NG PAM via phage-assisted continuous evolution. Nishimasu et al.21 used a rationally engineered spCas9 variant (spCas9-NG) for NG PAM recognition. Recently, Walton et al.22 generated the SpG variant, which is capable of recognizing NG PAM and a near PAM-less spCas9 variant (SpRY) by structure-guided mutagenesis. To increase the PAM availability of PEs, we developed PE2 variants using various spCas9 variants and successfully introduced more than 50 mutations in HEK293T cells using the PE2 variants.
Results
Prime editing at NGN PAM sites using PE2 variants
We constructed PE2 variants having a preference for non-canonical PAMs using structure-guided engineered spCas9 variants, including the VQR and VRQR variants for NGA PAM, and the VRER variant for NGCG PAM from Joung et al.,18,19 the NG variant for NG PAM from Nureki et al.,21 and both the SpG variant for NG PAM and SpRY variant for unconstrained PAM preferences from Kleinstiver et al.22 H840A mutations were further introduced in each variant to generate nickases, and PE2 variants were constructed by changing the wild-type spCas9-H840A nickase to these nickases. We designated these variants as PE2-VQR, PE2-VRQR, PE2-VRER, PE2-NG, PE2-SpG, and PE2-SpRY.
To compare the activity of PE2 variants with that of wild-type PE2, we designed pegRNAs, which can target 35 genomic sites bearing NGN PAM. The pegRNAs had the recommended length of primer binding site (PBS) and RT template (13-nt PBS and 11- to 14-nt RT templates) and were designed to introduce three types of mutations (insertions, deletions, and substitutions) at the target genomic sites. We constructed the pegRNAs more efficiently using the single-strand DNA assembly method (Supplemental materials and methods).23 We named the pegRNAs as follows: (1) multiple pegRNAs targeting the same gene were distinguished by a number after a hyphen (e.g., FANCF-1, FANCF-2), and (2) the intended mutation type and position of each pegRNA were listed next to the number (e.g., FANCF-1+5Cins, FANCF-2+5G>C). Using these pegRNAs, we examined the prime editing activity of six PE2 variants and wild-type PE2 in HEK293T cells (Figures 1A–1C and S1) by targeted deep sequencing, and we set the threshold of editing activity to above 0.5%. As expected, wild-type PE2 induced targeted mutations predominantly at the NGG PAM sites (active in 7 of 9 sites, up to 23.8% at the UBE3A-3+5G>C site) with some recognition of the NGA PAM sites (active in 4 of 9 sites, up to 22.9% at the VEGFA-4+1G>C site) and did not induce mutations at the NGC sites (active in 0 of 8 sites) or NGT sites (active in 0 of 9 sites). In contrast, the PE2-NG, PE2-SpG, and PE2-SpRY variants could edit NGN sites (PE2-NG, active in 24 of 34 sites; PE2-SpG, active in 26 of 35 sites; PE2-SpRY, active in 24 of 35 sites). In comparison with other PE2 variants, the PE2-SpG variant showed the highest activity at NGH (where H is A, C, or T) PAM sites (Figure 1C). The PE2-VQR and PE2-VRQR variants were both active at 5 of 7 NGA PAM sites. The PE2-VQR variant showed up to 32.7% activity at the VEGFA-2+5G>T site, and the PE2-VRQR variant exhibited up to 30.2% activity at the VEGFA-1+5G>C site (Figure S2). We also confirmed that the PE2-VRER variant showed a preference for NGCG PAM compared with NGG PAM (up to 30.9% activity at the VEGFA-3+5Gdel site) (Figure S3).
Figure 1.

Prime editing in HEK293T cells by PE2 variants
(A) Representative prime editing activities at the NGA, NGC, NGG, and NGT PAM target sites. (B) Summarized prime editing activities of each PE2 variant at eight target sites with NGG PAM. (C) Summarized prime editing activities of each PE2 variant at 26 target sites with NGH PAM. (D) Representative prime editing activities at a non-NGN PAM site (NAN, NCN, and NTN PAM). (E) Summarized prime editing activities of each PE2 variant at 23 target sites with NHN PAM. (F) Prime editing activities at the HEK4-6+1C>G site with pegRNAs of various lengths of PBS and RT templates. The PAM sequence is shown in parentheses. The numerical values of prime editing activities are listed in Table S1, and information for pegRNAs is described in Table S2. Mean ± SEM of n = 3 or n = 6 independent biological replicates in (A), (D), and (F). Mean ± SEM of the average prime editing frequency for each target site in (B), (C), and (E).
Prime editing at NHN PAM sites using PE2 variants
As the spCas9-SpRY variant can edit the genome in a near-PAMless manner, we investigated whether the PAM specificity of the PE2-SpRY variant was also relaxed. We additionally designed 23 pegRNAs targeting NHN PAM sites, which could introduce three types of mutations (insertions, deletions, and substitutions) at the target sites. The PE2-SpRY variant was active in 18 of 23 NHN PAM sites and induced prime editing up to 9.6% at the CUL3-3 +1C>G site, and other PE2 variants showed lower activity (PE2, active in 2 of 23 sites; PE2-NG, active in 5 of 23 sites; PE2-SpG, active in 4 of 23 sites) across NHN PAM sites (Figures 1D and 1E). Overall, we examined a total of 58 pegRNAs targeting NNN PAM sites and found that the PE2-SpRY variant could manipulate genome in a PAM-independent manner (active in 43 of 58 sites). Collectively, these results showed that previously developed spCas9 variants could be successfully integrated into prime editing toolkits to relax the canonical PAM preferences.
Optimization of the length of PBS and RT templates of pegRNAs
Although we chose target sequences with high levels of indels generated by the spCas9 nuclease, prime editing activity varied from 0.0% to 51.7%, and many sites showed less than 0.5% activity (Table S1). As previous studies described that the prime editing activity of wild-type PE2 is affected by the lengths of the PBS and RT templates, we examined whether the composition of pegRNA could affect the prime editing activity of PE2 variants.
Two pegRNAs (targeting the HEK4-6+1C>G site with NGC PAM and the VEGFA-4+1G>C site with NGA PAM) were selected and reconstituted to have various PBS and RT template lengths. At the HEK4-6+1C>G site, although wild-type PE2 was inactive with all pegRNAs, the PE2-NG, PE2-SpG, and PE2-SpRY variants were active and showed the highest prime editing activity (12.8%, 26.9%, and 21.4%, respectively) by pegRNAs with the 13-nt PBS and 14-nt RT templates (Figure 1F). Longer PBSs were preferred for all three PE2 variants (average activity of 3.29% with a 9-nt PBS and 16.6% with a 13-nt PBS); however, there was no preference in terms of the lengths of the RT templates. These preferences were also observed at the VEGFA-4+1G>C site (Figure S4). As prime editing activity depends on the lengths of the PBS and RT templates, optimization of pegRNAs is essential to maximize the activity of the PE2 variants.
Comparison of the off-target effect of PE variants compared to their nuclease
Previously, compared with Cas9 nuclease, wild-type PE2 has lower off-target effects at already known Cas9 off-target sites.15,24,25 Recently, off-target effects of PE2 were analyzed through the Cas9-H840 nickase-dependent manner, but since there have been no further advances in the technology to analyze PE2-dependent off-target effects, we chose several sites to validate the potential off-target effects of PE2 variants as follows: (1) HEK4+2G>T site and its off-target site, which is known as an off-target site for wild-type PE2; (2) FANCF-4+5Cins, EMX1-4+5G>C, and MECP2-3+1G>C sites and their off-target sites, which are known as off-target editing sites for the SpCas9-SpRY variant; and (3) CUL3-3+1C>G and HEK2-4 +1Cins sites, which showed high activity with the PE2-SpRY variant at NHN PAM sites, and their homologous sites (Table S3). We determined the mutation frequencies by targeted deep sequencing and calculated each specificity ratio, which means the relative mutation frequency of on-target sites compared to that of off-target sites, to compare the off-target effects of nucleases and PE variants (Figures 2A and 2B; Table S4). We found that the PEs including wild-type PE2, PE2-SpG, and PE2-SpRY induced much lower off-target editing compared with their nucleases (spCas9, spCas9-SpG, and spCas9-SpRY). For example, at the EMX1-4 +5G>C site and its off-target site, the PE2-SpG variant showed a specificity ratio above 124, which was 36.8-fold higher than the specificity ratio of the spCas9-SpG nuclease (3.37). The high specificities of the PEs may be attributed to the requirement of additional base pairing with the PBS and RT templates to prime edit the genome.
Figure 2.

Off-target analysis of PE2 variants
(A and B) Six on-target sites and their 10 off-target sites were examined to have mutations with nuclease (spCas9, spCas9-SpG, spCas9-SpRY) (A) and PEs (wild-type PE2, PE2-SpG, and PE2-SpRY) (B) in HEK293T cells (detailed information is listed in Table S3). The specific ratio, which means the relative mutation frequency of on-target sites compared to that of off-target sites, is listed in Table S4. The numerical values of the mutation frequencies are described in Table S1, and information for pegRNAs is described in Table S2. Mean ± SEM of n = 3 independent biological replicates.
Prime editing in PE3 and PE3b systems
Anzalone et al.15 demonstrated an increase in prime editing activity by inducing additional nicks on the non-edited strand, which was named PE3 or PE3b. To examine whether additional nicks affect the prime editing activity of PE2 variants, we constructed additional gRNAs to incorporate PE3, which can induce additional nicks distant from pegRNA-induced nicks, or PE3b, which can induce nicks on the non-edited strand only after the resolution of the edited strand flap. At the HEK2-2+1Gdel site, PE3 improved prime editing activity up to 2.8-fold (6.6%–18.7%), and PE3b improved prime editing activity up to 2.2-fold (6.6%–14.8%) (Figure 3A). At the CUL3-3+1C>G site, PE3 increased prime editing up to 2.9-fold (12.0%–34.4%), and at the HEK4-4+1G>C site, PE3b increased prime editing up to 2.1-fold (5.4%–11.4%) (Figures 3B and 3C). Interestingly, we could not detect any increase in indels at these three sites with the PE2-SpRY variant when using different amounts of gRNAs with PE3 or PE3b (Figure S5), and we speculated that the nicking activity of spCas9-H840A variants may affect the indel frequencies of PE3 or PE3b systems. Overall, we found that PE3 and PE3b could successfully increase the prime editing activity of PE2 variants without excess indel formation.
Figure 3.

Prime editing in HEK293T cells by PE3 and PE3b systems
(A–C) Comparison of prime editing and indel activities at the HEK2-2+1Gdel site (A), at the HEK4-4+1C>G site (B), and at the CUL3-3+1C>G site (C). The PAM sequences are shown in parentheses. The numerical values of the mutation frequencies are described in Table S1, and the information for pegRNAs is described in Table S2. Mean ± SEM of n = 3 independent biological replicates.
Introduction of the BRAF V600E mutation using the PE2-SpRY variant
We performed a direct comparison of wild-type PE2 and PE2 variants for inducing the same mutations. We chose four sites bearing the NHN PAM sequence, which were active with PE2 variants, and constructed additional pegRNAs targeting those sites for wild-type PE2. We compared their prime editing activities in HEK293T cells, and targeted deep sequencing showed the advantage of PE2 variants over wild-type PE2 (Figure S6). For example, at VEGFA-3 target sites, the +5G del mutation was located 15 nt away from the nick position of wild-type PE2 and was shown to be introduced more efficiently by PE2-SpG compared with wild-type PE2 (23.8% and < 0.05%, respectively).
BRAF V600E (which is caused by a T:A to A:T transversion mutation) is a well-known driver mutation in various cancers and diseases and a therapeutic target for anti-cancer drugs.26 Despite the broad ability of wild-type PE2 to introduce various pathogenic mutations, the distance between the pegRNA nick site and BRAF V600E target locus is 18 nt, which could not be efficiently targeted with wild-type PE2 (Figure 4A). To compare the BRAF V600E targeting efficiency of the wild-type PE2 and PE2-SpRY variant, we constructed pegRNAs with single-base resolution around the BRAF V600E target locus and examined their prime editing activity in HEK293T cells (Figure 4B). We found that PE2-SpRY improved prime editing activity up to 6.7-fold compared with the activity of wild-type PE2 (0.3%–1.8%); however, there was no significant increase in prime editing activity by the optimization of the lengths of PBS and RT templates (Figure S7). Then, we chose two active pegRNAs, BRAF-GAA and BRAF-AAA, and assessed the PE3 and PE3b systems; we found that PE3b significantly improved prime editing activity up to 11.8% with BRAF-AAA pegRNA and BRAF-PE3b-1 gRNA (Figures 4C and S8).
Figure 4.

The BRAF V600E mutation by the PE2-SpRY variant in HEK293T cells
(A) Schematic overview of prime editing for the BRAF V600E mutation. The target thymine nucleotide is highlighted in red, and the closest PAM sequence of wild-type PE2 (TGG) is indicated in blue, which can induce nick 18 nt away from that target thymine nucleotide. The 12 pegRNAs were designed to induce nicks of 1–6 nt in both directions from the target thymine by the PE2-SpRY variant. (B) Prime editing activities of PE2 and the PE2-SpRY variant at the BRAF V600E site. (C) Prime editing activity of the PE2-SpRY variant in PE3 and PE3b systems at the BRAF V600E site. The numerical values of the mutation frequencies are described in Table S1, and the information for pegRNAs is described in Table S2. Mean ± SEM of n = 3 independent biological replicates. (D) Comparison of prime editable pathogenic variants. Among 87,203 variants in the ClinVar database, targetable pathogenic variants using PE3b pegRNAs were analyzed by a computational pipeline, which was developed by Morris et al.27
Discussion
In summary, we generated various types of PE2 variants with PAM flexibility and analyzed the prime editing activity of the variants at more than 50 types of mutations. Compared with other PE2 variants, the PE2-SpG variant may be suitable for targeting the NGH PAM site (p values of 0.011 and 0.025 compared to the PE2-NG and PE2-SpRY variant, respectively). The PE2-SpRY variant induced prime editing without PAM restriction, albeit with reduced activity. In comparison with Cas9 nucleases or base editors, the wild-type PE2 is less dependent on a precisely positioned NGG PAM sequence for targeted genome editing due to flexibility in RT template design; however, as prime editing activities are highly dependent on the composition of pegRNAs, experimental optimization of the pegRNA is required to achieve efficient prime editing. Our PE2 variants could offer more optimal spacer sequences regardless of PAM restriction. Using the PE2-SpRY variant, we successfully introduced the clinically significant BRAF V600E mutation with more than 10% prime editing efficiency. Furthermore, we think that the reversion of the BRAF V600E mutation to the wild-type BRAF sequence could be possible by using the PE2 variant.
Overall, the PE2 variant could enhance prime editing systems by providing more options for spacer sequences and expanding the use of the PE3b strategy with PAM flexibility. The PE2-SpRY variant could offer increased prime editable sites per human pathogenic variant (covering 94.4% of pathogenic variants) with PAM flexibility and could also increase the number of PE3b applicable sites (up to 28.8 sites per pathogenic variant) for improving prime editing efficiency without unwanted mutations (Figures S9 and S10; Table S5). Recent studies are helpful for the design of pegRNAs and analysis of PE-meditated off-target effects.25,28 Our constructs have been shared to Addgene for the research community.
Materials and methods
Plasmid DNA and pegRNAs
To construct PE2 variants, we used pCMV-PE2 (Addgene plasmid #132775) plasmid DNA as vector DNA, and the coding sequences of spCas9 variants were obtained by PCR amplification from NG-ABEmax (NG variant, Addgene plasmid #124163), RTW3520 (VQR variant, Addgene plasmid #139990), RTW3160 (VRER variant, Addgene plasmid #139991), RTW3161 (VRQR variant, Addgene plasmid #139992), RTW4177 (SpG variant, Addgene plasmid #139998), and RTW4830 (SpRY variant, Addgene plasmid #139989). All of the PE2 variants used in this study were available from Addgene. The pegRNAs were cloned into the pU6-pegRNA-GG-acceptor (Addgene plasmid #132777), and the spacer sequences and RT template and PBS sequences are listed in Table S2.
Mammalian cell culture and transfection
HEK293T cells (ATCC CRL-11268) were maintained Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Welgene). Cells were not tested for mycoplasma contamination. HEK293T cells were seeded onto a TC-treated 96-well plate (Corning Life Sciences) at 3 × 104 cells per well density 1 day before transfection. Transfection was conducted using 400 ng of plasmids and 0.6 μL of Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocol at approximately 60% cell confluency. Unless otherwise specified, 300 ng of PE2 and 100 ng of pegRNA-encoding plasmids were used. In the PE3 and PE3b experiments, two different doses of plasmids encoding gRNAs were used: (1) 30 ng of gRNA with 270 ng of PE2 and 100 ng of pegRNA-encoding plasmids; and (2) 100 ng of gRNA with 200 ng of PE2 and 100 ng of pegRNA-encoding plasmids. The transfected cells were incubated at 37°C for 3 days, and genomic DNA was prepared by directed lysis of the cells using the lysis buffer (10 mM Tris-HCl with pH 7.5, 0.05% SDS, 100 μg/mL Proteinase K; QIAGEN). The cell lysate was incubated at 56°C for 30 min, followed by a 99°C 15-min incubation to inactive the Proteinase K. In off-target analysis experiments, 1.5 × 105 HEK293T cells were seeded onto TC-treated 24-well plate (Corning Life Sciences) and 1.5 μg of PE2 variants plasmids and 500 ng of pegRNA plasmids DNA were transfected, and genomic DNA was isolated using the DNeasy Blood & Tissue kit (QIAGEN) according to the manufacturer’s protocol.
Targeted deep sequencing and data analysis
The target sites were amplified by a total of three rounds of PCR and subjected to the Illumina MiniSeq or iSeq 100 sequencing system as previously described.29 Briefly, 3 μL of cell lysate or 1 μL of isolated genomic DNA was subjected to the first round of PCR, and then,1 μL of the first PCR product was used in the second round of PCR. The Illumina TruSeq HT dual index adaptor sequences were attached with index PCR primer pairs using 1 μL of second-round PCR product. The size of the PCR amplicon was confirmed in 2% agarose gel and the amplicons were subjected to 150-bp paired-end sequencing using the Illumina MiniSeq or iSeq 100 sequencing system. The paired-end reads were joined using the fastq-join tool (https://github.com/brwnj/fastq-join). Targeted deep sequencing analysis was performed using MAUND30 (https://github.com/ibs-cge/maund), and all results were confirmed by Cas-Analyzer (http://www.rgenome.net/cas-analyzer/).31 Substitution and indel frequencies were quantified as the percentage of total sequencing reads, and the threshold of editing activity was set to above 0.5%.
Analysis of targetable pathogenic variants in the ClinVar database
All possible pegRNAs for correcting pathogenic variants in the ClinVar database32 (https://ftp.ncbi.nlm.nih.gov/pub/clinvar/) were designed by using the pipeline (https://gitlab.com/sanjanalab/primeediting) developed by Morris et al.27 with modifications. We downloaded all variants in ClinVar (released date: August 10, 2020) and selected the variants with the “pathogenic” identifier. Only single base-pair substitutions and insertions and deletions of 10 base pairs or less were calculated as targeted variants. We designed and counted the number of all possible probes for PE2 and PE3b separately with either NGG, NGN (PAM for PE2-NG and PE2-SpG), or NNN (PAM for PE2-SpRY) for each pathogenic variant. The PBS end site was located at the nick site, 3 nt downstream of the PAM, and the target editing sites were able to be placed in any position of RT template but the last. The lengths of PBS and RT templates were fixed to 13 and 16 nt, respectively. The secondary sgRNAs for PE3b were designed separately and complementary with the edited strand of DNA sequence, which can induce a nick within 3 bp downstream of the target.
Data availability
All data supporting the findings of this study are available in the article, in Supplemental information, or from the corresponding author upon request. The deep sequencing data have been deposited in the NCBI BioProject: PRJNA675696. All plasmids used in this study are available through Addgene.
Acknowledgments
This work was supported by the National Research Foundation of Korea (grants 2020R1F1A1075508, 2017M3A9B4062419, and 2018R1A5A2020732) and the Ministry of Trade, Industry & Energy, Korea (20012445)
Author contributions
Y.K. supervised the research. J.K., A.-H.J., H.R.S., J.-E.S., and G.J. carried out experiments. J.K. and Y.K. performed data analysis. J.-K.Y. analyzed the ClinVar database. J.-K.Y. and J.-I.K. provided conceptual advice. Y.K. and J.K. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.02.022.
Supplemental information
References
- 1.Anzalone A.V., Koblan L.W., Liu D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020;38:824–844. doi: 10.1038/s41587-020-0561-9. [DOI] [PubMed] [Google Scholar]
- 2.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho S.W., Kim S., Kim J.M., Kim J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- 5.Hwang W.Y., Fu Y., Reyon D., Maeder M.L., Tsai S.Q., Sander J.D., Peterson R.T., Yeh J.R., Joung J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., van der Oost J., Regev A. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018;24:927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 9.Ihry R.J., Worringer K.A., Salick M.R., Frias E., Ho D., Theriault K., Kommineni S., Chen J., Sondey M., Ye C. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018;24:939–946. doi: 10.1038/s41591-018-0050-6. [DOI] [PubMed] [Google Scholar]
- 10.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishida K., Arazoe T., Yachie N., Banno S., Kakimoto M., Tabata M., Mochizuki M., Miyabe A., Araki M., Hara K.Y. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science. 2016;353:aaf9729. doi: 10.1126/science.aaf8729. [DOI] [PubMed] [Google Scholar]
- 12.Hess G.T., Frésard L., Han K., Lee C.H., Li A., Cimprich K.A., Montgomery S.B., Bassik M.C. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat. Methods. 2016;13:1036–1042. doi: 10.1038/nmeth.4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma Y., Zhang J., Yin W., Zhang Z., Song Y., Chang X. Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat. Methods. 2016;13:1029–1035. doi: 10.1038/nmeth.4027. [DOI] [PubMed] [Google Scholar]
- 14.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A⋅T to G⋅C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin Q., Zong Y., Xue C., Wang S., Jin S., Zhu Z., Wang Y., Anzalone A.V., Raguram A., Doman J.L. Prime genome editing in rice and wheat. Nat. Biotechnol. 2020;38:582–585. doi: 10.1038/s41587-020-0455-x. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y., Li X., He S., Huang S., Li C., Chen Y., Liu Z., Huang X., Wang X. Efficient generation of mouse models with the prime editing system. Cell Discov. 2020;6:27. doi: 10.1038/s41421-020-0165-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kleinstiver B.P., Prew M.S., Tsai S.Q., Topkar V.V., Nguyen N.T., Zheng Z., Gonzales A.P., Li Z., Peterson R.T., Yeh J.R. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481–485. doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z., Joung J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu J.H., Miller S.M., Geurts M.H., Tang W., Chen L., Sun N., Zeina C.M., Gao X., Rees H.A., Lin Z., Liu D.R. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018;556:57–63. doi: 10.1038/nature26155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishimasu H., Shi X., Ishiguro S., Gao L., Hirano S., Okazaki S., Noda T., Abudayyeh O.O., Gootenberg J.S., Mori H. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science. 2018;361:1259–1262. doi: 10.1126/science.aas9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walton R.T., Christie K.A., Whittaker M.N., Kleinstiver B.P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science. 2020;368:290–296. doi: 10.1126/science.aba8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A., 3rd, Smith H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 24.Schene I.F., Joore I.P., Oka R., Mokry M., van Vugt A.H.M., van Boxtel R., van der Doef H.P.J., van der Laan L.J.W., Verstegen M.M.A., van Hasselt P.M. Prime editing for functional repair in patient-derived disease models. Nat. Commun. 2020;11:5352. doi: 10.1038/s41467-020-19136-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim D.Y., Moon S.B., Ko J.H., Kim Y.S., Kim D. Unbiased investigation of specificities of prime editing systems in human cells. Nucleic Acids Res. 2020;48:10576–10589. doi: 10.1093/nar/gkaa764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karoulia Z., Gavathiotis E., Poulikakos P.I. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer. 2017;17:676–691. doi: 10.1038/nrc.2017.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morris J.A., Rahman J.A., Guo X., Sanjana N.E. Automated design of CRISPR prime editors for thousands of human pathogenic variants. bioRxiv. 2020 doi: 10.1101/2020.05.07.083444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim H.K., Yu G., Park J., Min S., Lee S., Yoon S., Kim H.H. Predicting the efficiency of prime editing guide RNAs in human cells. Nat. Biotechnol. 2021;39:198–206. doi: 10.1038/s41587-020-0677-y. [DOI] [PubMed] [Google Scholar]
- 29.Kweon J., Jang A.H., Kim D.E., Yang J.W., Yoon M., Rim Shin H., Kim J.S., Kim Y. Fusion guide RNAs for orthogonal gene manipulation with Cas9 and Cpf1. Nat. Commun. 2017;8:1723. doi: 10.1038/s41467-017-01650-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim D., Kim D.E., Lee G., Cho S.I., Kim J.S. Genome-wide target specificity of CRISPR RNA-guided adenine base editors. Nat. Biotechnol. 2019;37:430–435. doi: 10.1038/s41587-019-0050-1. [DOI] [PubMed] [Google Scholar]
- 31.Park J., Lim K., Kim J.S., Bae S. Cas-analyzer: an online tool for assessing genome editing results using NGS data. Bioinformatics. 2017;33:286–288. doi: 10.1093/bioinformatics/btw561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Landrum M.J., Chitipiralla S., Brown G.R., Chen C., Gu B., Hart J., Hoffman D., Jang W., Kaur K., Liu C. ClinVar: improvements to accessing data. Nucleic Acids Res. 2020;48(D1):D835–D844. doi: 10.1093/nar/gkz972. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available in the article, in Supplemental information, or from the corresponding author upon request. The deep sequencing data have been deposited in the NCBI BioProject: PRJNA675696. All plasmids used in this study are available through Addgene.