

Abstract
As the world energy crisis remains a long-term challenge, development and access to renewable energy sources are crucial for a sustainable modern society. Electrochemical energy conversion devices are a promising option for green energy supply, although the challenge associated with electrocatalysis have caused increasing complexity in the materials and systems, demanding further research and insights. In this field, scanning probe microscopy (SPM) represents a specific source of knowledge and understanding. Thus, our aim is to present recent findings on electrocatalysts for electrolysers and fuel cells, acquired mainly through scanning electrochemical microscopy (SECM) and other related scanning probe techniques. This review begins with an introduction to the principles of several SPM techniques and then proceeds to the research done on various energy-related reactions, by emphasizing the progress on non-noble electrocatalytic materials.
Investigation of electrocatalytic materials with scanning probe techniques (SECM, SICM, SECCM and AFM) for energy storage and conversion devices.
Introduction
In a time when our planet is suffering from environmental disasters caused by fossil fuel pollution, which put at risk the health of humankind,1,2 it is of primary importance to have renewable energy accessible worldwide.3 Among other environmentally friendly energy providers,4 fuel cells are green-energy conversion devices that convert chemical energy to electrical energy by oxidizing a fuel in the anode and reducing another chemical species at the cathode.5 Some of the fuels that can be utilized are formic acid,6 hydrogen sulfide,7 hydrazine,8 ethanol,9 methanol,10 ammonia11 and hydrogen.12 The latter is considered as one of the most unique fuels:13–16 besides several methodologies for hydrogen generation,17 an electrolyser is a device where H2 can be produced from water at the cathode, while O2 evolves at the anode.18 In a regenerative fuel cell, H2 and O2 are generated while the device functions in the electrolytic mode, and then these products are fed to the fuel cell in order to produce electricity by operating in the galvanic mode (Fig. 1).19
Fig. 1. Fuel cell and electrolyser reaction scheme in acidic media.

However, all these reactions require electrocatalysts that are based on noble metals, such as Pt-based materials for the oxygen reduction reaction (ORR), hydrogen oxidation reaction (HOR) and hydrogen evolution reaction (HER),20,21 along with IrO2 and RuO2 for the oxygen evolution reaction (OER) in acidic media electrolysers.22,23 This is one of the factors leading to a stagnation in fuel cell commercialization,24,25 which is motivating researchers to seek methods to study these reactions and alternative non-scarce and simpler electrocatalysts.26,27
Here, we review the work that has been done in recent 5 years by employing scanning probe electrochemical microscopy for studying electrocatalysts, with an emphasis on SECM as the most used method in this field within the scanning probe techniques. Initially, the working principle of the methods will be presented. Then, some recent studies of noble-metal-based electrocatalysts for the ORR, OER and HER are mentioned; however we emphasize the major trend of the field, i.e. the developments in non-noble materials and the experimental conditions used. Finally, some emerging SECM applications in electrocatalysis, namely in the hydrogen oxidation reaction (HOR) and carbon dioxide reduction reaction (CO2RR), will be presented briefly. To the best of our knowledge, similar treatment of this topic is not present in the literature since the reviews of Bertoncello28 and Polcari29 published in 2010 and 2016 respectively. Until now, a wide range of novel electroactive materials and ideas have emerged and will be discussed herein.
Rotating disk electrode (RDE) and rotating ring disk electrode (RRDE)
The investigation of electrocatalysts is mostly done through RDE and RRDE methods, based on a three-electrode cell where the working electrode (WE) is typically a disk glassy carbon electrode surrounded by an insulating material. The RRDE WE has an additional ring electrode (generally platinum) separated by GC through the insulating material.30 This ring electrode serves for the detection of intermediates that may be produced by the sample (often H2O2, detailed more in the ORR section) and works independently from GC.31 The working electrodes are subjected to rotations at certain speeds (convection). The higher the convection, the thinner the diffusion layer close to the electrode, leading to a steady state current.32 The diffusion within the layer aids the transport of the reactant to the catalyst.32 Hence, this apparatus is a convenient means for the quantitative study of electrode kinetics.
Scanning probe microscopy (SPM)
SPM techniques are a family of instruments which consist in the displacement of a probe in the vicinity of a substrate, thus providing spatially localized information of different nature, depending on the type of interaction between the probe and the sample. The possibility to use different sizes and materials of probes expands their application possibility in materials science, electrochemistry, biology, biochemistry and medicine. Besides probing a material, SPM can also be helpful for material alteration at the nanoscale, such as the usage of an AFM cantilever probe as a means for nanolithography.33 Here we will review the principles and application of some electrochemical scanning probe techniques such as SECM, SECM-AFM, SECM-SICM and SECCM in the study of electrocatalysts for fuel cells and electrolyzers.
Scanning electrochemical microscopy (SECM)
As an alternative to RDE and RRDE methods, scanning electrochemical microscopy is a pure diffusion-based technique and a type of scanning probe microscopy which enables the analysis of redox processes in samples by determining their spatially resolved chemical, electrochemical, and/or topographic specificities. This can be acquired by placing an ultra-microelectrode (UME, typically ≤25 μm) at a certain distance from the sample of interest and moving it across the sample (x, y direction), by keeping the UME still (fixed x, y, z), or by moving the UME only in the z direction.34 The UME is placed along with the sample, counter and reference electrode in a cell and controlled by using a potentiostat (Fig. 2). Electrochemical information is obtained from the (electro)chemical reactions occurring either at the UME, at the sample, or both. The tip-to-sample distance d and tip size have great impact on the spatial resolution.35 The d can be established by obtaining approach curves,36 which can then be compared with theory.37,38
Fig. 2. Illustration of the scanning electrochemical microscope set-up. Reproduced from ref. 39 with permission from the Royal Society of Chemistry, © 2015.

The microelectrodes commonly used in SECM display different phenomena from macroelectrodes typically used by the RDE method. While in flat macroelectrodes the diffusion of species occurs in a perpendicular manner, in microelectrodes hemispherical diffusion around the electrode occurs, promoting higher mass transfer and limiting current densities40 owing to the geometry (which can be diverse41) and the small size of the UME. In other words, smaller dimensions of the electrode permit a better sensitivity in measurements, and the absence of convection in SECM investigations simplifies the interpretation of results compared to hydrodynamic methods. A derived equation from Fick's law which expresses the diffusion flux of species is shown in eqn (1),42 where i represents the current intensity, n the transferred electrons, F the Faraday constant, A the electrode area, D the diffusion coefficient, t is the time, r0 the radius of UME and the last expression (4NFDC*r0) represents iss which is the steady state current intensity.
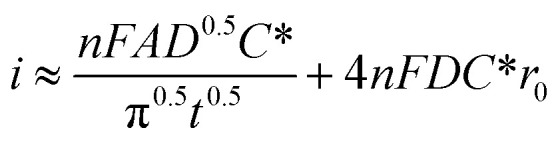 |
1 |
As the spatial resolution in SECM depends on the size of the electrode, the usage of nano-sized ones permits electrochemical studies in single nano-objects.43,44 This offers several opportunities; for example it allows the distinction of activity between different nano-objects in the same catalyst, which can lead to important indications for appropriate design of electroactive materials.45 Besides, the possibility to perform experiments with small gaps between the catalyst and the tip enables the detection of specifically short-lived intermediates (i.e. CO2˙−) that can be produced by the sample, paving the way to a better understanding of reaction mechanisms.46 Localized catalyst fabrication has been performed with SECM,47 along with the assessment of charge density,48 dissolution49 and diffusion50 of species. SECM lately was used even to determine the location where the OER occurs within the sample51 or in the formation of a solid electrolyte interphase (SEI) in lithium ion batteries.52 SECM has also found utility in H2O2 detection as the intermediate during the ORR and has some advantages compared to the traditional macroscopic method, RRDE. By using the latter, the amount of H2O2 detected at the ring versus the one really produced by the sample is ambiguous, since the amount of H2O2 close to the disk electrode may vary with the rotation of the electrode.53 Furthermore, catalyst layers studied with this method are often quite thick, in which case H2O2 can decompose within the layer before getting in contact with the ring, leading to lower amounts of intermediates detected.54 Aside from intermediates, the SECM detection of gases such as oxygen in OER investigations is rigorous. Even though generally the gas-evolving reactions are quite utile in industry, their in-depth electrochemical study with traditional methods can be a challenge considering that there is formation of bubbles, which may often block the electrodes and prevent the accurate measurement of current. The lower current densities under SECM conditions make the appearance of bubbles less of a problem. Moreover, determining the correct onset potential with a RDE may be questionable, since it can include other oxidation reactions that may occur in the catalyst.55 This can be rectified if the sample is studied by SECM, through measuring the anodic current generated at the electrocatalyst and the cathodic current of the UME, in which the reduction of the previously evolved O2 occurs.55,56 Furthermore, for the HER SECM is a convenient way to evaluate the faradaic efficiency of the system. The capability of SECM to map the electrocatalytic activity of a catalyst layer is of great importance, considering that the layers are often not homogeneous. The possibility to perform conductivity measurements57 with SECM is also an advantage, as it can ease the interpretation of the electrocatalytic activity results. Evidently, there are numerous appealing features of SECM which benefit electrocatalytic studies.
However, challenges associated with this technique are noteworthy as well, one of the most important ones being the separate measurement of topography and electrocatalytic activity, in other words the assessment of net electrochemical activity without topographic influences. It has been shown that theoretically these influences cannot be entirely removed.58 Nevertheless, many successful attempts were made for solving this issue by instrumental manipulations, such as the development of shear-force SECM (SF-SECM),59 intermittent-contact SECM (IC-SECM),60 hopping-intermittent-contact SECM (HIC-SECM),61 alternating-current SECM (AC-SECM),62 or the combination of SECM with SICM or AFM. When working with conventional SECM with a micro-scale d and tip, the topographic influences become somewhat less important compared to nanoscale measurements. As in any other SPM technique, SECM spatial resolution is mainly governed by the size of the probe used in the experiment, and the most straightforward way to suppress convolution (although sometimes experimentally challenging) is to decrease the size of the probe: moreover, maintaining a constant d is not possible in conventional SECM, limiting the usage of samples to very flat and aligned ones.63 Another drawback is the fact that the sample and tip have to be immersed in solution during SECM experiments, possibly for hours, which can lead to surface changes in the sample and tip.
Since the introduction of SECM by Bard et al. in 1989,64 a great deal of effort has been made by researchers to improve it and to expand its application possibilities. To name a few, an interesting approach for investigating the reactivity of ion transfer in liquid/membrane and liquid/liquid interfaces by means of SECM was introduced by Shao and Mirkin in 1998.65 Moreover, the first simultaneous measurements of topography and electrochemistry in a fluid were acquired by Macpherson and Unwin by combining AFM and SECM.66 Nanoscale measurements were performed as well, for which a detailed guide for nano-electrode fabrication with radii as small as 10 nm was presented by Katemann and Schuhmann.67 Furthermore, the widely used redox competition mode was developed by Eckhard et al.68 Owing to such advancements in the technique, nowadays the utilization of SECM enables researchers to obtain very useful information on a variety of materials for numerous applications.44,69–71
SECM modes
Depending on the purpose of experiment, several modes can be employed by SECM and they are reviewed in more detail by several groups, such as Bard et al.64 and Polcari et al.29 Herein, a rather brief presentation of the modes applied in electrocatalysis will be presented and illustrated (Fig. 3–5). Their application for the respecting reactions will be detailed in the rest of the review.
Fig. 3. Illustration of positive and negative feedback approach curves.

Fig. 4. Schematic representation of the main SECM modes. Substrate generation/tip collection (SG/TC), tip generation/substrate collection (TC/SC), redox competition (RC), and positive and negative feedback modes.

Fig. 5. Schematics of the proposed mechanism for the surface interrogation. (A) A reactive species is chemically or electrochemically adsorbed on the substrate upon a potential scan or step while the tip is at open circuit. (B) The substrate is put to open circuit, and the tip generates the titrant, which reacts at the surface of the substrate to support positive feedback at the same tip. (C) Upon consumption of the adsorbate at the substrate, the tip experiences negative feedback. (D) Expected current response at the tip following the events depicted in panels A–C for an arbitrary electrode setup. Reproduced from ref. 74 with permission from the American Chemical Society © 2008.

Feedback mode
Feedback mode is one of the most used ones in the field, described initially along with SECM itself in 1989. Let us consider an oxidized species O as a free mediator present in solution that is reduced to R by a polarized UME. At a large distance from the sample, the tip exhibits a constant diffusion-limited current iss, whose magnitude is proportional to the concentration of O and electrode size. As illustrated Fig. 4, if the sample is not reactive (an insulator) and the tip approaches it, the hemispherical diffusion will be disturbed and thus the diffusion of O to the tip will be hindered. This will lead to a decrease in the diffusion-limited tip current, known as the negative feedback effect.42 In the case of positive feedback, an electrochemically active sample (conductor) is approached by the tip. The substrate can thus oxidize the R formed at the tip back to O, thus increasing the concentration of O in the diffusion zone and increasing the tip current.42 The current vs. d curves obtained while the tip approaches a conductive or insulating substrate are called approach curves (Fig. 3). The concentration and diffusion coefficient of O do not influence the approach curve, as it includes dimensionless variables solely.34 The degree of current change is dictated by the activity of the sample, enabling the distinction between more and less active spots42 in the same substrate and the determination of d and the electrode radius a. The rg parameter (insulating part of the UME) has an influence on negative feedback curves as it hampers the diffusion.34 Through feedback mode one measures the current only at the tip, thus it is convenient to use in cases when the substrate cannot be polarized.
Generation/collection (GC) modes
While in feedback mode the redox mediator is already present in solution, which leads to a faradaic background current in the tip,42 in the generation/collection modes the mediator is produced at one of the electrodes. The generation of redox species at the sample, after which they diffuse to the tip and undergo electrochemical reactions, is known as sample generation/tip collection (SG/TC) mode.72 In this case, the current of the tip will provide information about the electrochemical activity of the sample. Conversely, the production of species at the tip and their diffusion in the sample to get oxidized or reduced is known as tip generation/sample collection (TG/SC) mode.73 Herewith, the sample current holds information about the sample local electrochemical activity at the tip location.42
Redox competition (RC) mode
In the Redox Competition (RC) mode,68 the sample and the tip undergo the same reaction, and they both compete for the same mediator. When the tip approaches the sample in line scans (x, y direction), due to the extensive consumption of the mediator by the sample as well, there is less mediator detected by the tip; hence there is a decrease in the tip current which is measured.68 The RC mode suppresses the background current issue generally encountered with TG/SC mode.68
Surface interrogation mode (SI-SECM)
The SI mode was introduced by Bard's group back in 2008.74 In contrast to the other modes, it is based on transient measurements. It consists in a titrant generation from a redox mediator through a SECM tip, which then undergoes a chemical reaction with certain species generated at the closely placed substrate. In other words, the generation and collection of the oxidized and reduced mediator species lead to variations in the electrochemical signals, which can be detected by the SECM tip. This leads to a transient positive feedback loop as long as the investigated species is being consumed.74
Scanning ion conductance microscopy (SICM)
This technique, introduced by Hansma et al.,75 is typically based on a bias application between a single-channel nanopipette electrode which is filled with an electrolyte and contains a quasi-reference counter electrode (QRCE) inside and in the bulk solution, in which case the ion flow between these electrodes produces an ion current (I) (Fig. 6).76,77 The flowing ionic current then depends on the nanopipette and electrolyte resistance.77 By detecting the current flow in the nanopipette with an applied bias, SICM can be a powerful tool for mapping spatial distributions of ionic fluxes. Hereby, the modulation of the probe distance or bias can provide information about topography and activity with a single channel probe, simultaneously.78 Besides topography measurements, SICM has also found utility in the determination of ion conductivity.79 Very recently a development has been made, where SICM can be used for generating local electrochemical impedance spectra, in which case the local capacitance and topography can be determined separately in one measurement.80 It's worth mentioning that the possibility of non-contact imaging with SICM allows the investigation of fragile samples that otherwise are investigated with difficulty by AFM for example.76 This being said, SICM has been especially useful in studying living systems, their morphology, physiological activity and subcellular structures.81 The temporal resolution once considered as a challenge has been improved by modulating its mechanical and software elements.82 SICM application has expanded to electrocatalysis as well thanks to its combination with SECM (SICM-SECM). In this way, one can take advantage of the possibility to record faradaic signals with SECM while at the same time avoiding its inferiority in topographic measurements.77 In contrast to conventional SECM, the d in SICM can be controlled,77 although it is only relatively specialized.83 Depending on the application, several SICM modes are possible and they are reviewed carefully elsewhere.76,84
Fig. 6. SICM set-up scheme. Reproduced from ref. 78 with permission from the American Chemical Society © 2016.

Scanning electrochemical cell microscopy (SECCM)
Introduced in 2010 by Ebejer et al.,85 SECCM is a technique based on an electrolyte-filled, dual or single-barred pipette, with a QRCE in every channel (Fig. 7).63 A droplet is formed at the end of the pipette when it is in contact with the sample. Once a potential is applied between the electrodes, ion and electron transfer occurs through the droplet meniscus. This results in modulations of conductance current (idc) which serves as a feedback signal for probe positioning. The pipette moves laterally across the surface at a certain d or in hopping mode, during which the current signals are recorded.63 This technique enables the imaging of electrochemistry, conductivity and topography of samples, simultaneously.86 The fact that only a small part of the sample is in contact with the solution very briefly makes SECCM an encouraging method for investigating samples sensitive to corrosion, passivation or surface fouling.76 SECCM has been used for performing microscale voltametric measurements, correlating the properties of specific parts of the substrate with the electron-transfer kinetics.87 Moreover, it has found utility in the fabrication of polymer nanostructures,88 in assessing localized capacitance,89 electrochemical activity,90 and ion transfer91 and even in nanoparticle landing experiments.92 One of the important advantages of this method is the reduced time needed for acquiring the images.93 SECCM is up-and-coming in the electrocatalysis area, as reactive sites can be detected explicitly through local electrochemical and structural analysis. Being introduced recently (in 2010), there is still room for SECCM development compared to analogue techniques.83 Many features of this method are explained thoroughly elsewhere.63,94
Fig. 7. SECCM schematic set-up for voltammetry and amperometry. (a) dual barrel, (b) single barrel probes (micro- or nano-pipets) filled with an electrolyte and QRCE(s) used as SECCM scanning probes; schematic of probe movement at a (c) constant tip distance from the surface, and (d) hopping scanning regime over the sample. Inset in (d) shows the E–t linear sweep voltammogram (LSV) and the corresponding current recorded each time the meniscus lands. Reproduced from ref. 95 with permission from Elsevier © 2020.

Atomic force microscopy (AFM)
AFM, introduced in 1986, is a scanning probe technique which measures attractive or repulsive interactions between a sharp tip on a flexible cantilever and a sample under investigation. After the cantilever approaches the surface, it gets deflected by the interactions with it in accordance with Hooke's law (Fig. 8).96 This deflection is measured thanks to a laser beam and a photodiode and holds information about the sample,96 while the sample or the tip are moved by a piezo scanner. There are certain possibilities of AFM operation, such as in contact, noncontact, static or dynamic mode for which a comprehensive introduction was made by Haugstad.97 This high resolution technique made it possible to image non-conducting samples in open air, something which was not achievable back then by its analogue scanning tunneling microscopy (STM).98–100 AFM is mostly used for assessing the surface topography, morphology, and roughness. Its application possibilities are excessive, to name a few: molecule generation, manipulation and characterization,101 study of the mechanical properties of cells,99 bubble–particle interactions,102 nanofabrication,103etc. AFM has been beneficial in electrocatalysis as well, for example in measuring the operando electrochemical potential of electrocatalysts thanks to the use of a conducting tip.100 When AFM features are combined with the ones of SECM (AFM-SECM), their features merge and allow topographical and electrochemical measurements with high resolution and with precise control of the probe tip position. AFM-SECM image resolution is competitive to that of SICM-SECM, although the probe of the former combination is costly and has a limited reliability and durability.83
Fig. 8. Scheme of the imaging mode in AFM. Reproduced from ref. 108 with permission from MDPI © 2017.

It's worth mentioning other SPM techniques with similar principles to AFM, such as Kelvin probe force microscopy (KPFM) and electrostatic force microscopy (EFM), which unlike AFM, are based on long range interactions and operate always in non-contact mode.104 These methods give access to work function (WF) and local charge phenomena,105 nowadays even with a nanometric resolution.106 Moreover, electrochemical and ionic phenomena can be determined by electrochemical strain microscopy (ESM), a technique whose working principle consists in detecting changes in electrochemical strains of the sample.107
Electrochemical reactions
The more extensively investigated electrocatalytic reactions are the ORR, OER and HER, which will be discussed in more detail herein. Noble electrocatalytic materials have been studied for these reactions, often as models to illustrate the potentiality of the technique. Even though such studies will be mentioned, this review focuses on discussing the findings on non-precious electrocatalysts. Although more limited, interesting SECM research has been reported on the CO2RR and HOR as well and they will be presented briefly at the end of this review.
Oxygen reduction reaction (ORR)
The selective and efficient reduction of oxygen has crucial importance for the proper functionality of fuel cells. O2 can be reduced either directly to water through a 4-electron transfer, or indirectly through a 2-electron transfer forming hydrogen peroxide as an intermediate. The reactions of both pathways in acidic and alkaline media are presented below as (2)–(7).109 The detailed mechanism of the ORR is rather convoluted and it depends on the electrocatalyst; however some proposed mechanisms are summarized by Nie et al.110
Acidic media
| Direct pathway: O2 + 4H++ 4e− → 2H2O | 2 |
| Indirect pathway: O2 + 2H+ + 2e− → H2O2 | 3 |
| H2O2 + 2H+ + 2e− → 2H2O | 4 |
Alkaline media
| Direct pathway: O2 + 2H2O + 4e− → 4OH− | 5 |
| Indirect pathway: O2 + H2O +2e− → HO2− + OH− | 6 |
| HO2− + H2O + 2e− → 3OH− | 7 |
With a thin layer of the catalyst, the calculation of the number of transferred electrons per O2 molecule namely n, through the RDE method, is typically made by using the Koutecky–Levich (K–L) equation (eqn (8)), where j = current density, jL = current density limited by diffusion, jK = kinetic current density, and ω = angular velocity and B is expressed as eqn (9), where F (Faraday constant) = 96 485 C mol−1, CO2 = bulk concentration of oxygen, DO2 = diffusion coefficient and v = kinematic viscosity of the electrolyte.111
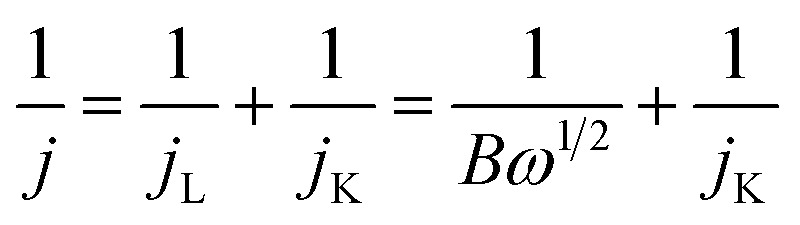 |
8 |
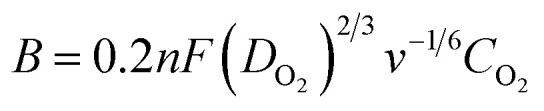 |
9 |
The indirect pathway would be troublesome for our purpose since the production of H2O2 lowers the overall efficiency. Moreover, it may degrade the membrane of the fuel cell112 or decompose the frequently used Nafion.113 Nevertheless, the quantification of the intermediate produced during the ORR aids in the determination of electrocatalyst selectivity. This can be done with the RRDE method, by evaluating the percentage of hydrogen peroxide through eqn (10) in which id is the current of the disk electrode, ir the current of the Pt ring and N the collection efficiency of the ring.114 This information can be used for evaluating the n as well (eqn (11)), which can be a convenient way to test the reliability of the results, by analyzing the consistency with the n evaluated by using the K–L equation.
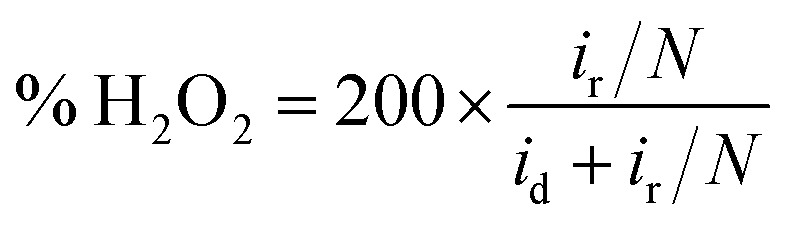 |
10 |
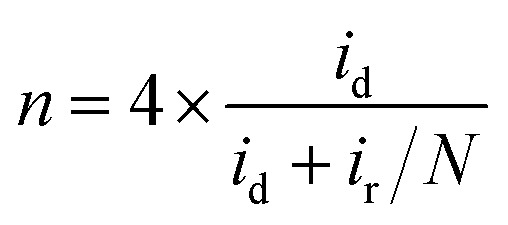 |
11 |
SECM has been very utile for studying the ORR. The tip generation/sample collection (TG/SC) mode is often used for ORR investigation, where the tip generates O2 and the sample collects it, thus measuring the sample current.115 This however leads to a high background current especially on large samples which compromises the sensitivity and resolution, leading to the development of Redox Competition (RC) mode.68 On the other hand, sample generation/tip collection (SG/TC) mode can be used for detecting H2O2 at the tip, while it is produced in the sample during the ORR.116 A simultaneous combination of these three modes was introduced by Eckhard and Schuhmann,117 enabling the determination of the activity and selectivity of electrocatalysts.
One of the main factors that predict the suitability of an electrocatalyst towards the ORR is the binding energy of O2 with the catalyst, which is often illustrated by volcano plots.118 The bond has to be strong enough for the oxygen to bind to the catalyst, but not too strong so it prohibits the removal of the intermediates.118 This kind of in-between bond strength is what puts noble metal electrocatalysts on top of the volcano curve, which makes them very suitable candidates for the ORR.119–125 However, these state-of-the art electrocatalysts are more costly; thus researchers have been developing various combinations to decrease the noble-metal loading, thus presumably decreasing the cost. For instance, Sun et al.126 have prepared alloys such as PdW nanoparticles supported on nitrogen and sulfur doped graphene (NSG) and demonstrated through RC-SECM a high electrocatalytic activity for the ORR. On the other hand, Pham Truong et al.127 have hosted the Pt catalyst onto a polymer brush ionic liquid (poly(IL)), which showed a promising electroactivity towards the ORR. Moreover, Kim et al.128 have explored nanowires of silver chloride and bromide (AgClNWs and AgBrNWs respectively) for their oxygen reduction activity in alkaline media, using SG/TC mode and an Au UME (12.7 μm in diameter) at a d of only 5 μm. The substrate used was GC loaded with the samples and compared with a bare Pt substrate. The results for AgClNWs presented an even better activity (higher current) than Pt.128
In recent years, priority has been more and more given to the investigation of non-noble electrocatalysts. For instance, the ORR electrocatalytic activity of cobalt sulfide (CoS2) relative to its morphological evolution was for the first time studied by Singh et al.129 A strong dependence between specifically exposed surfaces and ORR activity was previously found for several metal oxides, possibly because of differences in surface energies.130,131 In this work, octahedral CoS2 crystals with {111} and {220} planes were synthesized. SECM was employed for the visualization of local electrocatalytic activity using RC mode, a GC plate as the substrate, a Pt UME in 0.1 M perchloric acid (HClO4) and a tip-to-substrate distance of 10 μm. SECM results revealed a homogeneous distribution of ORR active sites throughout the catalyst. RDE and RRDE results on the other hand showed a higher ORR activity of {111} facets.
The same group analyzed manganese tungstate (MnWO4) with a bird-feather (BF) like morphology, acquired by optimizing the concentration of a structure directing agent (SDA), in this case hydrated trisodium citrate (TSC). The catalyst spots synthesized with different TSC concentrations were mapped through RC-SECM mode, in which case a good agreement with RDE results was found (Fig. 9). The latter figure shows the high alteration of ORR activity with changes in substrate potential, suggesting no significant contributions from topography. One can also recognize a superior activity for 10 mM TSC concentration, along with the homogeneous distribution of active sites (red area) at all applied substrate potentials. This catalyst exhibited good electrocatalytic activity towards the ORR in alkaline media (2 + 2e− pathway), a result acquired through SG/TC mode of SECM and RRDE methods as well.132
Fig. 9. ORR 3D RC-SECM images of MnWO4–BF synthesized at four TSC concentrations, namely 0, 5, 10 and 15 mM at different substrate potentials: (a) 0.80 V, (b) 0.55 V, (c) 0.25 V and (d) 0.15 V. The tip (Pt UME 10 μm in diameter) was held at 0.55 V at a 10 μm distance from the sample in a 0.1 M NaOH electrolyte, (e) RDE results for the studied TSC concentrations. Counter electrode: Pt coil and reference electrode: Ag/AgCl/3 M KCl (potentials converted to the RHE scale). Reproduced from ref. 132 with permission from the Royal Society of Chemistry © 2018.

The ORR investigation of nickel and cobalt-based oxides, namely NiO, Co3O4 and their combination (NCO) with a structure of NixCo3−xO4, was done by Sidhureddy et al.133 The measurements were conducted in alkaline media, by measuring the intermediate (HO2−) current through SECM in SG/TC mode, using a Pt UME of 10 μm in diameter, a GCE substrate and d = 2.0 μm. The images of current produced by HO2− production are shown in Fig. 10, where one can see that the combination of oxides (NCO-1) exhibits the lowest HO2− current and hence the highest electroactivity towards the ORR, which matches also their results obtained by the RDE method. This may be the outcome of a higher electrical conductivity of the NCO-1 in comparison to non-mixed oxides.134,135
Fig. 10. SECM tip current profile for the intermediate generated at (A) NiO, (B) Co3O4 and (C) NCO-1. Reproduced from ref. 133 with permission from the American Chemical Society © 2019.

Besides metal sulfides and oxides, bare metal structures have been studied as well for their ORR activity. For instance, Michalak et al.136 have examined copper nanostructures (CuNSs) by utilizing SECM in feedback mode with an Au UME of 100 μm in diameter and d = 30 μm. After the oxygen is generated at the Au UME, it diffuses to the sample, where it reduces again and forms OH−, which generates a positive feedback at the UME. The effect of sample topography can be neglected considering the 30 μm distance of the sample from the flat support. The ORR activity was assessed in different electrolytes and it was found that it is elevated in electrolytes with a higher concentration of chloride ions (Fig. 11). The presence of such ions seems to fasten the formation of crystalline CuNSs rather than amorphous and less active structures. Nevertheless, selectivity was not acquired, since the intermediate could not be oxidized (detected) at the Au tip,136 although this has been achieved by the previously mentioned work of Kim et al.128
Fig. 11. ORR images of CuNS microspots deposited on indium doped tin oxide (ITO), analyzed by SECM in feedback mode, at several support potentials (−0.3, −0.4 and −0.5 V) and various electrolytes with a tip potential of +1.6 V vs. Ag/AgCl.136 Reproduced from ref. 136 with permission from the Royal Society of Chemistry, © 2019.

Additionally, copper was studied for its ORR activity by Zhang et al.137 with the purpose of evaluating its corrosion properties. The investigation was carried out with SECM in TG/SC mode with which the amount of H2O2 was determined as well. The substrate used was Pt (62.5 μm in radius) with a determined collection efficiency for H2O2 of 83%. A Cu tip 12.5 μm in radius was utilized at d = 5 μm in 0.1 M sodium perchlorate (NaClO4) solution with two different pHs. The low amount of intermediate produced (20% in neutral and 10% in alkaline solution) suggested that the electron transfer occurred mostly through a four-electron transfer at both pHs.137
Some groups have studied electrocatalysts that are metal-free, such as Tiwari et al.138 who have evaluated the performance of nitrogen-bearing carbon spheres (NCSs) synthesized at 600, 800 and 900 °C (NCS-600, NCS-800 and NCS-900). The application of metal free carbon-based nanomaterials in electrocatalysis has emerged recently, thanks to their good conductivity, high surface area and stability.139 Exclusively the nitrogen-doped ones have the attention since the electron density of the sp2 carbon structure is rearranged, thus activating the reduction of O2.140 In this work, they determined the H2O2 produced during the ORR by the catalyst on a GC plate (working electrode 1, WE1), using a Pt tip 10 μm in diameter (working electrode 2, WE2) above the sample at d = 10 μm, through SG/TC mode of SECM in 1 M potassium hydroxide (NaOH) and a loading of 50 μg cm−2. The H2O2 current appears at the same time with the O2 reduction current and disappears when the reduction is complete. The four-electron pathway ORR was assumed considering that the oxidative current stays constant after it decreases to the baseline. The imaging of local activity was done in the RC mode which revealed a uniform distribution of the active sites throughout the NCS-800 catalyst as the best performing catalyst according to RRDE results. The better activity of NCS-800 (n = 3.8, K–L plots) was attributed to the higher percentage of C–C sp2 carbon in the structure (70%) compared to the other studied materials, which was determined by X-ray photoelectron spectroscopy (XPS) analysis.
A combination of carbon materials and metal-based compounds was made by Xin et al.,141 who have investigated nanostructured hybrids based on MoSe2 and reduced graphene oxide (rGO) nanosheets (MoSe2@rGO) in 0.1 M KOH. MoSe2 is a type of transition metal dichalcogenide (TMD) whose layered nanostructure exhibits a high surface area; however in its two-dimensional (2D) nanosheet form it has a tendency for agglomeration which compromises its capability for ORR activity.142,143 To surpass this drawback and the fact that MoSe2 has a low conductivity,144 they combined three-dimensional (3D) MoSe2 structures with highly conductive graphene-based materials. SECM measurements in RC mode with a Pt tip of 25 μm in diameter, glassy carbon electrode (GCE), d = 50 μm and sample loading of 510 μg cm−2 were performed by line scans in the XY plane and showed that the hybrid had the highest ORR activity compared to MoSe2, rGO and their physical mixture (MoSe2 + rGO), similar to the result acquired from the K–L plots (RDE method).
Similarly, Dobrzeniecka et al.112 have investigated ORR dependence on the loading (70–700 μg cm−2) of multiwalled carbon nanotubes (MWCNTs) and their composite with cobalt(ix) protoporphyrin (MWCNTs/CoP) in a 0.1 M phosphate buffer as the electrolyte. By utilizing a combination of redox competition and generation/collection modes of SECM,117 the authors were able to extract the number of electrons transferred (n), amount of H2O2 produced, and rate constants and then compare them with the results obtained from RDE and RRDE methods. They utilized a 25 μm diameter Pt UME and d = 30 μm at a rigid x–y grid position. The n was quantified based on the data gathered from the reduction of oxygen and oxidation of H2O2 using eqn (12) which is similar to eqn (11), except that here the background current is corrected. The collection efficiency (N) was considered 100%, based on the fact that the ORR and H2O2 currents did not change as a function of d.53
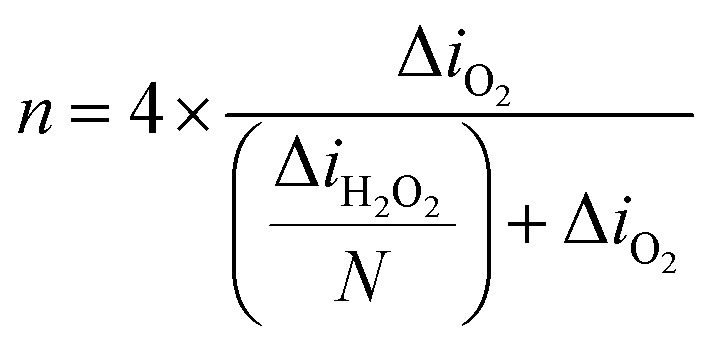 |
12 |
The results acquired from RDE and RRDE methods did not reveal very similar results to RC-SECM.112 For the same thickness layers, the RRDE method revealed a relatively larger value of n compared to the SECM results. This is interpreted by suggesting that in the RRDE method the intermediate may have decomposed or undergone other reactions before it arrived in the Pt-ring for detection.112 On the other hand, in SECM, the tip was positioned in a perpendicular manner only 30 μm above the sample and was more likely to collect a more realistic amount of H2O2 produced. Therefore, the lower H2O2 amount detected through the RRDE method may falsely lead to a higher number of electrons compared to the SECM investigation.112
Likewise, the superior ORR catalytic activity of a composite based on ZnCo2O4 and carbon nanotubes (ZnCo2O4/CNTs) compared to its individual components was demonstrated by Ma et al.145 by means of RC-SECM. A GCE was utilized as the substrate, Pt 25 μm as the UME, d = 40 μm and an alkaline electrolyte (0.1 M KOH). The high ORR activity of the composite was credited to the synergetic effects between Zn2+ and Co3+, and between CNTs and ZnCo2O4.145
The possibility to combine SECM with other methods is one of the reasons why it is such a proficient technique. For example, a simultaneous ORR and H2O2 measurement was made by Kolagatla et al.146 by joining SECM and AFM in one set-up and using SG/TC mode and an AFM-SECM dual electrode tip. The authors performed nanoscale measurements of platinum nanoparticles (Pt NPs) on a GC surface, by using an Au-coated SiO2 Pt tip (collection efficiency ≈ 70%) at only 4–8 nm from the substrate. They determined where activity is detected and found that the pixels corresponding to ORR activity are almost the same as the ones corresponding to the H2O2 generated (Fig. 12). Interestingly, considering the high amount of H2O2 produced by the particles, the authors concluded that the Pt NPs exhibited a two-electron pathway reduction of O2 and only half of the catalyst surface undergoes the ORR.146 This is highly contradictive to most results obtained through the RRDE method and to the fact that Pt materials are generally accepted as the state-of-the-art ORR electrocatalyst. However, such a set-up can be beneficial for investigating electrocatalytic activity relative to the surface morphology of electrocatalysts.
Fig. 12. Electrocatalytic current mapping through AFM-SECM on Pt NPs with an Au-c-Pt tip: (a) topography; (b and c) Z-profiles of the NPs marked ‘1’ and ‘2’ respectively; (d) ORR currents and (e) H2O2 currents of the same NPs in O2-saturated 0.1 M HClO4. Esubstrate = 0.70 V vs. the standard hydrogen electrode (SHE), Etip = 0.98 V vs. SHE. Tip size, ∼100 nm. Active pixel distribution of the (f) ORR and (g) H2O2 of Pt NPs marked as ‘1’ in black, ‘2’ in red and ‘3’ in blue. Reproduced from ref. 146 with permission from John Wiley and Sons © 2019.

Another innovative set-up was applied for ORR investigations by O'Connell et al.147 through combining SECM in SG/TC mode and scanning ion conductance microscopy (SECM-SICM). The authors demonstrated the utility of the technique by mapping the ORR activity in gold nanoparticles (Au NPs) deposited on GC by utilizing a twin-barrelled pipette, in which one barrel serves as a channel of ion conductance and the other is a solid electrode which carries out the faradaic process measurements. RRDE experiments revealed the highest H2O2 amount (67%) at 0.4 V. This is in accordance with what was observed from the SECM-SICM images shown in (Fig. 13A), where the highest peroxide generation is at 0.4 V. It was also observed that particles with a similar size determined by SICM exhibited different peroxide detection signals (Fig. 13B and C) which may indicate possible unlike crystal structures within the ensemble.147
Fig. 13. SECM-SICM images of (A) Au NP ensemble electrodes at several substrate potentials (E converted to the RHE scale in 0.1 M NaOH) where the SECM tip is polarized at 1 V vs. RHE for peroxide oxidation. Magnified area of a high density ensemble with surface biased at 0.3 V vs. RHE is shown in (B) SICM topography and (C) the corresponding peroxide detected with SECM. Rectangle mark represents the cluster of particles discussed in the text and the white line the cross section position for highlighting particles with different peroxide signals.147 Reproduced from ref. 147 with permission from the Royal Society of Chemistry, © 2015.

Besides the influence of heteroatom doping of carbon materials, certain defects in the structure also have been shown to modulate the electronic and catalytic properties, thus influencing the adsorption of intermediates on the catalyst.148 Actually, even non-doped but edge-defective carbon materials have shown promising results for the ORR.149 Nevertheless, studies on charge visualization of active sites have been lacking.
By using SICM as a charge-sensitive technique,150 the interdependence between surface charge and electrocatalytic activity was studied by Tao et al.151 in defective highly oriented pyrolytic graphite (HOPG). SICM data of pristine HOPG were acquired with a 0.5 μm nanopipette–HOPG distance. After the introduction of defects by plasma irradiation, the distance changed from 0.5 to 6.6 μm, indicating an increase in the surface charge of the sample (Fig. 14). ORR performance was better in the defective HOPG, with an onset potential of 0.745 V vs. the reversible hydrogen electrode (RHE) compared to 0.171 V in pristine HOPG in 0.1 M KOH.
Fig. 14. (a and b) Topography 2D/3D image and (c) respective profile perpendicular to the stripe of pristine HOPG. (d and e) 2D/RD topography image and (f) extracted profile perpendicular to the stripe of defective HOPG. The before and after plasma irradiation images (pristine and defective HOPG, respectively) correspond to the same sample region. Reproduced from ref. 151 with permission from John Wiley and Sons © 2019.

Oxygen evolution reaction (OER)
While the oxygen evolution reaction in acidic media is presented in Fig. 1, the reaction in alkaline media is expressed by eqn (13).152 The OER occurs via a 4-electron transfer, preferably through single-electron transfers at each of multi-step reactions through which it takes place.152 Several studies have been dedicated to unraveling the mechanisms of the OER in acidic and alkaline media in a variety of materials.153–157
| 4OH− → 2H2O + O2 + 4e− | 13 |
Lithium intercalated nickel phosphorus trisulfide (NiPS3) nanosheets of less than 1 nm in thickness were investigated in alkaline media for their OER onset potential by Konkena et al.,158 using SECM in SG/TC mode with a 800 nm diameter nanoelectrode and d = 4 μm. The onset potential has a value of 1.48 V vs. RHE where there is an abrupt rise in current, a value which corresponds well to the ones present in the literature at that time.159,160 The metallic-edge sites and defects available in the catalyst structure were associated with the promising OER activity, along with the high conductivity of NiPS3.158
An interesting and rapid approach for studying the OER in individual catalyst spots by SECM in SG/TC mode was introduced by Minguzzi et al.161 In order to hamper the expansion of the diffusion layer from the O2 produced, potential steps were applied with values that correspond to the OER activity and non-activity of the sample. Digital simulation was used in order to establish the conditions for preventing the overlap of diffusion layers that are developed at each spot.161 Then, using this approach, the authors studied the OER activity of a mixture containing SnO2–IrO2. It was found that the OER activity was higher with an increase in the percentage of IrO2 in the sample, a trend which was in agreement with the results in the literature for the same sample, thus proving the validity of the introduced approach. Besides, this way enabled the usage of a conventional SECM tip instead of the metal-shielded one162 and a shorter time frame for the total experiment.161
Surface-interrogation (SI-SECM) was employed by Ahn and Bard48 with the purpose of analyzing the active atoms in the surface of the well-known OER electrocatalyst CoPi and the reaction rate constants, by amperometric titrations of CoIII and CoIV with 1,1′-ferroceniumdimethanol (FcDM+) or K2IrCl6. Two Au electrodes were used as the substrate (where the catalyst is deposited) and collector (which detects the analyte), both with a size of a = 12.5 μm and separated by 1.7 μm from each other. The principle consists in applying positive potential pulses on the substrate which generate CoIII and CoIV and then scanning the tip to potentials which reduce FcDM+ to FcDM, so that the last one can be oxidized again from CoIII and CoIV generated previously at the substrate (Fig. 15). From the plateau in the acquired plots of charge density vs. substrate potential it was possible to extract the density of Co atoms that are available to water. The density of the active sites on the surface of the catalyst was found to be 11 Co per nm2 (comparable to the literature163), while the pseudo-first-order rate constants of both cobalt species with H2O were 0.19 s−1 for CoIII and >2 s−1 for CoIV.
Fig. 15. Scheme of the SI-SECM setup and the feedback response (on the right) representing the substrate detection of the analyte generated at the tip (FcDM+ reduction at the tip). The tip current is expressed in black and substrate current in red. The concentrations of FcDM and IrCl62− were 1.2 and 1.1 mM respectively. Reproduced from ref. 48 with permission from the American Chemical Society © 2015.

On the other hand, Kim et al.164 have used gold UMEs (a = 12.5 μm) separated by 3.8 μm from each other, to analyze the OER active iron atoms on hematite (α-Fe2O3) grown on F-doped tin oxide coated glass (FTO). The reaction was light-driven, so the catalyst thin film has a pinhole underneath the substrate in order to let the light in, while ferrocenemethanol (FcMeOH+/0) was used as the mediator. Initially, Fe3+ is present in the substrate, until it is subjected to light, after which Fe4+ is generated which is responsible for the evolution of O2. Afterwards, FcMeOH+ gets reduced to FcMeOH0 which then reduces Fe4+. As a result, the density of photo-active atoms was evaluated to be 18 Fe per nm2 at several substrate potentials and the pseudo-first order reaction rate constants evaluated by time-dependent titration were 0.03 to 0.19 s−1. In another study, active site densities of layered double hydroxides (LDHs) and amorphous Ni–Fe (oxy)hydroxides (Ni1−xFexOOH) were determined in situ through SECM by Barforoush et al.165 The authors found that the density of active sites is much higher in Ni0.8Fe0.2OOH LDH (4500 sites per nm2) than the rock salt Ni0.8Fe0.2 oxide (500 sites per nm2), owing to the presence of NiIV/FeIV formed by water and hydroxide below the electrode/electrolyte interface.
An issue related to the study of the OER is the appearance of surface oxides in metal electrodes before the onset of the OER; thus studying alteration in the composition and surface structure of the catalysts is important in this field.166 Arroyo-Currás and Bard166 studied the surface species (–OH(ads) and –H(ads)) formed at the surface of the catalyst (polycrystalline Ir UMEs) by using Fe(iii/ii)–TEA as a mediator at a very basic pH (2 M NaOH) through SI-SECM. The size of the used Ir substrate UME was a = 62.5 μm and the GC tip electrode a = 50 μm. In Fig. 16 in red, the first Ir oxide can be seen around −0.88 V and the oxide growth continues between −0.85 and −0.47 V, where OH– gets adsorbed on the substrate, in which case the coverage of the substrate is θ < 1. On the other hand, the first H(ads) can be seen in green around −0.70 V, followed by an increase in surface coverage forming a monolayer of hydrogen atoms (−0.70 to −1.10 V). In a certain potential region (∼120 mV), these two species coexist in the substrate surface, marked as pzc in Fig. 16, where the green and red plots overlap, assuming that these species are not quite free to move in the surface. In blue we can see the cyclic voltammogram (CV) of Ir, where one can recognize several small waves indicated by black arrows, which correspond well to a charge density increase, ascribed to transformation processes in the surface such as: dehydration of –OH(ads) to –O(ads) or oxide growth. This was assumed since in a potential range between −0.03 and 0.2 V the titration curve (red) is constant, excluding the possibility of any faradaic process occurring in that range. The charge density of one monolayer of the adsorbed species was quantitatively determined and the following results were found: Qθ=1,OH = 456 ± 2.0 μC cm−2 and Qθ=1,H = 224.2 ± 0.2 μC cm−2 for –OH(ads) and –H(ads) respectively.
Fig. 16. Q tip vs. E subs plots acquired by the interrogation of transients of the two adsorbed species on Ir (–OH(ads) and –H(ads)), the former being interrogated with 10 mM Fe(ii)–TEA (red dots) and the latter with 1 mM FcMeOH+(green dots). ΔEsub = 0.01 V and each dot represents an individual SI-SECM measurement. Blue line: CV of Ir UME in 2 M NaOH; v = 20 mV s−1. Black and green lines: Frumkin isotherm fits, using g′ = −6 and g′ = −2 respectively. Red line: order in which the data were obtained at ΔEsubs > 0.44 V. Reproduced from ref. 166 with permission from the American Chemical Society © 2015.

Another innovative way to take advantage of SECM is by combining it with Raman spectroscopy and perform spectroelectrochemical measurements, where a single Raman measurement is executed during all the applied potentials. Steimecke et al.167 have made an investigation of the OER electroactivity of nickel/iron (Ni/Fe) and Ni thin films by using such a combined set-up, which besides probing the local electrochemistry, also allows the extraction of information on the structure or oxidation state changes in the materials during the electrochemical reaction. The measurement is done in such a way that the SECM UME is placed in proximity above the substrate and the Raman probe is placed beneath the substrate (Fig. 17). SG/TC mode is used as one would expect, by using a 25 μm Pt UME deposited in ITO and in a 0.1 M KOH electrolyte.
Fig. 17. (a) Setup developed for the in situ measurements, the used electrodes (here WE1 is a thin film electrode) and the Raman microscope from below; (b) picture of the installed instrument. Reproduced from ref. 167 with permission from the American Chemical Society © 2017.

The recorded substrate potential, besides the OER, also corresponds to the oxidation of Ni(OH)2 to NiOOH and the generation of the latter was determined by the Raman double band at 475 and 557 cm−1. Besides NiOOH, Fe impurities also play a role in the OER performance of nickel oxide and hydroxide films.168,169 In Fig. 18 one can see the dependence of Fe impurity quantity on the I475/I557 ratio and onset potential, where the increase of Fe presence results in an increase of γ-NiOOH disorder. While up to 15% Fe percentage leads to a decrease in onset potential, higher percentages seem to have a contrary effect.
Fig. 18. (a) Raman spectra of all Ni and Ni/Fe samples at 0.63 V; (b) ratio of band intensity vs. applied potential. The SECM determined onset potential is marked for each sample by a square frame in the respective color, while the black arrow represents the increase of the Fe content with a lower I475/I557 band ratio. Reproduced from ref. 167 with permission from the American Chemical Society © 2017.

Bifunctional electrocatalysts for the ORR and OER
Separately, OER or ORR electrocatalysts find utility in many applications152,170–173 and several reviews have covered a diversity of materials that have been studied for their ORR174–178 and OER activity179–183 with hydrodynamic methods. However, for regenerative fuel cells a bifunctional electrocatalyst for both reactions is required.184 This is considered as a challenge, knowing that the ideal ORR electrocatalyst is not on top of the OER volcano as well, and vice versa.185
Nevertheless, many advancements have been made in this direction by utilizing SECM. For instance, Seiffarth et al.186 have synthesized a catalyst by combining an oxide Ni0.9Co0.1Fe2O4 with nitrogen doped carbon nanotubes (NCNTs) and investigated its electrocatalytic activity by utilizing a loading of only 20 μg cm−2. The applied pulse profile introduced in 2007 (ref. 117) was used to assess ORR activity, in an indium doped tin oxide (ITO) substrate and an UME of Pt 25 μm in diameter at d = 25 μm. The number of electrons was calculated by using eqn (12), and the collection efficiency was assumed to be 100%, considering that the variation of the tip-to-sample distance in a range of 10–15–35 μm did not significantly affect the ORR and H2O2 currents.53 The current densities of ORR and H2O2 oxidation are presented in Fig. 19A and B, along with the corresponding n values of the mixed oxide, the NCNTs, their combination and the heated combination. They all show roughly a four-electron transfer, while the slight decrease in the activity of the heated combination was associated with the reduction of FeNiCo oxides.186
Fig. 19. (A) Number of transferred electrons and (B) current densities acquired in RC-SECM mode for ORR and H2O2 oxidation, with a corrected baseline. (C) Tip currents while the tip was held at −0.60 V and a linear potential sweep was applied at the substrate. Reproduced from ref. 186 with permission from John Wiley and Sons © 2016.

The OER was analyzed by SECM in SG/TC mode, by applying a constant potential at the UME which reduced the oxygen produced by the electrocatalyst. The NCNT onset potential (estimated at −0.1 mA cm−2) is the most promising; however the current densities achieved by the mixed oxide and the combined electrocatalyst are the highest (Fig. 19C). As a conclusion, the combined catalyst exhibited the best results among its peers for both reactions. The voltage gap acquired from RDE experiments and SECM was 0.868 V and 0.773 V respectively. This difference in values exists supposedly because the measurement of current in SECM takes place at the UME, while in linear sweep voltammetry (LSV) measurement it takes place at the catalyst layer.186
By adopting a similar methodology to SECM, and hence sequential pulses for obtaining H2O2 and ORR currents and SG/TC mode for OER current, Chen et al.56 have studied nickel and cobalt-based oxides (NixOy and CoxOy respectively) embedded in nitrogen doped carbon (NC) for their bifunctional electroactivity. Normalized SECM images were obtained to illustrate oxygen reduction, oxygen evolution and hydrogen peroxide production. The ORR results suggest the best activity for CoxOy/NC considering that it has the lowest H2O2 and highest ORR current. It was concluded that the carbon supported oxides show generally good activity for both reactions, while NiO is more active for the OER than the ORR.
On the other hand, Chakrabarty et al.187 have demonstrated the bifunctional electroactivity of flower-like ZnCo2O4 grafted onto a reduced graphene oxide (rGO) sheet by studying both reactions with SECM in SG/TC mode at a mass loading of 150 μg cm−2, using an ITO substrate, a Pt 25 μm UME and 1 M KOH. The potential gap of rGO–ZnCo2O4 between the ORR and OER was 0.679 V, which was considered by the authors as a satisfactory value compared to the ones present in the literature. Besides the high conductivity provided by the rGO layer, the promising bifunctional activity was ascribed also to the mixed oxidation state of Co (2+ and 3+) ions in ZnCo2O4, along with the porous nature of the metal oxide which provides a high catalytic surface area.187
Similarly, three-dimensional nanosheet-structured composite materials based on carbon supports and metal oxides (NiCoO2/CNTs) were studied by Ma et al.188 by comparing the performance to that of up-to-date electrocatalysts. The RDE and RRDE results were supported by SECM measurements in RC-SECM mode for oxygen reduction and SG/TC mode for oxygen evolution. They used a GCE as the substrate, Pt 25 μm in diameter, 0.1 M KOH, d = 50 μm and sample loading of 510 μg cm−2. The SECM images confirm the results of RDE and RRDE methods, by proving a superior local activity of the composite compared to distinct NiCoO2 and CNTs, and a similar activity to the state-of-the-art electrocatalysts.188 The prevention of composite aggregation and the supply of extensive active sites by the carbon support may be the reason behind the good activity of NiCoO2/CNTs, along with the synergetic effect between Ni2+ and Co2+.188
Recently, the ORR/OER activity of cobalt-based metalloids (CoxB and CoxP) introduced into a nitrogen-doped carbon matrix (NC) was analyzed by Barwe et al.189 A GCE substrate was used, along with a Pt UME of 25 μm diameter at d = 12.5 μm and sample loading 500 μg cm−2. SG/TC mode was employed for H2O2 evaluation. It is worth mentioning that in some earlier studies,53,112 the collection efficiency was determined by evaluating the change in currents in a certain range of tip-to-substrate distances. Here, they established it by utilizing a sample electrode that reduces oxygen in a 2-electron pathway (Hg), by applying a constant potential of −0.4 V vs. Ag/AgCl 3 M KCl at the Hg and of 0.7 V at the UME. Then, the calculation was done using the tip and sample currents through the expression CE = −itip/isample which gave a value of 3.7 × 10−4. Afterwards, the number of electrons was calculated with the previously established eqn (12). The authors concluded that both CoxB/NC and CoxP/NC exhibited selective reduction of oxygen to OH− with a nearly 4-electron transfer and low overpotentials for both reactions. The ORR/OER round-trip voltage was determined to be 0.81 V. The good performance of the catalyst was linked to the pyridinic and pyrrolic nitrogens in the structure and to boron and phosphorus moieties combined with the Co ions and/or atoms.189
Even though carbon nanotubes are generally acknowledged for their high conductivity, their resistance can further be decreased by more than 50% if they form hybrids with graphene, due to enhanced effectiveness of charge tunneling.190 Lately, such a hybrid electrocatalyst based on graphene nanoflakes (GFs) and carbon nanotubes (CNTs) doped with heteroatoms such as N, Co and Mo (N–Co–Mo–GF/CNT) was studied for its ORR and OER activity by Tavakkoli et al.51 The OER catalytic activity was investigated by means of SECM in SG/TC mode in alkaline media on two different substrates, namely nickel (Ni) and glassy carbon (GC). When GC was used, the tip response (increase of ORR current) was almost simultaneous with the oxygen generation (OER onset potential), compared to the Ni substrate in which case the tip response was delayed (Fig. 20), indicating a more rapid release of oxygen by GC. This was confirmed as well by simulation studies at different layer thicknesses. In this way the authors were able to reveal that the OER at the Ni substrate occurred at the catalyst/substrate interface, while on GC it occurred close to the surface of the catalyst. The RDE measurements of the OER revealed an enhanced activity of the catalyst on the Ni substrate with an onset overpotential (ηOER,10) of ∼50 mV lower compared to the GC substrate. As for the ORR, the number of transferred electrons was evaluated by using a RDE and K–L equation and was close to 4 for the catalyst on all studied substrates. The promising bifunctional electrocatalytic activity was attributed to the synergistic effect between the N–C and M–C sites in the catalyst structure.51 Finally, this work demonstrates how SECM can be helpful in resolving the location where the OER occurs, and also how the substrate choice plays an important role in the activity of the electrocatalyst.
Fig. 20. (a) Scheme of the reaction where the Pt SECM tip approaches the electrocatalyst to observe the feedback from the sample and substrate, as well as the O2 diffusion from the substrate/catalyst interface to the tip. (b and c) SG/TC results, with the tip polarized at 0.3 V vs. RHE for driving the ORR, while the substrate was scanned in the OER region at 5 mV s−1. The tip was in proximity to N–Co–Mo–GF/CNT on (b) GC and (c) Ni substrates. Reproduced from ref. 51 with permission from American Chemical Society © 2020. https://pubs.acs.org/doi/abs/10.1021/acscatal.0c00352. Further permissions related to this material should be directed to the American Chemical Society.

Hydrogen evolution reaction (HER)
The hydrogen evolution reaction in acidic media is presented in Fig. 1 and the reaction in alkaline media is shown below:191
| 2H2O + 2e− → 2OH− + H2 | 15 |
In general, in acidic and alkaline media the HER can occur via two steps (reactions (16)–(18)).192–195 For instance, in alkaline media, first H2O which is adsorbed at the catalyst surface gets reduced to a hydrogen atom (Volmer step):
| H2O + e− → Hads + OH− | 16 |
Then, the Hads can couple with another Hads to form H2 which escapes the surface (Tafel step):
| Hads + Hads → H2 | 17 |
Or the Hads reacts with a H2O molecule to produce H2 (Heyrovsky step):
| Hads + H2O + e− → H2 + OH− | 18 |
One of the factors that can dictate the mechanism in which hydrogen evolves is the strength of the metal–hydrogen bond.196
The up-to date electrocatalysts for the HER occupying the top of the volcano plot are noble metals,196 which were recently studied by SECM, for example by Fernández and Zoski,197 who investigated noble nanoparticles for their HER performance in acidic media. They performed electro-deposition of Pt nanoparticles on gold nanoelectrode ensembles of UME dimensions, and studied them in SG/TC mode, through which they found matching results with polycrystalline Pt. Similar results were found as well for the Au nanoelectrode ensembles compared to polycrystalline Au.197
Moreover, nanocomposites based on palladium/titanium oxide (Pd/TiO2) and multi-walled carbon nanotubes (MWCNTs) were investigated by Valenti et al.198 for their HER activity by using SG/TC mode of SECM in a neutral pH electrolyte. As anticipated, the introduction of MWCNTs increases the HER activity of Pd/TiO2, possibly due to improved pairing of palladium electronic levels at the interface of TiO2/MWCNTs which exhibits enhanced surface states.198
However, as mentioned earlier, non-noble electrocatalysts are advantageous and they have been the focus of research for HER activity in the past few years. For instance, transition metal dichalcogenides have shown promising performance towards the HER,199–201 in which the disulfide-terminated edges play the role of catalytic sites for hydrogen evolution.200
Hyper thin one-dimensional (1D, wires) and two-dimensional (2D, discs) FeS2 nanostructures were studied for their HER activity at neutral pH by Jasion et al.202 in SG/TC mode of SECM, by polarizing the sample at a negative potential for hydrogen evolution and the tip at a positive hydrogen-oxidation potential. The substrate used was a GCE and a 200 μm Pt tip at d = 100 μm. The results were compared with those of a Pt substrate as the state-of-the art HER electrocatalyst and with those of a bare glassy carbon electrode (Fig. 21). The authors conclude that the HER occurred at the 2D FeS2 sample at an overpotential of less than 50 mV higher than that at the Pt catalyst. Moreover, faradaic efficiency which represents the ratio of the experimental quantity of hydrogen evolved and the theoretical quantity203 was determined for the discs by means of SG/TC mode, with a 200 μm Au substrate modified with the sample (FeS2 discs) and it was found to have a value of 92 ± 8%.202 The highlight of this work was the better HER activity of the 1D and 2D FeS2 nanostructures compared to the 3D cube structures.
Fig. 21. HER electroactivity maps acquired via SECM: (a) scheme of the SECM experiment (SG/TC mode), (b) the reactivity map of a glassy carbon electrode, (c) HER electrochemical reactivity map of Pt on glassy carbon and (d) HER electrochemical reactivity map of FeS2 discs on glassy carbon. Reproduced from ref. 202 with permission from the American Chemical Society © 2015.

A CoSx electrocatalyst obtained from metal–organic frameworks (MOFs) via an SECM tip-induced method was subsequently analyzed for its HER behavior by Liberman et al.47 SG/TC mode was used with a 10 μm Pt tip at d = 11.5 ± 0.5 μm in a ring-shaped ∼100 μm CoSx sample. The alike diameter of the patterned sample and the glass-coated tip leads to a high collection efficiency, needless of using an UME as a substrate. The HER mapping is presented in Fig. 22, whereas the shape of the sample pattern was confirmed as well by energy-dispersive X-ray spectroscopy (EDS). As a conclusion, the authors successfully combined localized catalyst fabrication and its consecutive SECM analysis, which is believed to be a useful approach for forthcoming screening of electrocatalysts and for the design of patterned arrays.47
Fig. 22. Hydrogen evolution reaction activity map of the as-prepared 60× localized electrochemical conversion pattern.47 Reproduced from ref. 47 with permission from the Royal Society of Chemistry, © 2020.

The higher HER activity of Mg(OH)2 coated on Mg, compared to pristine Mg, was demonstrated by Salleh et al.204 by using SECM in SG/TC mode (Fig. 23). A 25 μm diameter Pt tip was used for the detection (oxidation) of hydrogen, at a fixed tip-to-substrate distance of 30 μm in 0.1 M NaCl solution. The hydrogen evolution rate of the Mg(OH)2 coated Mg surface was determined to be ∼2 to 3 times higher compared to pristine Mg, a result which was compatible with the potentiodynamic polarization measurements. These results are associated with the activation of H2O self-dissociation in the presence of the adsorbed OH− groups.204 It's worth mentioning that the topographical alteration did not significantly affect the currents measured at the microelectrode, because the HOR is not a diffusion-controlled reaction on the Pt surface. The aforementioned outcome can be very important when it comes to the efficiency of systems that are based on Mg.205,206
Fig. 23. SECM HER image recorded from pristine Mg across to the Mg coated with a Mg(OH)2 surface. A normalization of microelectrode currents with respect to the average current of pristine Mg was done. The microelectrode was rastered with a sweep velocity of 50 μg s−1 and the electrochemical measurements were performed at steps of 100 μm per point. Reproduced from ref. 204 with permission from Elsevier © 2015.

A widely investigated catalyst for the hydrogen evolution reaction is molybdenum disulphide (MoS2),207–209 in which the edge sites play an active role in the HER,210 while the basal planes were considered inert211 until they were activated through creation of sulphur vacancies.144,212 Li et al.211 employed SECM in SG/TC mode in combination with multiphysics modelling in order to study the kinetics of S vacancies on monolayers of MoS2, using the following conditions: 0.1 M HClO4 electrolyte, 25 μm diameter Pt UME at d = ∼4 μm and d = ∼5 μm for SV-MoS2 (unstrained area) and V-MoS2 (strained area) respectively. The activities of strained and unstrained areas were compared and in both cases the SECM results show a fast decrease in tip current caused by H2 bubbles.211 Finally, it was concluded that tensile strain stimulates the kinetics of hydrogen evolution in S-vacancies in MoS2.211
Very recently, a thorough investigation of HER electroactivity in MoS2 was done by Sun et al.45 through nanoscale mapping with <20 nm spatial resolution. The HER activity of metallic (1T) and semiconducting (2H) phases within the MoS2 nanostructure was compared by employing SECM in SG/TC mode, in which case it was found that the metallic phase has superior HER activity compared to 2H, while within the 2H phase, the edges have higher activity (Fig. 24). These results might be an important indication for converting MoS2 into an only-metallic phase for an elevated HER activity.45
Fig. 24. Line profiles of the hydrogen evolution reaction obtained by using SECM in SG/TC mode in mixed-phase MoS2 nanosheets. Reproduced from ref. 45 with permission from the Royal Society of Chemistry © 2019.

An improvement in MoS2 activity can be obtained by incorporating it into matrices with high conductivity.213 This was done by Kumar et al.214 who combined MoS2 NPs of the T1 phase with graphene oxide (GO) or reduced graphene oxide (rGO) in which case both composites exhibited comparable HER activity to the state-of-the-art Pt/C.214
On the other hand, nickel foams with a karst landform structure were studied for their HER activity by Gao et al.215 through SG/TC mode, a Pt nanoelectrode and Si substrate. The SECM images indicate a high activity in the valley areas, which was confirmed by the topographical image as well.215 The same electrocatalyst exhibited good activity towards the OER as well and this performance is attributed to the karst landform structure, which seems to ease the mass diffusion and to provide sufficient catalytic sites.215
SECM was employed as well for the investigation of photo-generated hydrogen at a 1,2-dichloroethane/water (DCE/W) interface by Jedraszko et al.216 During the HER, decamethylruthenocene (DMRc) is formed as a by-product during the hydrogen evolution, and then serves as an electron donor, which permits a continuous HER.216 The SECM setup consisted of two Pt electrodes separated by only 50 μm from each other through a liquid/liquid interface, in order to ensure an efficient generation of DMRc by DMRc+ reduction. Close to the DCE/W interface, the bottom electrode served for uninterrupted regeneration of DMRc+, while the photogenerated hydrogen was captured at the upper electrode. Herein, SECM was used to demonstrate the regeneration of DMRs, which opens the possibilities for further development of biphasic systems towards H2 generation. The experimental details and the resulting tip current due to H2 oxidation in the presence and absence of light can be seen in Fig. 25.
Fig. 25. (A) Schematic representation of the used SECM setup for the study of DMRc regeneration with two Pt microelectrodes placed near immiscible electrolyte solutions (ITIES), where the blue double arrow represents the connection to the syringe pump that controls the position of the liquid/liquid interface. Anaerobic conditions are supplied by Ar, (B) schematic representation of the reaction and (C) current of the tip measured at 0.20 V vs. RHE, above the bottom Pt electrode and polarized at −0.18 V (a) exposed to UV light, (b) in darkness and (c) in darkness without substrate polarization. The organic phase contained 5 mmol dm−3 DMRc and 5 mmol dm−3 bis(triphenylphosphoranylidene)ammonium tetrakis(pentafluorophenyl)borate (BATB). The solution in which the Pt tip was immersed contained 0.1 M HClO4 and 5 mM lithium tetrakis(pentafluorophenyl)borate (LiTB). The distance between the microelectrodes was 50 μm and the tip lateral velocity was 50 μm s−1 Reproduced from ref. 216 with permission from Elsevier © 2018.

Bentley and Unwin217 demonstrated through studying the HER at MoS2 how SECCM is a powerful robust tool that can be used to obtain topographical and voltammetric data at a 50 nm spatial resolution, and potential-resolved movies of electrocatalytic performance, in just minutes. The working principle of this technique is illustrated in Fig. 26. The authors were able to obtain the specificities of the catalyst surface at a sub-10 nm scale. Moreover, {111} Au nanocrystals (AuNCs) were also investigated, displaying uniform HER activity up to a sub-single entity level.
Fig. 26. (a) Scheme of a nanoscale synchronous electrochemical/topographical map obtained by SECCM in voltammetric hopping mode by using a single channel nanopipette containing 100 mM H2SO4 and a GC support. The voltage was applied at the QRCE to control the WE potential (Eapp) while the WE current (isurf) was measured. The latter also served as a feedback signal for detecting meniscus-surface contact during the approach. Arrows show the movement of the nanopipette probe along the surface during scanning (inset, top-right). (b) Plots of (i) z-extension, (ii) Eapp and (iii) isurf during a ‘single hop’ at the AuNC substrate. Scan rate (v) = 10 V s−1 and data acquisition time (td) = 260 μs. Reproduced from ref. 217 with permission from the Royal Society of Chemistry © 2018.

Choi et al.218 took advantage of SECCM for evaluating the HER electrocatalytic activity of individual Au nanocubes (NCs, {100}) and nano-octahedra (ODs, {111}) both with edge lengths <100 nm, by using a dual barrel nanopipette (∼200 nm in outer diameter) and glassy carbon as a substrate. The authors established that performing CV through SECCM is an efficient way to investigate the HER activity of individual nanoparticles. The current results showed that the cubes have a higher performance compared to the octahedra and misshaped particles, a result which matched the macroscale measurements. This outcome may be valuable when it comes to effective future design of HER electrocatalysts.
A metal free material that has been studied for HER activity is also hexagonal boron nitride (h-BN), thanks to the possibility of electronic tunneling219 between its ultrathin layers and the underlying metal support substrate.220 The effect of a metal substrate on the electrocatalytic activity of 2D h-BN nanosheets was studied through SECCM in hopping mode by Liu et al.221 The authors investigated the grown h-BN nanosheets on Cu and on Au substrates, namely h-BN/Cu and h-BN/Au, respectively, by using a 0.1 M HClO4 filled nanopipette with a diameter from 150 to 300 nm. The exchange current evaluated by local voltammetry and Tafel analysis showed that h-BN/Au has superior HER electrocatalytic activity, easing the way to a proper future design of HER electrocatalysts.
Emerging topics
The electrochemical reduction of the greenhouse gas CO2 has attracted attention lately,222 since it can lead to the formation of beneficial chemicals.223–225 Some products that form during the CO2RR can be evaluated through the RRDE method;226–228 however a better collection efficiency may be achieved by SECM.229
The catalytic properties of boron-doped graphene (BG) for the reduction of CO2 to formate (FA) were studied by Sreekanth et al.230 SECM in SG/TC mode was utilized for identifying the electroactive product (FA) through a Pt UME, that forms during CO2 reduction in the GCE substrate in 0.1 M KHCO3. The oxidation of FA was spotted through cyclic voltammetry, with a sharp tip current peak at a substrate potential of −1.4 V vs. SCE. Control experiments were conducted to ensure that the signal is associated with the FA oxidation and not with CO oxidation as another potential product. Finally, the BG catalyst exhibited higher tip current during FA oxidation compared to the benchmark catalyst (Bi).230,231 The same group investigated the formation of CO and FA through bicarbonate reduction with SECM in SG/TC mode.229 Moreover, the good catalytic activity of silver nanoparticles and nanoclusters (Ag NPs and Ag NCs respectively) for bicarbonate reduction to FA was demonstrated by Arrocha-Arcos et al.232 by using SG/TC mode of SECM.
The first step of CO2 reduction is the one-electron transfer reaction of CO2˙− production, an intermediate which has a very short lifetime46 as it can be quickly protonated, dimerized, or reduced.233 Therefore, it has been a major challenge to capture it experimentally, leading to a lack of understanding of the overall CO2RR mechanism. Not long ago, this has been achieved by Kai et al.,46 who detected for the first time the CO2˙− intermediate through SECM in TG/SC mode, by making use of an Hg/Pt UME tip and small distances between the tip and the substrate (Au UME). The substrate current rose due to intermediate detection and the tip current increased due to the oxidation of some CO2˙− to CO2 at the sample which is fed back to the tip46 (Fig. 27).
Fig. 27. The black curve represents the current due to CO2˙− collection at the SECM substrate (an a = 12.5 μm Au UME) in DMF which contains 0.1 M TBAPF6. The tip potential was kept at Et = −2.8 V, while the substrate potential was swept from −2.1 V to −0.9 V. The tip current is represented by the red curve. Reproduced from ref. 46 with permission from the American Chemical Society © 2017.

Grain boundaries (GBs) in polycrystalline materials have been shown to have an effect on CO2RR electrocatalysis;234,235 however for a proper future design, a thorough understanding of the GB dependence in activity is necessary. Mariano et al.236 have studied the influence of GB on polycrystalline Au with big grain sizes on the CO2RR to CO. The authors used a ∼300 nm single-barrel pipette in hopping mode, by applying a fixed potential at the Au substrate. Current is recorded when the droplet contacts the sample, and then the pipette is moved away for the next measurement, allowing the recording of line scans. Through bulk electrochemistry the authors found that the CO2RR increases with the density of GBs, while SECCM was used to justify this result, in which case it was found that surface terminations are responsible for the elevated CO2RR (Fig. 28).
Fig. 28. Two approaches detailed in this work for the electrochemical characterization of defect effects on the CO2RR. (A) Bulk electrolysis of a well-defined polycrystalline Au electrode within a glass, two-compartment H cell and (B) SECCM using a ∼300 nm nanopipette electrochemical cell. Reproduced from ref. 236 with permission from the American Association for the Advancement of Science © 2017.

Nanoscale measurements can be helpful in cases when one needs to discriminate between the performance of an ensemble with that of individual nanoparticles.237 Such measurements were done by Kim et al.43 for the study of the hydrogen oxidation reaction (HOR) of Pt NPs electrodeposited on highly oriented pyrolytic graphite (HOPG), with a Pt nanoelectrode at d = 134 nm. To acquire the SECM images, H+ got reduced at the tip, while the H2 which was formed afterwards got oxidized at the sample (Fig. 29 left). In Fig. 29 (right) one can see the current of H2 oxidation at five distinct nanoparticles. Furthermore, as a result of elevated mass transfer due to nano-scale conditions, a HOR rate constant of k0eff ≥ 2 cm s−1 was acquired.43
Fig. 29. (left) Schematic representation of the electron transfer reactions in the substrate and sample by means of SECM and (right) SECM image of the electron transfer reaction of H+/H at five Pt nanoparticles in 2 mM HClO4 and 10 mM NaClO4 with the tip polarized at −1.0 V and the substrate at −0.4 V vs. a Pt quasi reference electrode (QRE). Pt nanotip was scanned at 200 nm s−1. Reproduced with permission from ref. 43 © 2016 American Chemical Society.

Conclusion and prospects
In this review we summarized recent investigations (mostly between 2015–2020) made for the ORR, OER and HER electrocatalysts, by employing SECM and other related techniques. The non-noble class of materials that seem to be frequently studied for the two former reactions are metal oxides on carbon supports, in which case the synergetic effects between the two components can lead to a superior electrocatalytic activity. As for the HER, metal sulfides seem to have attention as promising non-noble electrocatalysts. Generally, the local ORR activity was studied in RC-SECM mode and then compared to the RDE results. However, in some cases, the intermediate produced was quantified as well by SECM, where a combination of RC and generation/collection modes was used (pulsed profile). This approach, which allows the calculation of n, is not put into practice as much as one would have expected. Instead, the selectivity is mostly acquired from RDE and/or RRDE methods, and then the activity is somewhat qualitatively compared to RC-SECM results. The outcome of the different approaches is not always concordant, and thus additional research should be a prerequisite for more unambiguous conclusions regarding their compatibility and complementarity.
The gas-evolution reactions (OER and HER) are studied essentially in SG/TC mode. SI-SECM seems to have potential in the study of the OER, especially for mechanistic studies while Raman-SECM was used for OER in situ studies. Concerning the experimental conditions for all reactions, interestingly a Pt UME of 25 μm diameter was most frequently used, along with mainly an ITO or GCE substrate, at different tip-to-substrate distances. Since most experiments were performed in alkaline electrolytes, it would be interesting for further studies to conduct more experiments in acidic media, as it would expand the application possibilities in different kinds of fuel cells. The combined techniques such as SECM-AFM and SECM-SICM were utilized for ORR investigations, while SECCM found most utility in the analysis of the HER. Finally, the study of the CO2RR with SECM seems to be auspicious, while further SPM investigations of the HOR, especially in non-noble electrocatalysis, are highly encouraged. Clearly, SECM appears to be the leader within the SPM techniques for electrocatalytic investigations.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The authors acknowledge the SENTINEL project, funded by the European Union's Horizon 2020 Research and Innovation Program under the Marie Skłodowska-Curie grant agreement no. 812398, and the PEGASUS project, funded by the European Union's Horizon 2020 Research and Innovation Program FCH-01-2-2017, no. 779550.
Biographies
Biography
N. Limani.

Ndrina Limani received her BSc degree in Chemical Engineering from the University of Prishtina (2017) in Kosovo, after which she continued her master's studies abroad with a fully funded Erasmus+ scholarship. She earned a double MSc degree in Chemistry from the University of Porto in Portugal and Paris-Saclay University in France (2019). Presently, she is conducting her PhD studies as a Marie Curie Early-Stage-Researcher (SENTINEL-ITN) in the field of SECM and electrocatalysis at CEA-Saclay, under the supervision of Dr Renaud Cornut and Dr Bruno Jousselme.
Biography
A. Boudet.

Alice Boudet graduated in 2018 from the engineering school Grenoble INP-Phelma with a degree in electrochemistry and process engineering. She is now pursuing a PhD at Université Paris-Saclay in the laboratory LICSEN from CEA Saclay. She is working under the supervision of Dr Bruno Jousselme and Dr Renaud Cornut on SECM for the evaluation of the catalytic activity of PGM-free catalysts. Her work involves both experimental and modelling studies.
Biography
N. Blanchard.

Nicolas Blanchard received his BSc degree in Chemistry from the University of Sorbonne (2017) in France and his master's degree in materials chemistry from PSL University (2019). After a short experience at ICMPE in Paris Est on the electrochemical reduction of carbon dioxide, he is currently doing a PhD thesis in electrochemistry under the supervision of Renaud Cornut and Bruno Jousselme at LICSEN from CEA Saclay. His job is to study the transport of species within of fuel cell electrodes, using mainly electrochemical microscopy (SECM) and electrochemical impedance spectroscopy (EIS) techniques.
Biography
B. Jousselme.

Bruno Jousselme is a Senior Scientist in the LICSEN group at CEA Saclay LICSEN. Hired in 2006, he started new research programs on the synthesis of conjugated systems and surface functionalization for applications in molecular electronics and renewable energies. His motivations in the latter field are to develop cheap, stable and efficient nanomaterials for photo- and electro-catalysis based mainly on carbon nanotubes.
Biography
R. Cornut.

Renaud Cornut has been a researcher at LICSEN in CEA Saclay since 2011. He graduated in 2009 from Grenoble INP-Phelma with a degree in electrochemistry and numerical simulation, and did postdoctorate at UQAM 2009–2011 in Bio-SECM and lithium batteries. His current topics of interest are SECM and fuel cell electrochemistry.
References
- Perera F. Int. J. Environ. Res. Public Health. 2017;15:16. doi: 10.3390/ijerph15010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray H. B. Nat. Chem. 2009;1:7. doi: 10.1038/nchem.141. [DOI] [PubMed] [Google Scholar]
- Cook T. R. Dogutan D. K. Reece S. Y. Surendranath Y. Teets T. S. Nocera D. G. Chem. Rev. 2010;110:6474–6502. doi: 10.1021/cr100246c. [DOI] [PubMed] [Google Scholar]
- Owusu P. A. Asumadu-Sarkodie S. Cogent Eng. 2016;3:1167990. [Google Scholar]
- Carrette L. Friedrich K. A. Stimming U. Fuel Cells. 2001;1:5–39. doi: 10.1002/1615-6854(200105)1:1<5::AID-FUCE5>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Mikolajczuk-Zychora A. Borodzinski A. Kedzierzawski P. Mierzwa B. Mazurkiewicz-Pawlicka M. Stobinski L. Ciecierska E. Zimoch A. Opałło M. Appl. Surf. Sci. 2016;388:645–652. doi: 10.1016/j.apsusc.2016.02.065. [DOI] [Google Scholar]
- Aguilar L. Zha S. Cheng Z. Winnick J. Liu M. J. Power Sources. 2004;135:17–24. doi: 10.1016/j.jpowsour.2004.03.061. [DOI] [Google Scholar]
- Serov A. Kwak C. Appl. Catal., B. 2010;98:1–9. doi: 10.1016/j.apcatb.2010.05.005. [DOI] [Google Scholar]
- Lamy C. Rousseau S. Belgsir E. M. Coutanceau C. Léger J.-M. Electrochim. Acta. 2004;49:3901–3908. doi: 10.1016/j.electacta.2004.01.078. [DOI] [Google Scholar]
- Wasmus S. Küver A. J. Electroanal. Chem. 1999;461:14–31. doi: 10.1016/S0022-0728(98)00197-1. [DOI] [Google Scholar]
- Afif A. Radenahmad N. Cheok Q. Shams S. Kim J. H. Azad A. K. Renewable Sustainable Energy Rev. 2016;60:822–835. doi: 10.1016/j.rser.2016.01.120. [DOI] [Google Scholar]
- Thomas C. Int. J. Hydrogen Energy. 1998;23:507–516. doi: 10.1016/S0360-3199(97)00102-X. [DOI] [Google Scholar]
- Morales-Guio C. G. Stern L.-A. Hu X. Chem. Soc. Rev. 2014;43:6555. doi: 10.1039/C3CS60468C. [DOI] [PubMed] [Google Scholar]
- Bockris J. O. M. Int. J. Hydrogen Energy. 2013;38:2579–2588. doi: 10.1016/j.ijhydene.2012.12.026. [DOI] [Google Scholar]
- Hanley E. S. Deane J. Gallachóir B. Ó. Renewable Sustainable Energy Rev. 2018;82:3027–3045. doi: 10.1016/j.rser.2017.10.034. [DOI] [Google Scholar]
- Jiao Y. Zheng Y. Jaroniec M. Qiao S. Z. Chem. Soc. Rev. 2015;44:2060–2086. doi: 10.1039/C4CS00470A. [DOI] [PubMed] [Google Scholar]
- Turner J. A. Science. 2004;305:972–974. doi: 10.1126/science.1103197. [DOI] [PubMed] [Google Scholar]
- Freire C. Fernandes D. M. Nunes M. Abdelkader V. K. ChemCatChem. 2018;10:1703–1730. doi: 10.1002/cctc.201701926. [DOI] [Google Scholar]
- Aijaz A. Masa J. Rösler C. Xia W. Weide P. Botz A. J. R. Fischer R. A. Schuhmann W. Muhler M. Angew. Chem., Int. Ed. 2016;55:4087–4091. doi: 10.1002/anie.201509382. [DOI] [PubMed] [Google Scholar]
- Yu F.-Y. Lang Z.-L. Yin L.-Y. Feng K. Xia Y.-J. Tan H.-Q. Zhu H.-T. Zhong J. Kang Z.-H. Li Y.-G. Nat. Commun. 2020;11:490. doi: 10.1038/s41467-019-14274-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydova E. S. Mukerjee S. Jaouen F. Dekel D. R. ACS Catal. 2018;8:6665–6690. doi: 10.1021/acscatal.8b00689. [DOI] [Google Scholar]
- Lee Y. Suntivich J. May K. J. Perry E. E. Shao-Horn Y. J. Phys. Chem. Lett. 2012;3:399–404. doi: 10.1021/jz2016507. [DOI] [PubMed] [Google Scholar]
- Schmidt O. Gambhir A. Staffell I. Hawkes A. Nelson J. Few S. Int. J. Hydrogen Energy. 2017;42:30470–30492. doi: 10.1016/j.ijhydene.2017.10.045. [DOI] [Google Scholar]
- Offer G. J. Howey D. Contestabile M. Clague R. Brandon N. P. Energy Policy. 2010;38:24–29. doi: 10.1016/j.enpol.2009.08.040. [DOI] [Google Scholar]
- Wang J. Wang H. Fan Y. Engineering. 2018;4:352–360. doi: 10.1016/j.eng.2018.05.007. [DOI] [Google Scholar]
- Wang Y. Ruiz Diaz D. F. Chen K. S. Wang Z. Adroher X. C. Mater. Today. 2020;32:178–203. doi: 10.1016/j.mattod.2019.06.005. [DOI] [Google Scholar]
- Whiston M. M. Azevedo I. L. Litster S. Whitefoot K. S. Samaras C. Whitacre J. F. Proc. Natl. Acad. Sci. U. S. A. 2019;116:4899–4904. doi: 10.1073/pnas.1804221116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoncello P. Energy Environ. Sci. 2010;3:1620. doi: 10.1039/C0EE00046A. [DOI] [Google Scholar]
- Polcari D. Dauphin-Ducharme P. Mauzeroll J. Chem. Rev. 2016;116:13234–13278. doi: 10.1021/acs.chemrev.6b00067. [DOI] [PubMed] [Google Scholar]
- Jia Z., Yin G. and Zhang J., in Rotating Electrode Methods and Oxygen Reduction Electrocatalysts, Elsevier, 2014, pp. 199–229 [Google Scholar]
- Rotating electrode methods and oxygen reduction electrocatalysts, ed. W. Xing, G. Yin and J. Zhang, Elsevier, Amsterdam, 2014 [Google Scholar]
- Du C., Tan Q., Yin G. and Zhang J., in Rotating Electrode Methods and Oxygen Reduction Electrocatalysts, Elsevier, 2014, pp. 171–198 [Google Scholar]
- Salapaka S. M. Salapaka M. V. IEEE Contr. Syst. 2008;28:65–83. [Google Scholar]
- Zoski C. G. J. Electrochem. Soc. 2016;163:H3088–H3100. doi: 10.1149/2.0141604jes. [DOI] [Google Scholar]
- Lai S. C. S. Macpherson J. V. Unwin P. R. MRS Bull. 2012;37:668–674. doi: 10.1557/mrs.2012.146. [DOI] [Google Scholar]
- Scanning electrochemical microscopy, ed. A. J. Bard and M. V. Mirkin, CRC Press, Boca Raton, Fla, 2nd edn, 2012 [Google Scholar]
- Cornut R. Mayoral M. Fabre D. Mauzeroll J. J. Electrochem. Soc. 2010;157:F77. doi: 10.1149/1.3374340. [DOI] [Google Scholar]
- Cornut R. Poirier S. Mauzeroll J. Anal. Chem. 2012;84:3531–3537. doi: 10.1021/ac203047d. [DOI] [PubMed] [Google Scholar]
- Ventosa E. Schuhmann W. Phys. Chem. Chem. Phys. 2015;17:28441–28450. doi: 10.1039/C5CP02268A. [DOI] [PubMed] [Google Scholar]
- Heinze J. Angew. Chem., Int. Ed. Engl. 1993;32:1268–1288. doi: 10.1002/anie.199312681. [DOI] [Google Scholar]
- Senthamarai R. Rajendran L. J. Theor. Comput. Chem. 2008;07:205–219. doi: 10.1142/S0219633608003721. [DOI] [Google Scholar]
- Schuhmann W. and Bron M., in Polymer Electrolyte Membrane and Direct Methanol Fuel Cell Technology, Elsevier, 2012, pp. 399–424 [Google Scholar]
- Kim J. Renault C. Nioradze N. Arroyo-Currás N. Leonard K. C. Bard A. J. J. Am. Chem. Soc. 2016;138:8560–8568. doi: 10.1021/jacs.6b03980. [DOI] [PubMed] [Google Scholar]
- Kai T. Zoski C. G. Bard A. J. Chem. Commun. 2018;54:1934–1947. doi: 10.1039/C7CC09777H. [DOI] [PubMed] [Google Scholar]
- Sun T. Zhang H. Wang X. Liu J. Xiao C. Nanayakkara S. U. Blackburn J. L. Mirkin M. V. Miller E. M. Nanoscale Horiz. 2019;4:619–624. doi: 10.1039/C8NH00346G. [DOI] [Google Scholar]
- Kai T. Zhou M. Duan Z. Henkelman G. A. Bard A. J. J. Am. Chem. Soc. 2017;139:18552–18557. doi: 10.1021/jacs.7b08702. [DOI] [PubMed] [Google Scholar]
- Liberman I. He W. Shimoni R. Ifraemov R. Hod I. Chem. Sci. 2020;11:180–185. doi: 10.1039/C9SC04141A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn H. S. Bard A. J. J. Am. Chem. Soc. 2015;137:612–615. doi: 10.1021/ja511740h. [DOI] [PubMed] [Google Scholar]
- Davoodi A. Pan J. Leygraf C. Norgren S. Appl. Surf. Sci. 2006;252:5499–5503. doi: 10.1016/j.apsusc.2005.12.023. [DOI] [Google Scholar]
- Zhang J. Slevin C. J. Morton C. Scott P. Walton D. J. Unwin P. R. J. Phys. Chem. B. 2001;105:11120–11130. doi: 10.1021/jp004592j. [DOI] [Google Scholar]
- Tavakkoli M. Flahaut E. Peljo P. Sainio J. Davodi F. Lobiak E. V. Mustonen K. Kauppinen E. I. ACS Catal. 2020;10:4647–4658. doi: 10.1021/acscatal.0c00352. [DOI] [Google Scholar]
- Zampardi G. Ventosa E. La Mantia F. Schuhmann W. Chem. Commun. 2013;49:9347. doi: 10.1039/C3CC44576C. [DOI] [PubMed] [Google Scholar]
- Dobrzeniecka A. Zeradjanin A. Masa J. Puschhof A. Stroka J. Kulesza P. J. Schuhmann W. Catal. Today. 2013;202:55–62. doi: 10.1016/j.cattod.2012.03.060. [DOI] [Google Scholar]
- Hijazi I. Bourgeteau T. Cornut R. Morozan A. Filoramo A. Leroy J. Derycke V. Jousselme B. Campidelli S. J. Am. Chem. Soc. 2014;136:6348–6354. doi: 10.1021/ja500984k. [DOI] [PubMed] [Google Scholar]
- Maljusch A. Ventosa E. Rincón R. A. Bandarenka A. S. Schuhmann W. Electrochem. Commun. 2014;38:142–145. doi: 10.1016/j.elecom.2013.11.024. [DOI] [Google Scholar]
- Chen X. Botz A. J. R. Masa J. Schuhmann W. J. Solid State Electrochem. 2016;20:1019–1027. doi: 10.1007/s10008-015-3028-z. [DOI] [Google Scholar]
- Jindal A. Basu S. Aby C. P. RSC Adv. 2015;5:69378–69387. doi: 10.1039/C5RA10884E. [DOI] [Google Scholar]
- Sklyar O. Wittstock G. J. Phys. Chem. B. 2002;106:7499–7508. doi: 10.1021/jp020301q. [DOI] [Google Scholar]
- Ludwig M. Kranz C. Schuhmann W. Gaub H. E. Rev. Sci. Instrum. 1995;66:2857–2860. doi: 10.1063/1.1145568. [DOI] [Google Scholar]
- Mkelvey K. Edwards M. A. Unwin P. R. Anal. Chem. 2010;82:6334–6337. doi: 10.1021/ac101099e. [DOI] [PubMed] [Google Scholar]
- Lazenby R. A. MKelvey K. Unwin P. R. Anal. Chem. 2013;85:2937–2944. doi: 10.1021/ac303642p. [DOI] [PubMed] [Google Scholar]
- Gębala M. Schuhmann W. La Mantia F. Electrochem. Commun. 2011;13:689–693. doi: 10.1016/j.elecom.2011.04.010. [DOI] [Google Scholar]
- Ebejer N. Güell A. G. Lai S. C. S. McKelvey K. Snowden M. E. Unwin P. R. Annu. Rev. Anal. Chem. 2013;6:329–351. doi: 10.1146/annurev-anchem-062012-092650. [DOI] [PubMed] [Google Scholar]
- Bard A. J. Fan F. R. F. Kwak J. Lev O. Anal. Chem. 1989;61:132–138. doi: 10.1021/ac00177a011. [DOI] [Google Scholar]
- Shao Y. Mirkin M. V. J. Phys. Chem. B. 1998;102:9915–9921. doi: 10.1021/jp9828282. [DOI] [Google Scholar]
- Macpherson J. V. Unwin P. R. Anal. Chem. 2000;72:276–285. doi: 10.1021/ac990921w. [DOI] [PubMed] [Google Scholar]
- Katemann B. B. Schuhmann W. Electroanalysis. 2002;14:22–28. doi: 10.1002/1521-4109(200201)14:1<22::AID-ELAN22>3.0.CO;2-F. [DOI] [Google Scholar]
- Eckhard K. Chen X. Turcu F. Schuhmann W. Phys. Chem. Chem. Phys. 2006;8:5359. doi: 10.1039/B609511A. [DOI] [PubMed] [Google Scholar]
- Payne N. A. Stephens L. I. Mauzeroll J. Corrosion. 2017;73:759–780. doi: 10.5006/2354. [DOI] [Google Scholar]
- Bergner S. Vatsyayan P. Matysik F.-M. Anal. Chim. Acta. 2013;775:1–13. doi: 10.1016/j.aca.2012.12.042. [DOI] [PubMed] [Google Scholar]
- Amemiya S. Bard A. J. Fan F.-R. F. Mirkin M. V. Unwin P. R. Annu. Rev. Anal. Chem. 2008;1:95–131. doi: 10.1146/annurev.anchem.1.031207.112938. [DOI] [PubMed] [Google Scholar]
- Kucernak A. R. Chowdhury P. B. Wilde C. P. Kelsall G. H. Zhu Y. Y. Williams D. E. Electrochim. Acta. 2000;45:4483–4491. doi: 10.1016/S0013-4686(00)00504-1. [DOI] [Google Scholar]
- Leonard K. C. Bard A. J. J. Am. Chem. Soc. 2013;135:15890–15896. doi: 10.1021/ja407395m. [DOI] [PubMed] [Google Scholar]
- Rodríguez-López J. Alpuche-Avilés M. A. Bard A. J. J. Am. Chem. Soc. 2008;130:16985–16995. doi: 10.1021/ja8050553. [DOI] [PubMed] [Google Scholar]
- Hansma P. Drake B. Marti O. Gould S. Prater C. Science. 1989;243:641–643. doi: 10.1126/science.2464851. [DOI] [PubMed] [Google Scholar]
- Chen C.-C. Zhou Y. Baker L. A. Annu. Rev. Anal. Chem. 2012;5:207–228. doi: 10.1146/annurev-anchem-062011-143203. [DOI] [PubMed] [Google Scholar]
- Page A. Perry D. Unwin P. R. Proc. R. Soc. A. 2017;473:20160889. doi: 10.1098/rspa.2016.0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momotenko D. McKelvey K. Kang M. Meloni G. N. Unwin P. R. Anal. Chem. 2016;88:2838–2846. doi: 10.1021/acs.analchem.5b04566. [DOI] [PubMed] [Google Scholar]
- Payne N. A. Dawkins J. I. G. Schougaard S. B. Mauzeroll J. Anal. Chem. 2019;91:15718–15725. doi: 10.1021/acs.analchem.9b03907. [DOI] [PubMed] [Google Scholar]
- Shkirskiy V. Kang M. McPherson I. J. Bentley C. L. Wahab O. J. Daviddi E. Colburn A. W. Unwin P. R. Anal. Chem. 2020;92:12509–12517. doi: 10.1021/acs.analchem.0c02358. [DOI] [PubMed] [Google Scholar]
- Chen C.-C. Zhou Y. Baker L. A. Annu. Rev. Anal. Chem. 2012;5:207–228. doi: 10.1146/annurev-anchem-062011-143203. [DOI] [PubMed] [Google Scholar]
- Zhang J. Zhu T. Lang J. Fu W. Li F. Curr. Opin. Electrochem. 2020;22:178–185. doi: 10.1016/j.coelec.2020.06.001. [DOI] [Google Scholar]
- Shi X. Qing W. Marhaba T. Zhang W. Electrochim. Acta. 2020;332:135472. doi: 10.1016/j.electacta.2019.135472. [DOI] [Google Scholar]
- Bentley C. L. Edmondson J. Meloni G. N. Perry D. Shkirskiy V. Unwin P. R. Anal. Chem. 2019;91:84–108. doi: 10.1021/acs.analchem.8b05235. [DOI] [PubMed] [Google Scholar]
- Ebejer N. Schnippering M. Colburn A. W. Edwards M. A. Unwin P. R. Anal. Chem. 2010;82:9141–9145. doi: 10.1021/ac102191u. [DOI] [PubMed] [Google Scholar]
- Ebejer N. Güell A. G. Lai S. C. S. McKelvey K. Snowden M. E. Unwin P. R. Annu. Rev. Anal. Chem. 2013;6:329–351. doi: 10.1146/annurev-anchem-062012-092650. [DOI] [PubMed] [Google Scholar]
- Patten H. V. Lai S. C. S. Macpherson J. V. Unwin P. R. Anal. Chem. 2012;84:5427–5432. doi: 10.1021/ac3010555. [DOI] [PubMed] [Google Scholar]
- McKelvey K. O'Connell M. A. Unwin P. R. Chem. Commun. 2013;49:2986. doi: 10.1039/C3CC00104K. [DOI] [PubMed] [Google Scholar]
- Patten H. V. Meadows K. E. Hutton L. A. Iacobini J. G. Battistel D. McKelvey K. Colburn A. W. Newton M. E. Macpherson J. V. Unwin P. R. Angew. Chem., Int. Ed. 2012;51:7002–7006. doi: 10.1002/anie.201203057. [DOI] [PubMed] [Google Scholar]
- Miller T. S. Ebejer N. Güell A. G. Macpherson J. V. Unwin P. R. Chem. Commun. 2012;48:7435. doi: 10.1039/C2CC32890A. [DOI] [PubMed] [Google Scholar]
- Liu B. Shao Y. Mirkin M. V. Anal. Chem. 2000;72:510–519. doi: 10.1021/ac990771p. [DOI] [PubMed] [Google Scholar]
- Kleijn S. E. F. Lai S. C. S. Miller T. S. Yanson A. I. Koper M. T. M. Unwin P. R. J. Am. Chem. Soc. 2012;134:18558–18561. doi: 10.1021/ja309220m. [DOI] [PubMed] [Google Scholar]
- Momotenko D. Byers J. C. McKelvey K. Kang M. Unwin P. R. ACS Nano. 2015;9:8942–8952. doi: 10.1021/acsnano.5b02792. [DOI] [PubMed] [Google Scholar]
- Bentley C. L. Kang M. Unwin P. R. Curr. Opin. Electrochem. 2017;6:23–30. doi: 10.1016/j.coelec.2017.06.011. [DOI] [Google Scholar]
- Wahab O. J. Kang M. Unwin P. R. Curr. Opin. Electrochem. 2020;22:120–128. doi: 10.1016/j.coelec.2020.04.018. [DOI] [Google Scholar]
- Toca-Herrera J. L. ChemSusChem. 2019;12:603–611. doi: 10.1002/cssc.201802383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugstad G., Atomic Force Microscopy: Understanding Basic Modes and Advanced Applications, Wiley, 2012 [Google Scholar]
- Dufrêne Y. F. Ando T. Garcia R. Alsteens D. Martinez-Martin D. Engel A. Gerber C. Müller D. J. Nat. Nanotechnol. 2017;12:295–307. doi: 10.1038/nnano.2017.45. [DOI] [PubMed] [Google Scholar]
- Deng X. Xiong F. Li X. Xiang B. Li Z. Wu X. Guo C. Li X. Li Y. Li G. Xiong W. Zeng Z. J. Nanobiotechnol. 2018;16:102. doi: 10.1186/s12951-018-0428-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nellist M. R. Laskowski F. A. L. Qiu J. Hajibabaei H. Sivula K. Hamann T. W. Boettcher S. W. Nat. Energy. 2018;3:46–52. doi: 10.1038/s41560-017-0048-1. [DOI] [Google Scholar]
- Pavliček N. Gross L. Nat. Rev. Chem. 2017;1:0005. doi: 10.1038/s41570-016-0005. [DOI] [Google Scholar]
- Xing Y. Xu M. Gui X. Cao Y. Babel B. Rudolph M. Weber S. Kappl M. Butt H.-J. Adv. Colloid Interface Sci. 2018;256:373–392. doi: 10.1016/j.cis.2018.01.004. [DOI] [PubMed] [Google Scholar]
- Rangelow I. W. Kaestner M. Ivanov T. Ahmad A. Lenk S. Lenk C. Guliyev E. Reum A. Hofmann M. Reuter C. Holz M. J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom. 2018;36:06J102. [Google Scholar]
- Balke N. Bonnell D. Ginger D. S. Kemerink M. MRS Bull. 2012;37:633–637. doi: 10.1557/mrs.2012.141. [DOI] [Google Scholar]
- Sadewasser S. and Barth C., in Characterization of Materials, ed. E. N. Kaufmann, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2012 [Google Scholar]
- Jakob D. S. Wang H. Xu X. G. ACS Nano. 2020;14:4839–4848. doi: 10.1021/acsnano.0c00767. [DOI] [PubMed] [Google Scholar]
- Jesse S. Kumar A. Arruda T. M. Kim Y. Kalinin S. V. Ciucci F. MRS Bull. 2012;37:651–658. doi: 10.1557/mrs.2012.144. [DOI] [Google Scholar]
- Iturri J. Toca-Herrera J. Polymers. 2017;9:383. doi: 10.3390/polym9080383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khotseng L., in Electrocatalysts for Fuel Cells and Hydrogen Evolution - Theory to Design, ed. A. Ray, I. Mukhopadhyay and R. K. Pati, IntechOpen, 2018 [Google Scholar]
- Nie Y. Li L. Wei Z. Chem. Soc. Rev. 2015;44:2168–2201. doi: 10.1039/C4CS00484A. [DOI] [PubMed] [Google Scholar]
- Gao S. Fan H. Zhang S. J. Mater. Chem. A. 2014;2:18263–18270. doi: 10.1039/C4TA03558E. [DOI] [Google Scholar]
- Dobrzeniecka A. Zeradjanin A. R. Masa J. Blicharska M. Wintrich D. Kulesza P. J. Schuhmann W. Catal. Today. 2016;262:74–81. doi: 10.1016/j.cattod.2015.07.043. [DOI] [Google Scholar]
- Gewirth A. A. Thorum M. S. Inorg. Chem. 2010;49:3557–3566. doi: 10.1021/ic9022486. [DOI] [PubMed] [Google Scholar]
- Muthuswamy N. Buan M. E. M. Walmsley J. C. Rønning M. Catal. Today. 2018;301:11–16. doi: 10.1016/j.cattod.2017.03.045. [DOI] [Google Scholar]
- Fernández J. L. Bard A. J. Anal. Chem. 2004;76:2281–2289. doi: 10.1021/ac035518a. [DOI] [PubMed] [Google Scholar]
- Sánchez-Sánchez C. M. Bard A. J. Anal. Chem. 2009;81:8094–8100. doi: 10.1021/ac901291v. [DOI] [PubMed] [Google Scholar]
- Eckhard K. Schuhmann W. Electrochim. Acta. 2007;53:1164–1169. doi: 10.1016/j.electacta.2007.02.028. [DOI] [Google Scholar]
- Shao M. Liu P. Zhang J. Adzic R. J. Phys. Chem. B. 2007;111:6772–6775. doi: 10.1021/jp0689971. [DOI] [PubMed] [Google Scholar]
- Perales-Rondón J. V. Herrero E. Solla-Gullón J. Sánchez-Sánchez C. M. Vivier V. J. Electroanal. Chem. 2017;793:218–225. doi: 10.1016/j.jelechem.2016.12.049. [DOI] [Google Scholar]
- Fernández J. L. Imaduwage K. P. Zoski C. G. Electrochim. Acta. 2015;180:460–470. doi: 10.1016/j.electacta.2015.08.027. [DOI] [Google Scholar]
- Zhang Y. Wu X. Fu Y. Shen W. Zeng X. Ding W. J. Mater. Res. 2014;29:2863–2870. doi: 10.1557/jmr.2014.343. [DOI] [Google Scholar]
- Cho Y.-B. Lee C. Lee Y. J. Electrochem. Soc. 2015;162:H792–H798. doi: 10.1149/2.0841510jes. [DOI] [Google Scholar]
- Botz A. Clausmeyer J. Öhl D. Tarnev T. Franzen D. Turek T. Schuhmann W. Angew. Chem., Int. Ed. 2018;57:12285–12289. doi: 10.1002/anie.201807798. [DOI] [PubMed] [Google Scholar]
- Li W. Fan F.-R. F. Bard A. J. J. Solid State Electrochem. 2012;16:2563–2568. doi: 10.1007/s10008-012-1775-7. [DOI] [Google Scholar]
- Nørskov J. K. Rossmeisl J. Logadottir A. Lindqvist L. Kitchin J. R. Bligaard T. Jónsson H. J. Phys. Chem. B. 2004;108:17886–17892. doi: 10.1021/jp047349j. [DOI] [Google Scholar]
- Sun X. Li W. Mi H. Li Y. Zhang P. Ren X. Int. J. Hydrogen Energy. 2018;43:5530–5540. doi: 10.1016/j.ijhydene.2018.01.093. [DOI] [Google Scholar]
- Pham Truong T. N. Randriamahazaka H. Ghilane J. ACS Catal. 2018;8:869–875. doi: 10.1021/acscatal.7b03158. [DOI] [Google Scholar]
- Kim S. Lee S.-C. Lee C. Kim M. H. Lee Y. Nano Energy. 2018;48:134–143. doi: 10.1016/j.nanoen.2018.03.041. [DOI] [Google Scholar]
- Singh V. Tiwari A. Nagaiah T. C. J. Mater. Chem. A. 2018;6:22545–22554. doi: 10.1039/C8TA06710D. [DOI] [Google Scholar]
- Xiao J. Kuang Q. Yang S. Xiao F. Wang S. Guo L. Sci. Rep. 2013;3:2300. doi: 10.1038/srep02300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H. Zhu X. Li M. Tang Q. Sun G. Yang W. Electrochim. Acta. 2014;144:31–41. doi: 10.1016/j.electacta.2014.08.087. [DOI] [Google Scholar]
- Tiwari A. Singh V. Nagaiah T. C. J. Mater. Chem. A. 2018;6:2681–2692. doi: 10.1039/C7TA10380H. [DOI] [Google Scholar]
- Sidhureddy B. Prins S. Wen J. Thiruppathi A. R. Govindhan M. Chen A. ACS Appl. Mater. Interfaces. 2019;11:18295–18304. doi: 10.1021/acsami.8b22351. [DOI] [PubMed] [Google Scholar]
- Yan X. Li K. Lyu L. Song F. He J. Niu D. Liu L. Hu X. Chen X. ACS Appl. Mater. Interfaces. 2016;8:3208–3214. doi: 10.1021/acsami.5b10724. [DOI] [PubMed] [Google Scholar]
- Zheng F. Zhu D. Chen Q. ACS Appl. Mater. Interfaces. 2014;6:9256–9264. doi: 10.1021/am501512j. [DOI] [PubMed] [Google Scholar]
- Michalak M. Roguska A. Nogala W. Opallo M. Nanoscale Adv. 2019;1:2645–2653. doi: 10.1039/C9NA00166B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q. Liu P. Zhu Z. Zhang J. Cao F. Corros. Sci. 2020;164:108312. doi: 10.1016/j.corsci.2019.108312. [DOI] [Google Scholar]
- Tiwari A. Singh V. Mandal D. Nagaiah T. C. J. Mater. Chem. A. 2017;5:20014–20023. doi: 10.1039/C7TA05503J. [DOI] [Google Scholar]
- Shi Q. Peng F. Liao S. Wang H. Yu H. Liu Z. Zhang B. Su D. J. Mater. Chem. A. 2013;1:14853. doi: 10.1039/C3TA12647A. [DOI] [Google Scholar]
- Gong K. Du F. Xia Z. Durstock M. Dai L. Science. 2009;323:760–764. doi: 10.1126/science.1168049. [DOI] [PubMed] [Google Scholar]
- Xin S. Liu Z. Ma L. Sun Y. Xiao C. Li F. Du Y. Nano Res. 2016;9:3795–3811. doi: 10.1007/s12274-016-1249-9. [DOI] [Google Scholar]
- Wang T. Zhuo J. Chen Y. Du K. Papakonstantinou P. Zhu Z. Shao Y. Li M. ChemCatChem. 2014;6:1877–1881. doi: 10.1002/cctc.201402038. [DOI] [Google Scholar]
- Liao L. Zhu J. Bian X. Zhu L. Scanlon M. D. Girault H. H. Liu B. Adv. Funct. Mater. 2013;23:5326–5333. doi: 10.1002/adfm.201300318. [DOI] [Google Scholar]
- Li H. Tsai C. Koh A. L. Cai L. Contryman A. W. Fragapane A. H. Zhao J. Han H. S. Manoharan H. C. Abild-Pedersen F. Nørskov J. K. Zheng X. Nat. Mater. 2016;15:48–53. doi: 10.1038/nmat4465. [DOI] [PubMed] [Google Scholar]
- Ma L. Zhou H. Xin S. Xiao C. Li F. Ding S. Electrochim. Acta. 2015;178:767–777. doi: 10.1016/j.electacta.2015.08.060. [DOI] [Google Scholar]
- Kolagatla S. Subramanian P. Schechter A. ChemSusChem. 2019;12:2708–2714. doi: 10.1002/cssc.201900656. [DOI] [PubMed] [Google Scholar]
- O'Connell M. A. Lewis J. R. Wain A. J. Chem. Commun. 2015;51:10314–10317. doi: 10.1039/C5CC01640A. [DOI] [PubMed] [Google Scholar]
- Yan D. Li Y. Huo J. Chen R. Dai L. Wang S. Adv. Mater. 2017;29:1606459. doi: 10.1002/adma.201606459. [DOI] [PubMed] [Google Scholar]
- Shen A. Zou Y. Wang Q. Dryfe R. A. W. Huang X. Dou S. Dai L. Wang S. Angew. Chem. 2014;126:10980–10984. doi: 10.1002/ange.201406695. [DOI] [PubMed] [Google Scholar]
- Perry D. Paulose Nadappuram B. Momotenko D. Voyias P. D. Page A. Tripathi G. Frenguelli B. G. Unwin P. R. J. Am. Chem. Soc. 2016;138:3152–3160. doi: 10.1021/jacs.5b13153. [DOI] [PubMed] [Google Scholar]
- Tao L. Qiao M. Jin R. Li Y. Xiao Z. Wang Y. Zhang N. Xie C. He Q. Jiang D. Yu G. Li Y. Wang S. Angew. Chem. 2019;131:1031–1036. doi: 10.1002/ange.201810207. [DOI] [PubMed] [Google Scholar]
- Tahir M. Pan L. Idrees F. Zhang X. Wang L. Zou J.-J. Wang Z. L. Nano Energy. 2017;37:136–157. doi: 10.1016/j.nanoen.2017.05.022. [DOI] [Google Scholar]
- Liu Y. Cheng H. Lyu M. Fan S. Liu Q. Zhang W. Zhi Y. Wang C. Xiao C. Wei S. Ye B. Xie Y. J. Am. Chem. Soc. 2014;136:15670–15675. doi: 10.1021/ja5085157. [DOI] [PubMed] [Google Scholar]
- Song F. Hu X. Nat. Commun. 2014;5:4477. doi: 10.1038/ncomms5477. [DOI] [PubMed] [Google Scholar]
- Liang H. Meng F. Cabán-Acevedo M. Li L. Forticaux A. Xiu L. Wang Z. Jin S. Nano Lett. 2015;15:1421–1427. doi: 10.1021/nl504872s. [DOI] [PubMed] [Google Scholar]
- Suen N.-T. Hung S.-F. Quan Q. Zhang N. Xu Y.-J. Chen H. M. Chem. Soc. Rev. 2017;46:337–365. doi: 10.1039/C6CS00328A. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y. Sato E. Mater. Chem. Phys. 1986;14:397–426. doi: 10.1016/0254-0584(86)90045-3. [DOI] [Google Scholar]
- Konkena B. Masa J. Botz A. J. R. Sinev I. Xia W. Koßmann J. Drautz R. Muhler M. Schuhmann W. ACS Catal. 2017;7:229–237. doi: 10.1021/acscatal.6b02203. [DOI] [Google Scholar]
- Gong M. Li Y. Wang H. Liang Y. Wu J. Z. Zhou J. Wang J. Regier T. Wei F. Dai H. J. Am. Chem. Soc. 2013;135:8452–8455. doi: 10.1021/ja4027715. [DOI] [PubMed] [Google Scholar]
- Louie M. W. Bell A. T. J. Am. Chem. Soc. 2013;135:12329–12337. doi: 10.1021/ja405351s. [DOI] [PubMed] [Google Scholar]
- Minguzzi A. Battistel D. Rodríguez-López J. Vertova A. Rondinini S. Bard A. J. Daniele S. J. Phys. Chem. C. 2015;119:2941–2947. doi: 10.1021/jp510651f. [DOI] [Google Scholar]
- Minguzzi A. Alpuche-Aviles M. A. López J. R. Rondinini S. Bard A. J. Anal. Chem. 2008;80:4055–4064. doi: 10.1021/ac8001287. [DOI] [PubMed] [Google Scholar]
- Jiao F. Frei H. Angew. Chem., Int. Ed. 2009;48:1841–1844. doi: 10.1002/anie.200805534. [DOI] [PubMed] [Google Scholar]
- Kim J. Y. Ahn H. S. Bard A. J. Anal. Chem. 2018;90:3045–3049. doi: 10.1021/acs.analchem.7b04728. [DOI] [PubMed] [Google Scholar]
- Barforoush J. M. Seuferling T. E. Jantz D. T. Song K. R. Leonard K. C. ACS Appl. Energy Mater. 2018;1:1415–1423. doi: 10.1021/acsaem.8b00190. [DOI] [Google Scholar]
- Arroyo-Currás N. Bard A. J. J. Phys. Chem. C. 2015;119:8147–8154. doi: 10.1021/acs.jpcc.5b00106. [DOI] [Google Scholar]
- Steimecke M. Seiffarth G. Bron M. Anal. Chem. 2017;89:10679–10686. doi: 10.1021/acs.analchem.7b01060. [DOI] [PubMed] [Google Scholar]
- Klaus S. Cai Y. Louie M. W. Trotochaud L. Bell A. T. J. Phys. Chem. C. 2015;119:7243–7254. doi: 10.1021/acs.jpcc.5b00105. [DOI] [Google Scholar]
- Trotochaud L. Young S. L. Ranney J. K. Boettcher S. W. J. Am. Chem. Soc. 2014;136:6744–6753. doi: 10.1021/ja502379c. [DOI] [PubMed] [Google Scholar]
- Lu M. Kharkwal S. Ng H. Y. Li S. F. Y. Biosens. Bioelectron. 2011;26:4728–4732. doi: 10.1016/j.bios.2011.05.036. [DOI] [PubMed] [Google Scholar]
- Zhang C. Mahmood N. Yin H. Liu F. Hou Y. Adv. Mater. 2013;25:4932–4937. doi: 10.1002/adma.201301870. [DOI] [PubMed] [Google Scholar]
- Wu J. Park H. W. Yu A. Higgins D. Chen Z. J. Phys. Chem. C. 2012;116:9427–9432. doi: 10.1021/jp301644e. [DOI] [Google Scholar]
- Song F. Bai L. Moysiadou A. Lee S. Hu C. Liardet L. Hu X. J. Am. Chem. Soc. 2018;140:7748–7759. doi: 10.1021/jacs.8b04546. [DOI] [PubMed] [Google Scholar]
- Bezerra C. W. B. Zhang L. Lee K. Liu H. Marques A. L. B. Marques E. P. Wang H. Zhang J. Electrochim. Acta. 2008;53:4937–4951. doi: 10.1016/j.electacta.2008.02.012. [DOI] [Google Scholar]
- Yang L. Zhao Y. Chen S. Wu Q. Wang X. Hu Z. Chin. J. Catal. 2013;34:1986–1991. doi: 10.1016/S1872-2067(12)60713-X. [DOI] [Google Scholar]
- Higgins D. Zamani P. Yu A. Chen Z. Energy Environ. Sci. 2016;9:357–390. doi: 10.1039/C5EE02474A. [DOI] [Google Scholar]
- Jiang R. on Tung S. Tang Z. Li L. Ding L. Xi X. Liu Y. Zhang L. Zhang J. Energy Storage Mater. 2018;12:260–276. doi: 10.1016/j.ensm.2017.11.005. [DOI] [Google Scholar]
- Shao M. Chang Q. Dodelet J.-P. Chenitz R. Chem. Rev. 2016;116:3594–3657. doi: 10.1021/acs.chemrev.5b00462. [DOI] [PubMed] [Google Scholar]
- Jamesh M.-I. Sun X. J. Power Sources. 2018;400:31–68. doi: 10.1016/j.jpowsour.2018.07.125. [DOI] [Google Scholar]
- Wang H.-F. Tang C. Li B.-Q. Zhang Q. Inorg. Chem. Front. 2018;5:521–534. doi: 10.1039/C7QI00780A. [DOI] [Google Scholar]
- Li J. Zheng G. Adv. Sci. 2017;4:1600380. doi: 10.1002/advs.201600380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reier T. Nong H. N. Teschner D. Schlögl R. Strasser P. Adv. Energy Mater. 2017;7:1601275. doi: 10.1002/aenm.201601275. [DOI] [Google Scholar]
- Jamesh M. I. J. Power Sources. 2016;333:213–236. doi: 10.1016/j.jpowsour.2016.09.161. [DOI] [Google Scholar]
- Narayan S. R. Manohar A. Mukerjee S. Interface Magazine. 2015;24:65–69. doi: 10.1149/2.F06152if. [DOI] [Google Scholar]
- Busch M. Halck N. B. Kramm U. I. Siahrostami S. Krtil P. Rossmeisl J. Nano Energy. 2016;29:126–135. doi: 10.1016/j.nanoen.2016.04.011. [DOI] [Google Scholar]
- Seiffarth G. Steimecke M. Walther T. Kühhirt M. Rümmler S. Bron M. Electroanalysis. 2016;28:2335–2345. doi: 10.1002/elan.201600254. [DOI] [Google Scholar]
- Chakrabarty S. Mukherjee A. Su W.-N. Basu S. Int. J. Hydrogen Energy. 2019;44:1565–1578. doi: 10.1016/j.ijhydene.2018.11.163. [DOI] [Google Scholar]
- Ma L. Zhou H. Sun Y. Xin S. Xiao C. Kumatani A. Matsue T. Zhang P. Ding S. Li F. Electrochim. Acta. 2017;252:338–349. doi: 10.1016/j.electacta.2017.08.192. [DOI] [Google Scholar]
- Barwe S. Andronescu C. Engels R. Conzuelo F. Seisel S. Wilde P. Chen Y.-T. Masa J. Schuhmann W. Electrochim. Acta. 2019;297:1042–1051. doi: 10.1016/j.electacta.2018.12.047. [DOI] [Google Scholar]
- Liao Y. Mustonen K. Tulić S. Skákalová V. Khan S. A. Laiho P. Zhang Q. Li C. Monazam M. R. A. Kotakoski J. Lipsanen H. Kauppinen E. I. ACS Nano. 2019;13:11522–11529. doi: 10.1021/acsnano.9b05049. [DOI] [PubMed] [Google Scholar]
- Lasia A., in Handbook of Fuel Cells, ed. W. Vielstich, A. Lamm, H. A. Gasteiger and H. Yokokawa, John Wiley & Sons, Ltd, Chichester, UK, 2010 [Google Scholar]
- Gong M. Wang D.-Y. Chen C.-C. Hwang B.-J. Dai H. Nano Res. 2016;9:28–46. doi: 10.1007/s12274-015-0965-x. [DOI] [Google Scholar]
- Seh Z. W. Kibsgaard J. Dickens C. F. Chorkendorff I. Nørskov J. K. Jaramillo T. F. Science. 2017;355:eaad4998. doi: 10.1126/science.aad4998. [DOI] [PubMed] [Google Scholar]
- Zhang J. Li H. Guo P. Ma H. Zhao X. S. J. Mater. Chem. A. 2016;4:8497–8511. doi: 10.1039/C6TA01657J. [DOI] [Google Scholar]
- Walter M. G. Warren E. L. McKone J. R. Boettcher S. W. Mi Q. Santori E. A. Lewis N. S. Chem. Rev. 2010;110:6446–6473. doi: 10.1021/cr1002326. [DOI] [PubMed] [Google Scholar]
- Trasatti S. J. Electroanal. Chem. Interfacial Electrochem. 1972;39:163–184. doi: 10.1016/S0022-0728(72)80485-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández J. L. Zoski C. G. J. Phys. Chem. C. 2018;122:71–82. doi: 10.1021/acs.jpcc.7b08976. [DOI] [Google Scholar]
- Valenti G. Boni A. Melchionna M. Cargnello M. Nasi L. Bertoni G. Gorte R. J. Marcaccio M. Rapino S. Bonchio M. Fornasiero P. Prato M. Paolucci F. Nat. Commun. 2016;7:13549. doi: 10.1038/ncomms13549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z. Fang B. Bonakdarpour A. Sun A. Wilkinson D. P. Wang D. Appl. Catal., B. 2012;125:59–66. doi: 10.1016/j.apcatb.2012.05.013. [DOI] [Google Scholar]
- Faber M. S. Lukowski M. A. Ding Q. Kaiser N. S. Jin S. J. Phys. Chem. C. 2014;118:21347–21356. doi: 10.1021/jp506288w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran P. D. Tran T. V. Orio M. Torelli S. Truong Q. D. Nayuki K. Sasaki Y. Chiam S. Y. Yi R. Honma I. Barber J. Artero V. Nat. Mater. 2016;15:640–646. doi: 10.1038/nmat4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasion D. Barforoush J. M. Qiao Q. Zhu Y. Ren S. Leonard K. C. ACS Catal. 2015;5:6653–6657. doi: 10.1021/acscatal.5b01637. [DOI] [Google Scholar]
- Zou X. Zhang Y. Chem. Soc. Rev. 2015;44:5148–5180. doi: 10.1039/C4CS00448E. [DOI] [PubMed] [Google Scholar]
- Salleh S. H. Thomas S. Yuwono J. A. Venkatesan K. Birbilis N. Electrochim. Acta. 2015;161:144–152. doi: 10.1016/j.electacta.2015.02.079. [DOI] [Google Scholar]
- Zhang T. Tao Z. Chen J. Mater. Horiz. 2014;1:196–206. doi: 10.1039/C3MH00059A. [DOI] [Google Scholar]
- Kundu A. Gil J. H. Jang J. H. Lee H. R. Jung C. R. Ku B. S. Chae K. S. Int. J. Hydrogen Energy. 2010;35:10827–10832. doi: 10.1016/j.ijhydene.2010.07.032. [DOI] [Google Scholar]
- Nidola A. Schira R. Int. J. Hydrogen Energy. 1986;11:449–454. doi: 10.1016/0360-3199(86)90064-9. [DOI] [Google Scholar]
- Merki D. Hu X. Energy Environ. Sci. 2011;4:3878. doi: 10.1039/C1EE01970H. [DOI] [Google Scholar]
- Hinnemann B. Moses P. G. Bonde J. Jørgensen K. P. Nielsen J. H. Horch S. Chorkendorff I. Nørskov J. K. J. Am. Chem. Soc. 2005;127:5308–5309. doi: 10.1021/ja0504690. [DOI] [PubMed] [Google Scholar]
- Jaramillo T. F. Jorgensen K. P. Bonde J. Nielsen J. H. Horch S. Chorkendorff I. Science. 2007;317:100–102. doi: 10.1126/science.1141483. [DOI] [PubMed] [Google Scholar]
- Li H. Du M. Mleczko M. J. Koh A. L. Nishi Y. Pop E. Bard A. J. Zheng X. J. Am. Chem. Soc. 2016;138:5123–5129. doi: 10.1021/jacs.6b01377. [DOI] [PubMed] [Google Scholar]
- Ataca C. Ciraci S. Phys. Rev. B. 2012;85:195410. doi: 10.1103/PhysRevB.85.195410. [DOI] [PubMed] [Google Scholar]
- Li H. Yu K. Li C. Tang Z. Guo B. Lei X. Fu H. Zhu Z. Sci. Rep. 2015;5:18730. doi: 10.1038/srep18730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S. Sahoo P. K. Satpati A. K. ACS Omega. 2017;2:7532–7545. doi: 10.1021/acsomega.7b00678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X. Chen Y. Sun T. Huang J. Zhang W. Wang Q. Cao R. Energy Environ. Sci. 2020;13:174–182. doi: 10.1039/C9EE02380A. [DOI] [Google Scholar]
- Jedraszko J. Adamiak W. Nogala W. Girault H. H. Opallo M. J. Electroanal. Chem. 2018;819:101–106. doi: 10.1016/j.jelechem.2017.09.026. [DOI] [Google Scholar]
- Bentley C. L. Unwin P. R. Faraday Discuss. 2018;210:365–379. doi: 10.1039/C8FD00028J. [DOI] [PubMed] [Google Scholar]
- Choi M. Siepser N. P. Jeong S. Wang Y. Jagdale G. Ye X. Baker L. A. Nano Lett. 2020;20:1233–1239. doi: 10.1021/acs.nanolett.9b04640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britnell L. Gorbachev R. V. Jalil R. Belle B. D. Schedin F. Katsnelson M. I. Eaves L. Morozov S. V. Mayorov A. S. Peres N. M. R. Castro Neto A. H. Leist J. Geim A. K. Ponomarenko L. A. Novoselov K. S. Nano Lett. 2012;12:1707–1710. doi: 10.1021/nl3002205. [DOI] [PubMed] [Google Scholar]
- Gao M. Lyalin A. Taketsugu T. Int. J. Quantum Chem. 2013;113:443–452. doi: 10.1002/qua.24066. [DOI] [Google Scholar]
- Liu D.-Q. Tao B. Ruan H.-C. Bentley C. L. Unwin P. R. Chem. Commun. 2019;55:628–631. doi: 10.1039/C8CC08517J. [DOI] [PubMed] [Google Scholar]
- Duan X. Xu J. Wei Z. Ma J. Guo S. Wang S. Liu H. Dou S. Adv. Mater. 2017;29:1701784. doi: 10.1002/adma.201701784. [DOI] [PubMed] [Google Scholar]
- Costentin C. Robert M. Savéant J.-M. Chem. Soc. Rev. 2013;42:2423–2436. doi: 10.1039/C2CS35360A. [DOI] [PubMed] [Google Scholar]
- Olah G. A. Goeppert A. Prakash G. K. S. J. Org. Chem. 2009;74:487–498. doi: 10.1021/jo801260f. [DOI] [PubMed] [Google Scholar]
- Wang W.-H. Himeda Y. Muckerman J. T. Manbeck G. F. Fujita E. Chem. Rev. 2015;115:12936–12973. doi: 10.1021/acs.chemrev.5b00197. [DOI] [PubMed] [Google Scholar]
- Zhang J. Pietro W. J. Lever A. B. P. J. Electroanal. Chem. 1996;403:93–100. doi: 10.1016/0022-0728(95)04270-9. [DOI] [Google Scholar]
- Zhang F. Co A. C. J. Electrochem. Soc. 2020;167:046517. doi: 10.1149/1945-7111/ab7a80. [DOI] [Google Scholar]
- Wadas A. Rutkowska I. A. Bartel M. Zoladek S. Rajeshwar K. Kulesza P. J. Russ. J. Electrochem. 2017;53:1194–1203. doi: 10.1134/S1023193517100135. [DOI] [Google Scholar]
- Sreekanth N. Phani K. L. Chem. Commun. 2014;50:11143–11146. doi: 10.1039/C4CC03099K. [DOI] [PubMed] [Google Scholar]
- Sreekanth N. Nazrulla M. A. Vineesh T. V. Sailaja K. Phani K. L. Chem. Commun. 2015;51:16061–16064. doi: 10.1039/C5CC06051F. [DOI] [PubMed] [Google Scholar]
- Hori Y., in Modern Aspects of Electrochemistry, ed. C. G. Vayenas, R. E. White and M. E. Gamboa-Aldeco, Springer New York, New York, NY, 2008, vol. 42, pp. 89–189 [Google Scholar]
- Arrocha-Arcos A. A. Cervantes-Alcalá R. Huerta-Miranda G. A. Miranda-Hernández M. Electrochim. Acta. 2017;246:1082–1087. doi: 10.1016/j.electacta.2017.06.147. [DOI] [Google Scholar]
- Amatore C. Saveant J. M. J. Am. Chem. Soc. 1981;103:5021–5023. doi: 10.1021/ja00407a008. [DOI] [Google Scholar]
- Verdaguer-Casadevall A. Li C. W. Johansson T. P. Scott S. B. McKeown J. T. Kumar M. Stephens I. E. L. Kanan M. W. Chorkendorff I. J. Am. Chem. Soc. 2015;137:9808–9811. doi: 10.1021/jacs.5b06227. [DOI] [PubMed] [Google Scholar]
- Li C. W. Ciston J. Kanan M. W. Nature. 2014;508:504–507. doi: 10.1038/nature13249. [DOI] [PubMed] [Google Scholar]
- Mariano R. G. McKelvey K. White H. S. Kanan M. W. Science. 2017;358:1187–1192. doi: 10.1126/science.aao3691. [DOI] [PubMed] [Google Scholar]
- Lai S. C. S. Dudin P. V. Macpherson J. V. Unwin P. R. J. Am. Chem. Soc. 2011;133:10744–10747. doi: 10.1021/ja203955b. [DOI] [PubMed] [Google Scholar]