

Abstract
A photoredox-catalyzed oxidative decarboxylative coupling of small peptides is reported, giving access to a variety of N,O-acetals. They were used as intermediates for the addition of phenols and indoles, leading to novel peptide scaffolds and bioconjugates. Amino acids with nucleophilic side chains, such as serine, threonine, tyrosine and tryptophan, could also be used as partners to access tri- and tetrapeptide derivatives with non-natural cross-linking.
A photoredox approach for the generation of N-acyliminiums derived from peptides enabling diversification via Friedel–Crafts reactions.
1. Introduction
Bioactive peptides have gained a lot of attention in the past decade as therapeutic compounds interacting with receptors or inhibiting protein–protein interactions. The biological activity and metabolic stability of native peptides is usually improved by the introduction of unnatural amino-acids (UAA) and rigidifying elements.1 Moreover bioconjugation between peptides and other types of synthetic or natural bioactive molecules opens access to a broader chemical space. This is particularly interesting for drug discovery as it allows combining the best of two worlds, such as the higher target affinity of peptides and the better membrane permeability of small non polar synthetic compounds.2 This strategy can be used to finely tune the pharmacological properties of drugs.3 With the ambition to develop an efficient method for the structural diversification of peptides, we were interested in the functionalization of the C-termini of peptides, which can have a fundamental influence on their bioactivity.4 In addition, as the C-terminus is unique and free in most peptides, such an approach would be at the same time general and selective. Most methods for the modification of native peptides rely on the specific reactivity of nucleophilic residues present in amino acids such as cysteine and lysine, and examples of functionalization of the C-terminus remain scarce.5
Recently, decarboxylative coupling has emerged as a method of choice to reach this goal, either via one or two-electron processes (Scheme 1A). Concerning the latter, the oxidative decarboxylative conversion of carboxylic acids to alcohols under electrochemical conditions was already reported by Hofer and Moest in 1902.6 Seebach and coworkers later applied this method to amino acids and small peptides.7 In the presence of methanol or acetic acid, N,O-acetals were generated. Activation of the N,O-acetals to give versatile N-acyliminium intermediates I8 then allowed the addition of different nucleophiles (Grignard reagents, phosphites, allylsilanes, terminal alkyne or TMSCN). In 2020, the Malins group reported an extension of this approach to more complex peptides for the total synthesis of analogues of Biseokeaniamides A–C.9
Scheme 1. (A) Decarboxylative functionalization of peptides; (B) our photoredox strategy for the synthesis of bioconjugates.

The formation of N,O-acetals could also be achieved using chemical oxidants, in particular hypervalent iodine reagents. The Suárez group investigated the decarboxylative hydroxylation of proline derivatives under visible light using phenyliodine(iii) diacetate (PIDA) as oxidant.10 The strategy was later used by Kita and coworkers on non-cyclic amino-acids, but using heating instead of light to initiate the oxidation step.11 This method, often coupled with in situ functionalization of the acetals, was then applied to amino acids and small peptides.12
The advent of photoredox catalysis contributed to the development of new strategies for bioconjugation via one–electron processes (Scheme 1A).13 Starting from native peptides, C-terminal decarboxylative arylation,14 reduction,15 allylation,16 alkynylation,17 cyanation,18 azidation19 and Giese coupling20 have been described. Decarboxylative coupling reactions can alternatively be performed by activation of the carboxylic acid via the formation of a redox-active ester (RAE). Single Electron Transfer (SET) from a metal complex or from a photocatalyst then induces the decarboxylation.13d Using nickel complexes, the Baran group elegantly functionalized peptides through decarboxylative alkylation,21 alkenylation,22 alkynylation,23 Giese coupling24 and borylation.25 Moreover, photoredox catalyzed decarboxylative arylation,26 thio-27 and selenoarylation28 were also reported. The generation of an α-aminyl radical intermediate II was involved in all these processes. However, in the case of the cyanation methodology involving a hypervalent iodine reagent, computation indicated that the radical was further oxidized to form an iminium intermediate, constituting one of the rare cases of two-electron oxidation under photoredox catalysis.18
Decarboxylative photoredox catalyzed C–O bonds formation has been described only in a few examples of protected amino acids, and is often associated with overoxidation to give amides.29 To the best of our knowledge, the generation of peptide derived N,O-acetals from free carboxylic acids has never been reported using photoredox conditions. When considering that photocatalysis can proceed under milder conditions (room temperature, visible light, weaker oxidants) and requires less technical know-how than electrochemistry, such a method would be highly useful for synthetic and medicinal chemists requiring modified peptides. Furthermore, the method would be complementary to existing one-electron approaches: functional group tolerance would be lower, but access to versatile iminium intermediates would allow extensive diversification of the products with nucleophiles, leading to different types of bond formations.
Herein, we report a photoredox catalyzed oxidative-decarboxylative coupling of small peptides (Scheme 1B). This methodology allowed the synthesis of modified peptides with N,O-acetals at the C-terminus, which served as intermediates to introduce phenols and indoles via Friedel–Crafts reactions. The scope of nucleophiles included alcohols and electron-rich aromatic residues present in natural amino acids such as serine, threonine, tyrosine and tryptophan, giving access to new types of peptide cross-linking for the synthesis of non-natural tri- and tetrapeptides.
2. Results and discussions
2.1. Discovery and optimization of the reaction
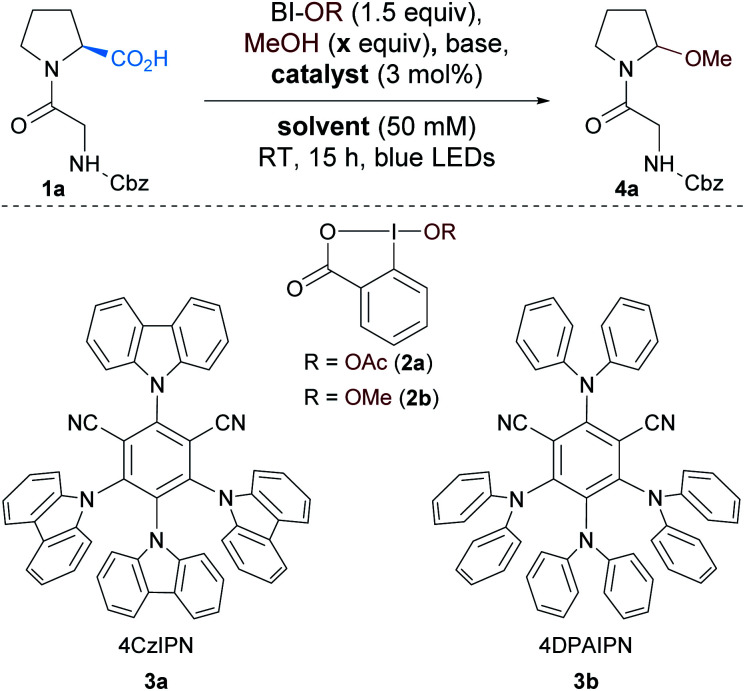
We started our study by exploring the oxidative decarboxylation of Cbz–Gly–Pro (1a) in presence of methanol as the model reaction (Table 1). The hypervalent iodine reagent acetoxybenziodoxolone (BI-OAc, 2a) was selected as oxidant for the optimization, due to its recent success in oxidative decarboxylative reactions.30
Optimization of the oxidative decarboxylation of dipeptides.
| |||||
|---|---|---|---|---|---|
| Entry | Solvent | Catalyst | x | Base (2 equiv.) | HPLC yielda (%) |
| 1b | DMF | 4CzIPN (3a) | 50 | K2HPO4 | 46 (18)c |
| 2b | DMF | Ru(bpy)3Cl2 | 50 | K2HPO4 | 59 |
| 3b | DMF | Ru(bpy)3Cl2 | 10 | K2HPO4 | 78 |
| 4 | DMF | Ru(bpy)3Cl2 | 10 | K2HPO4 | 82 |
| 5 | DMF | Ru(bpy)3Cl2 | 5 | K2HPO4 | >95 |
| 6 | DMF | Ru(bpy)3Cl2 | 2 | K2HPO4 | >95 |
| 7 | DMF | Eosin Y | 5 | K2HPO4 | 17 |
| 8d | DMF | Rhodamine B | 5 | K2HPO4 | 27 |
| 9d | DMF | Rose bengal | 5 | K2HPO4 | 35 |
| 10d | DMF | 4DPAIPN (3b) | 5 | K2HPO4 | 45 |
| 11 | MeCN | Ru(bpy)3Cl2 | 5 | K2HPO4 | >95 |
| 12 | MeCN | Ru(bpy) 3 Cl 2 | 5 | None | >95 (68) c |
| 13 | DCE | Ru(bpy) 3 Cl 2 | 5 | None | >95 |
| 14 e | MeCN | Ru(bpy) 3 Cl 2 | 0 | None | >95 (75) c |
Ratio of integration at 214 nm by RP-HPLC.
Concentration 10 mM.
Isolated yield on 0.3 mmol.
Green LEDs.
BI-OMe (2b) instead of BI-OAc (2a).
Based on our work on the alkynylation of peptides using organic photoredox catalysts,17 4CzIPN (2,4,5,6-tetra(9H-carbazol-9-yl)isophthalonitrile, 3a) was selected as photocatalyst and K2HPO4 as base. To our delight the desired methanol N,O-acetal 4a was formed with 50 equivalents of MeOH in DMF in 46% HPLC yield and 18% isolated yield (entry 1). Changing the photocatalyst to Ru(bpy)3Cl2 slightly increased the yield (entry 2).31 Decreasing the amount of MeOH in the reaction mixture and increasing the concentration had a beneficial impact on the yield (entries 3 and 4). Full conversion was observed with either 5 or 2 equivalents of methanol (entries 5 and 6). For practical reasons, 5 equivalents of methanol were employed for the rest of the optimization, but 2 equivalents only of alcohols will be used during the investigation of the scope of the reaction.
With those conditions in hand we decided to screen some organic dyes to accomplish a metal-free process. We thus selected organophotocatalysts with similar oxidative properties in the excited state as Ru(bpy)3Cl2 (RuII*/RuI: +0.77 V vs. SCE).32 However, neither Eosin Y (EY*/EY˙−: +0.83 V vs. SCE),33 rhodamine B (RhB*/RhB˙−: +0.84 V vs. SCE)34 nor rose bengal (RB*/RB˙−: +0.81 V vs. SCE)34 were suitable for the transformation and degradation of the dyes was observed (entries 7–9). Similarly, despite its promising redox properties, 4DPAIPN (2,4,5,6-tetrakis(diphenylamino)isophthalonitrile, 3b) (4DPAIN*/4DPAIN˙−: 0.90 V vs. SCE)17 only afforded the desired product in 45% yield (entry 10). Unfortunately DMF was not a suitable solvent for further functionalization of the N,O-acetals. Based on the reported use of N,O-acetals to generate N-acyliminiums in MeCN,35 we tested this solvent and full and clean conversion was observed (entry 11). A control experiment without base was attempted and full conversion to product 4a was also observed (entry 12). Under these conditions, N,O-acetal 4a could be isolated in 68% yield on a 0.3 mmol scale. A similar result was obtained in DCE (entry 13). Interestingly, the reaction was also working with methoxybenziodoxolone (BI-OMe, 2b) and the corresponding N,O-acetal 4a was obtained in 75% isolated yield (entry 14). A slightly higher yield was obtained with 2b but the necessity to pre-form the hypervalent iodine reagent prior to the reaction is not desirable for more complex alcohols. Finally, control experiments were carried out and only traces of the desired product were observed in the absence of light or catalyst.
2.2. Scope of the oxidative decarboxylation reaction
A robustness test with different protected amino acids present in the reaction mixture showed a moderate functional group tolerance,36 setting the basis for the investigation of the scope (Scheme 2). Previous works based on electrochemistry or chemical oxidation had focused mostly on the incorporation of simple alcohols as solvents.7 Our method was already efficient with only two equivalents of methanol, as well as allyl, propargyl and benzyl alcohols, to give N,O-acetals 4b–d in 64–98% yield (Scheme 2A). Functional groups such as cyanide, chloride and azide were well tolerated and the corresponding N,O-acetals 4e–g were obtained in 47–91% yield. The introduction of bioorthogonal terminal alkyne or azido groups is highly valuable as it allows to further functionalize the products. The more sterically hindered secondary alcohol (L)-menthol gave the N,O-acetal 4h in 35% yield.
Scheme 2. Scope of obtained N,O-acetals. Reactions performed on 0.3 mmol scale. Yields of isolated products are given. aCompound obtained with a low diastereoselectivity that could not be determined exactly due to the presence of rotamers.

The method could be extended to the alcohol-containing amino acids serine and threonine allowing to access a new type of cross-linked peptides 4i and 4j in 42% and 38% yield, respectively (Scheme 2B). Finally the conditions were successfully applied to other dipeptides (Cbz–Ala–Ala (1b), Cbz–Gly–Phe (1c) and Cbz–Pro–Val (1d)) with various alcohols and the N,O-acetals 4k–m were formed in 46–68% yields as mixtures of diastereoisomers in the case of 4k and 4m (Scheme 2C).
2.3. Functionalization of the N,O-acetals
During our studies on the formation of N,O-acetals we observed that in absence of alcohol the reaction was still taking place and N,OAc-acetals were generated. As we could not isolate these compounds, we wondered whether we could take advantage of their reactivity for the direct reaction with nucleophiles (Scheme 3).
Scheme 3. Scope of the one-pot arylation of dipeptides with phenols and indoles. Reactions performed on 0.3 mmol scale. Yields of isolated products are given. aCompound obtained with a low diastereoselectivity that could not be determined exactly due to the presence of rotamers.

Based on the report of White and coworkers,37 we examined the addition of p-cresol in presence of BF3·Et2O as Lewis acid. While only degradation was observed in MeCN, we were pleased to observe full conversion of Cbz–Gly–Pro (1a) to Friedel–Crafts product 5a in DCE (Scheme 3A(1), conditions A). After the one-pot two steps procedure 5a could be obtained in 62% yield with complete selectivity for the ortho position. Similarly, electron-rich para-methoxyphenol could be added to give 5b in 78% yield. Estrone was also compatible with the reaction conditions: a 7 : 3 mixture of two regioisomers of 5c was obtained in 63% yield with a preference for the position in para to the alkyl donating group. Para-methoxyphenol was successfully employed for the arylation of other dipeptides (Cbz–Gly–Phe (1c), Cbz–Pro–Val (1d) and Cbz–Ala–Ala (1b)) to give products 5d–f in 49–64% yield. Cbz–Pro–Gly (1e) could also be arylated at the C-terminal position. Electron-withdrawing groups such as bromine and fluorine were tolerated on the phenol with this more reactive iminium and the corresponding products 5g and 5h were obtained in 47% and 57% yield respectively. To our delight phenols in tyrosine were compatible with our method (Scheme 3A(2)). The addition of Cbz–Tyr–OMe and Cbz–Ala–Tyr–OMe led to the formation of unprecedented unnatural tri- and tetrapeptides 5i and 5j in 37% and 40% yield respectively.
We were then interested in adding indoles, which are also proteinogenic nucleophiles present in tryptophan. Wang and coworkers recently reported an asymmetric Friedel–Crafts reaction between indoles and N-acyl iminiums generated in situ by decarboxylation of RAE derived from amino acids.38 However, this transformation was not extended to peptides and prefunctionalization of the carboxylic acids was needed. Inspired by conditions reported by our group,35 a one-pot two steps arylation procedure directly from free carboxylic acids was developed using a slight excess of indoles and one equivalent of TFA (Scheme 3B(1), conditions B). Using Cbz–Gly–Pro (1a) as model substrate several indoles reacted preferentially at the C3-position. 1H-indole afforded 6a in 66% yield. A chlorine substituent in C6 position and a trifluoromethyl substituent in C7 position were tolerated and led to the formation of the desired products 6b and 6c in 66% and 50% yield respectively. 2-Methylindole could be introduced in 49% yield onto dipeptide 1a. When C3-substituted indoles were employed, N-addition was the major outcome. For instance, 3-methylindole led to the formation of 6e in 43% yield. C2-addition was also detected in the crude mixture (ratio N/C2-addition 2 : 1), but the corresponding product could not be isolated in pure form. Interestingly, melatonin, which is a hormone helping to regulate the sleep–wake cycle and which can be used for short-term treatment of insomnia, was compatible with the reaction conditions. Product 6f was obtained in 64% yield. Cbz–Gly–Phe (1c) and Cbz–Pro–Val (1d) could also be arylated with 1H-indole and the corresponding products 6g and 6h were obtained in 58% and 64% yields respectively. The tryptophan indoles of Cbz–Trp–OMe and Cbz–Val–Trp–OMe reacted via C–N bond formation to give the unprecedented unnatural tri- and tetrapeptides 6i and 6j in 57% and 50% yield respectively (Scheme 3B(2)).
Finally, we investigate the extension of our methodology to larger peptides (Scheme 4). Ac–Ala–Phe–Gly–Ala (7a) was selected as model substrate. By increasing the amount of oxidant and photocatalyst, indole could be added successfully to 7a. The desired modified tetrapeptide 8a was the only product observed by HPLC and was obtained in 66% yield (See ESI† for more details). Serine was also tolerated at the C-terminus and full conversion towards 8b was observed. A protected amide from asparagine and a protected amine from lysine were moderately tolerated leading to products 8c and 8d. Moreover, more complex unnatural peptides could be obtained by reaction with proteinogenic indoles from Cbz–Trp–OMe and Cbz–Val–Trp–OMe. For instance 8e and 8f were formed in moderate to good HPLC ratios. Protected aspartic acid, histidine and arginine were also submitted to the reaction conditions and led to the formation of arylated peptides but with lower HPLC ratios (<25%, see SI for more details).39 Unfortunately, the current conditions for phenols were not compatible with tetramers and would require further optimization .
Scheme 4. Preliminary results towards arylation of tetrapeptides. Reactions performed on 1 μmol scale. Relative HPLC ratios of the area of the product over remaining starting material and peptide side-products at 214 nm are given. Average of 3 independent trials. aCalibrated yield, see SI for details. bCompound 8c could not be fully separated from a side product by HPLC.

3. Conclusion
In summary, we have developed a photoredox-catalyzed oxidative decarboxylative strategy towards the introduction of diverse functional groups on peptides. Under the developed conditions, valuable alcohols were successfully introduced leading to structurally diverse N,O-acetals. Moreover, the N,O-acetals were also employed as key reactive intermediates for arylation with phenols and indoles. Bioconjugation of bioactive compounds such as estrone or melatonin with dipeptides was possible. Additionally, serine, threonine, tyrosine and tryptophan derivatives could be used as nucleophilic partners to give new types of cross-linked peptides. As a proof of concept for the arylation of larger peptides, indole and two tryptophan derivatives were successfully added to several tetrapeptides.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work is supported by the Swiss National Science Foundation (Grant No. 200020_182798), the NCCR Chemical Biology of the Swiss National Science Foundation and EPFL. The MS service from EPFL-ISIC is acknowledged for their support.
Electronic supplementary information (ESI) available: Experimental data. See DOI: 10.1039/d0sc06180h. Raw data for NMR IR and MS is available at https://zenodo.org, DOI: 10.5281/zenodo.4384417.
Notes and references
- (a) Fosgerau K. Hoffmann T. Drug Discov. Today. 2015;20:122–128. doi: 10.1016/j.drudis.2014.10.003. [DOI] [PubMed] [Google Scholar]; (b) Dang T. Süssmuth R. D. Acc. Chem. Res. 2017;50:1566–1576. doi: 10.1021/acs.accounts.7b00159. [DOI] [PubMed] [Google Scholar]; (c) Hamley I. W. Chem. Rev. 2017;117:14015–14041. doi: 10.1021/acs.chemrev.7b00522. [DOI] [PubMed] [Google Scholar]; (d) Henninot A. Collins J. C. Nuss J. M. J. Med. Chem. 2018;61:1382–1414. doi: 10.1021/acs.jmedchem.7b00318. [DOI] [PubMed] [Google Scholar]
- Craik D. J. Fairlie D. P. Liras S. Price D. Chem. Biol. Drug Des. 2013;81:136–147. doi: 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- (a) Ueda T. Biochim. Biophys. Acta Protein Proteonomics. 2014;1844:2053–2057. doi: 10.1016/j.bbapap.2014.06.008. [DOI] [PubMed] [Google Scholar]; (b) Drake P. M. Rabuka D. Curr. Opin. Chem. Biol. 2015;28:174–180. doi: 10.1016/j.cbpa.2015.08.005. [DOI] [PubMed] [Google Scholar]; (c) Cohen D. T. Zhang C. Fadzen C. M. Mijalis A. J. Hie L. Johnson K. D. Shriver Z. Plante O. Miller S. J. Buchwald S. L. Pentelute B. L. Nat. Chem. 2019;11:78–85. doi: 10.1038/s41557-018-0154-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S. Schiller M. R. Crit. Rev. Biochem. Mol. Biol. 2019;54:85–102. doi: 10.1080/10409238.2019.1586828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) deGruyter J. N. Malins L. R. Baran P. S. Biochemistry. 2017;56:3863–3873. doi: 10.1021/acs.biochem.7b00536. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rosen C. B. Francis M. B. Nat. Chem. Biol. 2017;13:697–705. doi: 10.1038/nchembio.2416. [DOI] [PubMed] [Google Scholar]
- Hofer H. Moest M. Justus Liebigs Ann. Chem. 1902;323:284–323. doi: 10.1002/jlac.19023230304. [DOI] [Google Scholar]
- (a) Renaud P. Seebach D. Angew. Chem., Int. Ed. 1986;25:843–844. doi: 10.1002/anie.198608431. [DOI] [Google Scholar]; (b) Seebach D. Charczuk R. Gerber C. Renaud P. Berner H. Schneider H. Helv. Chim. Acta. 1989;72:401–425. doi: 10.1002/hlca.19890720302. [DOI] [Google Scholar]; (c) Gerber C. Seebach D. Helv. Chim. Acta. 1991;74:1373–1385. doi: 10.1002/hlca.19910740702. [DOI] [Google Scholar]
- (a) Speckamp W. N. Moolenaar M. J. Tetrahedron. 2000;56:3817–3856. doi: 10.1016/S0040-4020(00)00159-9. [DOI] [Google Scholar]; (b) Vinogradov M. G. Turova O. V. Zlotin S. G. Russ. Chem. Rev. 2017;86:1–17. doi: 10.1070/RCR4628. [DOI] [PubMed] [Google Scholar]; (c) Wu P. Nielsen T. E. Chem. Rev. 2017;117:7811–7856. doi: 10.1021/acs.chemrev.6b00806. [DOI] [PubMed] [Google Scholar]
- Lin Y. Malins L. R. Chem. Sci. 2020;11:10752–10758. doi: 10.1039/D0SC03701J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Boto A. Hernández R. Suárez E. Tetrahedron Lett. 1999;40:5945–5948. doi: 10.1016/S0040-4039(99)01180-6. [DOI] [Google Scholar]; (b) Boto A. Hernández R. Suárez E. J. Org. Chem. 2000;65:4930–4937. doi: 10.1021/jo000356t. [DOI] [PubMed] [Google Scholar]
- Harayama Y. Yoshida M. Kamimura D. Wada Y. Kita Y. Chem.–Eur. J. 2006;12:4893–4899. doi: 10.1002/chem.200501635. [DOI] [PubMed] [Google Scholar]
- Selected examples: ; (a) Boto A. Gallardo J. A. Hernández R. Saavedra C. J. Tetrahedron Lett. 2005;46:7807–7811. doi: 10.1016/j.tetlet.2005.09.019. [DOI] [Google Scholar]; (b) Saavedra C. J. Hernández R. Boto A. Álvarez E. Tetrahedron Lett. 2006;47:8757–8760. doi: 10.1016/j.tetlet.2006.10.007. [DOI] [Google Scholar]; (c) Fan R. Li W. Wang B. Org. Biomol. Chem. 2008;6:4615. doi: 10.1039/B815227F. [DOI] [PubMed] [Google Scholar]; (d) Xu K. Wang Z. Zhang J. Yu L. Tan J. Org. Lett. 2015;17:4476–4478. doi: 10.1021/acs.orglett.5b02142. [DOI] [PubMed] [Google Scholar]; (e) Hernández D. Boto A. Guzmán D. Álvarez E. Org. Biomol. Chem. 2017;15:7736–7742. doi: 10.1039/C7OB02033C. [DOI] [PubMed] [Google Scholar]
- (a) Larionov V. A. Stoletova N. V. Maleev V. I. Adv. Synth. Catal. 2020;362:4325–4367. doi: 10.1002/adsc.202000753. [DOI] [Google Scholar]; (b) Aguilar Troyano F. J., Merkens K., Anwar K. and Gómez-Suárez A., Angew. Chem., Int. Ed., 10.1002/anie.202010157 [DOI] [PMC free article] [PubMed]; (c) King T. A., Mandrup Kandemir J., Walsh S. J. and Spring D. R., Chem. Soc. Rev., 10.1039/d0cs00344a [DOI] [PubMed]; (d) Malins L. R. Pept. Sci. 2018;110:e24049. doi: 10.1002/pep2.24049. [DOI] [Google Scholar]; (e) Hu C. Chen Y. Tetrahedron Lett. 2015;56:884–888. doi: 10.1016/j.tetlet.2014.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Bottecchia C. Noël T. Chem.–Eur. J. 2019;25:26–42. doi: 10.1002/chem.201803074. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Mondal S. Chowdhury S. Adv. Synth. Catal. 2018;360:1884–1912. doi: 10.1002/adsc.201800011. [DOI] [Google Scholar]
- (a) Maeda K. Saito H. Osaka K. Nishikawa K. Sugie M. Morita T. Takahashi I. Yoshimi Y. Tetrahedron. 2015;71:1117–1123. doi: 10.1016/j.tet.2014.12.075. [DOI] [Google Scholar]; (b) Lipp B. Nauth A. M. Opatz T. J. Org. Chem. 2016;81:6875–6882. doi: 10.1021/acs.joc.6b01215. [DOI] [PubMed] [Google Scholar]
- (a) Itou T. Yoshimi Y. Nishikawa K. Morita T. Okada Y. Ichinose N. Hatanaka M. Chem. Commun. 2010;46:6177. doi: 10.1039/C0CC01464H. [DOI] [PubMed] [Google Scholar]; (b) Cassani C. Bergonzini G. Wallentin C.-J. Org. Lett. 2014;16:4228–4231. doi: 10.1021/ol5019294. [DOI] [PubMed] [Google Scholar]
- Lang S. B. O'Nele K. M. Douglas J. T. Tunge J. A. Chem.–Eur. J. 2015;21:18589–18593. doi: 10.1002/chem.201503644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garreau M. Le Vaillant F. Waser J. Angew. Chem., Int. Ed. 2019;58:8182–8186. doi: 10.1002/anie.201901922. [DOI] [PubMed] [Google Scholar]
- Le Vaillant F. Wodrich M. D. Waser J. Chem. Sci. 2017;8:1790–1800. doi: 10.1039/C6SC04907A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcote D. C. Street-Jeakings R. Dauncey E. Douglas J. J. Ruffoni A. Leonori D. Org. Biomol. Chem. 2019;17:1839–1842. doi: 10.1039/C8OB02702A. [DOI] [PubMed] [Google Scholar]
- (a) Chu L. Ohta C. Zuo Z. MacMillan D. W. C. J. Am. Chem. Soc. 2014;136:10886–10889. doi: 10.1021/ja505964r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McCarver S. J. Qiao J. X. Carpenter J. Borzilleri R. M. Poss M. A. Eastgate M. D. Miller M. M. MacMillan D. W. C. Angew. Chem., Int. Ed. 2017;56:728–732. doi: 10.1002/anie.201608207. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bloom S. Liu C. Kölmel D. K. Qiao J. X. Zhang Y. Poss M. A. Ewing W. R. MacMillan D. W. C. Nat. Chem. 2018;10:205–211. doi: 10.1038/nchem.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin T. Cornella J. Li C. Malins L. R. Edwards J. T. Kawamura S. Maxwell B. D. Eastgate M. D. Baran P. S. Science. 2016;352:801–805. doi: 10.1126/science.aaf6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards J. T. Merchant R. R. McClymont K. S. Knouse K. W. Qin T. Malins L. R. Vokits B. Shaw S. A. Bao D.-H. Wei F.-L. Zhou T. Eastgate M. D. Baran P. S. Nature. 2017;545:213–218. doi: 10.1038/nature22307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. M. Qin T. Merchant R. R. Edwards J. T. Malins L. R. Liu Z. Che G. Shen Z. Shaw S. A. Eastgate M. D. Baran P. S. Angew. Chem., Int. Ed. 2017;56:11906–11910. doi: 10.1002/anie.201705107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin T. Malins L. R. Edwards J. T. Merchant R. R. Novak A. J. E. Zhong J. Z. Mills R. B. Yan M. Yuan C. Eastgate M. D. Baran P. S. Angew. Chem., Int. Ed. 2017;56:260–265. doi: 10.1002/anie.201609662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. Wang J. Barton L. M. Yu S. Tian M. Peters D. S. Kumar M. Yu A. W. Johnson K. A. Chatterjee A. K. Yan M. Baran P. S. Science. 2017;356:eaam7355. doi: 10.1126/science.aam7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Jin Y. Jiang M. Wang H. Fu H. Sci. Rep. 2016;6:20068. doi: 10.1038/srep20068. doi: 10.1038/srep20068. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cheng W.-M. Shang R. Fu Y. ACS Catal. 2017;7:907–911. doi: 10.1021/acscatal.6b03215. [DOI] [Google Scholar]; (c) Jin Y. Yang H. Fu H. Org. Lett. 2016;18:6400–6403. doi: 10.1021/acs.orglett.6b03300. [DOI] [PubMed] [Google Scholar]
- Jin Y. Yang H. Fu H. Chem. Commun. 2016;52:12909–12912. doi: 10.1039/C6CC06994K. [DOI] [PubMed] [Google Scholar]
- Jiang M. Yang H. Fu H. Org. Lett. 2016;18:1968–1971. doi: 10.1021/acs.orglett.6b00489. [DOI] [PubMed] [Google Scholar]
- Selected examples: ; (a) Inuki S. Sato K. Fujimoto Y. Tetrahedron Lett. 2015;56:5787–5790. doi: 10.1016/j.tetlet.2015.09.012. [DOI] [Google Scholar]; (b) Khan S. N. Zaman M. K. Li R. Sun Z. J. Org. Chem. 2020;85:5019–5026. doi: 10.1021/acs.joc.0c00312. [DOI] [PubMed] [Google Scholar]; (c) Shirase S. Tamaki S. Shinohara K. Hirosawa K. Tsurugi H. Satoh T. Mashima K. J. Am. Chem. Soc. 2020;142:5668–5675. doi: 10.1021/jacs.9b12918. [DOI] [PubMed] [Google Scholar]; (d) Faraggi T. M. Li W. MacMillan D. W. C. Isr. J. Chem. 2020;60:410–415. doi: 10.1002/ijch.201900130. [DOI] [Google Scholar]
- (a) Huang H. Zhang G. Chen Y. Angew. Chem., Int. Ed. 2015;54:7872–7876. doi: 10.1002/anie.201502369. [DOI] [PubMed] [Google Scholar]; (b) Li G.-X. Hu X. He G. Chen G. ACS Catal. 2018;8:11847–11853. doi: 10.1021/acscatal.8b04079. [DOI] [Google Scholar]
- Sakakibara Y. Cooper P. Murakami K. Itami K. Chem.–Asian J. 2018;13:2410–2413. doi: 10.1002/asia.201800529. [DOI] [PubMed] [Google Scholar]
- Tucker J. W. Stephenson C. R. J. J. Org. Chem. 2012;77:1617–1622. doi: 10.1021/jo202538x. [DOI] [PubMed] [Google Scholar]
- Hari D. P. König B. Chem. Commun. 2014;50:6688–6699. doi: 10.1039/C4CC00751D. [DOI] [PubMed] [Google Scholar]
- Shen T. Zhao Z.-G. Yu Q. Xu H.-J. J. Photochem. Photobiol. Chem. 1989;47:203–212. doi: 10.1016/1010-6030(89)87066-2. [DOI] [Google Scholar]
- Wang M. Waser J. Angew. Chem., Int. Ed. 2019;58:13880–13884. doi: 10.1002/anie.201907060. [DOI] [PubMed] [Google Scholar]
- From the reactive amino acids, arginine, serine and aspartic acid were well tolerated. The presence of methionine, cysteine, histidine and glycine led to moderate yields. Tyrosine, tryptophan and lysine were not tolerated. See ESI† for details
- Osberger T. J. Rogness D. C. Kohrt J. T. Stepan A. F. White M. C. Nature. 2016;537:214–219. doi: 10.1038/nature18941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M.-L. Shen Y. Wang P.-S. Org. Lett. 2019;21:2993–2997. doi: 10.1021/acs.orglett.9b00442. [DOI] [PubMed] [Google Scholar]
- A mass corresponding to an enamide obtained from deprotonation of the iminium intermediate was often observed in case of lower yields, but further studies will be needed to confirm this structural assignment and to identify other side products
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.