

Abstract
Biomolecular condensates are emerging as an efficient strategy developed by cells to control biochemical reactions in space and time by locally modifying composition and environment. Yet, local increase in protein concentration within these compartments could promote aberrant aggregation events, including the nucleation and growth of amyloid fibrils. Understanding protein stability within the crowded and heterogeneous environment of biological condensates is therefore crucial, not only when the aggregation-prone protein is the scaffold element of the condensates but also when proteins are recruited as client molecules within the compartments. Here, we investigate the partitioning and aggregation kinetics of the amyloidogenic peptide Abeta42 (Aβ-42), the peptide strongly associated with Alzheimer's disease, recruited into condensates based on low complexity domains (LCDs) derived from the DEAD-box proteins Laf1, Dbp1 and Ddx4, which are associated with biological membraneless organelles. We show that interactions between Aβ-42 and the scaffold proteins promote sequestration and local increase of the peptide concentration within the condensates. Yet, heterotypic interactions within the condensates inhibit the formation of amyloid fibrils. These results demonstrate that biomolecular condensates could sequester aggregation-prone proteins and prevent aberrant aggregation events, despite the local increase in their concentration. Biomolecular condensates could therefore work not only as hot-spots of protein aggregation but also as protective reservoirs, since the heterogenous composition of the condensates could prevent the formation of ordered fibrillar aggregates.
Biomolecular condensates sequester an aggregation-prone peptide and prevent its aggregation, showing that heterotypic interactions within the condensates can prevent the formation of amyloid fibrils, despite the local increase in concentration.
Introduction
The conversion of peptides and proteins into insoluble fibrillar aggregates known as amyloids is strongly associated with the onset and development of several neurodegenerative disorders, including Alzheimer's and Parkinson's disease.1–3
In many cases the formation of amyloids is driven by the presence of prion-like domains in the protein structures. Recently, it has been demonstrated that a similar class of intrinsically disordered protein sequences plays an important role in the formation of functional membraneless compartments in cells.4,5 Such sequences, defined as low complexity domains (LCDs), encode a variety of attractive intermolecular interactions that drive the formation of protein-rich condensates via liquid–liquid phase separation (LLPS). These condensates exhibit a series of attractive features and underlie several functions, some of them in connection with RNA metabolism and stress signaling.6–8
A drawback of this process is the inherent risk of aberrant aggregation events within the protein-rich phase, which can lead to the nucleation and growth of protein aggregates such as amyloid fibrils. This has been demonstrated for FUS,9 hnRNPA1,10 hnRNPA2,11 TDP-43,12 Huntingtin,13 Tau14 and α-synuclein,15 which are connected to amyotrophic lateral sclerosis, fronto temporal dementia, Huntington disease, Alzheimer's disease and Parkinson's disorder. Formation of amyloids and protein aggregates following liquid–liquid demixing has been reported also with short peptides16,17 and multi-domain proteins,18 and resembles features of the two-step nucleation process observed in protein crystallization.19 Liquid–liquid phase separated compartments could therefore represent only a metastable phase with respect to the thermodynamically more favored solid aggregates. This “ageing” of the condensantes is modulated by interactions between the proteins within the crowded environment of the condensates,4,20 and is highly affected by perturbations such as single-point mutations21 as well as by environmental factors such as shear.22
Most of the reported studies focused on the formation of protein aggregates from liquid droplets in which the aggregation-prone protein is one of the main scaffold molecules. However, amyloidogenic proteins could also represent client molecules that are recruited by biological condensates consisting of a variety of components. In this case, it is more challenging to predict the effect of the recruitment on the aggregation rate and the behavior of amyloidogenic proteins within the crowded and heterogeneous environment of the dense phase. Theoretical predictions illustrate that the local increase in concentration within the protein-rich droplets can drastically increase aggregation kinetics.23 Indeed, partitioning in polypeptide coacervates has been shown to accelerate the polymerization of actin.24
While the effect of local increase in concentration is easily appreciable, additional interactions between amyloidogenic proteins and the scaffold proteins of the condensates can potentially compete with the monomer–monomer interactions that drive amyloid formation. Moreover, the environment within the condensates differs from the continous phase and is often characterized by high viscosity and lower polarity.25,26
All together, these factors could either promote or inhibit biochemical reactions, including aggregation events. Complex biomolecular condensates could therefore work not only as hot-spots that accelerate amyloid formation but also as protective “safe reservoirs” which sequester amylogenic proteins and protect cells from aberrant aggregation events.
This competition of aggregation events in a multi-phase system is similar to the situation observed with the partitioning of proteins between a bulk phase and an interface. Weak adsorption on surfaces has been shown to inhibit aggregation kinetics for aggregation-prone peptides despite the local increase of concentration.27 In contrast, the adsorption of proteins with low tendency to form amyloids, together with possible conformational changes, often accelerates fibril formation.28–31 In this context, the surface offered by biomolecular condensates could be another factor that promotes or inhibits aggregation.
In this work, we investigate the partitioning and aggregation kinetics of the peptide Abeta42 (Aβ-42), the peptide strongly associated with Alzheimer's disease, within condensates formed by multi-domain proteins containing biological low complexity domains (LCDs). We selected the peptide Aβ-42 since its aggregation mechanism in homogenous solutions has been recently elucidated at the molecular level32–34 and represents therefore an ideal model system. We formed biomolecular condensates based on intrinsically disordered proteins derived from the biological DEAD-box proteins Laf-1,35 Dbp1 6 and Ddx4,36 which are associated with the formation of P-bodies in C. elegans, S. cerevisiae and humans, respectively, and represent well-studied low complexity domains.35,37 Following a strategy developed in our laboratory,18 the N-terminal region of Laf-1 or of Dbp1 (Dbp1N) were expressed in a multi-domain format consisting of a globular enzyme (adenylate kinase, AK) conjugated with the low complexity domains at the N- and C-terminus, defined in the following as Laf1-AK-Laf1 and Dbp1N-AK-Dbp1N. The LCD derived from Ddx4 will be indicated in the following simply as Ddx4. The full sequences of the proteins are reported in ESI Table S1.†
Results
We produced the peptide Aβ-42 following a well-established recombinant protocol that guarantees highly reproducible aggregation kinetics.33,38 We first characterised the partioning of this peptide into the protein-rich condensates by confocal fluorescence microscopy after tagging the peptide with the fluorescent dye Atto-488 (Fig. 1A–C). To this aim we introduced 10 μM condensate forming protein (Laf1-AK-Laf1, Dbp1N-AK-Dbp1N, or Ddx4) into a solution of 7 μM Aβ-42 containing 20% labelled peptide in 20 mM phosphate buffer at pH 8.0. We selected these buffer conditions to reproduce previous kinetic studies of Aβ-42.32,33 We observed formation of protein-rich condensates and recruitment of the peptide inside the compartments. From the fluorescence intensity values we estimated a partition coefficient (K = cinside/coutside) equal to 44.6 ± 7.0, 37.0 ± 5.3, 11.3 ± 1.2 for Laf1-AK-Laf1, Dbp1N-AK-Dbp1N and Ddx4, respectively. Since this approach may suffer from limitations, for instance due to quenching of the dye, for a more accurate estimation of the partition coefficient we designed a second experiment, in which we measured the concentration of Aβ-42 in the continuous phase by a kinetic assay (Fig. 1D). This kinetic assay is more accurate than absorbance measurements in determining low concentrations of Aβ-42 peptide. We formed condensates at very low volume fraction (using a concentration of Laf1-AK-Laf1 of 0.5 μM) to allow only partial recruitment of Aβ-42. We let the condensates sediment at the bottom of the well over several hours before introducing Aβ-42. After 15 minutes of equilibration, the supernatant was removed and the aggregation profile was monitored over time in a plate reader via thioflavin T (ThT) staining, which is a common reporter of the formation of β-sheet structures of amyloids.39 The Aβ-42 concentration was determined from the aggregation profiles by the known dependence of the aggregation rate on Aβ-42 concentration.32,40 From the known concentration of Laf1-AK-Laf1 in the dispersed phase (700 μM)25 and in the continuous phase (approximately 0.1 μM), we estimated the volume fraction of the dispersed phase (ΘD) equal to 0.06%. This corresponds to a partition coefficient of K = 1050 ± 40, much larger compared to the value estimated by fluorescence microscopy. We note, however, that a robust evaluation of K requires a more accurate measurement of the volume fraction of the dispersed phase, which is difficult to achieve at the very low values of ΘD used in this experiment. Higher values of ΘD leads to essentially full sequestration and complete inhibition of aggregation over the time scale of the experiments, impeding the analysis of the aggregation profiles (Fig. 1D).
Fig. 1. Sequestration of Aβ-42 into the condensates inhibits fibril formation. (A)–(C) Confocal fluorescence microscopy (top panels) and brightfield microscopy images (bottom panels) of a solution of 10 μM Laf1-AK-Laf1 (A), Dbp1N-AK-Dbp1N (B) or Ddx4 (C) with (left panels) and without (right panels) 7 μM Aβ-42 labelled with the dye Atto-488 in 20 mM phosphate buffer at pH 8.0. (D) Measurements of Aβ-42 concentration in the continuous phase by monitoring the aggregation profile of the supernatant solution obtained after removal of the dispersed phase. 3 μM Aβ-42 solution was incubated in the absence (black curve) and presence of 0.5 μM Laf1-AK-Laf1 (blue curve) or 10 μM Laf1-AK-Laf1 (dark blue circles). From the measured half-time, the concentration of Aβ-42 in the supernatant (msup) is estimated from a known calibration curve determined from data reported in the literature.32 (E) Aggregation profiles of 2 μM Aβ-42 solution in the absence (black symbols) and presence (blue symbols) of 10 μM Laf1-AK-Laf1.

Next, we investigated how the recruitment affects the formation of fibrils in the heterogeneous mixture by a combination of electron microscopy, size exclusion chromatography and ThT staining. We incubated samples in the absence and presence of the Laf1-AK-Laf1 condensates at room temperature both in a plastic and in a glass bottom multi-well plate, which led to different aggregation kinetics. We observed an increase of the intensity values of ThT fluorescence only in samples lacking condensates (Fig. 1E and violet bar in Fig. 2B). The ThT assay in the glass bottom multi-well plate was performed off-line by re-suspending the condensates by vigorous mechanical agitation before analysis.
Fig. 2. Inhibition of Aβ-42 fibril formation in the presence of the condensates (A) time evolution of the ThT fluorescence intensity of samples incubated in glass bottom plates for 3 days: 4 μM Aβ-42 solution without (violet) and with 20 μM Laf1-AK-Laf1 (orange). The dispersed phase was re-suspended by mechanical agitation before analysis. (B) Residual Aβ-42 monomer measured by size exclusion chromatography. Values correspond to the areas under the monomer peak in the chromatograms measured for freshly purified monomeric 4 μM Aβ-42 solution before incubation (“Before”) and after incubation without (−LL) and with (+LL) 20 μM Laf1-AK-Laf1 (“After”). (C) and (D) Representative TEM images of fibrils formed in homogeneous solutions of 4 μM Aβ-42 in the absence of Laf1-AK-Laf1 after 3 days (C), and of a solution of 4 μM Aβ-42 incubated with 20 μM Laf1-AK-Laf1 for 3 days, showing lack of fibrils (D).

To investigate whether or not Aβ-42 forms ThT-negative aggregates, we evaluated the amount of residual Aβ-42 monomer at the end of the incubation by size exclusion chromatography (Fig. 2B and ESI Fig. S1†). The samples incubated in the absence of condensates showed full monomer conversion, in agreement with the high value of fluorescence ThT intensity (Fig. 2A). In contrast, a residual high amount of Aβ-42 monomer was still present in samples incubated in the presence of protein-rich droplets, indicating that Aβ-42 remains largely monomeric within the condensates. The difference of residual Aβ-42 amount in the condensates with respect to the control is likely due to adsorption of condensates at the bottom of the plate, which trap Aβ-42 monomers.
In agreement with these results, after 3 days of incubation amyloid fibrils were observed by TEM analysis only in samples incubated in the absence of condensates (Fig. 2C, D and ESI Fig. S2†). In the presence of condensates fibrils were not observed even after 6 days of incubation (ESI Fig. S2†).
Overall, these results indicate that the biomolecular condensates sequester Aβ-42 monomers (Fig. 1) and inhibit their aggregation in the continuous phase by reducing their concentration (Fig. 1D). Since Aβ-42 remains monomeric within the condensates (Fig. 2B), the decrease of the aggregation rate in the continuous phase is not compensated by an increase of aggregation within the dense phase, leading to an overall reduction of the total aggregation rate (Fig. 1E and 2).
To further confirm this result, we monitored the kinetics of Aβ-42 amyloid formation by recording the time evolution of ThT fluorescence intensity in heterogeneous mixtures containing an increasing amounts of condensates. We incubated the samples in non-binding plates to compare our results with previous kinetic studies.32,33 In the absense of condensates, we observed the characteristic sigmoidal increase of ThT intensity over time, indicating the conversion of monomeric Aβ-42 into amyloids. Importantly, the presence of biomolecular condensates delayed the formation of amyloids in a dose dependent manner (Fig. 3A). Incubation with higher amounts of condensates fully inhibited aggregation over the entire observed time scale (data not shown).
Fig. 3. Condensates inhibit Aβ-42 fibril formation in a dose dependent manner. (A) Aggregation profiles of 4 μM Aβ-42 solution in the absence and presence of 0.25 μM, 0.5 μM and 1 μM Laf1-AK-Laf1 (from left to right). Solid lines represent model simulations. (B) From the analysis of the aggregation profiles in (A) we estimated the concentration of Aβ-42 in the continuous phase (ms) in the presence of increasing volume fractions of the dispersed phase (ΦD). Solid line indicates the fit based on the overall mass balance. (C) Aggregation profiles of solutions containing 5.5 μM, 5 μM, 4 μM and 3 μM Aβ-42 at constant 0.5 μM Laf1-AK-Laf1. Solid lines indicate model simulations. (D) Concentration of Aβ-42 in the continuous phase estimated from the model simulations in (C) versus initial Aβ-42 concentrations (m0Aβ). Solid line indicates fit based on the overall mass balance. (E) and (F) Confocal brightfield (left) and fluorescence (right) images of 2 μM Aβ-42 incubated with 5 μM Laf1-AK-Laf1 and 20 μM ThT after 30 min (E) and 150 min (F) of incubation. This experiment required a different multi-well plate compared to panels (A)–(C), leading to different aggregation kinetics (see Materials and methods).

To get insights into the mechanism underlying this inhibition, we analysed the ThT profiles measured at increasing volume fractions of condensates with a theoretical model of amyloid formation that considers molecular mechanisms of primary nucleation, secondary nucleation, and elongation.32,41 Model simulations were performed by fixing the set of kinetic parameters previously estimated in the literature42 and using the monomer concentration in the continuous phase as single fitting parameter (Fig. 3A). Using this approach, as a first approximation we were able to describe the inhibition effect of the condensates as a reduction of the effective initial concentration of Aβ-42 in the continuous phase (Fig. 3B). These results were confirmed by experiments performed at constant concentration of condensates and increasing concentrations of Aβ-42 (Fig. 3C and D). From the mass balance of Aβ-42 in the two phases we can express the Aβ-42 monomer concentration in the continuous phase as 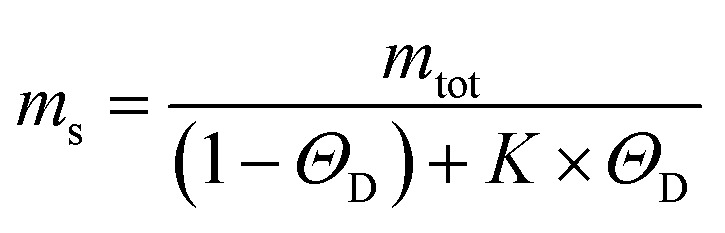 , where mtot is the total initial Aβ-42 monomer concentration, K the partition coefficient and ΘD the volume fraction of the dispersed phase, which was estimated as before.25 Based on this mass balance, from the data shown in Fig. 3B and D we estimated a partition coefficient equal to approximately 1500 and 1250, respectively.
, where mtot is the total initial Aβ-42 monomer concentration, K the partition coefficient and ΘD the volume fraction of the dispersed phase, which was estimated as before.25 Based on this mass balance, from the data shown in Fig. 3B and D we estimated a partition coefficient equal to approximately 1500 and 1250, respectively.
Although as a first approximation the inhibition effect can be explained with this simple model that takes into account the depletion of the effective initial monomer concentration (Fig. 3A–D), confocal microscopy analysis shows that at these very low volume fractions of disperse phase the real mechanism can be more complex, with aggregation potentially initiating at the interface of the condensates (Fig. 3E). Moreover, at these low volume fractions during later stages of the aggregation process fibrils interact with the condensates and disrupt their structures (Fig. 3F and ESI Fig. S3†). Therefore, the obtained values of the partition coefficients K should be taken with a lot of caution, since the interplay between protein aggregation and condensation can be more complex, in particular during the later stages of the process. Understanding these mechanisms can be important for the potential implications for the connection between aberrant protein aggregation and functional condensates in biology.
We further stress that the evaluation of K is also challenged by the very low values of ΘD required by the experiments, since higher values of ΘD leads to essentially full sequestration of Aβ-42 and complete inhibition of aggregation over several hours (Fig. 1E).
We repeated these experiments with biomolecular condensates composed of Dbp1N-AK-Dbp1N and Ddx4, and observed very similar results (Fig. 4 and ESI Fig. S4†). Also in these cases, the inhibition of aggregation in the presence of condensates can be explained in a first approximation as a reduction of the effective initial concentration of Aβ-42 (simulations in Fig. 4).
Fig. 4. Condensates composed of different LCDs inhibit Aβ-42 fibril formation. (A) Aggregation profiles of 3.75 μM Aβ-42 solution in the absence (black) and presence of 1 μM, 2 μM, 5 μM and 10 μM Dpb1N-AK-Dpb1N (from left to right). (B) Aggregation profiles of 5 μM Aβ-42 solution in the absence (black) and presence of 1 μM, 5 μM, 10 μM and 20 μM (from left to right) LCD derived from Ddx4. In both panels solid lines represent model simulations that consider a decrease in the effective initial monomer concentration.

These results indicate that the inhibitory effect can be obtained with different compositions of the condensates. Yet, to exclude that the recruitment of Aβ-42 is generic to any condensate, we tested the uptake of Aβ-42-Atto488 into compartments formed by synthetic polymers produced by reversible addition–fragmentation chain-transfer polymerization (RAFT) (see Materials and methods): a zwitterionic polymer exhibiting upper critical solution temperature (UCST) and a PEG-based polymer characterized by lower critical solution temperature (LCST). In contrast to the protein-based condensates, the Aβ-42-Atto488 peptide was excluded from these polymeric compartments (Fig. 5) and the aggregation profiles of Aβ-42 were not modified by the presence of these condensates.
Fig. 5. Polymeric condensates preferentially exclude Aβ-42 and do not affect fibril formation. (A) and (B) Brightfield (left) and fluorescence (right) confocal images of 1 μM Aβ-42 labelled with Atto-488 incubated with coacervates formed by the zwitterionic polymer (A) and by the LCST polymer (B). (C) Aggregation profiles of 5 μM Aβ-42 in the absence (black) and presence of 20 μM zwitterionic polymer (orange) and 20 μM LCST polymer (green) at 27.5 °C.

The inhibition of Aβ-42 aggregation within the biomolecular condensates can be due to the interactions between the scaffold proteins and Aβ-42, which drive the partitioning of Aβ-42 and can inhibit amyloid formation by competing with the peptide–peptide interactions. To identify whether interactions are related to the intrinsically disordered protein domain (Laf1) or to the globular domain (AK), we tested the effect of AK and Laf1 incubated individually with Aβ-42 under the same solution conditions. Both proteins delayed the aggregation rate of Aβ-42 in a similar way as Laf1-AK-Laf1 (ESI Fig. S5†), indicating that both domains interact with Aβ-42. Since Laf1-AK-Laf1 is positively charged and Aβ-42 carries a net negative charge at this pH value, we expect that electrostatic interactions play a major role in Aβ-42 sequestration. Indeed, we observed that addition of 150 mM NaCl abolished the inhibitory effect of AK due to charge screening (ESI Fig. S5F†). The effect of salt could not be investigated for Laf1-AK-Laf1 condensates because phase separation is reduced at 150 mM NaCl. From the approximated estimation of the partition coefficient (K = 1000–1500) within the condensates we calculated a stoichiometry of 3–7 Aβ-42 molecules per Laf1-AK-Laf1, which seems reasonable considering that both the two Laf1 regions (17 kDa each) and the AK domain (24 kDa) can interact with the Aβ-42 peptide (4.5 kDa) (ESI Fig. S5†).
Conclusion
While it is emerging that the formation of biomolecular condensates could promote fibril formation, we have shown that biomolecular condensates are capable to sequester the amyloidogenic peptide Aβ-42 and inhibit fibril formation despite the local increase in peptide concentration. This is likely due to the heterotypic interactions between the scaffold proteins and Aβ-42 that drive the recruitment of the peptide into condensates.
Since the strong sequestration of Aβ-42 peptides reduces their concentration in the continuous phase and Aβ-42 remains monomeric within the condensates, the decrease of the aggregation rate in the continuous phase is not compensated by an increase of aggregation rate within the dense phase, leading to an overall reduction of the total aggregation rate (Fig. 1E and 2).
At very low volume fractions of the disperse phase and only partial Aβ-42 recruitment, the situation is more complex. Aggregation starts in the continuous phase, potentially triggered by the interface of the condensates, and at higher monomer conversion the process of fibril formation disrupts the structure of the condensates.
These results show how the composition of biomolecular condensates is crucial to regulate not only the dynamic, stimulus-responsive formation of the condensates but also the recruitment of host proteins and the rate of corresponding biochemical reactions occurring within them. This control requires the encoding of multiple intermolecular interactions by the scaffold proteins, despite liquid–liquid phase separation processes could be triggered with much simpler polymers.
This work shows that heterotypic interactions within the condensates can prevent aberrant aggregation of host proteins despite the local increase in concentration.
Experimental
Aβ-42 expression and purification
Abeta42 (Aβ-42) was expressed and purified as described in Walsh et al.43 Briefly, Aβ-42 on pETSac35 plasmid was overexpressed in E. coli BL21-GOLD (DE3). The cultures, supplemented with 100 μg mL−1 ampicillin (Bio Grade, PanReac AppliChem, Darmstadt, Germany) were grown to OD 0.7 before inducing expression by addition of 0.5 mM isopropyl d-thiogalactopyranoside (Bio Grade, PanReac AppliChem, Darmstadt, Germany). Aβ-42 was overexpressed over 4 hours. Cells were spun down at 4500 rpm and re-suspended in working buffer (10 mM Tris, 1 mM EDTA buffer at pH 8.5). Inclusion bodies were recovered from lysed cells and solubilized upon addition of 8 M urea. Aβ-42 was purified from the supernatant with a combination of ion exchange chromatography on a DEAE resin (GE Healthcare, Chicago, IL) and size exclusion chromatography (SD 75 26/600, GE Healthcare, Chicago, IL). The collected fractions were lyophilised and stored at −20 °C.
Protein expression and purification
Proteins were expressed recombinantly in E. coli BL21-GOLD (DE3) cells. Bacterial cultures were induced at OD 0.7 with 0.5 mM isopropyl d-thiogalactopyranoside (Bio Grade, PanReac AppliChem, Darmstadt, Germany) and grown for an additional 16 h at 20 °C (Dpb1N-AK-Dpb1N, AK) or at 37 °C (Laf1-AK-Laf1, Laf1, Ddx4). Laf1-AK-Laf1 and Dpb1N-AK-Dpb1N were purified as described previously.25 Ddx4 was purified with the same protocol as for Laf1-AK-Laf1 and Dbp1N-AK-Dbp1N with the SEC-buffer for Ddx4 supplemented with 1 M NaCl. Laf1 was purified from inclusion bodies using immobilized-metal affinity chromatography according to a standard protocol and by size exclusion chromatography (SD 75 16/600, GE Healthcare, Chicago, IL) in 50 mM Tris (pH 7.5) buffer supplemented with 500 mM NaCl and 2 M urea. AK was purified using immobilized-metal affinity chromatography and size exclusion chromatography (SD 75 16/600, GE Healthcare, Chicago, IL) in 50 mM Tris (pH 7.5) buffer supplemented with 200 mM NaCl.
Synthesis of polymers
Materials
Di(ethylene glycol)methyl ether methacrylate (EG2MA, 95%), ethylene glycol methyl ether methacrylate (EGMA, 99%), 4,4′-azobis(4-cyanovaleric acid) (ACVA, ≥98%), 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPA, ≥97%), [2-(methacryloyloxy)ethyl]dimethyl-(3-sulfopropyl)ammonium hydroxide (SB, 97%), 2-(dimethylamino)ethyl methacrylate (DMAEMA, 98%), 1,3-propanediol cyclic sulfate (TMS, 98%) were purchased from Sigma-Aldrich (Darmstadt, Germany) and used as received. Sulfabetaine methacrylate (ZB) was synthesized according to a previously published protocol.44
Polymer synthesis and characterization
30EG2MA-10EGMA was synthesized via RAFT copolymerization of EG2MA and EGMA by following a previously published protocol.45 Briefly, 52 mg of CPA, 10 mg of ACVA, 1.05 g of EG2MA and 268 mg of EGMA were dissolved in 5.5 g of ethanol and poured in a round bottom flask. The mixture was purged with nitrogen for 20 minutes and left to react at 65 °C for 2 days under constant stirring. The final copolymer was purified via precipitation in 45 mL of isopropyl ether. The recovered reddish waxy solid was dried under vacuum. 30EG2MA-10EGMA number-averaged molecular weight (Mn) and dispersity (Đ) were evaluated via gel permeation chromatography (HPLC 1100, Agilent, Santa Clara, CA). Samples were dissolved at 4 mg mL−1 in THF and filtered through a 0.45 μm pore-size PTFE membrane. The separation was performed at a flow rate of 0.5 mL min−1 at room temperature with a guard and two Agilent PLgel 20 μm MIXED-ALS columns (Agilent, Santa Clara, CA). The reported values are relative poly(methyl methacrylate) standards (Mn = 6335 g mol−1, Đ = 1.24).
70SB-130ZB was synthetized via RAFT copolymerization of SB and ZB according to a previously reported protocol.44 Briefly, 2.88 g of SB, 7.14 g of ZB, 48 mg of CPA, and 13 mg of ACVA were dissolved in 35 mL of 20/80% v/v DMSO/1 M NaCl acetic buffer (pH = 4.5) and poured into a round bottom flask. The mixture was purged with nitrogen for 20 min and then heated to 65 °C for 24 h under constant stirring. The product was dialyzed against deionized water for 24 h with a dialysis membrane tube (Spectrapor, MWCO = 3500 Da, Repligen, Waltham, MA). The phase-separated polymer was collected at the bottom of the tubing and precipitated twice in 150 mL of acetone to obtain a red powder. 70SB-130ZB number-averaged molecular weight (Mn) and dispersity (Đ) were evaluated as described above. Samples were dissolved at 4 mg mL−1 in a 0.05 M Na2SO4/acetonitrile (80/20% v/v) solution and filtered through a 0.45 μm pore-size nylon membrane (Sigma-Aldrich, Darmstadt, Germany). The separation was performed at a flow rate of 0.5 mL min−1 at room temperature with a guard and two Suprema columns (particle size 10 mm and pore sizes 100 and 1000 Å, Polymer Standards Service, Mainz, Germany). The results are relative to polyethylene glycol standards (Mn = 19 925 g mol−1, Đ = 1.10).
Confocal microscopy
Fluorescence confocal microscopy was performed on a Leica TCS SP8 with a Hamamatsu Orca Flash 4.0 cMOS camera and an AOBS laser system (HyD Detector; Wetzlar, Germany). To observe sequestration (Fig. 1), samples were incubated in a glass bottom 384-well plate (MatriPlate 384Well Plate, Brooks Life Science Systems, Poway, CA). Since aggregation occurs over long time scales in these plates, to monitor changes over time (Fig. 3E and F) we incubated samples in a plastic bottom 384-well plate (MICROPLAE 384Well, μClear, Non Binding, Greiner Bio-One, Kremsmünster Austria). Images were recorded at λem = 520 nm after excitation at λex = 500 nm for Atto-488 and at λem = 485 nm after excitation at λex = 405 nm for ThT.
Transmission electron microscopy (TEM)
Transmission electron microscopy (TEM) images were acquired on a TFS Morgagni 268 microscope. 5 μL of samples were spotted on ultrathin carbon support films (Lacey Carbon Support Film Grids, 300 mesh, Gold, Agar Scientific, Essex, UK) for 30 s, washed with distilled water, and negative-stained with a 2% uranyl acetate solution.
Measurement of residual monomer amount by size exclusion chromatography
The amount of residual Aβ-42 monomer after incubation was measured by size exclusion chromatography on an Akta Explorer 100 (Ambersham Biosciences) equipped with a Superdex 75 column (10/30, GE Healthcare, Chicago, IL). Samples were injected using a 100 μL injection loop at a flow rate of 0.5 mL min−1. Chromatograms were recorded by UV absorbance at 215 nm.
Kinetic aggregation assays
The aggregation profiles were monitored by a well-established thioflavin (ThT) binding assay following the protocol described in Cohen et al.32 The stock solution of Aβ-42 peptide was freshly purified via size exclusion chromatography (SD 75, 10/300, GE Healthcare, Chicago, IL) in 20 mM phosphate buffer with 0.2 mM EDTA at pH 8.0 and kept on ice in low binding tubes. Samples were prepared and transferred into a 96-well non-binding plate (Corning Incorporated Life Sciences, Acton, MA) after addition of 20 μM ThT (Sigma-Merck KGaA, Darmstadt, Germany). The time evolution of the ThT fluorescence intensity was recorded on a plate reader (CLARIOstar, BMG Labtech, Offenburg, Germany) at λem = 457 nm and λem = 487 nm at 27.5 °C.
To determine the concentration of Aβ-42 in the continuous phase (Fig. 1) condensates were initially formed in the well and let sediment for 3 hours. Aβ-42 was introduced in the sample at a concentration of 3 μM. After waiting 15 minutes to allow equilibration, the supernatant was removed and analysed by the aggregation assay.
Kinetic model analysis
The aggregation profiles were simulated according to the analytical solution of the mass balance equation describing the time evolution of the total mass of fibrils (M) over time, as described in detail in Meisl et al.41 where the kinetic parameters B±, C±, κ, k∞, k̃∞ are based on the kinetic constants of elongation (k+), primary nucleation (kn) and secondary nucleation (k2) and the reaction order of primary nucleation (nc) and secondary nucleation (n2). These parameters were taken from Cohen et al.42 In all model simulation the initial monomer concentration in the continuous phase msol was the only free parameter. This was estimated by minimizing a least squared error function defined as
where the kinetic parameters B±, C±, κ, k∞, k̃∞ are based on the kinetic constants of elongation (k+), primary nucleation (kn) and secondary nucleation (k2) and the reaction order of primary nucleation (nc) and secondary nucleation (n2). These parameters were taken from Cohen et al.42 In all model simulation the initial monomer concentration in the continuous phase msol was the only free parameter. This was estimated by minimizing a least squared error function defined as 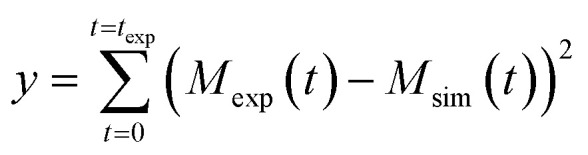 , where Mexp(t) and Msim(t) are the experimental and the simulated total fibril mass fraction at time t, respectively.
, where Mexp(t) and Msim(t) are the experimental and the simulated total fibril mass fraction at time t, respectively.
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
We gratefully acknowledge Dr Thomas Michaels and Dr Christoph Weber for scientific discussions, as well as ScopeM (ETH Zurich) for their support and assistance in this work. We kindly thank the Synapsis Foundation for Alzheimer's disease (Zurich), the Swiss National Science Foundation (grant 205321_179055) and the Claude and Giuliana Foundation for financial support.
Electronic supplementary information (ESI) available: Supplementary data including supplementary figures. See DOI: 10.1039/d0sc04395h
References
- Chiti F. Dobson C. M. Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- Eisenberg D. Jucker M. The Amyloid State of Proteins in Human Diseases. Cell. 2012;148(6):1188–1203. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles T. P. J. Vendruscolo M. Dobson C. M. The Amyloid State and Its Association with Protein Misfolding Diseases. Nat. Rev. Mol. Cell Biol. 2014;15(6):384–396. doi: 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- Peran I. Mittag T. Molecular Structure in Biomolecular Condensates. Curr. Opin. Struct. Biol. 2020;60:17–26. doi: 10.1016/j.sbi.2019.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.-M. Holehouse A. S. Pappu R. V. Physical Principles Underlying the Complex Biology of Intracellular Phase Transitions. Annu. Rev. Biophys. 2020;49(1):107–133. doi: 10.1146/annurev-biophys-121219-081629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondele M. Sachdev R. Heinrich S. Wang J. Vallotton P. Fontoura B. M. A. Weis K. DEAD-Box ATPases Are Global Regulators of Phase-Separated Organelles. Nature. 2019:144–148. doi: 10.1038/s41586-019-1502-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. Elbaum-Garfinkle S. Langdon E. M. Taylor N. Occhipinti P. Bridges A. A. Brangwynne C. P. Gladfelter A. S. RNA Controls PolyQ Protein Phase Transitions. Mol. Cell. 2015;60(2):220–230. doi: 10.1016/j.molcel.2015.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Treeck B. Protter D. S. W. Matheny T. Khong A. Link C. D. Parker R. RNA Self-Assembly Contributes to Stress Granule Formation and Defining the Stress Granule Transcriptome. Proc. Natl. Acad. Sci. U. S. A. 2018;115(11):2734–2739. doi: 10.1073/pnas.1800038115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A. Lee H. O. Jawerth L. Maharana S. Jahnel M. Hein M. Y. Stoynov S. Mahamid J. Saha S. Franzmann T. M. Pozniakovski A. Poser I. Maghelli N. Royer L. A. Weigert M. Myers E. W. Grill S. Drechsel D. Hyman A. A. et al., A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell. 2015;162(5):1066–1077. doi: 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]
- Molliex A. Temirov J. Lee J. Coughlin M. Kanagaraj A. P. Kim H. J. Mittag T. Taylor J. P. Phase Separation by Low Complexity Domains Promotes Stress Granule Assembly and Drives Pathological Fibrillization. Cell. 2015;163(1):123–133. doi: 10.1016/j.cell.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan V. H. Dignon G. L. Zerze G. H. Chabata C. V. Silva R. Conicella A. E. Amaya J. Burke K. A. Mittal J. Fawzi N. L. Mechanistic View of HnRNPA2 Low-Complexity Domain Structure, Interactions, and Phase Separation Altered by Mutation and Arginine Methylation. Mol. Cell. 2018;69(3):465–479. doi: 10.1016/j.molcel.2017.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babinchak W. M. Haider R. Dumm B. K. Sarkar P. Surewicz K. Choi J.-K. Surewicz W. K. The Role of Liquid-Liquid Phase Separation in Aggregation of the TDP-43 Low Complexity Domain. J. Biol. Chem. 2019;294(16):6306–6317. doi: 10.1074/jbc.RA118.007222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peskett T. R. Rau F. O'Driscoll J. Patani R. Lowe A. R. Saibil H. R. A Liquid to Solid Phase Transition Underlying Pathological Huntingtin Exon1 Aggregation. Mol. Cell. 2018;70(4):588–601. doi: 10.1016/j.molcel.2018.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow E. Eftekharzadeh B. Laskowski P. R. Dujardin S. Zoltowska K. M. Hyman A. A. Muller D. Hyman B. T. Wegmann S. Bennett R. E. Tepper K. Vanderburg C. Fan Z. MacKenzie D. Molliex A. M. Taylor J. P. Roe A. D. Kamath T. Commins C. et al., Tau Protein Liquid–Liquid Phase Separation Can Initiate Tau Aggregation. EMBO J. 2018;37(7):e98049. doi: 10.15252/embj.201798049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray S. Singh N. Kumar R. Patel K. Pandey S. Datta D. Mahato J. Panigrahi R. Navalkar A. Mehra S. Gadhe L. Chatterjee D. Sawner A. S. Maiti S. Bhatia S. Gerez J. A. Chowdhury A. Kumar A. Padinhateeri R. et al., α-Synuclein Aggregation Nucleates through Liquid–Liquid Phase Separation. Nat. Chem. 2020:1–12. doi: 10.1038/s41557-020-0465-9. [DOI] [PubMed] [Google Scholar]
- Levin A. Mason T. O. Adler-Abramovich L. Buell A. K. Meisl G. Galvagnion C. Bram Y. Stratford S. A. Dobson C. M. Knowles T. P. J. Gazit E. Ostwalds Rule of Stages Governs Structural Transitions and Morphology of Dipeptide Supramolecular Polymers. Nat. Commun. 2014;5:1–8. doi: 10.1038/ncomms6219. [DOI] [PubMed] [Google Scholar]
- Yan X. Yuan C. Levin A. Chen W. Xing R. Zou Q. Herling T. W. Challa P. K. Knowles T. P. J. Nucleation and Growth of Amino-Acid and Peptide Supramolecular Polymers Through Liquid-Liquid Phase Separation. Angew. Chem., Int. Ed. 2019;100190:2–10. doi: 10.1002/anie.201911782. [DOI] [PubMed] [Google Scholar]
- Faltova L. Küffner A. M. Hondele M. Weis K. Arosio P. Multifunctional Protein Materials and Microreactors Using Low Complexity Domains as Molecular Adhesives. ACS Nano. 2018;12(10):9991–9999. doi: 10.1021/acsnano.8b04304. [DOI] [PubMed] [Google Scholar]
- Galkin O. Pan W. Filobelo L. Hirsch R. E. Nagel R. L. Vekilov P. G. Two-Step Mechanism of Homogeneous Nucleation of Sickle Cell Hemoglobin Polymers. Biophys. J. 2007;93(3):902–913. doi: 10.1529/biophysj.106.103705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes M. P. Sawaya M. R. Boyer D. R. Goldschmidt L. Rodriguez J. A. Cascio D. Chong L. Gonen T. Eisenberg D. S. Atomic Structures of Low-Complexity Protein Segments Reveal Kinked β Sheets That Assemble Networks. Science. 2018;359(6376):698–701. doi: 10.1126/science.aan6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. Choi J. M. Holehouse A. S. Lee H. O. Zhang X. Jahnel M. Maharana S. Lemaitre R. Pozniakovsky A. Drechsel D. Poser I. Pappu R. V. Alberti S. Hyman A. A. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell. 2018;174(3):688–699. doi: 10.1016/j.cell.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y. Ruggeri F. S. Vigolo D. Kamada A. Qamar S. Levin A. Iserman C. Alberti S. George-Hyslop P. S. Knowles T. P. J. Biomolecular Condensates Undergo a Generic Shear-Mediated Liquid-to-Solid Transition. Nat. Nanotechnol. 2020;15(10):841–847. doi: 10.1038/s41565-020-0731-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber C. Michaels T. Mahadevan L. Spatial Control of Irreversible Protein Aggregation. Elife. 2019;8:1–27. doi: 10.7554/eLife.42315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall M. P. Srivastava S. Perry L. S. Kovar R. D. Gardel L. G. Tirrell V. M. Partitioning and Enhanced Self-Assembly of Actin in Polypeptide Coacervates. Biophys. J. 2018;114:1636–1645. doi: 10.1016/j.bpj.2018.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küffner A. M. Prodan M. Zuccarini R. Capasso Palmiero U. Faltova L. Arosio P. Acceleration of an Enzymatic Reaction in Liquid Phase Separated Compartments Based on Intrinsically Disordered Protein Domains. ChemSystemsChem. 2020;2(4):1–8. doi: 10.1002/syst.202000001. [DOI] [Google Scholar]
- Nott T. J. Craggs T. D. Baldwin A. J. Membraneless Organelles Can Melt Nucleic Acid Duplexes and Act as Biomolecular Filters. Nat. Chem. 2016;8(6):569–575. doi: 10.1038/nchem.2519. [DOI] [PubMed] [Google Scholar]
- Vácha R. Linse S. Lund M. Surface Effects on Aggregation Kinetics of Amyloidogenic Peptides. J. Am. Chem. Soc. 2014;136(33):11776–11782. doi: 10.1021/ja505502e. [DOI] [PubMed] [Google Scholar]
- Campioni S. Carret G. Jordens S. Nicoud L. Mezzenga R. Riek R. The Presence of an Air-Water Interface Affects Formation and Elongation of α-Synuclein Fibrils. J. Am. Chem. Soc. 2014;136(7):2866–2875. doi: 10.1021/ja412105t. [DOI] [PubMed] [Google Scholar]
- Galvagnion C. Buell A. K. Meisl G. Michaels T. C. T. Vendruscolo M. Knowles T. P. J. Dobson C. M. Lipid Vesicles Trigger α-Synuclein Aggregation by Stimulating Primary Nucleation. Nat. Chem. Biol. 2015;11(3):229–234. doi: 10.1038/nchembio.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linse S. Cabaleiro-Lago C. Xue W. F. Lynch I. Lindman S. Thulin E. Radford S. E. Dawson K. A. Nucleation of Protein Fibrillation by Nanoparticles. Proc. Natl. Acad. Sci. U. S. A. 2007;104(21):8691–8696. doi: 10.1073/pnas.0701250104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigolato F. Colombo C. Ferrari R. Rezabkova L. Arosio P. Mechanistic Origin of the Combined Effect of Surfaces and Mechanical Agitation on Amyloid Formation. ACS Nano. 2017;11(11):11358–11367. doi: 10.1021/acsnano.7b05895. [DOI] [PubMed] [Google Scholar]
- Cohen S. I. A. Linse S. Luheshi L. M. Hellstrand E. White D. A. Rajah L. Otzen D. E. Vendruscolo M. Dobson C. M. Knowles T. P. J. Proliferation of Amyloid-Beta 42 Aggregates Occurs Through a Secondary Nucleation Mechanism. Proc. Natl. Acad. Sci. U. S. A. 2013;110(24):9758–9763. doi: 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arosio P. Michaels T. C. T. Linse S. Mansson C. Emanuelsson C. Presto J. Johansson J. Vendruscolo M. Dobson C. M. Knowles T. P. J. Kinetic Analysis Reveals the Diversity of Microscopic Mechanisms Through Which Molecular Chaperones Suppress Amyloid Formation. Nat. Commun. 2016;7:10948. doi: 10.1038/ncomms10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaels T. C. T. Šarić A. Curk S. Bernfur K. Arosio P. Meisl G. Dear A. J. Cohen S. I. A. Dobson C. M. Vendruscolo M. Linse S. Knowles T. P. J. Dynamics of Oligomer Populations Formed During the Aggregation of Alzheimer's Aβ42 Peptide. Nat. Chem. 2020;12(5):445–451. doi: 10.1038/s41557-020-0452-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaum-Garfinkle S. Kim Y. Szczepaniak K. Chen C. C. Eckmann C. R. Myong S. Brangwynne C. P. The Disordered P Granule Protein LAF-1 Drives Phase Separation into Droplets with Tunable Viscosity and Dynamics. Proc. Natl. Acad. Sci. U. S. A. 2015;112:7189–7194. doi: 10.1073/pnas.1504822112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott T. J. Petsalaki E. Farber P. Jervis D. Fussner E. Plochowietz A. Craggs T. D. Bazett-Jones D. P. Pawson T. Forman-Kay J. D. Baldwin A. J. Phase Transition of a Disordered Nuage Protein Generates Environmentally Responsive Membraneless Organelles. Mol. Cell. 2015;57(5):936–947. doi: 10.1016/j.molcel.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne C. P. Eckmann C. R. Courson D. S. Rybarska A. Hoege C. Gharakhani J. Jülicher F. Hyman A. A. Germline P Granules Are Liquid Droplets That Localize by Controlled Dissolution/Condensation. Science. 2009;324(5935):1729–1732. doi: 10.1126/science.1172046. [DOI] [PubMed] [Google Scholar]
- Grigolato F. Arosio P. Sensitivity Analysis of the Variability of Amyloid Aggregation Profiles. Phys. Chem. Chem. Phys. 2019;21(3):1435–1442. doi: 10.1039/C8CP05904G. [DOI] [PubMed] [Google Scholar]
- Biancalana M. Koide S. Molecular Mechanism of Thioflavin-T Binding to Amyloid Fibrils. Biochim. Biophys. Acta, Proteins Proteomics. 2010;1804(7):1405–1412. doi: 10.1016/j.bbapap.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arosio P. Cukalevski R. Frohm B. Knowles T. P. J. Linse S. Quantification of the Concentration of A Beta 42 Propagons During the Lag Phase by an Amyloid Chain Reaction Assay. J. Am. Chem. Soc. 2014;136(1):219–225. doi: 10.1021/ja408765u. [DOI] [PubMed] [Google Scholar]
- Meisl G. Kirkegaard J. B. Arosio P. Michaels T. C. T. Vendruscolo M. Dobson C. M. Linse S. Knowles T. P. J. Molecular Mechanisms of Protein Aggregation from Global Fitting of Kinetic Models. Nat. Protoc. 2016;11(2):252–272. doi: 10.1038/nprot.2016.010. [DOI] [PubMed] [Google Scholar]
- Cohen S. I. A. Cukalevski R. Michaels T. C. T. Šarić A. Törnquist M. Vendruscolo M. Dobson C. M. Buell A. K. Knowles T. P. J. Linse S. Distinct Thermodynamic Signatures of Oligomer Generation in the Aggregation of the Amyloid-β Peptide. Nat. Chem. 2018;10(5):523–531. doi: 10.1038/s41557-018-0023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D. M. Thulin E. Minogue A. M. Gustavsson N. Pang E. Teplow D. B. Linse S. A Facile Method for Expression and Purification of the Alzheimer's Disease-Associated Amyloid β-Peptide. FEBS J. 2009;276(5):1266–1281. doi: 10.1111/j.1742-4658.2008.06862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sponchioni M. Rodrigues Bassam P. Moscatelli D. Arosio P. Capasso Palmiero U. Biodegradable Zwitterionic Nanoparticles with Tunable UCST-Type Phase Separation under Physiological Conditions. Nanoscale. 2019;11(35):16582–16591. doi: 10.1039/C9NR04311J. [DOI] [PubMed] [Google Scholar]
- Sponchioni M. Ferrari R. Morosi L. Moscatelli D. Influence of the Polymer Structure over Self-Assembly and Thermo-Responsive Properties: The Case of PEG-b-PCL Grafted Copolymers via a Combination of RAFT and ROP. J. Polym. Sci., Part A: Polym. Chem. 2016;54(18):2919–2931. doi: 10.1002/pola.28177. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.