

Abstract
Axon regeneration is limited in the central nervous system, which hinders the reconstruction of functional circuits following spinal cord injury (SCI). Although various extrinsic molecules to repel axons following SCI have been identified, the role of semaphorins, a major class of axon guidance molecules, has not been thoroughly explored. Here we show that expression of semaphorins, including Sema5a and Sema6d, is elevated after SCI, and genetic deletion of either molecule or their receptors (neuropilin1 and plexinA1, respectively) suppresses axon retraction or dieback in injured corticospinal neurons. We further show that Olig2+ cells are essential for SCI-induced semaphorin expression, and that Olig2 binds to putative enhancer regions of the semaphorin genes. Finally, conditional deletion of Olig2 in the spinal cord reduces the expression of semaphorins, alleviating the axon retraction. These results demonstrate that semaphorins function as axon repellents following SCI, and reveal a novel transcriptional mechanism for controlling semaphorin levels around injured neurons to create zones hostile to axon regrowth.
Keywords: axon dieback, corticospinal tract, Olig2, semaphorin, spinal cord injury
Introduction
Spinal cord injury (SCI) leads to severe deficits in essential motor, sensory, and autonomic functions. Axon regeneration is a fundamental step in reconstructing injured neural circuits to restore function, however, the ability to regrow injured axons is limited in the adult central nervous system (CNS) (Geoffroy and Zheng 2014). Two major factors have been demonstrated to suppress axonal growth: 1) extrinsic factors such as inhibitory molecules expressed in or around the lesion, and 2) intrinsic factors such as the lack of growth-related signaling molecules like PTEN in adult neurons (Liu et al. 2011; Silver et al. 2014). Although a large number of extrinsic factors have been identified (e.g., myelin-associated inhibitors, axon guidance molecules, and chondroitin sulfate proteoglycans [CSPGs]), experimental blockage of those molecules through the use of inhibitors or via genetic approaches have shown only limited or controversial effects on axon regeneration (Harel and Strittmatter 2006; Ueno and Yamashita 2008; Giger et al. 2010; Silver et al. 2014). While some of these approaches succeed in promoting axonal growth, their effects are moderate at best, suggesting that multiple factors and as of yet unidentified molecules might work in concert to inhibit axon regeneration in injured neurons. A greater understanding of the extrinsic molecular environment of the lesion will be required for the full reconstruction of injured spinal cord circuits.
The semaphorin family, consisting of 20 members in mammals, is thought to repel axonal growth of intact or injured neurons in the CNS (Pasterkamp 2012; Yoshida 2012). Although some semaphorins are expressed in CNS lesions (De Winter et al. 2002; O'Malley et al. 2014), it remains unclear whether they act to repel axons of injured neurons. For example, secreted class 3 semaphorins are expressed in CNS lesions (Pasterkamp et al. 1999; De Winter et al. 2002; Pasterkamp and Verhaagen 2006), but the role of Sema3A in axon repulsion after SCI is still unclear. A chemical inhibitor for Sema3A facilitated the regrowth of serotonergic raphespinal axons (Kaneko et al. 2006). However, genetic deletion of plexinA3 and plexin A4, receptors for class 3 semaphorins, did not induce regeneration (Lee et al. 2010a). Deletion of plexinA2, encoding a receptor for Sema6A, enhanced sprouting of corticospinal axons in a pyramidotomy model, but its effects post-SCI have not been examined (Shim et al. 2012). Similarly, expression of membrane-associated semaphorins such as Sema4D and Sema5A have been shown to be elevated after SCI and optic nerve injury, respectively (Moreau-Fauvarque et al. 2003; Goldberg et al. 2004). Their roles in axon regeneration, however, remain unclear. Thus, although previous studies implicate semaphorins as potential extrinsic axon inhibitors, this is not supported by genetic evidence, and the mechanisms causing the elevated expression of these semaphorins remain unknown.
In this study, we investigated the roles of semaphorins in axon repulsion after SCI, and found that the expression of multiple semaphorin family members was induced by Olig2, a basic helix-loop-helix (bHLH) transcription factor. Genetic evidence further showed that this molecular system creates an inhibitory environment for injured corticospinal axons following SCI.
Materials and Methods
Animals
Adult C57BL/6 J mice (wild-type [WT] control, Jackson laboratory), Emx1-Cre (#005628, Jackson laboratory), plexinA1 floxed (plexinA1f/f) (Yoshida et al. 2006), Sema6d−/− (Takamatsu et al. 2010), Nrp1 floxed (Nrp1 f/f) (a gift from Dr Chengua Gu, Harvard Medical School) (Gu et al. 2003), Sema5a−/− (a gift from Dr Alex Kolodkin, Johns Hopkins University) (Matsuoka et al. 2011), Olig2-CreER (Takebayashi et al. 2002), Olig2 floxed (Olig2 f/f) (Yue et al. 2006), and CAG-lox-stop-lox-tdTomato (lsl-tdTomato, Ai14, Jackson laboratory (Madisen et al. 2010)) mice were used in this study. Mutants were backcrossed with C57BL/6 J mice for at least five generations. Mice were maintained in a pathogen-free environment in accordance with protocols approved by the Institutional Animal Care and Use Committee of the Cincinnati Children’s Hospital Medical Center and Niigata University.
Two rhesus monkeys (Macaca mulatta) were studied for the primate portion of this project. One monkey was used as a control, while the other was as a primate SCI test subject (both 4 years old, 4.8 and 5.2 kg, respectively). The monkeys were housed in individual cages and placed on a 12-h light/dark cycle. The experimental protocol was approved by the Animal Welfare and Animal Care Committee of the Primate Research Institute, Kyoto University. All experiments were conducted in accordance with the Guidelines for Care and Use of Nonhuman Primates (Ver. 3, 2010) issued by the institute.
Spinal Cord Injuries (SCIs)
Adult female mice (8 weeks of age) were anesthetized with isoflurane. For a thoracic SCI, a laminectomy was first performed to expose the spinal cord, then dorsal hemisection (depth: 1.0 mm) was performed at T9–10 with a number 11 surgical blade to completely sever the dorsal and dorsolateral corticospinal tracts (CSTs) (Nakamura et al. 2011). To inflict a cervical SCI, a lateral hemisection on the right side (depth: 1.0 mm) was performed at the C6–7 border with a number 11 surgical blade. The dorsal muscle layers and skin were then sutured. They were placed in cages under a 12-h light/dark cycle and fed commercial pellets and water ad libitum. Bladders were manually expressed daily.
Surgery on the monkeys were conducted as previously reported with minor modifications (Nakagawa et al. 2015). Each monkey was sedated with a combination of ketamine hydrochloride (10 mg/kg, i.m.), xylazine hydrochloride (1 mg/kg, i.m.), and atropine (0.05 mg/kg, i.m.), and then anesthetized with sodium pentobarbital (25 mg/kg, i.v.). Upon full anesthetization, skin and axial muscles were dissected at the level of the C2 to T1 segments. Subsequently, laminectomy involving the C3 to C7 segments was performed, the dura mater was cut unilaterally, and the border region between the C6 and the C7 segments in the right side was lesioned with a number 11 surgical blade and a 27-gauge needle. In this SCI model, the dorsolateral funiculus was fully injured to remove the CST fibers that are located laterally. After SCI, the dura mater was sutured, a spongel (Astellas) was placed on it to stop the bleeding, and the skin and axial muscles were then sutured. Finally, the monkey was given an analgesic (Lepetan, Otsuka, i.m.; Indomethacin, Isei, 1 week, oral) and an antibiotic (Viccillin, Meiji Seika, for 4 days, i.m.).
In Situ Hybridization
Control and SCI mice were perfused transcardially with 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB). The brain and spinal cord were dissected and postfixed in the same fixatives overnight. The tissues were then cryopreserved in 30% sucrose/4% PFA for 24–48 h and embedded in Tissue-Tek OCT compound (Sakura Finetek). Serial 20-μm-thick sections were made with a cryostat and mounted on SuperFrost Plus slides (Fisher Scientific).
Digoxigenin-labeled riboprobes were prepared by in vitro transcription from the cDNA fragments encoding semaphorins, neuropilins, and plexins as previously described (Yoshida et al. 2006). In situ hybridizations were performed as described previously (Leslie et al. 2011). Signals were detected using alkaline phosphatase-conjugated antidigoxigenin antibodies (Roche Diagnostics) with nitroblue tetrazolium (NBT) and 5-bromo-4-chloro-3-indolyl phosphate (BCIP) as substrates for the color reaction. Some sections were further processed for immunohistochemistry.
Tissue preparation and in situ hybridizations of monkey brains and spinal cords were performed as described previously (Nakagawa et al. 2018). In brief, the control monkey and the test monkey subjected to SCI (day 7) underwent perfusion-fixation with 4% PFA under deep anesthesia. The brains and spinal cords were removed and postfixed in 4% PFA at 4°C overnight, and then immersed in a 30% sucrose solution containing 4% PFA at 4°C overnight. Forty-μm-thick sections were placed onto glass slides (Fisher Scientific). Digoxigenin-labeled riboprobes for plexin A1, neuropilin 1, sema3d, sema6d, and sema5a were used (GenoStaff). Signals were detected using alkaline phosphatase-conjugated antidigoxigenin antibodies (Roche Diagnostics) with NBT and BCIP for the chromatic reaction.
Quantitative Real-Time PCR
Total RNA was extracted from the T9–10 segment of the spinal cord (1.5-mm-long tissue including the lesion site) using TRIzol reagent (Invitrogen), and reverse transcribed for first-strand cDNA synthesis using the SuperScript III First-Strand Synthesis kit (Invitrogen). Real-time PCR was performed with oligonucleotide primer sets corresponding to the cDNA sequences of semaphorins, plexins, neuropilins, Olig2, and Gapdh (Supplementary Table 1). The 20-μl PCR reaction mixture contained 10 μL of Fast SYBR Green real-time PCR master mix (Applied Biosystems), 1 μL each of the sense and antisense primers (2 μM), and 1 μL of the cDNA sample was preheated at 95°C for 20 s and then treated with 40 amplification cycles (denaturation at 95°C for 3 s, annealing and extension at 60°C for 30 s) in a StepOnePlus™ real-time PCR system (Applied Biosystems). The relative intensity of the PCR product was calculated against Gapdh and the fold change relative to the control was evaluated.
Anterograde Tracing
Anterograde tracing was performed as described previously (Ueno et al. 2012). Six weeks after SCI, mice were anesthetized with isoflurane and placed on a stereotaxic frame. Small holes were made in the corresponding injection sites using a needle. To label the CST, biotinylated dextran amine (BDA; MW 10000; 10% in phosphate buffered saline [PBS]; Thermo Fisher), an anterograde tracer, was injected at four sites in the right and left hemispheres (depth, 0.5 mm: coordinates, 0.6–1.2 mm posterior, 0.8–1.4 mm lateral to bregma, 0.4 μL/site) using a Hamilton syringe tipped with a glass micropipette. After injections, scalps were sutured. Spinal cords were dissected 2 weeks later for histological analyses.
Trans-Synaptic Tracing With Pseudorabies Virus (PRV)
Bartha strain PRV614 (expressing RFP; 3.9 x 109 pfu/ml) (Banfield et al. 2003) and PRV152 viruses (expressing GFP; 4.9 × 109 pfu/ml; a gift from Dr Lynn Enquist, Princeton University) (Smith et al. 2000) were used as trans-synaptic and retrograde tracers (Ueno et al. 2016; Gu et al. 2017b; Ueno et al. 2018). Under anesthesia with isoflurane, a skin incision was made to expose the target muscle of the right hindlimb, the rectus femoris. PRV was injected into the muscle using a glass capillary (total 5 μL) and the skin was sutured. Animals were kept for 6 days, then sacrificed for histological analyses. In pilot studies, we determined that day 5 was the time-point at which PRVs trans-synaptically infected and expressed fluorescent proteins in third-order neurons of the sensorimotor cortex in control mice. However, we found that in WT SCI mice, 6 days were required to see PRV-labeled cells in the cortex. This may be due to limited viral uptake in SCI mice (Duale et al. 2009) or viral spread through other, less direct connections to fourth-order cerebral neurons.
AAV Injections
To delete the plexinA1 gene in plexinA1f/f mice, AAV1-Syn-EGFP-Cre (4.3 × 1012 GC/mL, 0.8 μL/site; Penn Vector Core) was injected into both sides of the hindlimb sensorimotor areas (AP –0.8 mm; ML 1.2 mm, from bregma; all at a depth of 0.5 mm) 2 weeks before SCI. The injections were performed as described in the anterograde tracing section.
Immunohistochemistry
The animals were perfused transcardially with 4% PFA at 7, 10, or 56 days after SCI. The spinal cord was dissected and postfixed in the same fixatives overnight. The tissue was then cryopreserved in 30% sucrose in PBS overnight and embedded in Tissue-Tek OCT compound (Sakura Finetek). Serial 20- or 50-μm-thick sections were made with a cryostat and mounted on SuperFrost Plus slides (Fisher Scientific).
For immunohistochemistry staining, sections were blocked with 1% bovine serum albumin or 5% skim milk in 0.3% Triton X-100 and PBS for 2 h and then incubated at 4°C overnight with the following primary antibodies: rabbit anti-Olig2 (1:500, Millipore, AB9610), mouse anti-GFAP (1:400; Sigma-Aldrich, G3893), goat anti-SOX9 (1:100, R&D, AF3075-SP), rabbit anti-SOX9 (1:500, Cell Signaling, 82 630), mouse anti-NeuN (1:500, Millipore, MAB377), rabbit anti-Iba1 (1:500; Wako, 019–19 741), rabbit anti-GFP (1:1000; Invitrogen, A11122), rat anti-GFP (1:1000; Nacalai, 04404–84), and rabbit anti-RFP (Rockland, 1:1000, 600-401-379). After washing with 0.1% Tween 20/PBS, the sections were incubated with the following secondary antibodies for 2 h at room temperature: Alexa Fluor 488, 568, or 647 donkey antirabbit, mouse, rat, or goat IgG (1:1000; Invitrogen).
For BDA labeling, sections were incubated with 0.3% Triton X-100 and PBS for 4 h, and then with Alexa Fluor 568- or 488-conjugated streptavidin (1:400; Invitrogen) for 2 h at room temperature. Images were acquired using a fluorescence microscope (Zeiss, AXIO IMAGER Z1 or Olympus BX51, DP71).
Behavioral Analyses
Hindlimb locomotor performance was analyzed by the Basso mouse scale (BMS) open-field score. The BMS was scored on days 3, 7, 14, 21, 28, 35, and 42 following injury to assess hindlimb motor function on stepping and coordination in movement. The motor performance was scored along 9 scales, with another 11 scales for subscores (Basso et al. 2006; Nakamura et al. 2011).
The grid walking test was performed to assess the accuracy of hind paw placement on the rungs of a grid during spontaneous exploration (Yoshimura et al. 2011). The mice were trained to walk on a wire grid (0.35-m long with 10-mm squares) before the surgery. During each test, we counted the number of foot slips in which a hind paw dropped below the grid plane in the first 50 steps. Tests were performed on days 7, 14, 21, 28, 35, and 42 after SCI, and the percentage of foot slips were calculated for each test.
Quantification of CST Dieback and Axonal Regrowth
To quantify the dieback and regeneration of CST axons, BDA-labeled pixel areas of CST fibers in the dorsal column were measured at each 100-μm distance bin from the lesion border in sagittal sections by ImageJ software (Yoshimura et al. 2011). The lesion border was defined as the border between the GFAP-positive and -negative areas, which correspond to the astroglial and fibrotic scars, respectively (Liu et al. 2008; Herrmann et al. 2010). Three to seven sections including BDA+ dorsal CST axons were assessed per animal. To normalize interanimal differences in tracing volumes, the BDA+ area at each distance was divided by the average BDA+ area in the rostral 500–600, 600–700, and 700–800-μm bins, and the value was defined as the “axonal index.”
Assessment of Scar Volume
Fibrotic and glial scar volumes were assessed in parasagittal GFAP-immunostained sections by ImageJ software. Fibrotic scars were defined as GFAP-negative lesion cores (Herrmann et al. 2010). The GFAP-negative area in each section 80-μm apart was measured, multiplied by 80 μm, and summed to estimate a fibrotic scar volume. To assess the area percentage of glial scar, the GFAP-positive pixel area within a 1.2 mm rostral and caudal distance from the lesion center was measured and divided by the total spinal cord area. Sections every 160-μm apart was evaluated and the average percentage was calculated.
Quantification of PRV+ Neurons
Serial 50-μm-thick coronal cortical sections were made and images of every other section were acquired by fluorescence microscopy. The positions of PRV-labeled cells along the mediolateral (ML) and anteroposterior (AP) axes (ML, 0 mm at midline; AP, 0 mm at the bregma (Paxinos and Franklin 2001)) were plotted using ImageJ software (Point picker, NIH), and the total number of PRV+ cells were counted.
Western Blot
The severed spinal cord (1.5 mm long including the lesion epicenter) was dissected and homogenized in lysis buffer (50 mM Tris–HCl (pH 8.0) with 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP-40, and a protease inhibitor cocktail (Roche)). After centrifugation at 12000 × g for 20 min at 4°C, the proteins were separated by SDS-PAGE and transferred to a PVDF membrane (Millipore). The membrane was blocked with 5% nonfat dry milk in PBS containing 0.05% Tween-20 and then incubated overnight at 4°C with either rabbit anti-Sema5A antibody (1:1000; a gift from Dr Alex Kolodkin, Johns Hopkins University) (Matsuoka et al. 2011), mouse anti-Sema6D antibody (1:1000; MBL) (Kang et al. 2018), rabbit anti-PlexinA1 antibody (1:5000) (Yoshida et al. 2006), rabbit anti-Olig2 antibody (1:500; Millipore) or mouse anti-beta actin antibody (1:4000; Sigma). After washing, the membrane was incubated with HRP-linked anti-mouse IgG antibody or anti-rabbit IgG antibody (1:5000; Santa Cruz or Cell Signaling Technology). Detection of antibody-bound proteins was performed with a Luminata Forte Western HRP Substrate (Millipore).
Tamoxifen Injections
Tamoxifen (Sigma, 20 mg/ml in corn oil) was injected for three consecutive days (0.1 mg/g bw, i.p.). Two weeks later, the animals were subjected to SCI, and tamoxifen was again injected at days 1 and 2 post-injury.
Analyses of ChIP-Sequencing (ChIP-seq) Data
ChIP-seq data of Olig2, histone H3K4me3, and H3K27Ac were acquired from different differentiation stages of rat oligodendrocytes (oligodendrocyte precursor cells (OPCs), immature oligodendrocytes (iOLs), and mature oligodendrocytes (mOL)), and peak binding positions were detected by MACS (model-based analysis of ChIP-seq), as described previously (Yu et al. 2013). The data were visualized with MochiView software. The detected peak positions of Olig2, histone H3K4me3 and H3K27Ac were compared, and putative binding sites of Olig2 within the sequences were analyzed using the CisBP database (http://cisbp.ccbr.utoronto.ca/; Weirauch et al. 2014). Conserved sequence regions among rat, mouse, rhesus monkey, and human were analyzed using VISTA-Point alignment software (http://genome.lbl.gov/vista/index.shtml; Frazer et al. 2004).
Statistical Analysis
Quantitative data are represented as the mean ± SEM. Statistical analyses were performed using Prism 6 (GraphPad). Differences among the groups were statistically analyzed by one-way ANOVA or two-way repeated measures ANOVA followed by Tukey’s test. Two experimental groups were compared by unpaired t-test or Mann–Whitney test. A P value of less than 0.05 was considered significant.
Results
Expression of Select Semaphorins Increases After SCI
We first examined the expression of all 20 semaphorins in the thoracic spinal cords of control and SCI mice. Using quantitative PCR, we found that expression of various semaphorins (Sema3a, Sema3b, Sema3c, Sema3d, Sema3g, Sema4c, Sema4d, Sema5a, Sema6a, and Sema6d) were elevated one week post-injury (Fig. 1A). We then performed in situ hybridizations to examine mRNA expression patterns within the lesion (Fig. 1B–U). Consistent with the PCR data, expression of Sema3d, 4c, 4d, 5a, 6a, and 6d were elevated in the cells accumulated around the lesion (Fig. 1 E, K, L, O, Q, and T). Sema3a, 3b, 3c, and 3g expression also appeared to increase, although the signals were relatively faint in the cells (Fig. 1 B–D and H). Expression levels of mRNA were highest one week post-injury, then gradually decreased 2 and 4 weeks after SCI (Supplementary Figs 1–3, 4A). The expression of Sema5a and 6d proteins were also increased after SCI (Supplementary Fig. 4B).
Figure 1.
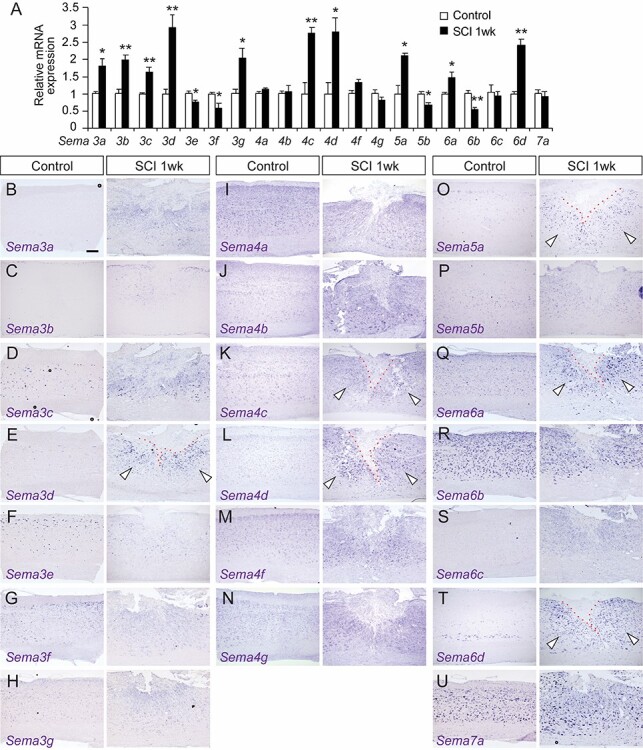
Expression of semaphorins in control and SCI mice. (A) Real-time PCR comparisons of semaphorin mRNA expression in the thoracic cord of control and SCI mice at 1 week post-injury. N = 3–4, **P < 0.01, *P < 0.05, unpaired t-test. (B–U) In situ hybridizations of Sema3a (B), 3b (C), 3c (D), 3d (E), 3e (F), 3f (G), 3 g (H), 4a (I), 4b (J), 4c (K), 4d (L), 4f (M), 4 g (N) 5a (O), 5b (P), 6a (Q), 6b (R), 6c (S), 6d (T), and 7a (U) mRNA in sagittal thoracic spinal cord sections (levels T9–10) from control and SCI mice at 1 week post-injury. Arrowheads indicate increased Sema expression around the lesion site, which is shown by the red dotted line. Scale bar, 200 μm.
Figure 4.
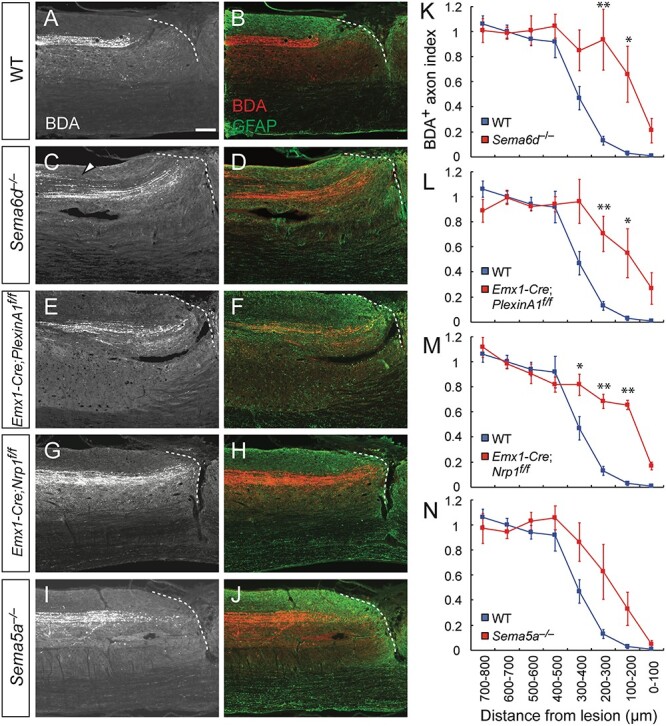
Genetic deletion of Sema6d, PlexinA1, or Nrp1 suppresses dieback of CST fibers after SCI in mice. (A–J) Representative sagittal views of the thoracic spinal cord showing BDA-labeled CST axons in the rostral area of the lesion in WT (A, B), Sema6d−/− (C, D), Emx1-Cre; PlexinA1f/f (E, F), Emx1-Cre; Nrp1f/f (G, H), and Sema5a−/− mice (I, J) at day 56 post-injury. BDA, white (left panel) and red (right panel); GFAP, green. Dotted lines represent the rostral lesion borders. The arrowhead in (C) indicates defasciculated CST fibers. Scale bar, 200 μm. (K–N) Quantification of CST axon amounts in the bins rostral to the lesion site in WT (n = 5, blue), Sema6d−/− (K, n = 5), Emx1-Cre; PlexinA1f/f (L, n = 5), Emx1-Cre; Nrp1f/f (M, n = 6), and Sema5a−/− mice (N, n = 7). **P < 0.01, *P < 0.05, two-way repeated measures ANOVA and Tukey’s test.
We then examined which cell types expressed semaphorins (i.e., neurons, astrocytes, oligodendrocytes, or microglia/macrophage). For this analysis, we selected Sema3d, 4c, 4d, 5a, 6a, and 6d, which showed clear cellular expression by in situ hybridization analysis. Interestingly, most of the elevated semaphorins (Sema3d, 4c, 4d, 5a, 6d) were highly expressed in Olig2+ cells one week post-SCI (Fig. 2A–E). A low percentage of NeuN+ neurons and Sox9+ astrocytes (Sun et al. 2017) also expressed those semaphorins (Fig. 2F and G), but the mRNA signal intensities were lower than those of Olig2+ cells. Only a few Iba1+ microglia/macrophages expressed the semaphorin mRNA, with weak signals.
Figure 2.

Expression of various semaphorins are elevated in Olig2+ cells after SCI. (A–E) Expression of Sema3d (A), 4c (B), 4d (C), 5a (D), and 6d (E) mRNA in Olig2+ cells (green) in the thoracic spinal cord at 1 week post-injury. Sema mRNAs were detected by in situ hybridization and Olig2 proteins were visualized by immunohistochemistry. Arrowheads indicate Sema+/Olig2+ cells. Scale bar, 50 μm. (F, G) Percentages of different cell types (Olig2+, NeuN+, Sox9+, and Iba1+ cells) in Sema3d+, 4c+, 4d+, 5a+, 6a+, and 6d+ cells (F) and percentages of Sema3d+, 4c+, 4d+, 5a+, 6a+, and 6d+ cells in each cell type (G).
Since semaphorins are potential axon repellents for injured corticospinal (CS) neurons, we investigated the expression of their receptors, plexins, and neuropilins, in the sensorimotor area of the cerebral cortex where layer V CS neurons are located (Supplementary Fig. 5). PlexinA1, A2, A4, and C1 were abundantly expressed in the sensorimotor cortex, including layer V neurons (Supplementary Fig. 5A, B, D, and J). They were expressed in the same level in control and SCI mice at 1 week post-SCI. In contrast, plexins A3, B1, B2, and B3 were only weakly expressed (Supplementary Fig. 5C, G–I). Neuropilin-1 (Nrp1) was also expressed highly in a subset of layer V neurons in control and SCI mice, whereas mRNA expression of neuropilin-2 (Nrp2) was very weak in layer V (Supplementary Fig. 5E, F). Quantitative PCR analyses also showed constant mRNA expression of plexins and Nrp1 at 1 week post-injury (Supplementary Fig. 5K). PlexinA1 protein expression also remained consistent after SCI (Supplementary Fig. 5L).
To investigate whether post-SCI elevation of semaphorins occurs in other species, we then examined the expression of Sema3d, 5a, and 6d and their receptors, PlexinA1 and Nrp1, in monkeys. In a unilateral cervical SCI model (Fig. 3A), the expression of PlexinA1 and Nrp1 were clearly detected in both sides of the motor cortex (M1 region), where transected and intact CS neurons were located (Fig. 3B, C). At the injury site, Sema3d expression was undetectable after the injury, suggesting that injury responses differ somewhat from mice (Fig. 3D). In contrast, cells expressing Sema5a and 6d appeared around the lesion site while low signals were observed in the cervical cord of the control monkey (Fig. 3E and F ). Moreover, mice with unilateral cervical SCIs showed elevated semaphorin expression similar as in the thoracic SCI (Supplementary Fig. 6). These data suggest that some semaphorin signaling could be a conserved axon inhibitor in primates.
Figure 3.
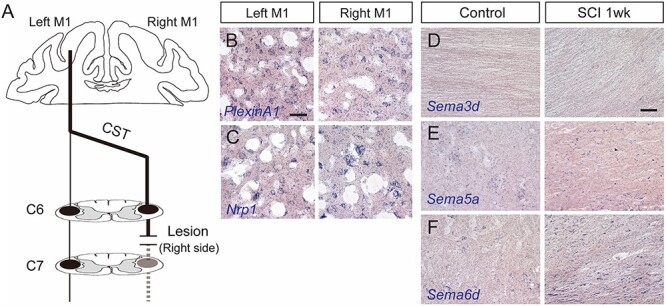
Expression of semaphorins and their receptors in rhesus monkeys. (A) Schema of the unilateral cervical SCI model in monkeys. (B, C) PlexinA1 (B) and Nrp1 (C) expression in the left (containing transected corticospinal [CS] neurons) and right (containing intact CS neurons) M1 regions at day 7 after SCI. Scale bar, 50 μm. (D–F) Expression of Sema3d (D), 5a (E), and 6d (F) in the cervical cord and the area around the lesion site of control (left panels) and SCI monkeys at day 7 post-injury (right panels).
Sema6d, PlexinA1, and Nrp1 Mutant Mice Showed Reduced Axon Dieback of Injured CSTs
To elucidate the role of semaphorins in axonal repulsion after SCI, we examined whether post-injury axonal length within the CST was altered in mutant mice. We used a dorsal hemisection thoracic SCI model to completely transect the dorsal and dorsolateral CSTs in WT and mutant mice (Nakamura et al. 2011; Yoshimura et al. 2011). Based on the elevated expression profiles of Sema3a–d, 4c-d, 5a, 6a, and 6d, we focused on Sema5a and Sema6d mutant mice as well as Emx1-Cre; plexinA1f/f and Emx1-Cre; Nrp1f/f mice for cerebral cortex-specific deletion of plexinA1, a receptor for Sema6D (Toyofuku et al. 2004; Yoshida et al. 2006), and Nrp1, a receptor for Sema3A–D (Sharma et al. 2012; Degenhardt et al. 2013), respectively. Since plexinB1 and B2, the receptors for Sema4D and Sema4C, respectively (Tamagnone et al. 1999; Deng et al. 2007; Maier et al. 2011), were not expressed clearly in layer V neurons (Supplementary Fig. 5G, H), their mutant mice were not analyzed in this study. BDA, an anterograde tracer, was injected into the hindlimb region of the sensorimotor cortex to label CS axons, and CS axon length was assessed at 8 weeks post-injury.
In post-SCI WT mice, CS axons exhibited marked dieback, retracting far from the lesion border (Fig. 4A and B) (Seif et al. 2007). In contrast, more BDA+ CST fibers were retained in the rostral ~300 μm area close to the lesion in Sema6d−/−, Emx1-Cre; plexinA1f/f, and Emx1-Cre; Nrp1f/f mice compared to WT mice (Fig. 4 C–H, K–M). The amount of CST fibers in Sema5a−/− mice also tended to be higher in the rostral area compared to that of WT mice (Fig. 4I, J, N; P = 0.0591). These results indicate that the genetic deletions of semaphorins enhance axonal regrowth and/or reduce axon dieback (or retraction).
To examine whether axon dieback was suppressed in the mutant mice, the rostro-caudal amount of BDA+ CST axons was assessed at day 10, during the subacute phase when axon dieback typically begins (Liu et al. 2010). In WT mice, CS axons started exhibiting axon retraction, although the degree of dieback at day 10 was lower than that at day 56 (Supplementary Fig. 7A, B, I; in comparison with Fig. 4A and B). In contrast, CST fibers remained close to the lesion border in Emx1-Cre; plexinA1f/f mice (P = 0.0298; Supplementary Fig. 7E, F, J). Sema6d−/− mice also showed a greater amount of CST fibers compared to that of WT mice (P = 0.0699; Supplementary Fig. 7C, D, I), whereas Emx1-Cre; Nrp1f/f mice did not show a significant difference in the amount of fibers at day 10 (P = 0.3252; Supplementary Fig. 7G, H, K). Together, these data suggest that plexinA1 and Sema6d mutant mice suppress dieback, while Nrp1 deletion suppresses dieback in later phases after initial retractions.
Although a number of CS axons were observed in the rostral border of the lesion, no BDA+ axons were found within the lesion core (GFAP− area) and very few, if any fibers were found across the lesion. We observed a few BDA+ axons passing through the ventral funiculus in a fraction of the mice tested (WT, 1/5 mice; Sema6d−/−, 2/5; Emx1-Cre; plexinA1f/f, 1/5; Emx1-Cre; Nrp1f/f, 2/6; Sema5a−/, 0/6). The axons might be ventral CST fibers, which is sometimes observed even in WT mice (Steward et al. 2008), or they could be a result of defects in axon elimination of the ventral CST in Sema6d−/− and Emx1-Cre; PlexinA1f/f mice (Gu et al. 2017a). We also found defasciculated CST fibers in the dorsal funiculus of Sema6d−/− mice (Fig. 4C, D), consistent with the phenotype seen in the optic nerve (Kuwajima et al. 2012). Taken together, the data indicate that mutant mice lacking semaphorin-related genes do not exhibit axon growth beyond the lesion site.
Since semaphorins are expressed around the lesion after SCI, they might affect the process of scar formation after the injury. However, the volumes of GFAP+ glial scars and GFAP− fibrotic scars were not altered across the genotypes (Supplementary Fig. 8). This suggests that the increased CST axon density in semaphorin mutants was not attributable to the altered formation of scar tissues.
Next, to test if connections between the cerebral cortex, sub-lesional spinal cord and muscles were promoted in semaphorin mutant mice, we injected PRV, a trans-synaptic retrograde viral tracer, into the hindlimb rectus femoris muscle at day 42 post-injury (Fig. 5A) (Ueno et al. 2016; Gu et al. 2017b). In WT mice, PRVs labeled a number of CS neurons in layer V of the hindlimb area of the motor cortex (data not shown) that dramatically decreased following SCI (Fig. 5B, H). Sema5a−/−, and Emx1-Cre; Nrp1f/f mice showed similar numbers as WT mice (Fig. 5F, G, L, M). In contrast, higher numbers of PRV+ neurons were observed in Emx1-Cre; plexinA1f/f and Sema6d−/− mice following injury compared to WT mice (Fig. 5C, D, I, J). Since both Sema6d and plexinA1 mutant mice maintain ventral/ventrolateral CSTs that are normally eliminated in WT mice during early postnatal development (Gu et al. 2017a), we decided to delete plexinA1 during the adult stage to allow for normal CST formation, permitting a more focused look at plexinA1 deletion on post-SCI recovery. This temporally targeted deletion was accomplished by injecting adeno-associated virus (AAV) expressing Cre (AAV-Cre) into the sensorimotor cortex of plexinA1f/f mice 2 weeks before SCI. In this case, these mice did not show any increases in PRV+ CS neurons (Fig. 5E, K), suggesting that the increased numbers of PRV+ neurons in Emx1-Cre; plexinA1f/f might be due to the maintained ventral and ventrolateral CS axons, which were spared in the dorsal thoracic injury.
Figure 5.
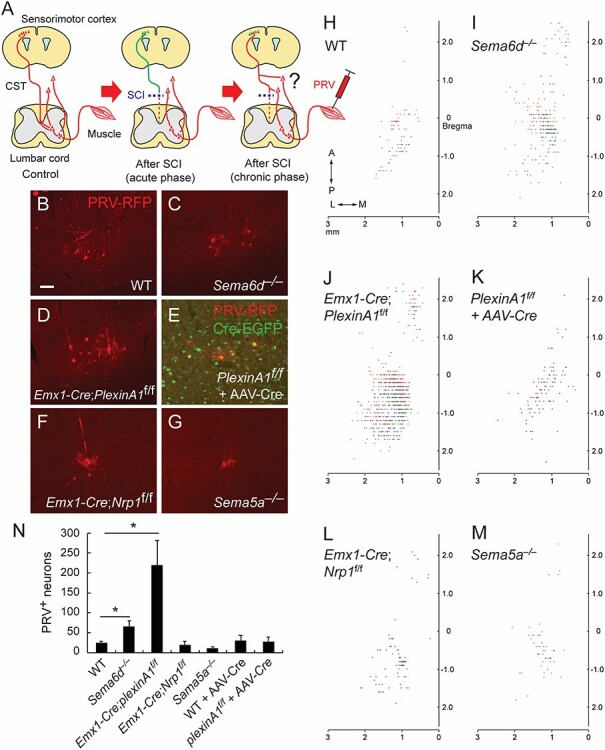
Connectivity between the hindlimb muscle and cerebral cortex following SCI in semaphorin mutant mice. (A) Schema of the PRV tracing experiment from the hindlimb muscle to examine whether cortical neurons form new connections with spared circuits that bypass the lesion to the lower spinal levels (right panel). (B–G) Representative images of PRV-labeled layer V neurons (red) in WT (B), Sema6d−/− (C), Emx1-Cre; PlexinA1f/f (D), PlexinA1f/f + AAV-Cre-EGFP (E), Emx1-Cre; Nrp1f/f (F), and Sema5a−/− mice (G) after SCI at day 42 + 6. Scale bar, 50 μm. (H–M) Top views of the cortical locations of PRV+ layer V neurons in the cerebral cortices of WT (H), Sema6d−/− (I), Emx1-Cre; PlexinA1f/f (J), PlexinA1f/f + AAV-Cre (K), Emx1-Cre; Nrp1f/f (L), and Sema5a−/− mice (M) after SCI. Plots of three representative animals are shown in red, blue, and green. (N) The number of PRV+ layer V neurons in WT (n = 5), Sema6d−/− (n = 6), Emx1-Cre; PlexinA1f/f (n = 7), Emx1-Cre; Nrp1f/f (n = 5), Sema5a−/− (n = 6), WT + AAV-Cre (n = 6), and Emx1-Cre; PlexinA1f/f + AAV-Cre mice (n = 7). *P < 0.05, Mann–Whitney tests or unpaired t-tests.
To examine the effects of PlexinA1 and Sema6D on axonal sprouting, we evaluated the amount of CS axon collaterals in the rostral part of the lesion in plexinA1 and Sema6d mutants (Supplementary Fig. 9). Although collateral fibers seemed to be slightly increased in Sema6d−/− and Emx1-Cre; plexinA1f/f mice, the differences were not statistically significant (P = 0.1493 and 0.1058, respectively; Supplementary Fig. 9B–E). Thus, our data indicate that semaphorins induce dieback (or retraction) of transected axons, but have limited effect on the suppression of axonal sprouting and rewiring after SCI.
We further evaluated motor recovery after SCI in Sema6d, PlexinA1, Nrp1, and Sema5a mutant mice. Using BMS scoring to assess hindlimb functions during locomotion, and grid walking tests to evaluate skilled motor functions, we observed that post-SCI WT mice exhibited severe motor deficits in the acute phase (days 3–7), but spontaneously recovered to mid-level functioning by day 28 (Supplementary Fig. 10) (Nakamura et al. 2011; Yoshimura et al. 2011). Semaphorin mutant mice did not show any improvement in scores over the same time period (Supplementary Fig. 10). This indicates that although inhibition of semaphorin signals could suppress axon dieback, it was not sufficient to reform functional circuits to enhance motor recovery over that of control mice. This finding is consistent with the results reported in several growth-promoting SCI models (Geoffroy et al. 2015).
Semaphorin Expression is Induced by Olig2 after SCI
Although a variety of axon repellent molecules have been reported to be expressed following injury, the mechanisms underlying their transcriptional controls are largely unknown. We explored the molecular triggers for semaphorin expression after SCI by investigating the possibility that Olig2, a bHLH transcription factor, regulates semaphorin gene expression, since most of the semaphorin genes were found to be expressed in Olig2+ cells (Fig. 2).
We began by examining Olig2 expression following thoracic SCI. Expression of the Olig2 protein was elevated at 1-week post-injury (Fig. 6A). Immunohistochemistry revealed abundant Olig2-expressing cells near the lesion (Fig. 6B), similar to those observed in brain injury models (Chen et al. 2008; Tatsumi et al. 2008). Since semaphorins were mainly expressed in Olig2+ cell lineages, we examined the anatomical interactions between Olig2+ cells and CST axons around the lesion. We observed a number of transected axons adjacent to accumulating Olig2+ cells, which were labeled by tdTomato in Olig2CreER/+; lox-stop-lox-tdTomato (lsl-tdTomato) mice injected with tamoxifen (Fig. 6C). These data indicate that Olig2+ cells are spatially located in areas where they could inhibit CST axon growth.
Figure 6.
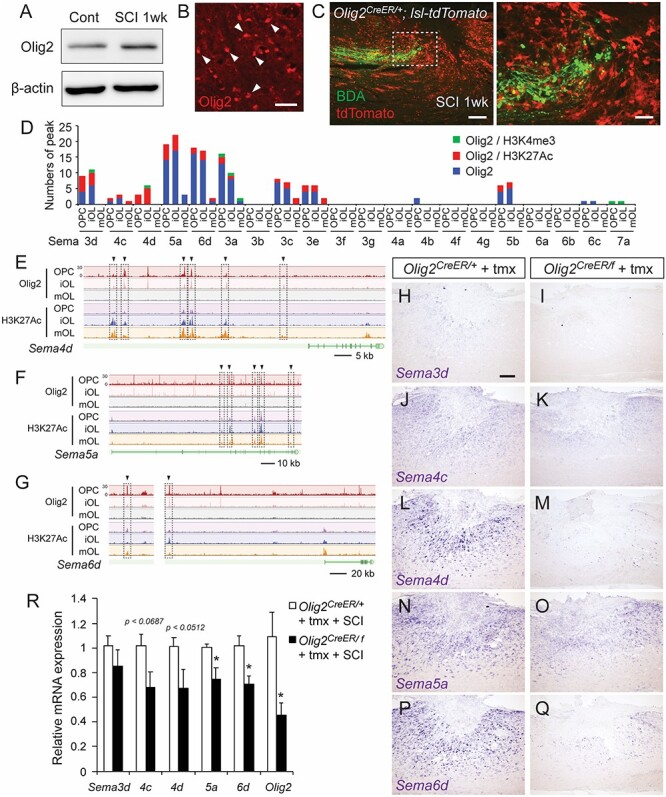
Olig2 is required for semaphorin expression following SCI. (A) Western blots of the Olig2 protein in the thoracic cords of control and SCI mice at 1 week post-injury. (B) A number of Olig2+ cells were seen around the lesion. Scale bar, 25 μm. (C) Transected BDA+ CS axons (green) in front of accumulating Olig2+ cells labeled with tdTomato (red), in the rostral area of the lesion in Olig2CreER/+; lsl-tdTomato mice at day 7 post-injury. Sagittal section (right panel shows a magnified view of the dotted box). (D) The number of Olig2-binding peaks in genomic regions of Sema genes in ChIP-seq data acquired from OPCs, iOLs, and mOLs (Yu et al. 2013). Green, peaks corresponding to H3K4me3 binding peaks; red, peaks corresponding to H3K27Ac; blue, other Olig2 binding peaks. (E–G) ChIP-seq data of Olig2 and H3K27Ac in Sema4d (E), Sema5a (F), and Sema6d (G) genomic regions. Arrowheads and dotted squares represent the binding peaks common to Olig2 and H3K27Ac. (H–Q) In situ hybridizations of Sema3d (H, I), 4c (J, K), 4d (L, M), 5a (N, O), and 6d (P, Q) mRNA in the thoracic cords of tamoxifen-injected control Olig2CreER/+ and Olig2CreER/f mice at 1 week post-SCI. (R) Comparison of levels of Sema3d, 4c, 4d, 5a, and 6d mRNA expression in the thoracic cords of tamoxifen-injected Olig2CreER/+ and Olig2CreER/f mice at 1 week post-SCI, assessed by real-time PCR. N = 6, *P < 0.05, unpaired t-test. Scale bars, 25 μm (B), 200 μm (C, left), 50 μm (C, right), 200 μm (H–Q).
Previous studies using ChIP-seq data have shown the locations of genomic Olig2 binding sites (Yu et al. 2013). We re-evaluated the ChIP-seq data and found that Olig2 bound to intergenic regions or introns of the semaphorin genes that were highly induced post-SCI (Fig. 6D–G; Sema3d, Sema4c, Sema4d, Sema5a, and Sema6d). Although most of the binding sites did not correspond to transcription start sites marked by histone H3K4me3, they were well correlated with the peak binding positions of activating histone H3K27Ac, which represents gene activation at enhancers (Strahl and Allis 2000; Martin and Zhang 2005). These data strongly suggest that Olig2 targets the genomic open enhancer regions of semaphorins to regulate their transcription rates.
To test whether Olig2 regulates semaphorin expression after SCI, we deleted the Olig2 gene using Olig2-CreER and Olig2-floxed mice with tamoxifen injections. Since CreER was inserted after the start codon in Olig2-CreER mice (Takebayashi et al. 2002), the Olig2-CreER allele deletes the Olig2 gene. In the Olig2 floxed allele, the Olig2 coding region is flanked by loxP sequences (Yue et al. 2006). Olig2CreER/f mice were thus treated with tamoxifen to induce conditional Olig2 knockout, while Olig2CreER/+ mice were used as controls. Five tamoxifen injections efficiently induced recombination in Olig2CreER/+; lox-stop-lox-tdTomato mice to express tdTomato in most Olig2+ cells (Fig. 6C). The recombination efficiency was likely lower in the flox sequence flanking the Olig2 gene, since tamoxifen injections decreased Olig2 expression in Olig2CreER/f spinal cord to only half the level of Olig2CreER/+ mice (Fig. 6R). In those mice, we first examined the expression of Sema3d, 4c, 4d, 5a, and 6d by in situ hybridization and RT-PCR. Among them, expression of Sema5a and Sema6d were significantly decreased, while Sema4c and 4d expression seemed to decrease with Olig2 deletion (P = 0.0687, 0.0512, respectively; Fig. 6H–R). These data strongly suggest that Olig2 regulates semaphorin expression after SCI.
Lastly, we examined the post-SCI axonal projections of CS fibers in Olig2-deleted mice. Compared to the substantial dieback of CS axons in control Olig2CreER/+ mice, a significant number of BDA+ CST fibers were retained in the rostral areas of the lesion (~400 μm) in tamoxifen-injected Olig2CreER/f mutant mice (Fig. 7A–E). Among the mutants, we observed one Olig2CreER/f mouse that contained numerous axons elongating caudal to the lesion (data not shown); however, it was difficult to confirm whether they were regenerating axons or axons evaded from the injury. We then looked for detour connections between the cerebral cortex and lower spinal cord by injecting PRV into the hindlimb muscle. However, no significant increase in PRV+ CS neurons was observed in Olig2CreER/f mice (Fig. 7F). Since Olig2 is reported to be involved in glial responses in brain injury models (Chen et al. 2008; Tatsumi et al. 2008), we also examined scar volumes after SCI. Although Olig2CreER/f mice showed a tendency to increase the volume of GFAP− fibrotic scar and to decrease that of GFAP+ glial scar, they were not statistically different (Supplementary Fig. 11).
Figure 7.
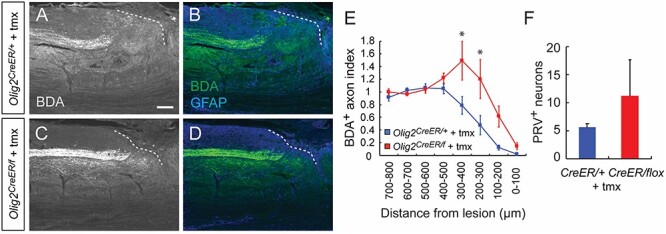
Genetic deletion of Olig2 prevents CS axon retraction after SCI. (A–D) Representative images of BDA+ CST axons in the rostral area of the lesion site in tamoxifen (tmx)-treated Olig2CreER/+; lsl-tdTomato (A, B) and Olig2CreER/f; lsl-tdTomato mice (C, D) at day 56 post-injury. BDA, white (left panel) and green (right panel); GFAP, blue. Sagittal view of the thoracic cord. Dotted lines represent the lesion borders. Scale bar, 200 μm. (E) Quantification of CST axon amounts in the thoracic cord rostral to the lesion in Olig2CreER/+; lsl-tdTomato and Olig2CreER/f; lsl-tdTomato mice. N = 5, *P < 0.05, two-way repeated measures ANOVA and Tukey’s test. (F) The number of PRV+ neurons in the cerebral cortex at day 42 + 6 post-injury in tamoxifen (tmx)-treated Olig2CreER/+; lsl-tdTomato (n = 5) and Olig2CreER/f; lsl-tdTomato mice (n = 5). Mann–Whitney test.
Taken together, these results reveal that transcriptional regulation of semaphorins by Olig2 is essential for semaphorin induction after SCI, and that the transcriptional machinery plays a role in forming a suppressive environment for axonal growth after SCI.
Discussion
In this study, we show that semaphorin expression is induced after SCI, mediating the retraction of injured neurons. In our knowledge, this study is the first comprehensive expression study on the semaphorin family members following SCI. We also present genetic evidence for their roles in repelling axons after SCI. Reducing the inhibitory environment by semaphorin deletion may provide a potential therapeutic avenue to aid in the reconstruction of neural circuits. We have further discovered a novel transcriptional mechanism involving Olig2 that regulates the expression of semaphorins. Although most SCI recovery studies have thus far focused on altering the expression levels of inhibitory molecules, targeting transcriptional machinery could be a novel therapeutic strategy to enhance regeneration after SCI. Importantly, since targeting a single inhibitory molecule has limited effects, controlling a transcriptional machinery may have the potential to prevent expression of multiple inhibitory molecules simultaneously, and exert additive effects on reduced axon retraction or axonal growth after SCI.
Previous studies have indicated that different cell types such as astrocytes, macrophage/microglia, and oligodendrocytes, produce inhibitory molecules for axonal growth (Silver et al. 2014). Oligodendrocytes produce myelin-associated proteins such as Nogo, OMgp, MAG, and EphrinB3, all of which have been shown to suppress axon regrowth (Lee et al. 2010a,b; Duffy et al. 2012; Geoffroy and Zheng 2014). The results of our study indicate that semaphorins, another family of molecules secreted from oligodendrocytes, can also act as inhibitors to injured axons. A recent study showed that NG2+ cells, the majority of which are OPCs, trap axons to inhibit axonal growth (Filous et al. 2014). The present study demonstrates another mechanism that Sema6d and potentially other semaphorins such as Sema3d, 4c, 4d and 5a act as molecular repellents to severed axons. The expression of semaphorins in oligodendrocytes is corroborated by data in gene expression databases of isolated neurons and glial cells (Zhang et al. 2014); however, it is unclear if post-SCI expression of semaphorins occurs in myelin contents or processes of prematurated oligodendrocytes and OPCs. Since each semaphorin is expressed in a subset of Olig2+ cells (Fig. 2G), it is possible that semaphorins are distributed in specific differentiation stages of oligodendrocytes. Premature oligodendrocytes continue to proliferate around lesion sites for at least 4 weeks (Hesp et al. 2015), and thus may act as a long-lasting barrier to axon growth. Sema6d is also expressed highly in Sox9+ astrocytes (Fig. 2F, G) and possibly in a subset of proliferating astrocytes expressing Olig2, which only appear in the early phase following injury (Chen et al. 2008; Tatsumi et al. 2008). Although genetic deletion of semaphorins did not seem to affect scar formation, they may have other effects on oligodendrocytes or astrocytes, such as in spatial distribution, proliferation, differentiation, and cellular interactions. These issues will be explored in future studies.
Axon dieback is the process in which axons retract from injury sites, which involves an acute degeneration process with axon fragmentation, followed by retraction phases (Kerschensteiner et al. 2005; Hill 2017). Based on the role of semaphorins in axon repulsion during development, semaphorins would be involved in the retraction phase of axon dieback after SCI. While we could not exclude the possibility that the absence of the semaphorin might also promote regrowth of some axons in the glial scar of dorsal funiculus, as well as reduction of axon retraction, we did not observe any marked increase in axon regeneration into and beyond the lesion core. The type and size of the lesion may influence the degree of regeneration (Zukor et al. 2013), but induction of axonal regrowth caudal to the lesion seems to be difficult in general, even when extrinsic factors are genetically manipulated. For example, genetic deletion of triple myelin-associated genes did not induce axon regeneration across the lesion site (Lee et al. 2010b). Since a number of different inhibitory molecules are known to affect axonal growth, it is possible that other molecules may still suppress regeneration even if a few molecules were deleted. Within the semaphorin family, we found multiple molecules expressed around the lesion site. We also found that CS axons never grew into the fibrotic scar although they extended into the glial scar. Fibrotic scars have been shown to exhibit strong barrier effects when Pten was deleted to increase the intrinsic regenerative capacity of the CST (Zukor et al. 2013) and when scar tissue was directly ablated genetically (Dias et al. 2018). Thus, additional methods that modulate scar tissue formation may be needed to overcome the strict inhibitory barrier to axon regrowth.
In the course of our study, we also discovered that Olig2 is a transcriptional regulator of semaphorin expression. Olig2 might collaborate with other transcriptional components such as Brg1 to exert transcription activation, which has been observed in cellular responses and differentiation (Yu et al. 2013); however, this mechanism might differ from the transcriptional repressor activity of Olig2, which was reported for cell fate decision during development (Novitch et al. 2001). Since Olig2 likely regulates a number of additional molecules (Yu et al. 2013), the scope of its effect on axon regeneration may extend well beyond that of semaphorin gene suppression. For instance, we found that Olig2 also bound to genomic regions of many other known inhibitory molecules (Efnb2, 3, a5, Slit1–3, Ntn1, Wnt5a, etc.). Thus, our study proposes a novel target for inhibiting dieback and/or promoting axon growth by focusing on transcriptional mechanisms that create inhibitory barriers around injured neural circuits. Caution must be exercised, however, as Olig2 deletion may have additional effects on the repair process. For example, Olig2 has also been reported to participate in glial scar formation (Chen et al. 2008). Thus, it will be valuable to explore additional transcriptional factors that control the formation of these inhibitory environments.
Although many axon repellent molecules are expressed following CNS injuries (Harel and Strittmatter 2006; Ueno and Yamashita 2008; Giger et al. 2010), the mechanisms underlying the upregulation of expression are poorly understood. Only a few studies have focused on transcriptional regulation, such as the Sox9-regulated expression of CSPG (McKillop et al. 2013). Further in-depth analyses of the upstream signal cascades of inhibitory molecules will likely illuminate new ways to manipulate the tissue environment proximal to an injury site in the effort to promote axon regeneration. Although it may be technically difficult to pharmaceutically inhibit transcription factors, one strategy may be to suppress expression of transcriptional regulators by shRNA in a temporally controlled manner. In addition to identifying semaphorins as novel axon repellents following SCI, our study highlighted transcriptional control machinery as an important, potentially far-reaching extrinsic mechanism that can impede axonal growth. Transcriptional regulatory molecules might be a promising target for therapeutic approaches to enhance axon regeneration after SCI.
Supplementary Material
Acknowledgments
We would like to thank C. Gu and A. Kolodkin for Nrp1 floxed mice, A. Kolodkin for Sema5a–/– mice and anti-Sema5a antibodies; L. Enquist and the Center for Neuroanatomy with Neurotropic Viruses (CNNV; NIH grant P40RR018604) at Princeton University for providing PRVs; H. Wang, Y. Deng, and L. Xu (Cincinnati Children’s Hospital Medical Center) for sharing mice and ChIP seq data; P. Thanh, A. Epstein, M. Sandy (Cincinnati Children’s Hospital Medical Center), T. Sato, S. Tsuboguchi, and K. Hoshina (Niigata University) for their technical assistance; T. Yamashita (Osaka University), K. Shibuki, and O. Onodera (Niigata University) for supporting materials; E. Hollis II (Burke Neurological Institute), J. Carmel (Columbia University), and J. Martin (CUNY School of Medicine) for critical reading of the manuscript.
Funding
This work was supported by NINDS-NS100772 and -NS093002, Craig Neilsen Foundation, and New Jersey Commission of Spinal Cord Research (Y.Y.); PRESTO (JST; JPMJPR13M8), AMED-CREST (JP19gm1210005), JSPS KAKENHI 17H04985, 17H05556, 17K19443, JSPS Postdoctoral Fellowships for Research Abroad, KANAE Foundation for the Promotion of Medical Science, Kato Memorial Bioscience Foundation, Grant-in-Aid from the Tokyo Biochemical Research Foundation, the Narishige Neuroscience Research Foundation, Ube Industries Foundation, Takeda Science Foundation, Japan Heart Foundation Research Grant, and Senri Life Science Foundation (M.U.).
References
- Banfield BW, Kaufman JD, Randall JA, Pickard GE. 2003. Development of pseudorabies virus strains expressing red fluorescent proteins: new tools for multisynaptic labeling applications. J Virol. 77:10106–10112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG. 2006. Basso mouse scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma. 23:635–659. [DOI] [PubMed] [Google Scholar]
- Chen Y, Miles DK, Hoang T, Shi J, Hurlock E, Kernie SG, Lu QR. 2008. The basic helix-loop-helix transcription factor olig2 is critical for reactive astrocyte proliferation after cortical injury. J Neurosci. 28:10983–10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Winter F, Oudega M, Lankhorst AJ, Hamers FP, Blits B, Ruitenberg MJ, Pasterkamp RJ, Gispen WH, Verhaagen J. 2002. Injury-induced class 3 semaphorin expression in the rat spinal cord. Exp Neurol. 175:61–75. [DOI] [PubMed] [Google Scholar]
- Degenhardt K, Singh MK, Aghajanian H, Massera D, Wang Q, Li J, Li L, Choi C, Yzaguirre AD, Francey LJ et al. 2013. Semaphorin 3d signaling defects are associated with anomalous pulmonary venous connections. Nat Med. 19:760–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng S, Hirschberg A, Worzfeld T, Penachioni JY, Korostylev A, Swiercz JM, Vodrazka P, Mauti O, Stoeckli ET, Tamagnone L et al. 2007. Plexin-B2, but not Plexin-B1, critically modulates neuronal migration and patterning of the developing nervous system in vivo. J Neurosci. 27:6333–6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias DO, Kim H, Holl D, Werne Solnestam B, Lundeberg J, Carlen M, Goritz C, Frisen J. 2018. Reducing pericyte-derived scarring promotes recovery after spinal cord injury. Cell. 173:153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duale H, Hou S, Derbenev AV, Smith BN, Rabchevsky AG. 2009. Spinal cord injury reduces the efficacy of pseudorabies virus labeling of sympathetic preganglionic neurons. J Neuropathol Exp Neurol. 68:168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy P, Wang X, Siegel CS, Seigel CS, Tu N, Henkemeyer M, Cafferty WB, Strittmatter SM. 2012. Myelin-derived ephrinB3 restricts axonal regeneration and recovery after adult CNS injury. Proc Natl Acad Sci USA. 109:5063–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filous AR, Tran A, Howell CJ, Busch SA, Evans TA, Stallcup WB, Kang SH, Bergles DE, Lee SI, Levine JM et al. 2014. Entrapment via synaptic-like connections between NG2 proteoglycan+ cells and dystrophic axons in the lesion plays a role in regeneration failure after spinal cord injury. J Neurosci. 34:16369–16384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. 2004. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32:W273–W279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoffroy CG, Lorenzana AO, Kwan JP, Lin K, Ghassemi O, Ma A, Xu N, Creger D, Liu K, He Z et al. 2015. Effects of PTEN and Nogo codeletion on corticospinal axon sprouting and regeneration in mice. J Neurosci. 35:6413–6428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoffroy CG, Zheng B. 2014. Myelin-associated inhibitors in axonal growth after CNS injury. Curr Opin Neurobiol. 27:31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giger RJ, Hollis ER, Tuszynski MH. 2010. Guidance molecules in axon regeneration. Cold Spring Harb Perspect Biol. 2:a001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JL, Vargas ME, Wang JT, Mandemakers W, Oster SF, Sretavan DW, Barres BA. 2004. An oligodendrocyte lineage-specific semaphorin, Sema5A, inhibits axon growth by retinal ganglion cells. J Neurosci. 24:4989–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Rodriguez ER, Reimert DV, Shu T, Fritzsch B, Richards LJ, Kolodkin AL, Ginty DD. 2003. Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev Cell. 5:45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Kalamboglas J, Yoshioka S, Han W, Li Z, Kawasawa YI, Pochareddy S, Li Z, Liu F, Xu X et al. 2017a. Control of species-dependent cortico-motoneuronal connections underlying manual dexterity. Science. 357:400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Serradj N, Ueno M, Liang M, Li J, Baccei ML, Martin JH, Yoshida Y. 2017b. Skilled movements require non-apoptotic Bax/Bak pathway-mediated corticospinal circuit reorganization. Neuron. 94:626–641 e624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harel NY, Strittmatter SM. 2006. Can regenerating axons recapitulate developmental guidance during recovery from spinal cord injury? Nat Rev Neurosci. 7:603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann JE, Shah RR, Chan AF, Zheng B. 2010. EphA4 deficient mice maintain astroglial-fibrotic scar formation after spinal cord injury. Exp Neurol. 223:582–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesp ZC, Goldstein EA, Miranda CJ, Kaspar BK, McTigue DM. 2015. Chronic oligodendrogenesis and remyelination after spinal cord injury in mice and rats. J Neurosci. 35:1274–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CE. 2017. A view from the ending: axonal dieback and regeneration following SCI. Neurosci Lett. 652:11–24. [DOI] [PubMed] [Google Scholar]
- Kaneko S, Iwanami A, Nakamura M, Kishino A, Kikuchi K, Shibata S, Okano HJ, Ikegami T, Moriya A, Konishi O et al. 2006. A selective Sema3A inhibitor enhances regenerative responses and functional recovery of the injured spinal cord. Nat Med. 12:1380–1389. [DOI] [PubMed] [Google Scholar]
- Kang S, Nakanishi Y, Kioi Y, Okuzaki D, Kimura T, Takamatsu H, Koyama S, Nojima S, Nishide M, Hayama Y et al. 2018. Semaphorin 6D reverse signaling controls macrophage lipid metabolism and anti-inflammatory polarization. Nat Immunol. 19:561–570. [DOI] [PubMed] [Google Scholar]
- Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T. 2005. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat Med. 11:572–577. [DOI] [PubMed] [Google Scholar]
- Kuwajima T, Yoshida Y, Takegahara N, Petros TJ, Kumanogoh A, Jessell TM, Sakurai T, Mason C. 2012. Optic chiasm presentation of Semaphorin6D in the context of Plexin-A1 and Nr-CAM promotes retinal axon midline crossing. Neuron. 74:676–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Chow R, Xie F, Chow SY, Tolentino KE, Zheng B. 2010a. Combined genetic attenuation of myelin and semaphorin-mediated growth inhibition is insufficient to promote serotonergic axon regeneration. J Neurosci. 30:10899–10904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Geoffroy CG, Chan AF, Tolentino KE, Crawford MJ, Leal MA, Kang B, Zheng B. 2010b. Assessing spinal axon regeneration and sprouting in Nogo-, MAG-, and OMgp-deficient mice. Neuron. 66:663–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie JR, Imai F, Fukuhara K, Takegahara N, Rizvi TA, Friedel RH, Wang F, Kumanogoh A, Yoshida Y. 2011. Ectopic myelinating oligodendrocytes in the dorsal spinal cord as a consequence of altered semaphorin 6D signaling inhibit synapse formation. Development. 138:4085–4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Lu Y, Lee JK, Samara R, Willenberg R, Sears-Kraxberger I, Tedeschi A, Park KK, Jin D, Cai B et al. 2010. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat Neurosci. 13:1075–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Tedeschi A, Park KK, He Z. 2011. Neuronal intrinsic mechanisms of axon regeneration. Annu Rev Neurosci. 34:131–152. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wang X, Lu CC, Kerman R, Steward O, Xu XM, Zou Y. 2008. Repulsive Wnt signaling inhibits axon regeneration after CNS injury. J Neurosci. 28:8376–8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR et al. 2010. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 13:133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier V, Jolicoeur C, Rayburn H, Takegahara N, Kumanogoh A, Kikutani H, Tessier-Lavigne M, Wurst W, Friedel RH. 2011. Semaphorin 4C and 4G are ligands of Plexin-B2 required in cerebellar development. Mol Cell Neurosci. 46:419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C, Zhang Y. 2005. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 6:838–849. [DOI] [PubMed] [Google Scholar]
- Matsuoka RL, Chivatakarn O, Badea TC, Samuels IS, Cahill H, Katayama K, Kumar SR, Suto F, Chedotal A, Peachey NS et al. 2011. Class 5 transmembrane semaphorins control selective mammalian retinal lamination and function. Neuron. 71:460–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKillop WM, Dragan M, Schedl A, Brown A. 2013. Conditional Sox9 ablation reduces chondroitin sulfate proteoglycan levels and improves motor function following spinal cord injury. Glia. 61:164–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau-Fauvarque C, Kumanogoh A, Camand E, Jaillard C, Barbin G, Boquet I, Love C, Jones EY, Kikutani H, Lubetzki C et al. 2003. The transmembrane semaphorin Sema4D/CD100, an inhibitor of axonal growth, is expressed on oligodendrocytes and upregulated after CNS lesion. J Neurosci. 23:9229–9239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Ninomiya T, Yamashita T, Takada M. 2015. Reorganization of corticospinal tract fibers after spinal cord injury in adult macaques. Sci Rep. 5:11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Ninomiya T, Yamashita T, Takada M. 2018. Treatment with the neutralizing antibody against repulsive guidance molecule-a promotes recovery from impaired manual dexterity in a primate model of spinal cord injury. Cereb Cortex. 29:561–572. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Fujita Y, Ueno M, Takai T, Yamashita T. 2011. Paired immunoglobulin-like receptor B knockout does not enhance axonal regeneration or locomotor recovery after spinal cord injury. J Biol Chem. 286:1876–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novitch BG, Chen AI, Jessell TM. 2001. Coordinate regulation of motor neuron subtype identity and pan-neuronal properties by the bHLH repressor Olig2. Neuron. 31:773–789. [DOI] [PubMed] [Google Scholar]
- O'Malley AM, Shanley DK, Kelly AT, Barry DS. 2014. Towards an understanding of semaphorin signalling in the spinal cord. Gene. 553:69–74. [DOI] [PubMed] [Google Scholar]
- Pasterkamp RJ. 2012. Getting neural circuits into shape with semaphorins. Nat Rev Neurosci. 13:605–618. [DOI] [PubMed] [Google Scholar]
- Pasterkamp RJ, Giger RJ, Ruitenberg MJ, Holtmaat AJ, De Wit J, De Winter F, Verhaagen J. 1999. Expression of the gene encoding the chemorepellent semaphorin III is induced in the fibroblast component of neural scar tissue formed following injuries of adult but not neonatal CNS. Mol Cell Neurosci. 13:143–166. [DOI] [PubMed] [Google Scholar]
- Pasterkamp RJ, Verhaagen J. 2006. Semaphorins in axon regeneration: developmental guidance molecules gone wrong. Philos Trans R Soc Lond B Biol Sci. 361:1499–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. 2001. The Mouse Brain in Stereotaxic Coordinates. Cambridge, MA: Academic Press. [Google Scholar]
- Seif GI, Nomura H, Tator CH. 2007. Retrograde axonal degeneration "dieback" in the corticospinal tract after transection injury of the rat spinal cord: a confocal microscopy study. J Neurotrauma. 24:1513–1528. [DOI] [PubMed] [Google Scholar]
- Sharma A, Verhaagen J, Harvey AR. 2012. Receptor complexes for each of the class 3 Semaphorins. Front Cell Neurosci. 6:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim SO, Cafferty WB, Schmidt EC, Kim BG, Fujisawa H, Strittmatter SM. 2012. PlexinA2 limits recovery from corticospinal axotomy by mediating oligodendrocyte-derived Sema6A growth inhibition. Mol Cell Neurosci. 50:193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J, Schwab ME, Popovich PG. 2014. Central nervous system regenerative failure: role of oligodendrocytes, astrocytes, and microglia. Cold Spring Harb Perspect Biol. 7:a020602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BN, Banfield BW, Smeraski CA, Wilcox CL, Dudek FE, Enquist LW, Pickard GE. 2000. Pseudorabies virus expressing enhanced green fluorescent protein: a tool for in vitro electrophysiological analysis of transsynaptically labeled neurons in identified central nervous system circuits. Proc Natl Acad Sci U S A. 97:9264–9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Zheng B, Tessier-Lavigne M, Hofstadter M, Sharp K, Yee KM. 2008. Regenerative growth of corticospinal tract axons via the ventral column after spinal cord injury in mice. J Neurosci. 28:6836–6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. 2000. The language of covalent histone modifications. Nature. 403:41–45. [DOI] [PubMed] [Google Scholar]
- Sun W, Cornwell A, Li J, Peng S, Osorio MJ, Su Wanga NA, Benraiss A, Lou N, Goldman SA, Nedergaard M. 2017. SOX9 is an astrocyte-specific nuclear marker in the adult brain outside the neurogenic regions. J Neurosci. 37:4493–4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamatsu H, Takegahara N, Nakagawa Y, Tomura M, Taniguchi M, Friedel RH, Rayburn H, Tessier-Lavigne M, Yoshida Y, Okuno T et al. 2010. Semaphorins guide the entry of dendritic cells into the lymphatics by activating myosin II. Nat Immunol. 11:594–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebayashi H, Nabeshima Y, Yoshida S, Chisaka O, Ikenaka K, Nabeshima Y. 2002. The basic helix-loop-helix factor olig2 is essential for the development of motoneuron and oligodendrocyte lineages. Curr Biol. 12:1157–1163. [DOI] [PubMed] [Google Scholar]
- Tamagnone L, Artigiani S, Chen H, He Z, Ming GI, Song H, Chedotal A, Winberg ML, Goodman CS, Poo M et al. 1999. Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell. 99:71–80. [DOI] [PubMed] [Google Scholar]
- Tatsumi K, Takebayashi H, Manabe T, Tanaka KF, Makinodan M, Yamauchi T, Makinodan E, Matsuyoshi H, Okuda H, Ikenaka K et al. 2008. Genetic fate mapping of Olig2 progenitors in the injured adult cerebral cortex reveals preferential differentiation into astrocytes. J Neurosci Res. 86:3494–3502. [DOI] [PubMed] [Google Scholar]
- Toyofuku T, Zhang H, Kumanogoh A, Takegahara N, Suto F, Kamei J, Aoki K, Yabuki M, Hori M, Fujisawa H et al. 2004. Dual roles of Sema6D in cardiac morphogenesis through region-specific association of its receptor, Plexin-A1, with off-track and vascular endothelial growth factor receptor type 2. Genes Dev. 18:435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M, Hayano Y, Nakagawa H, Yamashita T. 2012. Intraspinal rewiring of the corticospinal tract requires target-derived brain-derived neurotrophic factor and compensates lost function after brain injury. Brain. 135:1253–1267. [DOI] [PubMed] [Google Scholar]
- Ueno M, Nakamura Y, Li J, Gu Z, Niehaus J, Maezawa M, Crone SA, Goulding M, Baccei ML, Yoshida Y. 2018. Corticospinal circuits from the sensory and motor cortices differentially regulate skilled movements through distinct spinal interneurons. Cell Rep. 23. e1287:1286–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M, Ueno-Nakamura Y, Niehaus J, Popovich PG, Yoshida Y. 2016. Silencing spinal interneurons inhibits immune suppressive autonomic reflexes caused by spinal cord injury. Nat Neurosci. 19:784–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M, Yamashita T. 2008. Strategies for regenerating injured axons after spinal cord injury - insights from brain development. Biol: Targets Ther. 2:253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K et al. 2014. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 158:1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y. 2012. Semaphorin signaling in vertebrate neural circuit assembly. Front Mol Neurosci. 5:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Han B, Mendelsohn M, Jessell TM. 2006. PlexinA1 signaling directs the segregation of proprioceptive sensory axons in the developing spinal cord. Neuron. 52:775–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura K, Ueno M, Lee S, Nakamura Y, Sato A, Yoshimura K, Kishima H, Yoshimine T, Yamashita T. 2011. C-Jun N-terminal kinase induces axonal degeneration and limits motor recovery after spinal cord injury in mice. Neurosci Res. 71:266–277. [DOI] [PubMed] [Google Scholar]
- Yu Y, Chen Y, Kim B, Wang H, Zhao C, He X, Liu L, Liu W, Wu LM, Mao M et al. 2013. Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell. 152:248–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue T, Xian K, Hurlock E, Xin M, Kernie SG, Parada LF, Lu QR. 2006. A critical role for dorsal progenitors in cortical myelination. J Neurosci. 26:1275–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N et al. 2014. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 34:11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zukor K, Belin S, Wang C, Keelan N, Wang X, He Z. 2013. Short hairpin RNA against PTEN enhances regenerative growth of corticospinal tract axons after spinal cord injury. J Neurosci. 33:15350–15361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.