

Abstract
A mechanistically unique functionalization strategy for a benzylic C(sp3)–H bond has been developed based on the facile oxidation event of indole substrates. This novel pathway was initiated by efficient radical generation at the benzylic position of the substrate, with subsequent transition metal catalysis to complete the overall transformation. Ultimately, an aryl or an acyl group could be effectively delivered from an aryl (pseudo)halide or an acid anhydride coupling partner, respectively. The developed method utilizes mild conditions and exhibits a wide substrate scope for both substituted indoles and C(sp2)-based reaction counterparts. Mechanistic studies have shown that competitive hydrogen atom transfer (HAT) processes, which are frequently encountered in conventional methods, are not involved in the product formation process of the developed strategy.
A mechanistically distinct Ni-catalysed benzylic functionalization of indoles is developed by the facile oxidation of arenes. The method exhibits a wide substrate scope and pronounced chemoselectivity that cannot be accessed via known protocols.
Introduction
The development of synthetic strategies that allow direct functionalization of C–H bonds has received considerable attention from organic chemists because such methods are cost-effective and environmentally benign.1 Among the various modes of hydrocarbon modification, the activation of benzylic C(sp3)–H bonds is of increasing interest because of the abundance and popularity of arene-based building blocks in organic synthesis.2 Recently, the scope of this transformation has been dramatically expanded by the advent of transition metal catalysis (Scheme 1a).3 Despite significant advances in this area, major challenges associated with reactivity and/or selectivity are encountered regularly and remain an active target of investigation.4
Scheme 1. Strategies for benzylic C(sp3)–H functionalization.

Recently emerging radical-based technologies for benzylic C–H bond activation have contributed significantly to solving many of these problems (Scheme 1b). The expeditious introduction of benzylic radical intermediates to a metal center allows for the efficient generation of metal alkyl species, forming the foundation for subsequent functionalization.5 Conventionally, radical formation events at the benzylic position have been realized by hydrogen atom transfer (HAT),6 with recent advances in photoredox catalysis (PC) having greatly broadened the applicability of this method.7 However, the HAT process generally requires the use of a large excess of substrate due to its insufficient reactivity. More importantly, selectivity problems can arise in complicated systems, as the discrimination of C–H bonds with comparable bond strength is difficult.8 A potential alternative to HAT towards selective functionalization of a benzylic C–H bond is the employment of the sequential electron transfer–proton transfer (ET/PT) activity of arenes.9 This process involves chemoselective one-electron oxidation followed by deprotonation, and can preferentially generate a benzylic radical on the periphery of the arene ring. Although the ET/PT-based radical formation strategy has found a multitude of applications for organic synthesis,10,11 a synergistical merger with transition metal catalysis which can dramatically broaden the scope of benzylic C–H bond functionalization is underdeveloped.12,13
Herein, we present the successful interfacing of the ET/PT-driven indole-derived benzylic radical formation strategy with a Ni-catalyzed cross-coupling reaction for the construction of C(sp3)–C(sp2) bonds (Scheme 2a). This method exploits the facile oxidation of indoles by photoredox catalysis,14 and has thus been applied to selective radical generation at the benzylic position of the indole ring without the involvement of any HAT. Eventually, a convenient preparation of indole derivatives containing an aryl or an acyl group at the C-3 alkyl group, common structural motifs with important bioactivity, could be realized (Scheme 2b).
Scheme 2. Benzylic C(sp3)–H functionalization of indoles by dual catalysis.

Results and discussion
The reactivity of the combined catalytic systems was evaluated using 1,3-dimethyl-1H-indole (1a) and p-bromoacetophenone (2a) as substrates (Table 1).15 In the presence of an Ir-based photocatalyst, visible-light irradiation, and a NiCl2·glyme precatalyst with a 1,10-phenanthroline ligand, the desired benzylic arylation product (3aa) was formed in 83% yield (entry 1). The reaction was set up with the aid of a glovebox for the optimal results. However, it is noteworthy that the reactivity could be preserved even when the reaction was set up outside of a glovebox by using a standard Schlenk technique, or when the reaction was run under air (76% and 73%, respectively). These results demonstrate the robustness of the developed method. In contrast to the HAT-based dual catalytic systems, the use of excess radical precursor was not required, demonstrating the efficiency of the transformation. Control experiments indicated that the presence of both the photoredox system and the Ni catalyst was crucial for successful reaction (entries 2–4). In addition, the use of a non-nucleophilic organic base, viz. 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), and the incorporation of LiCl additive were also important for optimum performance (entries 5 and 6).16,17 In agreement with the mechanistic postulate involving radical generation, no arylation product was detected when a radical scavenger was present (entry 7).18 Finally, the reactivity could be extended to arylation with other types of aryl (pseudo)halides, among which aryl bromides and chlorides are the best substrates (entries 8–10).
Optimization of reaction conditionsa.
| ||
|---|---|---|
| Entry | Conditions | Yield (%) |
| 1 | As shown | 83 (76b/73c) |
| 2 | No photocatalyst | N.D. |
| 3 | No light | N.D. |
| 4 | No NiCl2·glyme | N.D. |
| 5 | No DBU | 10 |
| 6 | No LiCl | 54 |
| 7 | With TEMPO (3.0 equiv.) | N.D. |
| 8 | ArCl instead of ArBr | 76 |
| 9 | ArI instead of ArBr | 16 |
| 10d | ArOTf instead of ArBr | 36 |
Reaction conditions: 1a (0.15 mmol), 2a (0.10 mmol), [Ir] (1.0 mol%), NiCl2·glyme (5.0 mol%), L (7.5 mol%), DBU (0.10 mmol), LiCl (0.15 mmol), and DMA (0.050 M) irradiated with 34 W blue LEDs. Yields were determined by 1H NMR analysis using 1,1,2,2-tetrachloroethane as an internal standard.
The reaction was set up using standard Schlenk technique on the benchtop.
The reaction was carried out under ambient conditions.
Without LiCl. [Ir] = Ir(dFCF3ppy)2(dtbbpy)PF6. dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine. L = 1,10-phenanthroline. DMA = N,N-dimethylacetamide. TEMPO = (2,2,6,6-tetramethylpiperidin-1-yl)oxyl. N.D. = not detected.
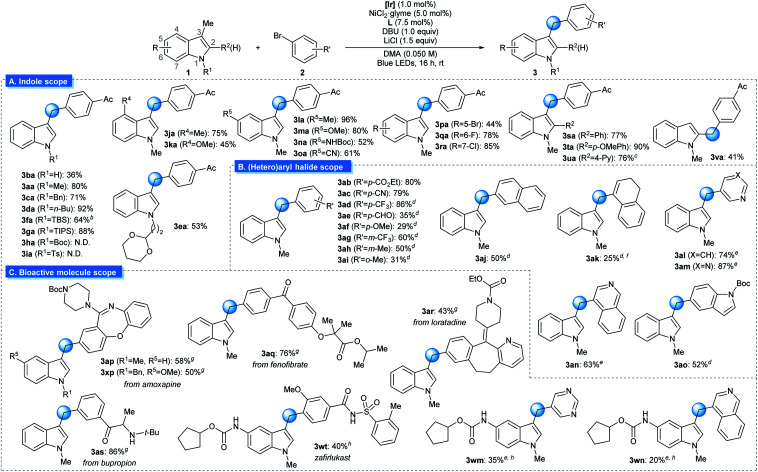
The generality of the reaction was examined under optimized conditions (Table 2A). Initially, the effect of varying the N1-substituent was assessed. Although an unmasked 3-methyl indole furnished the desired product in slightly reduced yield (3ba), indole derivatives with a variety of N1-alkyl substituents underwent the desired arylation smoothly (3aa, 3ca–3ea). Importantly, the placement of Si-based substituents, groups that can be conveniently removed after the targeted reaction, did not affect reactivity (3fa and 3ga). However, the presence of an electron-withdrawing group at N1 reduced reactivity, indicating the importance of the indole oxidation event (3ha and 3ia). Next, the substituent effects at other positions on the indole skeleton were explored. Various electron-donating (3ja–3na) and electron-withdrawing functional groups (3oa–3ra) at the C4–C8 positions of indole were found to be compatible with the optimized conditions. Moreover, halogen substituents, which can be utilized as handles for further functionalization via cross-coupling reactions, were tolerated (3pa–3ra). The sterically encumbered 2-(hetero)aryl indole derivatives could also be arylated at the 3-methyl group in synthetically useful yields (3sa–3ua). Finally, the protocol was applicable to the arylation of a 2-methyl indole derivative (3va). It should be noted that numerous functional groups that are highly susceptible to HAT-based activation remained intact, indicating the orthogonality of the discovered reactivity (3ca, 3ea, 3ka, 3ma, 3ta, and 3xp). Substrates based on other types of heterocycles, such as benzofuran, thiophene or pyrrole, were not suitable substrates for the transformation due to the inefficient oxidation process.14
Benzylic C(sp3)–H arylation of indoles by photoredox/nickel dual catalysisa.
|
Reaction conditions: 1 (0.30 mmol), 2 (0.20 mmol), [Ir] (1.0 mol%), NiCl2·glyme (5.0 mol%), L (7.5 mol%), DBU (0.20 mmol), LiCl (0.30 mmol), and DMA (0.050 M) irradiated with 34 W blue LEDs. All yields are isolated yields.
18% of the desilylated product was obtained during the course of the reaction.
The reaction was performed with 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) instead of [Ir] for 23 h.
4,4′-dimethoxy-2,2′-bipyridine instead of L.
The reaction was conducted at 53 °C.
The corresponding vinyl triflate was used as substrate.
NiCl2·glyme (10.0 mol%), 4,4′-dimethoxy-2,2′-bipyridine (15.0 mol%), DMA (0.033 M), and ArCl instead of ArBr.
NiCl2·glyme (10.0 mol%), L (15.0 mol%), and DMA (0.033 M). [Ir] = Ir(dFCF3ppy)2(dtbbpy)PF6. dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine. L = 1,10-phenanthroline. DMA = N,N-dimethylacetamide. N.D. = not detected.
Subsequently, the extent of the aryl (pseudo)halide coupling partners was investigated (Table 2B). Although highly electron-rich or sterically congested aryl halides underwent the targeted transformation with somewhat reduced productivity, a wide range of electron-neutral and electron-deficient arenes could be successfully used with minor modifications to the reaction conditions (3ab–3ai). In addition, coupling counterparts derived from an extended π-system (3aj), an alkenyl group (3ak), or heterocycles (3al–3ao) were also viable substrates.
The strength of the developed strategy was further illustrated by applying this method to the preparation of derivatives of bioactive molecules (Table 2C). Commercial drug molecules, containing a C–halogen bond, could be successfully employed as an aryl donor, demonstrating the robustness of the protocol (3ap–3as, 3xp). In addition, a novel synthetic pathway could be devised for the preparation of the complex drug molecule zafirlukast, which enabled late-stage unification of two parts of comparable complexity (3wt).19 Indeed, this strategy could be extended further to the facile preparation of zafirlukast derivatives using readily available aryl halides (3wm–3wn).
We then examined the mechanistic aspects of the transformation to investigate the potential involvement of HAT processes based on the generation of halogen radicals. First, the contribution of metal halide additive (i.e., LiCl) was examined by a Giese addition experiment (Scheme 3A).20 When the radical generation was attempted in the presence of a radical acceptor, the efficiency of the addition process was not affected by the presence of LiCl. Also, a fluorescence quenching study of the reaction system showed that LiCl is not the major electron transfer partner of the photocatalyst.21 Thus, it was concluded that LiCl was not involved in radical formation. Second, the possibility of the Ni-halide complex initiating radical formation was analyzed (Scheme 3B).7b,c,22 Upon exposure to the indole substrate and the photoredox system, an independently prepared Ni(ii) aryl halide complex ([Ni]-1) failed to deliver the expected arylation product (3ad). This result indicated that potential radical generation via HAT from the Ni complex did not lead to the desired pathway of product formation.
Scheme 3. Mechanistic studies.

Further insight could be gained from the dependence of the reaction outcome on the electronic nature of the substrates (Scheme 3C). Under the standard conditions, an electron-deficient indole substrate (1h), in stark contrast to the case of an electron-rich substrate (1a), showed virtually no reactivity towards arylation. In case a HAT process is operating, a comparable product yield should be observed, since the cross-coupling step is not significantly sensitive to the electronic properties of the arene substrate.23 Therefore, it is concluded that a HAT-driven radical generation is not predominantly involved in the overall transformation. This hypothesis was corroborated by a Stern–Volmer analysis of the indole substrates. The strong correlation between product yield and degree of fluorescence quenching suggested the involvement of the ET/PT-based process.
Based on the assembled information, a plausible catalytic cycle was proposed (Fig. 1). Initially, visible-light irradiation of the [IrIII] photocatalyst generates a potent excited-state oxidant [IrIII]* (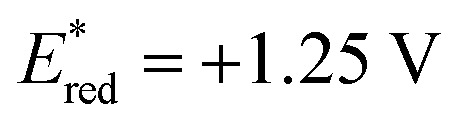 vs. Ag/AgCl in MeCN), which can oxidize the indole 1a (Eox = +1.00 V vs. Ag/AgCl in MeCN). The resulting radical cation (I) can be rapidly deprotonated to form the key benzylic radical intermediate (II). At this point, two possible downstream sequences of the radical intermediate to engage the Ni-based catalytic cycle were considered.24 In the path A, radical II can be intercepted by the ligand-bound Ni0 species (III) to generate an alkyl-Ni intermediate (IV), which in turn can afford a NiIII complex (V) via oxidative addition with an aryl halide. In the alternative route (path B), the radical addition is taking place to a NiII intermediate (VII), which is formed by direct oxidative addition of ligand-bound Ni0 species (III). Our mechanistic experiment suggests that a NiII complex, such as VII, is unlikely to be involved in the catalytic cycle: an independently prepared NiII oxidative addition complex ([Ni]-1, Scheme 3B) does not provide any of the desired cross-coupling product under the radical-generating conditions.25 Furthermore, it has been shown that the Ni0/NiI/NiIII pathway (path A) is energetically more favored over the Ni0/NiII/NiIII pathway (path B) in related systems.26 Therefore, path B was excluded from further considerations. Finally, subsequent reductive elimination from the NiIII complex (V) should furnish the desired product and a NiI–Br species (VI, Ered = −1.10 V vs. Ag/AgCl in DMF)27 which after single electron transfer by [IrII] (Ered = −1.32 V vs. Ag/AgCl in MeCN) will resume the dual catalytic cycle.
vs. Ag/AgCl in MeCN), which can oxidize the indole 1a (Eox = +1.00 V vs. Ag/AgCl in MeCN). The resulting radical cation (I) can be rapidly deprotonated to form the key benzylic radical intermediate (II). At this point, two possible downstream sequences of the radical intermediate to engage the Ni-based catalytic cycle were considered.24 In the path A, radical II can be intercepted by the ligand-bound Ni0 species (III) to generate an alkyl-Ni intermediate (IV), which in turn can afford a NiIII complex (V) via oxidative addition with an aryl halide. In the alternative route (path B), the radical addition is taking place to a NiII intermediate (VII), which is formed by direct oxidative addition of ligand-bound Ni0 species (III). Our mechanistic experiment suggests that a NiII complex, such as VII, is unlikely to be involved in the catalytic cycle: an independently prepared NiII oxidative addition complex ([Ni]-1, Scheme 3B) does not provide any of the desired cross-coupling product under the radical-generating conditions.25 Furthermore, it has been shown that the Ni0/NiI/NiIII pathway (path A) is energetically more favored over the Ni0/NiII/NiIII pathway (path B) in related systems.26 Therefore, path B was excluded from further considerations. Finally, subsequent reductive elimination from the NiIII complex (V) should furnish the desired product and a NiI–Br species (VI, Ered = −1.10 V vs. Ag/AgCl in DMF)27 which after single electron transfer by [IrII] (Ered = −1.32 V vs. Ag/AgCl in MeCN) will resume the dual catalytic cycle.
Fig. 1. Proposed reaction mechanism.

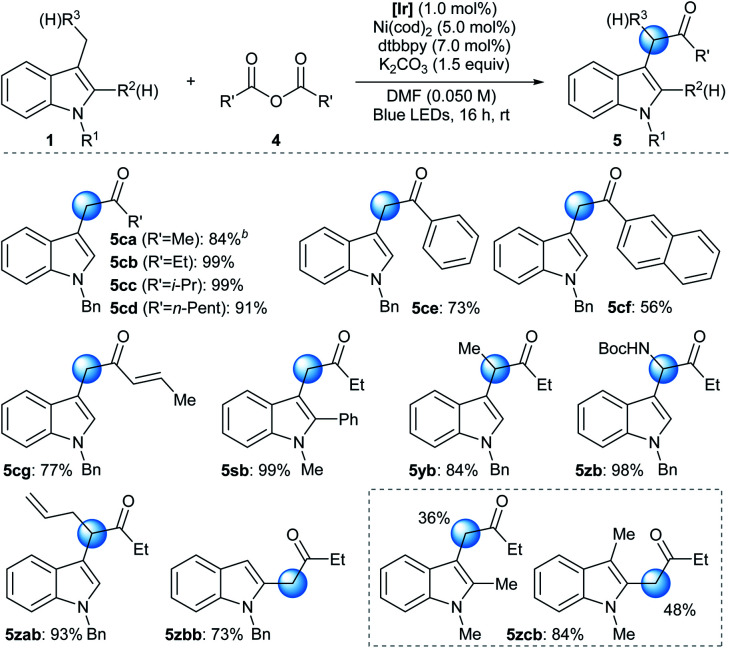
The developed reactivity could be further extended to the Ni-catalyzed acylation reaction using a symmetric acid anhydride as the acyl source (Table 3).28 Under slightly modified conditions involving the photocatalytic system, a Ni source, a bidentate ligand, and an inorganic base, N-alkyl indole derivatives underwent facile acylation reactions at the benzylic position.29 The strictly halide-free conditions further supported the involvement of the ET/PT process instead of HAT during radical generation. Acyl groups based on a variety of alkyl (5ca–5cd), aryl (5ce and 5cf), and alkenyl (5cg) groups were successfully installed. Analogously to the arylation reaction, synthetically useful yields could be obtained without the use of a glovebox when the standard Schlenk technique was applied or even when the reaction was carried out under ambient conditions (93% and 76% yield for 5cb, respectively). Moreover, the addition of an aryl substituent at the C2 position, which can pose significant steric congestion at the reaction center, did not affect reaction efficiency (5sb). Interestingly, a 3-ethyl indole derivative also underwent a facile acylation, furnishing a methine moiety at C3 (5yb). Furthermore, substrates containing a heteroatom-based substituent (5zb) or an allyl group (5zab) exhibited excellent reactivities towards the acylation reaction, demonstrating the pronounced robustness and chemoselectivity of the developed method, respectively.30 The protocol was also applied to the acylation of a 2-methyl indole derivative (5zbb). Finally, when multiple positions were available for acylation, a mixture of regioisomeric products was obtained (5zcb).31
Benzylic C(sp3)–H acylation of indoles by photoredox/nickel dual catalysisa.
|
Reaction conditions: 1 (0.20 mmol), 4 (0.24 mmol), [Ir] (1.0 mol%), Ni(cod)2 (5.0 mol%), dtbbpy (7.0 mol%), K2CO3 (0.30 mmol), and DMF (0.050 M) irradiated with 34 W blue LEDs. All yields are isolated yields.
The reaction was conducted in the absence of K2CO3. [Ir] = Ir(dFCF3ppy)2(dtbbpy)PF6. dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine. DMF = N,N-dimethylformamide.
Conclusions
In conclusion, a novel benzylic C(sp3)–H functionalization method for indoles, introducing C(sp2)-based functional groups, has been developed. This process is an outcome of the unprecedented combination of Ni catalysis and ET/PT-driven radical generation under photoredox conditions. The reaction exhibited extremely high efficiency under mild conditions, and was therefore applicable to the preparation of a wide range of indole products including complex drug molecule derivatives. Based on mechanistic investigations, it is evident that a conventionally employed HAT process was not involved in product formation. Rather, only the ET/PT process functioned as the major pathway. This distinct mechanistic pathway provides remarkable selectivity towards the activation of indole-derived benzylic C–H bonds over others. It is believed that the underdeveloped orchestration of ET/PT-controlled radical generation and other types of transition metal catalysis should provide a mild, robust, and efficient activation strategy for inert benzylic C(sp3)–H bonds. Moreover, the scope of the applicability should be expanded to other settings with the development of more efficient and selective arene oxidation protocols.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
Financial support was made by the National Research Foundation of Korea (NRF) funded by the Korean Government (MSIT, 2018R1C1B6008115). W. K. and J. K. were supported by Fellowship for Fundamental Academic Fields and NRF Grant funded by the Korean Government (NRF-2018-Global Ph.D. Fellowship Program, 2018H1A2A1060914), respectively.
Electronic supplementary information (ESI) available: Detailed experimental procedures, and spectroscopic data for all new compounds. See DOI: 10.1039/d0sc06666d
Notes and references
- For selected reviews, see: ; (a) Davies H. M. L. Du Bois J. Yu J.-Q. Chem. Soc. Rev. 2011;40:1855–1856. doi: 10.1039/C1CS90010B. [DOI] [PubMed] [Google Scholar]; (b) Gandeepan P. Müller T. Zell D. Cera G. Warratz S. Ackermann L. Chem. Rev. 2019;119:2192–2452. doi: 10.1021/acs.chemrev.8b00507. [DOI] [PubMed] [Google Scholar]; (c) Shilov A. E. Shul'pin G. B. Chem. Rev. 1997;97:2879–2932. doi: 10.1021/cr9411886. [DOI] [PubMed] [Google Scholar]
- For reviews, see: ; (a) Vanjari R. Singh K. N. Chem. Soc. Rev. 2015;44:8062–8096. doi: 10.1039/C5CS00003C. [DOI] [PubMed] [Google Scholar]; (b) Gulati U. Gandhi R. Laha J. K. Chem.–Asian J. 2020;15:3135–3161. doi: 10.1002/asia.202000730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a recent review, see: ; Yazaki R. Ohshima T. Tetrahedron Lett. 2019;60:151225. doi: 10.1016/j.tetlet.2019.151225. [DOI] [Google Scholar]
- For representative approaches, see: ; (a) Rosillo M. Domínguez G. Pérez-Castells J. Chem. Soc. Rev. 2007;36:1589–1604. doi: 10.1039/B606665H. [DOI] [PubMed] [Google Scholar]; (b) Rouquet G. Chatani N. Angew. Chem., Int. Ed. 2013;52:11726–11743. doi: 10.1002/anie.201301451. [DOI] [PubMed] [Google Scholar]; (c) Masuda Y. Ishida N. Murakami M. Chem.–Asian J. 2019;14:403–406. doi: 10.1002/asia.201801881. [DOI] [PubMed] [Google Scholar]; ; for recent examples, see: ; (d) Larsen M. A. Wilson C. V. Hartwig J. F. J. Am. Chem. Soc. 2015;137:8633–8643. doi: 10.1021/jacs.5b04899. [DOI] [PubMed] [Google Scholar]; (e) Takemoto S. Shibata E. Nakajima M. Yumoto Y. Shimamoto M. Matsuzaka H. J. Am. Chem. Soc. 2016;138:14836–14839. doi: 10.1021/jacs.6b08863. [DOI] [PubMed] [Google Scholar]; (f) Palmer W. N. Obligacion J. V. Pappas I. Chirik P. J. J. Am. Chem. Soc. 2016;138:766–769. doi: 10.1021/jacs.5b12249. [DOI] [PubMed] [Google Scholar]; (g) Sha S.-C. Tcyrulnikov S. Li M. Hu B. Fu Y. Kozlowski M. C. Walsh P. J. J. Am. Chem. Soc. 2018;140:12415–12423. doi: 10.1021/jacs.8b05143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For reviews, see: ; (a) Yi H. Zhang G. Wang H. Huang Z. Wang J. Singh A. K. Lei A. Chem. Rev. 2017;117:9016–9085. doi: 10.1021/acs.chemrev.6b00620. [DOI] [PubMed] [Google Scholar]; (b) Bosque I. Chinchilla R. Gonzalez-Gomez J. C. Guijarro D. Alonso F. Org. Chem. Front. 2020;7:1717–1742. doi: 10.1039/D0QO00587H. [DOI] [Google Scholar]
- For a review of HAT, see: ; (a) Mayer J. M. Acc. Chem. Res. 2011;44:36–46. doi: 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]; ; for recent examples, see:; (b) Zhang W. Wu L. Chen P. Liu G. Angew. Chem., Int. Ed. 2019;58:6425–6429. doi: 10.1002/anie.201902191. [DOI] [PubMed] [Google Scholar]; (c) Hu H. Chen S.-J. Mandal M. Pratik S. M. Buss J. A. Krska S. W. Cramer C. J. Stahl S. S. Nat. Catal. 2020;3:358–367. doi: 10.1038/s41929-020-0425-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a recent review, see: ; (a) Capaldo L. Ravelli D. Eur. J. Org. Chem. 2017:2056–2071. doi: 10.1002/ejoc.201601485. [DOI] [PMC free article] [PubMed] [Google Scholar]; ; for representative examples, see: ; (b) Heitz D. R. Tellis J. C. Molander G. A. J. Am. Chem. Soc. 2016;138:12715–12718. doi: 10.1021/jacs.6b04789. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shields B. J. Doyle A. G. J. Am. Chem. Soc. 2016;138:12719–12722. doi: 10.1021/jacs.6b08397. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shen Y. Gu Y. Martin R. J. Am. Chem. Soc. 2018;140:12200–12209. doi: 10.1021/jacs.8b07405. [DOI] [PubMed] [Google Scholar]; (e) Dewanji A. Krach P. E. Rueping M. Angew. Chem., Int. Ed. 2019;58:3566–3570. doi: 10.1002/anie.201901327. [DOI] [PubMed] [Google Scholar]; (f) Cheng X. Lu H. Lu Z. Nat. Commun. 2019;10:3549. doi: 10.1038/s41467-019-11392-6. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Krach P. E. Dewanji A. Yuan T. Rueping M. Chem. Commun. 2020;56:6082–6085. doi: 10.1039/D0CC01480J. [DOI] [PubMed] [Google Scholar]; (h) Ishida N. Masuda Y. Ishikawa N. Murakami M. Asian J. Org. Chem. 2017;6:669–672. doi: 10.1002/ajoc.201700115. [DOI] [Google Scholar]; (i) Kawasaki T. Ishida N. Murakami M. J. Am. Chem. Soc. 2020;142:3366–3370. doi: 10.1021/jacs.9b13920. [DOI] [PubMed] [Google Scholar]
- For selected examples of site-selectivity issues of HAT process, see: ; (a) Ide T. Barham J. P. Fujita M. Kawato Y. Egami H. Hamashima Y. Chem. Sci. 2018;9:8453–8460. doi: 10.1039/C8SC02965B. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Deng H.-P. Zhou Q. Wu J. Angew. Chem., Int. Ed. 2018;57:12661–12665. doi: 10.1002/anie.201804844. [DOI] [PubMed] [Google Scholar]; (c) Margrey K. A. Czaplyski W. L. Nicewicz D. A. Alexanian E. J. J. Am. Chem. Soc. 2018;140:4213–4217. doi: 10.1021/jacs.8b00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review, see: ; Cukier R. I. Nocera D. G. Annu. Rev. Phys. Chem. 1998;49:337–369. doi: 10.1146/annurev.physchem.49.1.337. [DOI] [PubMed] [Google Scholar]
- For benzylic functionalizations based on ET/PT, see: ; (a) Ohkubo K. Mizushima K. Iwata R. Souma K. Suzuki N. Fukuzumi S. Chem. Commun. 2010;46:601–603. doi: 10.1039/B920606J. [DOI] [PubMed] [Google Scholar]; (b) Pandey G. Pal S. Laha R. Angew. Chem., Int. Ed. 2013;52:5146–5149. doi: 10.1002/anie.201210333. [DOI] [PubMed] [Google Scholar]; (c) Zhou R. Liu H. Tao H. Yu X. Wu J. Chem. Sci. 2017;8:4654–4659. doi: 10.1039/C7SC00953D. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mazzarella D. Crisenza G. E. M. Melchiorre P. J. Am. Chem. Soc. 2018;140:8439–8443. doi: 10.1021/jacs.8b05240. [DOI] [PubMed] [Google Scholar]; (e) Lee B. J. DeGlopper K. S. Yoon T. P. Angew. Chem., Int. Ed. 2020;59:197–202. doi: 10.1002/anie.201910602. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Duhamel T. Muñiz K. Chem. Commun. 2019;55:933–936. doi: 10.1039/C8CC08594C. [DOI] [PubMed] [Google Scholar]; (g) Betori R. C. May C. M. Scheidt K. A. Angew. Chem., Int. Ed. 2019;58:16490–16494. doi: 10.1002/anie.201909426. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]; (h) Bloom S. McCann M. Lectka T. Org. Lett. 2014;16:6338–6341. doi: 10.1021/ol503094m. [DOI] [PubMed] [Google Scholar]; (i) Xu W. Wang W. Liu T. Xie J. Zhu C. Nat. Commun. 2019;10:4867. doi: 10.1038/s41467-019-12844-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Yu Y. Zhang L.-K. Buevich A. V. Li G. Tang H. Vachal P. Colletti S. L. Shi Z.-C. J. Am. Chem. Soc. 2018;140:6797–6800. doi: 10.1021/jacs.8b03973. [DOI] [PubMed] [Google Scholar]; (k) Zhang L. Hu X. Chem. Sci. 2020;11:10786–10791. doi: 10.1039/D0SC01445A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For ET/PT-based transformations of other classes of compounds, see: ; (a) Petronijević F. R. Nappi M. MacMillan D. W. C. J. Am. Chem. Soc. 2013;135:18323–18326. doi: 10.1021/ja410478a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Joe C. L. Doyle A. G. Angew. Chem., Int. Ed. 2016;55:4040–4043. doi: 10.1002/anie.201511438. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ahneman D. T. Doyle A. G. Chem. Sci. 2016;7:7002–7006. doi: 10.1039/C6SC02815B. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ohmatsu K. Nakashima T. Sato M. Ooi T. Nat. Commun. 2019;10:2706. doi: 10.1038/s41467-019-10641-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For seminal publications on the synergistic combination of transition metal catalysis and photoredox-mediated radical generation, see: ; (a) Tellis J. C. Primer D. N. Molander G. A. Science. 2014;345:433. doi: 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zuo Z. Ahneman D. T. Chu L. Terrett J. A. Doyle A. G. MacMillan D. W. C. Science. 2014;345:437. doi: 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To our best knowledge, a recent report by Hu describes the only example of ET/PT-based Ni catalysis using noncatalytic convergent paired electrolysis (ref. 10k). Mechanistically, however, involvement of the HAT process could not be ruled out in this case.
- Roth H. G. Romero N. A. Nicewicz D. A. Synlett. 2016;27:714–723. [Google Scholar]
- See the ESI for full optimization data (Table S1†)
- For additive effects in transition metal catalysis, see: ; Hong L. Sun W. Yang D. Li G. Wang R. Chem. Rev. 2016;116:4006–4123. doi: 10.1021/acs.chemrev.5b00676. [DOI] [PubMed] [Google Scholar]
- For discussions on LiCl-mediated enhancement of catalytic activity in cross-coupling reaction, see: ; (a) Sherwood J. Clark J. H. Fairlamb I. J. S. Slattery J. M. Green Chem. 2019;21:2164–2213. doi: 10.1039/C9GC00617F. [DOI] [Google Scholar]; . Also see: ; (b) Zhang C. Liu R. Xiang J. Kang H. Liu Z. Huang Y. J. Phys. Chem. B. 2014;118:9507–9514. doi: 10.1021/jp506013c. [DOI] [PubMed] [Google Scholar]
- The corresponding TEMPO adduct was detected in 22% yield. See the ESI for more details (Fig. S1 and S2†)
- (a) Goverdhan G. Reddy A. R. Himabindu V. Reddy G. M. Synth. Commun. 2013;43:498–504. doi: 10.1080/00397911.2011.603875. [DOI] [Google Scholar]; (b) Schierle S. Flauaus C. Heitel P. Willems S. Schmidt J. Kaiser A. Weizel L. Goebel T. Kahnt A. S. Geisslinger G. Steinhilber D. Wurglics M. Rovati G. E. Schmidtko A. Proschak E. Merk D. J. Med. Chem. 2018;61:5758–5764. doi: 10.1021/acs.jmedchem.8b00458. [DOI] [PubMed] [Google Scholar]; (c) Paladugu S. Mainkar P. S. Chandrasekhar S. ACS Omega. 2018;3:4289–4294. doi: 10.1021/acsomega.8b00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples associated with HAT processes mediated by added halide ions, see: ; (a) Rohe S. Morris A. O. McCallum T. Barriault L. Angew. Chem., Int. Ed. 2018;57:15664–15669. doi: 10.1002/anie.201810187. [DOI] [PubMed] [Google Scholar]; (b) Wang Z. Ji X. Han T. Deng G.-J. Huang H. Adv. Synth. Catal. 2019;361:5643–5647. doi: 10.1002/adsc.201901168. [DOI] [Google Scholar]
- See the ESI (Fig. S4 and S9†)
- (a) Hwang S. J. Anderson B. L. Powers D. C. Maher A. G. Hadt R. G. Nocera D. G. Organometallics. 2015;34:4766–4774. doi: 10.1021/acs.organomet.5b00568. [DOI] [Google Scholar]; (b) Hwang S. J. Powers D. C. Maher A. G. Anderson B. L. Hadt R. G. Zheng S.-L. Chen Y.-S. Nocera D. G. J. Am. Chem. Soc. 2015;137:6472–6475. doi: 10.1021/jacs.5b03192. [DOI] [PubMed] [Google Scholar]
- This is based on the nature of the benzylic HAT process, which is not significantly influenced by the electronics of the arene. See ref. 7f for a related example
- For a general review on the mechanisms of Ni-catalysed coupling reactions, see: ; Diccianni J. B. Diao T. Trends Chem. 2019;1:830–844. doi: 10.1016/j.trechm.2019.08.004. [DOI] [Google Scholar]
- An analogous stoichiometric experiment was utilised to identify the operating mechanism of a related reaction, see: ; Nakajima K. Nojima S. Nishibayashi Y. Angew. Chem., Int. Ed. 2016;55:14106–14110. doi: 10.1002/anie.201606513. [DOI] [PubMed] [Google Scholar]
- For related examples in support of our proposed reaction pathway, see: ; (a) Lee G. S. Won J. Choi S. Baik M.-H. Hong S. H. Angew. Chem., Int. Ed. 2020;59:16933–16942. doi: 10.1002/anie.202004441. [DOI] [PubMed] [Google Scholar]; (b) Yuan M. Song Z. Badir S. O. Molander G. A. Gutierrez O. J. Am. Chem. Soc. 2020;142:7225–7234. doi: 10.1021/jacs.0c02355. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Maity B. Zhu C. Yue H. Huang L. Harb M. Minenkov Y. Rueping M. Cavallo L. J. Am. Chem. Soc. 2020;142:16942–16952. doi: 10.1021/jacs.0c05010. [DOI] [PubMed] [Google Scholar]; (d) Tao X. Chen Y. Guo J. Wang X. Gong H. Chem. Sci. 2021;12:220–226. doi: 10.1039/D0SC05452F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Börjesson M. Moragas T. Martin R. J. Am. Chem. Soc. 2016;138:7504–7507. doi: 10.1021/jacs.6b04088. [DOI] [PubMed] [Google Scholar]
- For related examples of radical formation-based acylation reactions, see: ; (a) Amani J. Sodagar E. Molander G. A. Org. Lett. 2016;18:732–735. doi: 10.1021/acs.orglett.5b03705. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Amani J. Molander G. A. Org. Lett. 2017;19:3612–3615. doi: 10.1021/acs.orglett.7b01588. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Amani J. Alam R. Badir S. Molander G. A. Org. Lett. 2017;19:2426–2429. doi: 10.1021/acs.orglett.7b00989. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Amani J. Molander G. A. J. Org. Chem. 2017;82:1856–1863. doi: 10.1021/acs.joc.6b02897. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sun Z. Kumagai N. Shibasaki M. Org. Lett. 2017;19:3727–3730. doi: 10.1021/acs.orglett.7b01552. [DOI] [PubMed] [Google Scholar]; (f) Badir S. O. Dumoulin A. Matsui J. K. Molander G. A. Angew. Chem., Int. Ed. 2018;57:6610–6613. doi: 10.1002/anie.201800701. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Ackerman L. K. G. Martinez Alvarado J. I. Doyle A. G. J. Am. Chem. Soc. 2018;140:14059–14063. doi: 10.1021/jacs.8b09191. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h1) Levernier E. Corcé V. Rakotoarison L.-M. Smith A. Zhang M. Ognier S. Tatoulian M. Ollivier C. Fensterbank L. Org. Chem. Front. 2019;6:1378–1382. doi: 10.1039/C9QO00092E. [DOI] [Google Scholar]; . Also see ref. 7g and 11b.
- Other readily available and air-stable Ni(ii) salts showed synthetically useful level of reactivity (5 cases, 79% on average). However, optimal performance of the system was observed with Ni(cod)2. See the ESI for more details (Table S2†)
- For related examples of preferred activation of an allylic C–H bond over a benzylic C–H bond, see: ; (a) Cuthbertson J. D. MacMillan D. W. C. Nature. 2015;519:74–77. doi: 10.1038/nature14255. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li Y. Lei M. Gong L. Nat. Catal. 2019;2:1016–1026. doi: 10.1038/s41929-019-0357-9. [DOI] [Google Scholar]
-
It should be noted that the product ratio may not reflect the reactivity of each position due to potential 1,4-shift of a hydrogen atom. For a relevant review, see: ; Nechab M. Mondal S. Bertrand M. P. Chem.–Eur. J. 2014;20:16034–16059. doi: 10.1002/chem.201403951. [DOI] [PubMed] [Google Scholar];
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.